
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese vacancy-confined single-atom Ag in cryptomelane nanorods for efficient Wacker oxidation of styrene derivatives†
Hongling
Yang
a,
Xun
Zhang
c,
Yi
Yu
 c,
Zheng
Chen
d,
Qinggang
Liu
a,
Yang
Li
a,
Weng-Chon
Cheong
e,
Dongdong
Qi
f,
Zewen
Zhuang
a,
Qing
Peng
a,
Xin
Chen
*b,
Hai
Xiao
a,
Chen
Chen
*a and
Yadong
Li
a
c,
Zheng
Chen
d,
Qinggang
Liu
a,
Yang
Li
a,
Weng-Chon
Cheong
e,
Dongdong
Qi
f,
Zewen
Zhuang
a,
Qing
Peng
a,
Xin
Chen
*b,
Hai
Xiao
a,
Chen
Chen
*a and
Yadong
Li
a
aDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cchen@mail.tsinghua.edu.cn
bBeijing Advanced Innovation Center for Materials Genome Engineering, Institute of Solid State Chemistry, University of Science and Technology, Beijing 100083, China. E-mail: chenx@ustb.edu.cn
cSchool of Physical Science and Technology, Shanghai Tech University, Shanghai 201210, China
dCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China
eDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Taipa, Macau SAR 999078, China
fBeijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry, University of Science and Technology Beijing, Beijing 100083, China
First published on 16th March 2021
Abstract
Single-atom catalysts provide a pathway to elucidate the nature of catalytically active sites. However, keeping them stabilized during operation proves to be challenging. Herein, we employ cryptomelane-type octahedral molecular sieve nanorods featuring abundant manganese vacancy defects as a support, to periodically anchor single-atom Ag. The doped Ag atoms with tetrahedral coordination are found to locate at cation substitution sites rather than being supported on the catalyst surface, thus effectively tuning the electronic structure of adjacent manganese atoms. The resulting unique Ag–O–MnOx unit functions as the active site. Its turnover frequency reaches 1038 h−1, one order of magnitude higher than for previously reported catalysts, with 90% selectivity for anti-Markovnikov phenylacetaldehyde. Mechanistic studies reveal that the activation of styrene on the ensemble site of Ag–O–MnOx is significantly promoted, which can accelerate the oxidation of styrene and, in particular, the rate-determining step of forming the epoxide intermediate. Such an extraordinary electronic promotion can be extended to other single-atom catalysts and paves the way for their practical applications.
Introduction
Supported single-atom catalysts (SACs) have attracted considerable attention in the field of heterogeneous catalysis by virtue of their uniform active sites and coordination environment.1–11 It is critical to maintain the stability of SACs that have unambiguous active components under preparation and reaction conditions. Utilizing defects on supports to construct strong interaction between individual metal atoms and supports have become an effective approach to stabilizing SACs.12,13 Among them, metal cation vacancies on the surface of supports have great potential for anchoring single atoms, owing to their adjustable structure and multifarious electron and orbital distributions.14–16 Most recently, we reported a single-atom Pt catalyst anchored on the Ni2+ vacancy defects of Ni(OH)2 nanoboards.14 Although the developed catalyst exhibited high efficiency in diboration reactions, this method has limitations for other metals. Cryptomelane-type octahedral molecular sieve (OMS) nanorods feature ordered arrays of MnO6 moieties with shared edges and vertices, and could serve as an ideal support for anchoring single metal atoms, on account of their defect-rich nature, distinct porosity and tuneable electronic structure.17–21 So far, the cation vacancies on OMS-2 have not been reported, and utilizing the defect-rich OMS-2 to achieve a high metal-loading SAC has not yet been realized.The Wacker oxidation reaction, an atom-economic route for the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, is one of the most important processes in organic synthesis, because its anti-Markovnikov carbonyl products and their derivatives are value-added chemicals.22–27 The protocols developed thus far either depend on precious metals, but have low activities (turnover frequency <108 h−1), or generate Markovnikov products with no regiocontrol.28–32 Moreover, the nature of the active sites in heterogeneous catalysts remains elusive, as the multiple sites involved during the reaction may lead to unexpected synergies. Therefore, there is an urgent need to develop a robust heterogeneous catalyst for aldehyde-selective Wacker oxidation reactions. Notably, the active components in these reported heterogeneous catalysts for Wacker oxidation of styrene are all metal nanoparticles and clusters. Downsizing metal particles or clusters to single atoms is highly desirable to improve their catalytic activity and alter the product distribution. Therefore, the rational design of SACs would provide unexpected opportunities to obtain an ideal heterogeneous catalyst for target-oriented synthesis of anti-Markovnikov carbonyl compounds.
O bonds, is one of the most important processes in organic synthesis, because its anti-Markovnikov carbonyl products and their derivatives are value-added chemicals.22–27 The protocols developed thus far either depend on precious metals, but have low activities (turnover frequency <108 h−1), or generate Markovnikov products with no regiocontrol.28–32 Moreover, the nature of the active sites in heterogeneous catalysts remains elusive, as the multiple sites involved during the reaction may lead to unexpected synergies. Therefore, there is an urgent need to develop a robust heterogeneous catalyst for aldehyde-selective Wacker oxidation reactions. Notably, the active components in these reported heterogeneous catalysts for Wacker oxidation of styrene are all metal nanoparticles and clusters. Downsizing metal particles or clusters to single atoms is highly desirable to improve their catalytic activity and alter the product distribution. Therefore, the rational design of SACs would provide unexpected opportunities to obtain an ideal heterogeneous catalyst for target-oriented synthesis of anti-Markovnikov carbonyl compounds.
Herein, we report a catalyst with single Ag atoms periodically anchored on defect-rich cryptomelane-type octahedral molecular sieve (Ag1/OMS-2) nanorods via a simple thermal diffusion method. Although various strategies have been developed for preparing SACs, the construction of Ag SAC on cation vacancies of OMS-2 has never been achieved owing to the easy aggregation of Ag species. Aberration-corrected annular dark-field scanning transmission electron microscope (AC-HAADF-STEM) image simulation and theoretical calculations identify that the single Ag atoms occupy manganese vacancy sites and form strong Ag–O bonds with the neighbouring O atoms rather than being supported on the catalyst surface, which effectively modulates the Wacker oxidation activity. The turnover frequency (TOF) and the selectivity for phenylacetaldehyde can reach 1038 h−1 and 90%, respectively. To the best of our knowledge, this catalytic performance outperforms those of previously reported heterogeneous catalysts. Density functional theory (DFT) results indicate that single Ag atoms play a crucial role in promoting styrene activation, and lower the energy barrier for the rate-limiting step of formation of the epoxide intermediate. By contrast, the Ag nanoparticles supported on OMS-2 tend to over-oxidize the epoxide into benzaldehyde, leading to a lowered TOF and selectivity for phenylacetaldehyde (173 h−1 and 50%, respectively). Our method here is also applicable to other metals (Au, Ir, and Pd) and therefore is a general and robust fabrication route to SACs.
Results and discussion
The synthetic procedure for the catalyst is illustrated in Fig. 1a. To begin with, OMS-2 nanorods (that is, α-MnO2) were synthesized on a gram scale via a one-pot hydrothermal method.33,34 Subsequently, Ag species were anchored on the surface of the defective OMS-2 via two simultaneous redox and hydrolysis reactions.35 Namely, the chemical is etched onto the surface of OMS-2 nanorods by H2O2,36 thereby promoting the hydrolysis of AgNO3 to Ag(OH), and is further reduced into Ag nanoparticles, and then anchored onto the surface manganese-deficit defect sites of the OMS-2 nanorods. Next, the solid was dried at 120 °C overnight to obtain Ag NPs/OMS-2. After calcination in air at 500 °C, Ag1/OMS-2 was obtained through a thermal diffusion strategy.37 The defects in the OMS-2 nanorods were confirmed by positron annihilation spectroscopy, a well-established technique to determine defects in solids.38 The positron lifetime spectra of Ag1/OMS-2 and OMS-2 are similar (Fig. S1†), indicating that these two samples possess similar defect types. After analysing the spectra, we can see that Ag1/OMS-2 and OMS-2 feature three lifetime components (Table S1†). According to the two-state trapping model, the shortest lifetime component (τ1) could be attributed to the bulk lifetime of the samples, the longest lifetime (τ3) is due to the large voids (defect clusters) in the samples, and τ2 could be ascribed to the positron annihilation trapped at manganese vacancies,39,40 which implies the presence of manganese vacancies in these two samples. In addition to the lifetime of the positron, the relative intensity of positron lifetime provided more information on the concentration of the defects. Table S1† summarizes the lowered manganese vacancy concentration (I2) in the Ag1/OMS-2 sample in comparison to that of OMS-2, directly indicating that the single Ag atoms occupy the manganese vacancy sites rather than support on the catalyst surface.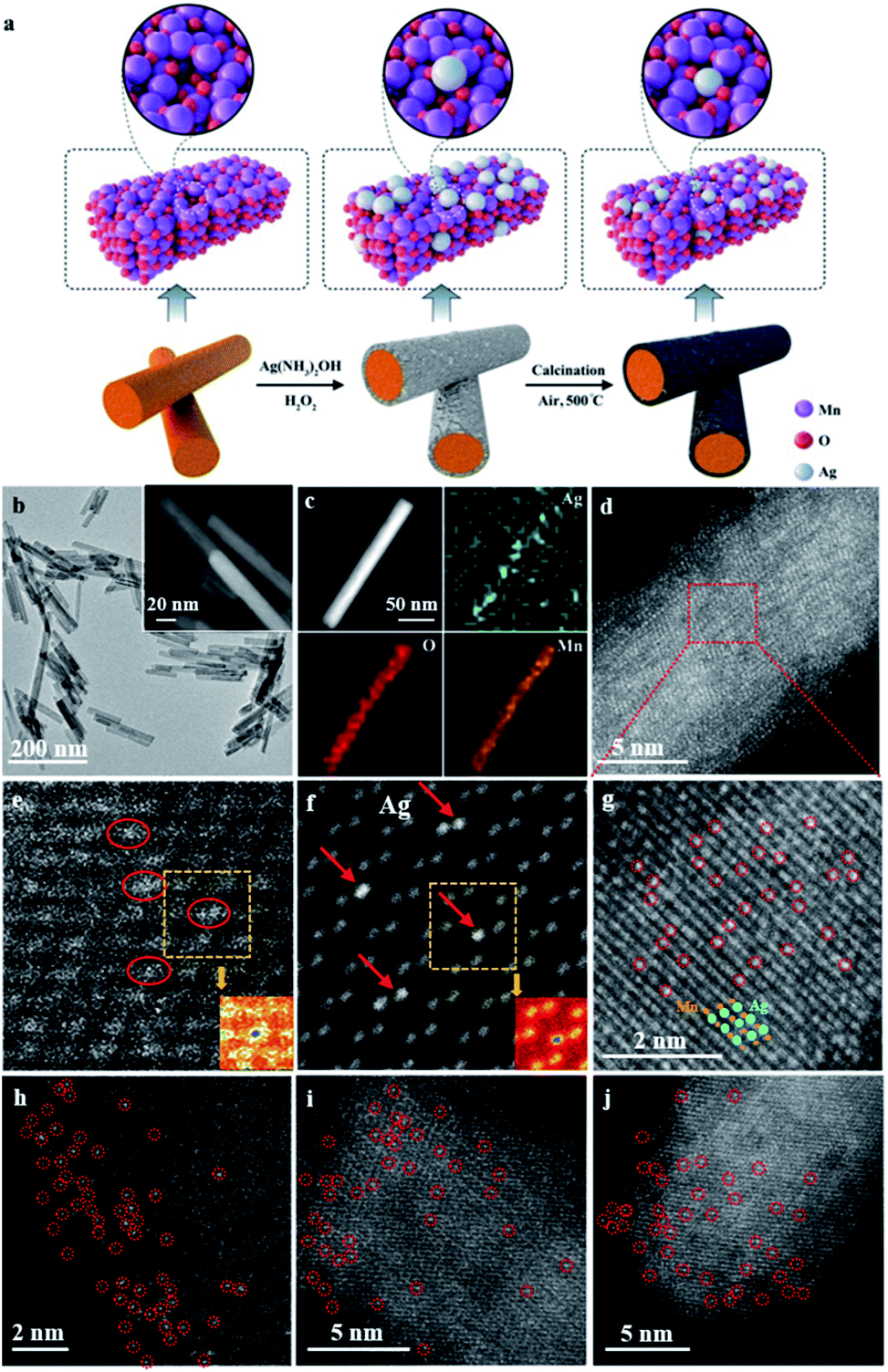 | ||
| Fig. 1 (a) Schematic illustration of the synthetic process. (b) TEM and magnified (inset) images of Ag1/OMS-2 nanorods. (c) HRTEM image and EDX elemental mapping. (d) AC-HAADF-STEM and (g) magnified AC-HAADF-STEM images of Ag1/OMS-2. (e) Experimental raw HAADF-STEM image and (f) simulated HAADF-STEM image using a model with seven Ag atoms replacing seven surface manganese atoms (inset: corresponding falsely colored images for better observation). (h–j) AC-HAADF-STEM images of Ir1/OMS-2, Au1/OMS-2, and Pd1/OMS-2, respectively; metal single atoms are highlighted with red circles. | ||
A uniform nanorod structure can be observed in the TEM and high-resolution TEM (HRTEM) images (Fig. 1b and its inset and S2†), which inherits the structure of OMS-2, with diameters of about 13 nm and lengths of 30–100 nm. HAADF-STEM and energy-dispersive X-ray spectroscopy (EDX) images reveal that Ag, O and Mn elements are evenly distributed over the entire OMS-2 structure, and no Ag nanoparticles are observed (Fig. 1c). The content of Ag is estimated to be approximately 2.5 wt% based on inductively coupled plasma optical emission spectrometry (ICP-OES) analysis. In the AC-HAADF-STEM images (Fig. 1d and g), high density bright dots (highlighted by red circles) corresponding to single Ag atoms are observed. As shown in the intensity profiles along the line (Fig. S3†), the single Ag atoms are much brighter than the manganese atoms. Moreover, these images also demonstrate that those small bright dots of Ag locate at the lattice walls of the manganese columns, rather than in the spacing in between. This indicates that the single Ag atoms are stabilized in the manganese-deficit defect sites by substitution instead of interstitial doping. This can be further verified by the following simulation. Fig. 1e and f, respectively, show the experimental HAADF-STEM image and the simulated HAADF-STEM image using a model with seven Ag atoms replacing seven surface manganese atoms. In the simulated image, the atom column containing Ag shows higher contrast than the nearby Mn–O atom columns, as Ag has a larger atomic number. The agreement between experiment and simulation provides clear evidence that the Ag atoms are anchored as substitutes of manganese. The detailed simulated procedure is summarized in the ESI.† In contrast, the simulated HAADF-STEM image using the model in which Ag atoms are in the interval between the manganese columns does not match the experimental image at all (Fig. S4†). Moreover, the surface area and pore size of the Ag1/OMS-2 catalyst were nearly identical to those of the pure OMS-2 support, indicating that the prepared Ag1/OMS-2 and OMS-2 materials have similar textural properties (Fig. S5 and Table S2†). This thermal diffusion strategy can be readily generalized to other elements, such as Ir, Au, and Pd. In addition, the atomically dispersed nature of the metal atoms throughout the OMS-2 support was demonstrated by the AC-HAADF-STEM images (Fig. 1h–j and S6†).
To further confirm that the single Ag atoms are dispersed in Ag1/OMS-2, and to unveil the changes in atomic and electronic structure of Ag1/OMS-2, we performed X-ray absorption near-edge structure spectroscopy (XANES), extended X-ray absorption fine structure spectroscopy (EXAFS), X-ray photoelectron spectrometry (XPS), and X-ray diffraction (XRD). The manganese K-edge XANES spectra of OMS-2 and Ag1/OMS-2 are quite similar (Fig. S7†), except that the absorption edge of Ag1/OMS-2 was shifted by about 0.5 eV to higher energy, indicating a slightly higher oxidation state of manganese in Ag1/OMS-2 compared with that in OMS-2, which can be ascribed to the occupation of manganese defects by Ag atoms.41,42 The oxidation state of manganese was further analysed by manganese 3s XPS (Fig. S8†), and the calculated average oxidation state of manganese increases from 3.39 to 3.53 after the introduction of Ag (Table S3†). The local atomic arrangements around manganese atoms in OMS-2 and Ag1/OMS-2 were revealed by the Fourier-transform (FT) magnitudes of k2-weighted EXAFS spectroscopy (Fig. S9†). The manganese EXAFS spectra for OMS-2 and Ag1/OMS-2 show three peaks in R-space spectra. The strongest peak at ∼1.5 Å is ascribed to the closest oxygen (Mn–O) in the MnO6 octahedra, whereas the second and third peaks, around 2.4 and 3.0 Å, originate mainly from the manganese in the edge-sharing (Mn–Mnedge) and corner-sharing (Mn–Mncorner) MnO6 octahedra, respectively.43,44 After the introduction of Ag, the magnitude of the FT peaks decreased, revealing the increased structural distortion due to the insertion of Ag into the OMS-2 structure and an increased amount of Mn4+ ions. In the XRD pattern of Ag1/OMS-2 (Fig. S10†), no peaks corresponding to Ag species (such as metallic Ag, Ag2O, and AgO) can be observed,45,46 indicating that Ag species are highly dispersed. The diffraction intensity of the OMS-2 is altered by the addition of Ag, and the reflections of the (220) and (310) planes become stronger, whereas those of the (301) and (411) planes are weaker. These changes may result from the influence of the structure alternation on diffraction intensity,33 assuming that the manganese-deficit vacancy defects are occupied by Ag atoms. The intensities of the (110), (200), and (002) planes are also substantially weakened, which probably originates from the rearrangement of the atoms in these planes induced by the addition of Ag, as Ag has a larger atomic radius than manganese.
Fig. 2a shows the XANES spectra of the Ag K-edge. The position of the absorption edge of Ag1/OMS-2 is located between those of Ag foil and AgO (Fig. 2a, inset), suggesting that the single Ag atom is positively charged and its oxidation state is between 0 and +2. The oxidation state of Ag was further analysed by XPS. As shown in Fig. 2b, the Ag 3d spectrum can be deconvoluted into two peaks centred at 367.8 and 373.8 eV, corresponding to 3d5/2 and 3d3/2 levels, respectively.47,48 The peak positions are between those of Ag(II) and Ag(0), indicating Ag atoms carry partially positive charge through electron transfer from the support to the Ag atoms, thus enhancing the metal–support interaction. Fig. 2c shows the FT k3-weighted EXAFS spectra of Ag1/OMS-2 and reference samples. The profile for Ag1/OMS-2 shows two main peaks at about 1.5 and 3.0 Å, associated with the first shell of Ag–O scattering and the higher shell of Ag–Mn, respectively. Compared with the FT profile of Ag foil, no scattering from the Ag–Ag contribution is observed, indicating the sole existence of atomically dispersed Ag in Ag1/OMS-2. The corresponding fits of the EXAFS spectra of Ag1/OMS-2 in R space and K space are exhibited in Fig. S11.† To further confirm this inference, a wavelet transform (WT) analysis of the Ag K-edge EXAFS oscillations was performed. In Fig. 2d, the WT contour plot of Ag1/OMS-2 in the first coordination shell shows one intensity maximum at around 4 Å−1, which is associated with the Ag–O contribution. However, for the higher coordination shell, the WT contour plot shows an additional maximum at around 5 Å−1, owing to the Ag–Mn contribution. In comparison to the WT contour plot of the Ag foil, there is no intensity maximum detected near 8.5 Å−1 (indexed to the Ag–Ag path), further confirming that Ag atoms are atomically dispersed.
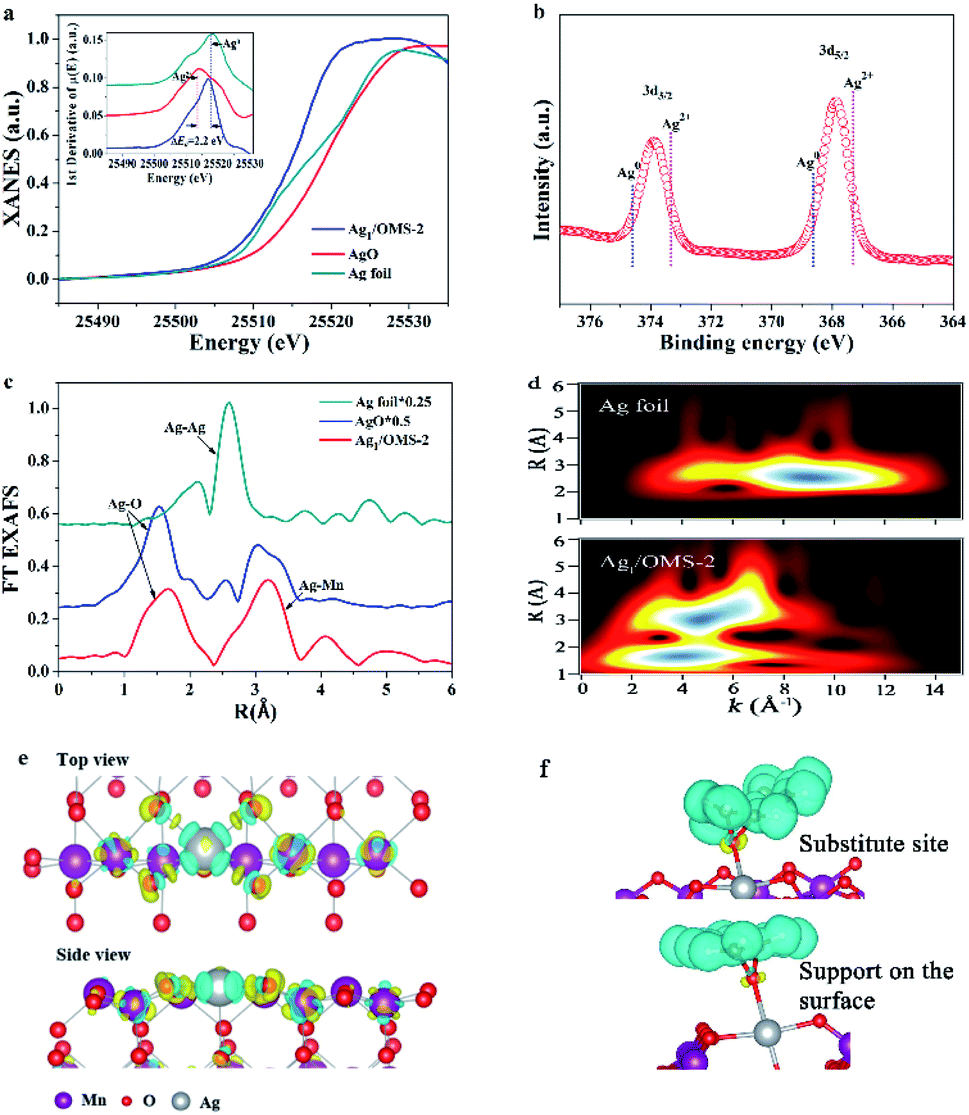 | ||
| Fig. 2 (a) Ag K-edge XANES spectra. Inset: first derivatives of the Ag K-edge XANES spectra. (b) XPS spectrum for Ag 3d of Ag1/OMS-2. (c) EXAFS FT spectra of Ag1/OMS-2 and reference. (d) WT for the EXAFS signals in Ag foil and Ag1/OMS-2. Charge density difference of (e) Ag1/OMS-2 with a plain view (upper image) and a side-on view (lower image), and (f) adsorbing styrene over an Ag atom in a substitution site of a manganese atom (upper image) and supported on the surface (lower image). Yellow and cyan regions represent electron accumulation and depletion, respectively. | ||
To get the precise atomic structure and explore the interaction between single-atom Ag and OMS-2 support, we carried out quantitative EXAFS curve fitting, and the results for Ag1/OMS-2 are listed in Table S4.† According to the fitting results, the Ag–O coordination number for Ag1/OMS-2 is 4, with a bond length of 2.06 Å, implying that each Ag atom is coordinated with four oxygen atoms in the OMS-2 nanorods. Moreover, the coordination number for the higher shell of Ag–Mn is 4.6 ± 0.7, with a bond length of 3.72 ± 0.02 Å. The possible structure of Ag1/OMS-2 was further elucidated by first-principles calculations, which can provide more insight into the interaction between manganese vacancies and single Ag atoms. The O-terminated (110) surface of OMS-2 was selected for the simulation model (Fig. S12†).49 For the OMS-2 with manganese vacancies, the manganese vacancy site and the fourfold hollow site of the oxygen atoms were confirmed to be the most stable adsorption sites for the single Ag atom. According to the results of charge density difference, single Ag atoms are fixed by the four top oxygen atoms near the manganese vacancy (Fig. 2e). In the simulation model, the bond lengths of Ag–Mn and Ag–O are 3.70 and 2.08 Å, respectively, which are in accord with the EXAFS results. The cyan region represents charge density depletion around a single Ag atom, which illustrates that the Ag is positively charged. The charge accumulation on Ag is |1.09e| based on the Bader charge analysis.
In addition, the Ag atom adsorbed onto the OMS-2 with manganese vacancies has a formation energy of −0.9 eV, indicating that this stable structure is readily available (Fig. S13,† inset). By contrast, for the Ag atom supported on the OMS-2 without manganese vacancies, it tends to locate at the site that has a slight deviation from the oxygen atoms locating at the fourfold hollow site. This is caused by the competition between the interaction between the Ag atom and the four top oxygen atoms and the electrostatic repulsion of the Ag and manganese atoms with positive charge (Fig. S14†). Based on these DFT calculation results and the XANES data, it can be inferred that the manganese vacancies play a critical role in stabilizing the isolated Ag atoms supported on OMS-2, through removing the spatial isolation between Ag atoms and O atom and thus promoting the charge transfer from OMS-2 to Ag. The total density of states and partial density of states (PDOS) of Ag1/OMS-2 were calculated to reveal the energy contribution from this atomic orbital hybridization. The PDOS of manganese atoms has more contributions, compared with those of O atoms, near the Fermi energy (Fig. S13†). The PDOS of the Ag atom mainly occupies the energy below the Fermi energy, thus the ensemble active site of Ag–O–MnOx is favourable for the activation of reactants.50Fig. 2f shows the charge density difference of adsorbing styrene over an Ag atom in the substitution site of a manganese atom and supported on the surface. After adsorbing the styrene molecule, the charge is more accumulated on the O atom, which was adsorbed by the Ag atom in the substitution site of manganese, indicating that this configuration promotes the rate-determining step of formation of styrene oxide.
The selective oxidation of styrene to aldehydes is one of the most important processes in organic synthesis. When we used Ag1/OMS-2 to catalyse the Wacker oxidation of styrene as a representative substrate (Fig. 3a), we found that the catalyst achieved a nearly 100% conversion of styrene, and produced almost exclusively the anti-Markovnikov oxidation product, phenylacetaldehyde (detected by gas chromatography-mass spectrometer (GC-MS), 1H nuclear magnetic resonance (NMR) and 13C NMR; Fig. S15–S19†). The calculated TOF of Ag1/OMS-2 reached as high as 1038 h−1. There is no evident metal Ag leaching in the solution, based on the ICP-OES result. The yield of phenylacetaldehyde catalysed by other SACs (Ru1/OMS-2, Rh1/OMS-2, Pd1/OMS-2, Ir1/OMS-2, Pt1/OMS-2, and Au1/OMS-2) are shown in Fig. S20.† Under the identical reaction conditions, no product was detected without any catalyst (Fig. 3b). We also assessed the catalytic performance of the pure OMS-2, and it gave a nearly 10% yield of phenylacetaldehyde, indicating that OMS-2 itself plays an important role in Wacker oxidation of styrene, which is consistent with previous reports.51 For comparison, a sample of Ag NPs/OMS-2 with an Ag loading of 2.5 wt% was prepared and tested, which offered a phenylacetaldehyde yield of merely 15% under the same conditions. Based on the reported literature,20 we conducted a controlled experiment to determine the reaction intermediates. The controlled experiment revealed that when we used styrene oxide as the reaction substrate, the complete transformation of styrene oxide to phenylacetaldehyde was realized under identical reaction conditions (Fig. S21†), indicating that styrene oxide is the intermediate for Wacker oxidation of styrene.
 | ||
| Fig. 3 (a) Scheme of Wacker oxidation of styrene. (b) Phenylacetaldehyde yield of different catalysts. (c) Recycling test of Ag1/OMS-2 for the catalytic Wacker oxidation of styrene. | ||
In practical production, catalyst recycling is an important issue in heterogeneous catalytic reactions.52 Therefore, we investigated the recyclability of our catalyst for the Wacker oxidation reaction by using styrene as a model substrate. The spent catalyst was separated, and then used after only washing with ethanol and drying, with no extra fresh catalyst being added. As shown in Fig. 3c, after five cycles, the activity and selectivity of Ag1/OMS-2 were well retained, suggesting a good recycling capability and potential for the Wacker oxidation of styrene in practical applications. Besides, the structures were unchanged with respect to those of the fresh samples, as identified by XRD and AC-HAADF-STEM (Fig. S22 and S23†). For the spent Ag1/OMS-2 catalyst, no Ag particles formed after the Wacker oxidation of styrene, which has been confirmed by EXAFS (Fig. S24†). Next, we examined the scope of alkenes for this Wacker oxidation reaction catalysed by Ag1/OMS-2, and the results are presented in Table S5.† When styrene substituted with electron-donating (Table S5,† entries 1–4) or electron-withdrawing groups (Table S5,† entries 5–9) were used, the desired anti-Markovnikov products were obtained in high yields (>90%). Furthermore, the catalysis comparison with reported works in the literature is summarized in Table S6.† Our Ag1/OMS-2 has both an outstanding TOF and a remarkable selectivity for phenylacetaldehyde, with low catalyst usage and simple preparation method.
To assess the activity of surface oxygen that participates in the styrene oxidation reaction, the electronic properties and the amount of adsorbed oxygen on OMS-2 and Ag1/OMS-2 were studied via XPS, temperature-programmed desorption of oxygen (O2-TPD), XANES, and Raman spectroscopy. The electronic density of oxygen was determined by the O 1s XPS, and an intensive peak with a shoulder at the high binding energy side is observed for OMS-2 and Ag1/OMS-2 catalysts in Fig. 4a. The main peak at about 528.5–530.5 eV and the broad one at 531.6 eV are assigned to surface lattice oxygen (Olat., Os2−) and adsorbed oxygen (Oads.), respectively. In addition, a discernible shift to lower binding energy for Ag1/OMS-2 is observed in comparison to OMS-2, which is commonly assigned to an increase of its negative charge. As displayed in Fig. 4a, the content of adsorbed oxygen on the surface of Ag1/OMS-2 (20.4%) is higher than that of OMS-2 (18.9%), suggesting that Ag1/OMS-2 is more conducive to generating active oxygen species, thereby promoting the oxidation of styrene.53–55 O2-TPD was conducted to identify the evolution of oxygen from the prepared materials (Fig. 4b). The desorption peak below 350 °C can be ascribed to the release of chemisorbed oxygen molecules and active surface oxygen, whereas the peak in the range 400–650 °C is attributed to the release of sub-surface lattice oxygen. The peak above 700 °C is due to the evolution of bulk lattice oxygen.56 The oxygen released at low temperature was considered as labile oxygen species, which were highly active for styrene oxidation. A lower oxygen-releasing temperature indicates a looser binding between the oxygen species and manganese atoms. In the case of Ag1/OMS-2, the desorption temperature was like that for OMS-2, and the amount of labile oxygen was higher than that of OMS-2, indicating that Ag1/OMS-2 contains much more loosely bound labile oxygen species than does OMS-2, which is in accordance with the O 1s XPS result. Therefore, the Ag1/OMS-2 deserves to give a better catalytic activity for styrene oxidation.
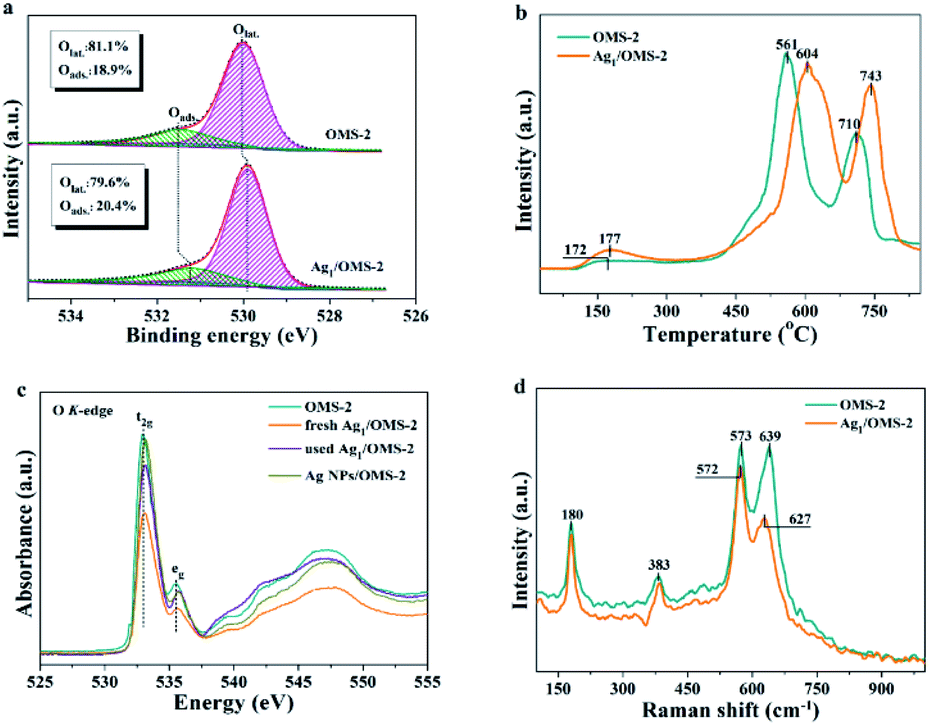 | ||
| Fig. 4 (a) O 1s XPS, (b) O2-TPD, (c) O K-edge XANES, and (d) Raman profiles of OMS-2 and Ag1/OMS-2. | ||
Fig. 4c shows the O K-edge XANES spectra for OMS-2, fresh and used Ag1/OMS-2, and Ag NPs/OMS-2. The broad peaks at ≥535 eV are attributed to the transition from O 1s to O 2p and the manganese 4sp orbital, whereas the characteristic peak at around 530 eV corresponds to the transition from O 1s to O 2p and the manganese 3d orbital.57 The peak feature in the O K-edge XANES for Ag NPs/OMS-2 is almost the same as that for the OMS-2 support, implying that the electronic structure of oxygen in the sample is unchanged by impregnating Ag nanoparticles. By contrast, the absorption peak intensity of fresh Ag1/OMS-2 decreases notably with respect to that of OMS-2, indicating that the introduction of Ag atoms induced electronic structural changes around oxygen ions but barely any change around the manganese ions, which might be the main origin for the increased mobility of active oxygen species after the introduction of Ag. In addition, the absorption peak intensities of the fresh Ag1/OMS-2 are significantly changed after styrene oxidation reaction, which further confirms that the active oxygen species are involved in the reaction. As shown in Fig. 4d, the Raman scattering spectra of OMS-2 and Ag1/OMS-2 both feature four main contributions at 180, 383, 573, and 639 cm−1. The Raman bands at around 180 and 383 cm−1 are assigned to the deformation vibration of Mn–O–Mn. The bands at around 639 and 573 cm−1 are attributed to the symmetric stretching vibration (Mn–O), which is perpendicular to the MnO6 group direction.58,59 It is very interesting to note that, compared with the Mn–O stretching vibration mode (639 cm−1) of OMS-2, a much weaker and a considerable downshift of the Mn–O stretching vibration can be observed for Ag1/OMS-2 (627 cm−1), suggesting that Ag incorporation increases the disorder of OMS-2 and induces more O vacancies in OMS-2,60 thus favouring the activity of the catalyst.
Further DFT calculations were carried out to unravel the reaction mechanism and to elucidate the fact that the Ag SAC has higher catalytic performance than Ag NPs. The atomic structure of Ag NPs is shown in Fig. S25.† The configuration of Ag1/OMS-2 has single Ag atoms surrounded by manganese atoms and four oxygen atoms with bond lengths of 3.70 and 2.08 Å, respectively. The styrene oxidation reaction catalysed by both Ag1/OMS-2 and Ag NPs/OMS-2 follows the E–I (epoxidation–isomerization) mechanism. As shown in Fig. 5, the styrene molecule first approaches the Ag1/OMS-2 catalyst via a non-planar configuration, where single Ag atoms play an important role in the adsorption and activation of styrene molecules (first step). Then, the styrene molecule connects to the active oxygen species derived from tert-butyl hydroperoxide (TBHP), forming an intermediate state of styrene oxide (second step). Subsequently, the hydride migration and rearrangement lead to the formation of the desired product, phenylacetaldehyde (third step). The energy barriers in the styrene oxide formation step are the highest, suggesting that the second step is the rate-determining step of this reaction. For the rate-limiting step, the Ag–O–MnOx moieties acting as active sites play a critical role in facilitating the formation of epoxide. Overall, in the first two elementary steps, the energy barriers over Ag1/OMS-2 are thermodynamically lower than those over Ag NPs/OMS-2. The energy barriers of the third step for Ag1/OMS-2 and Ag NPs/OMS-2 are similar, because the Lewis acidity of OMS-2 can make styrene oxide spontaneously isomerize into phenylacetaldehyde.61 The Lewis acidity has been verified by pyridine infrared spectroscopy (Fig. S26†). Besides, it is relatively difficult to break the epoxy ring to form benzaldehyde over Ag1/OMS-2, as the energy barrier of the third step is relatively high (Fig. S27†). Furthermore, the single Ag atoms substituting the manganese sites have lower energy barriers for Wacker oxidation than that of single Ag atoms supported on OMS-2, and pristine OMS-2 (Fig. S28 and S29†). These calculated results are in agreement with the superior catalytic performance for Ag1/OMS-2 observed in experiment.
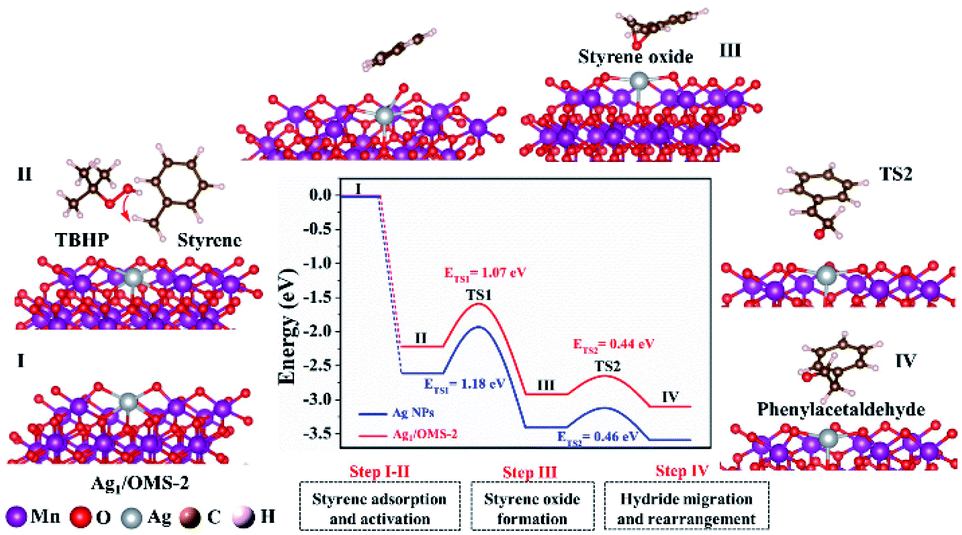 | ||
| Fig. 5 DFT calculation proposed a reaction pathway for the Wacker oxidation of styrene to phenylacetaldehyde on Ag1/OMS-2, and the calculated energy profiles for Ag1/OMS-2 (red line) and Ag NPs (blue line). | ||
Conclusions
In summary, we report a defect-rich OMS-2 nanorod-supported Ag SAC with remarkable performance towards anti-Markovnikov Wacker oxidation. This OMS-2 support with abundant manganese vacancy defects plays a key role in stabilizing the single Ag atoms via an enhanced charge-transfer mechanism. The TOF of the as-fabricated Ag1/OMS-2 reaches up to ∼1038 h−1, which surpasses those of conventional heterogeneous catalysts based on nanoparticles and clusters. In addition, no evident metal leaching or aggregation were found after the reaction. Combined with surface science, theoretical, and experimental data, the enhanced catalytic properties could be explained at the molecular level, which reveals the critical role of the location of single Ag atoms. Specifically, the single Ag atoms occupy the manganese vacancy sites of OMS-2 rather than being supported on the catalyst surface, and thus facilitate the activation of styrene and lower the energy barrier for the rate-limiting step of formation of the epoxide intermediate. Moreover, this is the first report of an SAC being used for the aldehyde-selective Wacker oxidation of styrene. Using the cation vacancy defects in supports to anchor single metal atoms could become an efficient approach to preparing these SACs on a large scale.Author contributions
H.Y. performed the experiments, collected and analysed the data, and wrote the paper. X.C., D.Q., and H.X. conducted the DFT calculations and analysis. X.Z. and Y.Y. obtained the AC-HAADF-STEM image simulation. Z.C., Q.L., and Y.L. discussed the catalytic process and gave useful suggestions. W.C. helped with the HRTEM characterization and analysis. Z.Z. helped with the X-ray structure characterization and analysis of the XAS data. Q.P. and Y.L. helped with data analyses and discussions. C.C. conceived the experiments, planned the synthesis, analysed results, and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (JQ18007), National Natural Science Foundation of China (21925202, 21872076 and 22008006), National Key R&D Program of China (2016YFA0202801, 2017YFA0700101), Tsinghua University Initiative Scientific Research Program and China Postdoctoral Science Foundation (2018M641321). We thank Tsinghua National Laboratory for Information Science and Technology and USTB MatCom of Beijing Advanced Innovation Center for Materials Genome Engineering for providing grid resources that have contributed to the calculation results. We acknowledge the support of Analysis Center of Tsinghua University for XPS measurements. Also, we thank Dr C. Zhang for help in preparing this article.Notes and references
- X. F. Yang, A. Q. Wang, B. T. Qiao, J. Li, J. Y. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- M. Z. Hu, J. Zhang, W. Zhu, Z. Chen, X. Gao, X. J. Du, J. W. Wan, K. B. Zhou, C. Chen and Y. D. Li, Nano Res., 2018, 11, 905–912 CrossRef CAS.
- J. Li, Y. D. Li and T. Zhang, Sci. China Mater., 2020, 63, 889–891 CrossRef.
- Y. B. Lu, J. M. Wang, L. Yu, L. Kovarik, X. W. Zhang, A. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. L. Xin and A. M. Karim, Nat. Catal., 2019, 2, 149–156 CrossRef CAS.
- C. Y. Xu, Y. T. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS.
- N. H. Fu, X. Liang, Z. Li, W. X. Chen, Y. Wang, L. R. Zheng, Q. H. Zhang, C. Chen, D. S. Wang, Q. Peng, L. Gu and Y. D. Li, Nano Res., 2020, 13, 947–951 CrossRef CAS.
- Q. Xu, C. X. Guo, S. B. Tian, J. Zhang, W. X. Chen, W. C. Cheong, L. Gu, L. R. Zheng, J. P. Xiao, Q. Liu, B. J. Li, D. S. Wang and Y. D. Li, Sci. China Mater., 2020, 63, 972–981 CrossRef CAS.
- Y. Y. Qian, D. D. Li, Y. L. Han and H. L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- H. L. Yang, Q. G. Liu, Y. Li, K. A. Sun, Z. Q. Chen, Q. Peng and C. Chen, ChemNanoMat, 2020, 6, 1647–1651 CrossRef CAS.
- X. J. Cui and F. Shi, Acta Phys.-Chim. Sin., 2021, 37, 2006080–2006095 Search PubMed.
- Q. H. Shang, N. F. Tang, H. F. Qi, S. Chen, G. L. Xu, C. T. Wu, X. D. Wang and Y. Cong, Chin. J. Catal., 2020, 41, 1812–1817 CrossRef CAS.
- Y. Q. Zhang, L. Guo, L. Tao, Y. B. Lu and S. Y. Wang, Small Methods, 2019, 3, 1800406–1800423 CrossRef.
- J. Deng, H. B. Li, J. P. Xiao, Y. C. Tu, D. H. Deng, H. X. Yang, H. F. Tian, J. Q. Li, P. J. Ren and X. H. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- J. Zhang, X. Wu, W. C. Cheong, W. X. Chen, R. Lin, J. Li, L. R. zheng, W. S. Yan, L. Gu, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Nat. Commun., 2018, 9, 1002–1010 CrossRef.
- D. Zhao, Z. Chen, W. J. Yang, S. J. Liu, X. Zhang, Y. Yu, W. C. Cheong, L. R. Zheng, F. Q. Ren, G. B. Ying, X. Cao, D. S. Wang, Q. Peng, G. X. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- H. W. Zhang and R. T. Lv, J. Materiomics, 2018, 4, 95–107 CrossRef.
- X. Chen, Y. F. Shen, S. L. Suib and C. L. O'Young, Chem. Mater., 2002, 14, 940–948 CrossRef CAS.
- C. K. King'ondu, N. Opembe, C. Chen, K. Ngala, H. Huang, A. Iyer, H. F. Garcés and S. L. Suib, Adv. Funct. Mater., 2011, 21, 312–323 CrossRef.
- J. Xie, L. Chen, W. F. Zhou, C. T. Au and S. F. Yin, J. Mol. Catal. A: Chem., 2016, 425, 110–115 CrossRef CAS.
- L. Yu, M. Sun, J. Yu, Q. Yu, Z. F. Hao and C. S. Li, Chin. J. Catal., 2008, 29, 1127–1132 CrossRef CAS.
- P. P. Wang, J. H. Duan, J. Wang, F. M. Mei and P. Liu, Chin. J. Catal., 2020, 41, 1298–1310 CrossRef CAS.
- S. C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges and F. H. Arnold, Science, 2017, 358, 215–218 CrossRef CAS PubMed.
- J. J. Dong, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 734–744 CrossRef CAS.
- K. E. Kim, J. M. Li, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 13179–13182 CrossRef CAS PubMed.
- B. Weiner, A. Baeza, T. Jerphagnon and B. L. Feringa, J. Am. Chem. Soc., 2009, 131, 9473–9474 CrossRef CAS PubMed.
- A. D. Chowdhury, R. Ray and G. K. Lahiri, Chem. Commun., 2012, 48, 5497–5499 RSC.
- G. F. Zhang, X. Q. Xie, Y. Wang, X. Wen, Y. Zhao and C. R. Ding, Org. Biomol. Chem., 2013, 11, 2947–2950 RSC.
- J. Chen and C. M. Che, Angew. Chem., Int. Ed., 2004, 43, 4950–4954 CrossRef CAS PubMed.
- P. L. Teo, Z. K. Wickens, G. B. Dong and R. H. Grubbs, Org. Lett., 2012, 14, 3237–3239 CrossRef CAS PubMed.
- J. Muzart, Tetrahedron, 2007, 63, 7505–7521 CrossRef CAS.
- J. A. Wright, M. J. Gaunt and O. B. Spencer, Chem.–Eur. J., 2006, 12, 949–955 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- Z. W. Huang, X. Gu, Q. Q. Cao, P. P. Hu, J. M. Hao, J. H. Li and X. F. Tang, Angew. Chem., Int. Ed., 2012, 124, 4274–4279 CrossRef.
- P. P. Hu, Z. W. Huang, Z. Amghouz, M. Makkee, F. Xu, F. Kapteijn, A. Dikhtiarenko, Y. X. Chen, X. Gu and X. F. Tang, Angew. Chem., Int. Ed., 2014, 126, 3486–3489 CrossRef.
- D. H. Xia, W. J. Xu, Y. C. Wang, J. L. Yang, Y. J. Huang, L. L. Hu, C. He, D. Shu, D. Y. C Leung and Z. H. Pang, Environ. Sci. Technol., 2018, 52, 13399–13409 CrossRef CAS PubMed.
- J. Chen, X. Chen, Z. Xu, W. J. Xu, J. J. Li, H. P. Jia and J. Chen, ChemistrySelect, 2016, 1, 4052–4056 CrossRef CAS.
- Y. X. Chen, Z. W. Huang, Z. Ma, J. M. Chen and X. F. Tang, Catal. Sci. Technol., 2017, 7, 4250–4258 RSC.
- S. Gao, B. C. Gu, X. C. Jiao, Y. F. Sun, X. L. Zu, F. Yang, W. G. Zhu, C. M. Wang, Z. M. Feng, B. J. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS.
- H. Kumar, A. Kaushik, P. A. Alvi, B. Dalela and S. Dalela, AIP Conf. Proc., 2018, 1953, 120049–120053 CrossRef.
- C. Xiao, X. M. Qin, J. Zhang, R. An, J. Xu, K. Li, B. X. Cao, J. L. Yang, B. J. Ye and Y. Xie, J. Am. Chem. Soc., 2012, 134, 18460–18466 CrossRef CAS PubMed.
- Y. X. Zhao, C. Chang, F. Teng, Y. F. Zhao, G. B. Chen, R. Shi, G. I. N. Waterhouse, W. F. Huang and T. R. Zhang, Adv. Energy Mater., 2017, 7, 1700005–1700011 CrossRef.
- A. Manceau, M. A. Marcus and S. Grangeon, Am. Mineral., 2012, 97, 816–827 CrossRef CAS.
- M. Jianga, C. Fua, J. Yanga, Q. Liub, J. Zhang and B. Suna, Energy Storage Mater, 2019, 18, 34–42 CrossRef.
- R. G. Zhang, X. Q. Yu, K. W. Nam, C. Ling, T. S. Arthur, W. Song, A. M. Knapp, S. N. Ehrlich, X. Q. Yang and M. Matsui, Electrochem. Commun., 2012, 23, 110–113 CrossRef CAS.
- Y. Yang, C. Q. Gong, S. Xiao, H. M. Gong, Q. Q. Wang and J. C. Zhong, Acta Phys.-Chim. Sin., 2006, 22, 791–796 CrossRef CAS.
- G. Q. Chang, X. Zheng, R. Y. Chen, X. Chen, L. Q. Chen and Z. Chen, Acta Phys.-Chim. Sin., 2008, 24, 1790–1796 CrossRef CAS.
- H. L. Yang, C. Y. Ma, X. Zhang, Y. Li, J. Cheng and Z. P. Hao, ACS Catal., 2018, 8, 1248–1258 CrossRef CAS.
- H. L. Yang, C. Y. Ma, Y. Li, J. H. Wang, X. Zhang, G. Wang, N. L. Qiao, Y. G. Sun, J. Cheng and Z. P. Hao, Chem. Eng. J., 2018, 347, 808–818 CrossRef CAS.
- W. X. Li, X. Y. Cui, R. Zeng, G. D. Du, Z. Q. Sun, R. K. Zheng, S. P. Ringer and S. X. Dou, Sci. Rep., 2015, 5, 8987–8994 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- R. Ghosh, X. F. Shen, J. C. Villegas, Y. S. Ding, K. Malinger and S. L. Suib, J. Phys. Chem. B, 2006, 110, 7592–7599 CrossRef CAS PubMed.
- Z. K. Zhao, H. L. Yang, Y. Li and X. W. Guo, Green Chem., 2014, 16, 1274–1281 RSC.
- H. W. Fan, T. W. Wu, Y. Y. Liu, H. Z. Wei, S. L. An and G. X. Jia, Chin. J. Struct. Chem., 2019, 38, 893–900 CAS.
- S. Z. Zhao, Y. F. Wen, X. J. Liu, X. Y. Pen, F. Lü, F. Y. Gao, X. Z. Xie, C. C. Du, H. H. Yi, D. J. Kang and X. L. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- M. Wang, M. Shen, X. X. Jin, J. J. Tian, M. L. Li, Y. J. Zhou, L. X. Zhang, Y. S. Li and J. L. Shi, ACS Catal., 2019, 9, 4573–4581 CrossRef CAS.
- M. Wang, M. Shen, X. X. Jin, J. J. Tian, M. L. Li, Y. J. Zhou, L. X. Zhang, Y. S. Li and J. L. Shi, Appl. Catal., B, 1996, 8, 405–415 CrossRef.
- Y. Kadoma, Y. Uchimoto and M. Wakihara, J. Phys. Chem. B, 2006, 110, 174–177 CrossRef CAS PubMed.
- G. X. Zhu, J. G. Zhu, W. J. Jiang, Z. J. Zhang, J. Wang, Y. F. Zhu and Q. F. Zhang, Appl. Catal., B, 2017, 209, 729–737 CrossRef CAS.
- M. Polverejan, J. C. Villegas and S. L. Suib, J. Am. Chem. Soc., 2004, 126, 7774–7775 CrossRef CAS PubMed.
- J. T. Hou, Y. Z. Li, L. L. Liu, L. Rena and X. J. Zhao, J. Mater. Chem. A, 2013, 1, 6736–6741 RSC.
- S. Sithambaram, L. P. Xu, C. H. Chen, Y. Ding, R. Kumar, C. Calvert and S. L. Suib, Catal. Today, 2009, 140, 162–168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed materials synthesis and characterization, XAFS measurement and analysis, STEM simulation details, computational details, extended figures, and tables (including the catalysis comparison with the reported literature). See DOI: 10.1039/d1sc00700a |
| This journal is © The Royal Society of Chemistry 2021 |