
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of amyloid-beta peptide aggregation via photoactivation of strained Ru(II) polypyridyl complexes†
Janaina C.
Bataglioli
a,
Luiza M. F.
Gomes
a,
Camille
Maunoir
a,
Jason R.
Smith
 a,
Houston D.
Cole
b,
Julia
McCain
c,
Tariq
Sainuddin
c,
Colin G.
Cameron
b,
Sherri A.
McFarland
*b and
Tim
Storr
*a
a,
Houston D.
Cole
b,
Julia
McCain
c,
Tariq
Sainuddin
c,
Colin G.
Cameron
b,
Sherri A.
McFarland
*b and
Tim
Storr
*a
aDepartment of Chemistry, Simon Fraser University, BC, Canada V5A-1S6. E-mail: tim_storr@sfu.ca
bDepartment of Chemistry and Biochemistry, University of Texas, Arlington, Texas, USA 76019. E-mail: sherri.mcfarland@uta.edu
cDepartment of Chemistry, Acadia University, Wolfville, Nova Scotia, Canada B4P 2R6
First published on 24th April 2021
Abstract
Alzheimer's disease (AD) is a chronic neurodegenerative disorder characterized by progressive and irreversible damage to the brain. One of the hallmarks of the disease is the presence of both soluble and insoluble aggregates of the amyloid beta (Aβ) peptide in the brain, and these aggregates are considered central to disease progression. Thus, the development of small molecules capable of modulating Aβ peptide aggregation may provide critical insight into the pathophysiology of AD. In this work we investigate how photoactivation of three distorted Ru(II) polypyridyl complexes (Ru1–3) alters the aggregation profile of the Aβ peptide. Photoactivation of Ru1–3 results in the loss of a 6,6′-dimethyl-2,2′-bipyridyl (6,6′-dmb) ligand, affording cis-exchangeable coordination sites for binding to the Aβ peptide. Both Ru1 and Ru2 contain an extended planar imidazo[4,5-f][1,10]phenanthroline ligand, as compared to a 2,2′-bipyridine ligand for Ru3, and we show that the presence of the phenanthroline ligand promotes covalent binding to Aβ peptide His residues, and in addition, leads to a pronounced effect on peptide aggregation immediately after photoactivation. Interestingly, all three complexes resulted in a similar aggregate size distribution at 24 h, forming insoluble amorphous aggregates as compared to significant fibril formation for peptide alone. Photoactivation of Ru1–3 in the presence of pre-formed Aβ1–42 fibrils results in a change to amorphous aggregate morphology, with Ru1 and Ru2 forming large amorphous aggregates immediately after activation. Our results show that photoactivation of Ru1–3 in the presence of either monomeric or fibrillar Aβ1–42 results in the formation of large amorphous aggregates as a common endpoint, with Ru complexes incorporating the extended phenanthroline ligand accelerating this process and thereby limiting the formation of oligomeric species in the initial stages of the aggregation process that are reported to show considerable toxicity.
Introduction
Alzheimer's disease (AD) is the most common form of dementia and is currently the 5th leading cause of death worldwide. An increase in life expectancy is expected to result in a sharp rise in the number of dementia cases in the next 30 years, with approximately 150 million people forecast to be living with dementia by 2050.1 The increased incidence of AD, and lack of effective treatment strategies, has stimulated an intense research effort to enhance our understanding of the pathophysiology of this disease and develop new therapeutics. The amyloid hypothesis was first proposed almost 25 years ago, and postulates that the progressive formation of oligomers and aggregates of the Aβ peptide is caused either by increased production or decreased clearance of Aβ, triggering a neurotoxic cascade in the brain.2–6 Proteolytic cleavage of the amyloid precursor protein (APP) affords the Aβ peptide in variable lengths (38 to 43 amino acids), with the fragment ending at position 40 (Aβ1–40) being the most abundant (∼90%) followed by 42 (Aβ1–42, ∼9%).7 Truncation at the N-terminus results in Aβ3(p)-n, Aβ4-n, and Aβ11(p)-n (where p refers to pyroglutamate), which are also components of amyloid plaques.8–10 Clinical trials of promising drugs targeting the amyloid pathway have so far failed, either due to off-target effects or a lack of efficacy.11,12 There is considerable debate as to when drug treatments should be initiated in AD, and drug trials targeting the amyloid pathway are now focused on healthy people at risk of AD.13,14Oxidative stress is widespread in AD, with early neuronal and pathological changes indicating oxidative damage.15 Fenton-type processes involving dysregulated redox-active metal ions (Cu, Fe), and metal-containing Aβ aggregates, are hypothesized to contribute to the production of reactive oxygen species (ROS) and resulting oxidative stress in AD.16–20 In addition, metal ion coordination to the Aβ peptide (Fe, Cu, Zn) alters its aggregation pattern, and thus it is hypothesized that metal-ion dysregulation and interaction with Aβ plays a significant role in AD development. A number of approaches for the prevention of metal-ion binding to Aβ have been developed, including the use of metal-binding agents,21–23 and metal complexes that target metal-binding residues,24–27 thereby modulating peptide aggregation. Due to the multifactorial nature of AD, it is likely that a multifunctional drug development strategy will be needed to effectively treat this disease. Metal complexes capable of modifying the Aβ peptide aggregation process, while also restricting adventitious metal ion binding to Aβ,28,29 limiting oxidative stress,30,31 inhibiting acetylcholine esterase (AChE) activity,32 and initiating peptide cleavage have received considerable attention.33,34 Work with Pt complexes has shown that in addition to covalent binding (most likely to Aβ peptide His residues), the incorporation of planar aromatic ligands can enhance non-covalent π–π interactions of metal complexes with the Aβ peptide, providing an additional mechanism to increase targeting.35–38 Commonly employed ligands to enhance π–π interactions include 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen), and cyclometalating ligands. Examples include a number of cyclometallated Rh and Ir complexes that have been reported to limit aggregation, and exhibit enhanced emissive properties when bound to the Aβ peptide.39,40 Recently, Lim et al. reported a series of cyclometallated Ir complexes that promote the photo-induced oxidation of Aβ in the presence of O2.41,42
Ru(II) polypyridyl complexes have found widespread application due to their interesting electrochemical, photophysical, and biological properties.43,44 In biology, these complexes have shown utility in DNA intercalation45 and protein binding.46 Ru(II) polypyridyl complexes can be photoactivated leading to the generation of ROS such as singlet oxygen (1O2) or ligand dissociation to afford a metal complex capable of binding to biological targets,47–50 or the release of biologically active ligands from the metal complex.51–55 A number of Ru(II) polypyridyl complexes have been reported to interact with the Aβ peptide via non-covalent π–π interactions. In the case of [Ru(bpy)3]2+, photoactivation in the presence of the Aβ peptide leads to amino acid oxidation and destabilization of peptide secondary structure via generation of 1O2.56 In elegant work, Martí and co-workers have shown that Ru(II) polypyridyl complexes with a specific extended planar aromatic ligand can also be used as sensitive fluorescent probes for amyloid fibrils57 and oligomers.58,59 Other Ru(II) polypyridyl complexes were shown to limit Aβ aggregation, inhibit AChE activity and protect against ROS.31,32
In this work we have investigated the interaction of a series of strained photoactivatable Ru(II) polypyridyl complexes with the Aβ peptide (Fig. 1). Photoactivation by visible light leads to loss of a 6,6′-dimethyl-2,2′-dipyridyl (6,6′-dmb) ligand, unmasking cis-exchangeable coordination sites capable of binding to biomolecules such as the Aβ peptide. We find that photoactivation is critical for modulating Aβ peptide aggregation, and in addition, the extended planar imidazo[4,5-f][1,10]phenanthroline ligand in Ru1 and Ru2 enhances Aβ peptide targeting in comparison to the bpy analogue Ru3.
 | ||
| Fig. 1 Photoactivatable Ru complexes (Ru1–3) used in this work. Visible light photoactivation leads to 6,6′-dimethyl–2,2′-dipyridyl (6,6′-dmb) ligand loss to unmask cis-exchangeable coordination sites for Aβ peptide binding. | ||
Results and discussion
Stability and photoactivation
Complexes Ru1 and Ru2 have been previously reported to dissociate one 6,6′-dimethyl-2,2′-dipyridyl (6,6′-dmb) ligand upon photoactivation,60,61 while the photochemical ligand dissociation process for Ru3 has not yet been reported. In addition, Ru1 and Ru2 present limited cytotoxicity in the dark and thus limited potential for off-target toxicity (EC50 = 37 μM in HL 60 cell line for Ru1 and >100 μM in SKMEL 28 cell line for Ru2),60,61 and these two complexes incorporate a planar aromatic [1,10]phenanthroline ligand, analogues of which have shown high affinity for amyloid aggregates.57–59,62 Thus we hypothesized that Ru1 and Ru2 would be good candidates for photoactivated binding to the Aβ peptide, while Ru3 would offer a suitable comparison that does not incorporate the extended [1,10]phenanthroline ligand. The sample preparation protocol for the Ru1–3 interaction experiments with the Aβ peptide is shown in Scheme 1.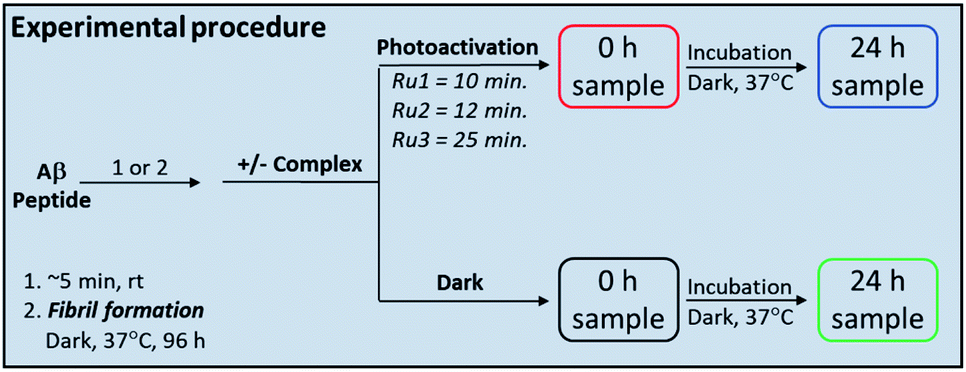 | ||
| Scheme 1 Sample preparation protocol for the Aβ peptide interaction experiments with Ru1–3. | ||
Under our conditions, Ru1 and Ru2 were stable in buffer solution over 24 h in the dark, however, Ru3 exhibited ca. 6% loss of a 6,6′-dmb ligand in the absence of light (Fig. S1 and S2†). As expected, Ru1–3 undergo photochemical ligand dissociation upon exposure to visible light (SOLLA 30W LED, 5.7 mW cm−2),60,61 with UV-vis measurements indicating reaction completion at 10 min, 12 min, and 25 min, respectively after initial light exposure (Fig. S3†). Although crystal structures and 3MC energies are unavailable at this time, it could be the case that the larger π-expansive ligands in Ru1 and Ru2 lead to larger distortions in the coordination geometries, which in turn suppresses rechelation and accelerates dissociation times in comparison to Ru3. Photoactivation produces very little singlet oxygen (1O2 yield 0.03 for Ru1,60 0.01 for Ru2,61 0.01 for Ru3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63) in comparison to [Ru(bpy)3]2+ (1O2 yield 0.56).64 The photodissociation was shown to be selective for dissociation of the 6,6′-dmb ligand for Ru1–3 as demonstrated by 1H NMR and ESI-MS (Fig. S4 and S5†). The MS spectra show peaks consistent with different ligands (H2O, DMSO, Cl−) occupying the coordination sites made available due to 6,6′-dmb photoejection. Interestingly, we observe a decrease in Ru(II) complex signals in the 1H NMR upon photoactivation, and the presence of a precipitate. These results are consistent with the relatively high concentration used in the NMR experiment (200 μM), the number of different complexes formed, and the formation of complexes with limited solubility (Fig. S6†).65–67 At lower concentrations (50–60 μM) no precipitate was observed vide infra, even after 24 h of incubation, however, at these lower concentrations the 1H NMR signals were not discernible. The photoactivation results show the availability of exchangeable coordination sites on Ru1–3 upon release of the 6,6′-dmb ligand for interaction with the Aβ peptide.
63) in comparison to [Ru(bpy)3]2+ (1O2 yield 0.56).64 The photodissociation was shown to be selective for dissociation of the 6,6′-dmb ligand for Ru1–3 as demonstrated by 1H NMR and ESI-MS (Fig. S4 and S5†). The MS spectra show peaks consistent with different ligands (H2O, DMSO, Cl−) occupying the coordination sites made available due to 6,6′-dmb photoejection. Interestingly, we observe a decrease in Ru(II) complex signals in the 1H NMR upon photoactivation, and the presence of a precipitate. These results are consistent with the relatively high concentration used in the NMR experiment (200 μM), the number of different complexes formed, and the formation of complexes with limited solubility (Fig. S6†).65–67 At lower concentrations (50–60 μM) no precipitate was observed vide infra, even after 24 h of incubation, however, at these lower concentrations the 1H NMR signals were not discernible. The photoactivation results show the availability of exchangeable coordination sites on Ru1–3 upon release of the 6,6′-dmb ligand for interaction with the Aβ peptide.
Binding of Ru1–3 to the Aβ peptide
We first evaluated the interaction of unactivated Ru1–3 with the Aβ peptide by 1H NMR and ESI-MS. For these initial studies we chose to use the hydrophilic Aβ1–16 peptide which includes most of the amino acids associated with metal ion binding (i.e. His6/13/14), and in addition, has a low propensity to aggregate. Incubation of one eq. of Ru1–3 with Aβ1–16 in the dark over 24 h resulted in no changes in NMR features suggesting no significant interaction in solution (Fig. 2 and S7–S10†). | ||
| Fig. 2 (A) 1H NMR spectra of Aβ1–16 (200 μM) in the presence of 1.0 eq. unactivated Ru1 showing no changes of peptide residue signals after 24 h of incubation. Samples were prepared in PBS buffer (0.01 M, pH 7.4) at 37 °C. * His,6 His13 and His.14 † Tyr.10 (B) ESI-MS of unactivated Ru1 + Aβ1–16 showing no evidence of adduct formation. Samples were prepared in NH4CO3 buffer (20 mM, pH 9.0). | ||
In addition, the ESI-MS data for unactivated Ru1–2 in the presence of Aβ1–16 show peaks for the intact complexes and the Aβ1–16 peptide, and no evidence of adduct formation or a ternary complex under the experimental conditions (Fig. 2 and S9†). Interestingly, while the 1H NMR of unactivated Ru3 with Aβ1–16 did not exhibit any changes to peptide residue signals over 24 h (Fig. S8†), the ESI-MS spectrum indicated species consistent with loss of the 6,6′-dmb ligand and adduct formation ([Ru3–Aβ1–16]2+; m/z = 1197.7) (Fig. S10†). This result is in agreement with the lower stability of unactivated Ru3 in solution and ca. 6% ligand loss measured by 1H NMR (Fig. S1†).
Upon addition of one eq. of Ru1 to Aβ1–16 and photoactivation for 10 min (SOLLA 30W LED, 5.7 mW cm−2) we observed the presence of free 6,6′-dmb in the 1H NMR, a shift in select peptide residues, and the loss of signals associated with the Ru(II) complex (Fig. 3). We attribute the loss of Ru(II) signals to the presence of multiple species bound to the peptide upon photoactivation and precipitation of the photodissociated complex at the 200 μM concentration. Interestingly, while the majority of the peptide residues do not shift upon photoactivation, the His resonance at 7.78 ppm shifts upfield to 7.71 ppm, overlapping with a free 6,6′-dmb signal at 7.71 ppm with a concomitant increase in integration value (Fig. 3). The data is consistent with binding of an Aβ His residue to photoactivated Ru1, and we hypothesize that there is likely no preference for any of the available His residues (His,6 His,13 His14).
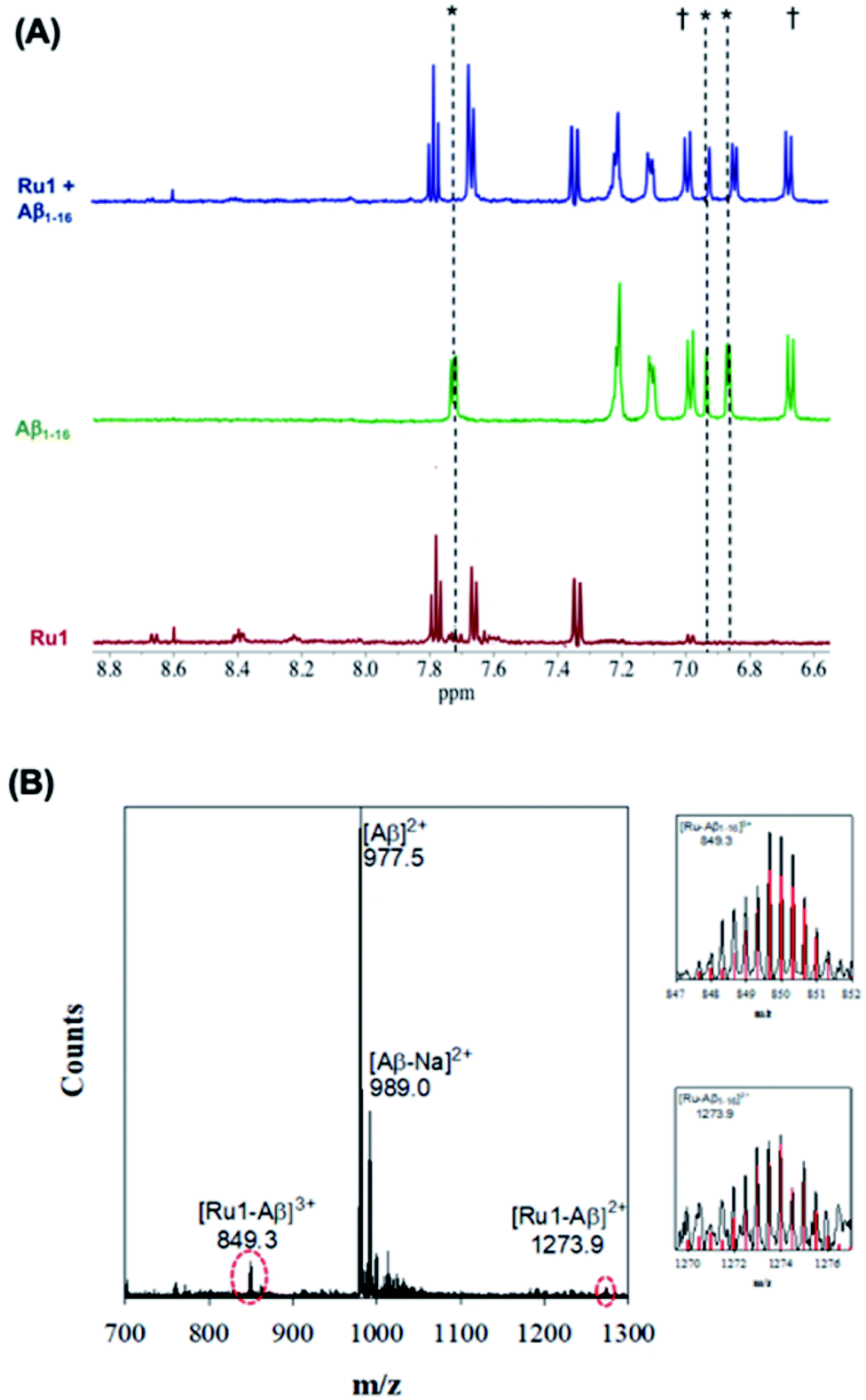 | ||
| Fig. 3 (A) 1H NMR spectra of photoactivated Ru1–Aβ1–16 (200 μM) showing His shifts immediately after photoactivation (10 min.). Samples were prepared in PBS buffer (0.01 M, pH 7.4) at 37 °C. * His,6 His13 and His.14 † Tyr.10 (B) ESI-MS of photoactivated Ru1 + Aβ1–16 showing evidence of adduct formation. Zoomed region shows the isotopic pattern of the detected adduct, and in red the theoretical isotopic pattern for the corresponding adduct. Samples were prepared in NH4CO3 buffer (20 mM, pH 9.0) and data was collected after 10 min of activation. | ||
Exposure of Aβ1–16 alone to the photolysis conditions (SOLLA 30W LED, 5.7 mW cm−2) did not shift any of the 1H NMR signals in comparison to Aβ1–16 in the absence of photolysis. An upfield shift of the His residue at 7.78 ppm is also observed for Ru2 upon photoactivation in the presence of Aβ1–16 (Fig. S7†), and our results are consistent with metal complex – Aβ binding reported for Ru(III) complexes,68 and for Pt(II) complexes reported by Guo et al.69 and Hureau et al.,29 indicating that the His residues are involved in the interaction of Ru1–2 with Aβ.
While photoactivated Ru3–Aβ1–16 samples exhibit free 6,6′-dmb ligand, no shifts of any peptide residues were observed after 24 h of incubation, even in the presence of 2 eq. of Ru3 (Fig. S8†). These results suggest that while Aβ His binding occurs for Ru1–2, Ru3 does not interact with the peptide in the same manner.
The interaction of photoactivated Ru1–3 with Aβ1–16 was further investigated via ESI-MS. In contrast to the MS spectrum of unactivated Ru1–Aβ1–16 (Fig. 2), photoactivated Ru1–Aβ1–16 indicates the formation of adducts [Ru1–Aβ1–16]3+ (m/z = 849.3) and [Ru1–Aβ1–16]2+ (m/z = 1273.9) (Fig. 3). The isotopic distribution confirms the presence of Ru in the adduct peaks, and the masses of the adducts are consistent with loss of the 6,6′-dmb ligand and coordination to the peptide. The ESI-MS data for Ru2 is similar to that for Ru1, indicating adduct formation upon photoactivation of the complex in the presence of the Aβ peptide (Fig. S8†). As detailed above, ESI-MS of unactivated Ru3 in the presence of Aβ1–16 indicates adduct formation, and upon photoactivation a number of adduct peaks are present including ([Ru3–Aβ1–16]3+; m/z = 798.5), ([Ru3–Cl–Aβ1–16]3+; m/z = 812.4), and ([Ru3–Aβ1–16]2+; m/z = 1197.5) (Fig. S10†). The latter peak is also observed in the unactivated ESI-MS spectrum. We also investigated the binding of unactivated and activated Ru1–3 with the longer length Aβ1–40 peptide, and the results are similar to that described for Aβ1–16 peptide, showing that adduct formation only occurs for the photoactivated complexes (Fig. S11†). Previous work by Park et al. using [Ru(bpy)3]2+ (ref. 56) and Lim et al. using cyclometallated Ir complexes41,42 show significant Aβ peptide oxidation upon photoactivation, however, we do not observe evidence of peptide oxidation in the photoactivation experiments by ESI-MS, in agreement with the low 1O2 quantum yields for Ru1–3. Unfortunately, our MS/MS fragmentation experiments were not successful in indicating the residue(s) responsible for peptide binding due to low signal to noise.
It is interesting to note that while Ru3 exhibits adduct formation in the ESI-MS spectrum, no significant His residue shifts (or any other peptide residue) were observed in the 1H NMR experiment. We speculate that while a significant amount of adduct forms in the photoactivation experiments for Ru1 and Ru2, comparatively less adduct forms for Ru3. We suggest a potential pre-organizing effect of the extended planar aromatic ligands for Ru1 and Ru2, which facilitates covalent binding upon photoactivation. The enhanced interaction of Aβ peptide aggregates with Ru(II) polypyridyl complexes incorporating extended planar aromatic ligands has been reported,57–59,62 and in addition, planar aromatic ligands enhance covalent adduct formation for Pt(II) complexes with both the Aβ peptide57–59,62 and DNA.70
Influence of Ru1–3 on Aβ peptide aggregation
Based on the promising binding data for photoactivated Ru1–2, and to a lesser extent Ru3, we investigated if the aggregation process of the Aβ peptide could be influenced by the complexes. The Aβ1–42 peptide was used in these studies due to the higher propensity for aggregation and toxicity.71–74 Using gel electrophoresis/western blotting we determined that unactivated Ru1–3 (1.0 eq. and 2.0 eq.) did not alter the aggregation pattern of the Aβ1–42 peptide (25 μM) relative to peptide alone at 24 h (Fig. 4A, C, and E). Even though a number of Ru(II) polypyridyl complexes have been reported to interact in a non-covalent manner with the Aβ peptide,57–59,62 and in some cases alter the aggregation process,32,56 unactivated Ru1–3 did not exhibit a sufficiently strong interaction to alter the peptide aggregation profile based on the gel electrophoresis experiment. | ||
| Fig. 4 Gel electrophoresis/western blot of Aβ1–42 (25 μM) and different concentrations of unactivated Ru1 (A), Ru2 (C), and Ru3 (E) in PBS buffer (0.01 M, pH 7.4) after 24 h of incubation at 37 °C. Lane 1: Aβ1–42; lane 2: Aβ1–42 + 1.0 eq. Ru(II) complex; lane 3: Aβ1–42 + 2.0 eq. Ru(II) complex. Influence of photoactivated Ru1 (B), Ru2 (D) and Ru3 (F) on the aggregation profile of Aβ1–42. Gel electrophoresis/western blot of 25 μM Aβ1–42 and 1.0 eq. of Ru1–3 in PBS buffer (0.01 M, pH 7.4) at incubation time points 0 h and 24 h, with agitation at 37 °C, using anti-Aβ antibody 6E10. Lane 1: Aβ1–42; lane 2: Aβ1–42 + 1.0 eq. Ru(II) complex. | ||
We next investigated the incubation of the Aβ1–42 peptide (25 μM) in the presence of photoactivated Ru1–3 (0.1 to 2.0 eq.) over 24 h. At 1.0 eq. of Ru1 and Ru2, peptide aggregation is significantly affected, resulting in the formation of higher molecular weight (MW) aggregates versus peptide alone, while Ru3 only shows a similar effect at 2.0 eq. (Fig. S12†). Based on these promising initial results we further studied the effect of the photoactivated complexes (1.0 eq.) on peptide aggregation immediately after photoactivation (0 h) and at the 24 h timepoint. At 0 h, Aβ1–42 in the absence of activated Ru complex is primarily present in solution in monomeric and dimeric forms (low MW species), with a range of higher MW species predominating at 24 h in agreement with previous reports (Fig. 4B, D, and F).33,75,76 The lack of observable dimer species at 0 h for Aβ1–42 alone in Fig. 4F in comparison to 4B and 4D is likely due to slightly different mixing/gel loading times. Photoactivation of Ru1 and Ru2 induced the formation of high MW aggregates immediately after photoactivation (t = 0 h, Fig. 4B and D), however, Ru3 did not exhibit induction of high MW species on the gel at the initial timepoint (Fig. 4F). The immediate formation of high MW aggregates for photoactivated Ru1–2, as opposed to Ru3, suggests that the greater degree of covalent binding observed by 1H NMR results in increased peptide aggregation. Interestingly, this immediate change from monomer/dimer to high MW aggregates for Ru1–2 limits the formation of oligomers in the ca. 15–30 kDa range, which are reported to exhibit significant toxicity.77–79
After 24 h of incubation, photoactivated Ru1–2 afford only high MW species (MW > 250 kDa) as observed on the gel (Fig. 4B and D). However, the aggregation pattern for Ru3 at 24 h appears qualitatively similar to peptide alone (Fig. 4F), again showing a significant difference in comparison to the results for Ru1–2. To investigate if the integrity of the peptide was compromised by the presence of the photoactivated Ru complexes (via oxidation and/or cleavage),41,42,56 a dot blot experiment was performed on the bulk sample (Fig. S13†). The peptide is recognized by the 6E10 antibody at the 24 h timepoint in all cases, showing that high MW aggregates formed in the presence of activated Ru1–2 do not penetrate the gel, and the lack of observable peptide aggregates on the gel is not due to oxidation/cleavage events restricting interaction with the 6E10 antibody. Overall, the gel electrophoresis results indicate that photoactivation, and covalent binding of Ru1–2 to the Aβ peptide, are necessary to observe substantial changes in the aggregation pattern, thus highlighting the role of the extended planar aromatic ligands of Ru1–2 in modulating Aβ peptide aggregation.
In order to further investigate the importance of both photoactivation and the extended aromatic ligand we studied the interaction of the Aβ peptide with the previously reported Ru(II) polypyridyl complex Ru(bpy)2CO3,80 which incorporates a labile κ2-carbonato ligand. Facile ligand exchange of the κ2-carbonato ligand provides a Ru(II) complex with two cis-exchangeable coordination sites, similarly to Ru1–3. This complex has been used previously to label peptides and proteins in the absence of photoactivation.81–83 The 1H NMR spectrum of Ru(bpy)2CO3 in the presence of Aβ1–16 did not show a shift of His residues (or any other shift, Fig. S14†), however ESI-MS data showed the formation of a peptide adduct indicating that the complex is able to bind to the peptide, similarly to results for photoactivated Ru3 (Fig. S15†). In addition, the Aβ1–42 peptide aggregation pattern in the presence of 0–2 eq. of Ru(bpy)2CO3 was investigated by gel electrophoresis and was unchanged in comparison to peptide alone at 0 h and 24 h (Fig. S16†). Thus, the binding and aggregation data for Ru(bpy)2CO3 are similar to that for photoactivated Ru3, providing further support for the importance of the extended hydrophobic [1,10]phenanthroline ligand in Ru1–2 in facilitating peptide binding and modulating the aggregation pathway.
While gel electrophoresis/western blotting revealed the presence of higher MW Aβ1–42 species and their size distribution, transmission electron microscopy (TEM) analysis allowed us to characterize larger, Aβ1–42 aggregates that are too large to penetrate into the gel matrix. Thus, the combination of these two methods provides a more complete picture of the Aβ1–42 aggregation pathway under different conditions.84,85 As expected, the TEM images did not show large aggregates for the Aβ1–42 sample at 0 h (Fig. 5), however, immediately after light activation, samples containing Ru1–2 showed large amorphous aggregates, while the sample containing Ru3 showed the presence of smaller aggregates (Fig. 5). Incubation of Aβ1–42 alone for 24 h led to the formation of both large amorphous aggregates and fibrillar species (Fig. 5 and S17†), which agrees with previous reports. Peptide in the presence of unactivated Ru1–3 showed similar aggregation morphology by TEM as Aβ1–42 alone. Incubation of Aβ1–42 for 24 h in the presence of photoactivated Ru1–3 affords similar sized amorphous aggregates (Fig. 5), indicating that the Ru(II) complexes inhibit fibrillization at 24 h. Our results show that upon photoactivation, Ru1 and Ru2 immediately promote changes in peptide aggregation via the formation of soluble high MW species and large amorphous aggregates. In contrast, Ru3 does not promote the formation of large amorphous aggregates immediately, however, similar-sized aggregates are observed by TEM after 24 h (Fig. 5). The TEM images are consistent with the gel electrophoresis results, highlighting that photoactivation is essential for modulation of peptide aggregation.
 | ||
| Fig. 5 TEM images of the morphology of Aβ1–42 aggregates at 0 h (photoactivated samples) and 24 h (for unactivated and photoactivated samples). Conditions: Aβ1–42 (25 μM), Ru1–3 (1.0 eq.) (scale bar 200 nm). | ||
To further analyze the change in peptide aggregation in the presence of the Ru(II) complexes, a bicinchoninic (BCA) assay was used to determine the total concentration of Aβ1–42 peptide in solution.86 Before measurement, the samples were centrifuged to remove insoluble aggregates using an established protocol.87 As expected, the results show a ca. 50% reduction in soluble peptide after 24 h for peptide alone, which is similar to the change in peptide concentration in the presence of unactivated Ru1–3 after 24 h (Fig. 6). Immediately after photoactivation, the concentration of soluble peptide is less than peptide alone for samples containing all three complexes, however, Ru1 and Ru2 display a slightly larger reduction in Aβ solubility in comparison to Ru3, which is consistent with the results obtained from the gel electrophoresis and TEM studies. Strikingly, in the presence of all three activated Ru(II) complexes, the peptide is almost completely precipitated at 24 h (Fig. 6), which is consistent with the large insoluble amorphous aggregates observed by TEM (Fig. 5), and gel electrophoresis of Ru1–2 where we observe that the species formed after 24 h incubation does not penetrate the gel (Fig. 4).
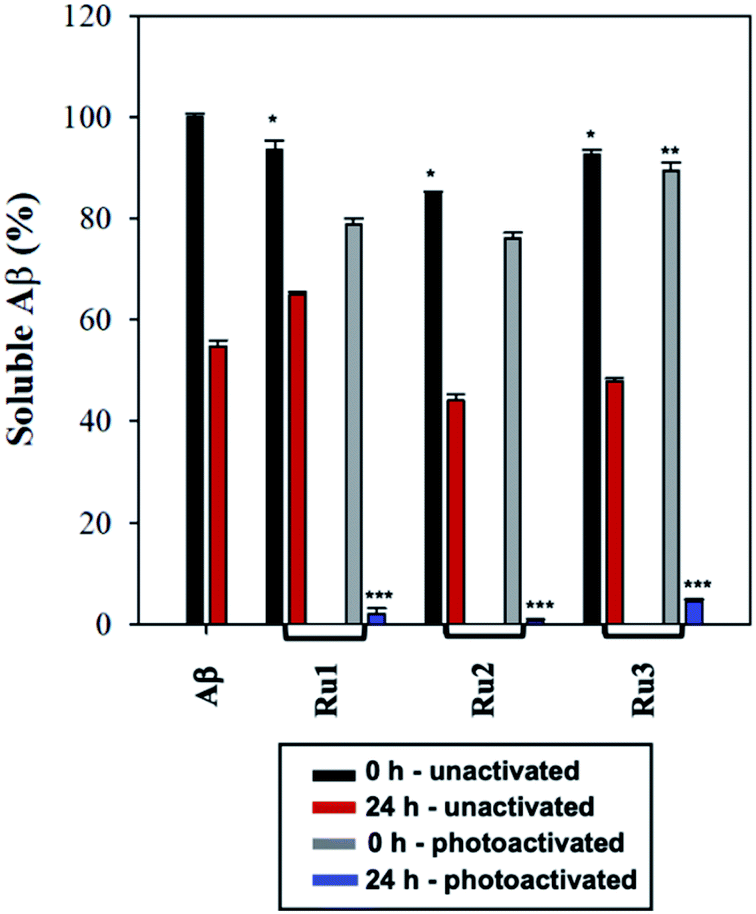 | ||
| Fig. 6 BCA assay of 60 μM Aβ1–42 in the presence of 1.0 eq. of Ru1–3 in PBS buffer (0.01 M, pH 7.4) at 0 h and 24 h with and without photoactivation. Samples were centrifuged at 14000g for 5 min prior to absorbance measurement. Statistically significant difference between: * Aβ1–42 0 h and all three unactivated complexes at 0 h (Ru1, p = 0.02; Ru2, p < 0.0001; Ru3, p = 0.0002); ** photoactivated Ru1–2 0 h compared to photoactivated Ru3 0 h (Ru3, p = 0.0003) (no statistical difference between photoactivated Ru1 and Ru2 at 0 h); and *** Aβ1–42 (24 h) and photoactivated Ru1–3 (24 h) (p < 0.0001). Calculated using 2-way ANOVA. | ||
Interestingly, the BCA results at 24 h show that photoactivated Ru3 also significantly decreases peptide solubility, even though the gel electrophoresis experiment still showed soluble high MW species. The morphology of the peptide aggregates formed in the presence of photoactivated Ru1–3 were similar as indicated by TEM, however we hypothesize that the aggregates formed in the presence of Ru3 are less stable, and susceptible to partial dissociation in the electrophoresis running buffer (containing 0.1% SDS).
The BCA results show that unactivated Ru1–3 do not have a large effect on peptide solubility, consistent with the gel electrophoresis and TEM analysis. Immediately after photoactivation, Ru1–2 lead to a significant change in the aggregation pattern of Aβ1–42 as shown by the TEM and gel electrophoresis data. These results highlight the importance of photoactivation for modulation of the peptide aggregation pathway. It is interesting to note that for Ru3, upon photoactivation, there is very little change in the aggregation pattern compared to Ru1–2, however, at 24 h the change in aggregation for Ru3 is similar to that for the other two complexes, resulting in large amorphous aggregates and significant precipitation of the peptide from solution.
Interaction of Ru1–3 with Aβ peptide fibrils
Based on the ability of the photoactivated Ru(II) complexes to modulate peptide aggregation in solution we questioned whether the complexes would interact with pre-formed peptide fibrils and if photoactivation would change the morphology/solubility of these ordered insoluble aggregates.We first investigated the binding of Ru1–3 with Aβ1–42 fibrils via Tyr10 fluorescence.36,88–90 As expected, Ru1–3 exhibited negligible photoluminescence in solution (Fig. S18†). To form Aβ1–42 fibrils, the monomeric peptide was incubated for 96 h and fibril formation was confirmed by TEM (Fig. S19†). Binding of the Ru complexes was compared to thioflavin T (ThT) as a positive control by employing a single-site binding model (Fig. 7 and S20†).57,69 The binding constant of ThT (Kd) under our conditions was determined to be 9.8 ± 1.4 μM, which is in agreement with published values ranging from 5 μM to 11 μM.91,92 Using the same protocol, the binding constants for Ru1 (2.6 ± 0.2 μM), Ru2 (3.2 ± 0.3 μM), and Ru3 (8.2 ± 0.4 μM) were obtained for the unactivated complexes (Fig. 7). The values for Ru1 and Ru2 compare well to the Kd of [Ru(bpy)2(dppz)]2+ (2.1 μM),57 while Ru3 displayed slightly weaker binding to Aβ1–42 fibrils. This data shows that the extended planar aromatic ligands for Ru1 and Ru2 lead to an enhanced interaction with Aβ1–42 fibrils, likely via hydrophobic interactions. While the fibril structure is obviously different in comparison to monomeric peptide in solution, the increased potential for hydrophobic interactions between Ru1–2 and the Aβ1–42 peptide may also pre-organize the complexes so that covalent binding occurs more readily to soluble peptide upon photoactivation.
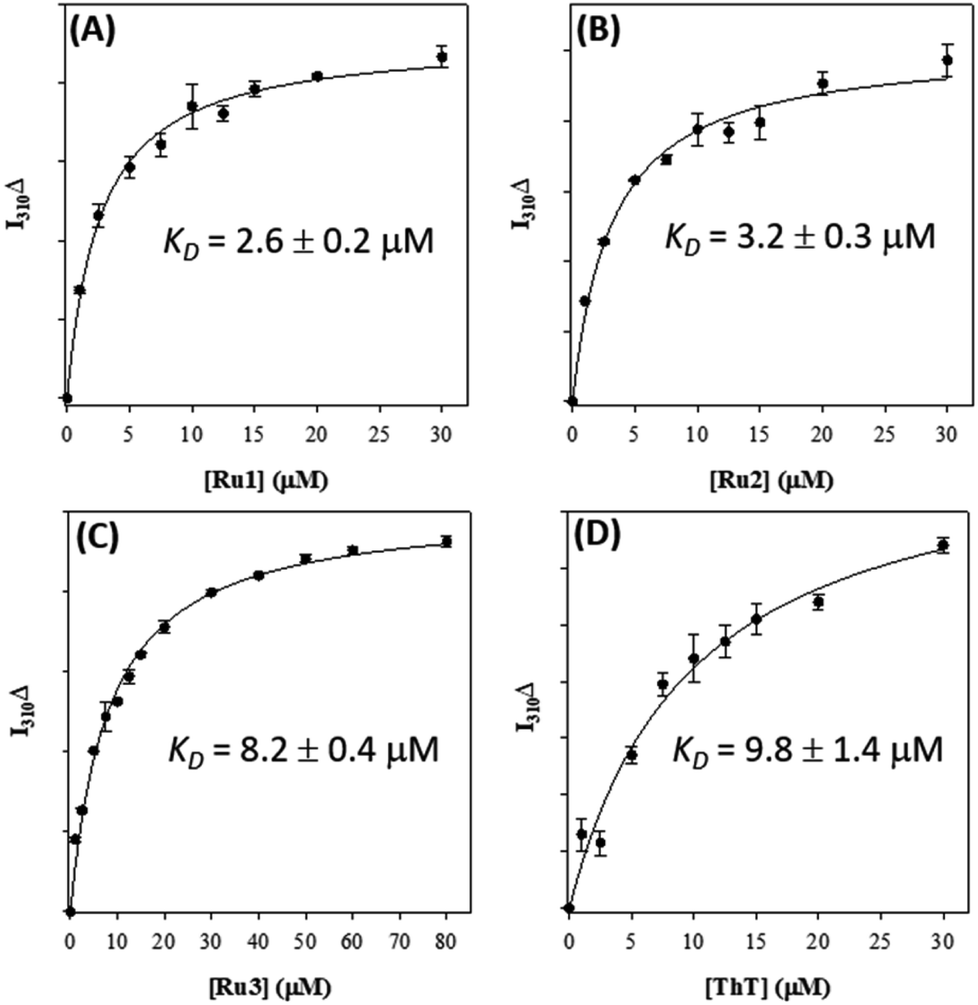 | ||
| Fig. 7 Binding constants of Ru1 (A), Ru2 (B), Ru3 (C), and ThT (D) with pre-formed Aβ1–42 fibrils (10 μM in PBS 0.01 M, pH 7.4) measured via change in Tyr fluorescence (λex/λem = 275/310 nm). | ||
To gain further insight into the interaction of Ru1–3 with Aβ1–42 fibrils we used molecular docking to visualize potential interactions between Ru1–3 and Aβ1–42 fibrils. We employed protein data bank structures (PDB) 2MXU93 and 5OQV94 as representative single and double symmetry fibril surfaces, and via flexible docking of Ru1–3 provide further information on the differential interactions of the complexes with Aβ fibrils. The 2MXU structure has a well-defined hydrophobic cleft and 12 β-strand filaments providing sufficient surface area for modeling the interactions with the Ru(II) complexes, however, the structure does not contain the Aβ1–10 region. In contrast, the 5OQV structure includes the complete Aβ1–42 peptide, though represents a much shorter length of only 5 strands. Note that as a dimer, binding sites on the other faces were essentially identical and omitted for clarity. In combination, docking with these two structures provides a broad representation of the interaction of Ru1–3 with Aβ fibrils. Our docking studies show that there is a shallow potential energy surface for fibril binding, with multiple binding sites effectively contributing to the overall binding affinity of Ru1–3 to the fibrils (Fig. 8 and S21–23†).
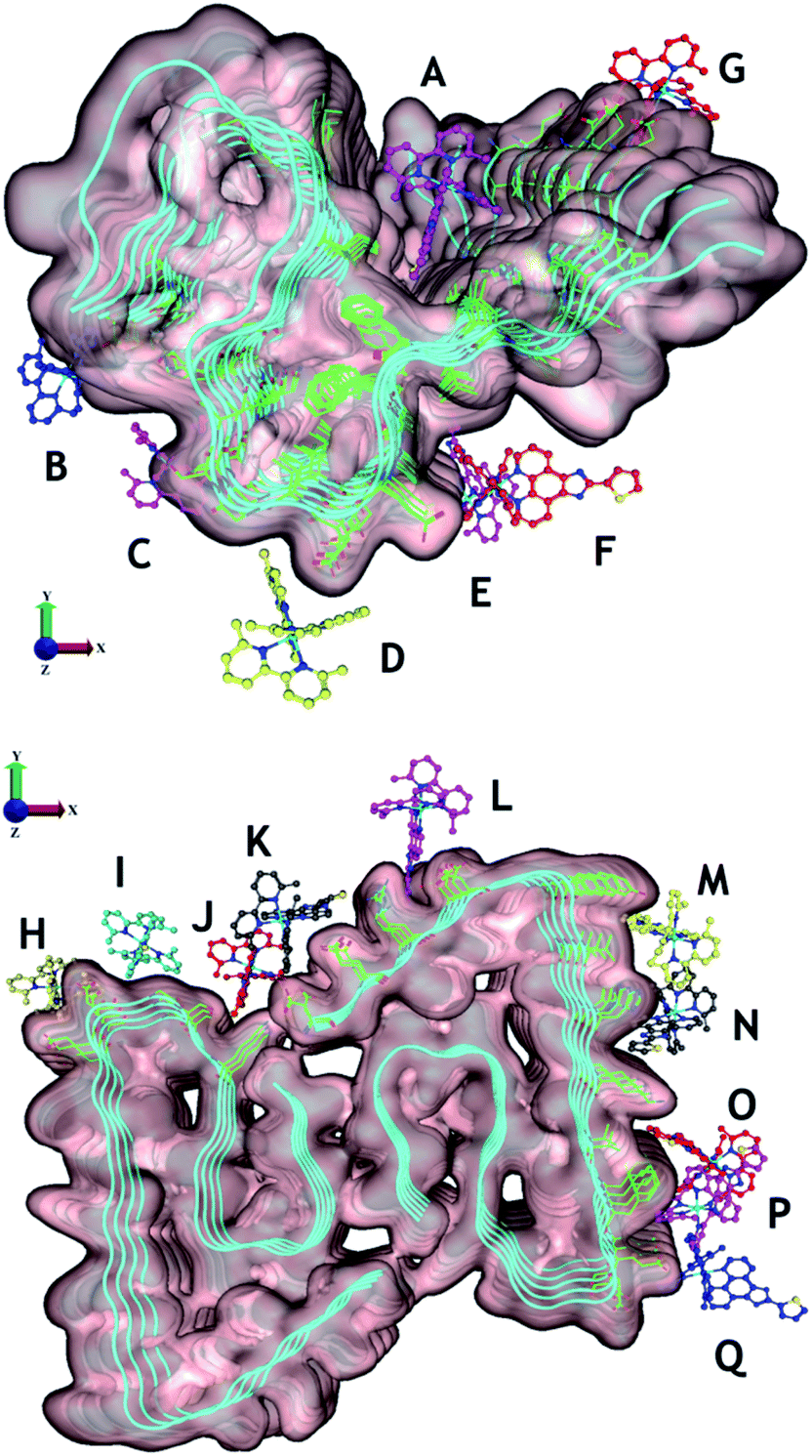 | ||
| Fig. 8 Potential binding sites of unactivated Ru1–3 to PDB structures 2MXU (top) and 5OQV (bottom). The observed binding sites can generally be characterised as those driven by electrostatic attraction between Ru and polar residues (e.g. B [C-terminus], D [Asp23], E and F [Glu22], G [Glu11], H [Glu,22 Asp23], I [Asp23], K [Asp,1 Glu3], and L [Asp7]), and/or driven by phenanthroline–fibril hydrophobic interactions. For clarity, only Ru1 results are shown with the different complex coloration indicating different binding sites. See Fig. S21–23† for further information on predicted binding interactions for Ru1–3. We excluded sites with a ligand interaction (docking score) below 5 kcal mol−1. | ||
Overall, Ru1–2 are predicted to have a higher binding affinity at a larger number of sites in comparison to Ru3, with the relative binding scores detailed in Table S1.† Significant electrostatic interactions between Ru1–3 and fibril carboxylate residues are predicted (especially at sites B, D, E–I, K, and L), and the more compact Ru3 complex allows for a closer approach and a more significant interaction at certain sites (e.g. sites F, I, O). However, enhanced hydrophobic interactions are predicted for Ru1–2 containing the extended phenanthroline ligand in comparison to Ru3, and these interactions occur at the majority of binding sites on the 2MXU and 5OQV fibril surfaces (Table S1,†Fig. 8 and S23†). The docking results provide further insight into the role of the extended hydrophobic ligands of unactivated Ru1–2 over Ru3 in the interaction with the peptide, which may facilitate covalent bond formation once the complexes are photoactivated.
In order to investigate if the Ru(II) complexes could change the morphology of insoluble Aβ1–42 fibrils, we incubated the unactivated and photoactivated activated complexes with pre-formed fibrils (see Scheme 1) and monitored for a change in morphology via TEM. Our binding studies show that the intact Ru1–2 complexes have a higher affinity for Aβ1–42 fibrils in comparison to Ru3 (vide supra), and thus we hypothesized that photoactivation of the Ru1–2 complexes (and possibly Ru3) may lead to alteration of aggregate morphology. As expected, incubation of Aβ1–42 for 96 h exclusively produced mature fibrillar structures (Fig. 9 and S19†). Remarkably, we observed that after addition of the Ru(II) complexes and photoactivation, an immediate aggregate morphology change from fibrillar to amorphous is observed for Ru1 and Ru2, yet a more gradual change is observed for Ru3 (Fig. 9). No further changes were observed over an additional 24 h incubation for Ru1–2, while for Ru3 the mixture of amorphous and fibrillar aggregates observed by TEM immediately after photoactivation changes to amorphous at 24 h (Fig. 9 and S24†). Photoactivation is necessary for morphology changes, as there was no difference between pre-incubated Aβ1–42 alone and Aβ1–42 incubated for additional 24 h in the presence of unactivated Ru1–3 (Fig. 9). Even though the intact complexes display a high affinity for Aβ1–42 fibrils, especially for Ru1–2 (Fig. 7), the non-covalent interaction does not in itself lead to a change in aggregate morphology.
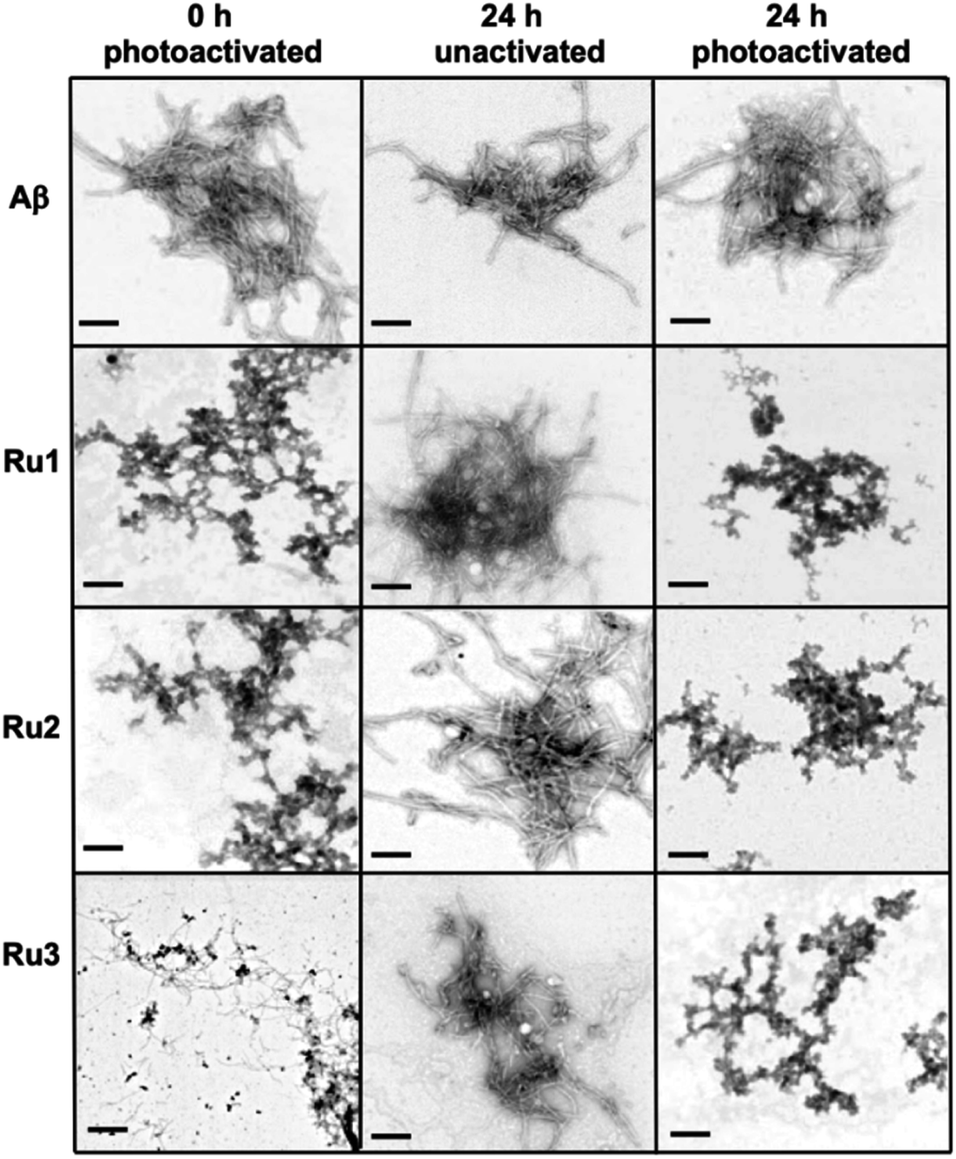 | ||
| Fig. 9 Influence of 1.0 eq. of Ru1–3 on the morphology of fibrillar Aβ1–42 (25 μM) at 0 h (photoactivated samples) and 24 h (for unactivated and photoactivated samples) (scale bar = 200 nm). | ||
Upon photoactivation, immediate changes to the peptide aggregate morphology are observed, with Ru1–2 exhibiting the most significant change in comparison to Ru3, in line with the measured binding affinities. However, all three photoactivated complexes exclusively afford amorphous aggregates at the 24 h timepoint.
Summary
This study demonstrates the ability of photoactivated Ru1–3 to target and modulate the aggregation pathway of the Aβ peptide. 1H NMR showed release of the 6,6′-dmb ligand and His residue shifts for Ru1 and Ru2, indicating that these residues are involved in the binding process. ESI-MS confirmed the release of the 6,6′-dmb ligand upon photoactivation, and also showed the presence of complex–peptide adducts for Ru1–3. Ru1–2, and to a lesser extent Ru3, significantly alter the Aβ1–42 aggregation process, with Ru1–2 promoting the formation of soluble high MW weight aggregates immediately after photoactivation. TEM analysis also shows the formation of large amorphous aggregates for Ru1–2, while the aggregates observed for Ru3 are considerably smaller. This immediate change for Ru1–2 upon photoactivation limits the formation of low MW peptide oligomers, suggesting that these complexes could bypass the formation of toxic oligomeric species.77–79 However, we have not investigated cellular toxicity at this time. After 24 h incubation, photoactivated Ru1–2 afford very little soluble Aβ1–42 as observed in the gel electrophoresis and BCA experiments, and TEM shows formation of large insoluble amorphous aggregates, in comparison to the presence of both fibrils and amorphous aggregates for peptide alone. Photoactivated Ru3 displays soluble aggregates in the 30–250 kDa size range at 24 h, however, the majority of the peptide has precipitated as indicated by the BCA assay, and large amorphous aggregates are observed by TEM, similarly for Ru1–2.All three Ru(II) complexes bind to fibrillar Aβ1–42, however, Ru1–2 display a higher affinity and molecular docking studies highlight the importance of the extended hydrophobic ligands in the interaction. Our results indicate that the complexes will be in close proximity to the peptide once the 6,6′-dmb ligand dissociates, likely favoring the formation of a covalent bond between the complexes and peptide. The docking experiments indicate that the extended hydrophobic ligands of Ru1 and Ru2 provide for an enhanced interaction with Aβ1–42 fibrils, indicating why Ru3 displays a weaker binding affinity. Extrapolating from the data obtained for Ru1–3 with fibrillar Aβ1–42 and the differences in structures of the complexes, we expect an enhanced interaction of Ru1–2 with the monomeric peptide in comparison to Ru3. Indeed, a number of similar Ru(II) complexes have been shown to interact with oligomeric species and not just fibrils.58,59 TEM images also demonstrated that the complexes are able to modify the morphology of mature fibrils after photoactivation, generating insoluble amorphous aggregates, with Ru1–2 able to induce this change much more quickly in comparison to Ru3.
In this proof-of-principle study we show that photoactivation of Ru1–3 is critical for modulating the Aβ1–42 aggregation process. The formation of amorphous aggregates in the presence of the photoactivated Ru(II) complexes is a common endpoint, either starting with monomeric peptide or fibrils. While visible light is incompatible with external activation of Ru1–3 due to limited tissue penetration, the recent development of near-infrared photoactivatable Ru complexes may provide an opportunity for this strategy in AD treatment moving forward.95–97 Overall, our results show that the extended hydrophobic ligands present in Ru1 and Ru2 enhance the peptide interaction, especially at early time points, facilitating the formation of a covalent adduct between the Ru(II) complexes and Aβ when samples are photoactivated.
Author contributions
JCB, SAM, and TimS designed the research project. JCB, LMFG, CM, JRS, HDC, JM, TariqS, and CGC completed experiments. JCB, JRS, SAM, and TimS analyzed data and wrote the manuscript.Conflicts of interest
S. A. M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council (NSERC) Discovery Grant RGPIN-2019-06749 (T. S.), a Michael Smith Career Investigator Award (T. S.), and Science without borders (CAPES – Proc. no. 0711/13-6, Brazil to L. M. F. G.). Compute Canada is thanked for access to computational resources. Dr Mariusz Mital is thanked for initial experiments. Research reported in this publication was supported in part by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under Award Number R01CA222227 (to SAM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- https://www.alz.co.uk/research/world .
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595 CrossRef CAS PubMed.
- D. J. Selkoe, Nat. Med., 2011, 17, 1693 CrossRef CAS.
- A. Abbott, Nature, 2008, 456, 161 CrossRef CAS PubMed.
- B. Zott, M. M. Simon, W. Hong, F. Unger, H. J. Chen-Engerer, M. P. Frosch, B. Sakmann, D. M. Walsh and A. Konnerth, Science, 2019, 365, 559 CrossRef CAS PubMed.
- R. Nortley, N. Korte, P. Izquierdo, C. Hirunpattarasilp, A. Mishra, Z. Jaunmuktane, V. Kyrargyri, T. Pfeiffer, L. Khennouf, C. Madry, H. Gong, A. Richard-Loendt, W. Huang, T. Saito, T. C. Saido, S. Brandner, H. Sethi and D. Attwell, Science, 2019, 365 Search PubMed.
- C. A. McLean, R. A. Cherny, F. W. Fraser, S. J. Fuller, M. J. Smith, K. Beyreuther, A. I. Bush and C. L. Masters, Ann. Neurol., 1999, 46, 860 CrossRef CAS PubMed.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A. U. S. A., 1985, 82, 4245 CrossRef CAS PubMed.
- M. Mital, N. E. Wezynfeld, T. Frączyk, M. Z. Wiloch, U. E. Wawrzyniak, A. Bonna, C. Tumpach, K. J. Barnham, C. L. Haigh, W. Bal and S. C. Drew, Angew. Chem., Int. Ed., 2015, 54, 10460 CrossRef CAS PubMed.
- C. Esmieu, G. Ferrand, V. Borghesani and C. Hureau, Chem.–Eur. J., 2021, 27, 1777–1786 CrossRef CAS PubMed.
- L.-K. Huang, S.-P. Chao and C.-J. Hu, J. Biomed. Sci., 2020, 27, 18 CrossRef CAS PubMed.
- M. Gold, Alzheimer's Dementia, 2017, 3, 402 CrossRef PubMed.
- J. Cummings, P. S. Aisen, B. DuBois, L. Frölich, C. R. Jack Jr, R. W. Jones, J. C. Morris, J. Raskin, S. A. Dowsett and P. Scheltens, Alzheimer's Res. Ther., 2016, 8, 39 CrossRef PubMed.
- K. Ritchie, C. W. Ritchie, K. Yaffe, I. Skoog and N. Scarmeas, Alzheimer's Dementia, 2015, 1, 122 CrossRef PubMed.
- D. H. Cho, T. Nakamura, J. Fang, P. Cieplak, A. Godzik, Z. Gu and S. A. Lipton, Science, 2009, 324, 102 CrossRef CAS PubMed.
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 67, 67–73 CrossRef PubMed.
- K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727 RSC.
- C. Cheignon, M. Jones, E. Atrián-Blasco, I. Kieffer, P. Faller, F. Collin and C. Hureau, Chem. Sci., 2017, 8, 5107 RSC.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS PubMed.
- A. K. Nath, A. Ghatak, A. Dey and S. G. Dey, Chem. Sci., 2021, 12, 1924 RSC.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836 RSC.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885 CrossRef CAS PubMed.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701 RSC.
- J.-M. Suh, G. Kim, J. Kang and M. H. Lim, Inorg. Chem., 2019, 58, 8 CrossRef CAS PubMed.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412 Search PubMed.
- H. Liu, Y. Qu and X. Wang, Future Med. Chem., 2018, 10, 679 CrossRef CAS PubMed.
- F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130 RSC.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404 RSC.
- L. M. F. Gomes, A. Mahammed, K. E. Prosser, J. R. Smith, M. A. Silverman, C. J. Walsby, Z. Gross and T. Storr, Chem. Sci., 2019, 10, 1634 RSC.
- D. E. S. Silva, M. P. Cali, W. M. Pazin, E. Carlos-Lima, M. T. Salles Trevisan, T. Venâncio, M. Arcisio-Miranda, A. S. Ito and R. M. Carlos, J. Med. Chem., 2016, 59, 9215 CrossRef CAS PubMed.
- N. A. Vyas, S. S. Bhat, A. S. Kumbhar, U. B. Sonawane, V. Jani, R. R. Joshi, S. N. Ramteke, P. P. Kulkarni and B. Joshi, Eur. J. Med. Chem., 2014, 75, 375 CrossRef CAS PubMed.
- J. S. Derrick, J. Lee, S. J. C. Lee, Y. Kim, E. Nam, H. Tak, J. Kang, M. Lee, S. H. Kim, K. Park, J. Cho and M. H. Lim, J. Am. Chem. Soc., 2017, 139, 2234 CrossRef CAS PubMed.
- J. Suh, S. H. Yoo, M. G. Kim, K. Jeong, J. Y. Ahn, M.-s. Kim, P. S. Chae, T. Y. Lee, J. Lee, J. Lee, Y. A. Jang and E. H. Ko, Angew. Chem., Int. Ed., 2007, 46, 7064 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813 CrossRef CAS PubMed.
- G. Ma, F. Huang, X. Pu, L. Jia, T. Jiang, L. Li and Y. Liu, Chem.–Eur. J., 2011, 17, 11657 CrossRef CAS PubMed.
- V. A. Streltsov, V. Chandana Epa, S. A. James, Q. I. Churches, J. M. Caine, V. B. Kenche and K. J. Barnham, Chem. Commun., 2013, 49, 11364 RSC.
- M. Turner, J. A. Platts and R. J. Deeth, J. Chem. Theory Comput., 2016, 12, 1385 CrossRef CAS PubMed.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917 RSC.
- L. Lu, H.-J. Zhong, M. Wang, S.-L. Ho, H.-W. Li, C.-H. Leung and D.-L. Ma, Sci. Rep., 2015, 5, 14619 CrossRef CAS PubMed.
- J. Kang, S. J. C. Lee, J. S. Nam, H. J. Lee, M.-G. Kang, K. J. Korshavn, H.-T. Kim, J. Cho, A. Ramamoorthy, H.-W. Rhee, T.-H. Kwon and M. H. Lim, Chem.–Eur. J., 2017, 23, 1645 CrossRef CAS PubMed.
- J. Kang, J. S. Nam, H. J. Lee, G. Nam, H.-W. Rhee, T.-H. Kwon and M. H. Lim, Chem. Sci., 2019, 10, 6855 RSC.
- T. Mede, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2018, 47, 7577 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC.
- A. Rilak Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727 CrossRef CAS PubMed.
- E. C. Glazer, Isr. J. Chem., 2013, 53, 391 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797 CrossRef CAS PubMed.
- K. Arora, M. Herroon, M. H. Al-Afyouni, N. P. Toupin, T. N. Rohrabaugh, L. M. Loftus, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2018, 140, 14367 CrossRef CAS PubMed.
- A. Li, R. Yadav, J. K. White, M. K. Herroon, B. P. Callahan, I. Podgorski, C. Turro, E. E. Scott and J. J. Kodanko, Chem. Commun., 2017, 53, 3673 RSC.
- T. N. Rohrabaugh, A. M. Rohrabaugh, J. J. Kodanko, J. K. White and C. Turro, Chem. Commun., 2018, 54, 5193 RSC.
- D. Havrylyuk, K. Stevens, S. Parkin and E. C. Glazer, Inorg. Chem., 2020, 59, 1006 CrossRef CAS PubMed.
- D. Havrylyuk, M. Deshpande, S. Parkin and E. C. Glazer, Chem. Commun., 2018, 54, 12487 RSC.
- G. Son, B. I. Lee, Y. J. Chung and C. B. Park, Acta Biomater., 2018, 67, 147 CrossRef CAS PubMed.
- N. P. Cook, M. Ozbil, C. Katsampes, R. Prabhakar and A. A. Martí, J. Am. Chem. Soc., 2013, 135, 10810 CrossRef CAS PubMed.
- B. Jiang, A. Aliyan, N. P. Cook, A. Augustine, G. Bhak, R. Maldonado, A. D. Smith McWilliams, E. M. Flores, N. Mendez, M. Shahnawaz, F. J. Godoy, J. Montenegro, I. Moreno-Gonzalez and A. A. Martí, J. Am. Chem. Soc., 2019, 141, 15605 CrossRef CAS PubMed.
- A. Aliyan, N. P. Cook and A. A. Martí, Chem. Rev., 2019, 119, 11819 CrossRef CAS PubMed.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, J. Inorg. Biochem., 2016, 158, 45 CrossRef CAS PubMed.
- J. Roque III, D. Havrylyuk, P. C. Barrett, T. Sainuddin, J. McCain, K. Colón, W. T. Sparks, E. Bradner, S. Monro, D. Heidary, C. G. Cameron, E. C. Glazer and S. A. McFarland, Photochem. Photobiol., 2020, 96, 327 CrossRef PubMed.
- A. Aliyan, B. Kirby, C. Pennington and A. A. Martí, J. Am. Chem. Soc., 2016, 138, 8686 CrossRef CAS PubMed.
- Analysis method reported here: J. Roque III, D. Havrylyuk, P. C. Barrett, T. Sainuddin, J. McCain, K. Colón, W. T. Sparks, E. Bradner, S. Monro, D. Heidary, C. G. Cameron, E. C. Glazer and S. A. McFarland, Photochem. Photobiol., 2020, 96(2), 327–339 CrossRef PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
- D. Hesek, Y. Inoue, S. R. L. Everitt, H. Ishida, M. Kunieda and M. G. B. Drew, J. Chem. Soc., Dalton Trans., 1999, 3701 RSC.
- J. L. Walsh and B. Durham, Inorg. Chem., 1982, 21, 329 CrossRef CAS.
- B. J. Coe, T. J. Meyer and P. S. White, Inorg. Chem., 1993, 32, 4012 CrossRef CAS.
- L. M. F. Gomes, J. C. Bataglioli, A. J. Jussila, J. R. Smith, C. J. Walsby and T. Storr, Front. Chem., 2019, 7, 1–13 CrossRef PubMed.
- X. Wang, X. Wang, C. Zhang, Y. Jiao and Z. Guo, Chem. Sci., 2012, 3, 1304 RSC.
- A. A. Almaqwashi, W. Zhou, M. N. Naufer, I. A. Riddell, Ö. H. Yilmaz, S. J. Lippard and M. C. Williams, J. Am. Chem. Soc., 2019, 141, 1537 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101 CrossRef CAS.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193 CrossRef CAS.
- Y. S. Gong, L. Chang, K. L. Viola, P. N. Lacor, M. P. Lambert, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10417 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172 CrossRef CAS PubMed.
- L. M. Gomes, R. P. Vieira, M. R. Jones, M. C. Wang, C. Dyrager, E. M. Souza-Fagundes, J. G. Da Silva, T. Storr and H. Beraldo, J. Inorg. Biochem., 2014, 139, 106 CrossRef CAS PubMed.
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636 RSC.
- F. G. De Felice, P. T. Velasco, M. P. Lambert, K. Viola, S. J. Fernandez, S. T. Ferreira and W. L. Klein, J. Biol. Chem., 2007, 282, 11590 CrossRef CAS PubMed.
- E. Y. Hayden and D. B. Teplow, Alzheimer's Res. Ther., 2013, 5, 60 CrossRef PubMed.
- W. L. Klein, J. Alzheimer's Dis., 2013, 33(Suppl 1), S49 Search PubMed.
- E. C. Johnson, B. P. Sullivan, D. J. Salmon, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1978, 17, 2211 CrossRef CAS.
- T. B. Karpishin, M. W. Grinstaff, S. Komar-Panicucci, G. McLendon and H. B. Gray, Structure, 1994, 2, 415 CrossRef CAS PubMed.
- K. Sigfridsson, M. Sundahl, M. J. Bjerrum and O. Hansson, J. Biol. Inorg. Chem., 1996, 1, 405 CrossRef CAS.
- D. S. Wuttke, M. J. Bjerrum, I. J. Chang, J. R. Winkler and H. B. Gray, Biochim. Biophys. Acta, 1992, 1101, 168 CAS.
- Y. Huang, H.-J. Cho, N. Bandara, L. Sun, D. Tran, B. E. Rogers and L. M. Mirica, Chem. Sci., 2020, 11, 7789 RSC.
- Y. Yi, Y. Lin, J. Han, H. J. Lee, N. Park, G. Nam, Y. S. Park, Y.-H. Lee and M. H. Lim, Chem. Sci., 2021, 12, 2456 RSC.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76 CrossRef CAS PubMed.
- Y. F. Mok and G. J. Howlett, Methods Enzymol., 2006, 413, 199 CAS.
- O. J. Rolinski, T. Wellbrock, D. J. S. Birch and V. Vyshemirsky, J. Chem. Phys. Lett., 2015, 6, 3116 CrossRef CAS.
- A. Alghamdi, V. Vyshemirsky, D. J. S. Birch and O. J. Rolinski, Methods Appl. Fluoresc., 2018, 6, 024002 CrossRef CAS PubMed.
- M. Amaro, D. J. S. Birch and O. J. Rolinski, Phys. Chem. Chem. Phys., 2011, 13, 6434 RSC.
- H. LeVine 3rd, Amyloid, 2005, 12, 5 CrossRef PubMed.
- M. Groenning, J. Chem. Biol., 2010, 3, 1 CrossRef PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499 CrossRef CAS PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116 CrossRef CAS PubMed.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 3262 CrossRef CAS PubMed.
- L. M. Lifshits, J. A. Roque III, P. Konda, S. Monro, H. D. Cole, D. von Dohlen, S. Kim, G. Deep, R. P. Thummel, C. G. Cameron, S. Gujar and S. A. McFarland, Chem. Sci., 2020, 11, 11740 RSC.
- M. H. Al-Afyouni, T. N. Rohrabaugh, K. F. Al-Afyouni and C. Turro, Chem. Sci., 2018, 9, 6711 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00004g |
| This journal is © The Royal Society of Chemistry 2021 |