
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization and reactivity study of non-heme high-valent iron–hydroxo complexes†
Kritika
Keshari
a,
Moumita
Bera
a,
Lucía
Velasco
b,
Sandip
Munshi
 c,
Geetika
Gupta
a,
Dooshaye
Moonshiram
*b and
Sayantan
Paria
*a
c,
Geetika
Gupta
a,
Dooshaye
Moonshiram
*b and
Sayantan
Paria
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: sparia@chemistry.iitd.ac.in
bInstituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA Nanociencia, Calle Faraday, 9, 28049 Madrid, Spain. E-mail: dooshaye.moonshiram@imdea.org
cSchool of Chemical Sciences, Indian Association for the Cultivation of Science, 2A & 2B Raja S. C. Mullick Road, Jadavpur, Kolkata 700032, India
First published on 27th January 2021
Abstract
A terminal FeIIIOH complex, [FeIII(L)(OH)]2− (1), has been synthesized and structurally characterized (H4L = 1,2-bis(2-hydroxy-2-methylpropanamido)benzene). The oxidation reaction of 1 with one equiv. of tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBAH) or ceric ammonium nitrate (CAN) in acetonitrile at −45 °C results in the formation of a FeIIIOH ligand radical complex, [FeIII(L˙)(OH)]− (2), which is hereby characterized by UV-visible, 1H nuclear magnetic resonance, electron paramagnetic resonance, and X-ray absorption spectroscopy techniques. The reaction of 2 with a triphenylcarbon radical further gives triphenylmethanol and mimics the so-called oxygen rebound step of Cpd II of cytochrome P450. Furthermore, the reaction of 2 was explored with different 4-substituted-2,6-di-tert-butylphenols. Based on kinetic analysis, a hydrogen atom transfer (HAT) mechanism has been established. A pKa value of 19.3 and a BDFE value of 78.2 kcal/mol have been estimated for complex 2.
Introduction
Activation of dioxygen at the FeII center in the presence of electrons and protons (or sacrificial substrates like α-keto acid) leads to the formation of transient high-valent Fen+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, which is a common mechanism for different heme and non-heme Fe enzymes. Such reaction intermediates have been identified in enzymes,1–3 as well as with synthetic model complexes.4–14 It has been implicated that Fen+O (n = 4 or 5) species further abstract a hydrogen atom from the substrate to form a high-valent Fe(n−1)–OH complex (and a substrate radical, Scheme 1), which performs versatile reactions such as hydroxylation,15 halogenation,16,17 ring closure,18 desaturation,8,15 decarboxylation,19etc. Probing such reactions spectroscopically is a challenging task as the formation of Fe–OH type species occurs in the rate-limiting step and the rebound of OH to the substrate radical happens concomitantly. Very recently, Solomon et al. identified an FeIII–OH intermediate in deacetoxycephalosporin C synthase, an α-KG-dependent non-heme Fe enzyme.20 A few structurally characterized FeIII–OH complexes are reported in the literature.21–26 However, very few demonstrate C–OH bond formation reactivity of α-KG-dependent enzymes. Goldberg reported mononuclear FeIII–OH/OCH3 complexes, which can react with carbon radicals (Ar3C˙) and form Ar3COH (or Ar3C(OCH3)) through the rebound mechanism.27–29 Fout has described an FeIII–OH complex, which also mimics the C–OH bond formation step of α-KG-dependent monooxygenases.30,31
O species, which is a common mechanism for different heme and non-heme Fe enzymes. Such reaction intermediates have been identified in enzymes,1–3 as well as with synthetic model complexes.4–14 It has been implicated that Fen+O (n = 4 or 5) species further abstract a hydrogen atom from the substrate to form a high-valent Fe(n−1)–OH complex (and a substrate radical, Scheme 1), which performs versatile reactions such as hydroxylation,15 halogenation,16,17 ring closure,18 desaturation,8,15 decarboxylation,19etc. Probing such reactions spectroscopically is a challenging task as the formation of Fe–OH type species occurs in the rate-limiting step and the rebound of OH to the substrate radical happens concomitantly. Very recently, Solomon et al. identified an FeIII–OH intermediate in deacetoxycephalosporin C synthase, an α-KG-dependent non-heme Fe enzyme.20 A few structurally characterized FeIII–OH complexes are reported in the literature.21–26 However, very few demonstrate C–OH bond formation reactivity of α-KG-dependent enzymes. Goldberg reported mononuclear FeIII–OH/OCH3 complexes, which can react with carbon radicals (Ar3C˙) and form Ar3COH (or Ar3C(OCH3)) through the rebound mechanism.27–29 Fout has described an FeIII–OH complex, which also mimics the C–OH bond formation step of α-KG-dependent monooxygenases.30,31
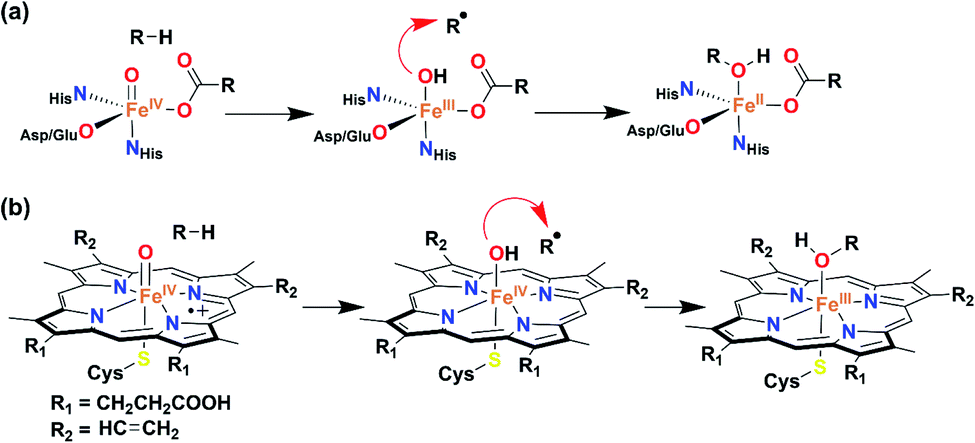 | ||
| Scheme 1 General mechanism for the hydroxide rebound of (a) α-keto glutarate-dependent oxygenase and (b) cytochrome P450. | ||
High-valent (Por)FeIV–OH species have been identified in compound II of cytochrome P450 and HRP-II, CcP.8 A computational investigation by Shaik revealed that (Por)FeIV–OH can also exist as a (Por˙+)FeIII–OH during the desaturation pathway of P450cam.32 Therefore, characterization and reactivity studies of high-valent Fe–OH complexes constitute an important topic of research to bioinorganic chemists. The artificial analogue of such species is rare and synthetically very challenging, as the mononuclear Fen+O complex has an inherent tendency to form thermodynamically more stable Fe–O–Fe-type complexes. Nonetheless, there remain only a few elusive analogues of FeIV–OH complexes reported in the literature.33–36 Borovik has described a protonated FeIVO complex of a tris-urea tripodal ligand, where the protonation likely occurred at the ligand backbone.37 Very recently, Nam et al., spectroscopically characterized and studied the reactivity of a ligand-protonated FeVO species of a Tetra-Amido Macrocyclic Ligand (TAML).38 Goldberg has reported a structurally characterized FeIV–OH complex of a corrole ligand, which was shown to participate in OH re-bound reactions similar to the enzymatic systems, the only reported functional model of compound II.33,36 Very recently, Hendrich described an FeIV–OHn (n = 1 or 2) of a TAML through various spectroscopic techniques.35
Herein, we report the synthesis, structural characterization, and reactivity study of a terminal FeIII–OH complex, [FeIII(L)(OH)]2− (1). One-electron oxidation of complex 1 with one equiv. of tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBAH) or ceric ammonium nitrate (CAN) results in the formation of an FeIII–OH ligand radical complex, [FeIII(L˙)(OH]− (2, Scheme 2) which is hereby characterized by UV-visible (UV-vis), 1H-Nuclear Magnetic Resonance (NMR), and X-ray absorption spectroscopy (XAS) techniques. The latter complex spontaneously reacts with Gomberg's dimer forming Ph3COH and mimics the C–OH bond formation step of Cpd II (Scheme 2).
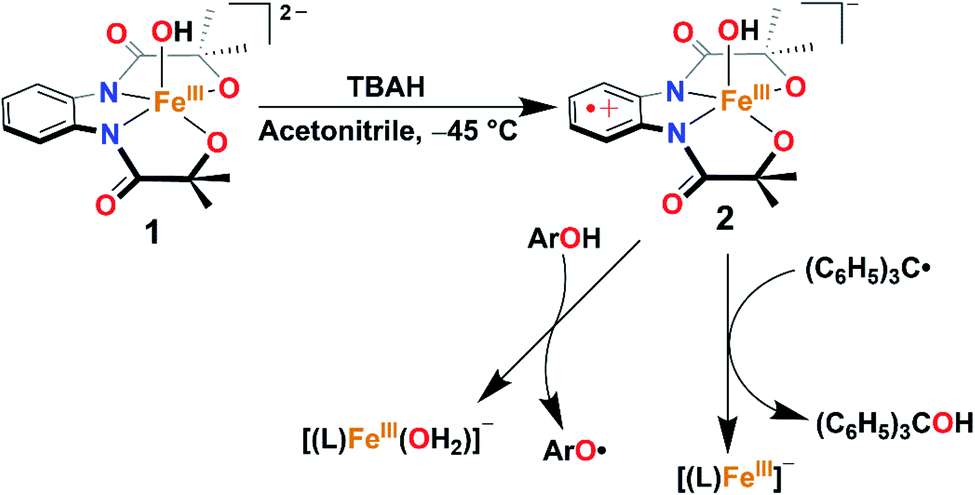 | ||
| Scheme 2 Formation and reactivity of 2 with Gomberg's dimer and phenol derivatives. | ||
Results and discussion
The tetradentate H4L ligand was synthesized according to the procedure described by Mayer.39 The FeIII complex was prepared by mixing equimolar amounts of H4L, Fe(ClO4)3·xH2O, and 4.5 equiv. of Me4NOH in methanol (for details, see the Experimental section). The complex was isolated as a yellow powder and characterized by routine spectroscopic techniques (Fig. S1–S6†). Analysis of single-crystal XRD data revealed the formation of a five-coordinate FeIII–OH complex (1), described in Fig. 1 and Tables S1 and S2.† A distorted square pyramidal geometry was found around the Fe center (τ5 = 0.158),40 coordinated to two amidate nitrogen and alkoxide oxygen donor atoms of the ligand in the equatorial plane. The Fe(1)–N(1) and Fe(1)–N(2) bond distances are 2.0684(18) and 2.0724(18) Å, respectively, which are much longer compared to the Fe–Neq. distances of [Fe(B*)(OH2)]− , (1.881(2) and 1.876(2) Å), where B* is a TAML.41 The Fe–O distances are shorter compared to the average Fe–N bond distances (Fe(1)–O(2), 1.9273(17), and Fe(1)–O(3), 1.9222(16) Å). The fifth position around iron is occupied by the OH− at a distance of 1.9093 (17) Å. The observed Fe–OH distance in 1 is comparable to that of non-hydrogen-bonded FeIII–OH complexes23,25 and shorter compared to the FeIII–OH2 distance in [Fe(B*)(OH2)]−, (2.097(2) Å).41 The Fe–OH distance of 1 is further very close to the Fe–OH distance (1.890(1) Å) observed in the pseudo-tetrahedral [Fe(ditox)3(OH)]− (ditox = tBu2(Me)CO−).42 Also, interestingly, the Fe center in 1 is significantly displaced (0.631 Å) toward OH− from the plane of coordinating nitrogen and oxygen atoms of the ligand, which is more displaced compared to that of [FeIII(TAML)(X)]n− complexes (X = Cl−, OH2, CN−).41 We speculate that due to the large displacement of Fe from the equatorial plane, hydroxide coordination to iron(III) is favoured. Complex 1 represents the first example of a structurally characterized FeIII–OH complex in a square pyramidal geometry. The 1H-NMR spectrum of 1 was measured in CD3CN at 298 K and shows paramagnetically shifted ligand proton resonances (Fig. S4†). The magnetic moment of 1 in CD3CN was measured at 25 °C by Evans' method43 and revealed μeff = 4.10 BM, which corresponds to an iron spin-state of S = 3/2 at room temperature (Fig. S5†).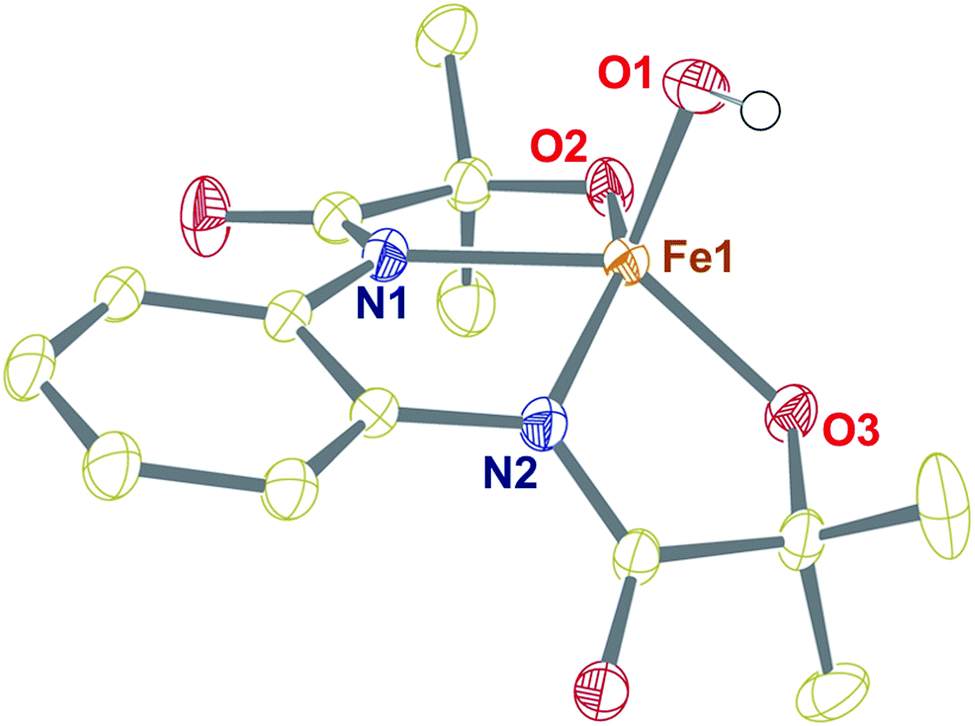 | ||
| Fig. 1 Crystal structure of complex 1 with 50% ellipsoid probability. Counter cations and all of the hydrogen atoms except those attached to O1 have been omitted for clarity. | ||
In contrast, the X-band EPR spectrum of complex 1 in acetonitrile at 100 K shows a rhombic spectrum having g values at 2.78, 2.21, and 1.36 (Fig. 2, inset), typical for low-spin iron(III) complexes having (dxy)2(dxz,dyz)3 electron configurations.44 We speculate that upon decreasing the temperature, the spin-state of 1 changes from S = 3/2 to S = 1/2.
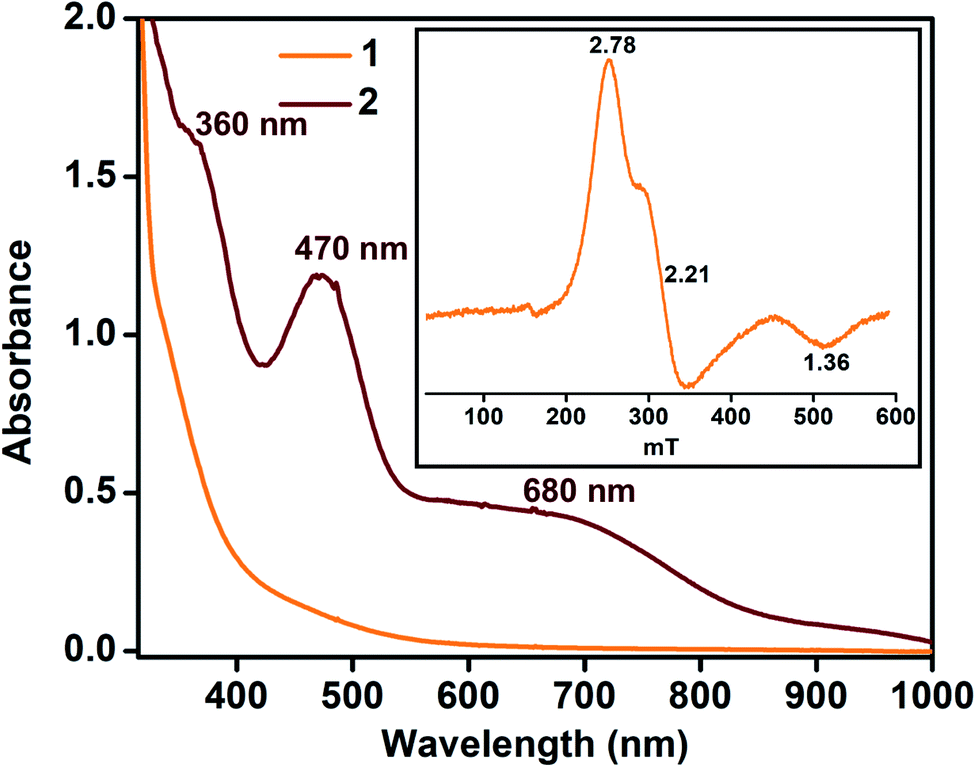 | ||
| Fig. 2 UV-vis spectra of 1 and 2 at −45 °C in acetonitrile (0.32 mM). Inset: EPR spectrum of complex 1 (0.5 mM) at 100 K in frozen acetonitrile. | ||
The redox properties of 1 were evaluated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in acetonitrile at 25 °C, which revealed a quasi-reversible redox process at E1/2 = 0.487 V (versus NHE), assigned to (L˙)FeIII/(L)FeIII (Fig. S7†).
Given the electrochemical properties, we envisioned that the chemical oxidation of 1 could form a high-valent Fe–OH complex. We utilized one-electron oxidizing agents like TBAH (EOX = 1.321 V versus NHE) or CAN (EOX = 1.61 V versus NHE) to oxidize 1. The reaction of 1 with TBAH was monitored by UV-vis spectroscopy in acetonitrile at −45 °C (Fig. 2). The addition of one equiv. of TBAH to 1 resulted in the formation of a new species 2 having absorbance maxima at 360 nm (5100 M−1 cm−1), 470 nm (3700 M−1 cm−1) and ca. 680 nm (1000 M−1 cm−1). A similar spectral feature was obtained upon the addition of one equiv. of CAN to the solution of complex 1 (Fig. S8†). We propose that such peaks originated from the π–π* transitions of the ligand radical.45 Formation of 2 was also monitored by 1H NMR spectroscopy. The NMR spectrum of 2, formed upon addition of TBAH to 1 in CD3CN, was measured at −30 °C and showed shifted peaks with the appearance of new spectral features, indicating the formation of a new intermediate complex (Fig. S9†).
In order to determine the spin-state of complex 2, solution magnetic moment was measured in CD3CN by Evans' method and revealed μeff = 3.05 μB, which corresponds to a spin-state of S = 1.46 The X-band EPR spectrum of 2 was further monitored in frozen acetonitrile at 100 K, which revealed a close to silent EPR spectrum, thus ascertaining its S = 1 spin state and the large zero-field splitting tensor (Fig. S10†).47
In order to determine the electronic and local structural conformations of 1 and 2, both complexes were studied in solution state by X-ray absorption near edge structure (XANES) and Extended X-ray absorption fine structure (EXAFS) spectroscopy (Fig. 3). Both 1 and 2 display a rising edge from 7120 to ∼7135 eV, corresponding to the 1s to 4p electronic transition. The distinct ligand versus metal-centered oxidation in complex 2 is confirmed by the missing shift of the rising edge towards the higher energy of the XANES spectra at 0.6 normalized absorption and at 7125.15 eV (Fig. 3A). By contrast, upon oxidation with 1 equiv. TBAH, changes at higher photon energies at ∼7133–7135 eV (Fig. 3A) and in the pre-edge region (Fig. 3A inset) are observed. The presence of pre-edge features corresponds to 1s to 3d quadrupole transitions and dipole excitations of the core electrons into the valence 3d states hybridized with ligand p orbitals.48,49 The changes observed in the pre-edge and rising edge features hereby reflect the variations and local symmetrical and geometrical differences expected between both complexes.50 The pre-edge peaks of complexes 1 and 2 were further fitted and yielded maximum peak energies of 7113.97 eV and 7113.71 eV, respectively. A pre-edge peak area of 19.3 units was, on one hand, obtained for complex 1 (Fig. 3B, Table S5†), which is close and consistent with that of previously studied five-coordinated ferric centers, demonstrating pre-edge areas of ∼17 units.51,52 The decreased pre-edge area of 16.1 units for complex 2 is however in sharp contrast to that of Fe(IV)oxo51,53 high-valent complexes, exhibiting typical areas of 41–52 units,51,53 thus confirming the lack of metal-centered oxidation.
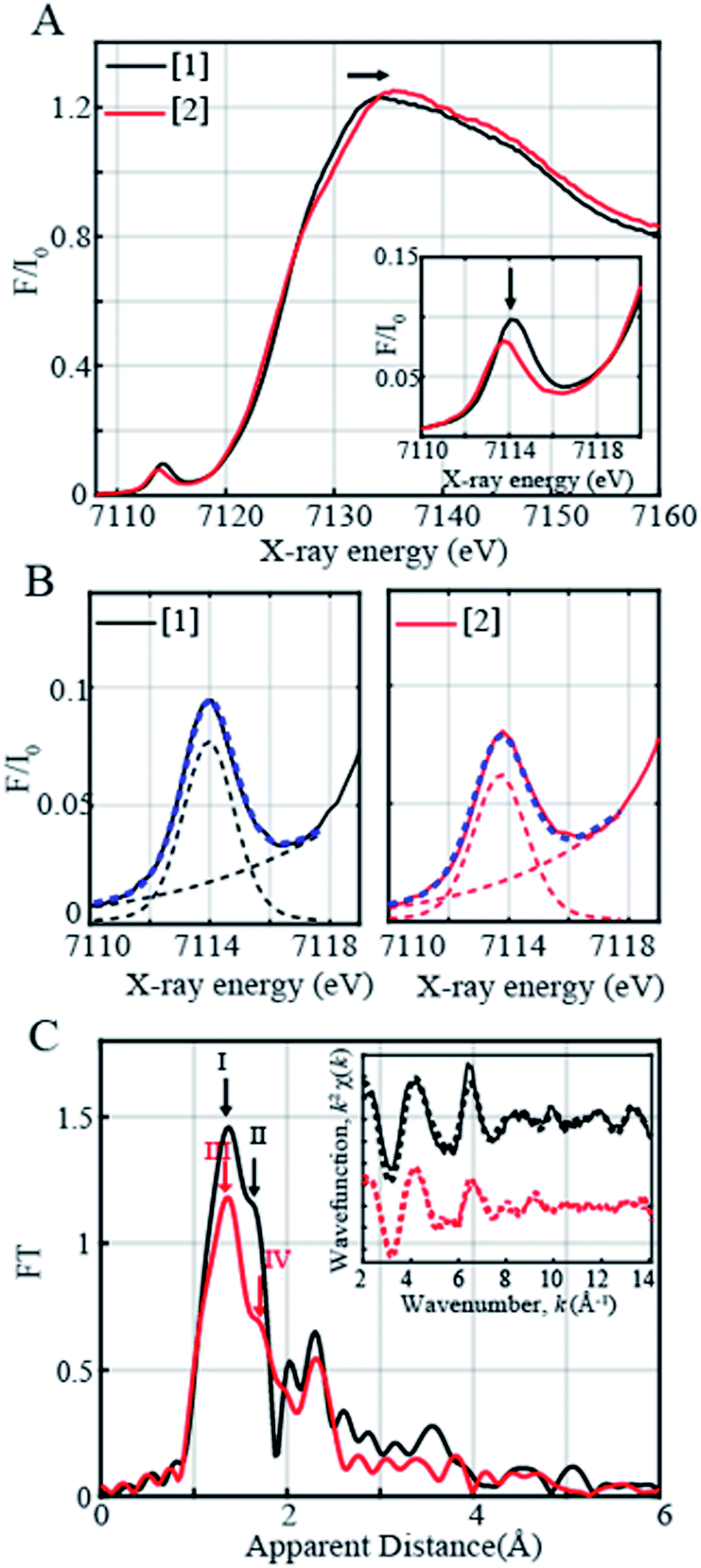 | ||
| Fig. 3 (A) Normalized Fe K-edge XANES spectra recorded at 20 K of 1 (5 mM Fe(III) complex shown in black) together with the ligand oxidized product, complex 2, shown in red. Complex 2 was generated with 1 equiv. of TBAH. (B) Zoom-in of the pre-edge regions of complexes 1 and 2 together with the respective fits shown in dashed blue. The black dashed and red dashed lines on the right and left panels correspond to the step and pseudo-Voigt functions used to fit the pre-edge peaks. (C) Fourier transforms of k2-weighted Fe EXAFS of 5 mM Fe(III) complex 1 (in black) and the ligand oxidized product, complex 2 (in red). Inset: Back Fourier transformed experimental (solid lines) and fitted (dashed lines) k2 [χ(k)] of complex 1 (solid black line) and its corresponding fit (Fit 3, Table S3) and complex 2 (solid red line) and its corresponding fit (Fit 6, Table S3). | ||
The EXAFS spectra of 1 and 2 are shown in Fig. 3C. Two prominent peaks are observed in complex 1's spectrum corresponding to the distinctive Fe–N and Fe–O bond distances. The EXAFS fits for the extraction of actual bond distances of the two complexes are shown in Fig. 3C inset, Table S3 and Fig. S11.† Analysis of EXAFS spectra in complex 1 in the solution state clearly resolved 3 Fe–O distances at 1.88 ± 0.01 Å and 2 Fe–N distances at 2.04 ± 0.01 Å, in close agreement with the obtained XRD data (Tables S3 and S4, Fig. S11†). Upon oxidation with 1 equiv. of TBAH, complex 2 demonstrates a prominent sharp peak denoted in red as III (Fig. 3C) and an elongated shoulder peak feature shown as Peak IV in the first coordination sphere (Fig. 3C). Analysis of the EXAFS region of complex 2 resolves 3 Fe–O distances at 1.89 ± 0.02 Å and comparatively lengthened Fe–N distances at 2.09 ± 0.02 Å (Table S3, Fig. S11†). The obtained local structural conformation not only ascertains the ligand-centered oxidation process in complex 2 but also additionally indicates that the Fe metal center likely lies slightly above the plane of the connected ligating atoms upon oxidation in 2 in comparison with 1. Here, it is important to note that the Fe–O distance observed in 2 is close to the Fe–O distance (1.857 Å) reported in [FeIV(ttppc)(OH)].33
The reactivity of complex 2 with Gomberg's dimer was examined by UV-vis spectroscopy in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 acetonitrile/THF (v/v) at −40 °C. The addition of 10-fold excess of Gomberg's dimer to 2 resulted in immediate decay of peaks at 470 and 680 nm (Fig. 4). The reaction products were further analyzed by gas chromatography-mass spectrometry (GC-mass) and 1H-NMR spectroscopy techniques, which confirmed the formation of Ph3COH with 80% yield. The source of oxygen atom in Ph3COH was evaluated by 18O labeling experiments. In the GC-mass spectrum, a peak was observed at m/z = 260.1 (Fig. S12†), which corresponds to the composition of [Ph3COH]+. This peak displays a two-mass unit shift to m/z = 262.1 (Fig. S13†) when the sample was prepared in the presence of [FeIII(L˙)(18OH)]− and Gomberg's dimer and confirms that the source of OH in the product is the Fe–OH moiety. The experiment demonstrated 75% incorporation of 18O into triphenylmethanol. The study thus shows that complex 2 mimics the reactivity of Cpd II and supports an alternative structure and reactivity pathway for Cpd II, as documented by Shaik.32
:2 acetonitrile/THF (v/v) at −40 °C. The addition of 10-fold excess of Gomberg's dimer to 2 resulted in immediate decay of peaks at 470 and 680 nm (Fig. 4). The reaction products were further analyzed by gas chromatography-mass spectrometry (GC-mass) and 1H-NMR spectroscopy techniques, which confirmed the formation of Ph3COH with 80% yield. The source of oxygen atom in Ph3COH was evaluated by 18O labeling experiments. In the GC-mass spectrum, a peak was observed at m/z = 260.1 (Fig. S12†), which corresponds to the composition of [Ph3COH]+. This peak displays a two-mass unit shift to m/z = 262.1 (Fig. S13†) when the sample was prepared in the presence of [FeIII(L˙)(18OH)]− and Gomberg's dimer and confirms that the source of OH in the product is the Fe–OH moiety. The experiment demonstrated 75% incorporation of 18O into triphenylmethanol. The study thus shows that complex 2 mimics the reactivity of Cpd II and supports an alternative structure and reactivity pathway for Cpd II, as documented by Shaik.32
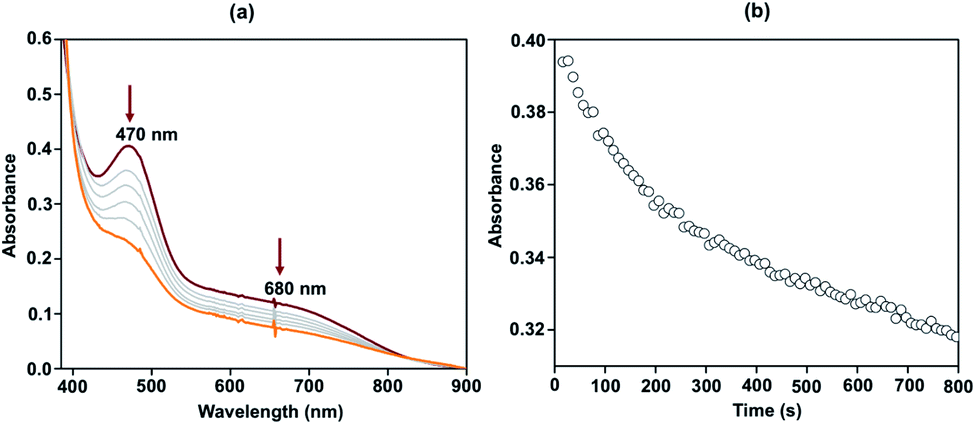 | ||
| Fig. 4 Change in the UV-vis spectrum of 2 (0.1 mM) upon addition of Gomberg's dimer in 3:2 acetonitrile/THF (v/v) at −40 °C (a) and time trace of the decay of 2 at 470 nm (b). | ||
The reaction of 2 was further explored with different 4-X-2,6-di-tert-butylphenols (X = –OCH3, –OCH3 (D), –CH3, –CH2CH3, –C(CH3)3, and –H) in acetonitrile at −25 °C. Addition of one equiv. of 4-methoxy-2,6-di-tert-butylphenol (4-OMe-DTBP) to 2 resulted in immediate decomposition of peaks at 470 and 680 nm in the UV-vis spectrum with concomitant appearance of a new peak at 405 nm, corresponding to the formation of a 4-methoxy-2,6-di-tert-butylphenoxyl radical (Fig. 5a).
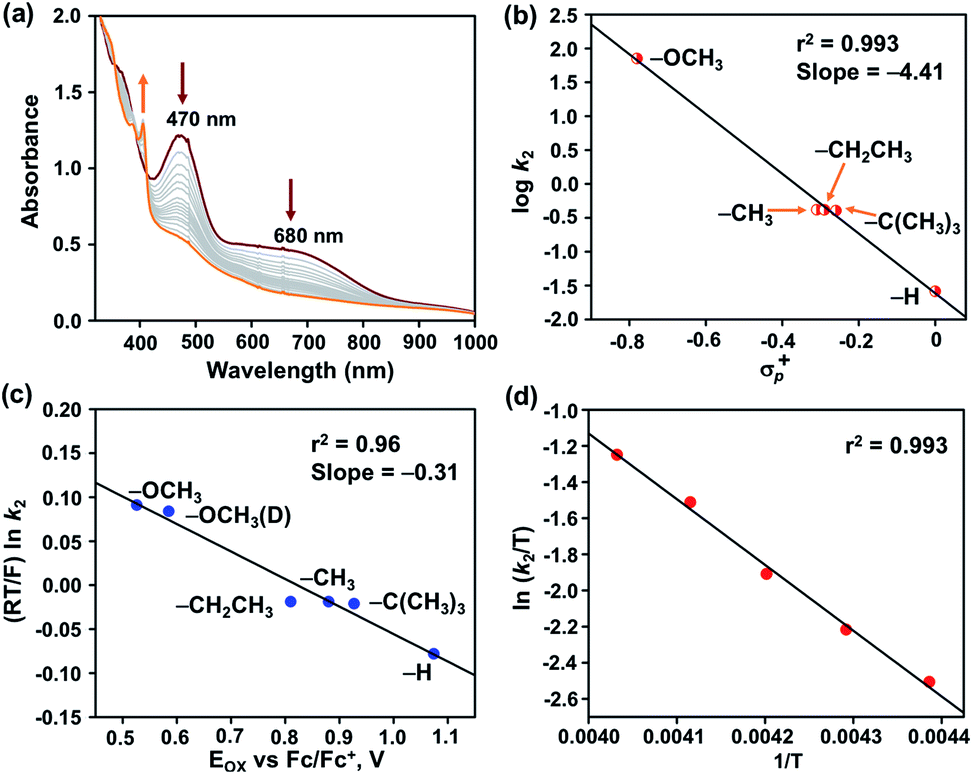 | ||
| Fig. 5 (a) Change in the UV-vis spectrum of 2 (0.32 mM) upon addition of one equiv. of 4-OMe-2,6-DTBP in acetonitrile at −45 °C. A plot of logk2vs. σp+ (b) and (RT/F) lnk2vs. EOX (vs. Fc/Fc+) of 4-X-2,6-di-tert-butylphenols (c). (d) Eyring plot for the reaction of 2 with 4-OMe-2,6-DTBP over 228–248 K. | ||
The second-order rate constant of the reaction was obtained from the slope of a plot of (A0 − A)/C0(A − A∞) vs. time (t) and yielded kH2 = 71.15 M−1 s−1 (Fig. S14†). The formation of 88% phenoxyl radical was additionally confirmed by EPR spectroscopy (Fig. S15†).
The reaction of 2 was subsequently explored in the presence of 4-OMe-DTBP-D, which yielded a kD2 value of 50.8 M−1 cm−1 in acetonitrile at −25 °C, thus displaying a kinetic isotope effect (KIE) of 1.4. The reactivity of 2 was further explored with 2,6-di-tert-butyl-4-methylphenol (Fig. S17–19†), 2,6-di-tert-butyl-4-ethylphenol (Fig. S20–22†), 2,4,6-tri-tert-butylphenol (Fig. S23–26†) and 2,6-di-tert-butylphenol (Fig. S27–29†) under pseudo-first-order reaction conditions resulting in k2 values of 0.417, 0.416, 0.375, and 0.026 M−1 cm−1, respectively. A plot of logk2versus O–H bond dissociation energy of phenols follows a linear correlation and resulted in a slope of −0.77 (Table S9 and Fig. S30†). A Hammett correlation plot was obtained by plotting logk2versus σp+ of 4-X-2,6-di-tert-butylphenols (Fig. 5b and Table S10†) and yielded a value of σ+ = −4.4, which hereby indicates the strong electrophilic nature of 2. It is important to remark that a σ+ value of −2.6(8) was reported for [FeIV(ttpc)(OH)]33 for similar reactions (ttppc = 5,10,15-tris(2,4,6-triphenyl)-phenyl corrole), which indicates a greater charge separation in the transition state of the reaction of 2 with O–H bonds. The σ+ value of 2 is higher than those of [FeIV(O)(TMC)(X)]n+ complexes for the reaction with phenolic O–H bonds (X = N3, σ+ = −1.5; X = CF3COO−, σ+ = −2.3; X = CH3CN, σ+ = −3.2; TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane).54 Furthermore, the Marcus correlation of the reaction of 2 with different 4-X-2,6-di-tert-butylphenols was explored. A plot of (RT/F) lnk2versus Eox of the phenols follows a linear correlation and resulted in a slope of −0.31 (Fig. 5c and Table S11†). The obtained value is close to the values reported for the hydrogen atom transfer reactions of [FeIV(ttpc)(OH)] (slope = −0.19(6)) and [MnIV(ttpc)(OH)] (slope = −0.12) with p-substituted 2,6-di-tert-butyl phenols.33
Eyring analysis of the reaction of 2 with 4-OMe-2,6-DTBP was also performed over a temperature range of 248 to 228 K (Table S7† and Fig. 5d). Activation enthalpy, ΔH# = 7.23 ± 0.28 kcal mol−1, and activation entropy, ΔS# = −20.54 ± 1.6 cal mol−1 K−1, were estimated from the slope and intercept of a plot of ln(k2/T) vs. 1/T, respectively. Such a large negative activation entropy suggests a well-ordered transition state in the rate-determining step. Although no similar values of high-valent Fe–OH complexes are available for comparison purposes, our results are close to the activation parameters reported for the hydrogen atom transfer type reaction of 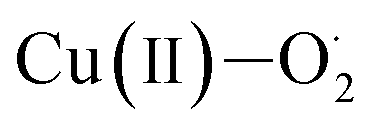 with 4-OMe-2,6-DTBP and other Cu(III), Mn(V) and Ru(VI) complexes (Table S8†).55 The activation parameters of 2 are very close to the values reported for the HAT type reaction of FeIVO complexes with 1,4-cyclohexadiene or xanthene.56,57 Based on the linear correlations obtained between the rates of the reaction of 2 and the BDE of phenols, the value of KIE, activation parameters of the reaction of 2 with 4-OMe-DTBP, and Marcus plot analysis, we propose that the cleavage of the O–H bond by 2 occurs in the rate-determining step.
with 4-OMe-2,6-DTBP and other Cu(III), Mn(V) and Ru(VI) complexes (Table S8†).55 The activation parameters of 2 are very close to the values reported for the HAT type reaction of FeIVO complexes with 1,4-cyclohexadiene or xanthene.56,57 Based on the linear correlations obtained between the rates of the reaction of 2 and the BDE of phenols, the value of KIE, activation parameters of the reaction of 2 with 4-OMe-DTBP, and Marcus plot analysis, we propose that the cleavage of the O–H bond by 2 occurs in the rate-determining step.
We additionally attempted to estimate the pKa value of 2. However, no spectral change was observed upon addition of equiv. amount of 2-aminopyridine (pKa = 14.3) or triethylamine (pKa = 18.82) to 2. Reversible deprotonation of 2 with pyrrolidine (pKa = 19.56)58 allowed us to estimate a pKa value of ∼19.3 for 2 (Fig. S31 and Scheme S1†). This value is higher than that of the FeIIIOH (pKa = 16.5(0.4)) complex stabilized by secondary coordination sphere hydrogen bonding interactions reported by Fout et al.30 Based on the obtained pKa and E0 value of the (L˙)FeIII/(L)FeIII redox couple, we determined the O–H bond dissociation free energy (BDFE) of 78.2 kcal mol−1 of 2 (eqn (1), CH = 54.9 kcal mol−1 in acetonitrile) from the thermodynamic square scheme described in Scheme 3.59 The BDFE value is further verified experimentally as 2 spontaneously reacts with different 4-substituted-2,6-di-tert-butylphenols having similar BDFE values.
| BDFE(O–H) = 1.37pKa + 23.06E0 + CH | (1) |
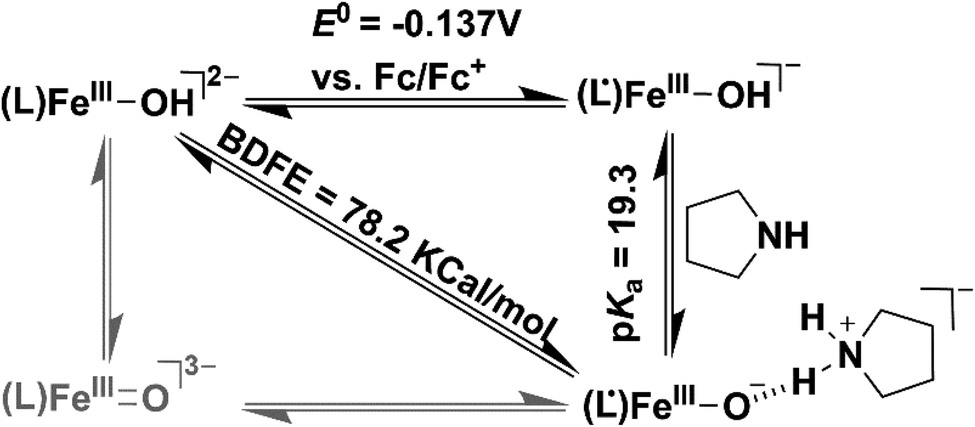 | ||
| Scheme 3 Thermodynamic square scheme for the determination of BDFE of 2. | ||
The experimentally obtained BDFE value of 2 is higher than those of FeIII–OH (70 kcal mol−1),30 FeIIIO (66 kcal mol−1),60 and MnIIIO (72 kcal mol−1)61 complexes, but lower than those of FeIVO complexes (87 kcal mol−1).62
Conclusions
The mechanism of hydroxide rebound or hydrogen atom transfer reactions of Fe–OH species in heme and non-heme enzymes is of burgeoning interest to bioinorganic chemists. Theoretical investigations demonstrated that (Por)FeIV–OH (compound II) can also exist as its electromer, (Por˙+)FeIII–OH, in P450cam. In this study, we have isolated and thoroughly characterized FeIII–OH complexes at two different redox levels, [FeIII(L)OH]2− (1) and [FeIII(L˙)OH]− (2). The reaction of 2 with Gomberg's dimer was explored, which resulted in the spontaneous formation of (C6H5)3COH which is a functional mimic of Cpd II of cytochrome P450, and the reaction further supports the possible existence of compound II as a FeIII porphyrin radical cation complex. An in-depth kinetic investigation of the reaction of 2 was performed towards O–H atom abstraction reactions with different 4-X-2,6-di-tert-butylphenols, and the study revealed a HAT type reaction mechanism, analogous to different heme and non-heme enzymes. The BDFE value of 2 was determined, which is lower than those of FeIVO complexes. Further reactivity studies of Fe–OH complexes are in progress in our laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP acknowledges Science and Engineering Research Board (ECR/2017/002433) and IIT Delhi for funding. D. M. acknowledges funding from the Severo Ochoa Excellence program (SEV-2016-0686) from the Instituto IMDEA Nanociencia, the Acciones de Dinamización “Europa Investigacion” grant (EIN2019-103399), and the Spanish Ministerio de Ciencia, Innovacion y Universidades grant (PID2019-111086RA-I00). The research leading to part of the results was further supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. We also thank Dr Wolfgang Caliebe for help with experiments at the Petra P64 beamline at DESY, a member of the Helmholtz Association (HGF) (Germany). The DFT calculations (SI) were performed in the Scientific Computing Center-Autonomous University of Madrid (CatDesignProject, PI: D. Moonshiram), thanks to CPU time and other resources granted by the institution. We acknowledge Prof. Kaushik Ghosh, IIT Roorkee for helping with EPR measurements. We thank Prof. Kuntal Manna, IIT Delhi for helping with GC-mass measurements. KK acknowledges MHRD for the fellowship. MB thanks SERB for the Inspire fellowship. GG thanks IIT Delhi for the Institute Post-Doctoral Fellowship.Notes and references
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS.
- S. Sinnecker, N. Svensen, E. W. Barr, S. Ye, J. M. Bollinger Jr, F. Neese and C. Krebs, J. Am. Chem. Soc., 2007, 129, 6168–6179 CrossRef CAS.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger Jr and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS.
- M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS.
- Z. Chen and G. Yin, Chem. Soc. Rev., 2015, 44, 1083–1100 RSC.
- A. J. Jasniewski and L. Que, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS.
- S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS.
- M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS.
- C. Panda, A. Sarkar and S. Sen Gupta, Coord. Chem. Rev., 2020, 417, 213314 CrossRef CAS.
- T. J. Collins and A. D. Ryabov, Chem. Rev., 2017, 117, 9140–9162 CrossRef CAS.
- S. Kal and L. Que, JBIC, J. Biol. Inorg. Chem., 2017, 22, 339–365 CrossRef CAS.
- L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh and C. L. Drennan, Nature, 2006, 440, 368–371 CrossRef CAS.
- M. Srnec and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 2396–2407 CrossRef CAS.
- E. Tamanaha, B. Zhang, Y. Guo, W.-c. Chang, E. W. Barr, G. Xing, J. St. Clair, S. Ye, F. Neese, J. M. Bollinger and C. Krebs, J. Am. Chem. Soc., 2016, 138, 8862–8874 CrossRef CAS.
- J. L. Grant, C. H. Hsieh and T. M. Makris, J. Am. Chem. Soc., 2015, 137, 4940–4943 CrossRef CAS.
- S. Goudarzi, S. R. Iyer, J. T. Babicz Jr, J. J. Yan, G. H. J. Peters, H. E. M. Christensen, B. Hedman, K. O. Hodgson and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5152–5159 CrossRef CAS.
- S. Ogo, S. Wada, Y. Watanabe, M. Iwase, A. Wada, M. Harata, K. Jitsukawa, H. Masuda and H. Einaga, Angew. Chem., Int. Ed., 1998, 37, 2102–2104 CrossRef CAS.
- S. Ogo, R. Yamahara, M. Roach, T. Suenobu, M. Aki, T. Ogura, T. Kitagawa, H. Masuda, S. Fukuzumi and Y. Watanabe, Inorg. Chem., 2002, 41, 5513–5520 CrossRef CAS.
- R. Celenligil-Cetin, P. Paraskevopoulou, R. Dinda, R. J. Staples, E. Sinn, N. P. Rath and P. Stavropoulos, Inorg. Chem., 2008, 47, 1165–1172 CrossRef CAS.
- J. Mukherjee, R. L. Lucas, M. K. Zart, D. R. Powell, V. W. Day and A. S. Borovik, Inorg. Chem., 2008, 47, 5780–5786 CrossRef CAS.
- H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024–10035 CrossRef CAS.
- S. A. Cook, J. W. Ziller and A. S. Borovik, Inorg. Chem., 2014, 53, 11029–11035 CrossRef CAS.
- V. Yadav, J. B. Gordon, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2019, 141, 10148–10153 CrossRef CAS.
- T. M. Pangia, C. G. Davies, J. R. Prendergast, J. B. Gordon, M. A. Siegler, G. N. L. Jameson and D. P. Goldberg, J. Am. Chem. Soc., 2018, 140, 4191–4194 CrossRef CAS.
- V. Yadav, R. J. Rodriguez, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2020, 142, 7259–7264 CrossRef CAS.
- M. J. Drummond, C. L. Ford, D. L. Gray, C. V. Popescu and A. R. Fout, J. Am. Chem. Soc., 2019, 141, 6639–6650 CrossRef CAS.
- Z. Gordon, T. J. Miller, C. A. Leahy, E. M. Matson, M. Burgess, M. J. Drummond, C. V. Popescu, C. M. Smith, R. L. Lord, J. Rodriguez-Lopez and A. R. Fout, Inorg. Chem., 2019, 58, 15801–15811 CrossRef CAS.
- W. Lai, H. Chen, S. Cohen and S. Shaik, J. Phys. Chem. Lett., 2011, 2, 2229–2235 CrossRef CAS.
- J. P. T. Zaragoza, T. H. Yosca, M. A. Siegler, P. Moenne-Loccoz, M. T. Green and D. P. Goldberg, J. Am. Chem. Soc., 2017, 139, 13640–13643 CrossRef CAS.
- J. P. T. Zaragoza, D. C. Cummins, M. Q. E. Mubarak, M. A. Siegler, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2019, 58, 16761–16770 CrossRef CAS.
- A. C. Weitz, M. R. Mills, A. D. Ryabov, T. J. Collins, Y. Guo, E. L. Bominaar and M. P. Hendrich, Inorg. Chem., 2019, 58, 2099–2108 CrossRef CAS.
- D. C. Cummins, J. G. Alvarado, J. P. T. Zaragoza, M. Q. Effendy Mubarak, Y.-T. Lin, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2020, 59, 16053–16064 CrossRef CAS.
- E. A. Hill, A. C. Weitz, E. Onderko, A. Romero-Rivera, Y. Guo, M. Swart, E. L. Bominaar, M. T. Green, M. P. Hendrich, D. C. Lacy and A. S. Borovik, J. Am. Chem. Soc., 2016, 138, 13143–13146 CrossRef CAS.
- S.-S. Xue, X.-X. Li, Y.-M. Lee, M. S. Seo, Y. Kim, S. Yanagisawa, M. Kubo, Y.-K. Jeon, W.-S. Kim, R. Sarangi, S. H. Kim, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2020, 142, 15305–15319 CrossRef CAS.
- T. J. Collins and S. W. Gordon-Wylie, J. Am. Chem. Soc., 1989, 111, 4511–4513 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. Chanda, D.-L. Popescu, F. Tiago de Oliveira, E. L. Bominaar, A. D. Ryabov, E. Muenck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606–619 CrossRef CAS.
- M. B. Chambers, S. Groysman, D. Villagran and D. G. Nocera, Inorg. Chem., 2013, 52, 3159–3169 CrossRef CAS.
- S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 CAS.
- C. T. Watson, S. Cai, N. V. Shokhirev and F. A. Walker, Inorg. Chem., 2005, 44, 7468–7484 CrossRef CAS.
- F. N. Penkert, T. Weyhermueller, E. Bill, P. Hildebrandt, S. Lecomte and K. Wieghardt, J. Am. Chem. Soc., 2000, 122, 9663–9673 CrossRef CAS.
- T. Kojima, F. Ogishima, T. Nishibu, H. Kotani, T. Ishizuka, T. Okajima, S. Nozawa, Y. Shiota, K. Yoshizawa, H. Ohtsu, M. Kawano, T. Shiga and H. Oshio, Inorg. Chem., 2018, 57, 9683–9695 CrossRef CAS.
- A residual signal corresponding to complex 1 was observed. Quantification of that signal revealed ca. 15% of unreacted complex 1.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- K. E. Loeb, T. E. Westre, T. J. Kappock, N. Mitic, E. Glasfeld, J. P. Caradonna, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 1901–1915 CrossRef CAS.
- R. Sarangi, Coord. Chem. Rev., 2013, 257, 459–472 CrossRef CAS.
- A. Chanda, X. Shan, M. Chakrabarti, W. C. Ellis, D. L. Popescu, F. Tiago de Oliveira, D. Wang, L. Que, T. J. Collins, E. Muenck and E. L. Bominaar, Inorg. Chem., 2008, 47, 3669–3678 CrossRef CAS.
- P. Chandrasekaran, S. C. E. Stieber, T. J. Collins, L. Que Jr, F. Neese and S. DeBeer, Dalton Trans., 2011, 40, 11070–11079 RSC.
- S. Pattanayak, A. J. Jasniewski, A. Rana, A. Draksharapu, K. K. Singh, A. Weitz, M. Hendrich, L. Que, A. Dey and S. Sen Gupta, Inorg. Chem., 2017, 56, 6352–6361 CrossRef CAS.
- V. Sastri Chivukula, J. Lee, K. Oh, J. Lee Yoon, J. Lee, A. Jackson Timothy, K. Ray, H. Hirao, W. Shin, A. Halfen Jason, J. Kim, L. Que Jr, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS.
- J. Y. Lee, R. L. Peterson, K. Ohkubo, I. Garcia-Bosch, R. A. Himes, J. Woertink, C. D. Moore, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 9925–9937 CrossRef CAS.
- V. Dantignana, J. Serrano-Plana, A. Draksharapu, C. Magallon, S. Banerjee, R. Fan, I. Gamba, Y. Guo, L. Que, M. Costas and A. Company, J. Am. Chem. Soc., 2019, 141, 15078–15091 CrossRef CAS.
- C. Kupper, B. Mondal, J. Serrano-Plana, I. Klawitter, F. Neese, M. Costas, S. Ye and F. Meyer, J. Am. Chem. Soc., 2017, 139, 8939–8949 CrossRef CAS.
- I. Kaljurand, A. Kuett, L. Soovaeli, T. Rodima, V. Maeemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS.
- A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC.
- R. Gupta and A. S. Borovik, J. Am. Chem. Soc., 2003, 125, 13234–13242 CrossRef CAS.
- S. K. Barman, J. R. Jones, C. Sun, E. A. Hill, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2019, 141, 11142–11150 CrossRef CAS.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, J. Am. Chem. Soc., 2013, 135, 17090–17104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2018882. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc07054h |
| This journal is © The Royal Society of Chemistry 2021 |