
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chameleonic macrocyclic peptide with drug delivery applications†
Colton D.
Payne
 a,
Bastian
Franke
a,
Mark F.
Fisher
b,
Fatemeh
Hajiaghaalipour
a,
Courtney E.
McAleese
a,
Angela
Song
a,
Carl
Eliasson
a,
Jingjing
Zhang
b,
Achala S.
Jayasena
b,
Grishma
Vadlamani
b,
Richard J.
Clark
a,
Rodney F.
Minchin
a,
Joshua S.
Mylne
b and
K. Johan
Rosengren
*a
a,
Bastian
Franke
a,
Mark F.
Fisher
b,
Fatemeh
Hajiaghaalipour
a,
Courtney E.
McAleese
a,
Angela
Song
a,
Carl
Eliasson
a,
Jingjing
Zhang
b,
Achala S.
Jayasena
b,
Grishma
Vadlamani
b,
Richard J.
Clark
a,
Rodney F.
Minchin
a,
Joshua S.
Mylne
b and
K. Johan
Rosengren
*a
aThe University of Queensland, School of Biomedical Sciences, Brisbane, QLD 4072, Australia. E-mail: j.rosengren@uq.edu.au
bThe University of Western Australia, School of Molecular Sciences, The ARC Centre of Excellence in Plant Energy Biology, Crawley, WA 6009, Australia
First published on 11th April 2021
Abstract
Head-to-tail cyclized peptides are intriguing natural products with unusual properties. The PawS-Derived Peptides (PDPs) are ribosomally synthesized as part of precursors for seed storage albumins in species of the daisy family, and are post-translationally excised and cyclized during proteolytic processing. Here we report a PDP twice the typical size and with two disulfide bonds, identified from seeds of Zinnia elegans. In water, synthetic PDP-23 forms a unique dimeric structure in which two monomers containing two β-hairpins cross-clasp and enclose a hydrophobic core, creating a square prism. This dimer can be split by addition of micelles or organic solvent and in monomeric form PDP-23 adopts open or closed V-shapes, exposing different levels of hydrophobicity dependent on conditions. This chameleonic character is unusual for disulfide-rich peptides and engenders PDP-23 with potential for cell delivery and accessing novel targets. We demonstrate this by conjugating a rhodamine dye to PDP-23, creating a stable, cell-penetrating inhibitor of the P-glycoprotein drug efflux pump.
Introduction
Macrocyclic and disulfide-rich peptides have generated significant interest for the development of peptide drugs because of their unique stability.1–3 Naturally occurring macrocyclic peptides ranging in size from 5–74 residues and incorporating up to three disulfide bonds have been discovered in bacteria,4 fungi,5 mammals6 and plants.7 One recently discovered family of ribosomally synthesized macrocycles are the PawS-Derived Peptides (PDPs) from the daisy family Asteraceae.8 The prototypic PDP, Sunflower Trypsin Inhibitor-1 (SFTI-1), was discovered in seeds of the common sunflower Helianthus annuus.9 The sequence for SFTI-1 is buried within a precursor for a seed storage albumin called Preproalbumin with SFTI-1 (PawS1).10 SFTI-1 is excised and head-to-tail cyclized during post-translational albumin processing by an asparaginyl endopeptidase.11Based on their distribution, the PDP family is over 18 million years old12 and smaller, related peptides pre-date them by another ∼25 million years.13,14 Like SFTI-1, all PDPs are only found in seeds and are encoded by precursor genes that additionally encode a seed storage albumin. To date, 22 unique PDP sequences have been identified,10,12,15,16 with 14 confirmed in planta. Nineteen PDPs are confirmed or predicted to possess a cyclic backbone, where a proto-N-terminal glycine and a proto-C-terminal aspartate are joined by a transpeptidation reaction performed by an asparaginyl endopeptidase. The N-terminal Gly is absolutely conserved among all PDPs, as is the C-terminal Asp for cyclic PDPs. All reported PDPs are ‘stapled’ by one disulfide bond, which creates a constrained stable structure.8
Natural macrocycles have inspired medicinal chemists working in peptide drug design and engineering applications. Strategies include cyclization of the peptide backbone in linear peptides with desirable bioactivity to increase stability.17,18 Alternatively, a bioactive motif can be ‘grafted’ i.e. incorporated into the backbone of stable cyclic peptide scaffolds.18,19 The grafting concept has been applied to a number of macrocycles, including both SFTI-1 and the larger cyclotides, and used in disease models for cancer,20,21 multiple sclerosis,22 or to improve proangiogenic activity.23 Rather than introducing new bioactivities, the inherent protease inhibitory activity of SFTI-1 can be tuned to specifically target different proteases.24,25
Peptides can also be conjugated to small molecule drugs and act as carriers. Typically, conjugates are focused on overcoming common small molecule issues, such as poor solubility, metabolism and off-target effects due to unfavorable distribution.26 Peptide–drug conjugates tested in clinical trials include somatostatin and analogues bound to isotopes 90Y, 111In, and 177Lu for targeted radiotherapy,27 gonadotropin-releasing hormone bound to doxorubicin for the treatment of ovarian and endometrial cancers,28 as well as angiopep-2 bound to paclitaxel for the treatment of glioblastoma, lung and ovarian cancers.29 The latter conjugate allows paclitaxel to cross the blood–brain barrier but prevents P-glycoprotein (P-gp) efflux.29
Here we describe the identification, chemical synthesis, structural characterization, and a drug conjugation application of PDP-23, a unique PDP twice the size of SFTI-1 and the first macrocyclic peptide described with two disulfide bonds. PDP-23 was identified by de novo transcriptomics in seeds of the common zinnia (Zinnia elegans). Chemical synthesis and NMR spectroscopy revealed PDP-23 self-associates to form a unique symmetrical homodimeric structure. The fold is resistant to temperature and enzymatic degradation. However, the quaternary structure disassociates in membrane-mimicking environments, and PDP-23 can penetrate cells. The combination of different loop lengths, multiple turns and a chameleonic structure offer novel opportunities for therapeutic design. This was demonstrated by attaching the small molecule 5/6-carboxy-tetramethyl-rhodamine, a substrate of the multidrug resistance transporter P-gp,30 to the PDP-23 scaffold, creating a cell-penetrating inhibitor of P-gp.
Experimental section
RNA-seq and transcriptome assembly for Z. elegans
Seeds of Z. elegans (Zinnia Early Wonder Mixed, Mr. Fothergill's, Australia) were ground under liquid nitrogen with glass beads to a fine powder. Total RNA from approximately 0.3 mL of frozen tissue powder was extracted as previously described.31 Contaminating genomic DNA was removed using DNase and the total RNA sample was further purified with a NucleoSpin RNA Clean-up kit (Macherey-Nagel). Sequencing was then performed on an Illumina HiSeq 1500 instrument as 101 bp single read runs.The de novo transcriptome assemblies were performed as previously described.32,33 We used stringent conditions for trimming and filtering of RNA-seq data. Quality trimming and filtering was done using the FASTX toolkit (hannonlab.cshl.edu/fastx_toolkit/) with raw reads trimmed with a minimum quality threshold of 30, implying a base call accuracy of 99.9%, and minimum length of 50. Trimmed reads were then filtered with a quality threshold of 30 and the percentage of bases that match the quality threshold was set to 90. De novo transcriptomes were assembled using CLC Genomics Workbench 8.5.1 (Qiagen). Filtered reads were assembled with five different word sizes (i.e. 23, 30, 40, 60, 64), keeping all other parameters as default. We used tBLASTn to search the transcriptome of Z. elegans using the PawS1 sequence (GenBank accession FJ469150) as a query. Then the filtered raw reads were mapped back to the assembled contig encoding PDP-23 with the following default settings: mismatch cost: 10, insertion cost: 10, deletion cost: 10, length fraction: 1, similarity fraction: 1.
LC-MS analysis of the Z. elegans seed peptide extract
A small amount of dry seed peptide extract was dissolved in HPLC-grade solvent consisting of 5% (v/v) acetonitrile 0.1% formic acid (v/v) in water (Honeywell). After centrifuging for 20 min at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g to remove any solids present, a 4 μL aliquot of the solution was dispensed onto a 96-well plate for LC-MS analysis. Two microliters of the sample were injected into a G4240-62010 HPLC chip (Agilent Technologies) made up of a 160 nL enrichment column (Zorbax 300SB-C18 5 μm) and a 150 mm × 75 μm analysis column (Zorbax 300SB-C18 5 μm). Separation was achieved using an 1100 series nano/capillary HPLC system (Agilent) with an elution gradient from 5% acetonitrile 0.1% formic acid to 95% acetonitrile 0.1% formic acid over 15 min at a flow rate of 0.3 μL min−1. Peptides were ionized by electrospray ionization and the ions were introduced into a 6510 Q-TOF mass spectrometer (Agilent). Spectra were analyzed with MassHunter Workstation Qualitative Analysis software version B.06.00 (Agilent).
000 × g to remove any solids present, a 4 μL aliquot of the solution was dispensed onto a 96-well plate for LC-MS analysis. Two microliters of the sample were injected into a G4240-62010 HPLC chip (Agilent Technologies) made up of a 160 nL enrichment column (Zorbax 300SB-C18 5 μm) and a 150 mm × 75 μm analysis column (Zorbax 300SB-C18 5 μm). Separation was achieved using an 1100 series nano/capillary HPLC system (Agilent) with an elution gradient from 5% acetonitrile 0.1% formic acid to 95% acetonitrile 0.1% formic acid over 15 min at a flow rate of 0.3 μL min−1. Peptides were ionized by electrospray ionization and the ions were introduced into a 6510 Q-TOF mass spectrometer (Agilent). Spectra were analyzed with MassHunter Workstation Qualitative Analysis software version B.06.00 (Agilent).
Extraction of the macrocycle PDP-23 from Z. elegans seeds
Z. elegans seeds were purchased from either Mr. Fothergill's (Zinnia Early Wonder Mixed) or from Royston Petrie (Lilliput Mix). To extract peptide-containing material, the seeds (100 g) were soaked in liquid nitrogen and blended to a fine meal using a food processor. A volume of 25 mL tissue powder was mixed with 160 mL of methanol containing 0.05% (v/v) trifluoroacetic acid (TFA) and 160 mL of dichloromethane containing 0.05% (v/v) TFA and shaken for 70 min at room temperature. After mixing, the mixture was filtered through a Whatman (No. 1) filter paper. Phase separation was achieved by adding 80 mL of 0.05% (v/v) TFA in water. The top aqueous layer containing the peptide was collected, residual dichloromethane was removed under vacuum and the peptide lyophilized. Lyophilized crude mix was dissolved in 80 mL of 0.05% (v/v) TFA in water, sonicated briefly and vortexed before centrifugation at 3260 × g for 10 min. Crude separation was achieved by using a RP-18 Strata column (500 mg/6 mL, Phenomenex). The following buffers were used sequentially for crude separation: 10% acetonitrile 0.05% (v/v) TFA (first elution); 30% acetonitrile 0.05% (v/v) TFA (second elution); 90% acetonitrile 0.05% (v/v) TFA (third elution). Each elution sample was analyzed by MALDI-TOF MS to determine the monoisotopic mass of PDP-23.RP-HPLC purification of PDP-23
The lyophilized crude sample mix was dissolved in buffer A and split in three for further separation by RP-HPLC on a Shimadzu Prominence (Rydalmere, Australia) using an analytical Grace Vydac C18 column (250 mm × 4.6 mm, 5 μm) and an Agilent Zorbax 300SB C18 column (150 mm × 2.1 mm, 5 μm) at a 1% gradient and a flow rate of 1 mL min−1 and 0.3 mL min−1 respectively. Buffer A consisted of 0.05% TFA and buffer B consisted of 90% acetonitrile 0.05% TFA. HPLC fractions were lyophilized and stored at −20 °C.Sequencing of PDP-23
The PDP-23 enriched extract was dissolved in 30 μL 0.1 M ammonium bicarbonate (pH 8.3). A volume of 2 μL 200 mM dithiothreitol was added and incubated for 30 min at 60 °C to reduce the disulfide bonds. To alkylate the cysteine thiol groups, 4 μL of 500 mM iodoacetamide was added and the mixture incubated at room temperature in the dark for 10 min. To digest PDP-23 for sequencing by mass spectrometry, 1 μL of a 0.1 μg μL−1 stock solution of chymotrypsin (Roche Diagnostics) was added at 37 °C for 3 h. To quench the reaction prior to spectrometric analysis, 10 μL of 1% formic acid was added.The digested PDP-23 enriched extract was analyzed by LC-MS/MS on a Shimadzu Prominence Nano HPLC (Rydalmere, Australia) coupled to a 5600 TripleTOF™ mass spectrometer (SCIEX) equipped with a nano electrospray ion source as previously described.34 For this analysis TFA was substituted with 0.1% formic acid in buffers A and B. In brief, 5 μL of digested PDP-23 enriched extract was injected and desalted. For peptide separation, a linear gradient of 2–40% buffer B over 44 min with a 500 nL min−1 flow rate was used, followed by a steeper gradient from 40–80% over 2 min and a hold at 80% for 2 min. The column was subsequently washed by increasing the concentration of B to 98% over 0.1 min and holding it at 98% for 1.9 min prior to a return to 2% B for re-equilibration (16 min). The ionspray voltage was set to 2400 V, declustering potential 100 V, curtain gas flow 25, nebuliser gas 112 and interface heater at 150 °C. The mass spectrometer was set to acquire TOF-MS data over the mass range 350–1800 for 250 ms followed by 20 full scan product ion spectra over the mass range 80–1400 with a maximum accumulation time of 250 ms in Information Dependent Acquisition mode. The 20 most intense ions observed in the TOF-MS scan exceeding a threshold of 120 counts and a charge state of +2 to +5 were set to trigger the acquisition of product ion MS/MS spectra. The data were acquired and processed using Analyst TF 1.6 software (SCIEX). ProteinPilot™ software 4.0.0.0 (SCIEX), with the paragon algorithm, was used as the MS/MS ion search engine.35 The custom-built database contained the sequence of PDP-23 and the search parameter settings were cysteine modification with iodoacetamide and chymotrypsin as the digestion enzyme.
Peptide synthesis
All three possible disulfide isomers for PDP-23 as well as the PDP-23 D28K analogue were assembled on 2-chlorotrityl chloride resin by Fmoc-based solid phase peptide synthesis using a CS336X peptide synthesizer (CSBio). To facilitate selective disulfide bond formation the side chains of CysII-CysIV (isomer I), CysI-CysIV (isomer II) and CysIII-CysIV (isomer III) were protected with acetamidomethyl groups and the other pair of Cys side chains were protected with trityl groups. Peptides were cleaved from resin with 1% TFA in dichloromethane and lyophilized. For backbone cyclization side chain protected peptide was dissolved in dimethylformamide to a concentration of 2 mM and 1 molar equivalent of O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-hexafluorphosphate was added before gradually adding 10 molar equivalents of N,N-diisopropylethylamine. The solution was stirred for 6 h at room temperature before being lyophilized. Global side chain deprotection was achieved by dissolving the lyophilized peptide in 30 mL of TFA, triisopropylsilane, 2,2′-(ethylenedioxy)diethanethiol and water in a ratio of 97:1:1:1, respectively. After stirring for 2 h at room temperature, TFA was removed under vacuum and the peptide was precipitated in ice-cold diethyl ether, filtered, and dissolved in 50% acetonitrile with 0.05% TFA. Residual diethyl ether was removed under vacuum and the peptide lyophilized. The crude peptide was purified by RP-HPLC on a Shimadzu Prominence (Rydalmere, Australia) using a preparative Phenomenex Jupiter C18 column (250 mm × 21.2 mm, 10 μm) at a 1% gradient and a flow rate of 8 mL min−1. Electrospray ionization mass spectrometry confirmed the molecular mass of the reduced peptides. Peptides were lyophilized before oxidization.
Disulfide bonds were formed either randomly and regioselectively. Random disulfide bond formation was conducted by dissolving peptide (0.25 mg mL−1) in 0.1 M ammonium bicarbonate buffer (pH 8.3) with 2 mM reduced glutathione and 0.05 mM oxidized glutathione and stirred for 6 h. To form the first disulfide bond between unprotected cysteines using regioselective strategies, peptide (0.25 mg mL−1) was dissolved in 0.1 M ammonium bicarbonate buffer (pH 8.3) with 2 mM reduced glutathione, stirred for 24 h at room temperature and then purified by RP-HPLC on a Shimadzu Prominence (Rydalmere, Australia) using a semi-preparative Grace Vydac C18 column (250 mm × 10 mm, 10 μm) at a 1% gradient and a flow rate of 3 mL min−1. MALDI-TOF MS confirmed the molecular mass of the partly oxidized peptides.
The second disulfide bond was formed between the acetamidomethyl-protected cysteine residues by dissolving the peptide in water containing 0.05% TFA at a concentration of 0.5 mg mL−1. A saturated iodine solution was slowly added until the mixture became dark yellow in color and this was incubated at 23 °C for 2 h in the dark. The reaction was quenched by adding ascorbic acid until the mixture became colorless again. The fully oxidized peptides were purified by RP-HPLC on a Shimadzu Prominence (Rydalmere, Australia) using a semi-preparative Grace Vydac C18 column (250 mm × 10 mm, 10 μm) at a 1% and 0.5% gradient and a flow rate of 3 mL min−1. MALDI-TOF MS confirmed the molecular mass of the fully oxidized peptides to have a theoretical molecular mass of [M + H]+ 3109.24.
Peptide labeling
The PDP-23 D28K analogue was labeled with ATTO 488 (Sigma-Aldrich, Missouri, USA) as follows: PDP-23 D28K (5 mg) was dissolved in 0.1 M sodium bicarbonate (pH 8.3) to a concentration of 2 mg mL−1. Then a pre-prepared solution of ATTO 488 NHS ester (10 mg mL−1 in dimethyl sulfoxide (DMSO)) was added to the peptide solution to obtain a molar ratio of 2:1 (dye:peptide). The solution was stirred at 23 °C for 2 h in the dark. The conjugation solution was diluted 10-fold and RP-HPLC was performed to filter and purify the labeled peptide from salt, excess dye, and side-reaction products. The PDP-23-ATTO488 conjugate was successfully collected showing the correct m/z of 924.89 [M + 4H]4+ and 1232.86 [M + 3H]3+ indicating the correct total mass of 3695.58 Da. The conjugation of 5/6-carboxy-tetramethyl-rhodamine to the PDP-23 D28K analogue was conducted as follows. PDP-23 D28K (∼5 mg) was dissolved in 0.1 M sodium phosphate to a concentration of 1 mg mL−1 and pH adjusted to 8.75. To this peptide solution a pre-prepared solution containing 10 molar equivalents of NHS-5/6-carboxy-tetramethyl-rhodamine at a concentration of 10 mg mL−1 in DMSO was added. The solution was stirred at room temperature overnight, protected from light. The conjugation solution was diluted 10-fold and RP-HPLC was performed to desalt, remove excess dye and purify the peptide–drug conjugate from side-reaction products. The PDP-23-rhodamine was successfully collected showing the correct m/z of 884.8 [M + 4H]4+ and 1179.6 [M + 3H]3+ indicating the correct total mass of 3536.49 Da.
NMR spectroscopy
For analysis in water PDP-23 was dissolved in 550 μL 90:10 H2O/D2O (v/v) at pH ∼4 to a final concentration of 1.25 mg mL−1. Acetonitrile samples were prepared with ∼1 mg mL−1 PDP-23 in 550 μL of 80:20 (v/v) H2O/CD3CN. For studies in micelles PDP-23 (∼1 mg) was dissolved in 550 μL of 100 mM deuterated sodium dodecyl sulfate (SDS) or 12 mM deuterated dodecylphosphocholine (DPC) in 90:10 H2O/D2O. Standard homonuclear two-dimensional (2D) datasets were recorded at 298 K on 600, 700 or 900 MHz Bruker Avance III spectrometers equipped with cryoprobes, and processed using Topspin 4.0.3 (Bruker Biospin). Mixing times of 80 ms for the TOCSY and 200 ms for the NOESY were used. Additional 13C and 15N HSQC (Heteronuclear Single Quantum Coherence Spectroscopy) spectra were recorded at natural abundance. Temperature coefficients were determined at temperatures ranging from 288 to 308 K. Temperature stability studies were performed at temperatures ranging from 298 to 363 K. 13C HSQC data were recorded in 80:20 D2O/CD3CN (v/v) or 100% D2O to minimize overlap of Hα–Cα resonances with the residual water. Data were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) at 0.0 ppm.
Spectral assignment and structure calculations
The recorded TOCSY, NOESY, 13C HSQC and 15N HSQC spectra were used for manual chemical shift assignments using sequential assignment strategies in CARA.36 The structure of PDP-23 was calculated from inter-proton distance restraints generated from the peak volumes of NOESY cross peaks and dihedral ϕ (C−1-N-CA-C) and ψ (N-CA-C-N+1) backbone angles were generated by Torsion Angle Likelihood Obtained from Shift and sequence similarity (TALOS-N). A set of 50 initial structures was generated using CYANA 3.9837 and final structures were annealed and refined in explicit water within CNS 1.21 using protocols from the RECOORDscript database.38,39 From the 50 calculated structures, a set of 20 structures with no violations, low energy and best MolProbity score was chosen.40 In the case of the symmetrical homodimer structure of PDP-23, the symmetrical homodimer function in CYANA 3.98 was used to generate 50 dimeric structures wherein each monomer was restrained to the other based on the determined intermolecular NOEs observed in the NOESY spectra. These structures were then further refined in explicit water using CNS 1.21. Final structures, restraints and chemical shift data have been submitted to the PDB and BMRB and given the following accession codes, respectively: PDP-23 in water 7L51 and 30827; PDP-23 in acetonitrile/water 7L53 and 30828; PDP-23 in SDS micelles 7L54 and 30829; PDP-23 in DPC micelles 7L55 and 30830.
Serum stability
PDP-23 was mixed with human pooled male serum (Sigma-Aldrich) to a final concentration of 50 μg mL−1 and incubated at 37 °C. Aliquots (100 μL) were taken at different time points (0, 2, 4, 6 and 24 h) and quenched with 100 mM ammonium acetate, pH 3 (900 μL), followed by incubation on ice for 30 min. To separate the peptide from the serum components, Oasis HLB 3 cc 60 mg cartridges (Waters) were activated with 6 mL methanol washes, preconditioned with 3 mL 70% acetonitrile, 1% formic acid and equilibrated with 3 mL 1% formic acid. The serum samples were then loaded onto the column and further washed with 3 mL 5% acetonitrile, 1% formic acid before elution with 35% acetonitrile, 1% formic acid.41 Samples were lyophilized and re-dissolved in 100 μL 1% formic acid before LC-MS/MS analysis on an API2000 (SCIEX) and quantification by MultiQuant (SCIEX). The serum stability tests were performed in triplicate and peptide stability was calculated using a non-linear fit of one phase decay in GraphPad Prism 6.Simulated intestinal fluid stability
Simulated intestinal fluid (SIF) was prepared according to U.S Pharmacopeia specification. Briefly, 68 mg KH2PO4 was dissolved in 250 μL MilliQ water. To this solution, 770 μL 0.2 M NaOH and 5 mL of MilliQ water were added, mixed with 100 μg of pancreatin from porcine pancreas 8× U.S.P (Sigma Aldrich). This mixture was adjusted to pH 6.8 ± 0.1 with 0.2 M HCl or 0.2 M NaOH and diluted with water to a final volume of 10 mL. SIF was pre-incubated for 15 min at 37 °C, before addition of 50 μg peptide to 500 μL SIF. Samples were taken at different time points (0, 0.083, 0.33, 1, 2, 4, 6 and 24 h). Each SIF aliquot (50 μL) was quenched with 50 μL 4% TFA. Samples were analyzed by RP-HPLC on an analytical Grace Vydac C18 column (2.1 mm × 150 mm, 5 μm) using a linear aqueous acetonitrile gradient containing 0.05% TFA at a flow rate of 0.3 mL min−1. PDP-23 was detected by recording the sample absorbance at 214 nm and quantified by the peak area relative to the peak area at time point 0 min.Simulated gastric fluid stability
Simulated gastric fluid (SGF) was prepared according to U.S Pharmacopeia specification. In brief, 20 mg of NaCl and 32 mg of pepsin from porcine gastric mucosa (Sigma Aldrich) were dissolved in 10 mL of 70 μL of HCl in water (final pH 1.2). SGF was pre-incubated for 15 min at 37 °C, before addition of 50 μg peptide to 500 μL SGF. Samples were taken at different time points (0, 0.083, 0.33, 0.66, 1, 4 and 24 h). Each SGF aliquot (50 μL) was quenched with 50 μL 0.2 M Na2CO3. Samples were analyzed by RP-HPLC on an analytical Grace Vydac C18 column (2.1 mm ×150 mm, 5 μm) using a linear aqueous acetonitrile gradient containing 0.05% TFA and a flow rate of 0.3 mL min−1. PDP-23 was detected by recording the sample absorbance at 214 nm and quantified by the peak area relative to the peak area at time point 0 min.Cell culture for PDP-23 toxicity assays
Human cervical cancer (HeLa) cells were used to assess the toxicity of PDP-23. The medium used for culture was Eagle's minimum essential medium supplemented with 10% fetal calf serum (FCS), 50 U mL−1 penicillin and 50 μg mL−1 streptomycin. Cells were incubated at 37 °C in 5% CO2. PDP-23 was evaluated for cellular toxicity at concentrations up to 32 μM using the standard 3-(4,5,-dimethyl-2-thazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay.42Cell uptake assays
The uptake of PDP-23 was visualized using confocal microscopy. CHO cells were seeded on sterile coverslips, placed into a 24-well culture plate at a density of 105 cells per well and incubated overnight. The medium was then removed and the cells incubated at 37 °C in fresh medium containing ATTO 488-labeled PDP-23-D28K at a concentration of 5 μM for 2 h. This medium was then removed and the cells washed with cold PBS. The cells were then treated with fresh medium containing 5 μg mL−1 wheat germ agglutinin conjugated with Alexa-633 and 4′,6-diamidino-2-phenylindole (DAPI) and kept at 4 °C for 30 min. The cells were then washed with cold PBS prior to fixing the cells by incubating them with 4% paraformaldehyde for 15 min at 4 °C. The cells were again washed with cold PBS and the sample mounted in an antifade medium (VECTASHIELD® Antifade Mounting Medium). Microscopy was then performed using a DMi8 SP8 inverted microscope (Leica).Cell culture and chemical preparation for PDP-23-rhodamine conjugate toxicity assays
KB-3-1 (sensitive) and KB-V-1 (resistant) cells were grown in poly-L-lysine-coated T75 flasks at 37 °C with 5% CO2 and maintained in RPMI 1640 medium, supplemented with 10% FCS, 2 mM glutamine and 100 U mL−1/100 μg mL−1 penicillin/streptomycin. Daunorubicin and verapamil were dissolved in DMSO and diluted in RPMI medium to produce working stocks, with the final solvent concentration not exceeding 1%. PDP-23-5/6-carboxy-tetramethyl-rhodamine (159 μg) was reconstituted in 1.5 mL of RPMI to a concentration of 30 μM, which was further diluted to produce working stocks.Lysate preparation and western blot
KB-3-1-and KB-V-1 cells were plated at a density of 300000 cells per well of a 12-well plate in 1 mL of the RPMI medium and incubated overnight. Cells were then lysed in Laemmli's buffer and heated for 5 min at 95 °C. Lysates were electrophoresed at 200 V for 45 min on a 7.5% SDS-acrylamide gel and transferred at 350 mA for 1 h to a nitrocellulose membrane. Blocking was performed for 1 h at RT in 5% skim milk/PBS with 0.05% Tween-20 (PBST). The membrane was washed thrice with PBST for 15 min and rocked overnight at 4 °C with P-gp primary antibody. The membrane was washed again and rocked with rabbit horseradish peroxidase-conjugated secondary antibody for 1 h at RT. After another set of washes, P-gp expression was detected via enhanced chemiluminescence (ECL). The loading control was generated by repeating the steps post blocking using an α-tubulin primary antibody and a mouse horseradish peroxidase-conjugated secondary antibody.
Daunorubicin sensitivity determination
Cells were seeded in a 96-well plate at a density of 10000 cells per well in 100 μL of RPMI medium and incubated overnight at 37 °C. Cells were treated with DMSO or one of nine concentrations of daunorubicin for 24 h. Medium was removed and cells incubated with the DNA-binding fluorescent dye reagent (CyQuant NF Proliferation assay kit) in Hank's Balanced Salt Solution, supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and 35 mg mL−1 NaHCO3, for 30 min at 37 °C. Fluorescence was measured at 485ex/520em. Data were normalized to the untreated control.
For PDP-23-rhodamine conjugate toxicity assays, cells were plated in a 96-well plate as described above. Cells were treated with positive controls of 30 μM verapamil supplemented with 1 μM daunorubicin, as well as daunorubicin 1 μM alone. Cells were additionally treated with PDP-23-rhodamine at concentrations of 1, 3, 10 or 30 μM supplemented with 1 μM daunorubicin. Negative controls included untreated cells as well as cells treated with 30 μM verapamil only. Toxicity was assessed using the CyQuant NF Proliferation assay protocol as above. Data were normalized to the untreated control.
Results
Discovery of PDP-23 by de novo transcriptomics
The assembly of whole de novo transcriptomes of dry seeds has been shown to be an effective way to discover members of the PDP family.12,33 A mature PDP sequence can be predicted based on conserved flanking residues that are necessary for processing.10,12,31 While searching a Zinnia elegans transcriptome for the PawS1 genes that encode PDPs, we found a sequence appearing to encode an unusually large PDP with 28 amino acid residues and the amino acid sequence GFCWHHSCVPSGTCADFPWPLGHQCFPD. A detailed analysis of the RNA-seq reads confirmed the sequence with an average depth of coverage of 696-fold at each nucleotide position, giving high confidence (Fig. S1†). The theoretical monoisotopic mass of the linear reduced peptide is 3130.29 Da. However, for all reported PDPs where the residue at the C-terminal processing site is an Asp, it is joined head-to-tail with Gly1 in a cleavage-coupled intramolecular transpeptidation reaction,11 and the four cysteine residues are expected to form two disulfide bonds. Thus, the expected monoisotopic mass for a processed cyclic and oxidized peptide is 3108.24 Da. Analyzing seed extracts by LC-MS, we detected a series of [M + 3H]3+ ions whose mass matched the prediction (Fig. 1), indicating this peptide does indeed exist. It was named PDP-23, in keeping with previous nomenclature.12,15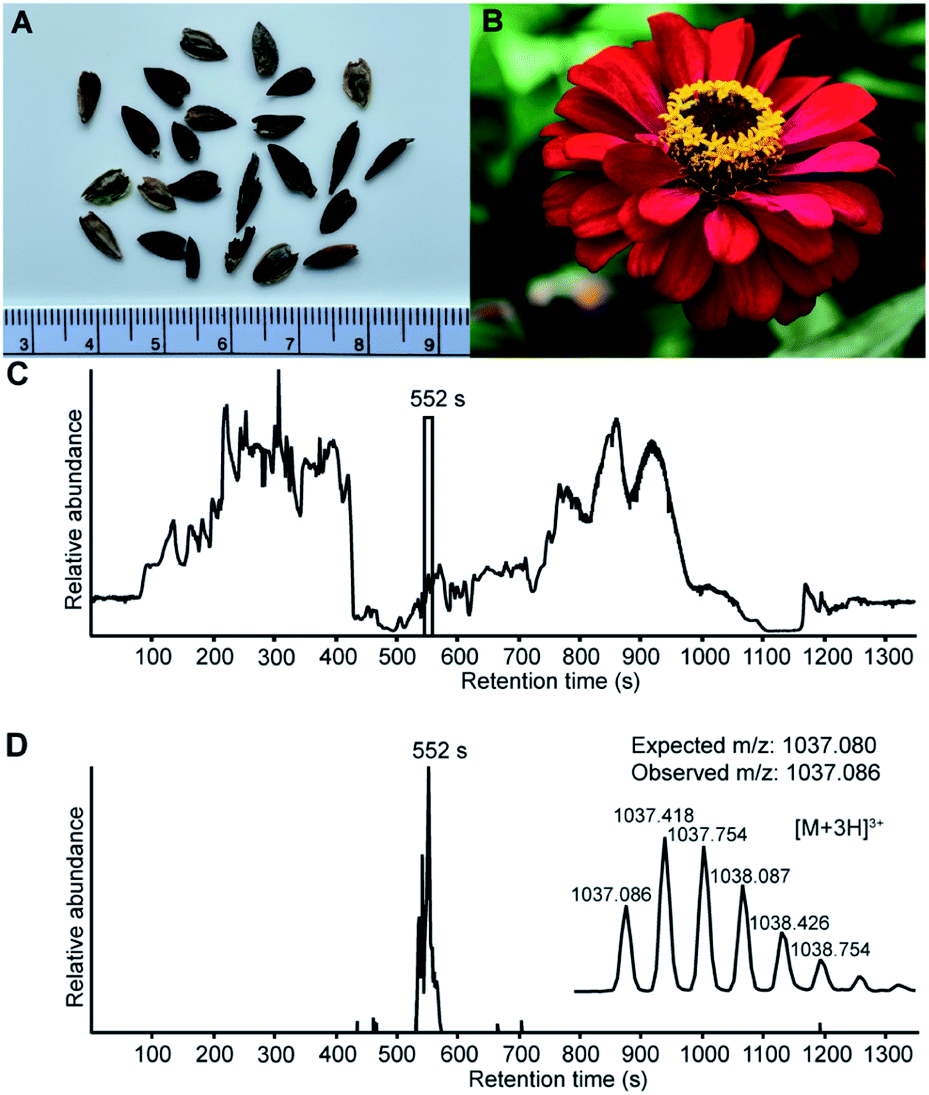 | ||
| Fig. 1 LC-MS analysis confirm that Zinnia elegans seeds contain the cyclic, 28 amino acid, two disulfide bond peptide PDP-23. (A) Zinnia elegans seeds, ruler placed for scale. (B) Image of a Zinnia elegans flower. (C) Total ion chromatogram of the seed extract marked with the retention time of PDP-23, confirming its presence at the protein level. (D) Extracted ion chromatogram (XIC) for PDP-23 showing the [M + 3H]3+ (m/z) state of the observed mass-to-charge ratio and its isotopic peak envelope. | ||
Extraction of PDP-23 and LC-MS/MS sequencing
To confirm the sequence of PDP-23, the peptide was extracted from seeds, partially purified using RP-HPLC and the monoisotopic mass confirmed by MALDI-TOF MS. The PDP-23 enriched extract was reduced, alkylated and digested with chymotrypsin, and the cleaved mixture was subjected to sequencing by LC-MS/MS. The spectra were searched against a custom-built peptide database and three peptide fragments that cover the entire sequence of PDP-23 (HHSCVPSGTCADFPWPL, GHQCFPDGFCW, and HHSCVPSGTCADFPWPLGHQCFPDGFCW), including the macrocyclization junction between Asp28 and Gly1, were identified. Each MS/MS spectrum was assigned b- and y-ion series (Fig. S2†). As mass spectrometry cannot readily distinguish between Gln/Lys, to confirm the presence of Gln24 we reduced, alkylated and digested the PDP-23 enriched extract with trypsin. No tryptic fragments were identified, consistent with the PDP-23 sequence not containing any Lys or Arg residues. MS also does not distinguish between Leu/Ile residues, but the transcriptomic raw reads strongly support a Leu residue at position 21 (Fig. S1†) and successful cleavage by chymotrypsin to generate the fragment HHSCVPSGTCADFPWPL also supports this assertion.Peptide synthesis and purification of PDP-23 disulfide isomers
PDP-23 was found to be of very low abundance in seeds. Thus, to get material to further characterize this peptide we turned to chemical synthesis. With two disulfide bonds, three different disulfide connectivities are theoretically possible (Fig. 2A). All three possible PDP-23 isomers were synthesized separately using solid phase Fmoc chemistry, cyclized in solution, and oxidized by regioselective disulfide bond formation. Each isomer was purified by RP-HPLC and the monoisotopic mass confirmed by MALDI-TOF MS (Fig. 2B). Isomers I and II showed a monodispersed peak shape, whereas isomer III eluted with a tailing shoulder. To confirm that the three synthesized isomers possess the same purity, sequence and cyclic backbone, they were reduced, alkylated and analyzed by RP-HPLC (Fig. 2C). The analytical RP-HPLC trace showed identical elution at 40.55 min for all three reduced and alkylated isomers. Comparing the elution times of the oxidized peptides with the PDP-23 enriched seed extract using analytical RP-HPLC identified the earliest eluting isomer, isomer III, as the closest match to the enriched extract (Fig. S3A†), however it was not possible to conduct co-elution studies as PDP-23 remained only a minor component of the extract and could not be purified from more abundant components. MALDI-TOF MS analysis highlighted the low abundance of PDP-23 within the enriched extract at a mass-to-charge ratio of 3109.35+, along with a number of more abundant masses (Fig. S3B†). Additionally, we attempted to randomly fold PDP-23 to determine if isomer III was the preferentially adopted disulfide bond configuration. Randomly folding PDP-23 resulted in two major products, isomers II & III, with a slight preference towards isomer III (Fig. S4†).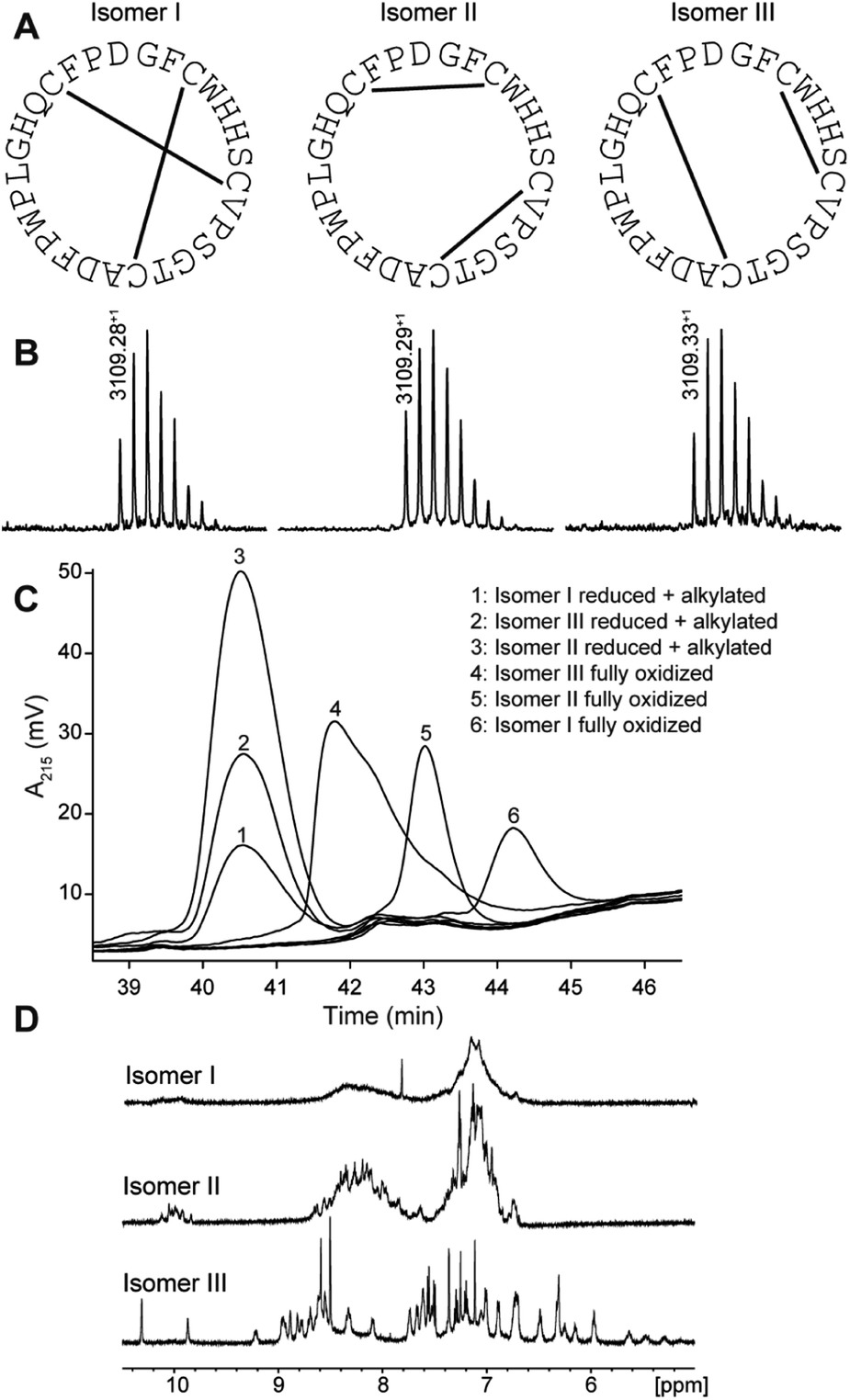 | ||
| Fig. 2 Characterization of synthetic disulfide isomers of PDP-23. (A) Cysteine connectivity of the three synthesized isomers with corresponding sequence. (B) MALDI-TOF MS showing a well-distributed experimental isotopic peak envelope with an observed monoisotopic mass-to-charge ratio of 3109.28+ for isomer I, 3109.29+ for isomer II and 3109.33+ for isomer III. (C) Analytical RP-HPLC trace of synthesized isomers in reduced and alkylated and fully oxidized state. The isomers eluted in the following order: isomer III (41.78 min), isomer II (43.02 min) and isomer I (44.22 min). (D) One-dimensional (1D) 1H NMR spectra of the three fully oxidized isomers. | ||
NMR spectroscopy studies of PDP-23
Solution NMR spectroscopy was used to study all three synthetic isomers to gain further insight into the likely native conformation. Isomer III showed excellent dispersion in the 1D 1H NMR spectrum recorded in water, indicating a folded, highly structured peptide, whereas isomers I and II showed poor dispersion consistent with disorder (Fig. 2D). In addition, isomer I showed very broad lines consistent with aggregation and isomer II generated multiple signals from the Trp indole protons suggesting multiple conformational states. Clearly the disulfide array of isomer III is structurally favored. All PDPs that have been isolated from seeds have been shown to be highly structured, whereas misprocessed and unstructured forms tend to be degraded,11 which strongly supports isomer III being the native form. Extensive NMR data were recorded for isomer III and assigned manually using sequential assignment strategies. The high-quality datasets allowed for the complete assignment of backbone and side chain resonances using TOCSY and NOESY spectra (Fig. S5†).43 HSQC data were recorded at natural abundance, allowing assignments of the 13C and 15N backbone and side chain resonances based on 1H assignments. PDP-23 contains four proline residues with Pro20 adopting a cis conformation, whilst Pro10, Pro18, and Pro27 all adopt a trans conformation, based on NOE patterns and 13C chemical shifts. No additional spin systems suggesting conformational inhomogeneity were identified.To determine the three-dimensional (3D) structure of PDP-23, structural restraints were derived from the NMR data. These included inter-proton distances based on NOEs, dihedral angles based on chemical shifts, and hydrogen bonds based on temperature coefficients. Intriguingly, after considerable efforts, it was realized that a 3D structure consistent with all data could not be computationally generated. A number of unambiguous NOEs were unable to be satisfied by the structures generated. For example, NOEs were observed from the Hβs of Trp4, the Hβs of Ser7, and the Hγ protons of Val9 to the Hδ methyl protons of Leu21. However, the distances between these protons all fell well outside the possible range of the NOE (∼5 Å) in the calculated models. The only possible explanation was that these are intermolecular NOEs, and as there is no resonance duplication PDP-23 must exist in solution as a symmetric multimer.
Structure determination of PDP-23 in monomeric and dimeric forms
In an attempt to separate the multimeric PDP-23 into individual monomers we explored adding organic solvent. Recording NMR spectra of PDP-23 in a solution of 80:20 H2O/CD3CN revealed a significant change in dispersion and peak line widths (Fig. S6†). The CD3CN data showed narrower lines, while overall peak dispersion was decreased, in particular in the aromatic region, indicating a change in the structure. Re-recording and re-assigning all 2D NMR revealed significant changes in the chemical shifts of numerous protons, and also in the NOE patterns observed. Critically, key NOEs identified as intermolecular were no longer present and residues involved experienced chemical shift deviations > 0.15 ppm for numerous protons. However, the secondary Hα chemical shifts, which are sensitive indicators of secondary structure, are remarkably similar between PDP-23 in water and in 80:20 H2O/CD3CN (Fig. S7†). Thus, as a whole, the backbone and secondary structure of PDP-23 remains largely the same.
Structural restraints were derived from the NMR data recorded in H2O/CD3CN and 3D structures calculated using simulated annealing. In contrast to the calculations performed using the aqueous data, this resulted in structures fully consistent with all observed NOEs. The ensemble of the 20 best models chosen to represent the monomeric structure of PDP-23 in 80:20 H2O/CD3CN is shown in Fig. 3A. PDP-23 comprises two double stranded anti-parallel β-sheets. A single disulfide bond bridges each β-sheet and the backbone is stabilized by a large number of hydrogen bonds (Fig. 3B). The structure is V-shaped with polar side chains being surface exposed, whereas the hydrophobic residues Phe2, Trp4, Val9, Phe17, Leu21, and Phe26 create a hydrophobic core sandwiched between the sheets on the inside of the ‘V’. In addition to the two anti-parallel β-sheets, several well-defined turns were identified: a type I′ turn comprising residues Trp4-Ser7, a type II turn comprising residues Pro10-Thr13, a type VIa1 turn comprising residues Pro18-Leu21 and a type I turn comprising residues Phe26-Gly1. Three of the turns feature proline residues, which may be key features for creating bends in the backbone.
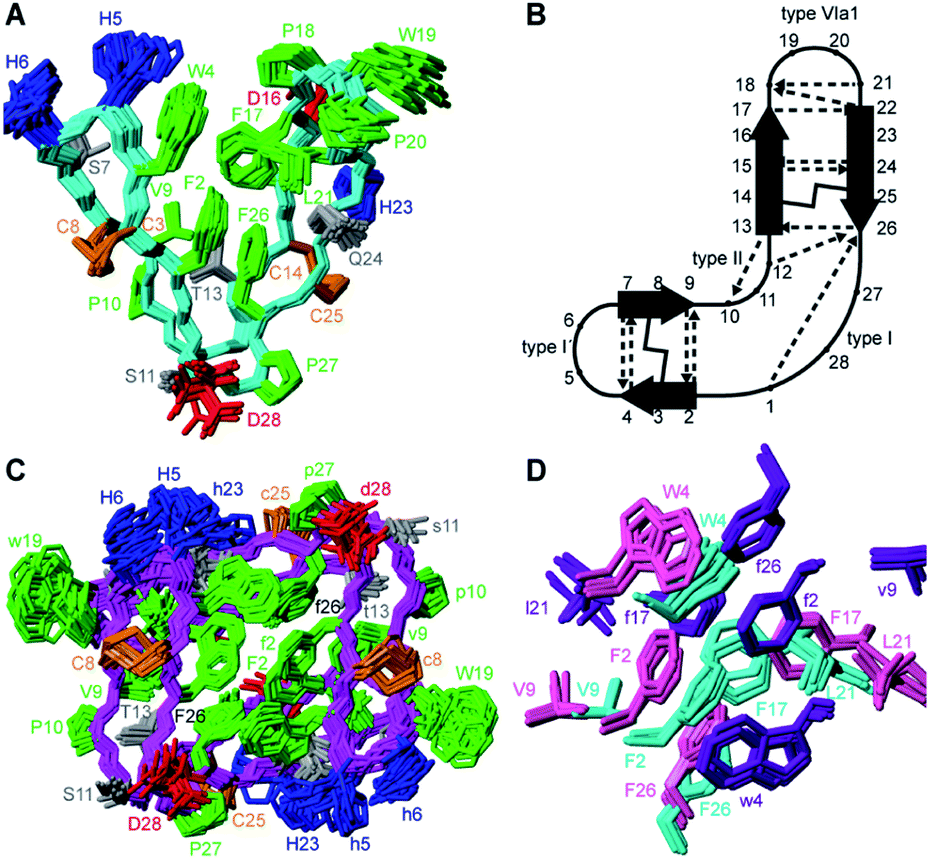 | ||
| Fig. 3 3D structure of PDP-23 in monomeric and dimeric forms. (A) Superposition of the structural ensemble of the PDP-23 monomer in 80:20 H2O/CD3CN. The backbone is shown in cyan, with side chains colored according to types: disulfides in orange, hydrophobic in green, basic in blue, acidic in red, and polar in grey. Residues are labeled with numbers and one letter amino acid codes. (B) Schematic representation of PDP-23 showing the two anti-parallel β-sheets, disulfide bonds (solid lines), turns and hydrogen bonds (dashed arrows). (C) Superposition of the structural ensemble of the PDP-23 dimer in water. The same coloring is used as in panel A but the cyclic backbone is displayed in magenta. Upper and lower case lettering are used to distinguish between the two PDP-23 molecules. (D) Comparison of the hydrophobic core regions of monomeric and dimeric PDP-23. Monomeric PDP-23 is shown in cyan and dimeric PDP-23 in magenta and purple. | ||
Returning to the NMR data recorded in water, we used the structural restraints derived to instead calculate a dimeric structure, enforcing symmetry whilst still allowing ambiguity in that NOEs could be assigned as intra- or intermolecular contacts. The generated structures of a dimer were able to satisfy all of the observed NOEs. A large number of NOEs including the aforementioned contacts between the Trp4, Ser7, Val9 and Leu21 were confirmed to result from intermolecular cross relaxation at the interface between two PDP-23 molecules. The structures were again refined and the best 20 models chosen to represent the dimeric structure of PDP-23 in water (Fig. 3C). The structural statistics of PDP-23 as both a monomer and a dimer highlight the high quality of the structures and the agreement with experimental data (Table S1†). Both structures are well defined with backbone RMSDs of 0.43 and 0.72 Å for the monomer and dimer, respectively. Comparing the dimer and monomer structures, as suggested by the secondary shifts, the backbone is very similar and identical hydrogen bonds are present in both forms. The differences are related to the V-shape which opens up in the dimer, increasing the distance between the loops from ∼13 to ∼21 Å. This allows the creation of a larger combined hydrophobic core arranging the two ‘Vs’ into a square prism shape. The cores of both the monomer and the dimer contain the same residues. However, the larger number of aromatic residues in the dimer's core and packing variations explain the differences in chemical shifts of these residues (Fig. 3D).
Structure determination of PDP-23 in micelles
PDP-23 can change structure based on conditions, because the disulfides only brace the individual elements of secondary structure and not their relative position in the tertiary fold. Consequently, we speculated that the large number of hydrophobic amino acids could allow it to interact with membranes. To mimic this situation, we prepared NMR samples containing SDS or DPC micelles. Exposure of PDP-23 to SDS micelles resulted in significant spectral changes. Line broadening occurred, consistent with an increase in correlation time as a result of binding to the micelles, and the signal dispersion was reduced. Comparing secondary chemical shifts revealed that the β-sheet structure is largely retained, with similar patterns of secondary Hα chemical shifts (Fig. S7†). Some side chain resonances, however, drastically changed chemical shift (Fig. S8† and Table S2†), and many side chain-to-side chain NOEs, particularly involving aromatic residues, disappeared. Importantly, no intermolecular NOEs are present. Thus, we conclude that, rather than forming a dimer, the hydrophobic face of PDP-23 is buried in the SDS micelles. Structures were calculated, and although less defined than in solution they indicate that the structure is largely maintained in the presence of SDS micelles, but it is more open, similar to the structure seen in the dimeric form (Fig. 4A). This aligns with the 3–4 nm diameter of an SDS micelle,44 which would allow for a PDP-23 molecule to expose its hydrophobic core to the micelle interior by slightly opening the V-shape, whilst retaining the polar groups in solution or around the polar lipid headgroups.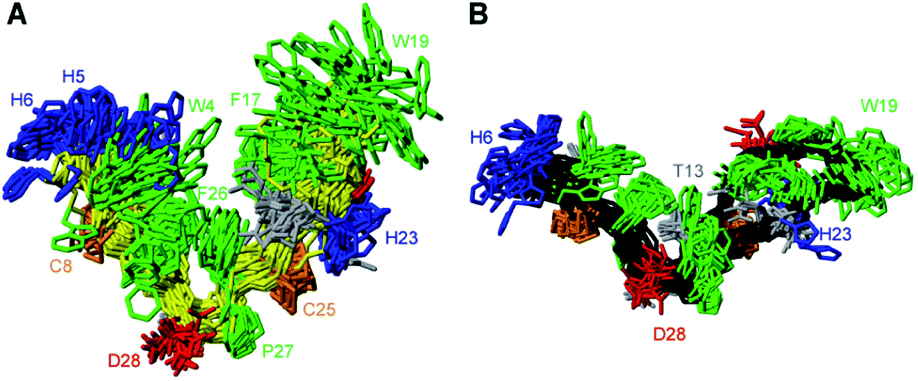 | ||
| Fig. 4 3D structure of PDP-23 in micelles. (A) Superposition of the structural ensemble of PDP-23 when exposed to SDS micelles. The same coloring is used as in Fig. 3, but the backbone is displayed in yellow. (B) Superposition of the structural ensemble of PDP-23 when exposed to DPC micelles. The same coloring is used as in Fig. 3 but the backbone is displayed in black. | ||
A similar trend occurred when exposing PDP-23 to DPC micelles, which are larger than SDS micelles at ∼6.4–7.2 nm in diameter.45 Again, line broadening occurred, in this case for numerous resonances, which created issues with separating spin systems from one another as well as from the noise. Many chemical shift changes in the hinge region and of the aromatic resonances (Figure S8 and Table S2†), as well as disappearance of NOEs, including all intermolecular NOEs, were similar to what was observed in the SDS data. The patterns of secondary Hα chemical shifts were again retained (Fig. S7†) and structure calculations using the DPC data indicate that the secondary structure is retained, while the peptide now presents as an almost flat surface, exposing all hydrophobic residues (Fig. 4B). The distance between the two loops has extended to ∼32 Å. To analyze differences in hydrogen bonding, amide temperature coefficients were determined under all conditions. These confirmed that the hydrogen bonding network within the sheets is retained in all structures. In contrast the hydrogen bonds stabilizing the turns at the bottom of the ‘V’ from the amide protons of Thr13 and Gly1 to the carbonyls of Pro10 and Phe26, respectively, are lost in the micelle structures (Fig. S9†). This is consistent with the opening of the hinge.
Thermal and enzymatic stability of PDP-23
As stability is a key feature among macrocyclic peptides, we assessed the thermal stability of PDP-23 in water using NMR spectroscopy. 1D 1H spectra were recorded at temperature intervals from 298 K to 363 K (Fig. S10†). PDP-23 was highly stable at elevated temperatures with the downfield Hα protons characteristic of the β-sheet being observed at temperatures up to 363 K. The chemical shift separation of the indole protons of the two tryptophan residues was also retained, however chemical shift changes in the aromatic region were consistent with changes in the hydrophobic core. The symmetrical homodimer quaternary fold was confirmed at temperatures up to 318 K using NOESY data. However, increasing the temperature further to 328 K caused the key dimer NOEs to disappear, indicating a likely dissociation of the dimer. After a decrease in temperature back to 298 K the 1D 1H NMR spectrum was found to be identical to the spectrum recorded prior to heating, showing a complete reversibility of any unfolding or dissociation.Given the potential of cyclic peptides as drugs, we also wanted to evaluate the stability of PDP-23 in biological settings. We exposed the peptide to serum as well as simulated gastric and intestinal fluids. PDP-23 was found to be highly stable with more than 50% remaining intact after 24 h incubation in either serum or gastric fluid. The half-life in simulated intestinal fluid was found to be less, possibly due to reduction of the disulfides (Fig. 5).
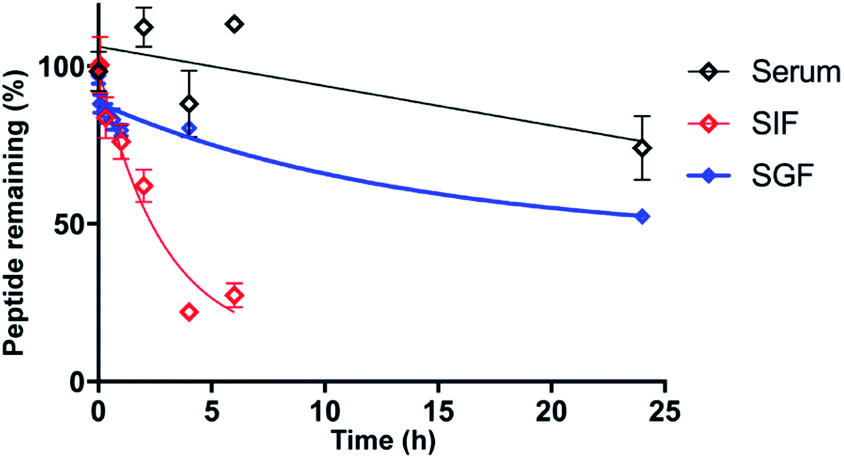 | ||
| Fig. 5 Stability of PDP-23. Non-linear regression fit of the decay of PDP-23 over time when exposed to human serum (black), simulated gastric fluid (blue) and simulated intestinal fluid (red). All data are presented as mean ± SEM, n = 3. | ||
PDP-23 is non-toxic and can penetrate cells
Given PDP-23's ability to insert into micelles we considered it may interfere with membrane integrity and be toxic to cells. To investigate the cytotoxicity of PDP-23 an MTT assay was conducted using HeLa cells. Cells were exposed to concentrations of PDP-23 ranging from 0.5 to 32 μM for 48 h at 37 °C, but no significant cytotoxic activity was observed (Fig. S11†). Fluorescence microscopy was used to investigate the ability of PDP-23 to internalize into cells. PDP-23 was synthesized with Asp28 replaced by a Lys (PDP-23 D28K) to allow labeling with ATTO 488 and incubated with CHO cells. The distinct green fluorescence of ATTO 488 labeled PDP-23 was found internalized in green puncta within CHO cells, suggesting endosomal uptake. Furthermore, weak diffuse green coloring of the cytosol suggested the peptide is able to reach the cytoplasm (Fig. 6).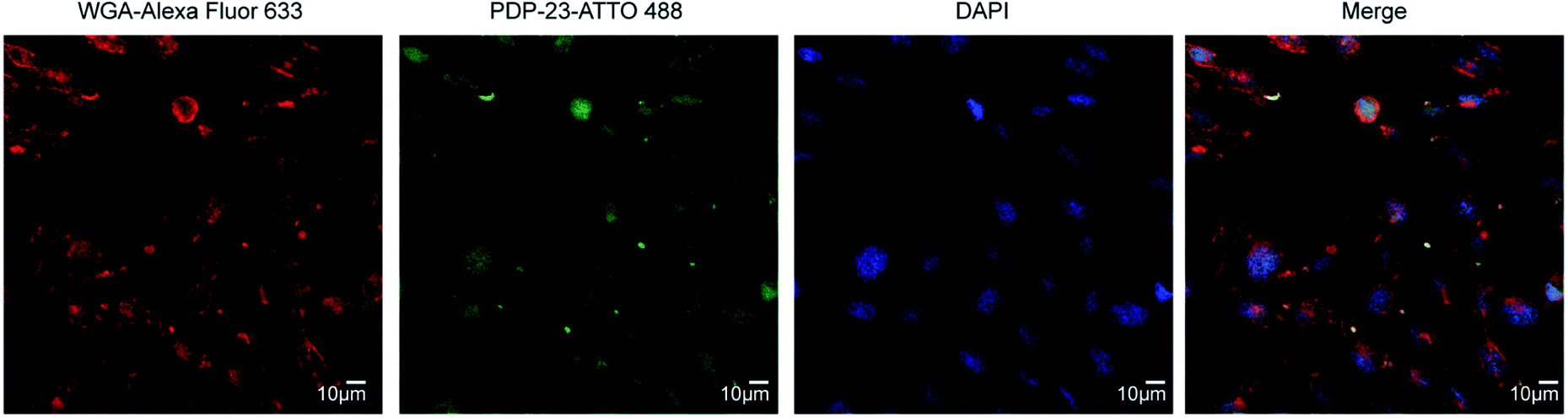 | ||
| Fig. 6 Internalization of PDP-23 in CHO cells. PDP-23 labeled with ATTO 488 (5 μM concentration) was incubated with the cells for 2 h at 37 °C. The plasma membrane was labeled with WGA-Alexa 633 as seen in red and the nucleus labeled with DAPI as seen in blue. The labeled PDP-23 is visible in green. The magnification used was 63×. | ||
PDP-23 as a scaffold for therapeutic drugs
PDP-23, due to its stability, lack of toxicity and ability to penetrate cells, was thought to be an excellent candidate as a scaffold for drug development. A substrate of the multidrug exporter P-gp, the dye 5/6-carboxy-tetramethyl-rhodamine, was conjugated to PDP-23 D28K. We speculated that the conjugate would be able to enter cells and while the rhodamine would interact with P-gp, the large peptide would prevent efflux. Thus, we tested PDP-23-rhodamine as an inhibitor of P-gp by exposing it in combination with daunorubicin, a well-established chemotherapeutic and substrate of P-gp,46 to the cancer cell line KB-3-1 and its derivative KB-V-1. The KB-V-1 cells overexpress P-gp whilst the P-gp expression in KB-3-1 cells was undetectable by Western blot (Fig. 7A). Consequently, the KB-V-1 cells displayed significantly decreased sensitivity to daunorubicin compared to the sensitive KB-3-1 cells, due to rapid export via P-gp (Fig. 7B). Addition of PDP-23-rhodamine in conjunction with daunorubicin, significantly (P ≤ 0.002) increased cell death in the KB-V-1 cells in a dose-dependent manner, demonstrating the restoration of sensitivity of the KB-V-1 cells to daunorubicin (Fig. 7C). However, PDP-23-rhodamine did not significantly increase cell death in the KB-3-1 cells, indicating the selective effect of the conjugate on the P-gp overexpressing KB-V-1 cells. These results are consistent with what was observed for the classical P-gp inhibitor verapamil.47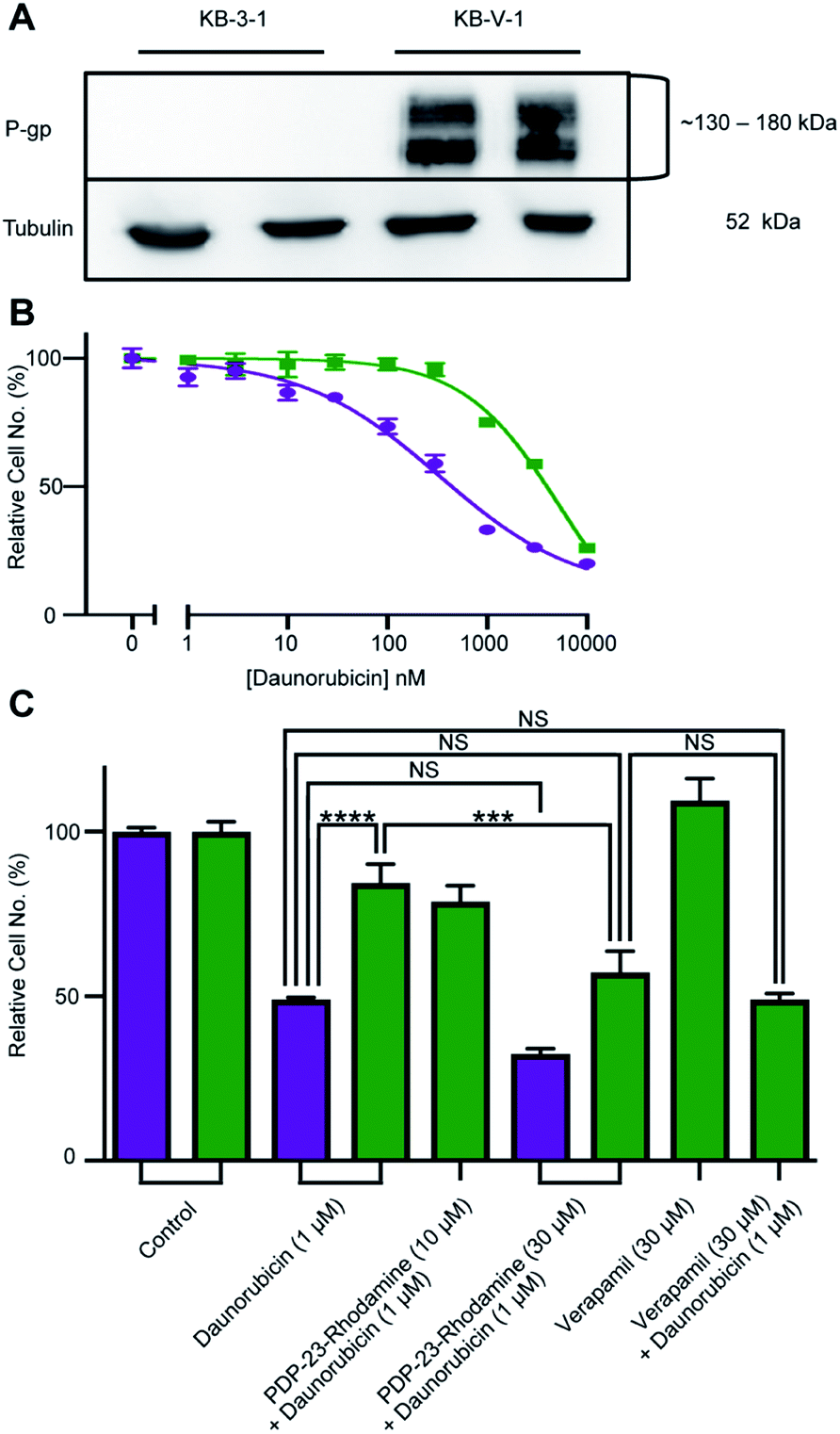 | ||
| Fig. 7 Reversal of daunorubicin resistance when dosed in combination with the PDP-23-rhodamine conjugate in P-gp-overexpressing cells. (A) Western blot of KB-3-1 and KB-V-1 cells confirming P-gp overexpression in the KB-V-1 cell line. (B) Dose–response curve for daunorubicin with KB-3-1 (purple) and KB-V-1 (green) cell lines. Cells were treated for 24 h with a range of nine concentrations of the drug. Cell populations determined via quantification of DNA with CyQuant Cell Proliferation Assay Kit and normalized to the untreated control. All data are means ± SEM, n = 4. (C) Comparison of toxicity of a single concentration of daunorubicin (1 μM) in sensitive (KB-3-1, purple) and resistant (KB-V-1, green) cells, in the presence or absence of the PDP-23-rhodamine conjugate. Verapamil (30 μM), a P-gp inhibitor, is included as a control for P-gp inhibition. Cell populations were determined via quantification of DNA with the CyQuant Cell Proliferation Assay Kit and normalized to an untreated control. All data are mean ± SEM, n = 4; significance determined by two-way ANOVA (****p < 0.0001), (***p < 0.001). | ||
Discussion
In this study, PDP-23 was identified by transcriptomics and confirmed in Z. elegans by MS/MS sequencing and MALDI-TOF MS analysis. At 28 residues, PDP-23 is twice the size of typical PDPs and has double the number of cysteine residues. The presence of multiple disulfide bonds presents a challenge when describing novel isolated peptides, as confirming the disulfide connectivity is not trivial. Even when substantial amounts of material are available, allowing large-scale extraction and purification, identification of disulfide bonds may require detailed NMR spectroscopy or chemical approaches.48 Despite there being sufficient PDP-23 for MS/MS sequencing, the overall abundance of PDP-23 is low and severe chromatographic overlap with other peptidic compounds prevented isolation of a pure native sample. Instead, the three possible disulfide isomers were synthesized by solid phase peptide synthesis and studied by chromatography and NMR spectroscopy. Isomer III was the earliest eluting variant and showed excellent peak dispersion in the 1D 1H NMR spectrum, consistent with a well-structured peptide. In contrast the NMR data for isomers I and II showed poor dispersion, conformational inhomogeneity and aggregation. Comparing the RP-HPLC elution times of the different synthetic isomers with the native peptide was a challenge without a pure native sample. The elution time of isomer III showed the best match with the elution time of the enriched PDP-23 extract, and this together with its ordered structure strongly suggests that it represents the native form. The XIC peak for PDP-23 (Fig. 1) is unusually broad and does not show a uniform peak, which likely reflects the disassociation of the symmetrical homodimer during elution with acetonitrile, consistent with the elution profile with a tailing shoulder observed for synthetic PDP-23 isomer III.The structural investigation of PDP-23 uncovered different folds of the peptide depending on the environment. In an environment with organic co-solvent, PDP-23 adopts a monomeric structure consisting of an elongated cyclic oval shape with two β-hairpins. A single disulfide bond, supported by an extensive hydrogen bond network, bridges each β-sheet. These loops then enclose a hydrophobic core to form a tertiary V-shaped structure (Fig. 3A). In an aqueous environment, PDP-23 forms a symmetrical homodimer through the self-association of two PDP-23 molecules by opening the ‘V’ and interlocking the hydrophobic cores. The quaternary structure of the symmetrical homodimer is roughly a square prism with the hinge of each monomer forming one of two planar vertices and the turn at the top of each loop create a stacked vertex (Fig. 3B). The PDP-23 dimer is able to disassociate and interact with micelles by exposing the hydrophobic core of each monomer, while maintaining the β-sheets of each loop. The amount of separation required of the two loops to expose the hydrophobic core of the PDP-23 monomer to a micelle appears to be dependent on the size of the micelle. This is illustrated by the difference in the structural fold observed between PDP-23 when exposed to either SDS or DPC micelles. The dimer also appears to dissociate into individual monomers at increased temperature (>50 °C), but again retains a high degree of structure and folds back into a dimer when the temperature is lowered even after heating to 90 °C. The adaptability of the structure indicates an innate ability of PDP-23 to conform and stabilize its molecular structure depending on the environment it is exposed to.
PDP-23 represents a new structural class of peptides. Previously described PDPs that are distributed throughout the sunflower family are mostly head-to-tail cyclic peptides that are bridged by a single disulfide bond, including the prototypic member SFTI-1.10,12,15,16 From a structural perspective PDP-23 has more in common with the much larger cyclotides that are found scattered throughout the plant kingdom (Fig. 8).49 These are also head-to-tail cyclized peptides that are rich in β-sheet structure and tight turns. However, the cyclotides have six conserved cysteines adopting a knotted arrangement of three disulfide bonds.49 Unlike PDP-23, in the cyclotides the disulfide bonds are buried in the core leaving all hydrophobic residues on the outside. Due to its cysteine connectivity (CysI-CysII and CysIII-CysIV), PDP-23 can be considered as adopting a disulfide-laddered structure. A disulfide-ladder motif is the signature of θ-defensins, which are cyclic peptides originally discovered in the leukocytes of rhesus monkeys.6 The θ-defensins comprise only a single β-sheet that is stabilized by three disulfide bonds and are straight in contrast to the V-shaped PDP-23. Intriguingly PDP-23 shares some structural details with both θ-defensins and cyclotides. The Trp19-Pro20 turn of PDP-23 is identical to a turn seen in kalata B1, which due to the cis peptide bond has been used to define the Möbius cyclotide sub-class. At the other end of PDP-23 the His5-His6 type 1′ turn and its stabilizing disulfide is conformationally akin to the θ-defensin RTD-1 (Fig. 8). Whilst disulfide-laddered peptides are rare, cysteine knotted counterparts are not only found in plants but are quite common in nature and are present in various spider and cone snail venoms.50,51 A degree of self-association has been reported for both cyclotides and θ-defensins under NMR conditions but this neither involves structural changes nor leads to stable defined multimeric forms.52,53 Importantly, we believe PDP-23 is unique among disulfide-rich peptides in that the disulfides do not directly cross-brace and lock in the tertiary fold, only the secondary structure, allowing for potential rearrangement of the position of the two hairpins. The folding around a hydrophobic core is instead more reminiscent of larger proteins.
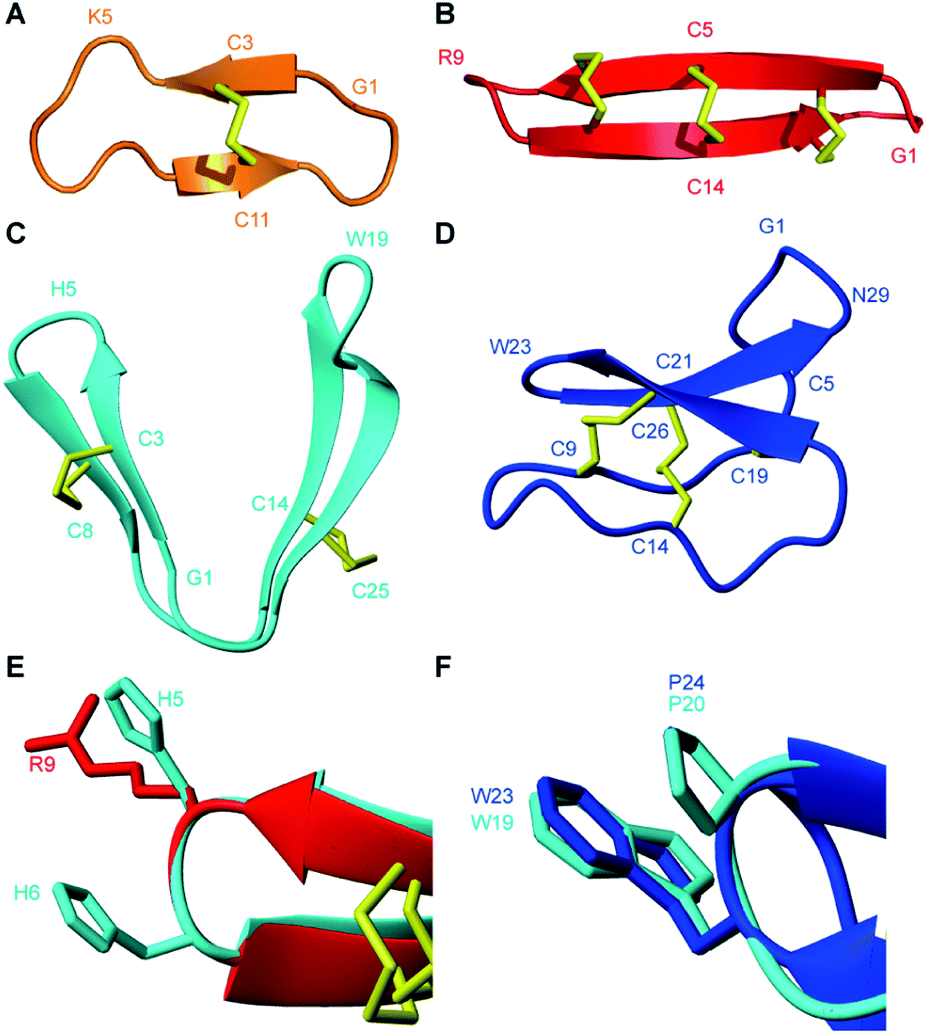 | ||
| Fig. 8 Three-dimensional structures of naturally occurring cyclic peptides. (A) SFTI-1 isolated from the seeds of the common sunflower Helianthus annuus, PDB code: 1JBL.59 (B) θ-defensin found in the leukocytes of rhesus macaques Macaca mulatta, PDB code: 2LYF.60 (C) PDP-23 isolated from the seeds of Zinnia elegans. (D) Kalata B1 isolated from the leaves of Oldenlandia affinis, PDB code: 1NB1.61 (E) Overlay of type I′ turns from θ-defensin RTD-1 (red) and PDP-23 (cyan). Residues in the turn region are shown in stick format and are numbered according to their position in the sequence. (F) Overlay of type VIa1 turns from kalata B1 (blue) and PDP-23 (cyan). Residues in the turn region are shown in stick format and are numbered according to their position in the sequence. | ||
Cyclization and the additional structural constraint imposed by disulfide bonds have been shown to provide resistance to degradation by proteases.54,55 PDP-23 is the first naturally occurring constrained bi-disulfide peptide with a cyclic backbone, which opens up new possibilities in terms of grafting due to its different loop sizes. The smaller PDPs usually contain β-type turn loops four residues in length,16 whereas the larger cyclotides contain six loops typically between one and eight residues.56 PDP-23 has a large loop between CysIII-CysIV containing ten amino acids that may host a larger functional loop. Hydrophobic surface patches have been described for various cyclotides, including the prototypic kalata B1, and they are involved in membrane-binding interactions in conjunction with residues that selectively bind to phosphatidylethanolamine phospholipids.57,58 In contrast, PDP-23 buries its hydrophobic side chains in the core, but is able to expose them in membrane environments. This chameleonic behavior may offer advantages in permeability, and we show that PDP-23 is able to internalize into cells without adverse toxic effects. Furthermore, the ability of PDP-23 to spatially adjust positions of turns offers new possibilities for modifying multiple sites.
The physiological relevance of PDP-23, and the vast majority of PDPs, in seeds remains an enigma. The potent trypsin inhibitor SFTI-1 has been proposed to have evolved as defense against gramnivore insects, given plants lack trypsin.12 Sequences highly homologous to SFTI-1 also inhibit trypsin, but most PDPs do not show protease inhibition and PDP-23 does not contain any required lysine or arginine residues within its sequence. In this study PDP-23 was instead investigated as a potential scaffold for small molecule therapeutics using conjugation chemistry. Attaching a rhodamine dye to PDP-23 yielded a conjugate that was able to demonstrate significant inhibition of P-gp by restoring the sensitivity of KB-V-1 cells to daunorubicin. 5/6-carboxy-tetramethyl-rhodamine has been previously reported to be a substrate of the P-gp transporter.30 However, specific inhibition of P-gp has not been reported. The creation of a P-gp inhibitor out of a small molecule substrate of the transporter not only demonstrates the ability of the PDP-23 scaffold to be used as a carrier of small molecule payloads for intracellular targets, it also presents a novel way of overcoming drug resistance. This application will be particularly useful for small molecule therapeutics that are easily broken down in vivo as well as for chemotherapeutics where resistance to the drug is achieved by efflux.
Conclusions
In conclusion we report the identification, characterization and potential applications of PDP-23, a peptide approximately twice the size of most PDPs and the first reported macrocyclic peptide with two disulfides. It folds into a remarkably optimized fold comprising extensive secondary, tertiary and quaternary structure stabilized by disulfide bonds, hydrogen bonds, and a hydrophobic core. The fold is highly resistant to proteolysis, but has a unique chameleonic character, allowing it to adapt to different environments. PDP-23 represents a novel scaffold for protein engineering, offering multiple turn sites for grafting of bioactive epitopes and conjugation of small molecules. Importantly, unlike other commonly used scaffolds these sites are not spatially confined, PDP-23 is not toxic and internalizes into cells. We highlighted this by creating an inhibitor of P-gp and show PDP-23 is capable of potentiating chemotherapeutics against resistant cancer cells.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work, B. F. and G. V. were supported by Australian Research Council (ARC) grants DP120103369 and DP190102058 to J. S. M. and K. J. R. C. D. P. was supported by a UQ Postgraduate Research Award. M. F. F. was supported by an Australian Postgraduate Award and a Bruce and Betty Green Postgraduate Research Scholarship. C. E. M. was supported by an Australian Postgraduate Award. J. Z., A. S. J. and F. H. were supported by International Postgraduate Research Scholarships and an Australian Postgraduate Award. K. J. R. and J. S. M. were supported by ARC Future Fellowships FT130100890 and FT120100013 respectively.Notes and references
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. Pulavarti, Q. Kaas, A. Eletsky, P. S. Huang, W. A. Johnsen, P. J. Greisen, G. J. Rocklin, Y. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Nature, 2016, 538, 329–335 CrossRef CAS PubMed.
- C. E. Correnti, M. M. Gewe, C. Mehlin, A. D. Bandaranayake, W. A. Johnsen, P. B. Rupert, M. Y. Brusniak, M. Clarke, S. E. Burke, W. De Van Der Schueren, K. Pilat, S. M. Turnbaugh, D. May, A. Watson, M. K. Chan, C. D. Bahl, J. M. Olson and R. K. Strong, Nat. Struct. Mol. Biol., 2018, 25, 270–278 CrossRef CAS PubMed.
- C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS PubMed.
- C. González, G. M. Langdon, M. Bruix, A. Gálvez, E. Valdivia, M. Maqueda and M. Rico, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11221–11226 CrossRef PubMed.
- J. Vetter, Toxicon, 1998, 36, 13–24 CrossRef CAS.
- Y.-Q. Tang, J. Yuan, G. Ösapay, K. Ösapay, D. Tran, C. J. Miller, A. J. Ouellette and M. E. Selsted, Science, 1999, 286, 498–502 CrossRef CAS PubMed.
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Biochemistry, 1995, 34, 4147–4158 CrossRef CAS PubMed.
- B. Franke, J. S. Mylne and K. J. Rosengren, Nat. Prod. Rep., 2018, 35, 137–146 RSC.
- S. Luckett, R. S. Garcia, J. J. Barker, A. V. Konarev, P. R. Shewry, A. R. Clarke and R. L. Brady, J. Mol. Biol., 1999, 290, 525–533 CrossRef CAS PubMed.
- J. S. Mylne, M. L. Colgrave, N. L. Daly, A. H. Chanson, A. G. Elliott, E. J. McCallum, A. Jones and D. J. Craik, Nat. Chem. Biol., 2011, 7, 257–259 CrossRef CAS PubMed.
- K. Bernath-Levin, C. Nelson, A. G. Elliott, A. S. Jayasena, A. H. Millar, D. J. Craik and J. S. Mylne, Chem. Biol., 2015, 22, 571–582 CrossRef CAS PubMed.
- A. G. Elliott, C. Delay, H. Liu, Z. Phua, K. J. Rosengren, A. H. Benfield, J. L. Panero, M. L. Colgrave, A. S. Jayasena, K. M. Dunse, M. A. Anderson, E. E. Schilling, D. Ortiz-Barrientos, D. J. Craik and J. S. Mylne, Plant Cell, 2014, 26, 981–995 CrossRef CAS PubMed.
- A. S. Jayasena, M. F. Fisher, J. L. Panero, D. Secco, K. Bernath-Levin, O. Berkowitz, N. L. Taylor, E. E. Schilling, J. Whelan and J. S. Mylne, Mol. Biol. Evol., 2017, 34, 1505–1516 CrossRef CAS PubMed.
- M. F. Fisher, J. Zhang, N. L. Taylor, M. J. Howard, O. Berkowitz, A. W. Debowski, B. Behsaz, J. Whelan, P. A. Pevzner and J. S. Mylne, Plant Direct, 2018, 2, e00042 CrossRef PubMed.
- B. Franke, A. S. Jayasena, M. F. Fisher, J. E. Swedberg, N. L. Taylor, J. S. Mylne and K. J. Rosengren, Biopolymers, 2016, 106, 806–817 CrossRef CAS PubMed.
- A. G. Elliott, B. Franke, D. A. Armstrong, D. J. Craik, J. S. Mylne and K. J. Rosengren, Amino Acids, 2017, 49, 103–116 CrossRef CAS PubMed.
- R. J. Clark, H. Fischer, L. Dempster, N. L. Daly, K. J. Rosengren, S. T. Nevin, F. A. Meunier, D. J. Adams and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13767–13772 CrossRef CAS PubMed.
- R. J. Clark, J. Jensen, S. T. Nevin, B. P. Callaghan, D. J. Adams and D. J. Craik, Angew. Chem., 2010, 49, 6545–6548 CrossRef CAS PubMed.
- B. Gaurav, M. Vikram Khipple, D. B. Christopher, M. G. Jason, J. H. Peta, C. Olivier, W. B. Garry, V. S. R. K. P. Surya, K. Quentin, E. Alexander, H. Po-Ssu, A. J. William, G. Per Jr, J. R. Gabriel, S. Yifan, W. L. Thomas, W. Andrew, A. R. Stephen, X. Xianzhong, P. C. Lauren, B. Richard, M. O. James, C. Evangelos, E. C. Colin, S. Thomas, J. C. David and B. David, Nature, 2016, 538, 329–335 CrossRef PubMed.
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, J. Am. Chem. Soc., 2013, 135, 11623–11633 CrossRef CAS PubMed.
- C. D'Souza, S. T. Henriques, C. K. Wang, O. Cheneval, L. Y. Chan, N. J. Bokil, M. J. Sweet and D. J. Craik, Biochemistry, 2016, 55, 396–405 CrossRef PubMed.
- C. K. Wang, C. W. Gruber, M. Cemazar, C. Siatskas, P. Tagore, N. Payne, G. Sun, S. Wang, C. C. Bernard and D. J. Craik, ACS Chem. Biol., 2014, 9, 156–163 CrossRef CAS PubMed.
- L. Y. Chan, S. Gunasekera, S. T. Henriques, N. F. Worth, S. J. Le, R. J. Clark, J. H. Campbell, D. J. Craik and N. L. Daly, Blood, 2011, 118, 6709–6717 CrossRef CAS PubMed.
- S. J. de Veer, J. E. Swedberg, M. Akcan, K. J. Rosengren, M. Brattsand, D. J. Craik and J. M. Harris, Biochem. J., 2015, 469, 243–253 CrossRef CAS PubMed.
- C. Y. Li, S. J. de Veer, A. M. White, X. Chen, J. M. Harris, J. E. Swedberg and D. J. Craik, J. Med. Chem., 2019, 62, 3696–3706 CrossRef CAS PubMed.
- R. He, B. Finan, J. P. Mayer and R. D. DiMarchi, Molecules, 2019, 24, 1855 CrossRef CAS PubMed.
- M. de Jong, R. Valkema, F. Jamar, L. K. Kvols, D. J. Kwekkeboom, W. A. P. Breeman, W. H. Bakker, C. Smith, S. Pauwels and E. P. Krenning, Semin. Nucl. Med., 2002, 32, 133–140 CrossRef PubMed.
- S. Westphalen, G. Kotulla, F. Kaiser, W. Krauss, G. Werning, H. P. Elsasser, A. Nagy, K. D. Schulz, C. Grundker, A. V. Schally and G. Emons, Internet J. Oncol., 2000, 17, 1063–1069 CAS.
- A. Régina, M. Demeule, C. Ché, I. Lavallée, J. Poirier, R. Gabathuler, R. Béliveau and J. P. Castaigne, Br. J. Pharmacol., 2008, 155, 185–197 CrossRef PubMed.
- C. Loetchutinat, C. Saengkhae, C. Marbeuf-Gueye and A. Garnier-Suillerot, Eur. J. Biochem., 2003, 270, 476–485 CrossRef CAS PubMed.
- J. S. Mylne, L. Y. Chan, A. H. Chanson, N. L. Daly, H. Schaefer, T. L. Bailey, P. Nguyencong, L. Cascales and D. J. Craik, Plant Cell, 2012, 24, 2765–2778 CrossRef CAS PubMed.
- D. Garcia-Seco, Y. Zhang, F. J. Gutierrez-Mañero, C. Martin and B. Ramos-Solano, BMC Genomics, 2015, 16, 5 CrossRef CAS PubMed.
- A. S. Jayasena, D. Secco, K. Bernath-Levin, O. Berkowitz, J. Whelan and J. S. Mylne, Plant Methods, 2014, 10, 34 CrossRef PubMed.
- B. Franke, M. L. Colgrave, J. S. Mylne and K. J. Rosengren, J. Proteomics, 2016, 147, 177–186 CrossRef CAS PubMed.
- I. V. Shilov, S. L. Seymour, A. A. Patel, A. Loboda, W. H. Tang, S. P. Keating, C. L. Hunter, L. M. Nuwaysir and D. A. Schaeffer, Mol. Cell. Proteomics, 2007, 6, 1638–1655 CrossRef CAS PubMed.
- R. L. J. Keller, The computer aided resonance assignment tutorial, CANTINA Verlag, 1 edn., 2004 Search PubMed.
- P. Güntert, Methods Mol. Biol., 2004, 278, 353–378 Search PubMed.
- A. T. Brunger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef CAS PubMed.
- A. J. Nederveen, J. F. Doreleijers, W. Vranken, Z. Miller, C. A. E. M. Spronk, S. B. Nabuurs, P. Güntert, M. Livny, J. L. Markley, M. Nilges, E. L. Ulrich, R. Kaptein and A. M. J. J. Bonvin, Proteins, 2005, 59, 662–672 CrossRef CAS PubMed.
- V. B. Chen, r. W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- M. A. Hossain, L. M. Haugaard-Kedström, K. J. Rosengren, R. A. Bathgate and J. D. Wade, Org. Biomol. Chem., 2015, 13, 10895–10903 RSC.
- J. van Meerloo, G. J. Kaspers and J. Cloos, Methods Mol. Biol., 2011, 731, 237–245 CrossRef CAS PubMed.
- C. I. Schroeder and K. J. Rosengren, in Snake and Spider Toxins: Methods and Protocols, ed. A. Priel, Springer US, New York, NY, 2020, pp. 129–162, DOI:10.1007/978-1-4939-9845-6_7.
- G. Duplâtre, M. F. Ferreira Marques and M. da Graça Miguel, J. Phys. Chem., 1996, 100, 16608–16612 CrossRef.
- E. Sikorska, D. Wyrzykowski, K. Szutkowski, K. Greber, E. A. Lubecka and I. Zhukov, J. Therm. Anal. Calorim., 2016, 123, 511–523 CrossRef CAS.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- A. K. Nanayakkara, C. A. Follit, G. Chen, N. S. Williams, P. D. Vogel and J. G. Wise, Sci. Rep., 2018, 8, 967–1018 CrossRef PubMed.
- U. Göransson and D. J. Craik, J. Biol. Chem., 2003, 278, 48188–48196 CrossRef PubMed.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, J. Mol. Biol., 1999, 294, 1327–1336 CrossRef CAS PubMed.
- P. K. Pallaghy, K. J. Nielsen, D. J. Craik and R. S. Norton, Protein Sci., 1994, 3, 1833–1839 CrossRef CAS PubMed.
- R. S. Norton and P. K. Pallaghy, Toxicon, 1998, 36, 1573–1583 CrossRef CAS PubMed.
- N. L. Daly, Y. K. Chen, K. J. Rosengren, U. C. Marx, M. L. Phillips, A. J. Waring, W. Wang, R. I. Lehrer and D. J. Craik, Biochemistry, 2007, 46, 9920–9928 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, P. J. Harvey and D. J. Craik, Biopolymers, 2013, 100, 453–460 CrossRef CAS PubMed.
- M. L. Colgrave and D. J. Craik, Biochemistry, 2004, 43, 5965–5975 CrossRef CAS PubMed.
- J. Wang, V. Yadav, A. L. Smart, S. Tajiri and A. W. Basit, Mol. Pharm., 2015, 12, 966–973 CrossRef CAS PubMed.
- J. F. Hernandez, J. Gagnon, L. Chiche, T. M. Nguyen, J. P. Andrieu, A. Heitz, T. Trinh Hong, T. T. Pham and D. Le Nguyen, Biochemistry, 2000, 39, 5722–5730 CrossRef CAS PubMed.
- S. T. Henriques, Y.-H. Huang, K. J. Rosengren, H. G. Franquelim, F. A. Carvalho, A. Johnson, S. Sonza, G. Tachedjian, M. A. R. B. Castanho, N. L. Daly and D. J. Craik, J. Biol. Chem., 2011, 286, 24231–24241 CrossRef CAS PubMed.
- S. T. Henriques, Y.-H. Huang, M. A. R. B. Castanho, L. A. Bagatolli, S. Sonza, G. Tachedjian, N. L. Daly and D. J. Craik, J. Biol. Chem., 2012, 287, 33629–33643 CrossRef CAS PubMed.
- M. L. Korsinczky, H. J. Schirra, K. J. Rosengren, J. West, B. A. Condie, L. Otvos, M. A. Anderson and D. J. Craik, J. Mol. Biol., 2001, 311, 579 CrossRef CAS PubMed.
- A. C. Conibear, K. J. Rosengren, P. J. Harvey and D. J. Craik, Biochemistry, 2012, 51, 9718–9726 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, J. Biol. Chem., 2003, 278, 8606–8616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00692d |
| This journal is © The Royal Society of Chemistry 2021 |