
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tellura-Baeyer–Villiger oxidation: one-step transformation of tellurophene into chiral tellurinate lactone†‡
Shitao
Wang
,
Chaoxian
Yan
,
Wenlong
Zhao
,
Xiaolan
Liu
,
Cheng-Shan
Yuan
,
Hao-Li
Zhang
 and
Xiangfeng
Shao
*
and
Xiangfeng
Shao
*
State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Tianshui Southern Road 222, Lanzhou 730000, China. E-mail: shaoxf@lzu.edu.cn
First published on 12th March 2021
Abstract
Baeyer–Villiger (BV) oxidation is a fundamental organic reaction, whereas the hetero-BV oxidation is uncharted. Herein, a tellura-BV oxidation is discovered. By oxidizing a tellurophene-embedded and electron-rich polycycle (1) with mCPBA or Oxone, an oxygen atom is inserted into the Te–C bond of the tellurophene to form tellurinate lactone mono-2. This reaction proceeds as follows: (i) 1 is oxidized to the tellurophene Te-oxide form (IM-1); (ii) IM-1 undergoes tellura-BV oxidation to give mono-2. Moreover, the hybrid trichalcogenasumanenes 7 and 8 are, respectively, converted to tellurinate lactones mono-9 and mono-10 under the same conditions, indicating that tellura-BV oxidation shows high chemoselectivity. Due to the strong secondary bonding interactions between the Te![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups on tellurinate lactones, mono-2, mono-9, and mono-10 are dimerized to form U-shaped polycycles 2, 9, and 10, respectively. Notably, mono-2, mono-9, mono-10, and their dimers show chirality. This work enables one-step transformation of tellurophene into tellurinate lactone and construction of intricate polycycles.
O groups on tellurinate lactones, mono-2, mono-9, and mono-10 are dimerized to form U-shaped polycycles 2, 9, and 10, respectively. Notably, mono-2, mono-9, mono-10, and their dimers show chirality. This work enables one-step transformation of tellurophene into tellurinate lactone and construction of intricate polycycles.
Introduction
The Baeyer–Villiger (BV) oxidation was discovered more than a century ago,1,2 and great efforts were devoted to making BV oxidation catalytic, along with high chemo-, regio-, and stereoselectivity.3–7 In BV oxidation, the C–C bond adjacent to the carbonyl is opened and an oxygen atom is inserted to form C–O–C (see Scheme 1a for a typical example). The BV oxidation is an efficient way to transform ketones into esters and/or lactones and is employed to synthesize intricate chemicals.3–7 A variety of oxidants can be used to perform BV oxidation, such as meta-chloroperbenzonic acid (mCPBA), H2O2, Oxone, and so forth.8–10 According to the reaction mechanism of BV oxidation, which involves an intermediate known as the Criegee adduct,11 one may speculate whether it is possible to achieve hetero-BV oxidation. For instance, do organic compounds containing sulfoxide, selenoxide, or telluroxide undergo BV-type oxidation to form the corresponding esters and/or lactones?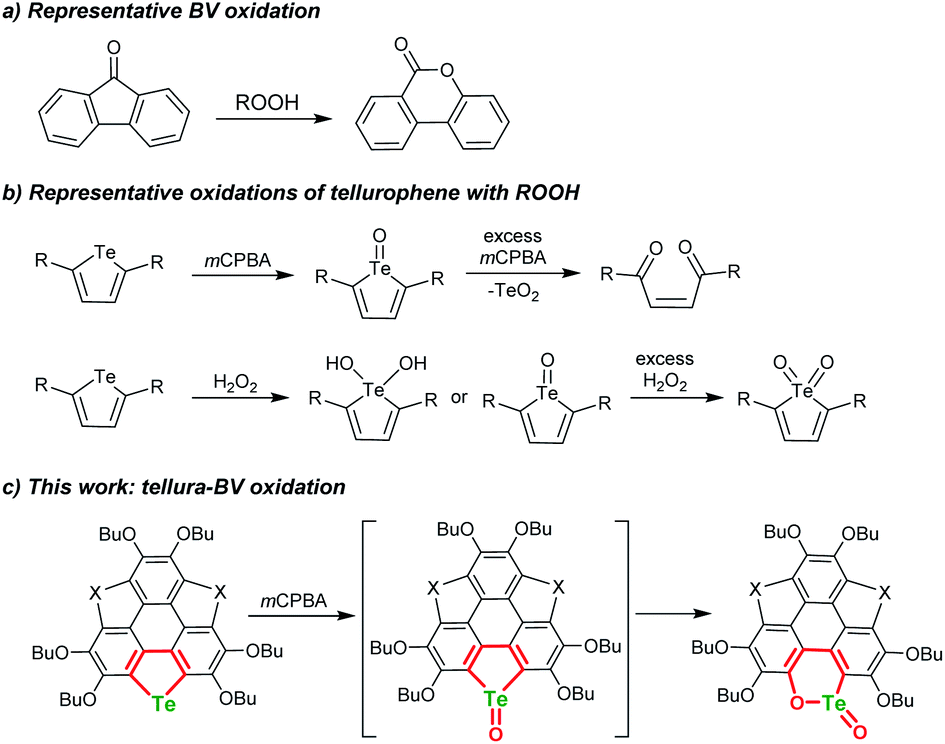 | ||
| Scheme 1 Schematic illustration of BV oxidation, oxidation of tellurophene by ROOH, and tellura-BV oxidation herein. | ||
Chalcogens (O, S, Se, and Te) have drawn tremendous interest in chemistry and materials science. Tellurium is the heaviest member of this family and shows maverick chemistry.12–17 The emerging development of tellurium compounds is apparent from their application in catalysis,18–21 coordination,22–26 and optoelectronic materials.27–34 The low Pauling electronegativity and high polarisability of tellurium lead to strong secondary bonding interactions (SBIs) and/or hypervalent states.12,35–38 Meanwhile, embedding Te onto the π-scaffolds of organotellurium compounds will narrow the HOMO–LUMO energy gap,39–42 produce intermolecular Te⋯X SBIs in the solid state,43–45 and alter their chemical behavior.46–52 Recently, Seferos's group performed the oxidation of tellurophenes with peroxides and found that they can be converted to various states including dihydroxy tellurophenes, telluroxides and tellurones (Scheme 1b).53,54 Further oxidation of tellurophene Te-oxide with mCPBA resulted in ring cleavage to form but-2-ene-1,4-dione instead of tellurinate lactone.53
In BV oxidation, the migration step is rate-determining in most cases and electron-donating substituents in the migrating group will promote the rearrangement8–10,55 Herein, with the intention of exploring hetero-BV oxidation, we chose tellurophene-embedded and electron-rich polycycles as substrates. By oxidizing them with mCPBA or Oxone, we discovered a tellura-BV oxidation (Scheme 1c). To the best of our knowledge, this kind of reaction has never been reported so far. This work enables the one-step conversion of tellurophene into chiral tellurinate lactone. Moreover, this reaction shows excellent chemoselectivity, i.e., only tellura-BV oxidation selectively occurs even if telluro-, seleno-, and thiophene moieties coexist in the substrates.
Results and discussion
An electron-rich polycycle 1 containing a tellurophene moiety was prepared from 2,3,6,7,10,11-hexabutoxytriphenylene (HBT) via the dilithiation and insertion of tellurium in one-pot (see the ESI‡).56–58 Upon oxidation of 1 by mCPBA (2 equiv., condition a) in CH2Cl2 at room temperature (r.t.), compound 2 was obtained in a yield of 52% (Scheme 2). The yield of 2 remained almost unchanged when excess mCPBA was used. On the other hand, compound 2 was harvested with a yield up to 82% by oxidizing 1 with Oxone (2 equiv., condition b) in a mixed solvent of THF–deionized water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). It is worth noting that the yield of 2 was distinctly lowered when tap water was used during the oxidation of 1 with Oxone. The reason is that the residual Cl− in tap water is converted to chlorine in the presence of Oxone,59,60 and chlorine will react with the tellurophene on 1 to form a hypervalent adduct.46–49,57
:1, v/v). It is worth noting that the yield of 2 was distinctly lowered when tap water was used during the oxidation of 1 with Oxone. The reason is that the residual Cl− in tap water is converted to chlorine in the presence of Oxone,59,60 and chlorine will react with the tellurophene on 1 to form a hypervalent adduct.46–49,57
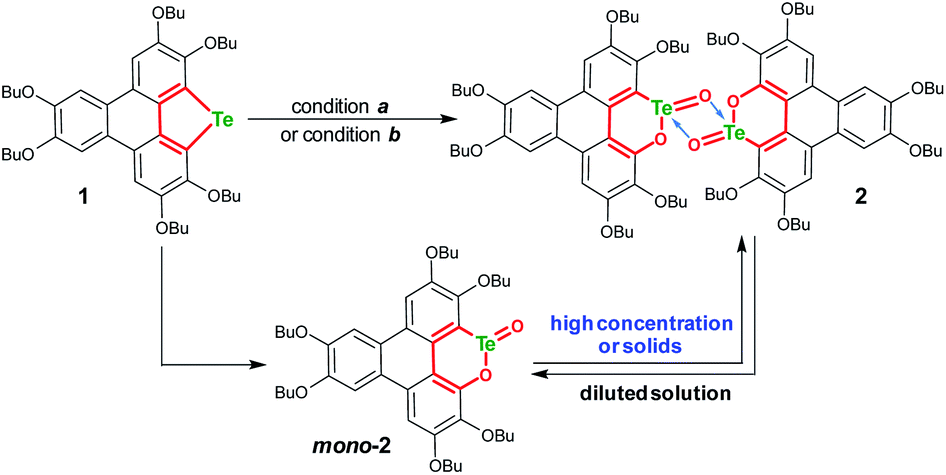 | ||
| Scheme 2 Transformation of 1 into dimeric tellurinate lactone 2. Reagents and conditions: (a) mCPBA (2 equiv.), CH2Cl2, r.t., 4 h, and yield 52%; (b) Oxone (2 equiv.), THF–deionized H2O (4:1, v/v), r.t., 4 h, and yield 82%. | ||
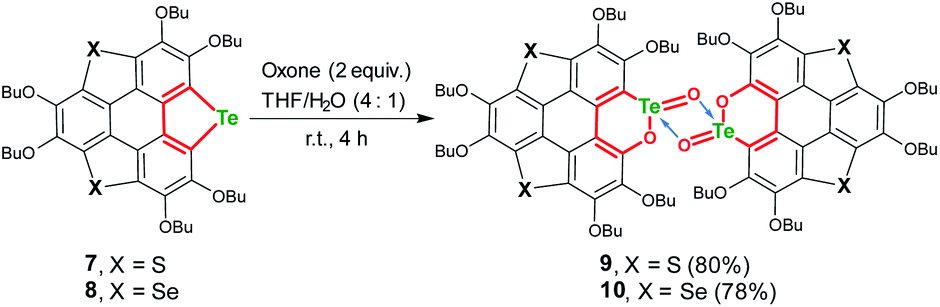
The structure of 2 was also determined through X-ray single crystal structure analysis. Referring to the crystal structure of 2 (Fig. 1), the tellurophene ring on 1 was opened and an oxygen atom was inserted to form six-member rings (A and B moieties). The bond lengths for the newly formed Te–O and O–C bonds in the A and B moieties are 2.02(1) and 1.36(1) Å, respectively (Fig. 1a). The A and B moieties are connected with a “Te2O2” four-member ring. The tellurium atoms in the “Te2O2” unit take the so-called four-center ten electron (10-E-4) hypervalent state.12 The “Te2O2” four-member ring is nearly perpendicular to the A and B moieties. As a result, compound 2 adopts a U-shaped conformation (Fig. 1b). According to the molecular structure of compound 2, this compound is non-centrosymmetric, and therefore it is a chiral molecule. However, a pair of enantiomers coexists in its crystal.
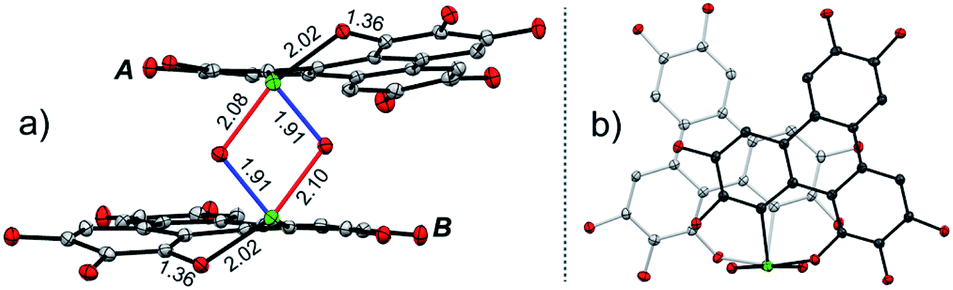 | ||
| Fig. 1 Crystal structure of 2, with n-Bu groups and hydrogen atoms omitted for clarity. (a) and (b) are, respectively, projected perpendicular and parallel to the “Te2O2” four-member ring. The selected bond lengths are shown in units of Å. The C, O, and Te atoms are, respectively, shown in grey, red, and green. | ||
There are two kinds of Te–O bonds in the “Te2O2” four-member ring of 2 with bond lengths of 1.91(1) and 2.09(1) Å (Fig. 1a). The former is close to the TeO bond length (1.89(1) Å) in PhTeO2 (ref. 61) and (C6F5)2TeO,62 and the latter is longer than the Te–O bond (1.97 Å) in X[(R2TeO)nTeR2]X.61,63 It was reported that the Te–X bond (where X is more electronegative than Te) is more polar in TeX, leading to stronger SBIs.12 For instance, the telluroxides usually exist as dimers owing to the strong intermolecular O → Te SBIs.64–67 Therefore, compound 2 can be regarded as a dimer of mono-2 formed through the intermolecular SBIs between the TeO bonds (Scheme 2). Accordingly, the two kinds of Te–O bonds in “Te2O2” can be reasonably assigned to TeO (1.91(1) Å) and O → Te (2.09(1) Å).
Consistent with the above deduction, we observed the ionic peak of mono-2 (C42H58O8Te + H+, 821.3257) in the high resolution mass spectrum (HRMS) of 2 as shown in Fig. 2a. This result implies that the dimerization of mono-2 to form 2 would be reversible under certain conditions. In this context, we carried out 1H NMR spectroscopic investigation of 2 at different concentrations (Fig. 2b). The transformation between mono-2 and 2 occurs indeed. It is found that compound 2 exists in the solution with a concentration higher than 4 × 10−4 mol L−1. As the concentration decreases, mono-2 gradually appears. When the concentration is below 8 × 10−5 mol L−1, compound 2 completely transforms into mono-2. On the other hand, the single crystals of 2 were harvested by slowly evaporating the solution of mono-2 (c ≤ 8 × 10−5 mol L−1). Therefore, mono-2 is the intrinsic product for the oxidation of 1 under condition a or b.
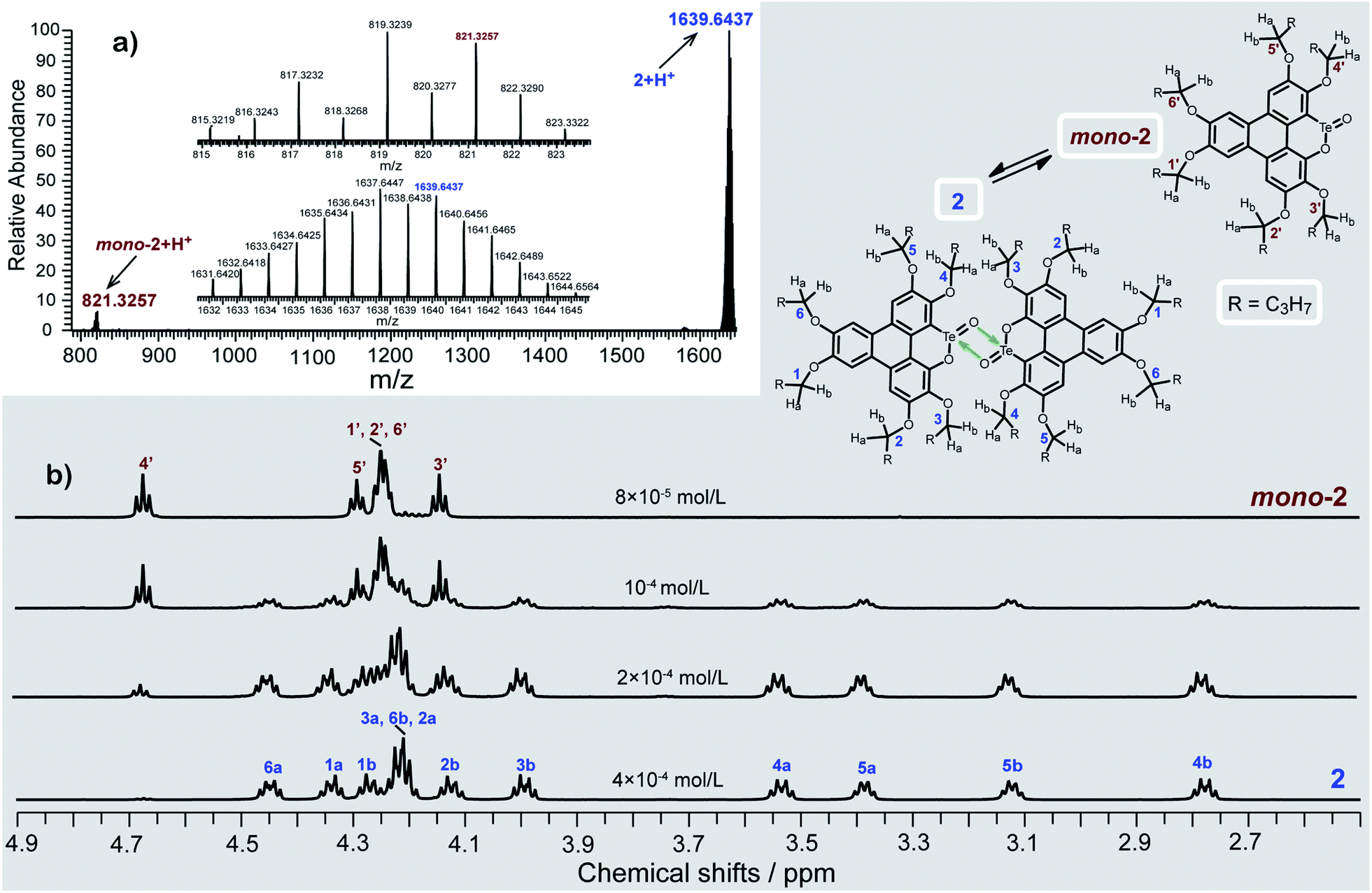 | ||
| Fig. 2 (a) HRMS spectrum and (b) variable-concentration 1H NMR spectra (n-butoxy region) in CDCl3 (600 MHz) of 2. The protons of 2 (n-butoxy region) are assigned with the assistance of two-dimensional NMR spectroscopy (Fig. S2–S6 and Tables S2, S3 in the ESI‡). | ||
The product mono-2 possesses a tellurinate lactone moiety, which would have originated from the oxidation of tellurophene Te-oxide. The formation of tellurinate lactone can be rationalized with a tellura-BV oxidation mechanism. We have proposed a mechanism for this transformation by employing mCPBA as an oxidant (Scheme 3). The tellurophene on 1 is first oxidized by mCPBA to afford a tellurophene Te-oxide intermediate IM-1.53 Owing to the highly polarized TeO bond,12 the IM-1 is easily attacked by mCPBA to form an intermediate IM-2, which resembles the Criegee adduct in BV oxidation. Finally, IM-2 undergoes a rearrangement to afford mono-2. Then mono-2 is dimerized in the concentrated solution and/or in the solid state to give 2. The reaction of 1 with Oxone would also follow the similar procedure. The high yield of 2 under condition b can be attributed to the acceleration of the rearrangement step, because the electron-deficient leaving group will facilitate the rearrangement.3–7
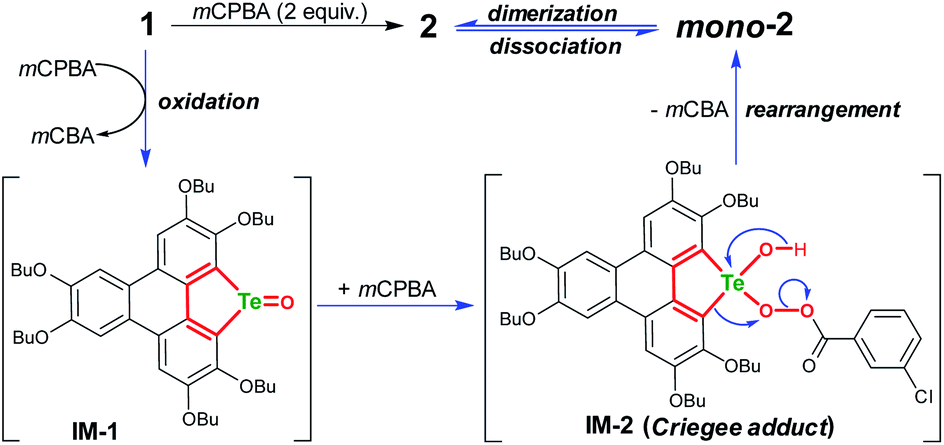 | ||
| Scheme 3 Proposed mechanism for converting 1 to 2via tellura-BV oxidation. | ||
To gain more insight into the mechanism, an attempt was made to identify the key intermediate IM-1 along the reaction pathway. It is known that tellurophene can be transformed into its Te-oxide form by oxidation with mCPBA.53 Thus, we performed the oxidation of compound 1 with 1 equivalent of mCPBA (Scheme 4). However, IM-1 cannot be isolated from the reaction mixture. Instead, compound 3 was obtained and its structure was confirmed by crystallographic analysis (Fig. 3). Because the TeO bond is prone to attack by nucleophiles,53 compound 3 would be generated via the IM-1 intermediate as follows: (i) compound 1 is oxidized by mCPBA to give IM-1, and mCPBA is reduced to meta-chlorobenzonic acid (mCBA); (ii) the IM-1 is attacked by mCBA to form an intermediate IM-3; (iii) the dehydration between two IM-3 molecules affords 3.68,69 This means that IM-1 is indeed formed when 1 is oxidized by mCPBA.
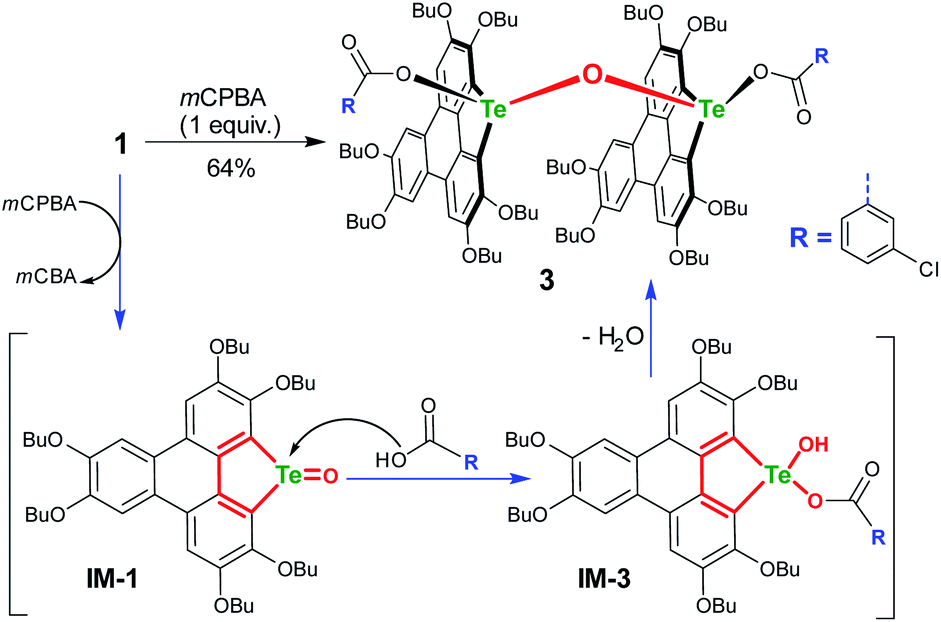 | ||
| Scheme 4 Oxidation of 1 with mCPBA (1 equiv.) in CH2Cl2 at r.t. | ||
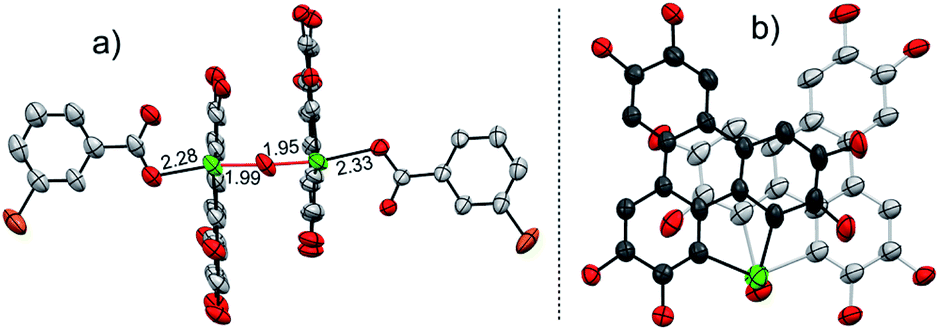 | ||
| Fig. 3 Crystal structure of 3, with n-Bu groups and hydrogen atoms omitted for clarity. (a) and (b) are, respectively, projected perpendicular and parallel to the HBT ring. The mCBA moieties are omitted in (b). The Cl atoms are shown in orange. | ||
Referring to the crystal structure of 3, the Te–O bonds (2.28–2.33 Å) between mCBA ester and Te are much longer than the typical Te–O single bond (1.97 Å),61 implying that the dissociation of these Te–O bonds would occur to recover IM-1 under certain conditions. Actually, the HRMS spectrum of 3 shows only the ionic peak of IM-1 (C42H58O7Te + H+, 805.3340). We also found that 3 was transformed into 2 when excess mCPBA was added. The transformation of 3 into 2 is therefore interpreted as follows: (i) the mCBA ester groups on 3 are hydrolyzed under acidic conditions (herein, mCPBA), followed by breaking of the Te–O–Te bridge to form IM-1; (ii) the IM-1 undergoes tellura-BV oxidation and successive dimerization to afford 2.
We then studied the electronic effect on tellura-BV oxidation. Employing a similar synthetic approach to that of compound 1, a polycycle containing a tellurophene moiety (compound 4) was prepared from phenanthrene. In comparison with 1, compound 4 doesn't possess the electron-rich substituents. As shown in Scheme 5, oxidation of 4 with excess mCPBA didn't afford tellurinate lactone 6, and only 5 was isolated. The formation of 5 is attributed to the transesterification during the purification of crude products via column chromatography, where the eluent contains formic acid. Along with previous reports,53,54 the present result further indicates that electron-donating substituents are essential for tellura-BV oxidation.
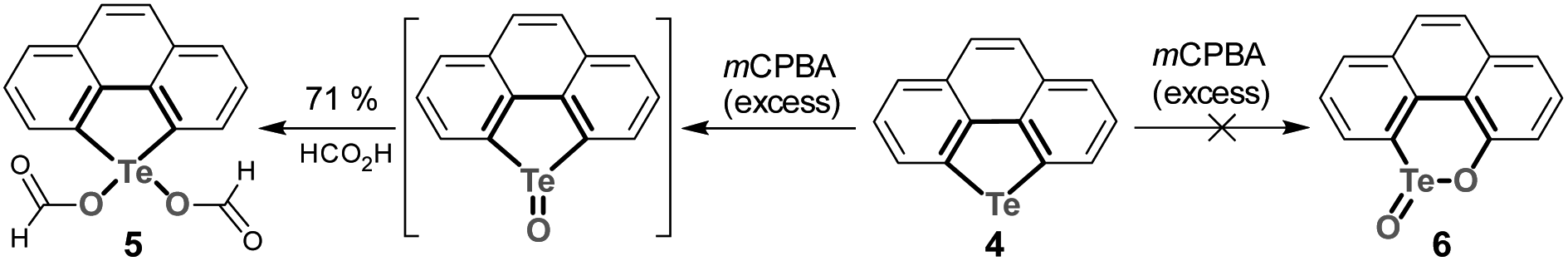 | ||
| Scheme 5 Oxidation of 4 with excess mCPBA in CH2Cl2. | ||
To further understand tellura-BV oxidation, we intend to clarify the chemoselectivity of this reaction. Considering that electron-donating substituents are required for tellura-BV oxidation, trichalcogenasumanenes (TCSs) which bear butoxy groups on the flank benzene rings would be suitable substrates. Previously, we achieved the synthesis of such TCSs56–58 and disclosed their chemical reactivities under different conditions.70–76 Particularly, the oxidative cleavage of flank benzene on trithia- and triselenasumanene was observed by using Oxone as the oxidant.70 To figure out the chemoselectivity of tellura-BV oxidation, we took TCSs 7 and 8 as substrates, which contain tellurophene and thio-/selenophene moieties (Scheme 6).58 Upon oxidation by Oxone (2 equiv.), compounds 7 and 8 were, respectively, converted to 9 and 10 which had similar structures to 2. Notably, the ring-opening of flank benzene and/or oxidation at S/Se atoms of 7/8 was not observed under these conditions. In diluted solution, the “Te2O2” four-member rings on 9/10 are dissociated to afford mono-9/mono-10, the same as that of 2. These results indicate that tellura-BV oxidation shows high chemoselectivity, since the chemical reactivities of hybrid TCSs are dominated by heavier chalcogenium, as we have concluded previously.58
 | ||
| Scheme 6 Conversion of TCSs 7/8 into dimeric tellurinate lactones 9/10. | ||
The structures of 9 and 10 were also determined by X-ray single crystal structure analyses. The crystal structures of 9 and 10 show the same features, and Fig. 4 depicts the crystal structure of 10 as a representative example. The molecular structure of 10 is reminiscent of that of 2. Two mono-10 are dimerized via a “Te2O2” four-member ring to form a non-centrosymmetric U-shaped structure. The tellurium atoms on the “Te2O2” four-member ring take the “10-E-4” hypervalent state (Fig. 4a). Two kinds of Te–O bonds in “Te2O2” are observed with bond lengths of 1.91(1) and 2.07(2) Å, respectively, assigned to TeO and O → Te. Compound 10 forms helical arrays in the crystal with various intermolecular atomic contacts between neighboring molecules (Fig. 4c), i.e., Te⋯C (3.44–3.70 Å), Te⋯O (2.95–3.33 Å), Te⋯Te (4.06–4.09 Å), and O⋯O (2.85–3.02 Å). While 10 is a chiral molecule, a pair of enantiomers coexists in its crystal.
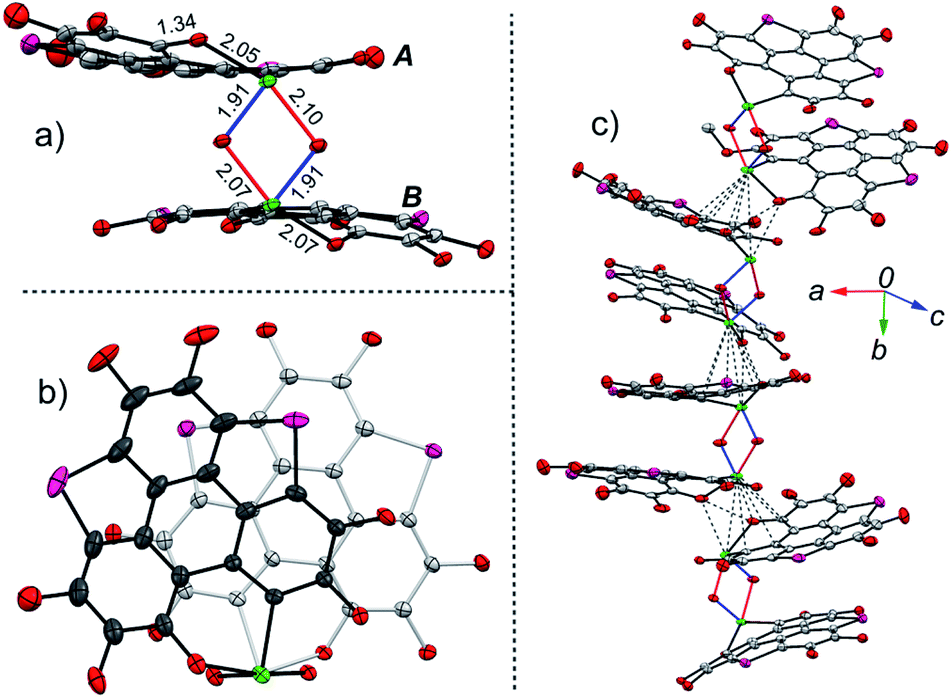 | ||
| Fig. 4 Crystal structure of 10, with n-Bu groups and hydrogen atoms omitted for clarity: (a) and (b) are molecular structures, respectively, projected perpendicular and parallel to the “Te2O2” four-member ring; (c) packing motif with intermolecular contacts shown in grey dashed lines. The Se atoms are shown in pink. | ||
It is worth noting that 2, 9, and 10 show good thermal stability at a decomposition temperature higher than 200 °C (Fig. S1 and Table S1 in the ESI‡). 2, 9, and 10 are solvable in organic solvents such as CH2Cl2, CHCl3, toluene, and THF, whereas they show poor solubility in methanol.
We elucidated the influence on electronic states by converting tellurophene into tellurinate lactone and its dimerized form. Herein, we take 8, mono-10, and 10 as representative examples, and those for the 1 and 7 series show similar phenomena. Theoretical studies reveal that the HOMO energy levels remain almost unchanged for 8, mono-10, and 10. The LUMO energy levels of mono-10 and 10 are the same, and both are clearly lower than that of 8 (Fig. 5a). This is caused by the electron-withdrawing unit (TeO) in mono-10 and 10. Consequently, the HOMO–LUMO energy gaps (Eg) of mono-10 (Eg = 3.75 eV) and 10 (Eg = 3.78 eV) are narrower than that of 8 (Eg = 4.19 eV). Consistent with the theoretical study, the UV-Vis absorption spectra in CH2Cl2 for mono-10 (c = 1 × 10−5 mol L−1) and 10 (c = 5 × 10−4 mol L−1) are almost identical (Fig. 5b). The compounds mono-10 and 10 exhibit broad absorption bands at 382–440 nm originating from the HOMO–LUMO transition, which is red-shifted compared to that of 8 (368–414 nm). The compounds mono-10 and 10 are non-emissive at r.t., but they show emissions at 77 K (Fig. 5c), ascribable to phosphorescence. The emission of 10 is clearly red-shifted compared to that of mono-10, which would be due to the excimer emission since the two π-frameworks of 10 are arranged face-to-face (see Fig. 4b). Similar phenomena are also observed for 2 and 9 (Fig. S26 and S27‡).
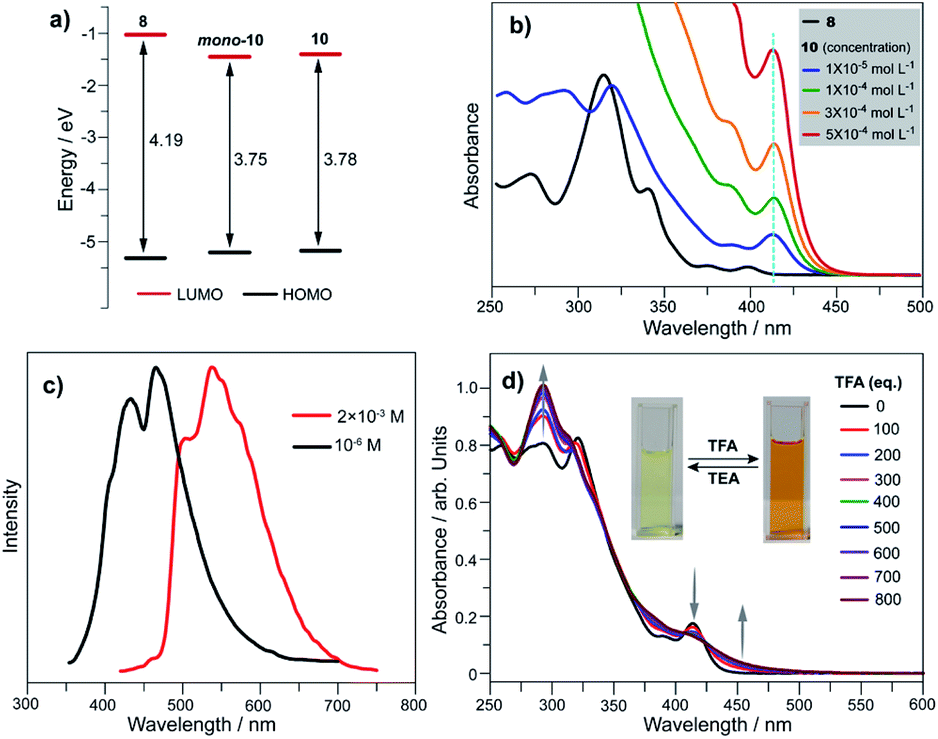 | ||
| Fig. 5 (a) Calculated energy levels for 8, mono-10, and 10; (b) variable-concentration UV-Vis absorption spectra of 10 in CH2Cl2, along with those of 8 (c = 1 × 10−5 mol L−1) for comparison. The cyan dashed line is a guide to the eye for the absorption maximum at long wavelength band. (c) The emission spectra of mono-10 (c = 1 × 10−6 mol L−1) and 10 (c = 2 × 10−3 mol L−1) in THF at 77 K; (d) UV-Vis absorption spectra of 10 in CH2Cl2 upon titration with TFA. | ||
Referring to Fig. 5d, the UV-Vis absorption of 10 is distinctly changed upon adding trifluoroacetic acid (TFA). Acidification of 10 also leads to a clear down-field shift of 125Te NMR (Fig. 6a). On the other hand, the original absorption bands and 1H NMR spectra of 10 will be restored when the acidified solution of 10 is neutralized by triethylamine (TEA) as shown in Fig. S25.‡ Thus, it is a reversible process and a proposed mechanism is shown in Fig. S25.‡ Variations in absorption spectra and 1H NMR of 10 are also observed in the presence of HCl (3 N, aqueous). However, the original spectra of 10 are not recovered by neutralization. 10 was quantitatively converted into a new product when it was reacted with HCl (3 N, aqueous). Single crystal structure analysis reveals that the tellurinate lactone moiety of 10 is destroyed and two Cl atoms are attached onto the Te atom to give compound 12 (Fig. 6b). The mechanism for the formation of 12 is shown in Scheme 7.
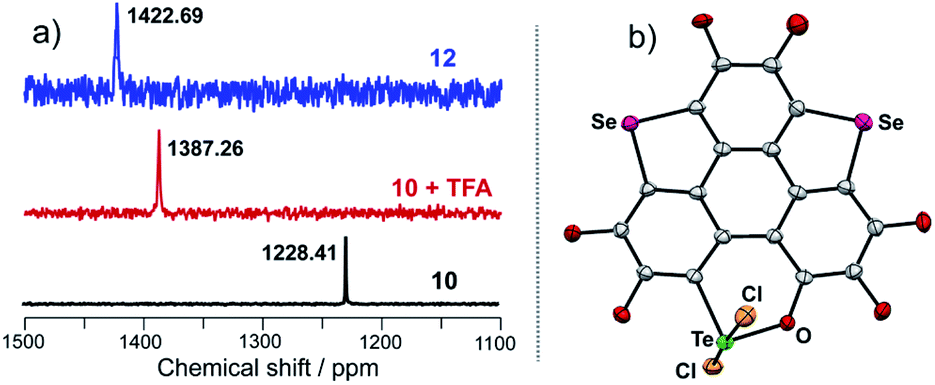 | ||
| Fig. 6 (a) 125Te NMR of 10, 12, and 10 + TFA measured in CDCl3, where chemical shifts are recorded using 125Te NMR of diphenyl ditelluride (δ = 420 ppm) as an external reference; (b) crystal structure of 12 with n-Bu groups omitted. | ||
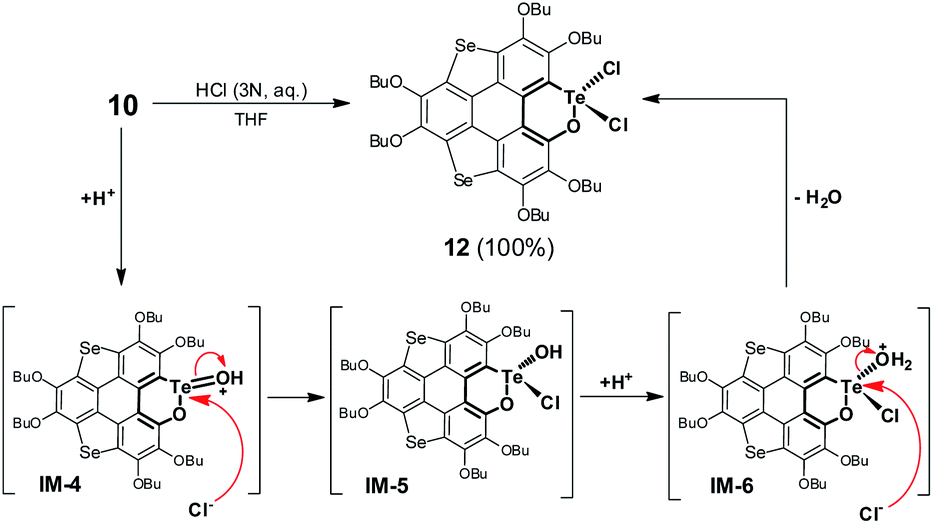 | ||
| Scheme 7 Transformation of 10 into 12 upon reaction with aqueous HCl. | ||
2, 9, and 10 possess chirality, whereas they are obtained as enantiomers. We separated the enantiomers of 2, 9, and 10via chiral HPLC on a Daicel CHIRALPAK IF (IF00CE-SL018) column (eluent: hexane/CH2Cl2 = 50/50). Fig. 7a shows the chiral chromatogram of 10 as a typical example. Two fractions of 10 have retention times of 3.4 min and 5.3 min, and are isolated with >93% ee. Circular dichroism (CD) spectra of the two fractions show a mirror symmetry (Fig. 7b), indicating that they have opposite chirality. According to the positive and negative Cotton effects at 378 nm, the two fractions are denoted as (+)-10 and (−)-10. Based on the molecular structure of 10 in a single crystal, we performed CD spectra simulation via TD-DFT calculation at the TD-PBE0/Def2-SVP level, where the butyl groups on (+)-10 and (−)-10 were replaced with methyl groups to give (+)-10′ and (−)-10′. The simulated CD spectra of (+)-10′ and (−)-10′ are in good agreement with the experimental results, and give the structure of each isomer as shown in Fig. 7c.
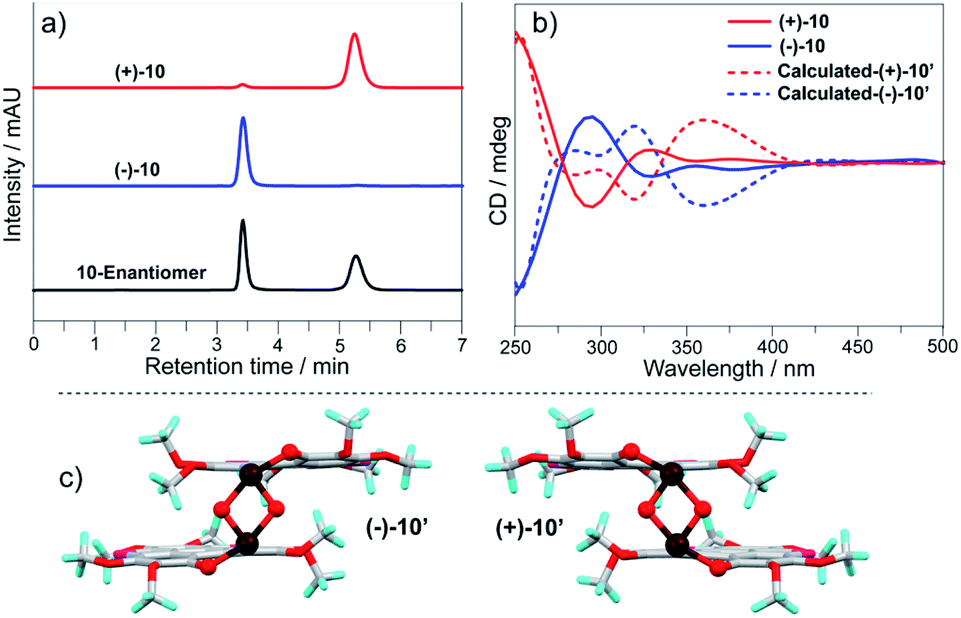 | ||
| Fig. 7 (a) Chiral chromatograms of 10; (b) CD spectra of (+)-10/(−)-10 in CH2Cl2 (c = 6 × 10−4 mol L−1) along with the simulated ones; (c) molecular structures of (+)-10′ and (−)-10′. | ||
Conclusions
In summary, we have discovered a tellura-BV oxidation which enables the one-step transformation of tellurophene into the chiral tellurinate lactone. Owing to the strong SBIs between TeO groups, the resulting tellurinate lactone is dimerized via the formation of a hypervalent “Te2O2” four-member ring in the solid state. This reaction shows good chemoselectivity, i.e., tellura-BV oxidation selectively occurs when the telluro-, seleno-, and thiophene moieties coexist in the substrates. It was also found that electron-donating substituents are essential for tellura-BV oxidation. Employing tellura-BV oxidation as a vital step, the hybrid trichalcogenasumanenes are transformed into the U-shaped polycycles. Further investigation will be directed to the development of stereoselective tellura-BV oxidation as well as the asymmetric synthesis and application of the U-shaped polycycles. Such work is ongoing in our laboratory.
Author contributions
S. W. conducted the synthesis, crystal structure analysis, and most of the measurements. C. Y. carried out the theoretical calculation. W. Z. supported the synthesis. X. L. contributed to the 125Te NMR measurement. C.-S. Y. contributed to the analysis of NMR spectra (including 2D NMR). H.-L. Z. contributed to the analysis and discussion on optical spectra. X. S. conceived the ideas and designed the project. S. W. and X. S. wrote the paper. All authors approved the final version of the manuscript.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
The authors acknowledge the grant from the National Natural Science Foundation of China (21871119, 21522203) and the National Key R&D Program of China (2017YFA0204903). We greatly appreciate Mr Fengming Qi at SKLAOC for the measurement of 125Te NMR.Notes and references
- A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625 CrossRef.
- A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1900, 33, 858 CrossRef CAS.
- G. R. Krow, in Organic Reaction: The Baeyer-Villiger Oxidation of Ketones and Aldehydes, ed. L. A. Paquette, John Wiley & Sons, Inc., 1993, vol. 43 Search PubMed.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 1999, 737 CrossRef.
- G. J. Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105 CrossRef CAS.
- L. Zhou, L. Lin, X. Liu, and X. Feng, in Molecular Rearrangements in Organic Synthesis: Baeyer-Villiger Oxidation/Rearrangement in Organic Synthesis, ed. C. M. Christian, 1st edn, John Wiley & Sons, Inc., 2016, vol. 2 Search PubMed.
- G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198 CrossRef PubMed.
- A. Berkessel, M. R. M. Andreae, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 2002, 41, 4481 CrossRef CAS PubMed.
- S.-I. Murahashi, S. Ono and Y. Imada, Angew. Chem., Int. Ed., 2002, 41, 2366 CrossRef CAS.
- M. Uyanik, D. Nakashima and K. Ishihara, Angew. Chem., Int. Ed., 2012, 51, 9093 CrossRef CAS PubMed.
- R. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127 CrossRef CAS.
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725 RSC.
- G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182 RSC.
- A. Nordheider, J. D. Woollins and T. Chivers, Chem. Rev., 2015, 115, 10378 CrossRef CAS PubMed.
- L. C. Roof and J. W. Kolis, Chem. Rev., 1993, 93, 1037 CrossRef CAS.
- Z. He, Y. Yang, J. W. Liu and S. H. Yu, Chem. Soc. Rev., 2017, 46, 2732 RSC.
- T. G. Chasteen and R. Bentley, Chem. Rev., 2003, 103, 1 CrossRef CAS PubMed.
- Z. Dong, X. Li, K. Liang, S. Mao, X. Huang, B. Yang, J. Xu, J. Liu, G. Luo and J. Shen, J. Org. Chem., 2007, 72, 606 CrossRef CAS PubMed.
- Z. Z. Huang and Y. Tang, J. Org. Chem., 2002, 67, 5320 CrossRef CAS PubMed.
- I. D. Rettig, J. Van, J. B. Brauer, W. Luo and T. M. McCormick, Dalton Trans., 2019, 48, 5665 RSC.
- M. Oba, M. Endo, K. Nishiyama, A. Ouchi and W. Ando, Chem. Commun., 2004, 1672 RSC.
- A. Rahaman, G. C. Lisensky, D. A. Tocher, M. G. Richmond, G. Hogarth and E. Nordlander, J. Organomet. Chem., 2018, 867, 381 CrossRef CAS.
- A. K. Singh and S. Sharma, Coord. Chem. Rev., 2000, 209, 49 CrossRef CAS.
- V. K. Jain and R. S. Chauhan, Coord. Chem. Rev., 2016, 306, 270 CrossRef CAS.
- S. Wang, C. Yan, J. Shang, W. Wang, C. Yuan, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2019, 58, 3819 CrossRef CAS PubMed.
- I. D. Sadekov, A. A. Maksimenko, A. G. Maslakov and V. I. Minkin, J. Organomet. Chem., 1990, 391, 179 CrossRef CAS.
- M. Kaur, D. S. Yang, K. Choi, M. J. Cho and D. H. Choi, Dyes Pigm., 2014, 100, 118 CrossRef CAS.
- A. A. Soares-Paulino, L. Giroldo, G. Celante, E. Oliveira, S. M. Santos, R. F. Mendes, F. A. Almeida Paz, A. M. Fioroto, P. V. Oliveira, S. H. P. Serrano, C. Lodeiro and A. A. DosSantos, Dyes Pigm., 2019, 160, 208 CrossRef CAS.
- T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254 CrossRef CAS PubMed.
- P. F. Li, T. B. Schon and D. S. Seferos, Angew. Chem., Int. Ed., 2015, 54, 9361 CrossRef CAS PubMed.
- G. Li, L. Xu, W. Zhang, K. Zhou, Y. Ding, F. Liu, X. He and G. He, Angew. Chem., Int. Ed., 2018, 57, 4897 CrossRef CAS PubMed.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096 CrossRef CAS PubMed.
- G. Li, B. Zhang, J. Wang, H. Zhao, W. Ma, L. Xu, W. Zhang, K. Zhou, Y. Du and G. He, Angew. Chem., Int. Ed., 2019, 58, 8468 CrossRef CAS PubMed.
- E. I. Carrera and D. S. Seferos, Macromolecules, 2015, 48, 297 CrossRef CAS.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357 CrossRef CAS PubMed.
- N. W. Alcock and W. D. Harrison, J. Chem. Soc., Dalton Trans., 1982, 1421 RSC.
- J. Beckmann, D. Dakternieks, A. Duthie, N. A. Lewcenko and C. Mitchell, Angew. Chem., Int. Ed., 2004, 43, 6683 CrossRef CAS PubMed.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010 CrossRef CAS PubMed.
- A. Aprile, K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Inorg. Chem., 2015, 54, 4934 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314 CrossRef CAS PubMed.
- A. Muranaka, S. Yasuike, C. Y. Liu, J. Kurita, N. Kakusawa, T. Tsuchiya, M. Okuda, N. Kobayashi, Y. Matsumoto, K. Yoshida, D. Hashizume and M. Uchiyama, J. Phys. Chem. A, 2009, 113, 464 CrossRef CAS PubMed.
- M. Planells, B. C. Schroeder and I. McCulloch, Macromolecules, 2014, 47, 5889 CrossRef CAS.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed.
- R. D. McCullough, G. B. Kok, K. A. Lerstrup and D. O. Cowan, J. Am. Chem. Soc., 1987, 109, 4115 CrossRef CAS.
- A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Org. Lett., 2009, 11, 1487 CrossRef CAS PubMed.
- E. I. Carrera, A. E. Lanterna, A. J. Lough, J. C. Scaiano and D. S. Seferos, J. Am. Chem. Soc., 2016, 138, 2678 CrossRef CAS PubMed.
- E. I. Carrera and D. S. Seferos, Dalton Trans., 2015, 44, 2092 RSC.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 3542 CrossRef CAS PubMed.
- E. I. Carrera, T. M. McCormick, M. J. Kapp, A. J. Lough and D. S. Seferos, Inorg. Chem., 2013, 52, 13779 CrossRef CAS PubMed.
- A. Alka, V. S. Shetti and M. Ravikanth, Dalton Trans., 2019, 48, 4444 RSC.
- L. Latos-Grażyński, E. Pacholska, P. J. Chmielewski, M. M. Olmstead and A. L. Balch, Angew. Chem., Int. Ed., 1995, 34, 2252 CrossRef.
- M. Abe, M. R. Detty, O. O. Gerlits and D. K. Sukumaran, Organometallics, 2004, 23, 4513 CrossRef CAS.
- E. I. Carrera and D. S. Seferos, Organometallics, 2017, 36, 2612 CrossRef CAS.
- T. M. McCormick, E. I. Carrera, T. B. Schon and D. S. Seferos, Chem. Commun., 2013, 49, 11182 RSC.
- J. A. Berson and S. Suzuki, J. Am. Chem. Soc., 1959, 81, 4088 CrossRef CAS.
- X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535 CrossRef CAS PubMed.
- S. Wang, X. Li, X. Hou, Y. Sun and X. Shao, Chem. Commun., 2016, 52, 14486 RSC.
- S. Wang, J. Shang, C. Yan, W. Wang, C. Yuan, H.-L. Zhang and X. Shao, Org. Chem. Front., 2019, 6, 263 RSC.
- G. Zhao, L. Liang, C. H. E. Wen and R. Tong, Org. Lett., 2019, 21, 315 CrossRef CAS PubMed.
- J. Ren and R. Tong, Org. Biomol. Chem., 2013, 11, 4312 RSC.
- N. W. Alcock and W. D. Harrison, J. Chem. Soc., Dalton Trans., 1982, 709 RSC.
- D. Naumann, W. Tyrra, R. Herrmann, I. Pantenburg and M. S. Wickleder, Z. Anorg. Allg. Chem., 2002, 628, 833 CrossRef CAS.
- J. Beckmann, D. Dakternieks, A. Duthie, F. Ribot, M. Schürmann and N. A. Lewcenko, Organometallics, 2003, 22, 3257 CrossRef CAS.
- K. Srivastava, A. Panda, S. Sharma and H. B. Singh, J. Organomet. Chem., 2018, 861, 174 CrossRef CAS.
- A. Gupta, R. Deka, A. Sarkar, H. B. Singh and R. J. Butcher, Dalton Trans., 2019, 48, 10979 RSC.
- J. Beckmann, J. Bolsinger, P. Finke and M. Hesse, Angew. Chem., Int. Ed., 2010, 49, 8030 CrossRef CAS PubMed.
- J. Beckmann, P. Finke, M. Hesse and B. Wettig, Angew. Chem., Int. Ed., 2008, 47, 9982 CrossRef CAS PubMed.
- P. Singh, A. K. S. Chauhan, R. J. Butcher and A. Duthie, Polyhedron, 2013, 62, 227 CrossRef CAS.
- P. Thavornyutikarn and W. R. McWhinnie, J. Organomet. Chem., 1973, 50, 135 CrossRef CAS.
- X. Li, Y. Zhu, J. Shao, L. Chen, S. Zhao, B. Wang, S. Zhang, Y. Shao, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2015, 54, 267 CrossRef CAS PubMed.
- X. Hou, Y. Zhu, Y. Qin, L. Chen, X. Li, H.-L. Zhang, W. Xu, D. Zhu and X. Shao, Chem. Commun., 2017, 53, 1546 RSC.
- Y. Sun, X. Li, C. Sun, H. Shen, X. Hou, D. Lin, H.-L. Zhang, C. A. Di, D. Zhu and X. Shao, Angew. Chem., Int. Ed., 2017, 56, 13470 CrossRef CAS PubMed.
- X. Hou, J. Sun, Z. Liu, C. Yan, W. Song, H.-L. Zhang, S. Zhou and X. Shao, Chem. Commun., 2018, 54, 10981 RSC.
- X. Hou, Y. Sun, L. Liu, S. Wang, R. Geng and X. Shao, Chin. Chem. Lett., 2016, 27, 1166 CrossRef CAS.
- D. Li and X. Shao, Synlett, 2020, 31, 1050 CrossRef CAS.
- W. Wang and X. Shao, Org. Biomol. Chem., 2021, 19, 101 RSC.
Footnotes |
| † Dedicated to Prof. Zhongli Liu on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, 1H-NMR, 13C-NMR spectra, HRMS, IR, single crystal structure, UV, CD, theoretical calculations and TGA. CCDC 2035868, 2035866, 2035865, 2035863, 2064765 and 2064766. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00397f |
| This journal is © The Royal Society of Chemistry 2021 |