
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly selective decarboxylative deuteration of carboxylic acids†
Nian
Li
a,
Yunyun
Ning
a,
Xiaopeng
Wu
a,
Jin
Xie
 *ab,
Weipeng
Li
*a and
Chengjian
Zhu
*ac
*ab,
Weipeng
Li
*a and
Chengjian
Zhu
*ac
aState Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: xie@nju.edu.cn; lwp1989@nju.edu.cn; cjzhu@nju.edu.cn
bAdvanced Catalytic Engineering Research Center of the Ministry of Education, Hunan University, Changsha 410082, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai 200032, China
First published on 3rd March 2021
Abstract
In this paper, we report a mild and practical method for precise deuteration of aliphatic carboxylic acids by synergistic photoredox and HAT catalysis. The reaction delivers excellent D-incorporation (up to 99%) at predicted sites even in substrates bearing reactive C–H bonds or versatile functional groups. The use of a recirculation reactor with a peristaltic pump supports a scalable preparative ability (up to 50 mmol) under very mild reaction conditions. The practical and precise deuteration of readily available complex carboxylic acids makes this protocol promising for the preparation of deuterium-labelled compounds.
Introduction
Deuterium-containing organic molecules are being used increasingly in synthetic chemistry, medicinal chemistry and organic materials.1 For example, deutetrabenazine, the first deuterated drug (Austedo) approved in 2017 by the FDA, was developed in part because deuterated drugs can potentially affect the metabolic sites and thus improve the pharmacokinetic properties. To further accelerate the optimization of lead candidates, the development of practical and scalable synthetic methods to precisely incorporate deuterium atoms into different kinds of organic skeletons is very meaningful.2 The most reliable strategies for deuterium incorporation rely on the metalation of the corresponding organic halides3 or of reactive C–H4 bonds to generate organometallic derivatives for subsequent deuteration. Recent efforts have focused on transition metal catalyzed H/D exchange.5 However, most of these methods generally suffer from poor functional group compatibility and uncontrolled chemoselectivity, particularly when the substrates possess weak chemical bonds or competing reactive reaction sites (Fig. 1a).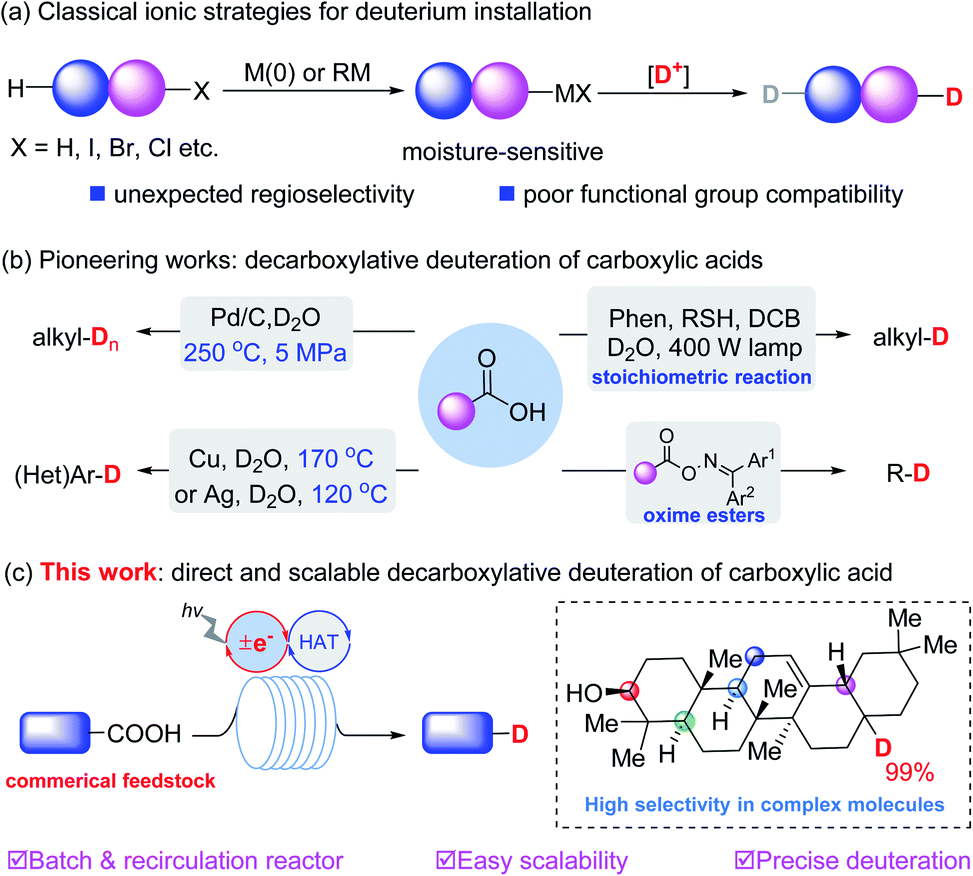 | ||
| Fig. 1 Classical deuteration strategies and decarboxylative deuteration approaches. (a) Classical ionic strategies. (b) Pioneering works. (c) This work. | ||
Carboxylic acids are common commercially available feedstocks.6 Decarboxylative strategies enable carboxylic acids to serve as traceless linkers with which to construct C–C and C–X bonds (X = F, Cl, N3).7 In view of the diverse skeletons of carboxylic acids, this type of reaction could significantly enhance the development of a useful library of deuterated organic molecules. In contrast with well-developed radical dehalogenative deuteration of organic halides,8 decarboxylative deuteration represents an elegant access to deuterated molecules from commercially available starting materials. For example, in 2004, Oshima et al.9a reported Pd-catalyzed decarboxylative deuteration of aliphatic carboxylic acids at 250 °C and 5 MPa. In 2010, Hatanaka et al.10 employed a 400 W high-pressure mercury lamp and a stoichiometric photosensitizer for the deuteration of alkyl carboxylic acids. Later, Gooßen et al.9b disclosed Cu or Ag-catalyzed decarboxylative deuteration of (hetero)aromatic carboxylic acids at elevated temperatures. Recently, deuteration of oxime carboxylate was developed by Glorius and coworkers.11
Despite these great efforts, the development of a scalable, practical and site-specific decarboxylative deuteration strategy is still highly desired. Pursuant to our previous work on synergistic catalysis,12 we designed a new strategy to achieve precise deuteration with D2O of a wide array of readily available acids via synergistic thiol and photoredox catalysis (Fig. 1c), efficiently affording structurally diverse deuterated molecules with up to 99% deuterium incorporation. We have explored the practicality of the method via recirculation reactors, developing a protocol which is easily scalable (up to 50 mmol) in laboratories.
Results and discussion
The proposed mechanism of decarboxylative deuteration is illustrated in Fig. 2a. Upon absorbing visible light, the photo-excited PC* with a high redox potential can accept an electron from a carboxylate anion and generate an alkyl radical (4) by decarboxylation.13 Due to the stronger BDE of the O–D bond (118 kcal mol−1) in D2O than that of normal C(sp3)–H bonds (ca. 100–110 kcal mol−1),14 direct hydrogen atom transfer between an alkyl radical (4) and D2O is thermodynamically unfavorable.15 Taking advantage of the pioneering work on radical deuteration,12a,b we used a thiol as the HAT catalyst. In the presence of excess D2O, it will undergo rapid H/D exchange to form RS–D because its pKa value will be lower than that of water (pKa = 32).16 Subsequently, the alkyl radical (4) will enter into a HAT process with R–SD (5) to furnish the desired product (3) and a thiyl radical (6). This can further accept an electron from the photocatalytic system to complete the HAT catalysis and photoredox cycle.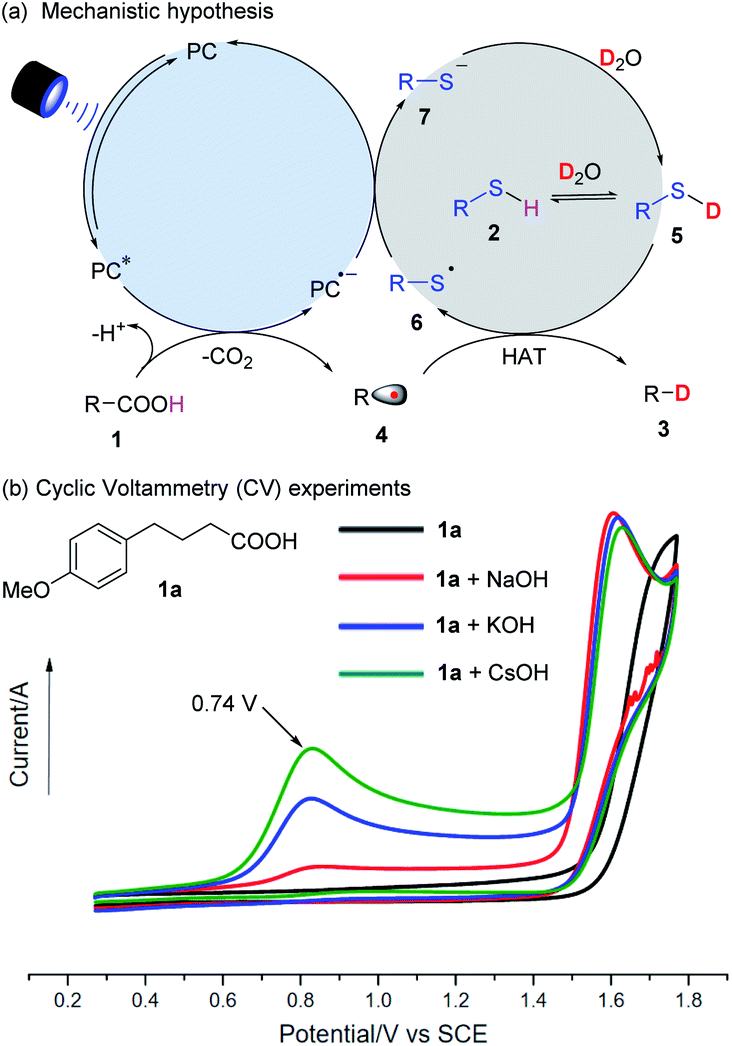 | ||
| Fig. 2 (a) Mechanistic hypothesis. (b) Cyclic Voltammetry (CV) experiments. | ||
As shown in Fig. 2b, the alkyl carboxylic acid (1a) does not have an oxidation potential in the range of 0–2 V, but a significant peak at 0.74 V vs. SCE (the oxidation potential of a carboxylate) is seen when a stoichiometric inorganic base, especially CsOH is added. It was presumed that the use of CsOH could benefit the formation of cesium carboxylate, which can undergo single electron oxidation and be decarboxylated. Radical trapping experiments with 2,2,6,6-tetramethylpiperidinooxy (TEMPO) and EPR results showed that the radical pathway was highly likely, consistent with the proposed mechanism shown in Fig. 2a.
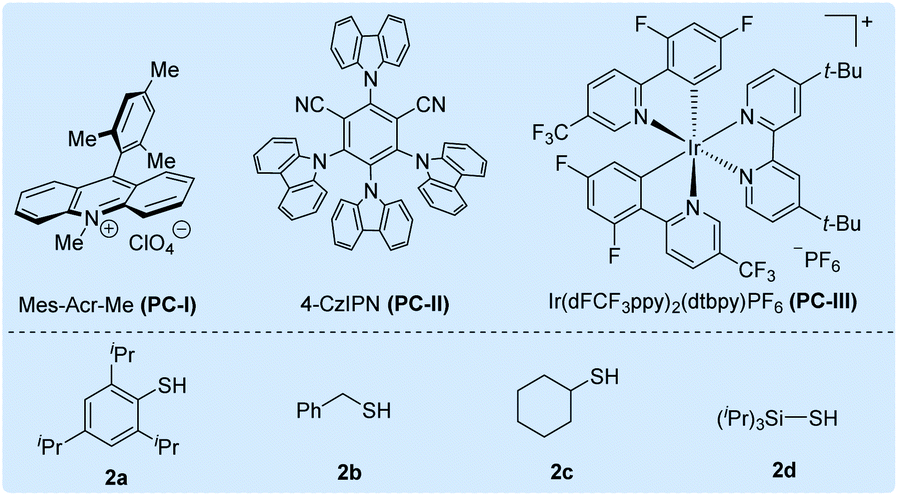
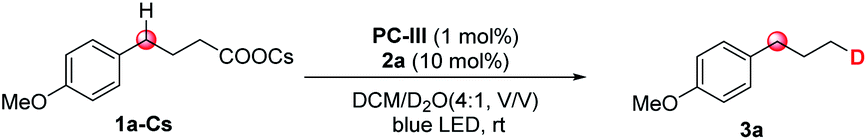
Initially, we selected cesium 4-(4-methoxyphenyl)butanoate (1a-Cs) as a model substrate, and it is readily available from the corresponding carboxylic acid in almost quantitative yield (Table 1). The optimized conditions include the use of 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [PC-III, 1/2Ered(*IrIII/IrII) = +1.21 V vs. SCE]17 as the photocatalyst, thiol (2a) as the HAT catalyst, DCM/D2O as the mixed solvent and irradiation of blue LEDs at ambient temperature (entry 1). The reaction delivered the desired 1-methoxy-4-(propyl-3-d)benzene (3a) in 75% yield and with up to 96% deuterium incorporation. The substrate cesium 4-(4-methoxyphenyl)butanoate (1a) has a weak benzylic C–H site, but the site remained intact during the decarboxylative deuteration. To our knowledge, C–H sites of this type are challenging in H/D exchange strategies.5c If sodium or potassium salts were used instead of cesium carboxylate, it was found that the D-incorporation was notably decreased (entries 2 and 3). However, the formation of the corresponding cesium carboxylate in situ from a carboxylic acid and CsOH gives a moderate yield and D-incorporation (entry 3). The use of other photocatalysts such as PC-I or PC-II and thiols (2b–2c) reduces the reaction efficiency (entries 5–9). Control experiments suggest that the photocatalyst, thiol HAT catalyst and light irradiation are key factors contributing to the success of the reaction (entries 10 and 11). On the other hand, we found that aliphatic acids such as 2-(4-(benzyloxy)phenyl)acetic acid (1b), which are stabilized by heteroatomic or aryl substitution, can directly react under modified optimized conditions when using Mes-Acr-Me (PC-I) as shown in Fig. 3 (see the ESI† for optimization details). This can precede the precise decarboxylative deuteration and afford the desired product (3b) in 97% yield and 95% D-incorporation. We envisioned that the strong oxidative ability of photoexcited *Mes-Acr-Me [1/2Ered(*PC/PC˙−) = +2.08 V vs. SCE]18 would facilitate the SET oxidation of carboxylic acid in the presence of an organic base. Interestingly, we found that the initial reaction rate of this decarboxylative deuteration was heavily light-dependent since a dramatically decreased rate was observed when only one LED lamp was used rather than two lamps (see the ESI† for details). This encouraged us to use a recirculation reactor to address the efficiency of the reaction.
|
|
|||
|---|---|---|---|
| Entry | Variation of standard conditions | Yieldb | D-Inc.c |
a
1a-Cs (0.2 mmol), PC-III (1 mol%), 2a (10 mol%), DCM/D2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v; 2 mL, 110 eq. of D2O), 45 W blue LEDs, room temperature, 18 h.
b Measured by GC using acetophenone as the internal standard due to the high volatility.
c Deuterium incorporation was determined by HRMS-ESI.
d Sodium 4-(4-methoxyphenyl)butanoate.
e Potassium 4-(4-methoxyphenyl)butanoate.
f CsOH (2.0 equiv.) as the base. DCM = dichloromethane, N.D. = not detected. :1, v/v; 2 mL, 110 eq. of D2O), 45 W blue LEDs, room temperature, 18 h.
b Measured by GC using acetophenone as the internal standard due to the high volatility.
c Deuterium incorporation was determined by HRMS-ESI.
d Sodium 4-(4-methoxyphenyl)butanoate.
e Potassium 4-(4-methoxyphenyl)butanoate.
f CsOH (2.0 equiv.) as the base. DCM = dichloromethane, N.D. = not detected.
|
|||
| 1 | None | 75% | 96% |
| 2 | With 1a-Nad | 75% | 77% |
| 3 | With 1a-Ke | 75% | 83% |
| 4 | 4-(4-Methoxyphenyl)butanoic acid and CsOHf | 57% | 60% |
| 5 | PC-I instead of PC-III | 4% | — |
| 6 | PC-II instead of PC-III | 3% | — |
| 7 | 2b instead of 2a | Trace | — |
| 8 | 2c instead of 2a | 23% | 96% |
| 9 | 2d instead of 2a | 14% | 96% |
| 10 | Without PC-III or light | N.D. | — |
| 11 | Without 2a | 5% | — |
|
|||
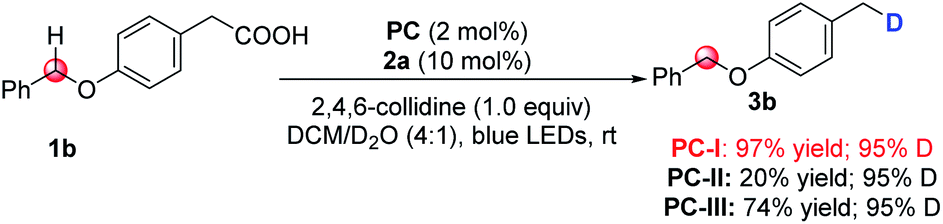 | ||
| Fig. 3 Modified standard conditions for benzylic acids. Reaction conditions: 1b (0.2 mmol), Mes-Acr-Me PC-I (2 mol%), 2a (10 mol%), 2,4,6-collidine (1.0 equiv.), DCM/D2O (4:1, v/v; 2 mL, 110 eq. of D2O), 45 W blue LEDs, ambient temperature, 10 h. Yield of the isolated product is shown. The deuterium incorporation was determined by 1H NMR analysis. | ||
With the optimized decarboxylative deuteration conditions in hand, we investigated the scope of the aliphatic carboxylic acids. As shown in Fig. 4, a wide variety of primary, secondary and tertiary carboxylic acid derivatives can smoothly undergo the decarboxylative deuteration, affording the products (3c–3s) in moderate to good yields with excellent (up to 99%) D-incorporation. The benzylic and α-heteroatom carboxylic acids (3c–3e, 3g–3k, 3n, 3q and 3r) can be directly employed under optimized reaction conditions A with Mes-Acr-Me (PC-I) as the photocatalyst. The reactive benzylic and α-amino C–H bonds, for which H/D exchange under photocatalysis has been reported,2a were not structurally affected by the reaction. The unactivated cesium carboxylates were employed under optimized conditions B, affording the desired products (3f, 3l, 3m, 3o, 3p and 3s) in satisfactory yields and with good D-incorporation. As shown in Fig. 4, the decarboxylative deuteration protocol has good functional group compatibility. Several relatively sensitive but versatile functional groups, such as a weak benzylic C–H bond (3c, 3d, 3l–3n and 3q), an α-amino C–H bond (3d, 3l–3n, 3q and 3u), heteroaryl (3j and 3k), boronic ester (3e), ketone (3o) or strained small rings (3r), survived the conditions well, which suggests promising applications in synthetic and medicinal chemistry. The synthetic robustness of the reaction was further illustrated by the decarboxylative deuteration of complex carboxylic acids such as erdosteine (3u), ambrisentan (3y), gemfibrozil (3aa) and oleanic acid (3cc). The resulting deuterated products (3t–3ee) are difficult to prepare from other available starting materials, such as organic halides which are usually prepared from acids by reduction and subsequent halogenation under harsh conditions. However, aromatic acids are unsuccessful examples in this decarboxylative protocol possibly because the radical decarboxylation of aromatic substrates is a kinetically less favoured pathway.11
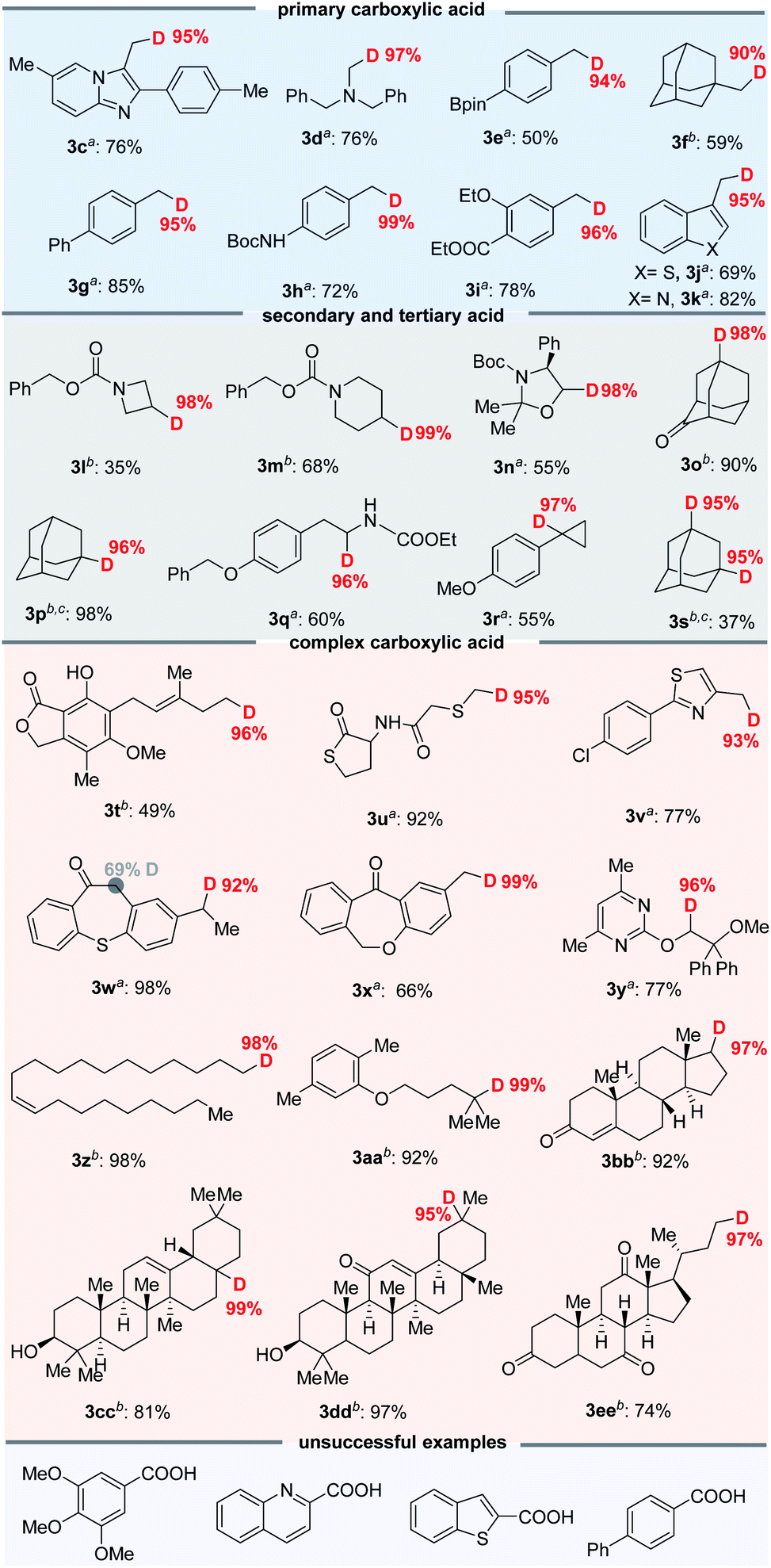 | ||
| Fig. 4 Scope of the decarboxylative deuteration. aStandard conditions A: aliphatic acids 1 (0.2 mmol), PC-I (2 mol%), 2a (10 mol%), 2,4,6-collidine (1.0 equiv.), DCM/D2O (4:1, v/v, 2 mL), blue LEDs, 10–20 h. bStandard conditions B: cesium carboxylate (0.2 mmol), PC-III (1 mol%), 2a (10 mol%), DCM/D2O (4:1, v/v, 2 mL, 110 eq. of D2O), 45 W blue LEDs, 10–20 h. cThese products are readily volatile and yields are determined by GC. Isolated yields are shown and the deuterium incorporation ratio was determined by 1H NMR spectroscopy (<95% D) or HRMS (>95% D). | ||
Although several important radical deuteration syntheses of various compounds have been established, a scalable and practical strategy remains a problem.8 Flow chemistry has significant advantages in terms of scalability, safety and productivity, and has been recently applied in photocatalyzed organic transformations.19 In general, the mesoscale two-phase photoreaction could be improved through the use of a flow reactor setup, allowing for a larger illumination area and liquid–liquid interfacial area. Consequently, we sought to develop scalable decarboxylative deuteration in flow reactors. However, we immediately found that the use of a continuous-flow micro-tubing reactor employing a syringe pump to deliver the reaction liquid decreased the D-incorporation ratio of products, such as 3b, to less than 40%. Inspired by the well-established continuous-flow technology, we designed a batch recirculation reactor with a peristaltic pump to transfer the reaction mixture (the detailed setup is shown in the ESI†); the enhanced mixing effect and mass transfer between two phases of our equipment improved the interfacial area20 of DCM and D2O, and this increased the D-incorporation ratio of 3b from 37% to 95% (see the ESI† for details).
As shown in Fig. 5, the decarboxylative deuteration assisted by our recirculation arrangement could be scaled up with 5.0 mmol of carboxylic acid. A wide range of aliphatic carboxylic acids are effective substrates, affording the desired products (3ff–3uu) in acceptable yields without decreasing the D-incorporation or the chemoselectivity. Furthermore, although the cesium carboxylate is not soluble well in recirculation reactors, it can also furnish the decarboxylation product (3hh) in 71% yield and 97% D-incorporation upon addition of 18-crown-6 as an additive with 0.5 mol% photocatalyst. The use of recirculation reactors can allow for 50.0 mmol scale-up ability to produce 3b (8.6 g) and 3vv (13.0 g) without compromising the reaction efficiency and D-incorporation (>95%). With this protocol, we were able to easily prepare 18 kinds of different deuterated compounds on a 2–50 mmol scale from readily available starting materials under mild reaction conditions.
 | ||
| Fig. 5 Scalable decarboxylative deuteration of carboxylic acids. Standard conditions: 1 (5.0 mmol), PC-I (2 mol%), 2a (10 mol%), 2,4,6-collidine (1.0 equiv.), DCM/D2O (4:1, v/v, 25 mL), blue LEDs, peristalsis rate = 30 mL min−1, 8–16 h. aCesium carboxylate (2.0 mmol), PC-III (0.5 mol%), 2a (5 mol%), 18-crown-6 (1.0 equiv.), DCM/D2O (4:1, v/v, 10 mL), blue LEDs, peristalsis rate = 80 mL min−1, 9 h. bYields are determined by GC due to the volatility. c1 (50.0 mmol), PC-I (1 mol%), 2a (5 mol%), 2,4,6-collidine (1.0 equiv.), DCM (80 mL), D2O (22.0 equiv., 20.9 mL), 45 W blue LEDs, peristalsis rate = 80 mL min−1, 16 h. | ||
Conclusions
In conclusion, we have developed a scalable, practical and precise decarboxylative deuteration of aliphatic carboxylic acids via synergistic photoredox and HAT catalysis with D2O as the deuterium source. A great number of structurally diverse deuterated organic molecules have been obtained with up to 99% D-incorporation from commercially available carboxylates, including a wide range of complex carboxylic acids. We have also explored this method on a preparative scale with a recirculation reactor. This provides a scalable and efficient method for the synthesis of deuterated compounds.Author contributions
J. X., W. L. and N. L. conceived and designed the project. N. L., Y. N. and X. W. performed, analyzed and discussed the experimental data. J. X. wrote the manuscript with input from all authors and discussed the manuscript with C. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22001117, 21971108, 21971111 and 21732003), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190006 and BK20190285), and the Foundation of Advanced Catalytic Engineering Research Center of the Ministry of Education. Shan Gao, Yubo Pang, Chenglong Ji, and Rehanguli Ruzi are warmly acknowledged for reproducing experimental procedures for products 3b, 3z, 3kk and 3hh.References
- (a) T. G. Gant, J. Med. Chem., 2014, 57, 3595 CrossRef CAS PubMed; (b) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758 CrossRef CAS PubMed.
- Recent examples: (a) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS PubMed; (b) H. Geng, X. Chen, J. Gui, Y. Zhang, Z. Shen, P. Qian, J. Chen, S. Zhang and W. Wang, Nat. Catal., 2019, 2, 1071 CrossRef CAS; (c) D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513 CrossRef CAS PubMed; (d) X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020, 59, 13962 CrossRef CAS PubMed; (e) J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS PubMed; (f) N. Zhu, M. Su, W. M. Wan, Y. Li and H. Bao, Org. Lett., 2020, 22, 991 CrossRef CAS; (g) T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467 CrossRef CAS PubMed; (h) M. Wang, Y. Zhao, Y. Zhao and Z. Shi, Sci. Adv., 2020, 6, eaba0946 CrossRef CAS PubMed; (i) X. Wang, M. H. Zhu, D. P. Schuman, D. Zhong, W. Y. Wang, L. Y. Wu, W. Liu, B. M. Stoltz and W. B. Liu, J. Am. Chem. Soc., 2018, 140, 10970 CrossRef CAS PubMed; (j) Y. Kuang, H. Cao, H. Tang, J. Chew, W. Chen, X. Shi and J. Wu, Chem. Sci., 2020, 11, 8912 RSC; (k) R. Zhou, J. Li, H. W. Cheo, R. Chua, G. Zhan, Z. Hou and J. Wu, Chem. Sci., 2019, 10, 7340 RSC.
- Selected examples of ionic dehalogenative deuteration: (a) D. C. Harrowven, I. L. Guy and M. I. T. Nunn, Chem. Commun., 2004, 1966 RSC; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009 CrossRef CAS PubMed; (c) W. F. Bailey and J. J. Patricia, J. Organomet. Chem., 1988, 352, 1 CrossRef CAS; (d) M. Yus, Chem. Soc. Rev., 1996, 25, 155 RSC; (e) H. J. Reich, J. Org. Chem., 2012, 77, 5471 CrossRef CAS PubMed.
- Recent reviews: (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022 CrossRef CAS PubMed; (b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS PubMed; (c) T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401 RSC; (d) M. Valero and V. Derdau, J. Labelled Compd. Radiopharm., 2020, 63, 266 CrossRef CAS PubMed.
- Selected recent examples of transition metal catalyzed H/D exchange: (a) M. Valero, R. Weck, S. Gussregen, J. Atzrodt and V. Derdau, Angew. Chem., Int. Ed., 2018, 57, 8159 CrossRef CAS PubMed; (b) L. Neubert, D. Michalik, S. Bahn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla and M. Beller, J. Am. Chem. Soc., 2012, 134, 12239 CrossRef CAS PubMed; (c) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS PubMed.
- (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- Selected examples of decarboxylation: (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (b) J. He, G. Chen, B. Zhang, Y. Li, J.-R. Chen, W.-J. Xiao, F. Liu and C. Li, Chem, 2020, 6, 1149 CrossRef CAS; (c) J. Xuan, Z. G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (d) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed; (e) H.-D. Xia, Z.-L. Li, Q.-S. Gu, X.-Y. Dong, J.-H. Fang, X.-Y. Du, L.-L. Wang and X.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 16926 CrossRef CAS PubMed; (f) X.-G. Liu, C.-J. Zhou, E. Lin, X.-L. Han, S.-S. Zhang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 13096 CrossRef CAS PubMed; (g) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed; (h) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412 CrossRef CAS PubMed; (i) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed.
- Selected examples of recent radical deuteration: (a) C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G. H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q. H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef PubMed; (b) Y. Dong, Y. Su, L. Du, R. Wang, L. Zhang, D. Zhao and W. Xie, ACS Nano, 2019, 13, 10754 CrossRef CAS PubMed; (c) C. Liu, S. Han, M. Li, X. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527 CrossRef CAS PubMed.
- (a) S. Matsubara, Y. Yokota and K. Oshima, Org. Lett., 2004, 6, 2071 CrossRef CAS PubMed; (b) L. Gooßen, M. Rudzki, A. Alcalde-Aragonés, W. Dzik and N. Rodríguez, Synthesis, 2012, 44, 184 CrossRef.
- T. Itou, Y. Yoshimi, K. Nishikawa, T. Morita, Y. Okada, N. Ichinose and M. Hatanaka, Chem. Commun., 2010, 46, 6177 RSC.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514 CrossRef CAS PubMed.
- (a) M. Zhang, X. A. Yuan, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312 CrossRef CAS PubMed; (b) W. Xu, W. Wang, T. Liu, J. Xie and C. Zhu, Nat. Commun., 2019, 10, 4867 CrossRef PubMed; (c) W. Xu, J. Ma, X. A. Yuan, J. Dai, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 10357 CrossRef CAS PubMed; (d) C. Zhu, J. Dong, X. Liu, L. Gao, Y. Zhao, J. Xie, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 12817 CrossRef CAS PubMed; (e) N. Zhou, X.-A. Yuan, Y. Zhao, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3990 CrossRef CAS PubMed; (f) M. Zhang, J. Xie and C. Zhu, Nat. Commun., 2018, 9, 3517 CrossRef PubMed; (g) J. Han and J. Xie, Chem, 2020, 6, 1053 CrossRef CAS; (h) R. Ruzi, K. Liu, C. Zhu and J. Xie, Nat. Commun., 2020, 11, 3312 CrossRef CAS PubMed; (i) Q. Liao, W. Xu, X. Huang, C. Ke, Q. Zhang, K. Xi and J. Xie, Sci. China: Chem., 2020, 63, 707 CrossRef CAS.
- (a) Q. Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L. Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS PubMed; (b) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340 CrossRef CAS PubMed; (c) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205 CrossRef CAS PubMed.
- E. T. Denisov and V. E. Tumanov, Russ. Chem. Rev., 2005, 74, 825 CrossRef CAS.
- B. Ruscic, A. F. Wagner, L. B. Harding, R. L. Asher, D. Feller, D. A. Dixon, K. A. Peterson, Y. Song, X. Qian, C.-Y. Ng, J. Liu, W. Chen and D. W. Schwenke, J. Phys. Chem. A, 2002, 106, 2727 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS.
- Selected examples of continuous-flow photoreactions: (a) B. Cai, H. W. Cheo, T. Liu and J. Wu, Angew. Chem., Int. Ed. DOI:10.1002/anie.202010710; (b) P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766 CrossRef CAS; (c) H. Seo, A. Liu and T. F. Jamison, J. Am. Chem. Soc., 2017, 139, 13969 CrossRef CAS PubMed; (d) D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557 CrossRef CAS PubMed; (e) N. J. W. Straathof, Y. Su, V. Hessel and T. Noël, Nat. Protoc., 2016, 11, 10 CrossRef CAS PubMed; (f) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085 CrossRef CAS PubMed; (g) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078 CrossRef CAS PubMed.
- D. Cambie, C. Bottecchia, N. J. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00528f |
| This journal is © The Royal Society of Chemistry 2021 |