
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective access to constitutionally identical, orientationally isomeric calix[6]arene-based [3]rotaxanes by an active template approach†
Margherita
Bazzoni
 a,
Leonardo
Andreoni
b,
Serena
Silvi
*b,
Alberto
Credi
cd,
Gianpiero
Cera
a,
Andrea
Secchi
*a and
Arturo
Arduini
*a
a,
Leonardo
Andreoni
b,
Serena
Silvi
*b,
Alberto
Credi
cd,
Gianpiero
Cera
a,
Andrea
Secchi
*a and
Arturo
Arduini
*a
aDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università di Parma, Parco Area delle Scienze 17/A, I-43124 Parma, Italy. E-mail: arturo.arduini@unipr.it; andrea.secchi@unipr.it
bDipartimento di Chimica “G. Ciamician”, Università di Bologna, Via Selmi 2, I-40126 Bologna, Italy. E-mail: serena.silvi@unibo.it
cIstituto per la Sintesi Organica e la Fotoreattività, Consiglio Nazionale delle Ricerche, Via Gobetti 101, I-40129 Bologna, Italy
dDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale del Risorgimento 4, I-40136 Bologna, Italy
First published on 1st April 2021
Abstract
Tris(phenylureido)calix[6]arene is endowed with unique properties that make it a valuable macrocyclic component for the synthesis of mechanically interlocked molecules. Its three-dimensional and intrinsically nonsymmetric structure is kinetically selective toward two processes: (i) in apolar media, the threading of bipyridinium based axle-like components takes place exclusively from the upper rim; (ii) SN2 alkylation reactions of a pyridylpyridinium precursor engulfed in the cavity occur selectively at pyridylpyridinium nitrogen atom located at the macrocycle upper rim (active template synthesis). Here we exploit such properties to prepare two series of [3]rotaxanes, each consisting of three sequence isomers that arise from the threading of two identical but nonsymmetric wheels on a symmetric thread differing only for the reciprocal orientation of the macrocycles. The features of the calix[6]arene and the active template synthetic approach, together with a careful selection of the precursors, enabled us to selectively synthesise the [3]rotaxane sequence isomers of each series with fast kinetics and high yields.
Introduction
A challenging task for the bottom-up construction of molecular-level working devices, designed to perform programmed functions, is to organise in space the functional groups and binding sites responsible for their performance with the highest possible precision and in a predetermined manner.1–4 In this context, mechanically interlocked molecules (MIMs) such as rotaxanes are among the most extensively studied platforms.4,5 In typical instances, their synthesis relies on forming a pseudorotaxane precursor between a macrocycle and an axle-type component, guided by molecular recognition. The insertion of bulky substituents (stoppers) at the axle's termini eventually leads to a rotaxane formation. This threading and capping synthetic strategy, defined as a passive template,6 allows the synthesis of MIMs with sophisticated molecular architectures.7In more recent times, Leigh and co-workers introduced a new approach, termed as active template approach, in which a metal ion, complexed to the macrocycle, both pivots the organisation of the axle precursors around the macrocyclic cavity and mediates the formation of the covalent bonds between them to create the mechanical link.8 Starting from this seminal work, a growing number of different metal-catalysed reactions have been exploited for the realisation of several otherwise inaccessible MIMs.9–12
It nevertheless appears that the increase of the structural complexity parallels the difficulties in governing/addressing the proper relative position and orientation of their active components. This issue is particularly relevant when the components are nonsymmetric13–15 and heteroditopic,16–18 and constitutes a serious obstacle towards the functional exploitation of such valuable structural features. In this regard, among the plethora of macrocycles so far employed for the construction of molecular devices based on MIMs, calixarenes represent a recent and unusual example of a three-dimensional, nonsymmetric and heteroditopic host.19,20 We have extensively investigated the tris-(N-phenylureido)calix[6]arene (CX) (Fig. 1) as a macrocyclic component for the construction of MIMs.21,22 It was evidenced that, in low polarity media, nonsymmetric N,N′-dialkylbispyridinium (viologen) salts can thread the calix[6]arene annulus exclusively with their shorter alkyl chain and through the macrocycle upper rim, thus yielding oriented pseudorotaxane complexes. Therefore, by adopting the passive template synthesis, the preparation of rotaxanes characterised by the univocal orientation of the calixarene rims with respect to the dumbbell was achieved (Fig. 1a).23–27
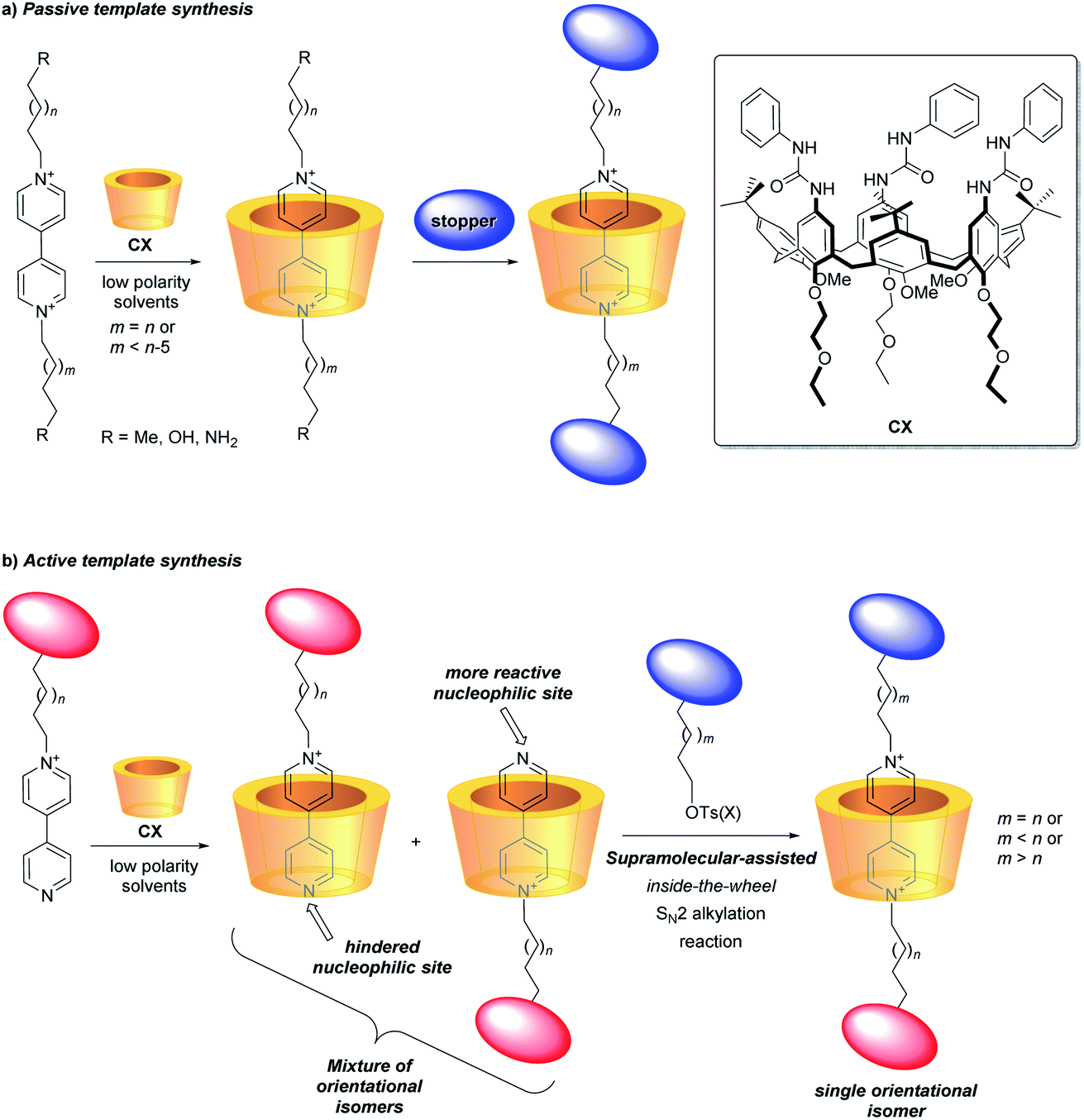 | ||
| Fig. 1 Graphical representation of the synthesis of [2]rotaxanes based on a calix[6]arene wheel (CX), for the sake of simplicity represented as a yellow cartoon, and bipyridinium (viologen) axles using either the passive (a) or the active (b) template synthesis. | ||
More recently, efficient synthetic routes to pseudorotaxanes and rotaxanes through a metal-free active template approach were devised.28–30 In this context, we have exploited the engulfment of positively charged pyridylpyridinium salts inside the π-rich cavity of CX for the selective synthesis of orientational isomers of calix[6]arene-based [2]rotaxanes (Fig. 1b).31,32 As a consequence of its inclusion, the bound salt exposing its neutral nitrogen to the upper rim of the calixarene experiences a faster reactivity towards an SN2 reaction, leading to the formation of oriented pseudorotaxanes and rotaxanes with faster kinetics and higher yields compared to the classic threading and capping strategy (passive template approach).33–37 This supramolecular-assisted strategy could thus expand the scope for the efficient synthesis of calix[6]arene-based MIMs with strict control on the mutual orientation of their nonsymmetric molecular components (Fig. 1b).26,32,38
Motivated by the general interest in higher-order rotaxanes as prospective nanodevices,39–45 and by our expertise in the synthesis of calix[6]arene-based oriented (pseudo)rotaxanes and catenanes,46,47 here we report on the preparation and properties of two series of calix[6]arene-based [3]rotaxanes configurational isomers (Fig. 2). Their common structural features are two calix[6]arene wheels threaded by a dumbbell terminating with two identical stoppers, in which a C12 alkyl chain spans two viologen units. The two series are distinguished by the length (C6 or C12) of the linkers connecting each viologen unit to its diphenylacetyl stopper. Within the two series, three distinct orientational isomers characterised by the different reciprocal orientation of the two calix[6]arenes that surround the two viologen units of the dumbbell were devised. Orientational isomers indicated as UU and LL present the two calix[6]arene wheels facing each other through their upper or lower rims, respectively. In the UL isomers, the two wheels are instead oriented head-to-tail along with the dumbbell (Fig. 2). Due to their structural complexity, these molecular systems will be hereafter described with apposite labels like R6UU, in which R denotes a rotaxane compound, while the number indicates the length of the alkyl chain linkers between the viologen units and the stoppers, and the subscripts point out the reciprocal orientation of the two wheels as described above (Fig. 2). The synthesis and the complete structural characterisation of the novel six [3]rotaxanes will be discussed, along with their electrochemical properties.
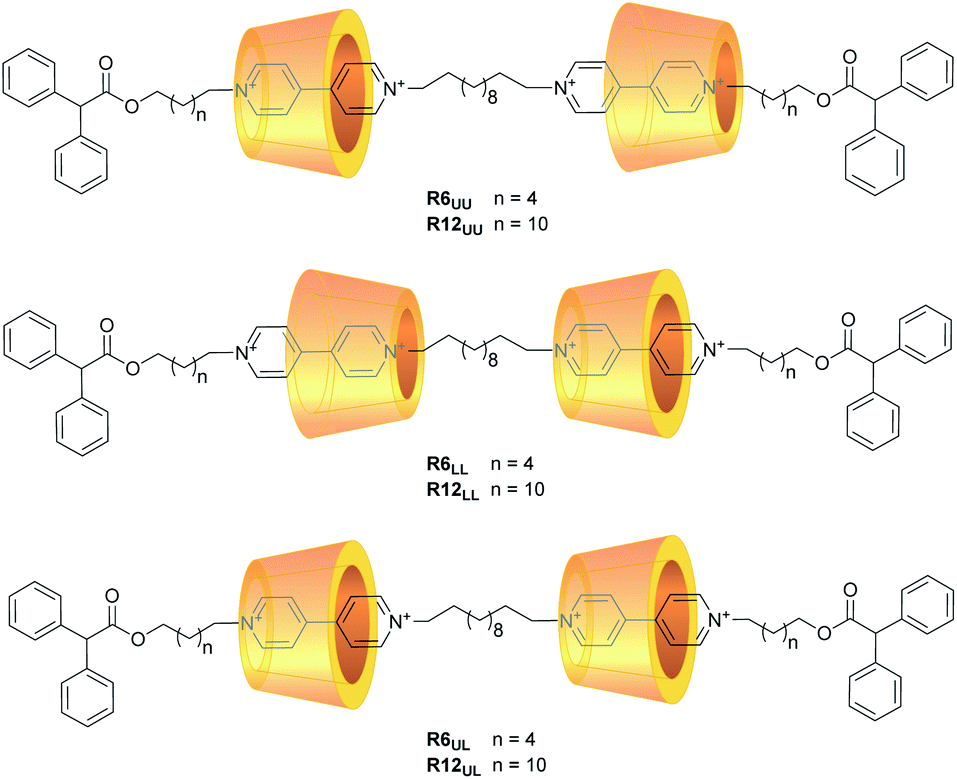 | ||
| Fig. 2 Schematic representation of the [3]rotaxane orientational isomers object of this study. The labels UU, UL and LL stand for upper–upper, upper–lower and lower–lower, and denote which rims of the two calixarene wheels are proximal in the rotaxane. The tosylate counteranions have been omitted for clarity. | ||
It should be emphasised that our compounds address the problem of sequence isomerism in [3]rotaxanes48–50 by taking advantage of the inherent orientation of the calixarene wheel along the main axis of the rotaxane.51 Neri and co-workers recently reported the elegant synthesis of calixarene-based [3]rotaxanes with UU and LL arrangements by a passive template synthesis.52,53 In these systems, a “through annulus” ring inversion of the two wheels, enabled by acid–base stimuli, allowed the observation of all three orientational isomers. In such compounds, however, the three isomers were not isolated. The present study aims at demonstrating that our active template synthesis affords (i) full control of the geometrical arrangement of the calix[6]arene wheels encircling the axle, and (ii) the preparation with high yields of sophisticated interlocked structures, otherwise inaccessible by classic threading and capping strategies (passive template synthesis).
Results and discussion
As mentioned in the Introduction (Fig. 1b), we have recently developed a very efficient one-pot synthetic method that allows us to synthesise several types of oriented [2]rotaxane regardless of the length of the arms of the resulting viologen-based dumbbell.31,32 The dumbbell is formed inside the calix[6]arene reactor by joining two sub-components: a stoppered pyridylpyridinium precursor and a stoppered alkylating agent. It is important to point out that the sole [2]rotaxane isolated in this one-pot reaction will always have the stopper of the pyridylpyridinium precursor at the calix[6]arene lower rim (red stopper in Fig. 1b). The necessary synthetic precursors for the synthesis of the [3]rotaxanes depicted in Fig. 2 – hereafter indicated as dumbbell components (DC1–5) – were appropriately chosen like pieces of a molecular Meccano by considering that in low polarity solvents: (i) mono-stoppered viologen-based axles always thread the cavity of CX from its upper rim, and (ii) the alkyl chain initially present on the pyridylpyridinium precursors will be in proximity of the lower rim of the wheels in the resulting [3]rotaxane.The synthesis of DC1–5 was summarised in Scheme 1 (see ESI† for the experimental details). The free dumbbells 7a,b were also synthesised for comparison. The first [3]rotaxane orientational isomers prepared with the supramolecular-assisted approach were of the UU-type, R6UU and R12UU, which differ for the length of the outer dumbbell arms. In reactions {A} of Scheme 1, CX was equilibrated in toluene with the appropriate dumbbell components DC-2 (4a for R6 and 4b for R12) and DC-3 using a CX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DC-2:DC-3 = 2.5:2.4:1 stoichiometric ratio. The resulting mixture was heated at 65 °C for at least seven days. In these two runs, [3]rotaxanes R6UU and R12UU were obtained in 62% and 43% yield, respectively, after chromatographic separation. No other orientational isomers were separated from the reaction mixtures. The preparation of the LL-type isomers R6LL and R12LL was carried out similarly, but using the dumbbell components DC-1 (3a for R6 and 3b for R12) and DC-4 as starting reagents (see reactions {C} in Scheme 1). These two [3]rotaxanes were isolated in 50% (R6LL) and 38% (R12LL) yields. Once again, no other orientational isomers were separated from the respective reaction mixtures.
:DC-2:DC-3 = 2.5:2.4:1 stoichiometric ratio. The resulting mixture was heated at 65 °C for at least seven days. In these two runs, [3]rotaxanes R6UU and R12UU were obtained in 62% and 43% yield, respectively, after chromatographic separation. No other orientational isomers were separated from the reaction mixtures. The preparation of the LL-type isomers R6LL and R12LL was carried out similarly, but using the dumbbell components DC-1 (3a for R6 and 3b for R12) and DC-4 as starting reagents (see reactions {C} in Scheme 1). These two [3]rotaxanes were isolated in 50% (R6LL) and 38% (R12LL) yields. Once again, no other orientational isomers were separated from the respective reaction mixtures.
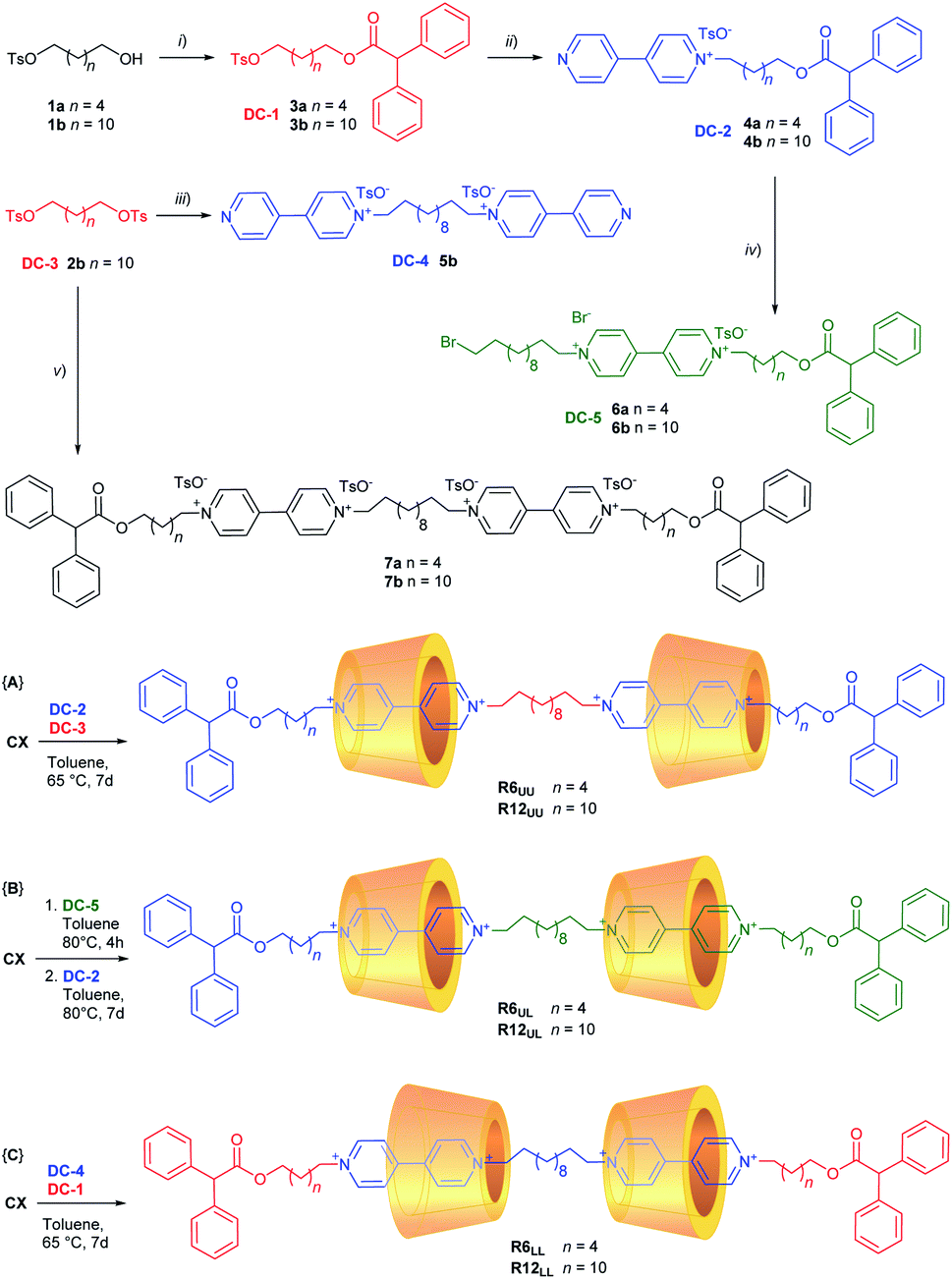 | ||
| Scheme 1 Reagents and conditions: (i) Ph2CHCOCl, Et3N, DCM, rt, 8 h; (ii) 4,4′-bipy (2 eq.), CH3CN, reflux, 24 h; (iii) 4,4′-bipy (2.5 eq.), CH3CN, reflux, 24 h; (iv) 1,12-dibromododecane (5 eq.), CH3CN, reflux, 7 d; (v) 4a,b, CH3CN, sealed tube, 100 °C, 7 d. In the cartoons of the [3]rotaxane structures, the counteranions have been omitted for clarity. | ||
For the synthesis of the third series of [3]rotaxane orientational isomers, R6UL and R12UL, in which both the nonsymmetric calix[6]arene wheels are oriented in the same direction with respect to the thread, a slightly different synthetic sequence was adopted (see reactions {B} in Scheme 1). DC-5 (6a for R6 and 6b for R12) was initially suspended in toluene at 80 °C with a twofold stoichiometric excess of CX to give a [2]pseudorotaxane orientational isomer in which the stoppered arm of the viologen-based axle is located at the wheel upper rim.‡ The formation of such complex was witnessed by the deep red colour assumed by the now homogeneous solution after a few hours of stirring. The resulting pseudorotaxane complex was not isolated but immediately reacted with DC-2 (4a for R6 and 4b for R12) for seven days at 80 °C. In this way, the supramolecular-assisted reaction occurs between the protruding ω-bromo termini of DC-5, already confined in CX, and a new complex resulting from the association of DC-2 and the excess of CX already present in the solution. After workup and chromatographic separation, the desired [3]rotaxanes R6UL and R12UL were obtained in 42% and 40% yield, respectively. Analysis of the crude reaction mixture did not allow us to identify unambiguously if any other stereoisomers were formed in the reaction. However, after careful chromatography, only the target geometric isomer was observed in fractions identified as containing [3]rotaxane by LC-MS. As the same eluent system was used to isolate R6UU and R6LL, this provides strong evidence for the stereoselectivity of the reaction.
The identity of the isolated compounds was initially investigated through HR-MS measurements. The recorded spectra (Fig. S25–S30†) show molecular peaks (triply and quadruply charged) with an isotopic distribution in agreement with the structure of the two [3]rotaxanes series. The 1H NMR spectra in CDCl3 of the [3]rotaxanes sharing the same dumbbell 7a and isolated from the reactions {A–C} are shown in the stack plot of Fig. 4. As expected, the spectra of the three [3]rotaxanes are not superimposable. However, the two spectra of R6UU (Fig. 4a) and R6LL (Fig. 4c) have a similar pattern, both showing two well-resolved doublets, with geminal coupling, for the “axial” (4.4 ppm) and “equatorial” (3.5 ppm) diastereotopic protons of the calix[6]arene wheels bridging methylene groups (see black circles in Fig. 4a and c). On the contrary, the spectrum of R6UL, isolated from reaction {B}, shows at ∼3.5 ppm (see white circles in Fig. 4b) two overlapped doublets suggesting the head-to-tail arrangement of the calixarene macrocycles with respect to the dumbbell.
To assess the structure of these MIMs, extensive NMR experiments were carried out to identify the isolated spin systems of the inner chain and external arms and their position with respect to the rims of the calix[6]arene macrocycles. The proton signals arising from the internal (α to ζ) and external (1 to 6) alkyl chains of the dumbbell in R6UU were unequivocally assigned through a series of 1D and 2D NMR experiments. Initially, the 1H NMR spectra of R6UU, the empty wheel CX, and the free dumbbell 7a were gathered in the stack plot of Fig. 3 (for the protons labelling, see the molecular sketches in Fig. 3 and 4). From previous studies carried out on calix[6]arene-based [2]rotaxanes,54–56 it is known that the methine proton (labelled as 0) of the diphenylacetic stoppers yields a diagnostic singlet at ca. 5 ppm. In R6UU, this signal is found at 5.07 ppm (see Fig. 3b and 4a). In the rotaxanes, several signals of the dumbbell are upfield-shifted (cf.Fig. 3b and c) because of its inclusion inside the cavities of CX. Through a 2D HSQC experiment (Fig. S3†), we were able to identify the resonance of the methylene groups (1) adjacent to the diphenyl acetic stopper. Starting from this signal and thanks to a 2D TOCSY experiment (Fig. S5†), the whole spin system of the external C6 alkyl chain in R6UU was recognised. The same approach allows us to identify the signals from β to δ of the internal alkyl chain starting from the well-recognisable α resonance at 3.2 ppm. In this respect, it is worth noticing the complexation induced shift (CIS) experienced by the signals of the two pairs of methylene protons (α) and (6), which are the linking units between the encapsulated viologen units and the dumbbell internal and external alkyl chains, respectively. In the free dumbbell 7a, they resonate as two close triplets at 4.72 and 4.67 ppm (Fig. 3c), while in R6UU, because of their encapsulation in the wheels, they give rise to two upfield-shifted resonances at 3.2 (Δδ = +1.4) and 3.7 (Δδ = +1) ppm, respectively (Fig. 3b). Since such complexation only leads to a single set of shifted signals for the methylene protons (α) and (6), it is very reasonable to assume that both viologen units experience the same magnetic environment due to the head-to-head orientation of the wheels. Very similar behaviour was observed for the two pairs of protons (¥,#) and ($,§) of the viologen units. Because of the symmetry, in the spectrum of the free 7a (Fig. 3c), the signals of these protons' pairs are overlapped. However, in R6UU, these viologen units experience an anisotropic magnetic environment exerted by the nonsymmetric calixarene cavities. Thus, their signals are split into four upfield-shifted resonances at 7.67 (¥), 7.32 (§), 6.67 (#) and 6.17 ($) ppm (see purple lines in Fig. 3b and c).
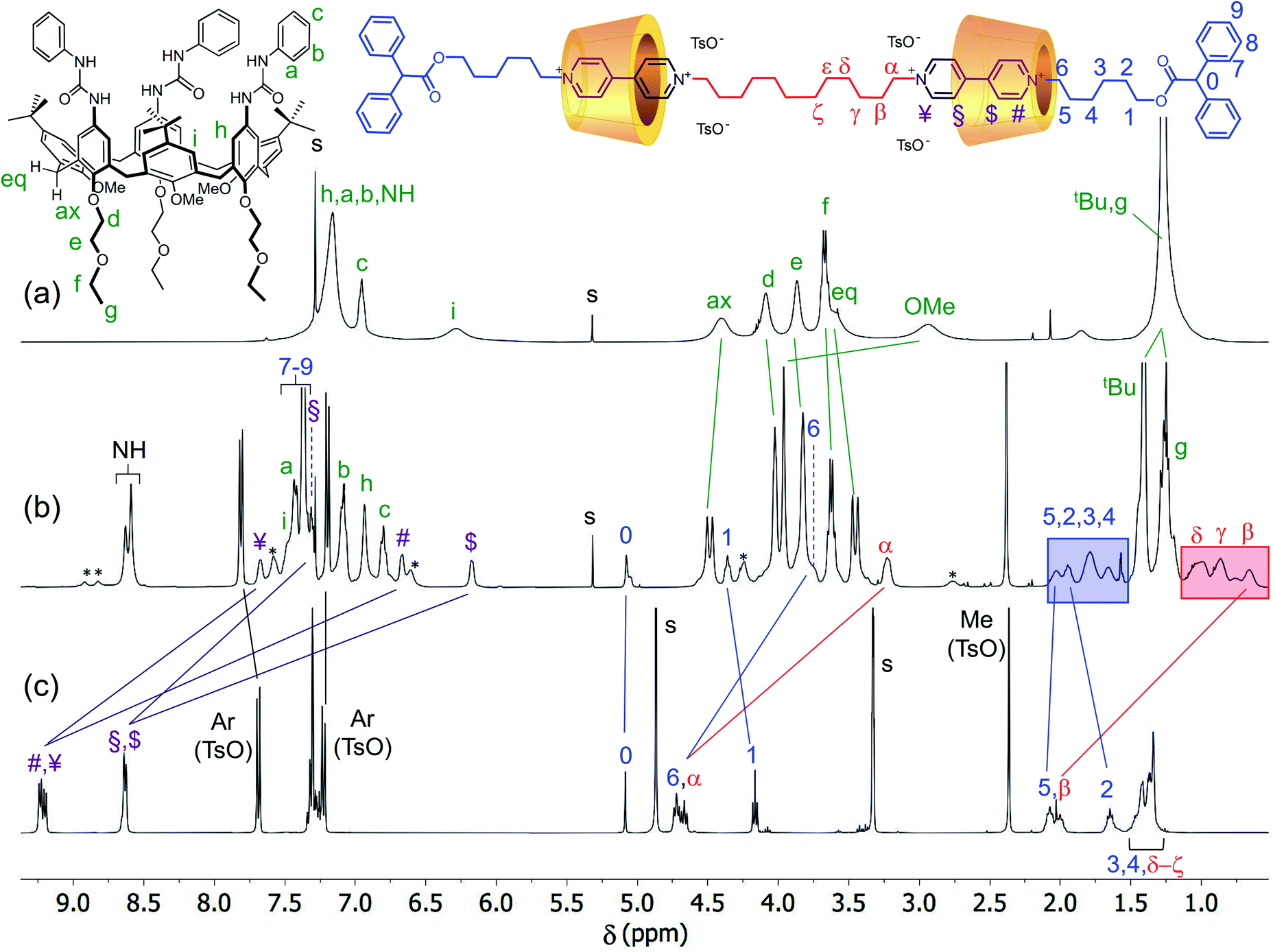 | ||
| Fig. 3 1H NMR stack plot (400 MHz, CDCl3) of (a) wheel CX, (b) [3]rotaxane R6UU, and (c) dumbbell 7a. For solubility reasons, spectrum (c) was taken in CD3OD. The most representative resonances are labelled according to the molecular sketches placed above the stack plot, while their complexation induced shifts are indicated with solid lines; the unlabeled signals marked with an asterisk in the spectrum (b) belong to a rotamer of R6UU in the partial cone (paCo) conformation; see text and ESI† for further details. | ||
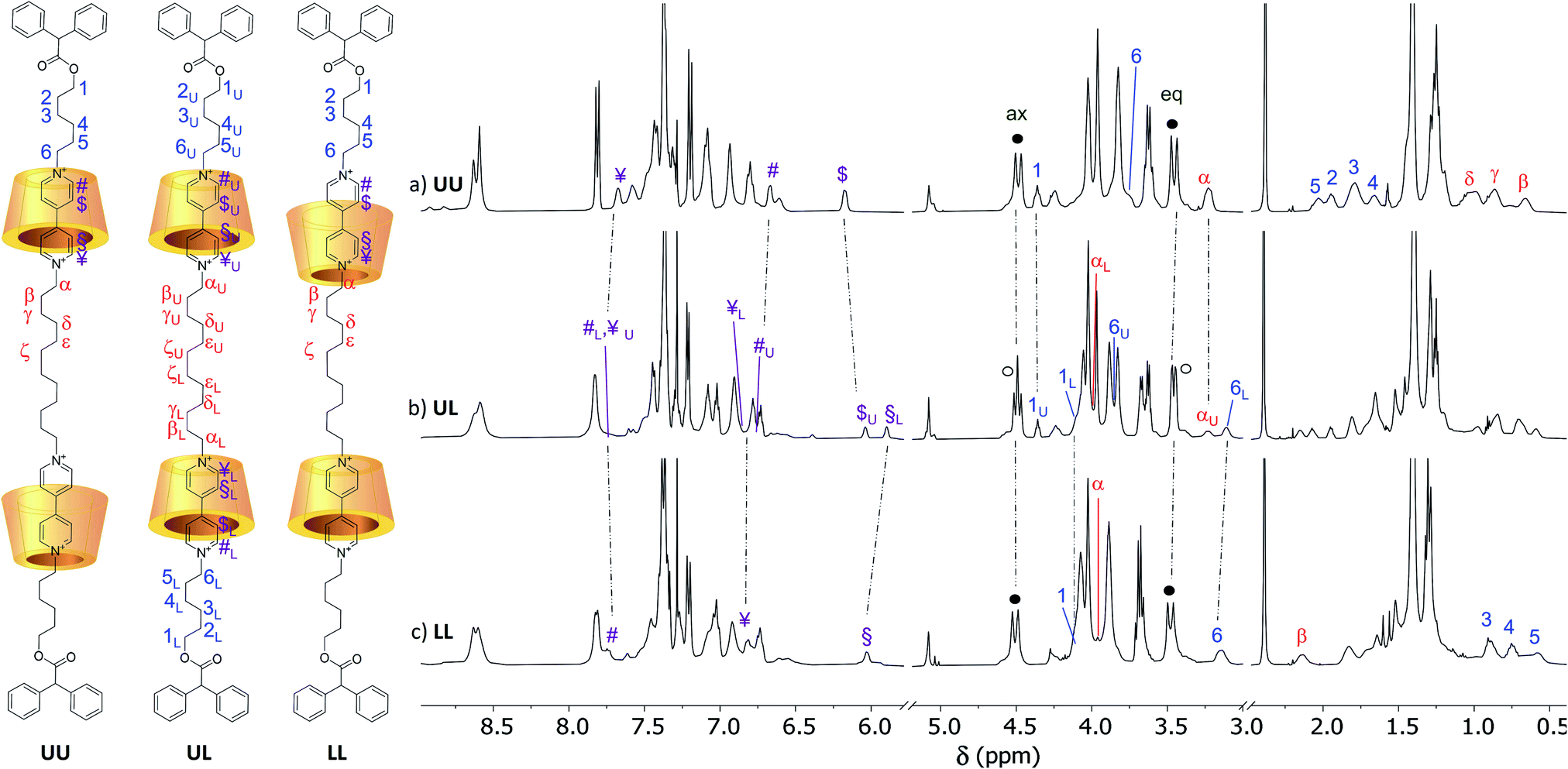 | ||
| Fig. 4 1H NMR stack plot (400 MHz, CDCl3) of the [3]rotaxanes sharing dumbbell 7a, isolated from the supramolecular-assisted reactions {A–C} of Scheme 1, and corresponding to (a) R6UU, (b) R6UL and (c) R6LL, respectively. The U and L subscript labels of the spectrum (b) indicate that the corresponding protons' chemical shifts coincide with those found for the UU and LL orientational isomers, respectively. In the sketch of the three [3]rotaxanes on the left, the tosylate counteranions have been omitted for clarity. | ||
Moving forward along the dumbbell's internal and external alkyl chains, up to the methylene groups (δ) and (2), different but very diagnostic shielding effect of the calixarene cavities could be detected. The shielding effect is still very significant for the methylene protons of the internal C12 alkyl chain (Δδ = +1.4, +0.6, and +0.4 ppm for β, γ and δ, respectively, see the red-shaded box in Fig. 3). In comparison, it is negligible for the methylene protons (5) [Δδ = +0.05 ppm] of the C6 external chains. A small deshielding effect was instead observed for the methylene (4) to (2) of the same chain (see the blue-shaded box in Fig. 3). Such an effect can be accounted for by considering that, in the postulated structure of R6UU, the methylene group (5), with the rest of the chain, protrudes from the macrocycles lower rim. Thus, the dumbbell external alkyl chains suffer the deshielding effect of ethylethyloxy alkyl chains present on the calix[6]arenes lower rim.§ To confirm these findings, we also carried out a 2D ROESY experiment that, through the analysis of the resulting cross-peaks, revealed the spatial proximity between the protons of the inner alkyl spacer (α to δ) and the aromatic protons (b and c) of the phenylureas at the macrocycles upper rim (Fig. 5). Analogous results were obtained for the [3]rotaxane R12UU (Fig. S21†), endowed with longer C12 external arms. This fact confirms that this synthetic strategy operates regardless of the length of the threads' external arms, and it is exclusively affected by the nature of the heteroditopic calix[6]arene wheel.
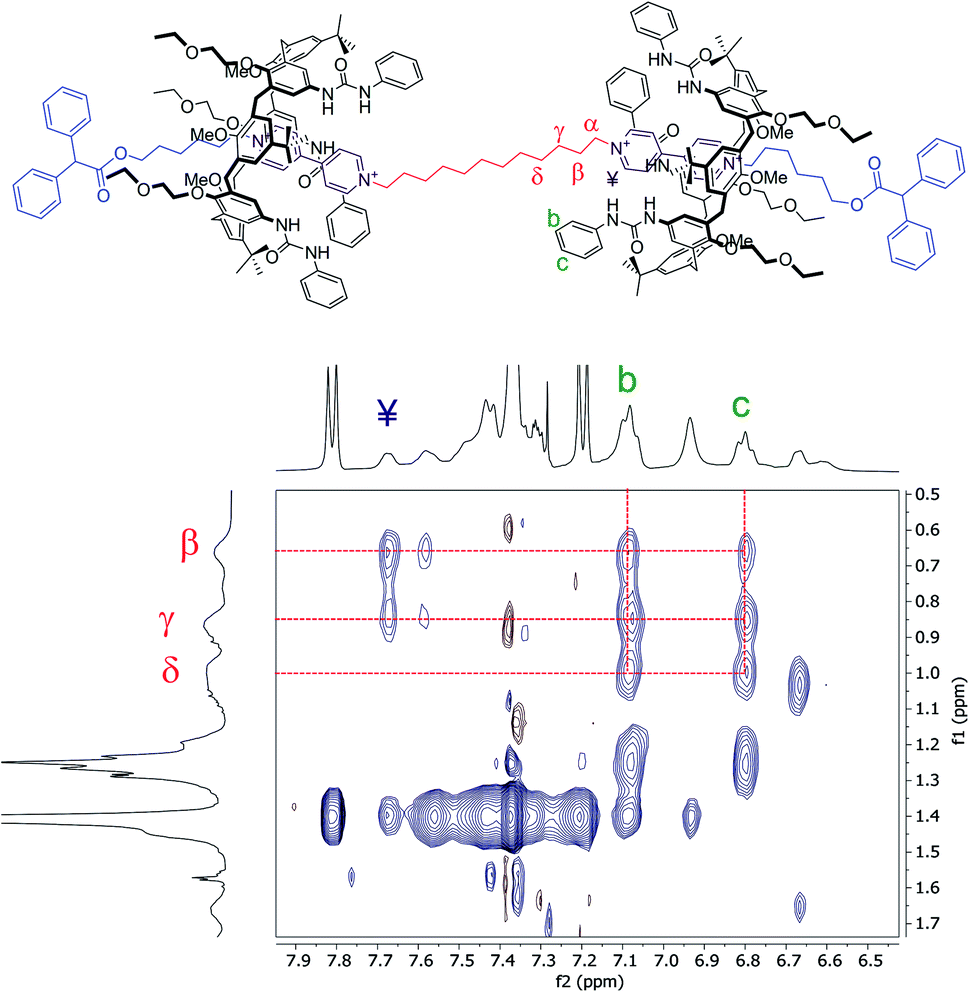 | ||
| Fig. 5 Expansion of a 2D ROESY spectrum (400 MHz, CDCl3, ROESY SL = 200 ms) of R6UU. Analysis of the cross-peaks shows the spatial proximity between the protons of the internal C12 alkyl chain (α–δ) and those of the phenylurea groups (b and c) present at the upper rim of the calix[6]arene macrocycles (for the full spectrum, see ESI†). | ||
It is worth noticing that some weak resonances in the spectrum of R6UU, highlighted in Fig. 3b with asterisks, are in chemical exchange with the dumbbell signals (6), (1), (#) and (¥). These exchanging signals were identified through a series of selective ROESY and NOESY experiments (see Fig. S7†). The weak and exchanging resonances are due to a rotamer in the partial cone (paCo) conformation, likely generated by the rotation of one of the wheel aromatic rings bearing the p-tert-butyl group (see the sketch in Fig. S7†).¶
As we did for the UU-type series of [3]rotaxanes, 2D HSQC and TOCSY (see ESI†) experiments were carried out on the samples of the LL-type series (R6LL and R12LL) to identify the whole spin system of the external C6 alkyl chain in these [3]rotaxanes. It is important to note that in these [3]rotaxane isomers, the signals of the external alkyl chains are significantly more upfield-shifted than for R6UU and R12UU. As an example, protons of methylene (6) in R6 resonate at ∼3.8 ppm for the UU isomer, while the same protons resonate at ∼3.2 ppm in the spectrum of LL (Δδ ∼ 0.6 ppm) (cf.Fig. 4a and c). Such higher shielding effect is even more evident for the protons labelled as (2–5) that in the spectrum of the UU isomer are found between 2.2 and 1.5 ppm (Fig. 4a), while in the LL isomer are at very high fields (1 to 0.5 ppm, Fig. 4c). These findings agree with the hypothesised structure of R6LL in which the external C6 arms endure the shielding effect of the phenylureas present on the wheels upper rim. A 2D ROESY experiment (Fig. S13†) confirmed these findings since it allowed us to identify several dipolar couplings witnessing the spatial proximity between the dumbbell methylene groups (3–5) with the wheels phenylurea groups. Since in R6LL the wheels adopt a tail-to-tail orientation, the signals of the C12 inner alkyl spacer (α–ζ) are downfield-shifted with respect to the same resonance in R6UU.
The 1H NMR spectra of R6UL and R12UL (see Fig. 4b and S31b†) orientational isomers evidence no matching with the corresponding UU (see Fig. 4a and S31a†) and LL-type (see Fig. 4c and S31c†) [3]rotaxane orientational isomers.|| Most importantly, several of their NMR signals are split into two separated resonances because of the two wheels' head-to-tail arrangement. In this regard, the splitting of the signals relative to the wheels bridging methylene groups was briefly discussed. The spectra of these compounds also show two distinct resonances for the N+CH2 groups sitting at the upper rim of the wheels. Thanks to a series of 1D TOCSY experiments (Fig. S20†), it was possible to assign the signal resonating at 3.25 ppm to the methylene αU while the one resonating at 3.13 ppm to 6L (Fig. 4b). Indeed, the external alkyl chain spin system starting from 6L unequivocally leads to a signal resonating at 4.09 ppm and relative to the OCH2 group 1L. Similarly, a second C6 alkyl chain starting from the OCH2 (1U) resonating at 4.36 ppm and leading to (6U) at 3.85 ppm was identified.
UV-visible spectroscopic and electrochemical measurements
The UV-visible spectroscopic characterisation of the six [3]rotaxanes was performed in CH2Cl2. All the compounds show the typical absorption features of calixarene–bipyridinium based rotaxanes,26,55i.e., an intense band in the UV region and a weak broad band in the visible region of the absorption spectrum around 450 nm (Fig. S32†). As expected, the molar absorption coefficients of these two bands are roughly doubled with respect to parent [2]rotaxanes on account of the presence of twice as many chromophoric units. The spectral shapes are very similar and almost superimposable, regardless of the length of the alkyl chains and the orientation of the calixarene rings (Fig. S32†).By a closer inspection of the UV band maximum, however, a slight shift can be observed for the R6 series (Fig. S33†). The band is red-shifted moving from the UU to the UL and LL isomers. This behaviour would suggest an effect of the relative orientation of the calixarene wheels, which can be detected only for the series with the shorter axle. Indeed, the band is at higher energy when the three urea moieties face each other and at lower energy when they point toward the stopper units. As the wavelength shift is comparable with the resolution of our instrument, we prefer not to speculate on this observation. However, it cannot be excluded that in the shorter rotaxanes, the bipyridinium encapsulation is slightly dependent on the wheel orientation because of the presence of a nearby stopper. For comparison, the absorption spectra of the model dumbbells 7a,b were collected (Fig. S34†): unfortunately, these two compounds are mostly insoluble in CH2Cl2, and their absorption coefficient could not be measured. Nevertheless, a comparison of the shape of the spectra confirms that the length of the chains has no effect on the optical properties of these compounds.
The electrochemical characterisation of the rotaxanes and the dumbbell model compounds was performed by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in CH2Cl2; the data are gathered in Table S2.† Compounds 7a and 7b show two reduction processes, respectively assigned to the first and second monoelectronic reduction of the two bipyridinium units, thus suggesting that they are independent and non-interacting. Both processes are slightly shifted to more negative potential values (ca. 50 mV) with respect to parent bipyridinium-based axle compounds.57 This observation could be related to the scarce solubility of the compounds and the formation of small aggregates: indeed, the quality of the CV curves is poor, showing the presence of spurious peaks, possibly related to partial precipitation of the compounds during the measurements.
The electrochemical behaviour of the six [3]rotaxanes is very similar (Fig. 6): all the compounds show two reduction processes, at more negative potential values with respect to model bipyridinium dumbbells and in analogy to parent [2]rotaxanes.26,55 The presence of only two processes, like in the dumbbell model compounds, suggests that the two bipyridinium units are equivalent and non-interacting with each other. The shift of the first process is ascribed to the charge transfer interaction between the bipyridinium units and the calixarene macrocycles, which stabilises the dicationic bipyridinium moieties. The shift of the second reduction process suggests that the calixarenes and the monoreduced bipyridinium sites are still interacting, as observed for related [2]rotaxanes.26,55 In cyclic voltammetric experiments, both reduction curves show some degree of electrochemical irreversibility, with peak-to-peak separation >100 mV at the explored scan rates (from 0.1 to 1 V s−1). Overall, the [3]rotaxanes show the same electrochemical features as the parent [2]rotaxanes. Apparently, no effect of the chain length and the relative orientation of the wheels could be detected. Small effects, like those observed in the absorption spectra, could be expected, but unfortunately, the poor reversibility of the processes prevented a more detailed analysis.
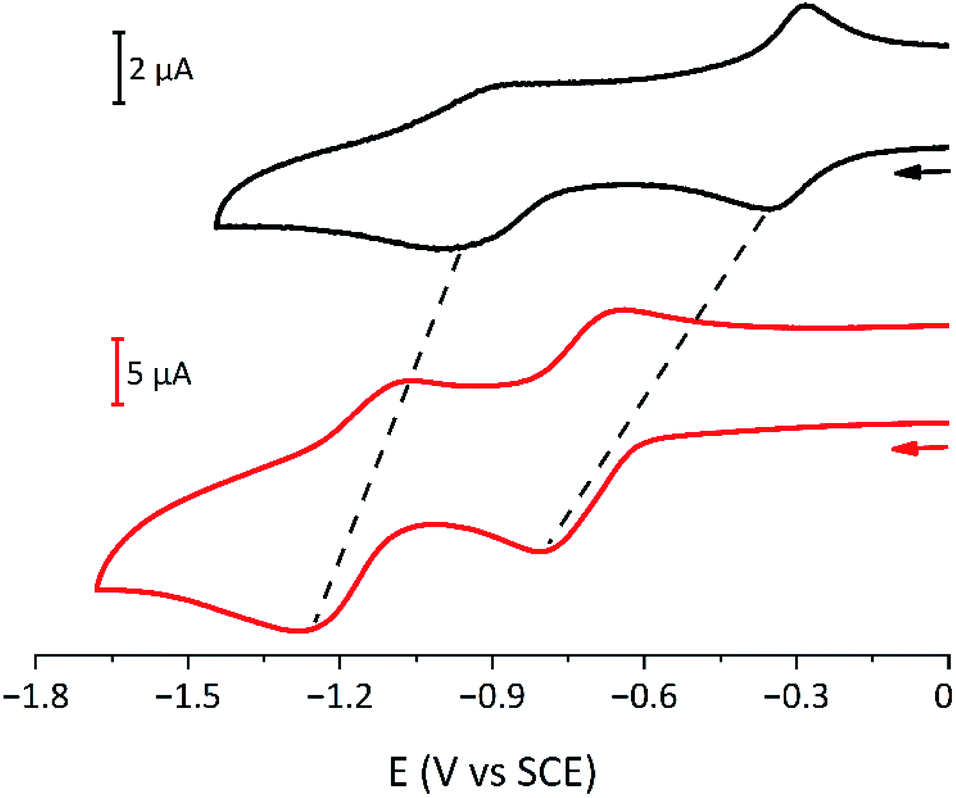 | ||
| Fig. 6 Cyclic voltammograms of dumbbell 7a (black line; 1 × 10−4 M) and [3]rotaxane R6UU (red line, 2 × 10−4 M) in CH2Cl2 at room temperature. A 100-fold excess of TBAPF6 was employed as supporting electrolyte. | ||
Conclusions
We have described a methodology to prepare [3]rotaxanes comprising two identical heteroditopic nonsymmetric calix[6]arene macrocycles with full control of their mutual orientation. The method, based on an active template effect exerted by the calixarene cavity on the axle subcomponents, ensures large flexibility to the axle structure. Sets of three isomeric [3]rotaxanes that are constitutionally identical but structurally non-equivalent, owing to the different relative orientations of the calixarene wheels, have been prepared and characterised for the first time. Two series of [3]rotaxanes that differ for the length of the axle components have been synthesised, demonstrating the versatility of the approach. Its success stems from the fact that the programmed isomer is obtained selectively and efficiently, thus avoiding the need for isolation procedures that, according to our observations, would be particularly problematic. To the best of our knowledge, this is the only example in which the three sequence isomers of [3]rotaxanes made of identical axle and wheel components originating from the intrinsic asymmetry of the wheel were selectively synthesised and isolated.The development of made-to-order synthetic approaches to MIMs that contain nonsymmetric components assembled with a predetermined mutual orientation is key to explore the virtually unlimited possibilities that the mechanical bond can offer to stereochemistry and its applications. The realisation of directional motion in molecular machines and the use of sequences of threaded molecular wheels to store information at the nanoscale are only two examples of this kind. Various functional elements – e.g., stoppers, spacers and recognition sites – can be introduced in the dumbbell-shaped component. In principle, the strategy discussed here could also be extended to make higher-order rotaxanes with more than two identical nonsymmetric wheels threaded in any desired sequence of relative orientations.
Author contributions
M. B. – investigation, data interpretation, writing & review; L. A. – investigation, data interpretation, & writing; S. S. – data interpretation, writing, review, supervision & editing; A. C. – review, supervision & funding acquisition; G. C. – writing & review; A. S. – data interpretation, original draft, writing, review, supervision & editing; A. A. – conceptualization, writing, review, supervision & funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Centro Interdipartimentale di Misure of the University of Parma for NMR and MS measurements. This work was supported by the European Union's Horizon 2020 research and innovation program (ERC AdG “Leaps” 692981 and FET-OPEN “Magnify” 801378) and the Italian Ministry of University and Research (PRIN 20173L7W8K and FARE R16S9XXKX3). This work has been carried out within the COMP-HUB Initiative, funded by the “Departments of Excellence” program of the Italian Ministry for Education, University and Research (MIUR, 2018–2022).Notes and references
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Artificial molecular motors, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- Molecular Machines and Motors: Recent Advances and Perspectives, ed. A. Credi, S. Silvi and M. Venturi, Springer International Publishing, 2014 Search PubMed.
- Molecular Machines, ed. T. R. Kelly, Springer-Verlag, Berlin Heidelberg, 2005, vol. 262 Search PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, New Jersey, 2017 Search PubMed.
- Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999 Search PubMed.
- K. E. Griffiths and J. F. Stoddart, Template-directed synthesis of donor/acceptor [2]catenanes and [2]rotaxanes, Pure Appl. Chem., 2008, 80, 485–506 CAS.
- T. J. Hubin and D. H. Busch, Template routes to interlocked molecular structures and orderly molecular entanglements, Coord. Chem. Rev., 2000, 200–202, 5–52 CrossRef CAS.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. Mc Burney, Active metal template synthesis of rotaxanes, catenanes and molecular shuttles, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, Active Metal Template Synthesis of [2]Catenanes, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed.
- Y. Sato, R. Yamasaki and S. Saito, Synthesis of [2]Catenanes by Oxidative Intramolecular Diyne Coupling Mediated by Macrocyclic Copper(I) Complexes, Angew. Chem., Int. Ed., 2009, 48, 504–507 CrossRef CAS PubMed.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, Catalytic “Click” Rotaxanes: A Substoichiometric Metal-Template Pathway to Mechanically Interlocked Architectures, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- S. Saito, E. Takahashi and K. Nakazono, Synthesis of [2]Rotaxanes by the Catalytic Reactions of a Macrocyclic Copper Complex, Org. Lett., 2006, 8, 5133–5136 CrossRef CAS PubMed.
- A. Hashidzume, H. Yamaguchi and A. Harada, Cyclodextrin-Based Rotaxanes: From Rotaxanes to Polyrotaxanes and Further to Functional Materials, Eur. J. Org. Chem., 2019, 2019, 3344–3357 CrossRef CAS.
- T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, Kinetic control of threading of cyclodextrins onto axle molecules, J. Am. Chem. Soc., 2005, 127, 12186–12187 CrossRef CAS PubMed.
- A. J. Baer and D. H. Macartney, Orientational isomers of α-cyclodextrin [2]semi-rotaxanes with asymmetric dicationic threads, Org. Biomol. Chem., 2005, 3, 1448–1452 RSC.
- J. R. Romero, G. Aragay and P. Ballester, Ion-pair recognition by a neutral [2]rotaxane based on a bis-calix[4]pyrrole cyclic component, Chem. Sci., 2016, 8, 491–498 RSC.
- S. Saha, S. Santra and P. Ghosh, [2]Pseudorotaxane Composed of Heteroditopic Macrobicycle and Pyridine N-Oxide Based Axle: Recognition Site Dependent Axle Orientation, Org. Lett., 2015, 17, 1854–1857 CrossRef CAS.
- M. Semeraro, A. Arduini, M. Baroncini, R. Battelli, A. Credi, M. Venturi, A. Pochini, A. Secchi and S. Silvi, Self-Assembly of Calix[6]arene-Diazapyrenium Pseudorotaxanes: Interplay of Molecular Recognition and Ion-Pairing Effects, Chem.–Eur. J., 2010, 16, 3467–3475 CrossRef CAS.
- A. Arduini, R. Ferdani, A. Pochini, A. Secchi and F. Ugozzoli, Calix[6]arene as a Wheel for Rotaxane Synthesis, Angew. Chem., Int. Ed., 2000, 39, 3453–3456 CrossRef CAS.
- G. De Leener, D. Over, C. Smet, D. Cornut, A. G. Porras-Gutierrez, I. López, B. Douziech, N. Le Poul, F. Topić, K. Rissanen, Y. Le Mest, I. Jabin and O. Reinaud, “Two-Story” Calix[6]arene-Based Zinc and Copper Complexes: Structure, Properties, and O2 Binding, Inorg. Chem., 2017, 56, 10971–10983 CrossRef CAS.
- A. Arduini, G. Orlandini, A. Secchi, A. Credi, S. Silvi and M. Venturi, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer International Publishing, Cham, 2016, pp. 761–781 Search PubMed.
- G. Cera, A. Arduini, A. Secchi, A. Credi and S. Silvi, Chem. Rec., 2021, 21 DOI:10.1002/tcr.202100012.
- G. Orlandini, L. Casimiro, M. Bazzoni, B. Cogliati, A. Credi, M. Lucarini, S. Silvi, A. Arduini and A. Secchi, Synthesis and properties of a redox-switchable calix[6]arene-based molecular lasso, Org. Chem. Front., 2020, 7, 648–659 RSC.
- V. Zanichelli, L. Dallacasagrande, A. Arduini, A. Secchi, G. Ragazzon, S. Silvi and A. Credi, Electrochemically Triggered Co-Conformational Switching in a [2]Catenane Comprising a Nonsymmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle, Molecules, 2018, 23, 1156–1168 CrossRef PubMed.
- V. Zanichelli, M. Bazzoni, A. Arduini, P. Franchi, M. Lucarini, G. Ragazzon, A. Secchi and S. Silvi, Redox-Switchable Calix[6]arene-Based Isomeric Rotaxanes, Chem.–Eur. J., 2018, 24, 12370–12382 CrossRef CAS PubMed.
- V. Zanichelli, G. Ragazzon, A. Arduini, A. Credi, P. Franchi, G. Orlandini, M. Venturi, M. Lucarini, A. Secchi and S. Silvi, Synthesis and Characterisation of Constitutionally Isomeric Oriented Calix[6]arene-Based Rotaxanes, Eur. J. Org. Chem., 2016, 2016, 1033–1042 CrossRef CAS.
- G. Orlandini, V. Zanichelli, A. Secchi, A. Arduini, G. Ragazzon, A. Credi, M. Venturi and S. Silvi, Synthesis by ring closing metathesis and properties of an electroactive calix[6]arene [2]catenane, Supramol. Chem., 2016, 28, 427–435 CrossRef CAS.
- C. Tian, S. D. P. Fielden, G. F. S. Whitehead, I. J. Vitorica-Yrezabal and D. A. Leigh, Weak functional group interactions revealed through metal-free active template rotaxane synthesis, Nat. Commun., 2020, 11, 744 CrossRef CAS PubMed.
- S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Pérez-Saavedra and I. J. Vitorica-Yrezabal, Spontaneous Assembly of Rotaxanes from a Primary Amine, Crown Ether and Electrophile, J. Am. Chem. Soc., 2018, 140, 6049–6052 CrossRef CAS PubMed.
- G. De Bo, G. Dolphijn, C. T. McTernan and D. A. Leigh, [2]Rotaxane Formation by Transition State Stabilization, J. Am. Chem. Soc., 2017, 139, 8455–8457 CrossRef CAS PubMed.
- G. Orlandini, G. Ragazzon, V. Zanichelli, A. Secchi, S. Silvi, M. Venturi, A. Arduini and A. Credi, Covalent capture of oriented calix[6]arene rotaxanes by a metal-free active template approach, Chem. Commun., 2017, 53, 6172–6174 RSC.
- V. Zanichelli, G. Ragazzon, G. Orlandini, M. Venturi, A. Credi, S. Silvi, A. Arduini and A. Secchi, Efficient active-template synthesis of calix[6]arene-based oriented pseudorotaxanes and rotaxanes, Org. Biomol. Chem., 2017, 15, 6753–6763 RSC.
- C. Gaeta, P. L. Manna, M. D. Rosa, A. Soriente, C. Talotta and P. Neri, Supramolecular Catalysis with Self-Assembled Capsules and Cages: What Happens in Confined Spaces, ChemCatChem DOI:10.1002/cctc.202001570.
- D. Chakraborty and P. K. Chattaraj, Bonding, Reactivity, and Dynamics in Confined Systems, J. Phys. Chem. A, 2019, 123, 4513–4531 CrossRef CAS PubMed.
- X. Hou, C. Ke and J. F. Stoddart, Cooperative capture synthesis: yet another playground for copper-free click chemistry, Chem. Soc. Rev., 2016, 45, 3766–3780 RSC.
- Y. Voloshin, I. Belaya and R. Krämer, in The Encapsulation Phenomenon: Synthesis, Reactivity and Applications of Caged Ions and Molecules, ed. Y. Voloshin, I. Belaya and R. Krämer, Springer International Publishing, Cham, 2016, pp. 419–498 Search PubMed.
- B. W. Purse and J. Rebek, Functional cavitands: Chemical reactivity in structured environments, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10777–10782 CrossRef CAS PubMed.
- A. Inthasot, M.-D. Dang Thy, M. Lejeune, L. Fusaro, O. Reinaud, M. Luhmer, B. Colasson and I. Jabin, Supramolecular Assistance for the Selective Monofunctionalization of a Calix[6]arene Tris-carboxylic Acid-Based Receptor, J. Org. Chem., 2014, 79, 1913–1919 CrossRef CAS PubMed.
- H.-Y. Zhou, Q.-S. Zong, Y. Han and C.-F. Chen, Recent advances in higher order rotaxane architectures, Chem. Commun., 2020, 56, 9916–9936 RSC.
- K. Yamamoto, R. Nameki, H. Sogawa and T. Takata, Macrocyclic Dinuclear Palladium Complex as a Novel Doubly Threaded [3]Rotaxane Scaffold and Its Application as a Rotaxane Cross-Linker, Angew. Chem., Int. Ed., 2020, 59, 18023–18028 CrossRef CAS PubMed.
- Y. Akae, H. Sogawa and T. Takata, Effective Synthesis and Modification of α-Cyclodextrin-Based [3]Rotaxanes Enabling Versatile Molecular Design, Eur. J. Org. Chem., 2019, 2019, 3605–3613 CrossRef CAS.
- L.-S. Zheng, J.-S. Cui and W. Jiang, Biomimetic Synchronized Motion of Two Interacting Macrocycles in [3]Rotaxane-Based Molecular Shuttles, Angew. Chem., Int. Ed., 2019, 58, 15136–15141 CrossRef CAS PubMed.
- H. V. Schröder, A. Mekic, H. Hupatz, S. Sobottka, F. Witte, L. H. Urner, M. Gaedke, K. Pagel, B. Sarkar, B. Paulus and C. A. Schalley, Switchable synchronisation of pirouetting motions in a redox-active [3]rotaxane, Nanoscale, 2018, 10, 21425–21433 RSC.
- K. Zhu, G. Baggi and S. J. Loeb, Ring-through-ring molecular shuttling in a saturated [3]rotaxane, Nat. Chem., 2018, 10, 625–630 CrossRef CAS PubMed.
- L. Ma, S. Wang, C. Li, D. Cao, T. Li and X. Ma, Photo-controlled fluorescence on/off switching of a pseudo[3]rotaxane between an AIE-active pillar[5]arene host and a photochromic bithienylethene guest, Chem. Commun., 2018, 54, 2405–2408 RSC.
- G. Cera, M. Bazzoni, A. Arduini and A. Secchi, Ion-Pair Selective Conformational Rearrangement of Sulfonamide Calix[6]arene-Based Pseudorotaxanes, Org. Lett., 2020, 22, 3702–3705 CrossRef CAS PubMed.
- M. Bazzoni, F. Terenziani, A. Secchi, G. Cera, I. Jabin, G. De Leener, M. Luhmer and A. Arduini, Tuning the Fluorescence Through Reorientation of the Axle in Calix[6]arene-Based Pseudorotaxanes, Chem.–Eur. J., 2020, 26, 3022–3025 CrossRef CAS PubMed.
- A. W. H. Ng, C.-C. Yee and H. Y. Au-Yeung, Radial Hetero[5]catenanes: Peripheral Isomer Sequences of the Interlocked Macrocycles, Angew. Chem., Int. Ed., 2019, 58, 17375–17382 CrossRef CAS PubMed.
- H. Y. Au-Yeung and A. W. H. Ng, Mechanical Interlocking of Macrocycles in Different Sequences, Synlett, 2020, 31, 309–314 CrossRef CAS.
- A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, Sequence Isomerism in [3]Rotaxanes, J. Am. Chem. Soc., 2010, 132, 4954–4959 CrossRef CAS PubMed.
- H.-X. Wang, S.-Z. Hu, Q. Shi and C.-F. Chen, Complexation between a triptycene-derived oxacalixarene and π-extended viologens: linker-length-dependent orientation of the macrocycles in pseudo[3]rotaxanes, Org. Biomol. Chem., 2016, 14, 10481–10488 RSC.
- C. Talotta, C. Gaeta, T. Pierro and P. Neri, Sequence stereoisomerism in calixarene-based pseudo[3]rotaxanes, Org. Lett., 2011, 13, 2098–2101 CrossRef CAS PubMed.
- C. Talotta, C. Gaeta and P. Neri, Stereoprogrammed direct synthesis of calixarene-based [3]rotaxanes, Org. Lett., 2012, 14, 3104–3107 CrossRef CAS PubMed.
- A. Arduini, F. Ciesa, M. Fragassi, A. Pochini and A. Secchi, Selective Synthesis of Two Constitutionally Isomeric Oriented Calix[6]arene-Based Rotaxanes, Angew. Chem., Int. Ed., 2005, 44, 278–281 CrossRef CAS PubMed.
- A. Arduini, R. Bussolati, A. Credi, A. Pochini, A. Secchi, S. Silvi and M. Venturi, Rotaxanes with a calix[6]arene wheel and axles of different length. Synthesis, characterisation, and photophysical and electrochemical properties, Tetrahedron, 2008, 64, 8279–8286 CrossRef CAS.
- A. Arduini, R. Bussolati, A. Credi, G. Faimani, S. Garaudée, A. Pochini, A. Secchi, M. Semeraro, S. Silvi and M. Venturi, Towards controlling the threading direction of a calix[6]arene wheel by using nonsymmetric axles, Chem.–Eur. J., 2009, 15, 3230–3242 CrossRef CAS PubMed.
- A. Credi, S. Dumas, S. Silvi, M. Venturi, A. Arduini, A. Pochini and A. Secchi, Viologen-Calix[6]arene Pseudorotaxanes. Ion-Pair Recognition and Threading/Dethreading Molecular Motions, J. Org. Chem., 2004, 69, 5881–5887 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and HR-MS spectral characterisation, UV/Vis and electrochemical data for the series of synthesised calix[6]arene-based [3]rotaxanes. See DOI: 10.1039/d1sc00279a |
| ‡ In weakly polar solvents, monostoppered viologen-based axles like 6a,b always thread the cavity of CX from its upper rim giving rise to orientational [2]pseudorotaxane isomers, see e.g. ref. 56. |
| § In a recent study, we demonstrated that the spontaneous threading of a ω-hydroxy endings tetracationic dumbbell inside two wheels, followed by a stoppering reaction, lead to the formation of R6UU as the only product (passive template synthesis). The comparison of the spectra of this product with those of R6UU from the active template synthesis {A} indirectly confirmed the goodness of the active template approach to obtain calix[6]arene-based [n]rotaxane structures with full control of the geometry of the components; M. Bazzoni, G. Orlandini, G. Cera, A. Secchi and A. Arduini, unpublished results. |
| ¶ The formation of paCo rotamers in calix[6]arene-based [2]pseudorotaxanes was already verified using both trisulfonamidocalix[6]arenes (see ref. 46) and CX (M. Bazzoni, G. Cera, A. Secchi and A. Arduini, unpublished results). |
| || For the full NMR characterisation of R6UL, see ESI.† |
| This journal is © The Royal Society of Chemistry 2021 |