
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-control of cap methylation and mRNA translation via genetic code expansion of Ecm1†
Dennis
Reichert
ab,
Henning D.
Mootz
 a and
Andrea
Rentmeister
*ab
a and
Andrea
Rentmeister
*ab
aDepartment of Chemistry, Institute of Biochemistry, University of Münster, Correnstr. 36, 48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
bCells in Motion Interfaculty Center, University of Münster, 48149 Münster, Germany
First published on 8th February 2021
Abstract
Gene expression is tightly regulated in all domains of life, with post-transcriptional regulation being more pronounced in higher eukaryotes. Optochemical and optogenetic approaches enable the actuation of many underlying processes by light, which is an excellent tool to exert spatio-temporal control. However, light-mediated control of eukaryotic mRNA processing and the respective enzymes has not been reported. We used genetic code expansion to install a photo-caged tyrosine (Y) in the active site of the cap methyltransferase Ecm1. This enzyme is responsible for guanine N7 methylation of the 5′ cap, which is required for translation. Substituting Y284 with the photocaged ortho-nitrobenzyl-tyrosine (ONBY) almost completely abrogated the methylation activity of Ecm1. Irradiation with light removed the ONB group, restoring the native tyrosine and Ecm1 activity, yielding up to 97% conversion of the minimal substrate GpppA within 60 min after activation. Using luciferase- and eGFP-mRNAs as reporters, we could show that light actuates translation by inducing activation of Ecm1 ONBY284 in a eukaryotic in vitro translation system.
Introduction
Gene expression is tightly regulated and comprises all steps of the central dogma of molecular biology.1,2 To investigate and understand the underlying mechanisms or interfere with them for therapeutic purposes, it is necessary to manipulate the individual processes with high precision. Light can be controlled with high spatio-temporal precision and proves useful to dissect individual processes if they can be rendered light-susceptible. Optogenetic and optochemical methods have emerged as versatile approaches for controlling biological activity by light.3,4 In order to control gene expression, photosensitive proteins were coupled to the CRISPR-Cas9 system and RNA-binding proteins.5,6 Similarly, genetic code expansion was used to regulate the activity of enzymes involved in gene expression, most notably polymerases7 and Cas9.8 In a different set of approaches, direct optical control of gene expression was achieved by introducing photo-caging groups at nucleotides9,10 and nucleic acids,11–14 by chemical or chemo-enzymatic methods. Most of these approaches have focused on the powerful regulation by transcription and RNA-induced silencing, and few on translation.15,16In eukaryotes, mRNA is heavily processed, including 5′ capping, splicing, and polyadenylation. While the importance of post-transcriptional regulation in higher eukaryotes is well-known, optogenetic and optochemical approaches for controlling the respective processes are scarce.17,18 The chemical or chemo-enzymatic preparation of photocaged RNA to achieve direct optical control of RNA-guided processes (siRNAs, miRNAs, or derivatives, most notably morpholinos)19–22 is a powerful approach, but tedious for long RNAs that are not directly accessible by chemical synthesis. The photocaging groups are at least stoichiometric (i.e. one or more per RNA molecule) to achieve the desired effect and require sufficient irradiation for removal.
Therefore, we anticipated that instead of photocageing the RNA molecule, it would be advantageous to photocage the responsible RNA-modifying enzyme for controlling mRNA processing steps by light.
The mRNA from in vitro transcription could then be used directly, circumventing any chemical or chemo-enzymatic treatment. Furthermore, the RNA-modifying enzyme is catalytic, meaning that photo-deprotection of one enzyme would actuate modification of multiple mRNA molecules. Short irradiation – to generate catalytic amounts of active RNA-modifying enzyme – would be sufficient, which is desirable to reduce potential photo-damage of RNA.
Fundamental processing steps of eukaryotic mRNA are 5′ capping, splicing, and polyadenylation. Capping consists of the consecutive action of three enzymes, a triphosphatase, a guanylyltransferase, and a guanine N7 methyltransferase (MTase).23 The cap is required for translation initiation as it interacts directly with the eukaryotic translation initiation factor (eIF4E).24 Methylation of the cap structure on N7 of the guanine base is essential for its recognition by cap-binding proteins.25 The eIF4E discriminates between methylated and unmethylated caps by a remarkable factor of >100.26,27 We and others showed that cap methylation is critical for the translation of eukaryotic mRNA,28–32 suggesting that this final step of cap formation might be suitable to bring translation under control of light. In this work, we thus aimed to control the function of the RNA 5′ cap by triggering site-specific enzymatic methylation by light.
To achieve this, we chose the guanine N7 MTase Ecm1 that can be efficiently produced in E. coli and that we used previously to generate functional 5′ caps.30,32–35 We sought to inhibit the activity of Ecm1 by incorporating sterically demanding but photo-cleavable groups in the active site. To generate such “caged” proteins, the genetic code can be expanded, allowing for the site-specific incorporation of non-natural “caged” amino acids into proteins.36–38 Irradiation should then lead to photo-deprotection and release the active MTase (Scheme 1). If this process can be carried out in the presence of an unmethylated mRNA (GpppG-mRNA), the irradiation would trigger the translation of the respective transcript without requiring the installation of photo-caging groups at the mRNA.
 | ||
| Scheme 1 Concept of light-triggered cap methylation and mRNA translation. Genetic code expansion is used to install the non-natural amino acid ONBY, a tyrosine with a photo-caging (PC) group in the active site of the cap methyltransferase Ecm1. Without methylation, mRNA with a GpppG-cap is not translated, yielding low luciferase activity. Irradiation with UV-light removes the PC group and activates Ecm1, resulting in cap-methylated mRNA and efficient translation. | ||
Results and discussion
To engineer light-inducible methyltransferase activity, we sought to introduce a non-natural amino acid with a photo-cleavable group in the substrate binding pocket of Ecm1. Cap-binding is often realized by stacking interactions in the active site of the enzyme.39 Sequence alignment of guanine N7 cap MTases from different organisms revealed three highly conserved tyrosines (Fig. 1A), denoted Y145, Y212, and Y284 in Ecm1. All of them are located in the active site, pointing towards the substrate (Fig. 1B). Y284 directly coordinates the cap guanosine at the N3G position, whereas Y145 and Y212 – together with E225 – contribute to hydrogen bonding networks interacting with the N1 and O6 position, respectively (Fig. 1C). All three residues are essential for Ecm1 activity, as established in previous studies.40,41 We therefore chose to harness these favorably placed tyrosines with a photo-cleavable ortho-nitrobenzyl group42 to block substrate binding and Ecm1 activity. To incorporate o-nitrobenzyl tyrosine (ONBY) site-specifically in Ecm1, we used amber stop codon suppression and a previously reported tyrosyl–tRNA synthetase from Methanococcus jannaschii (MjTyrRS) with its cognate MjtRNATyr.42 Ecm1 variants ONBY145, ONBY212, and ONBY284 were recombinantly produced in E. coli K12 UT5600, which minimizes the potential cytoplasmic reduction of the nitro to an amino group and therefore allows higher photo-deprotection yield of ONBY.43 As expected, full-length variants Ecm1 ONBY145, ONBY212, and ONBY284 were only obtained when the amino acid ONBY was added, according to an analysis of the crude lysate by PAA-gel and western blot (Fig. 2A). For the purified Ecm1 variants, the mass spectrometric analysis by ESI-TOF mass spectrometry detected a mass peak predominantly for the ONBY-containing Ecm1 and only a minor peak for the uncaged protein (Fig. 2B and S3†). These data show that the incorporation of ONBY was successful and efficient for all three variants. Furthermore, ONBY in Ecm1 remained stable during expression and purification. | ||
| Fig. 1 Identification of suitable incorporation sites. (A) Sequence alignment of mRNA cap guanine N7 methyltransferases from selected eukaryotic organisms. Three regions involved in substrate binding are shown. (B) Crystal structure of Ecm1 showing conserved regions and tyrosine residues important for cap guanine binding (PDB 1RI2). Water molecules are indicated as red spheres. (C) Schematic overview of guanine cap and SAM coordination in Ecm1. Dashed lines indicate hydrogen bonds. Conserved residues with an asterisk. | ||
 | ||
Fig. 2 Characterization Ecm1 ONBY variants (A) SDS-PAA gel showing samples of the crude cell lysate from E. coli cells grown in the presence or absence of ONBY. Arrow points to the expected length of Ecm1. Lower panel: the presence of full-length protein was confirmed by western blots using an anti-His antibody. (B) LC/MS analysis of purified Ecm1 variants. Deconvoluted protein mass (calc. 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 365.36 Da) was detected, confirming the incorporation of ONBY at the respective positions. (C) Reaction catalyzed by Ecm1. The 5′ cap analog GpppA (1a) is the minimal substrate, SAM (2a) is the cosubstrate. (D) Time courses of the reaction shown in (C) with Ecm1 WT or indicated ONBY variants. Data and error bars show average and standard deviations from biotransformations using two enzyme batches, each performed in duplicates (n = 4). (E) Model of Ecm1 with ONBY or natural tyrosine (Y) at position 284 before (top) or after irradiation (bottom), in complex with GTP and SAM (shown in sticks). Dashed lines indicate potential hydrogen bonds (based on PDB 1RI2). 365.36 Da) was detected, confirming the incorporation of ONBY at the respective positions. (C) Reaction catalyzed by Ecm1. The 5′ cap analog GpppA (1a) is the minimal substrate, SAM (2a) is the cosubstrate. (D) Time courses of the reaction shown in (C) with Ecm1 WT or indicated ONBY variants. Data and error bars show average and standard deviations from biotransformations using two enzyme batches, each performed in duplicates (n = 4). (E) Model of Ecm1 with ONBY or natural tyrosine (Y) at position 284 before (top) or after irradiation (bottom), in complex with GTP and SAM (shown in sticks). Dashed lines indicate potential hydrogen bonds (based on PDB 1RI2). | ||
With the purified Ecm1 variants in hand, we investigated how ONBY at different positions affects the enzymatic activity. To this end, we performed biotransformations of the 5′ cap analog GpppA (1a) with S-adenosyl-L-methionine (AdoMet or SAM, 2a) and Ecm1, yielding m7GpppA (1b) (Fig. 2C). HPLC analysis at different time points showed that the wild type enzyme (WT) led to almost complete conversion (91%) of 1a within 120 min (Fig. 2C), whereas all Ecm1 variants with ONBY showed diminished activity. Variants ONBY145 and ONBY212 led to 64% and 23% conversion, respectively (Fig. 2C) under otherwise identical conditions. In particular, substituting Y284 by ONBY almost completely abrogated Ecm1 activity—only 4% of GpppA were converted. To compare the relative activity of the Ecm1 variants, we determined the kinetic parameters for the cap analog 1a (Fig. S2†) and calculated the kcat/KM values with respect to wild type Ecm1 (100%). All Ecm1 variants exhibited significantly reduced relative activity, ranging from 8–47%, confirming the trend observed in the conversion (rel. activity: ONBY284 < ONBY212 < ONBY145). This observation can be rationalized based on the Ecm1 crystal structure (Fig. 2E and S1†). Here, ONBY at position 284 extends into the substrate binding pocket, replacing the substrate GTP and explaining the pronounced effect of this substitution. Taken together, these data show that substituting the tyrosines in the active site is a valid strategy for abrogating the activity of a cap MTase.
Next, we investigated whether the “caged” activity in Ecm1-ONBY variants could be resurrected by UV light. When we irradiated purified Ecm1 ONBY variants with light using an UV-A LED (3 W, λmax = 365 nm) for 2 min, we found that the o-nitrobenzyl group (ONB) was removed entirely according to ESI-TOF mass spectrometry analysis (Fig. 3A and S3†). To keep potential photo-damage in a biological setup as low as possible, we optimized the irradiation conditions and chose to irradiate for 1 min, which yields 80% of deprotection (Fig. 3B and S4†).
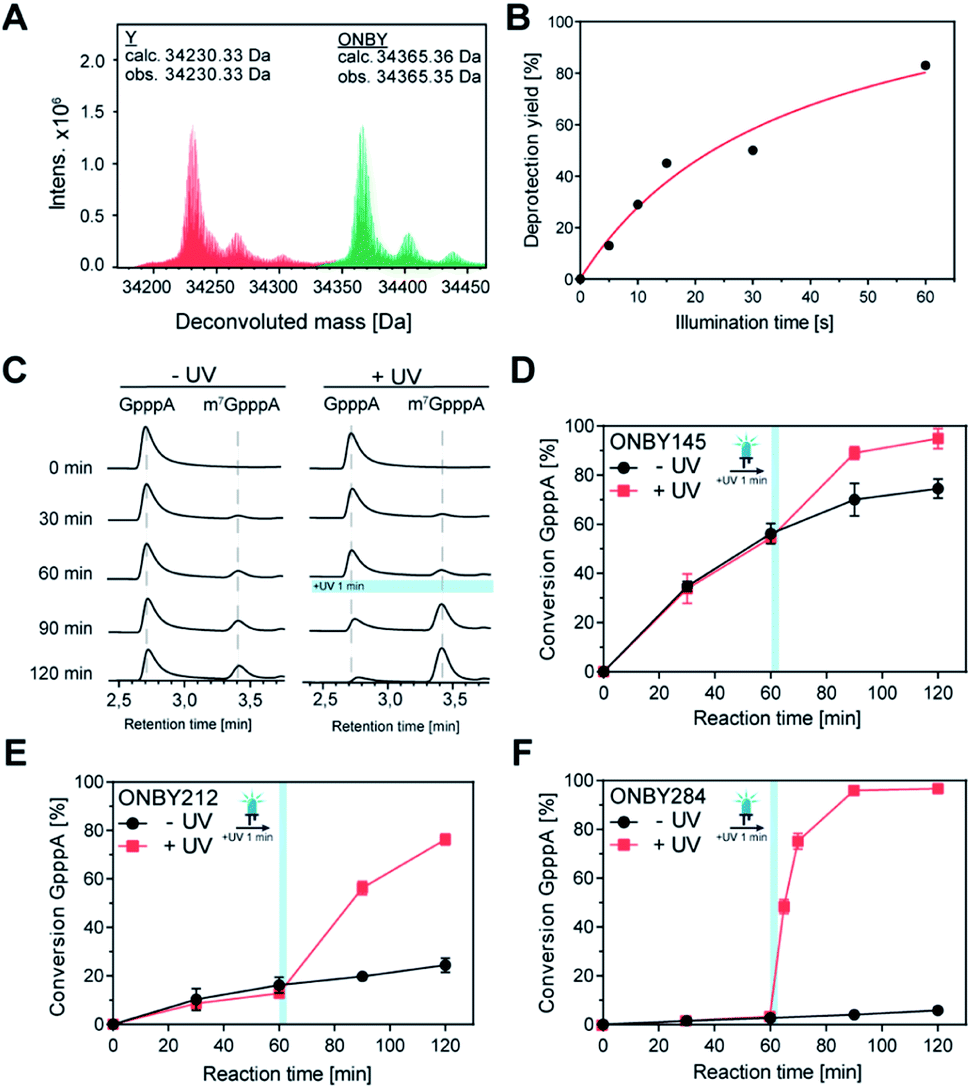 | ||
| Fig. 3 Light-induced activation of Ecm1 ONBY variants. (A) LC/MS analysis of representative purified Ecm1 variant (ONBY284). After irradiation with UV light (365 nm), the monoisotopic mass of the wild type enzyme was detected 34230.33 Da (calc. 34230.33 Da). (B) Photo-deprotection depending on irradiation time for Ecm1 ONBY212. (C) HPLC analysis of the light-induced reaction of Ecm1 ONBY212. Reactions were either kept in the dark (−UV) or irradiated after 60 min (+UV) for 1 min. (D–F) Time-courses of light-induced activation of Ecm1 variants ONBY145, ONBY212, and ONBY284, respectively. Data and error bars show the average and standard deviation of three individual experiments (n = 3). | ||
To assess whether photo-deprotection of Ecm1 will restore the MTase activity, we performed the biotransformations as described above, split them into two parts, and – after 60 min reaction time – irradiated one half for 1 min at 365 nm. HPLC analyses showed little or no formation of m7GpppA (1b) in the absence of UV light and a significant increase starting after irradiation for Ecm1 ONBY212 (Fig. 3C) as well as ONBY145 and ONBY284 (Fig. S5†). Time-dependent measurements were performed for all three Ecm1 variants before and after irradiation (Fig. 3D–F and S5†). The activity was increased after irradiation for all three variants, confirming that Ecm1 activity can be resurrected. To our delight, variant Ecm1 ONBY284 showed a remarkable turn-on effect, yielding almost complete conversion (97%) 1 hour after irradiation, whereas the sample that was kept in the dark showed only 5% conversion. For ONBY145 and ONBY212, we observed conversions of 95% vs. 74% and 76% vs. 24%, respectively. This comparison indicated an activation factor (active/inactive) of 19.4 fold for ONBY284, 3.2-fold for ONBY212, and 1.3-fold for ONBY145, respectively.
Encouraged by these results and knowing that guanine N7 methylation of the cap is crucial for efficient translation, we were curious to determine whether cap methylation could be used to trigger the translation of mRNAs. To test this, we had to develop an assay for in situ methylation of long capped mRNAs and detection of translation output. To this point, we could show that Ecm1 can be used to introduce the guanine N7 methylation to produce a cap-4 RNA, which displays seven distinguished additional methylations within the first four nucleotides35 and modified dinucleotide caps (e.g.1b), which can be applied for in vitro transcription. In a next step, these can be used to assess the effect of cap modifications on translation in a quantitative in vitro translation assay (IVTL).30 However, the in situ modification of long mRNAs with unmethylated cap (GpppG-mRNA) to start their translation had not been assessed.
To test whether long mRNAs can be enzymatically modified using wild type Ecm1, we enzymatically transferred a 4-azido-but-2-enyl moiety to a luciferase-mRNA (GpppG-FLuc) using AbSAM (2c, Fig. S7A†), as a cosubstrate.30 The azido group was then conjugated with a fluorescent dye, using DBCO-SRB in a strain-promoted alkyne–azide cycloaddition (SPAAC) (Fig. S6A†). As a negative control, an mRNA with an anti-reverse cap analog (ARCA 1e, Fig. S6†) – which is already methylated and should not be a substrate of Ecm1 – was produced and subjected to the same procedure. In-gel analysis showed a fluorescent band for Ecm1-treated GpppG-mRNA, indicating that a long GpppG-capped mRNA can be efficiently modified by Ecm1 (Fig. S7B†). As expected, the ARCA-capped mRNA was not labeled.
Next, we modified the GpppG-FLuc-mRNA using wild type Ecm1 and SAM (2a) and tested the effect on translation in a eukaryotic cell free expression system from Reticulocyte lysate (Fig. S8A and B†). We used a dual luciferase assay to compare the translation efficiency of differently capped FLuc-mRNAs and referenced signal to ARCA-capped-RLuc-mRNA. In the absence of Ecm1, the luciferase activity of GpppG-FLuc-mRNA was 10% of the positive control m7GpppG-FLuc, containing completely methylated cap (Fig. S8B†). This is in the range of the negative control, ApppG-FLuc-mRNA (7%), which does not undergo cap-dependent translation and indicates the background signal. To our delight, we observed that methylation by Ecm1 drastically increased the translation of GpppG-FLuc-mRNA, yielding even 90% of the luciferase activity of the positive control (Fig. S8B†). We validated the cap modification after digestion of the mRNA by nuclease P1 using LC-QQQ-MS. After enzymatic modification by Ecm1, 65% m7GpppG were detected in the reaction sample, whereas only GpppG was detected in the control, treated with denatured Ecm1 (Fig. S8C†). Furthermore, we were able to extend our approach to other reporter systems using fluorescent proteins (Fig. S9†). We could show that the protein levels produced in the translation mix are comparable for enzymatically modified GpppG-eGFP and m7GpppG-eGFP control mRNA (Fig. S9†).
Finally, we combined both GpppG-FLuc and ARCA-RLuc with wild type Ecm1 and SAM (2a) in the Reticulocyte lysate and monitored if in situ methylation of the reporter mRNA leads to increased translation (Fig. 4A). Analyzing samples at different time points clearly showed that in situ methylation of GpppG-capped-mRNA increased translation (Fig. 4B). After 90 min a 16-fold higher luminescence signal was observed in comparison to the ApppG-FLuc signal or the control with inactivated Ecm1 (Fig. 4B). These data show that Ecm1 can modify GpppG-capped mRNA and that this in situ methylation directly increases translation.
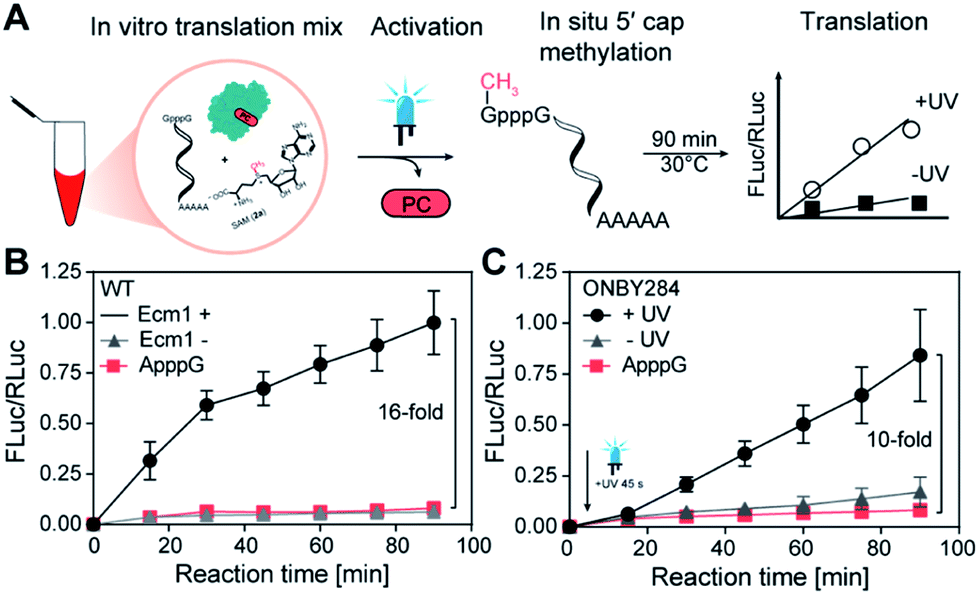 | ||
| Fig. 4 Light-controlled cap methylation and translation of FLuc-mRNA. (A) Scheme illustrating light-dependent in situ methylation of GpppG-FLuc mRNA. (B) Effect of in situ methylation of GpppG-mRNA with Ecm1 on translation. Time course of FLuc luminescence shows the translation of GpppG-FLuc-mRNAs in the presence/absence of Ecm1 or ApppG-FLuc-mRNA as control. Values are normalized to RLuc signal from translation of ARCA-RLuc-mRNA. (C) GpppG-FLuc-mRNA incubated with Ecm1 ONBY284. Samples irradiated for 45 s after 2 min reaction time (+UV) or non-irradiated (−UV) are shown in comparison to ApppG-FLuc control. Data and error bars show average and standard deviation from four independent experiments (n = 4). | ||
With this assay in hand, we wanted to test the effect of light on the translation of GpppG-mRNAs in the presence of Ecm1 variants with ONBY. We chose Ecm1 variant ONBY284, which was almost completely inactive and regained high activity after light-induced cleavage of the ONB group. We performed the IVTL assay, as described above. However, now we irradiated 2 min after mixing all reaction components, including Ecm1 ONBY284 and the Reticulocyte lysate (Fig. 4C). Analysis of samples taken at different time points showed a 10-fold increase in translation when the lysate had been exposed to UV light (365 nm, 45 s) (Fig. 4C). Importantly, when kept in the dark, the sample containing Ecm1 ONBY284 yielded only a low luminescence signal, slightly above the negative control containing ApppG-Fluc-mRNA. The slight difference between the Ecm1 ONBY284 containing samples kept in the dark and the negative control can be attributed to the background MTase activity, as observed in the studies with purified components (Fig. 3D–F and S5†). Taken together, the incorporation of ONBY at position Y284 is suitable for light-controlled cap methylation and actuation of translation.
Conclusions
In summary, a light-activated mRNA cap MTase from E. cuniculi (Ecm1) was successfully developed using a genetically encoded caged tyrosine amino acid (ONBY). Amber stop codon suppression in combination with ONBY has already proven successful in activating several fundamental processes in molecular biology by light. However, to the best of our knowledge, this is the first example of optochemical activation of an mRNA-processing enzyme and a new approach to trigger translation without requiring chemical modification of nucleic acids.We developed a dual luciferase assay to quantitatively assess the effect of in situ cap methylation of eukaryotic mRNAs on translation. This assay showed a 16-fold difference in the presence and absence of Ecm1—in line with the difference obtained for positive and negative control (i.e. mRNAs with m7GpppG- vs. ApppG-cap).
We identified and tested possible target sites for incorporation of ONBY in Ecm1 and observed different effects on the catalytic activity of the enzyme. In Ecm1 ONBY284, the activity was almost completely abrogated, resulting in only 4% of conversion in a 2 h reaction. After irradiation, the MTase activity immediately increased to yield (1) nearly complete conversion in 1 h (97%) – a 20-fold turn-on effect for the substrate GpppA – or (2) a 10-fold turn-on effect for translation if performed with mRNA.
Our approach capitalizes on the fundamental impact of cap methylation on recognition by cap-binding proteins and demonstrates the potential of controlling methylation.25 Many cap-interacting proteins and RNA-modifying enzymes use π-stacking and hydrogen bonding for substrate recognition and contain tyrosine residues directly in the active site.39 For instance, a crystal structure of the catalytic core of the human m6A writer complex comprising METTL3 and METTL14 also reveals tyrosine residues essential for the coordination of the acceptor adenine binding in the METTL3 active site.44 We therefore think that our approach can be applied to numerous RNA-modifying enzymes and prove useful to dissect the intricate mRNA-processing steps using light as a tool that can be controlled with high spatio-temporal precision. In addition, DNA N-MTases share a highly conserved Asn/Asp/Ser-Pro-Tyr/Phe motif, suggesting that our strategy can be readily transferred to other MTases.45,46
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank A. M. Lawernce-Dörner, Dr W. Dörner and S. Wulff for technical assistance and protein LC/MS analyses. Dr F. Höhn and the technical workshop for providing and building custom-made LED boxes. Z. Yilmaz & B. Jedlitzke are acknowledged for helpful discussions during writing of the manuscript. The pEVOL plasmid encoding tyrosyl–tRNA synthetase/tRNA pair was kindly provided by Peter G. Schultz Lab. A. R. gratefully acknowledges funding by the DFG (SFB858, SFB1459, RE2796/6-1, RE2796/7-1). D. R. is a member of CiM-IMPRS, the joint graduate school of the Cells-in-Motion Interfaculty Center (CiM), University of Münster, Germany and the International Max Planck Research School – Molecular Biomedicine, Münster, Germany.Notes and references
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33, 245–254 CrossRef CAS.
- G. S. Wilkie, K. S. Dickson and N. K. Gray, Trends Biochem. Sci., 2003, 28, 182–188 CrossRef CAS.
- A. Gautier, C. Gauron, M. Volovitch, D. Bensimon, L. Jullien and S. Vriz, Nat. Chem. Biol., 2014, 10, 533–541 CrossRef CAS.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS.
- A. M. Weber, J. Kaiser, T. Ziegler, S. Pilsl, C. Renzl, L. Sixt, G. Pietruschka, S. Moniot, A. Kakoti, M. Juraschitz, S. Schrottke, L. L. Bryant, C. Steegborn, R. Bittl, G. Mayer and A. Moglich, Nat. Chem. Biol., 2019, 15, 1085–1092 CrossRef CAS.
- C. Renzl, A. Kakoti and G. Mayer, Angew. Chem., Int. Ed., 2020, 59, 22414–22418 CrossRef CAS.
- J. Hemphill, C. J. Chou, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2013, 135, 13433–13439 CrossRef CAS.
- W. Y. Zhou and A. Deiters, Angew. Chem., Int. Ed., 2016, 55, 5394–5399 CrossRef CAS.
- S. Bohacova, Z. Vanikova, L. Postova Slavetinska and M. Hocek, Org. Biomol. Chem., 2018, 16, 5427–5432 RSC.
- Z. Vanikova, M. Janouskova, M. Kambova, L. Krasny and M. Hocek, Chem. Sci., 2019, 10, 3937–3942 RSC.
- V. Mikat and A. Heckel, RNA, 2007, 13, 2341–2347 CrossRef CAS.
- L. M. Ceo and J. T. Koh, ChemBioChem, 2012, 13, 511–513 CrossRef CAS.
- W. Y. Zhou, W. Brown, A. Bardhan, M. Delaney, A. S. Ilk, R. R. Rauen, S. I. Kahn, M. Tsang and A. Deiters, Angew. Chem., Int. Ed., 2020, 59, 8998–9003 CrossRef CAS.
- A. Ovcharenko, F. P. Weissenboeck and A. Rentmeister, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202013936.
- S. Ogasawara, ACS Chem. Biol., 2017, 12, 351–356 CrossRef CAS.
- D. Y. Zhang, C. Y. Zhou, K. N. Busby, S. C. Alexander and N. K. Devaraj, Angew. Chem., Int. Ed., 2018, 57, 2822–2826 CrossRef CAS.
- J. Hemphill, Q. Y. Liu, R. Uprety, S. Samanta, M. Tsang, R. L. Juliano and A. Deiters, J. Am. Chem. Soc., 2015, 137, 3656–3662 CrossRef CAS.
- P. Vogel, A. Hanswillemenke and T. Stafforst, ACS Synth. Biol., 2017, 6, 1642–1649 CrossRef CAS.
- I. A. Shestopalov, S. Sinha and J. K. Chen, Nat. Chem. Biol., 2007, 3, 650–651 CrossRef CAS.
- D. D. Young, H. Lusic, M. O. Lively, J. A. Yoder and A. Deiters, ChemBioChem, 2008, 9, 2937–2940 CrossRef CAS.
- S. Yamazoe, I. A. Shestopalov, E. Provost, S. D. Leach and J. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 6908–6911 CrossRef CAS.
- Y. Wang, L. Wu, P. Wang, C. Lv, Z. J. Yang and X. J. Tang, Nucleic Acids Res., 2012, 40, 11155–11162 CrossRef CAS.
- S. Shuman, Prog. Nucleic Acid Res. Mol. Biol., 2001, 66, 1–40 CAS.
- N. Sonenberg and A. G. Hinnebusch, Cell, 2009, 136, 731–745 CrossRef CAS.
- J. Marcotrigiano, A. C. Gingras, N. Sonenberg and S. K. Burley, Cell, 1997, 89, 951–961 CrossRef CAS.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS.
- C. J. Brown, I. McNae, P. M. Fischer and M. D. Walkinshaw, J. Mol. Biol., 2007, 372, 7–15 CrossRef CAS.
- E. Darzynkiewicz, J. Stepinski, I. Ekiel, C. Goyer, N. Sonenberg, A. Temeriusz, Y. X. Jin, T. Sijuwade, D. Haber and S. M. Tahara, Biochemistry, 1989, 28, 4771–4778 CrossRef CAS.
- A. Cai, M. Jankowska-Anyszka, A. Centers, L. Chlebicka, J. Stepinski, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, Biochemistry, 1999, 38, 8538–8547 CrossRef CAS.
- J. M. Holstein, L. Anhauser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS.
- F. Soukarieh, M. W. Nowicld, A. Bastide, T. Poyry, C. Jones, K. Dudek, G. Patwardhan, F. Meullenet, N. J. Oldham, M. D. Walkinshaw, A. E. Willis and P. M. Fischer, Eur. J. Med. Chem., 2016, 124, 200–217 CrossRef CAS.
- L. Anhauser, N. Klocker, F. Muttach, F. Masing, P. Spacek, A. Studer and A. Rentmeister, Angew. Chem., Int. Ed., 2020, 59, 3161–3165 CrossRef.
- F. Muttach, F. Masing, A. Studer and A. Rentmeister, Chem.–Eur. J., 2017, 23, 5988–5993 CrossRef CAS.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhauser and A. Rentmeister, Chem. Sci., 2018, 9, 531 RSC.
- J. Leiter, D. Reichert, A. Rentmeister and R. Micura, ChemBioChem, 2020, 21, 265–271 CrossRef CAS.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS.
- A. R. Nodling, L. A. Spear, T. L. Williams, L. Y. P. Luk and Y. H. Tsai, Essays Biochem., 2019, 63, 237–266 CrossRef.
- J. Wang, Y. Liu, Y. J. Liu, S. Q. Zheng, X. Wang, J. Y. Zhao, F. Yang, G. Zhang, C. Wang and P. R. Chen, Nature, 2019, 569, 509–513 CrossRef CAS.
- I. Topisirovic, Y. V. Svitkin, N. Sonenberg and A. J. Shatkin, Wiley Interdiscip. Rev.: RNA, 2011, 2, 277–298 CrossRef CAS.
- C. Fabrega, S. Hausmann, V. Shen, S. Shuman and C. D. Lima, Mol. Cell, 2004, 13, 77–89 CrossRef CAS.
- S. Zheng, S. Hausmann, Q. Liu, A. Ghosh, B. Schwer, C. D. Lima and S. Shuman, J. Biol. Chem., 2006, 281, 35904–35913 CrossRef CAS.
- A. Deiters, D. Groff, Y. H. Ryu, J. M. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728–2731 CrossRef CAS.
- J. K. Bocker, W. Dorner and H. D. Mootz, Chem. Commun., 2019, 55, 1287–1290 RSC.
- P. Sledz and M. Jinek, Elife, 2016, 5, e18434 CrossRef.
- K. Goedecke, M. Pignot, R. S. Goody, A. J. Scheidig and E. Weinhold, Nat. Struct. Biol., 2001, 8, 121–125 CrossRef CAS.
- T. Malone, R. M. Blumenthal and X. Cheng, J. Mol. Biol., 1995, 253, 618–632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00159k |
| This journal is © The Royal Society of Chemistry 2021 |