
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganoruthenium-catalyzed chemical protein synthesis to elucidate the functions of epigenetic modifications on heterochromatin factors†
Naoki
Kamo
 a,
Tomoya
Kujirai
b,
Hitoshi
Kurumizaka
b,
Hiroshi
Murakami
c,
Gosuke
Hayashi
*c and
Akimitsu
Okamoto
*ad
a,
Tomoya
Kujirai
b,
Hitoshi
Kurumizaka
b,
Hiroshi
Murakami
c,
Gosuke
Hayashi
*c and
Akimitsu
Okamoto
*ad
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: okamoto@chembio.t.u-tokyo.ac.jp
bLaboratory of Chromatin Structure and Function, Institute for Quantitative Biosciences, The University of Tokyo, Bunkyo-ku, Tokyo 113-0032, Japan
cDepartment of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: hayashi@chembio.nagoya-u.ac.jp
dResearch Center for Advanced Science and Technology, The University of Tokyo, Meguro-ku, Tokyo 153-8904, Japan
First published on 22nd March 2021
Abstract
The application of organometallic compounds for protein science has received attention. Recently, total chemical protein synthesis using transition metal complexes has been developed to produce various proteins bearing site-specific posttranslational modifications (PTMs). However, in general, significant amounts of metal complexes were required to achieve chemical reactions of proteins bearing a large number of nucleophilic functional groups. Moreover, syntheses of medium-size proteins (>20 kDa) were plagued by time-consuming procedures due to cumbersome purification and isolation steps, which prevented access to variously decorated proteins. Here, we report a one-pot multiple peptide ligation strategy assisted by an air-tolerant organoruthenium catalyst that showed more than 50-fold activity over previous palladium complexes, leading to rapid and quantitative deprotection on a protein with a catalytic amount (20 mol%) of the metal complex even in the presence of excess thiol moieties. Utilizing the organoruthenium catalyst, heterochromatin factors above 20 kDa, such as linker histone H1.2 and heterochromatin protein 1α (HP1α), bearing site-specific PTMs including phosphorylation, ubiquitination, citrullination, and acetylation have been synthesized. The biochemical assays using synthetic proteins revealed that the citrullination at R53 in H1.2 resulted in the reduced electrostatic interaction with DNA and the reduced binding affinity to nucleosomes. Furthermore, we identified a key phosphorylation region in HP1α to control its DNA-binding ability. The ruthenium chemistry developed here will facilitate the preparation of a variety of biologically and medically significant proteins containing PTMs and non-natural amino acids.
Introduction
Synthetic methodology for homogeneously modified proteins has been highly demanded to elucidate the biological significance of posttranslational modifications (PTMs) through biochemical and structural analysis. Total chemical protein synthesis has streamlined the preparation of target proteins bearing site-specific PTMs and functional molecules.1–5 For example, histone proteins with various PTMs have been chemically synthesized to understand the roles of PTMs.6 In the process of chemical protein synthesis, a target protein is divided into several peptide segments that can be synthesized by solid-phase peptide synthesis (SPPS).7 To assemble the peptide segments, many researchers have employed native chemical ligation (NCL) that utilizes the chemoselectivity between N-terminal cysteine (Cys) and C-terminal thioester to form amide bonds between divided segments.8 However, assembly of multiple peptide segments to obtain medium-size proteins (e.g., >20 kDa) requires multiple reactions and purification steps, which causes low overall yields and time-consuming works and therefore limits the access to target proteins bearing various patterns of PTMs. To address this issue, one-pot multiple peptide ligation strategies to eliminate the intermediate purification steps, including a kinetically controlled strategy9–11 and a protecting group-removal strategy, have been developed,12 although these methods have been mainly exploited for the assembly of three segments to afford small proteins.Organometallic compounds have been applied for chemical reactions on proteins, such as decaging13,14 and bioconjugation utilizing Suzuki–Miyaura coupling,15 Cys arylation through C–S reductive elimination using palladium (Pd) complexes,16 or Tsuji–Trost allylations.17,18 Recently, Brik and coworkers paved the way for the application of palladium (Pd) complexes for total chemical protein synthesis.19–28 They exploited several protecting groups that were labile by specific Pd complexes for chemoselective masking and one-pot ligation of peptide segments.21,24 Our group employed allyloxycarbonyl (alloc) groups for the protection of N-terminal Cys, which were removed with Pd/3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt (TPPTS) complexes.29 The one-pot peptide ligation using Pd/TPPTS complex allowed us to synthesize histone H2AX efficiently. However, the repetitive metal-mediated decaging reactions to ligate multiple peptide segments necessitated considerable amounts of metal complexes especially under NCL conditions containing an excess amount of thiol compounds that significantly degrade metal complexes.29 Generally, decaging or cross-coupling reactions on protein surfaces under aqueous conditions require excessive amounts of metal complexes13,14 probably because nucleophilic functional groups on proteins such as thiol groups30 would readily coordinate to metal complexes. Moreover, some metal complexes (e.g., Pd/TPPTS complexes) are very unstable under aerobic conditions, and these organometallic compounds required strict deoxygenated conditions. Furthermore, a high loading of transition metal complexes often cause protein aggregations,31 which interferes with the whole synthetic procedure to obtain target proteins. To overcome these problems, a highly active organometallic compound that minimizes the metal loading has been demanded in chemical protein synthesis to produce a variety of proteins of interest with modifications and accelerate biological research.
In this study, we focused on organoruthenium complexes to achieve decaging reactions on polypeptides in catalytic manners. After our screening of metal complexes, we discovered air-tolerant ruthenium (Ru) complexes that showed more than 50-fold catalytic activity compared with previous Pd complexes for the decaging reaction and only 20 mol% of an organoruthenium catalyst allowed the deprotection of the alloc groups on polypeptides under NCL conditions. The new one-pot multiple peptide ligation strategy, including consecutive more than five reaction steps using the Ru catalyst, was applied for the total chemical synthesis of linker histone H1.2 (size: 22 kDa) and heterochromatin protein 1α (HP1α) (size: 22 kDa). Our new streamlined synthetic method afforded homogeneous two kinds of H1.2s and six kinds of HP1α bearing different patterns of PTMs including phosphorylation, ubiquitination, citrullination, and acetylation. Finally, we investigated the effects of those PTMs decorated in heterochromatin factors on chromatosome formations or the binding ability toward DNA to elucidate the functions of the epigenetic marks.
Results and discussion
Exploration of organoruthenium complexes
Under NCL conditions involving excess MPAA, the metal complexes lost activity rapidly because of its sulfur poisoning effect. Therefore, it was important to increase the stability of the metal complexes under NCL conditions to achieve catalytic reactions on peptides or proteins. To accomplish this goal, we focused on Ru complexes rather than Pd complexes. Electron-rich Pd (10 outer electrons) forms a strong bond with thiol moieties because of the π-back donation from Pd to the empty relatively low-energy d orbitals of the sulfur.32 On the other hand, electron-deficient Ru (8 outer electrons) favors ionic interactions to fill its d orbital and does not form a strong coordination bond with soft nucleophiles.33 Moreover, electron-rich Pd is more sensitive to electronic modification by sulfur poisoning, which causes significant changes in catalytic activity, than electron-poor Ru.33 When we screened trivalent phosphine ligands for Pd, we could not discover more active ligands for the alloc removal than TPPTS (Fig. S1†). Although the Pd loading on proteins could be reduced by the addition of magnesium ion,21,34 these results suggested that it would be difficult to deprotect the alloc groups on peptides under NCL conditions with Pd complexes in catalytic manners. Meanwhile, some researchers reported that organoruthenium complexes tolerated sulfur poisoning and catalyzed the allyl transfer to thiol moieties,35 but these Ru complexes had not been applied for chemical reactions on biomolecules. To test whether organoruthenium complexes could sustain the catalytic activity for the removal of the alloc group on a peptide under NCL conditions and air atmosphere, we prepared Cp*Ru(cod)Cl (Ru-1: Cp* = η5-pentamethylcyclopentadienyl, cod = η4-1,5-cyclooctadiene), [Cp*Ru(QA)allyl]PF6 (Ru-2: QA = 2-quinolinecarboxylate), [CpRu(QA)allyl]PF6 (Ru-3: Cp = η5-cyclopentadienyl), [CpRu(QA-NMe2)allyl]PF6 (Ru-4: QA-NMe2 = 4-(N,N-dimethylamino)-2-quinolinecarboxylate) bearing N,O-bidentate ligand (Table 1).36–38 Next, we tested the alloc deprotection on peptide 1 with 10 mol% of metal complexes under NCL conditions (Table 1), and the conversion yields were calculated based on high-performance liquid chromatography (HPLC). In previous research, 2 equiv. of Pd/TPPTS complex was required to attain quantitative removal of the alloc groups under NCL conditions.29 When the amount of the Pd complex was reduced to 10 mol%, the conversion yield reached to only 12% after 3 h (Table 1, entry 1), indicating that the TON of Pd/TPPTS complexes under NCL conditions was 1.2. In contrast, Ru-1 and Ru-2 sustained the catalytic activity even in the presence of a high concentration of MPAA (entries 2 and 3) under air atmosphere, and quantitative removal of the alloc groups was observed after 2 h upon the treatment with 10 mol% Ru-2 (entry 3). Furthermore, only 5 mol% of Ru-3 or Ru-4 rapidly catalyzed the alloc deprotection, and the reaction reached completion within 10 min (entries 6 and 7). We did not observe any side reactions, such as the transfer of the allyl groups to nucleophilic functional groups at side chains of peptide 1 (Fig. S2†). The slow reaction rate of Ru-2 compared to Ru-3 could be explained by the sterically hindered Cp* disturbing nucleophilic attack of MPAA toward π-allyl Ru complex, which was already reported.38 We should note that the activity of the Ru catalysts diminished in the presence of TCEP (entries 8–11); however, oxidized TCEP after the reduction of the disulfide bond of MPAA dimer did not affect the activity of Ru-4 (entry 12, Fig. S3†). We screened other Ru complexes for the alloc deprotection under NCL conditions. We replaced the N,O-ligand with an N,N-ligand, and synthesized [Cp(η2-bipy)(η3-allyl)RuIV][PF6]2 (Ru-5: bipy = bipyridine) and [Cp(η2-phen-NMe2)(η3-allyl)RuIV][PF6]2 (Ru-6: phen = phenanthroline).39 However, the catalytic activities of Ru-5 and Ru-6 were much lower than those of Ru-3 or Ru-4 (entries 13 and 14) probably because of those rigid structures.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Metal complexes | Quantity (mol%) | Additive | Conversion yieldsa [%] | ||||
| 10 min | 30 min | 1 h | 2 h | 3 h | ||||
| a The conversion yields were calculated from peak areas analyzed by HPLC (Fig. S2). b TCEP was completely consumed for the reduction of MPAA disulfide dimer before the addition of Ru-4 as shown in Fig. S3. | ||||||||
| 1 | Pd/TPPTS | 10 | — | 11 | 11 | 12 | 12 | 12 |
| 2 | Ru-1 | 10 | — | 28 | 56 | 76 | 86 | 94 |
| 3 | Ru-2 | 10 | — | 41 | 76 | 92 | >95 | — |
| 4 | Ru-3 | 10 | — | >95 | — | — | — | — |
| 5 | Ru-4 | 10 | — | >95 | — | — | — | — |
| 6 | Ru-3 | 5 | — | >95 | — | — | — | — |
| 7 | Ru-4 | 5 | — | >95 | — | — | — | — |
| 8 | Ru-3 | 5 | TCEP (3 mM) | 65 | 67 | 67 | 70 | 72 |
| 9 | Ru-3 | 5 | TCEP (10 mM) | <5 | <5 | 7 | — | — |
| 10 | Ru-4 | 5 | TCEP (3 mM) | 55 | 67 | 69 | 75 | 78 |
| 11 | Ru-4 | 5 | TCEP (10 mM) | 8 | 9 | 10 | — | — |
| 12 | Ru-4 | 5 | TCEP (5 mM)b | >95 | — | — | — | — |
| 13 | Ru-5 | 5 | — | <5 | <5 | <5 | <5 | 6 |
| 14 | Ru-6 | 5 | — | 7 | 10 | 14 | 17 | 17 |
| 15 | Ru-7 | 5 | — | 19 | 40 | 60 | 76 | 80 |
| 16 | Ru-8 | 5 | — | <5 | 18 | 47 | 64 | 71 |
| 17 | Ru-9 | 5 | — | 18 | 36 | 53 | 73 | 76 |
| 18 | Ru-10 | 5 | — | 93 | >95 | — | — | — |
| 19 | Ru-10 | 5 | TCEP (3 mM) | 50 | 61 | 66 | 68 | 68 |
| 20 | Ru-10 | 5 | TCEP (10 mM) | <5 | <5 | 11 | — | — |
Previous research showed that the rate-determining step of the alloc removal using Ru-3 or Ru-4 was the oxidative addition of the allyl groups toward Ru when strong nucleophiles, such as thiol moieties, were employed.38 To accelerate the uncaging of the alloc groups and further improve the stability under NCL conditions, we focused on P,O-bidentate ligands to replace N,O-ligands, and increase the electron density of Ru complexes. Bruneau and coworkers reported Ru complexes coordinated by o-diphenylphosphino benzoate (o-DPPBz) or diphenylphosphinobenzene sulfonate (o-DPPBS).40 We synthesized [Cp*Ru(κ2-o-DPPBz)allyl]PF6 (Ru-7) and [Cp*Ru(κ2-o-DPPBS)allyl]PF6 (Ru-8) (Table 1) and tested those catalytic activities. There was no significant difference between Ru-7 and Ru-8 (entries 15 and 16) for the alloc deprotection of peptide 1 under NCL conditions. To reduce the electron density of the phosphine ligand and enhance the nucleophilic attack of MPAA toward π-allyl Ru complexes, we synthesized Ru complexes coordinated by o-DPPBz bearing CF3 groups (Ru-9), but no improvement of these Ru complexes over Ru-7 was observed (entry 17). Considering the effective deprotection of the alloc group using Ru-3 compared to Ru-2 (entries 3 and 4), we newly synthesized [CpRu(κ2-o-DPPBz)allyl]PF6 (Ru-10). The deprotection efficiency was dramatically improved by replacing Cp* with Cp, and the reaction reached completion within 30 min (entry 18), although the catalytic activity of Ru-10 was also reduced in the presence of TCEP (entries 19 and 20). We concluded that organoruthenium catalysts coordinated by Cp and X,O-bidentate (X = N or P) ligand efficiently catalyzed the removal of the alloc group under NCL conditions.
Calculation of TON of Ru catalysts
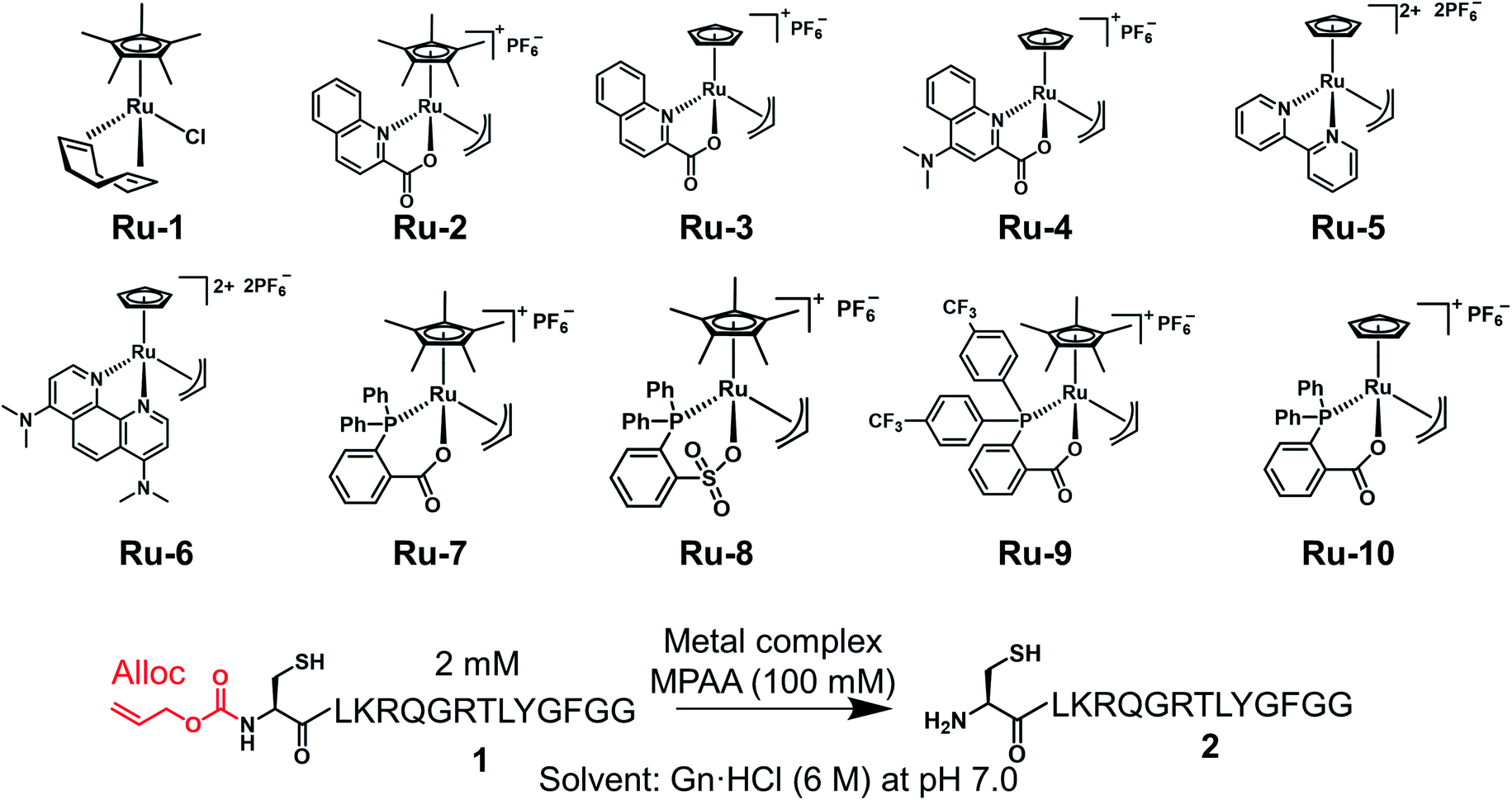
We examined TON and turnover frequency (TOF) of Ru-3, Ru-4, and Ru-10, which removed the alloc groups quantitatively with 5 mol% catalysts. We reduced the amount of each Ru complex to 1 mol% and tested the removal of the alloc group of peptide 1 under NCL conditions (Fig. 1). Ru-3 (TOF: 19 min−1) and Ru-4 (TOF: 20 min−1) showed similar TOF value, but the TON of Ru-4 (TON: 70) was slightly higher compared to Ru-3 (TON: 56), because of the NMe2 group in Ru-4, which increased the electron density of Ru and its stability against excess MPAA. The reaction rate using Ru-10 (TOF: 6 min−1) as a catalyst was slower than Ru-3 or Ru-4 due to the steric hindrance of the P,O-bidentate ligand of Ru-10, but Ru-10 sustained its catalytic activity for a longer time because of its stability and showed similar catalytic activity (TON: 72) to that of Ru-4 and higher than that of Ru-3. We further calculated the electrophilicity of each Ru catalyst based on density functional theory (DFT). The lowest unoccupied molecular orbital (LUMO) level of o-DPPBz was more than 0.5 eV higher than QA and QA-NMe2 (−1.44 eV vs. −2.15 eV vs. −1.94 eV) (Fig. S5†). This higher π-accepting ability of QA or QA-NMe2 led to the higher electrophilicity of Ru-3 or Ru-4 and the attack of MPAA toward π-allyl Ru complexes became faster than Ru-10. To summarize, compared to the activity of Pd/TPPTS complexes (TON: 1.2), Ru-4 and Ru-10 showed more than 50-fold catalytic activity, and catalytically removed the alloc groups of peptides under aqueous conditions and even in the presence of 5000 equiv. of MPAA that normally poisoned organometallic catalysts.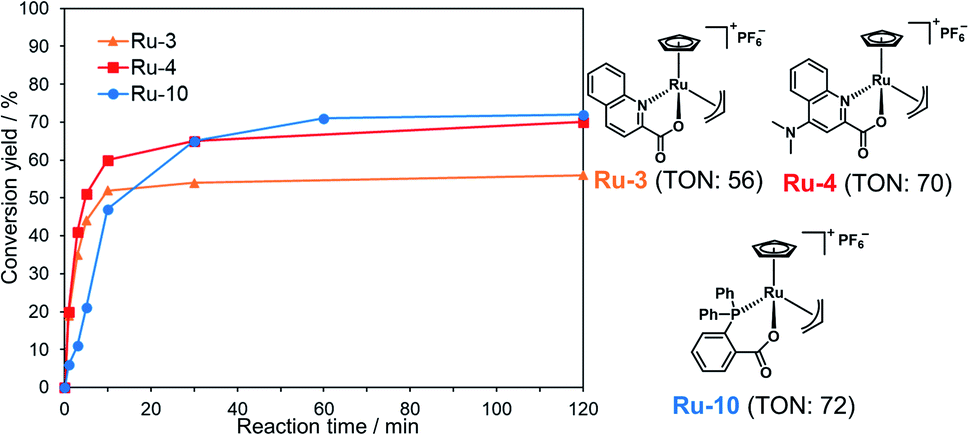 | ||
| Fig. 1 Calculation of TON and TOF of 1 mol% Ru-3, Ru-4, or Ru-10 for the deprotection of the alloc group of peptide 1 (2 mM) in NCL buffer [Gn·HCl (6 M), NaH2PO4 (200 mM), MPAA (100 mM, 5000 equiv. against the Ru catalyst) at pH 7.0]. The conversion yields under different conditions are shown in Table S1.† | ||
To further explore the difference between Ru-4 and Ru-10, we added these Ru complexes to a solution containing peptide 1 without MPAA, a scavenger for π-allyl Ru complexes (Fig. S6†). When 10 mol% of Ru-4 was tested, peptide 1 was completely consumed within 30 min, and single or double allyl transfer to functional groups of peptides was observed. Based on MS/MS analysis of each HPLC peak (Fig. S7†), it was found that the allyl groups were transferred to amine groups or thiol groups of N-terminal Cys, not to other amino acids, such as Lys, Tyr, or Thr. When 10 mol% of Ru-10 was examined, the starting material remained for over 1 h reaction, and the allyl transfer to peptide 1 barely occurred (Fig. S6†). These results indicated that Ru-4 was more reactive with nucleophiles than Ru-10 because of its more electrophilic π-allyl Ru complex, which presented the possibility of side reactions, such as allyl transfer to the functional groups of peptides viaRu-4. However, in the presence of excess MPAA (pKa 6.6),41 this strong nucleophile attacked π-allyl Ru complex before peptides and suppressed the undesired reactions as observed by comparing HPLC data shown in Fig. S2 and S6.†
Inactivation by MPAA to form a binuclear Ru complex
To achieve one-pot peptide ligation by repeating the cycles of NCL and decaging, the deactivation of metal complexes just before the subsequent NCL was a critical step.29 Therefore, we investigated the deactivation of Ru-4 or Ru-10 under NCL conditions (Fig. S8†). 200 mol% of Pd/TPPTS complex against peptide 1 was rapidly deactivated by 100 mM MPAA and lost activity within 10 min (Table S2,† entry 1). In contrast, 10 mol% of Ru-4 and Ru-10 against peptide 1 showed stability under NCL conditions (entries 2 and 3), and Ru-10 was more stable than Ru-4 because of its bulky P,O-bidentate ligand. We concluded that the greater stability of these Ru complexes over Pd complexes contributed to the catalytic activity for the removal of the alloc groups of peptides. By NMR and MALDI-TOF mass analysis, the half-sandwich Ru complex reacted with MPAA to form a binuclear Ru complex,42 which might be inactive for the alloc removal (Fig. S9 and S10†). Moreover, we observed the detachment of the bidentate ligand from Ru through the ligand exchange with MPAA.To summarize, the alloc deprotection of peptides based on the Tsuji–Trost reaction mechanism catalyzed by the Ru catalysts under NCL conditions proceeded very fast, and the reaction reached completion within 10 min. Along with the deprotection, the Ru catalyst slowly lost the catalytic activity through ligand exchange with MPAA to form a binuclear complex, enabling the subsequent NCL with alloc-protected peptides (Scheme 1). Newly synthesized Ru-10 coordinated by P,O-ligands showed the highest catalytic activity and the greatest stability under NCL conditions among tested Ru complexes. However, considering the facile deactivation by MPAA, we decided to employ Ru-4 to attain total chemical synthesis of proteins through one-pot multiple peptide ligation.
 | ||
| Scheme 1 Simultaneous catalytic alloc deprotection and deactivation of Ru-4. | ||
Chemical synthesis of histone H1.2 assisted by an organoruthenium catalyst
To demonstrate the utility of Ru-4 catalyst for chemical protein synthesis, we selected linker histone H1.2 as a target protein. This protein interacts with nucleosomes to form chromatosomes and contributes to the formation of a higher-order chromatin structure.43–46 Previous papers reported that various PTMs can be decorated on H1.2, and some of these modifications play roles in chromatin decondensation.47 However, it had been challenging to achieve the chemical synthesis of H1.2 compared to core histone proteins (<140 amino acids), because of its large size (212 amino acids). Therefore, we aimed to establish a synthetic method to obtain H1.2 through one-pot multiple peptide ligation using Ru-4.48As shown in Fig. 2A, Cys mutations for NCL reactions were incorporated at Ala sites (A48C, A68C, A100C, A150C) and divided into five peptide segments of H1.2 (peptides 3, 4, 6, 8, and 10), which were synthesized by 9-fluorenylmethyloxycarbonyl SPPS (Fmoc-SPPS) (Fig. S11†). The N-terminal Cys of peptides 4, 6, and 8 were protected by the alloc groups, and the one-pot five-segment ligation using Ru-4 was performed. All the NCL reactions and the removal of alloc groups were conducted in NCL buffer (6.0 M guanidinium chloride, 200 mM NaH2PO4, 100 mM MPAA) at 37 °C.
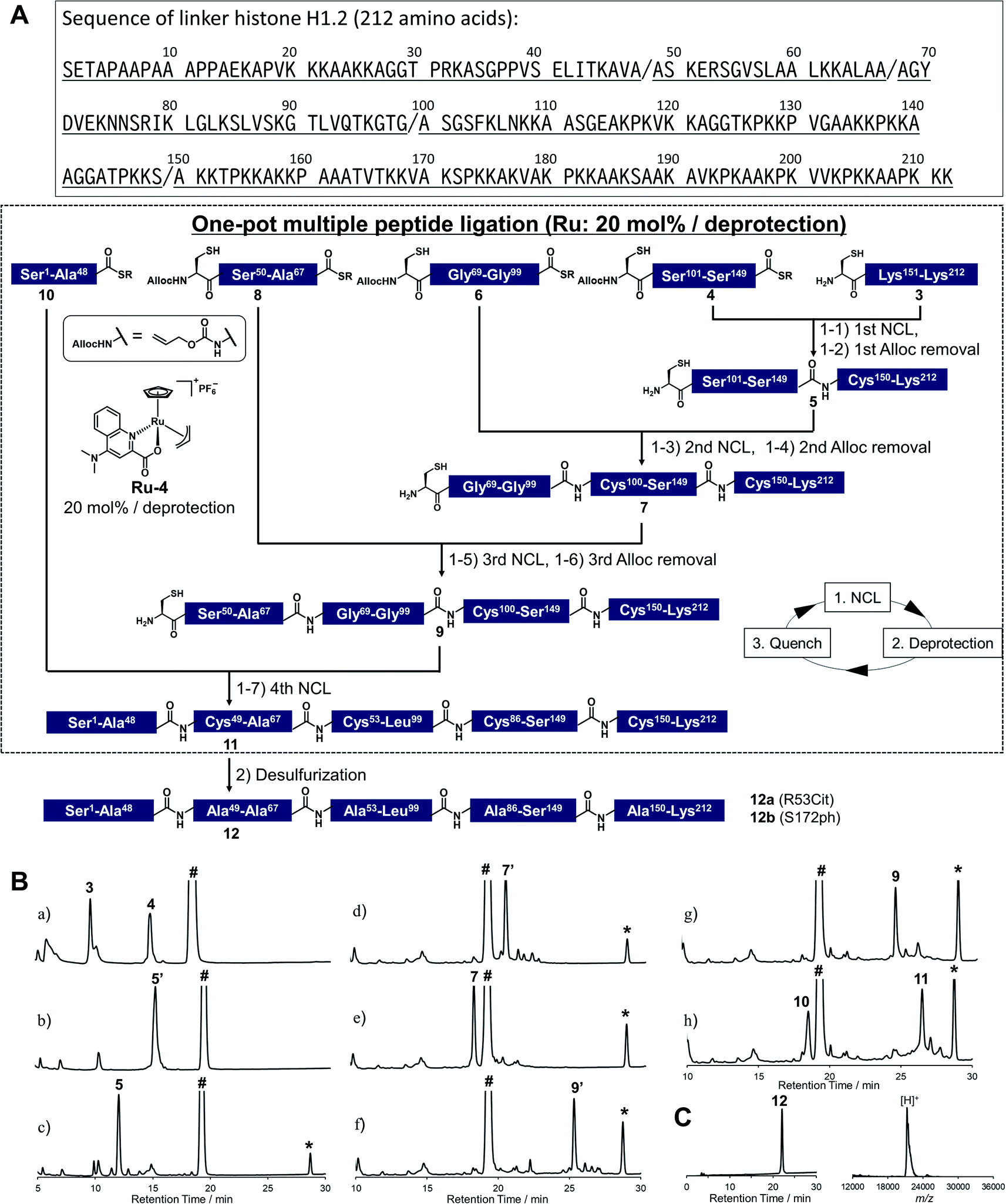 | ||
Fig. 2 Chemical synthesis of linker histone H1.2 using the Ru catalyst. (A) Synthetic strategy. / represents the cleavage sites. (1) NCL condition: peptides (2 mM), MPAA (100 mM) in denaturing buffer at pH 7.0, 37 °C. Removal condition: Ru-4 (20 mol%), 37 °C, 30 min. (2) Peptide (0.8 mM), TCEP (300 mM), GSH (150 mM), VA-044 (20 mM) in denaturing buffer at pH 7.0, 37 °C. (B) Reaction tracking of the one-pot ligation involving catalytic amount of Ru-4 (20 mol%) for the alloc deprotection by analytical HPLC (gradient: 10–46% for 30 min) at 220 nm. Compounds 5′, 7′, and 9′ are the alloc-protected peptides 5, 7, and 9, respectively. # = MPAA. * = allyl-transferred MPAA. (a) 1st NCL (t = 2 min). (b) 1st NCL (t = 2 h). (c) 1st deprotection. (d) 2nd NCL (t = 2 h). (e) 2nd deprotection. (f) 3rd NCL (t = 2 h). (g) 3rd deprotection. (h) 4th NCL (t = 2 h). (C) HPLC profile (left, gradient 20–50% for 30 min) and MALDI-TOF mass spectrum (right) of purified peptide 12. Calculated mass of 12 [M + H]+: 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 276.6; mass found [M + H]+: 21277.8. 276.6; mass found [M + H]+: 21277.8. | ||
Peptides 3 and 4 (1.1 equiv. relative to peptide 3) were dissolved in NCL buffer, and the NCL reaction reached completion within 2 h to afford peptide 5 (Fig. 2B). Ru-4 (0.2 equiv.) was added to the reaction solution, and the mixture was stirred for 30 min to remove the alloc group and fully deactivate Ru-4. After completion, a small amount of TCEP solution was added to reduce disulfide bonds and be consumed completely to minimize the effect on the next alloc removal with Ru-4 (Fig. S3†). Powdered peptide 6 was then added to initiate the second NCL reaction. These operations were repeated for the second deprotection and the third NCL and deprotection (Fig. 2B).
Finally, powdered peptide 10 was added to initiate the fourth NCL reaction. HPLC analysis showed a major peak corresponding to the desired ligated peptide 11 (Fig. 2B) after seven consecutive reaction steps. The reaction product was purified by HPLC, and 2.52 mg of peptide 11 was obtained in 23% yield from 3 (Fig. S13†). Compared to the previous one-pot ligation method with Pd/TPPTS complex,29 the total amount of metal complexes was reduced to 0.6 equiv. (Pd: more than 6.0 equiv.), and strict deoxygenated conditions established by argon bubbling were not required during the procedure. The mutated Cys residues were converted into original Ala residues by free-radical desulfurization49 to afford 1.34 mg of full-length H1.2 (12) almost quantitatively (Fig. 2C). Analysis by inductively coupled plasma mass spectrometry (ICP-MS) showed that the quantity of Ru attached to synthesized H1.2 was only 0.000037% (w/w) (Table S3†), and the amount of metal was much lower compared to the Pd content on previously synthesized histone H2AX (0.00096%) produced by one-pot ligation using Pd/TPPTS complexes.29
Next, we decided to incorporate PTMs into H1.2. As candidates of modifications, we chose citrullination of 53rd Arg (R53Cit) and phosphorylation of 172nd Ser (S172ph). It was reported that the R53 site was modified to Cit by peptidyl arginine deiminase 4 (PAD4), which regulates pluripotency, and the chromatin structure became decondensed and cells were reprogrammed.50 S172ph was observed specifically during interphase and mitosis and might have an influence on the stability of chromatosome.51 Therefore, we tried to investigate the effects of these PTMs on the binding affinity of H1.2 for nucleosome by creating homogeneously modified proteins. To synthesize modified H1.2, we prepared a peptide segment of H1.2 bearing R53Cit (8a) or S172ph (3a) by Fmoc-SPPS (Fig. S10†). According to Fig. 2A, the divided peptide segments were assembled in a one-pot manner with the Ru catalyst (Fig. S13†). After completion of all peptide ligations and purification, the Cys residues were converted into Ala residues. As a result, we accomplished the chemical synthesis of citrullinated H1.2 (12a) and phosphorylated H1.2 (12b) at mg scale (Fig. S14†). Thus, our facile and rapid synthetic method helped the efficient chemical synthesis of three types of full-length H1.2.
Reconstitution of chromatosomes
We examined the formation of chromatosomes using our synthetic H1.2 (Fig. 3A). The recombinant H1.2 or synthetic H1.2 (12) was mixed with Nap1, a histone chaperone to suppress aggregation during the reconstitution of chromatosomes,52 and each mixture was incubated at 37 °C. The nucleosome solution was then added to each mixture to reconstitute chromatosomes. The synthetic H1.2 (12) formed chromatosomes in a similar way to recombinant H1.2 (Fig. S16†), suggesting that synthetic H1.2 was refolded properly for chromatosomes formations.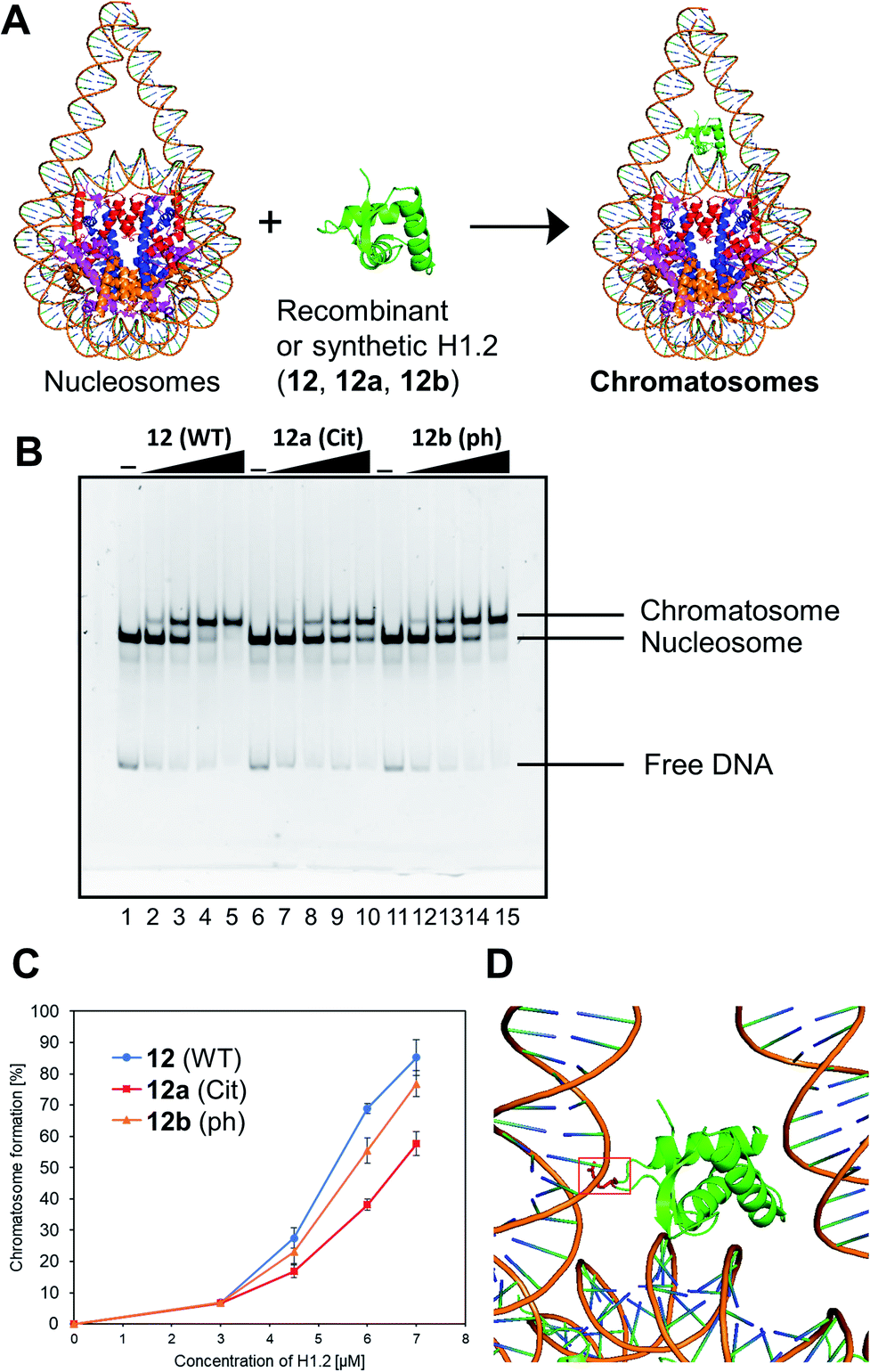 | ||
| Fig. 3 Gel band shift assay for reconstitution of chromatosomes. (A) Schematic representation of the reconstitution of chromatosomes. (B) Representative gel image of the H1.2 binding assay. Increasing amounts of wt H1.2, H1.2 bearing R53Cit, or H1.2 bearing S172ph (0 μM: lanes 1, 6 and 11; 0.3 μM: lanes 2, 7 and 12; 0.45 μM: lanes 3, 8 and 13; 0.6 μM: lanes 4, 9 and 14; 0.7 μM: lanes 5, 10 and 15) were mixed with nucleosomes (0.1 μM) in the presence of Nap1 (0.3 μM). After incubation at 37 °C, the complexes were detected by nondenaturing 5% PAGE with ethidium bromide staining. (C) Quantification of EMSAs using synthetic H1.2s. (D) Position of R42 (red) of X. laevis histone H1.0b, which is identical to R53 of human H1.2, in chromatosomes (PDB: 5NL0).53 | ||
Next, we investigated the effect of R53Cit or S172ph on the binding affinity for nucleosome by electrophoretic mobility shift assay (EMSA) (Fig. 3B). There was a slight difference between 12 and 12b with phosphorylation, but 12a with citrullination reduced its binding affinity for nucleosomes, and its conversion yield was reduced by approximately 30% compared to nonmodified H1.2 (Fig. 3C). R53 interacts with the phosphate backbone of the minor groove of linker DNA in chromatosome (Fig. 3D).53
We concluded that H1.2 with R53Cit reduced its binding ability toward nucleosomes because of the lack of electrostatic interaction between Arg and DNA, which would contribute to the relaxation of chromatin and reprogramming of cells.50 By our research using homogeneously modified H1.2, the direct effects of PTMs of H1.2 were examined.
Chemical synthesis of HP1α with Ru-4
To broaden the applicability of the organoruthenium-catalyzed chemical protein synthesis, we selected heterochromatin protein 1α (HP1α) as a second target. This protein contains two globular domains, a chromo domain (CD) and a chromo shadow domain (CSD), which are connected by a hinge region (HR).54–57 CD recognizes the trimethylation of Lys 9 within histone H3 (H3K9me3), a hallmark of transcriptionally silenced chromatin.58 CSD is a domain responsible for the self-dimerization of HP1α, and also provides the interface to interact with proteins bearing the PXVXL/I binding motif, such as shugoshin 1 (Sgo1).59 In the presence of H3K9me3, CD interacts with this modification and CSD forms intermolecular bridging, which condenses chromatin to form transcriptionally silenced states.60 HP1α was reported to be extensively decorated with PTMs,61 and these PTMs affected the interaction pattern, distribution, and localization of HP1α.57 Therefore, the elucidation of the functions of PTMs on HP1α was essential to understand the behaviour of dynamic chromatin.In accordance with Fig. 4A, Cys mutations for NCL reactions were incorporated at Ala sites (A98C, A129C, A153C), and full-length HP1α was divided into five peptide segments (peptides 13, 14, 15, 16, 20), and each segment was synthesized by Fmoc-SPPS (Fig. S17†). C-terminal Asp 58, bearing thioesters of peptide 20, was protected by the allyl group to inhibit hydrolysis of thioesters and isomer formation via intramolecular cyclization.62,63 The N-terminal Cys of peptides 14 and 15 were protected with the alloc groups, and acetamidomethyl (acm) groups were introduced at N-terminal Cys of peptide 16 (C59) and inner Cys of peptides 13 (C160). Furthermore, to improve the solubility of peptide 14, we installed a solubilizing tag bearing phenylacetamidomethyl (phacm) group at C133 site, which was removed in the same deprotection method as acm groups.22
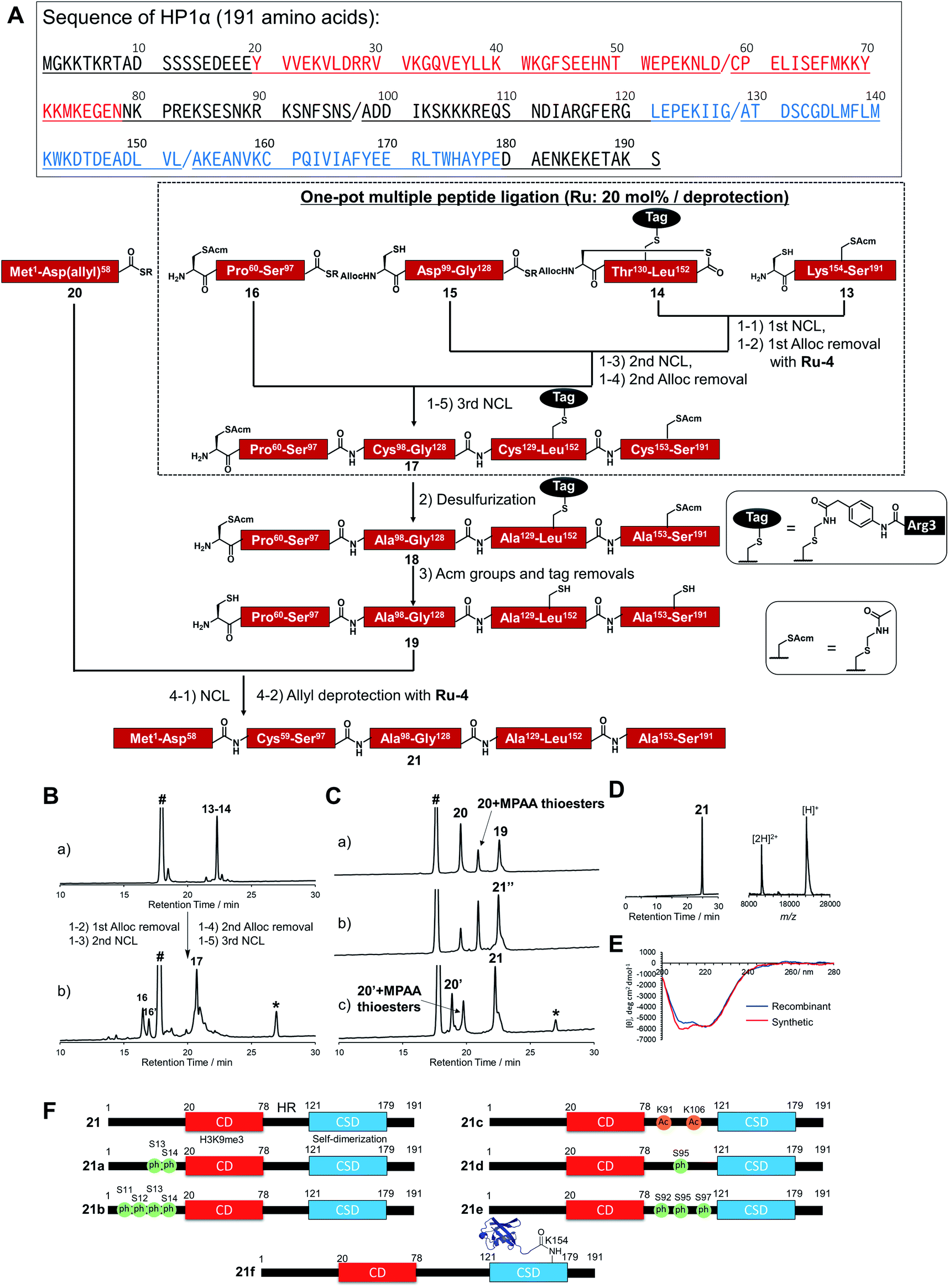 | ||
| Fig. 4 Chemical synthesis of HP1α using the Ru catalyst. (A) Synthetic strategy. / represents the cleavage cites. Red region represents chromodomain, underlined region represents hinge region, and blue region represents chromo shadow domain of HP1α. (1) NCL condition: peptides (2 mM), MPAA (100 mM) in denaturing buffer at pH 7.0, 37 °C. Removal condition: Ru-4 (20 mol%), 37 °C, 30 min. (2) Peptide (0.8 mM), TCEP (300 mM), GSH (150 mM), VA-044 (20 mM) in denaturing buffer at pH 7.0, 37 °C, overnight. (3) Peptide (0.5 mM), AgOAc (30 mM), H2O/AcOH (1:1), 37 °C, overnight. (4) NCL condition: peptides (1.5 mM), MPAA (100 mM) in denaturing buffer at pH 7.0, 37 °C. Removal condition: Ru-4 (20 mol%), 37 °C, 30 min. (B) Reaction tracking of the one-pot ligation with a catalytic amount of Ru-4 (20 mol%) for the alloc deprotection by analytical HPLC (gradient: 15–51% for 30 min) at 220 nm. (a) 1st NCL (t = 4 h). (b) 3rd NCL after overnight reaction. (C) Reaction tracking of NCL between peptides 19 and 20, followed by the allyl removal with Ru-4 (20 mol%) by analytical HPLC (gradient: 15–51% for 30 min) at 220 nm. (a) NCL (t = 3 min). (b) NCL (t = 1.5 h). (c) Allyl deprotection with Ru-4. 21′′ = 21 bearing allyl-protected Asp 58. (D) HPLC profile (gradient 15–51% for 30 min) and MALDI-TOF mass spectrum of purified peptide 21. Calculated mass of 21 [M + H]+: 22225.7; mass found [M + H]+: 22226.0. (E) CD spectrum of 21. (F) Chemically synthesized HP1α with PTMs in this study. 21: HP1α without PTMs. 21a and 21b: HP1α with PTMs at N-terminus. 21c, 21d, and 21e: HP1α with PTMs at HR. 21f: HP1α with Ub at K154 at CSD. | ||
Peptides 13, 14, 15, and 16 were assembled in a one-pot manner using Ru-4 catalyst (Fig. 4B). The removal of the alloc groups proceeded with only 20 mol% of Ru-4, and, after three NCL steps and two deprotection steps, the desired intermediate peptide 17 was obtained in 35% isolated yield (Fig. S18 and S19†). Free-radical desulfurization was then performed to convert mutated Cys residues into original Ala residues (Fig. S20†). To remove the acm groups and solubilizing tag, we first employed sodium tetrachloropalladate (Na2PdCl4).21,64 As a result, after the addition of this Pd complex (5.0 equiv. toward peptide 18), some aggregates appeared in the reaction solution, which led to low recovery rates after HPLC purification (Fig. S21†). Therefore, we used silver acetate (AgOAc) instead of Na2PdCl4.65 This time, aggregates were not observed after the overnight reaction and the desired product 19 was isolated in 62% yield (Fig. S22†). Finally, peptide 20 was ligated with peptide 19, and the reaction reached completion after 2 h (Fig. 4C). When we successively added Pd/TPPTS complex to remove the ally group at Asp 58, some aggregates appeared again. When we analyzed the aggregation of full-length HP1α in the presence of Pd or Ru by dynamic light scattering (DLS), the catalytic amount of Ru did not contribute to the formation of the aggregates (Fig. S24†). Therefore, Ru-4 was used for deprotection of the allyl group, and the desired full-length HP1α (21) was obtained without aggregation in 53% isolated yield (Fig. 4D). The measurement of Ru content by ICP-MS showed that 0.00011% of Ru was attached on 21 (Table S3†). The circular dichroism spectrum of 21 was measured after refolding (Fig. 4E), and the ellipticity around 208–218 nm of 21 was lower than that of recombinant HP1α, indicating that the antiparallel β sheet of 21 was more stabilized compared to commercially available recombinant HP1α. To summarize the chemical synthesis of HP1α, it would be difficult to apply Pd complexes for HP1α due to the aggregation probably caused by intermolecular bridging of Cys of HP1α by Pd complexes and hydrophobic interactions among CSDs. These results emphasized that the choice of an adequate metal complex was crucial to produce a target protein.
Preparation of a diversity of HP1α with different patterns of PTMs
We then tried to create HP1α bearing specific PTMs. First, we focused on phosphorylations decorated at Ser residues of the N-terminal tail of HP1α (S11, S12, S13, S14),66 which increase its binding affinity toward H3K9me3 and disturb the interaction between DNA and basic patches in HR of HP1α (residues 89–91 and residues 104–107).67 However, the effect of the number of phosphorylated Ser at the N-terminal tail had not been investigated. Moreover, it was reported that Ser residues of HR in mouse HP1α are phosphorylated in cells.66,68 S95ph mediated by NDR1 kinase is required for mitotic progression and Sgo1 binding to mitotic centromeres,69 and Aurora B kinase also mediates mitotic phosphorylation of HP1α at HR (mainly at S92).70 To investigate the effect of the phosphorylation at HR on the DNA binding affinity of HP1α, phosphorylated HP1α was prepared by enzymatic methods.66,70 However, the possibility of phosphorylations at other Ser sites by kinases could not be excluded and homogeneous HP1α bearing site-specific phosphorylations were required to investigate the precise property of each phosphorylation site. On the other hand, although the responsible enzymes had not been discovered, Lys 91 and Lys 106 in basic areas of HP1α were reported as decorated with acetylation,61 but the influence of acetylation on DNA binding was still unclear. To construct homogeneously phosphorylated or acetylated HP1α, we newly synthesized peptides 15a, 16a, 16b, 16c, 20a, and 20b by Fmoc-SPPS (Fig. S17†). By following the synthetic scheme shown in Fig. 4A, we constructed double phosphorylated (S13ph, S14ph) (21a) and quadruple phosphorylated (S11ph, S12ph, S13ph, S14ph) (21b) HP1α at the N-terminus, singly phosphorylated (S95ph) (21c) and triple phosphorylated (S92ph, S95ph, S97ph) (21d) HP1α at HR and double acetylated (K91ac, K106ac) (21e) HP1α at HR (Fig. 4F).We also focused on the PTMs decorated at CSD of HP1α. Ubiquitination (Ub) at K154 promotes the degradation of HP1α through the autophagy pathway to enable efficient DNA repair.71 K154 is located at the dimerization interface of CSDs, and we hypothesized that steric hindrance of Ub might disturb the dimerization of CSD and interaction with proteins bearing the PXVXL/I binding motif to relax chromatin structures. To create HP1α with Ub at K154, we synthesized peptides 22 and 23 (Fig. S26†). Peptide 22, which contained the C-terminal region of Ub linked by isopeptide linkage at K154 of HP1α, was prepared in a similar way to previous research (Scheme S2†).72 To achieve one-pot synthesis by combining the convergent method, peptide 24, bearing acyl hydrazide at its C-terminus, was also synthesized. Peptides 14, 22, 23, and 16–24 were assembled in a one-pot manner using Ru-4 to obtain peptide 25 (Fig. S27 and S28†), and, after desulfurization followed by the removal of the acm groups and the solubilizing tag (Fig. S29 and S30†), peptide 27 was ligated with peptide 20 followed by the allyl deprotection with Ru-4 to afford HP1α ubiquitinated at K154 (21f) (>30 kDa) (Fig. 4F and S31†). Finally, we obtained six kinds of full-length HP1α bearing the different patterns of PTMs. We note that these proteins were rapidly prepared owing to one-pot ligation of divided peptide segments using only catalytic amounts of organoruthenium complexes.
Fluorescence anisotropy assay of synthesized HP1α
After refolding of chemically synthesized HP1α by dialysis, we first tested the interaction of the CD of HP1α with H3K9me3 peptide bearing fluorescein at its N-terminus (residues 1–20) by fluorescence anisotropy assay. The binding affinity of 21 [dissociation constant (Kd): 23 μM] was almost the same as that of recombinant HP1α (Kd: 19 μM) (Fig. 5A and S32†), indicating that the CD of synthetic HP1α properly recognized the H3K9me3 mark. We further examined the binding of Sgo1 peptide (residues 446–466) bearing fluorescein toward dimerized interfaces of HP1α CSDs. 21 showed a similar Kd value (Kd: 0.47 μM) toward Sgo1 peptide as the recombinant one (Kd: 0.51 μM). These results indicated that our chemically synthesized HP1α was properly refolded, and recognized H3K9me3 modification and PXVXL/I binding motif through CD and dimerized CSDs, respectively. Next, we investigated the influence of Ub at K154 of HP1α on the binding ability toward the Sgo1 peptide. The Kd value of 21f was increased to 1.87 μM, suggesting that the affinity was reduced to 3.9-fold compared to canonical HP1α. We reasoned that Ub modification might disturb the intermolecular dimerization of HP1α CSDs probably because of the steric hindrance of Ub, which was elucidated for the first time by our study. It was envisaged that HP1α CSD might further reduce its affinity toward its CSD by being further attached with Ub chains.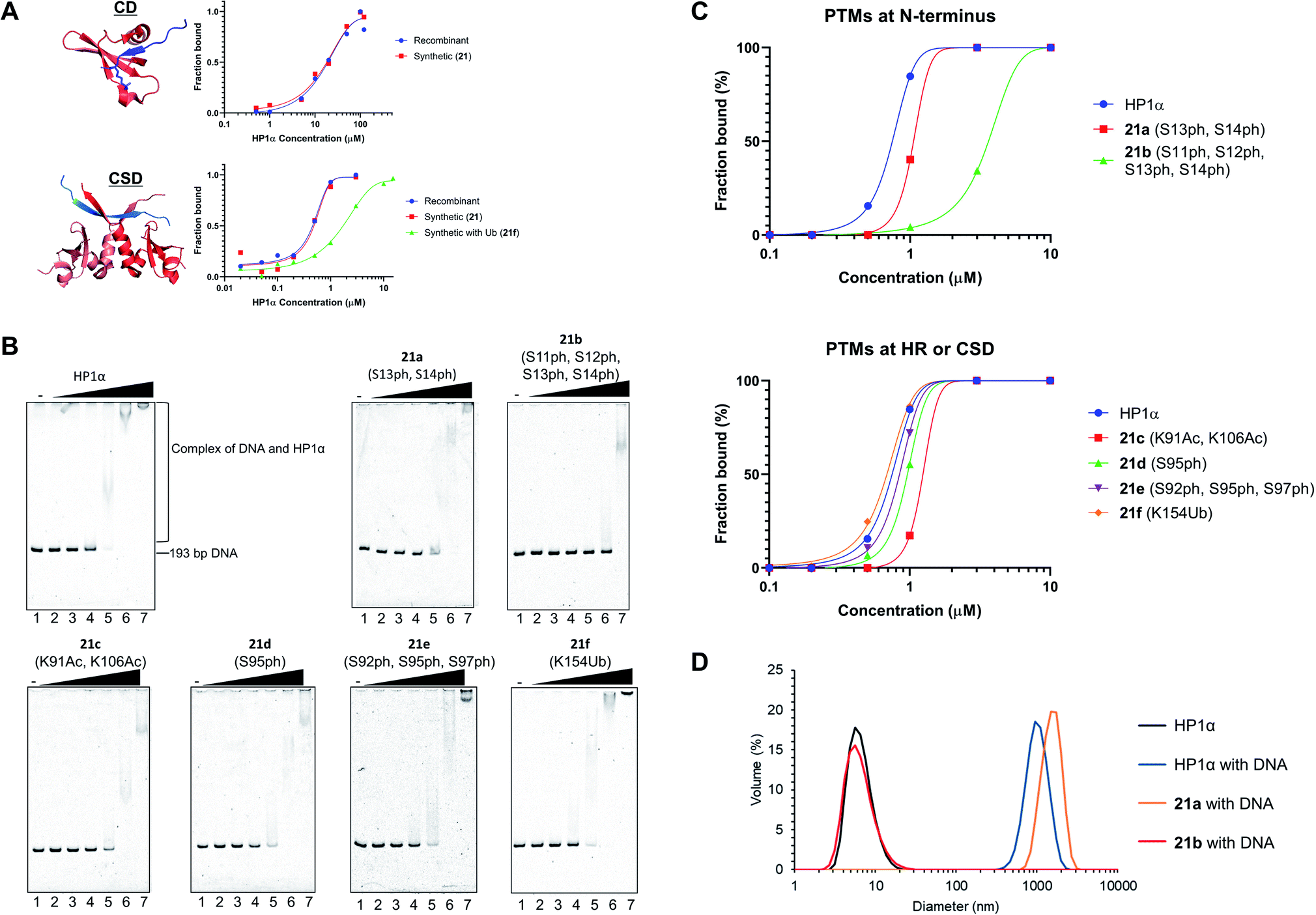 | ||
| Fig. 5 Biochemical assays of chemically synthesized HP1αs. (A) Fluorescence anisotropy assay. Binding of the CD of recombinant HP1α or synthetic HP1α (21) with fluorescein-label H3 peptide bearing K9me3 (red: CD of HP1α, blue: N-terminal tail of histone H3 with K9me3, PDB: 3FDT) and binding of the CSD of recombinant HP1α, 21, or 21f with fluorescein-label Sgo1 peptide (red: CSD of HP1β, blue: Sgo1 peptide, PDB: 3Q6S). (B) EMSAs using chemically synthesized HP1α with PTMs. Representative gel images of the binding of HP1α toward DNA. Various concentrations of HP1α were incubated with 193-bp DNA. The complexes were analyzed by 8% native-PAGE and visualized with SYBR Gold. (C) Quantification of the EMSAs using HP1α with PTMs at b N-terminus, c HR or CSD. (D) DLS measurement of complexes composed of 193-bp DNA (50 nM) and modified HP1α (10 μM). | ||
Investigation of DNA binding ability of modified HP1α
To investigate the relationship between PTMs installed at the N-terminus, HR, or CSD and DNA-binding ability of HP1α, EMSAs were performed using 193-bp DNA. In this assay, HP1α without PTMs efficiently bound DNA, and its Kd value was 0.75 μM (Fig. 5B and C), which was consistent with the previous results.6621a, containing doubly phosphorylated Ser residues at its N-terminus, showed a slightly higher Kd value compared to normal HP1α (Kd: 1.32 μM), but 21b, containing quadruple phosphorylation, significantly lowered the DNA-binding ability of HP1α (Kd: 4.67 μM). This result indicated that the number of phosphorylated Ser residues at the N-terminus of HP1α had an impact on its DNA-binding ability. We also evaluated the other PTMs on HR or CSD. 21c, with double acetylation of Lys at HR, slightly lowered its DNA-binding ability (Kd: 1.79 μM), and these PTMs were found to be little influential. Surprisingly, 21d and 21e with Ser phosphorylation at HR had little influence on the affinity of HP1α toward DNA [Kd: 0.95 μM (21d), Kd: 0.82 μM (21e)], although basic patches on HR of HP1α were the main responsible sites for the interaction with DNA.67 We found that the binding affinity of 21f with Ub at CSD was almost identical to that of normal HP1α (Kd: 0.71 μM).Next, we analyzed the particle size of complexes composed of DNA and HP1α by DLS. HP1α without PTMs formed large particles with DNA, and its diameter was around 1000 nm (Fig. 5D). 21a also formed large particles around 1000 nm diameter with DNA. However, 21b dramatically inhibited the formation of complexes and we could not observe large particles, which would be caused by the lower DNA-binding ability of 21b. Although HP1α possesses an acidic patch at its N-terminal (residues 15–19), HP1α has a higher binding ability toward DNA than other HP1 homologs. However, through the decoration of phosphorylations at S11–S14, HP1α significantly reduces its binding affinity by the extended acidic patch. Moreover, the number and location of phosphorylation were critical to control its DNA-binding ability, which would affect the behavior of HP1α in the nucleus. These significant discoveries in epigenetics were clarified by H1.2s and HP1αs with site-specific PTMs prepared through our new synthetic method using the organoruthenium catalyst.
Conclusions
We have developed a highly streamlined methodology for the chemical synthesis of various decorated proteins utilizing an air-tolerant organoruthenium catalyst. After the screening of organoruthenium complexes, we clarified that Ru complexes coordinated by Cp and X,O-bidentate (X = N or P) ligand efficiently removed the alloc group in catalytic manners under NCL conditions, and that the TONs of Ru-4 and Ru-10 exceeded 70 even in the presence of 5000 equiv. of MPAA, which could not be achieved with other current metal complexes. The organoruthenium catalyst was applied for total chemical protein synthesis to produce linker histone H1.2 and HP1α with two and six different modification patterns and our technology afforded a large protein above 30 kDa (ubiquitinated HP1α). Especially for the synthesis of HP1α, by selecting appropriate metal complexes, we prevented the generation of aggregates during the synthetic procedure. Through the analysis of chromatosomes reconstituted by using three types of synthetic H1.2s via EMSAs, we discovered that R53Cit modification contributed to the reduction of the binding affinity of H1.2 for nucleosomes. Furthermore, we revealed that four consecutive phosphorylations at S11–S14 of HP1α extended the acidic patch at its N-terminus, significantly reduced its DNA-binding ability, and disrupted the formation of complexes with DNA. With homogeneously modified proteins synthesized by using the Ru catalyst, the biological significance of the epigenetic marks on heterochromatin factors was elucidated. We believe that the streamlined chemical synthesis of proteins containing PTMs and other functional molecules with an organoruthenium catalyst would be a key for the elucidation of the mystery of biological phenomena and the creation of new therapeutic proteins.Author contributions
N. K. performed all experiments, conceived and designed the project, and wrote the manuscript. T. K. performed reconstitution of chromatosomes. T. K., H. K., H. M. assisted in the writing of the manuscript and ESI.† A. O. and G. H. supervised the project and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI 18H03931, 18H05504, and 19K22245 (A. O.), 18K05313, 19H05287, and 20H04704 and JST PRESTO JPMJPR19K6 (G. H.). N. K. was supported by a Research Fellow of the Japan Society for the Promotion of Science 19J13608. This work was also supported by JSPS KAKENHI JP18H05534, and the Platform Project for Supporting Drug Discovery and Life Science Research (BINDS) from Japan Agency for Medical Research and Development (AMED) under Grant Number JP20am0101076 and JST ERATO Grant Number JPMJER1901 (H. K.) and AMED under Grant Numbers 20he0622010h0001 (H. M.). We thank Dr Nozomu Suzuki (Nagoya University) to kindly allow us to measure CD spectra of recombinant and synthetic HP1αs. We also thank Dr Hiromi Takiguchi and Mr Tomohiro Kato for the measurement of Ru contents on synthetic proteins by using ICP-MS.References
- S. B. H. Kent, Protein Sci., 2018, 28, 313–328 CrossRef PubMed.
- S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- J. W. Bode, Acc. Chem. Res., 2017, 50, 2104–2115 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- K. Nakatsu, G. Hayashi and A. Okamoto, Curr. Opin. Chem. Biol., 2020, 58, 10–19 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- D. Bang, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- R. E. Thompson, X. Liu, N. Alonso-García, P. J. B. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS PubMed.
- C. Zuo, B. Zhang, B. Yan and J. S. Zheng, Org. Biomol. Chem., 2019, 17, 727–744 RSC.
- E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710–7729 RSC.
- M. Jbara, E. Eid and A. Brik, J. Am. Chem. Soc., 2020, 142, 8203–8210 CrossRef CAS PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- T. Schlatzer, J. Kriegesmann, H. Schröder, M. Trobe, C. Lembacher-Fadum, S. Santner, A. V. Kravchuk, C. F. W. Becker and R. Breinbauer, J. Am. Chem. Soc., 2019, 141, 14931–14937 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644–10655 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
- S. K. Maity, G. Mann, M. Jbara, S. Laps, G. Kamnesky and A. Brik, Org. Lett., 2016, 18, 3026–3029 CrossRef CAS PubMed.
- M. Jbara, S. Laps, S. K. Maity and A. Brik, Chem.–Eur. J., 2016, 22, 14851–14855 CrossRef PubMed.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 3154 CrossRef PubMed.
- S. Laps, H. Sun, G. Kamnesky and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 5729–5733 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity and A. Brik, Eur. J. Org. Chem., 2020, 3128–3132 CrossRef CAS.
- G. B. Vamisetti, G. Satish, P. Sulkshane, G. Mann, M. H. Glickman and A. Brik, J. Am. Chem. Soc., 2020, 142, 19558–19569 CrossRef CAS PubMed.
- S. Laps, G. Satish and A. Brik, Chem. Soc. Rev., 2021, 50, 2367–2387 RSC.
- N. Kamo, G. Hayashi and A. Okamoto, Angew. Chem., Int. Ed., 2018, 57, 16533–16537 CrossRef CAS PubMed.
- L. S. Hegedus and R. W. McCabe, Catalyst Poisoning, Marcel Dekker, New York, 1984 Search PubMed.
- B. G. Poulson, K. Szczepski, J. I. Lachowicz, L. Jaremko, A.-H. Emwas and M. Jaremko, RSC Adv., 2020, 10, 215–227 RSC.
- B. Koning, A. Meetsma and R. M. Kellogg, J. Org. Chem., 1998, 63, 5533–5540 CrossRef CAS.
- A. Kolpin, G. Jones, S. Jones, W. Zheng, J. Cookson, A. P. E. York, P. J. Collier and S. C. E. Tsang, ACS Catal., 2017, 7, 592–605 CrossRef CAS.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC.
- T. Kondo, Y. Morisaki, S. Uenoyama, K. Wada and T. Mitsudo, J. Am. Chem. Soc., 1999, 121, 8657–8658 CrossRef CAS.
- S. Tanaka, P. K. Pradhan, Y. Maegawa and M. Kitamura, Chem. Commun., 2010, 46, 3996–3998 RSC.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- M. D. Mbaye, B. Demerseman, J. L. Renaud, L. Toupet and C. Bruneau, Angew. Chem., Int. Ed., 2003, 42, 5066–5068 CrossRef CAS PubMed.
- B. Sundararaju, M. Achard, B. Demerseman, L. Toupet, G. V. M. Sharma and C. Bruneau, Angew. Chem., Int. Ed., 2010, 49, 2782–2785 CrossRef CAS PubMed.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- K. Mashima, H. Kaneyoshi, S. Kaneko, A. Mikami, K. Tani and A. Nakamura, Organometallics, 1997, 16, 1016–1025 CrossRef CAS.
- S. W. Harshman, N. L. Young, M. R. Parthun and M. A. Freitas, Nucleic Acids Res., 2013, 41, 9593–9609 CrossRef CAS PubMed.
- S. P. Hergeth and R. Schneider, EMBO Rep., 2015, 16, 1439–1453 CrossRef CAS PubMed.
- T. W. Flanagan and D. T. Brown, Biochim. Biophys. Acta, Gene Regul. Mech., 2016, 1859, 468–475 CrossRef CAS PubMed.
- A. Izzo and R. Schneider, Biochim. Biophys. Acta, Gene Regul. Mech., 2016, 1859, 486–495 CrossRef PubMed.
- D. V. Fyodorov, B. R. Zhou, A. I. Skoultchi and Y. Bai, Nat. Rev. Mol. Cell Biol., 2018, 19, 192–206 CrossRef CAS PubMed.
- Recently, Hong et al. reported the chemical synthesis of linker histone H1.2 through sequential ligations on the solid phase. Z. Z. Hong, R. R. Yu, X. Zhang, A. M. Webb, N. L. Burge, M. G. Poirier and J. J. Ottesen, bioRxiv, 2019 DOI:10.1101/661744.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- M. A. Christophorou, G. Castelo-Branco, R. P. Halley-Stott, C. S. Oliveira, R. Loos, A. Radzisheuskaya, K. A. Mowen, P. Bertone, J. C. R. Silva, M. Zernicka-Goetz, M. L. Nielsen, J. B. Gurdon and T. Kouzarides, Nature, 2014, 507, 104–108 CrossRef CAS PubMed.
- B. Sarg, W. Helliger, H. Talasz, B. Förg and H. H. Lindner, J. Biol. Chem., 2006, 281, 6573–6580 CrossRef CAS PubMed.
- K. Shintomi, M. Iwabuchi, H. Saeki, K. Ura, T. Kishimoto and K. Ohsumi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8210–8215 CrossRef CAS PubMed.
- J. Bednar, I. Garcia-Saez, R. Boopathi, A. R. Cutter, G. Papai, A. Reymer, S. H. Syed, I. N. Lone, O. Tonchev, C. Crucifix, H. Menoni, C. Papin, D. A. Skoufias, H. Kurumizaka, R. Lavery, A. Hamiche, J. J. Hayes, P. Schultz, D. Angelov, C. Petosa and S. Dimitrov, Mol. Cell, 2017, 66, 384–397 CrossRef CAS PubMed.
- C. Maison and C. Almouzni, Nat. Rev. Mol. Cell Biol., 2004, 5, 296–305 CrossRef CAS PubMed.
- D. Canzio, A. Larson and G. J. Narlikar, Trends Cell Biol., 2014, 24, 377–386 CrossRef CAS PubMed.
- G. Nishibuchi and J. Nakayama, J. Biochem., 2014, 156, 11–20 CrossRef CAS PubMed.
- A. Kumar and H. Kono, Biophys. Rev., 2020, 12, 387–400 CrossRef PubMed.
- J. S. Becker, D. Nicetto and K. S. Zaret, Trends Genet., 2016, 32, 29–41 CrossRef CAS PubMed.
- J. Kanga, J. Chaudharya, H. Donga, S. Kima, C. A. Brautigamb and H. Yu, Mol. Biol. Cell, 2011, 22, 1181–1190 CrossRef PubMed.
- S. Machida, Y. Takizawa, M. Ishimaru, Y. Sugita, S. Sekine, J. Nakayama, M. Wolf and H. Kurumizaka, Mol. Cell, 2018, 69, 385–397 CrossRef CAS PubMed.
- G. LeRoy, J. T. Weston, B. M. Zee and N. L. Young, Mol. Cell. Proteomics, 2009, 8, 2432–2442 CrossRef CAS PubMed.
- N. Kamo, G. Hayashi and A. Okamoto, Chem. Commun., 2018, 54, 4337–4340 RSC.
- M. Jbara, E. Eid and A. Brik, Org. Biomol. Chem., 2018, 16, 4061–4064 RSC.
- N. Kamo, G. Hayashi and A. Okamoto, Org. Lett., 2019, 21, 8378–8382 CrossRef CAS PubMed.
- T. Durek, V. Y. Torbeev and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4846–4851 CrossRef CAS PubMed.
- K. Hiragami-Hamada, K. Shinmyozu, D. Hamada, Y. Tatsu, K. Uegaki, S. Fujiwara and J. Nakayama, Mol. Cell. Biol., 2011, 31, 1186–1200 CrossRef CAS PubMed.
- G. Nishibuchi, S. Machida, A. Osakabe, H. Murakoshi, K. Hiragami-Hamada, R. Nakagawa, W. Fischle, Y. Nishimura, H. Kurumizaka, H. Tagami and J. Nakayama, Nucleic Acids Res., 2014, 42, 12498–12511 CrossRef CAS PubMed.
- E. Minc, Y. Allory, H. J. Worman, J.-C. Courvalin and B. Buendia, Chromosoma, 1999, 108, 220–234 CrossRef CAS PubMed.
- A. Chakraborty, K. V. Prasanth and S. G. Prasanth, Nat. Commun., 2014, 5, 3445 CrossRef PubMed.
- G. Nishibuchi, S. Machida, R. Nakagawa, Y. Yoshimura, K. Hiragami-Hamada, Y. Abe, H. Kurumizaka, H. Tagami and J. Nakayama, J. Biochem., 2019, 165, 433–446 CrossRef CAS PubMed.
- S. Chen, C. Wang, L. Sun, D.-L. Wang, L. Chen, Z. Huang, Q. Yang, J. Gao, X.-B. Yang, J.-F. Chang, P. Chen, L. Lan, Z. Mao and F.-L. Sun, Mol. Cell. Biol., 2015, 35, 406–416 CrossRef PubMed.
- S. Tang, L. J. Liang, Y. Y. Si, S. Gao, J. X. Wang, J. Liang, Z. Mei, J. S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 13333–13337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00731a |
| This journal is © The Royal Society of Chemistry 2021 |