
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor–acceptor–acceptor-type near-infrared fluorophores that contain dithienophosphole oxide and boryl groups: effect of the boryl group on the nonradiative decay†
Yoshiaki
Sugihara‡
a,
Naoto
Inai‡
a,
Masayasu
Taki
 b,
Thomas
Baumgartner
c,
Ryosuke
Kawakami
d,
Takashi
Saitou
d,
Takeshi
Imamura
d,
Takeshi
Yanai
*ab and
Shigehiro
Yamaguchi
*ab
b,
Thomas
Baumgartner
c,
Ryosuke
Kawakami
d,
Takashi
Saitou
d,
Takeshi
Imamura
d,
Takeshi
Yanai
*ab and
Shigehiro
Yamaguchi
*ab
aDepartment of Chemistry, Graduate School of Science, Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo, Chikusa, Nagoya, 464-8602, Japan. E-mail: yanait@chem.nagoya-u.ac.jp; yamaguchi@chem.nagoya-u.ac.jp
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya, 464-8602, Japan
cDepartment of Chemistry, York University, 4700 Keele St., Toronto, ON M3J 1P3, Canada
dDepartment of Molecular Medicine for Pathogenesis, Graduate School of Medicine, Ehime University, Shitsukawa, Toon City, Ehime 791-0295, Japan
First published on 25th March 2021
Abstract
The use of donor–π–acceptor (D–π–A) skeletons is an effective strategy for the design of fluorophores with red-shifted emission. In particular, the use of amino and boryl moieties as the electron-donating and -accepting groups, respectively, can produce dyes that exhibit high fluorescence and solvatochromism. Herein, we introduce a dithienophosphole P-oxide scaffold as an acceptor–spacer to produce a boryl- and amino-substituted donor–acceptor–acceptor (D–A–A) π-system. The thus obtained fluorophores exhibit emission in the near-infrared (NIR) region, while maintaining high fluorescence quantum yields even in polar solvents (e.g. λem = 704 nm and ΦF = 0.69 in CH3CN). A comparison of these compounds with their formyl- or cyano-substituted counterparts demonstrated the importance of the boryl group for generating intense emission. The differences among these electron-accepting substituents were examined in detail using theoretical calculations, which revealed the crucial role of the boryl group in lowering the nonradiative decay rate constant by decreasing the non-adiabatic coupling in the internal conversion process. The D–A–A framework was further fine-tuned to improve the photostability. One of these D–A–A dyes was successfully used in bioimaging to visualize the blood vessels of Japanese medaka larvae and mouse brain.
Introduction
Organic π-conjugated compounds with a donor–π–acceptor (D–π–A) framework have attracted substantial attention due to their wide range of applications.1–8 The absorption and emission properties of D–π–A compounds can be tuned by structural modifications of the individual components. By using an appropriate combination of donor, π-spacer, and acceptor moieties, even near-infrared (NIR) emission can be attained. Such NIR-emissive compounds are promising materials for NIR-emissive OLEDs,9,10 biosensing applications,11 and deep-tissue bioimaging given the high biopermeability of the NIR light.12,13Even though various D–π–A-type fluorophores that exhibit NIR emission have been developed so far, their fluorescence quantum yields (ΦF) tend to decrease drastically in polar solvents. However, D–π–A compounds that contain a diarylboryl moiety as the electron-accepting group represent one exception to this trend.5–8 In 1972, Williams and co-workers reported a simple p-(dimesitylboryl)-substituted triphenylamine as the first example of a boron-based D–π–A-type fluorophore.14 Since then, boryl-substituted D–π–A dyes have been extensively studied in order to explore their potential utility as nonlinear optical materials,15,16 two-photon-emissive materials,17 anion sensors,18 and bioimaging.19 These compounds often exhibit high ΦF values even in polar media, despite the significant red-shift of their emission bands.5c Two moieties in this type of molecular frameworks can be modified to obtain NIR emission: the aryl group on the boron atom and the π-spacer. For example, in 2015, Marder and co-workers reported D–π–A compounds with an electron-withdrawing perfluorophenyl or 3,5-(CF3)2C6H3 group at the para position of the aryl group on the boron atom (Fig. 1).20 In combination with the strong donor julolidine, the compounds showed NIR fluorescence in CH3CN with maximum emission wavelengths (λem) around 745 nm. On the other hand, in 2014, Zhao and co-workers reported a fluorophore that employed 2,1,3-benzothiadiazole as additional electron-accepting π-spacer, emitting in the far-red to NIR region (λem = 669 nm) in CH3CN (Fig. 1).21 Misra and co-workers achieved a further red-shifted emission (λem = 692 nm; ΦF = 0.27 in CH2Cl2) by insertion of acetylene spacers.22 Such donor–acceptor–acceptor (D–A–A)-type structures should thus be beneficial for achieving red-shifted emission.
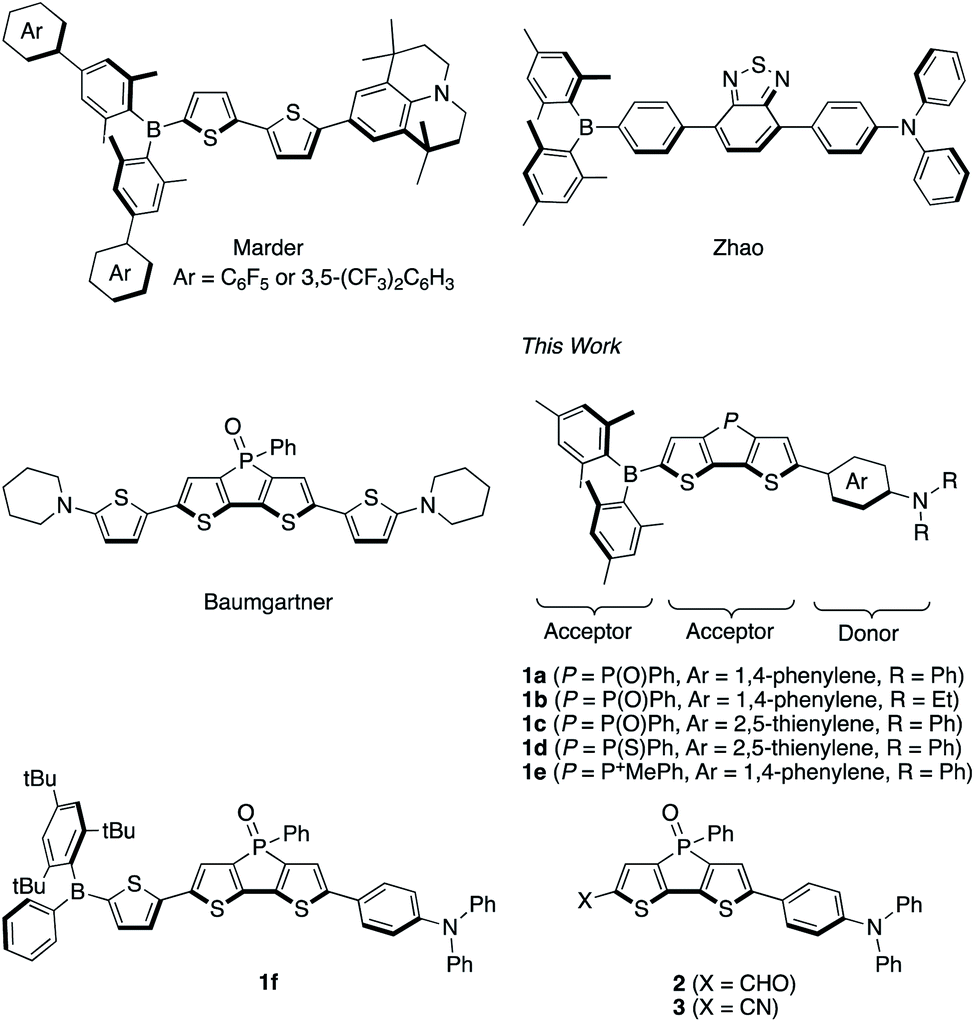 | ||
| Fig. 1 Representative boryl-substituted D–π–A dyes and dithienophosphole P-oxide derivatives and boryl-substituted D–A–A type dyes and relevant compounds studied in this work. | ||
To develop NIR-fluorescent dyes, we have focused our attention on the modification of the π-spacer by introducing a phosphine oxide group. We envisioned that the P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Ph group could bathochromically shift the emission by lowering the LUMO energy level due to the σ*–π* interaction23,24 as well as the inherent inductive electron-withdrawing effect. The profound effect of introducing a phosphine oxide group has already been documented for several fluorescent dyes. For example, fluorescein, a xanthene dye, exhibits a fluorescence maximum at 510 nm, while phospha-fluorescein, a phosphine-oxide-containing fluorescein analogue, exhibits an emission maximum of 656 nm.25 This example aptly illustrates the substantial impact of the phosphine oxide group on the electronic structure.
O)Ph group could bathochromically shift the emission by lowering the LUMO energy level due to the σ*–π* interaction23,24 as well as the inherent inductive electron-withdrawing effect. The profound effect of introducing a phosphine oxide group has already been documented for several fluorescent dyes. For example, fluorescein, a xanthene dye, exhibits a fluorescence maximum at 510 nm, while phospha-fluorescein, a phosphine-oxide-containing fluorescein analogue, exhibits an emission maximum of 656 nm.25 This example aptly illustrates the substantial impact of the phosphine oxide group on the electronic structure.
In this context, dithieno[3,2-b:2′,3′-d]phosphole P-oxide represents an attractive scaffold. Various dithienophosphole derivatives have already been developed by expanding its π-skeleton or modifying the P-aryl groups.26,27 While dithienophosphole P-oxide itself exhibits an emission maximum at 453 nm in CH2Cl2, its emission wavelength can be red-shifted by introducing electron-donating triphenylamine moieties at its termini.28 The incorporation of the stronger donor moiety aminothiophene further red-shifts the emission to 657 nm with a moderate fluorescence quantum yield (ΦF = 0.39) in CH2Cl2 (Fig. 1).29
In this article, we report the design and synthesis of boryl-substituted D–A–A-type fluorophores, which contain a dithienophosphole P-oxide scaffold as an additional acceptor moiety, as highly emissive NIR-fluorescent dyes (Fig. 1). The phosphine oxide group was expected to red-shift the emission band, improve the photostability of the dye, and afford a more rigid molecular structure, which would suppress the nonradiative decay process and thus improve the quantum yield. A series of the D–A–A type dyes 1a–1f that bear various aryl groups on the boron atom, phosphorus-substitution patterns, and electron-donating moieties was synthesized, and their substituent effects were studied in-depth in order to accomplish intense NIR emission and high photostability. A comparative study with other D–A–A type analogues 2 and 3 with different acceptor units in place of the boryl group (Fig. 1) revealed the important role of a terminal boryl group to suppress the non-radiative decay process, resulting in the high fluorescence quantum yields, according to excited-state theoretical calculations. To demonstrate the utility of such dyes, 1c was employed for fluorescence imaging in vivo.
Results and discussion
Compounds 1a–1f were synthesized using 2,6-dibromodithienophosphole P-oxide 428a as the key precursor (Scheme 1). The crucial step in the synthesis of 1a–1e is the mono-functionalization of 4. Its reduction with trichlorosilane, followed by treatment with 1.1 equiv. of n-BuLi produced mainly the mono-lithiated product, which was successively treated with Mes2BF. Further oxidation with pyridinium chlorochromate (PCC) or sulfur afforded 5 or 6 in 24% and 18% overall yield, respectively. Suzuki–Miyaura cross-coupling of 5 or 6 with the corresponding amino-substituted arylboronic acid or boronic ester afforded 1a–1d in moderate yields. The phosphonium derivative 1e was obtained by the reduction of 1a, followed by methylation of the P centre with MeOTf. Subsequent recrystallization from a hexane/CHCl3 mixed solvent afforded 1e.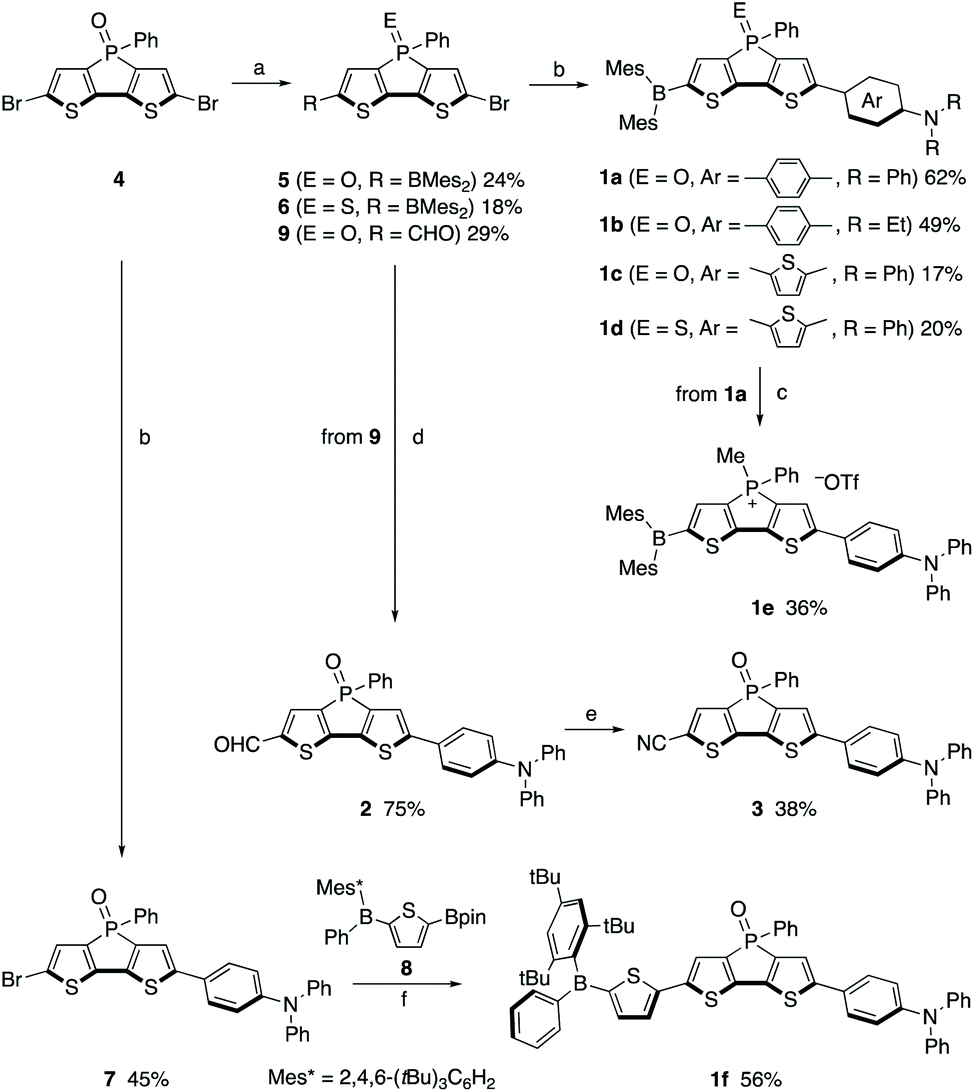 | ||
| Scheme 1 Synthesis of compounds 1a–1f, 2, and 3. Reagents and conditions: (a) (1) HSiCl3, toluene, rt; (2) n-BuLi, THF, −78 °C; (3) Mes2BF or DMF, THF, −78 °C to rt; (4) PCC, S8, or H2O2, CH2Cl2, rt; (b) 4-(Ph2N)C6H4B(OH)2, 4-(Et2N)C6H4B(OH)2, or 2-(Ph2N)-5-(pin)B-thiophene, Pd(PPh3)4, K2CO3, toluene, 110 °C; (c) (1) HSiCl3, toluene, rt; (2) MeOTf, CH2Cl2, rt; (d) (1) ethylene glycol, TsOH·H2O, benzene, 100 °C, (2) 4-(Ph2N)C6H4B(OH)2, Pd(PPh3)4, Na2CO3, toluene/H2O, (3) HClaq; (e) H2NOH·HCl, TsOH·H2O, MgSO4, toluene, 120 °C; (f) Pd(PPh3)4, Na2CO3, toluene/H2O, 110 °C. | ||
For the synthesis of 1f, bearing a bulky tri(t-butyl)phenyl group on the boron atom, mono-arylation of the key precursor 4 was employed. Specifically, the Suzuki–Miyaura cross-coupling of 4 with 1.0 equiv. of diphenylaminophenylboronic acid furnished 7, which was subsequently coupled with borylthienylboronic ester 8, obtained in situ from the direct borylation of the corresponding thienylborane precursor using an Ir catalyst, to generate 1f (for details, see the ESI†). Compounds 1a–1f are sufficiently stable to be handled in air without any special precautions.
The formyl- and cyano-substituted analogues 2 and 3 were synthesized as reference compounds (vide infra) via the mono-lithiation of 4; i.e., following the reduction of 4 and its lithiation with 1.1 equiv. of n-BuLi, DMF was added as the formyl source. Further oxidation of the phosphine moiety with H2O2 afforded 9. After protection of the formyl group, Suzuki–Miyaura cross-coupling and subsequent deprotection of the acetal moiety afforded 2. Transformation of the formyl group in 2 into a cyano group using hydroxylamine afforded 3.
Photophysical properties of D–A–A dye 1a
Initially, we evaluated the photophysical properties of 1a, summarized in Table 1, and the UV-vis absorption and fluorescence spectra of 1a in various solvents are shown in Fig. 2a. Regardless of the solvent used, 1a showed absorption maxima (λabs) at 458–466 nm with molar absorption coefficients >30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. In contrast, the fluorescence spectra of 1a exhibited significant solvatochromism. While 1a showed an emission maximum (λem) at 532 nm with a Stokes shift of 3037 cm−1 in nonpolar cyclohexane, a significant bathochromic shift (λem = 665 nm) with a large Stokes shift (6607 cm−1) was observed in polar CH3CN. This result demonstrates that 1a exhibits a strong intramolecular charge-transfer (ICT) character in the excited state, which is a typical feature of D–π–A fluorophores. The ICT character of 1a is also reflected in a large dipole moment in the excited state (μE = 20.6 D), which was estimated using the Lippert–Mataga equation (Fig. S8 and Table S2†). Notably, 1a maintained a high fluorescence quantum yield in polar solvents (e.g., in CH3CN: ΦF = 0.59) despite its large μE value. This behaviour differs significantly from that commonly seen in D–π–A-type fluorescent dyes.
000 M−1 cm−1. In contrast, the fluorescence spectra of 1a exhibited significant solvatochromism. While 1a showed an emission maximum (λem) at 532 nm with a Stokes shift of 3037 cm−1 in nonpolar cyclohexane, a significant bathochromic shift (λem = 665 nm) with a large Stokes shift (6607 cm−1) was observed in polar CH3CN. This result demonstrates that 1a exhibits a strong intramolecular charge-transfer (ICT) character in the excited state, which is a typical feature of D–π–A fluorophores. The ICT character of 1a is also reflected in a large dipole moment in the excited state (μE = 20.6 D), which was estimated using the Lippert–Mataga equation (Fig. S8 and Table S2†). Notably, 1a maintained a high fluorescence quantum yield in polar solvents (e.g., in CH3CN: ΦF = 0.59) despite its large μE value. This behaviour differs significantly from that commonly seen in D–π–A-type fluorescent dyes.
| Compound | Solvent | λ abs (nm) | ε (104 M−1 cm−1) | λ em (nm) | Stokes shift (cm−1) | Φ F | k r (108 s−1) | k nr (108 s−1) |
|---|---|---|---|---|---|---|---|---|
| a Only the longest absorption maximum wavelengths are shown. b Absolute fluorescence quantum yields were determined by a calibrated integrating sphere system within ±3% error. c Not determined due to poor solubility. | ||||||||
| 1a | Cyclohexane | 458 | 3.56 | 532 | 3037 | 0.81 | 2.6 | 0.62 |
| Toluene | 466 | 3.43 | 566 | 3791 | 0.90 | 2.8 | 0.31 | |
| CHCl3 | 466 | 3.18 | 597 | 4709 | 0.90 | 2.3 | 0.25 | |
| CH2Cl2 | 466 | 3.32 | 626 | 5485 | 0.88 | 2.1 | 0.28 | |
| CH3CN | 462 | 3.45 | 665 | 6607 | 0.59 | 1.5 | 1.0 | |
| 1b | Cyclohexane | 470 | 3.26 | 540 | 2758 | 0.81 | 2.3 | 0.54 |
| CH3CN | 487 | 3.28 | 695 | 6145 | 0.72 | 1.5 | 0.59 | |
| 1c | Cyclohexane | 486 | 3.14 | 570 | 3032 | 0.65 | 1.9 | 0.99 |
| CH3CN | 477 | 2.77 | 704 | 6760 | 0.67 | 1.5 | 0.74 | |
| 1d | Cyclohexane | 476 | 3.07 | 567 | 3372 | 0.86 | 2.4 | 0.39 |
| CH3CN | 475 | 2.77 | 699 | 6848 | 0.69 | 1.5 | 0.67 | |
| 1e | Cyclohexane | 488 | —c | 617 | 4284 | 0.62 | n.d. | n.d. |
| CH3CN | 480 | 2.67 | 748 | 7464 | 0.07 | n.d. | n.d. | |
| 1f | Cyclohexane | 470 | 4.47 | 550 | 3095 | 0.44 | 2.0 | 2.6 |
| CH3CN | 474 | 4.70 | 651 | 5736 | 0.71 | 1.8 | 0.72 | |
| 2 | Cyclohexane | 463 | 2.44 | 537 | 2976 | 0.60 | 1.5 | 1.0 |
| CH3CN | 464 | 2.70 | 709 | 7447 | 0.08 | 0.94 | 11 | |
| 3 | Cyclohexane | 453 | —c | 548 | 3827 | 0.59 | 1.4 | 0.98 |
| CH3CN | 452 | 2.46 | 681 | 7440 | 0.26 | 0.95 | 2.7 | |
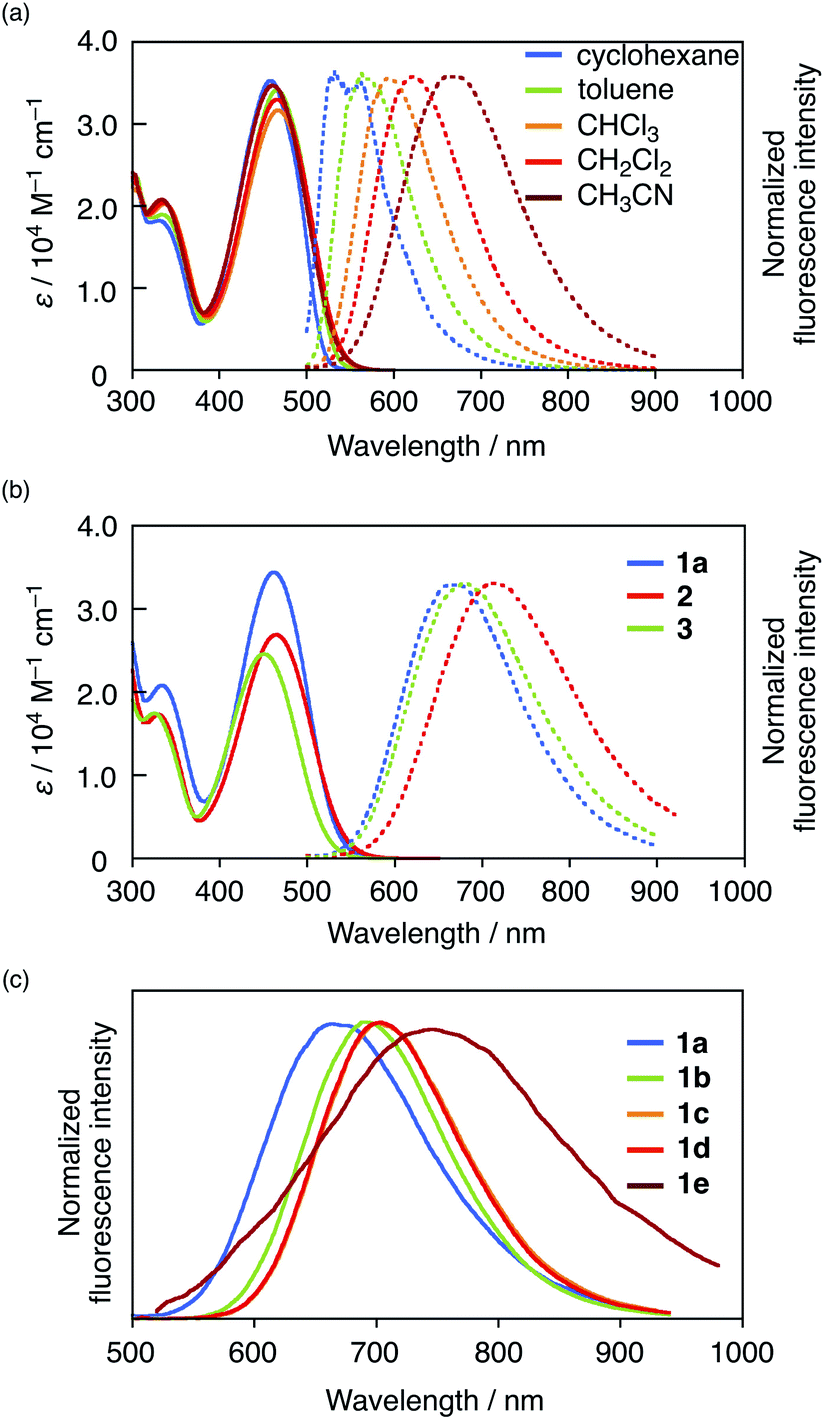 | ||
| Fig. 2 UV-vis absorption and emission spectra of (a) 1a in various solvents, (b) 1a, 2, and 3 in CH3CN, and (c) emission spectra of 1a–1e in CH3CN. | ||
Effects of the boryl group on the photophysical properties
To investigate the origin of the high fluorescence quantum yield of 1a, especially in polar solvents, its photophysical properties were compared to those of reference compounds 2 and 3, which contain a formyl and cyano group instead of the boryl group, respectively. While cyano analogue 3 showed a maximum emission wavelength slightly longer than that of 1a in CH3CN, formyl analogue 2 exhibited a more red-shifted emission (Δλ = 44 nm; Fig. 2b). Moreover, 2 and 3 also showed significant solvent effects, i.e., large dipole moments (2: μE = 23.8 D; 3: μE = 21.0 D) were estimated using the Lippert–Mataga equation (Fig. S8†), which suggests strong ICT character in the excited state, akin to that of 1a. However, their fluorescence quantum yields decreased drastically in polar solvents such as CH3CN (2: ΦF = 0.08; 3: ΦF = 0.26), which is typical for D–π–A-type dyes.To gain further insight into the features of boryl-substituted fluorescent dye 1a, we examined its excited-state dynamics in terms of its radiative (kr) and nonradiative (knr) decay rate constants from the excited singlet state (S1). These values are determined by the ΦF values and the fluorescence lifetimes τ. For 1a, 2, and 3, the kr values decrease with increasing solvent polarity (Table 1). Boryl derivative 1a exhibits a higher kr value than 2 or 3 in CH3CN, which is at least partially responsible for its higher ΦF value. Moreover, 1a shows the lowest knr value in CHCl3, which increases slightly in CH3CN. Importantly, the knr value of 1a in CH3CN is 1.0 × 108 s−1, while those of 2 and 3 are beyond 108 s−1. These results indicate that not only the higher kr value, but also the suppressed knr value are responsible for the high fluorescence quantum yield of 1a in polar solvents.
Theoretical examination of the effects of the boryl group
A theoretical analysis of the photophysical properties of the diarylboryl and dithienophosphole-based D–A–A type molecules was carried out using time-dependent density functional theory (TD-DFT). As the characteristic effects of the boryl group in 1a on ΦF were experimentally observed in comparison with reference compounds 2 and 3, this computational study was focused on the elucidation of the role of the boryl group in increasing ΦF. The solvent effects were considered using the polarizable continuum model (PCM) to verify their impact on ΦF. The calculations were designed to identify the major factors that determine kr and knr (for the details of the computational calculations, see the ESI†).First, the radiative transition relevant to the kr value was examined based on the TD-DFT results in S0 and S1 (Fig. 3). Boryl derivative 1a exhibits a slightly higher oscillator strength (f = 1.19) for the Franck–Condon transition from S0 to S1 relative to 2 (f = 1.02) and 3 (f = 0.97). This result is consistent with the fact that 1a exhibits a ∼50% higher molar absorption coefficient than 2 and 3. Notably, the f value for the vertical transition from S1 to S0 for 1a with the optimized S1 geometry is also higher than those for 2 and 3 (1a: f = 1.42; 2: 1.34; 3: 1.26, Fig. S13†). As kr is proportional to ν2f, where ν is the wavenumber of the emission, the higher f value in 1a should be partially responsible for the higher kr value relative to those of 2 and 3.
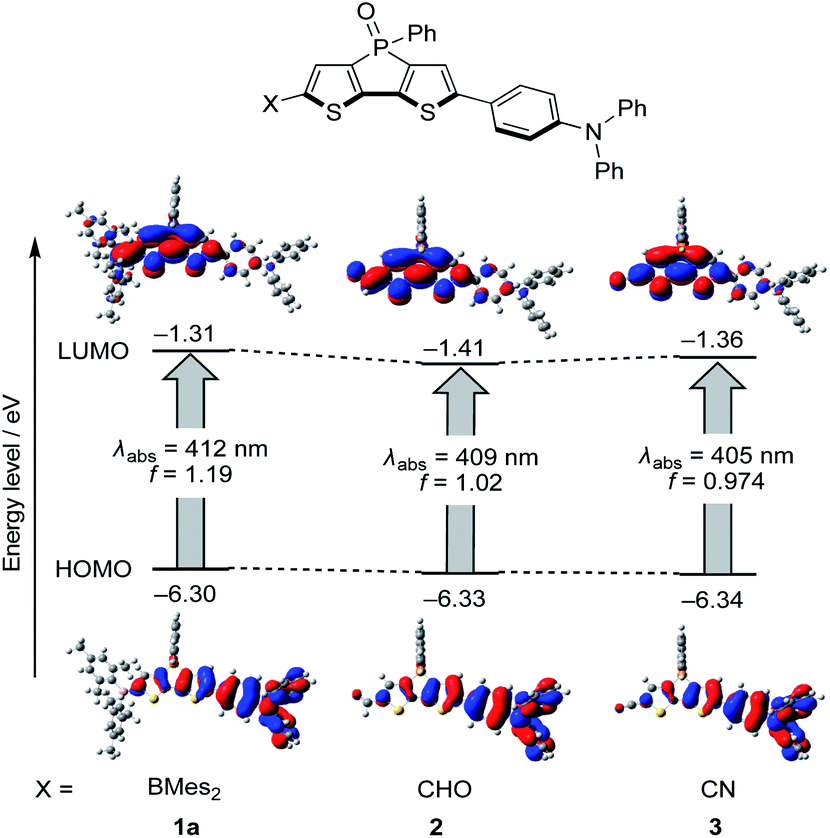 | ||
| Fig. 3 Energy diagrams for the frontier orbitals of 1a, 2, and 3 in CH3CN using the optimized ground state structures. Calculations were carried out at the CAM-B3LYP/6-31G(d) level of theory including the PCM model. | ||
Next, we examined the non-radiative transition relevant to the knr value, which may occur through various decay processes. The possible pathways include the internal conversion (IC) from S1 to S0 and/or the intersystem crossing (ISC) to the triplet states.30 We performed rate-constant calculations on the S1 → S0 IC process and the S1 → T2 ISC process; hereafter, their rates are denoted as kIC and kISC, respectively. With the relaxed S1 geometry, S1 and T2 lie closely in energy and largely away from other states, which were thus neglected (Fig. S14†). The kIC and kISC of 1a in CH3CN were estimated to be 1.4 × 108 and 1.0 × 107 [s−1], respectively, using MOMAP-2020A program31 (for details, see the ESI†). This implies that the main nonradiative decay pathway of these dyes in CH3CN is the S1 → S0 IC.
To discuss the relative trend of the experimentally obtained knr values among 1a, 2, and 3, which are assumed to be mainly kIC, the normal-mode contribution of the nonadiabatic coupling (NAC) calculated with the S1-optimized geometry was analysed as shown in Fig. 4a. For all compounds, the normal mode with a frequency of ca. 1570 cm−1 was found to have one of the largest NAC values (Fig. S15†). These modes were attributed mainly to the C5–C6 and C7–C8 stretching vibration in the bithiophene moiety of the dithienophosphole scaffold (for the atom labelling, see Fig. 4b). Because the largest geometry changes occur in the C5–C6 bond length between the optimized structures of S0 and S1, while they are nearly identical among 1a, 2, and 3 (Fig. S16†), this quinoidal mode should thus make the largest contribution to kIC through large nonadiabatic interstate coupling.32
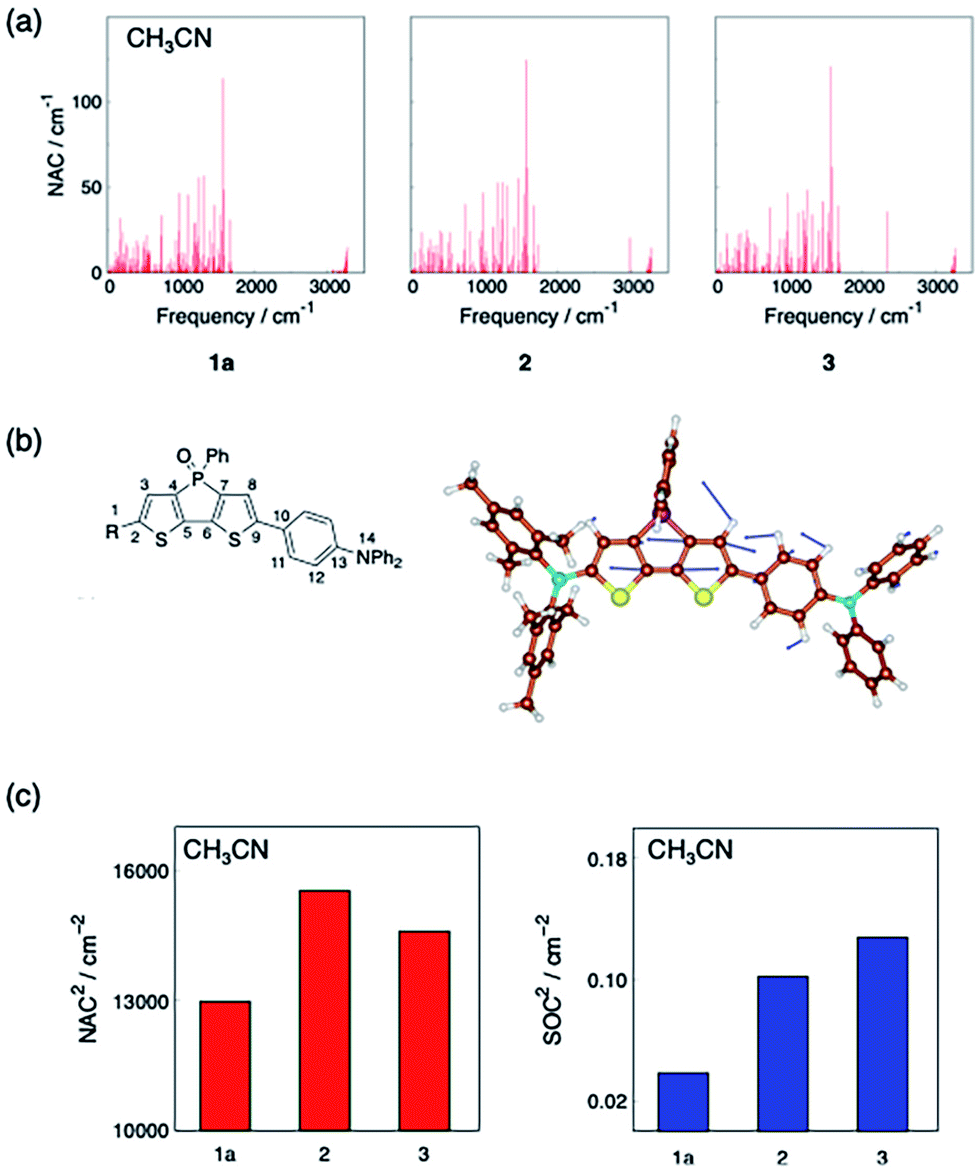 | ||
| Fig. 4 (a) Computed NAC and frequencies along with the normal modes of 1a, 2 and 3 in CH3CN. (b) A representative vibrational mode with large NAC of 1a in CH3CN, which has a large contribution of the C5–C6 and C7–C8 stretching and a frequency of 1572 cm−1; the representative vibrational modes of the other molecules are shown in Fig. S15.† (c) Comparison of the square of the largest NAC in Fig. 4a (left) and the square of the SOC between S1 and T2 (right) among 1a, 2 and 3 in CH3CN. Quantum mechanical calculations were conducted at the (TD-)CAM-B3LYP/6-31G(d) level of theory using Gaussian 16 Rev. B.01 program.33 SOC were computed using PySOC program.34 | ||
The NAC values along with the quinoidal mode were calculated to be 113.9 cm−1, 124.6 cm−1 and 120.8 cm−1 in CH3CN for 1a, 2, and 3, respectively. Assuming that this mode is the promoting mode of the S1 → S0 IC and vibrational terms are comparable among these dye molecules, the kIC should be proportional to the square of NAC along with this mode. The square of these NAC values provides the following relation: 1a < 3 < 2 (Fig. 4c). This trend qualitatively matches the relative magnitude of the experimental knr among the three compounds in CH3CN. For the CH3CN solutions, we can thus deduce that the substituent-dependence of kIC, which almost coincides with knr, is determined by the NACs associated with the quinoidal vibration.
The spin–orbit coupling (SOC) between S1 and T2 was also computed. In the procedure here, SOC was treated as a constant. This means that kISC is proportional to the square of SOC when vibrational terms are comparable among these dye molecules. Compound 1a has a smaller square of the SOC than the other molecules (Fig. 4c), which implies that 1a has smaller kISC than the others.
Considering that the experimentally obtained knr for 2 was about 10 times larger than that for 1a, our prediction might underestimate the interstate interaction of 2. This seemingly arises from the limitation of our model, where the terms that can be important in the calculation of kISC with small direct SOC (see the ESI† for details) were neglected.
Thus, the question of why boryl derivative 1a exhibits a larger ΦF (= kr/(kr + knr)) compared to 2 and 3 can be partially addressed by our theoretical model, which shows that 1a in S1 undergoes decay processes with a larger radiative decay rate constant kr and a smaller non-radiative decay rate constant knr. The latter can be rationalized in terms of a suppression of the nonadiabatic IC process with the smaller NACs and the ISC process with smaller SOC.
Structural modification to achieve NIR emission
To accomplish a more red-shifted emission, we modified the donor moiety, π-spacer, and/or the phosphorus moiety of the D–A–A framework (Fig. 2c). The photophysical properties of the corresponding compounds 1b–1e are summarized in Table 1 (for the full data in various solvents, see Table S1†). Compound 1b, which contains ethyl groups in place of phenyl groups on the amino moiety, and 1c, which contains a thiophene π-spacer, exhibited more red-shifted emission bands than 1a in CH3CN due to their enhanced electron-donating character (1b: λem = 695 nm; 1c: λem = 699 nm). Compound 1d, in which the PO bond of 1c is replaced with a PS bond, exhibited a comparable emission to that of 1c. Although 1e, with a quaternized P centre, showed the longest wavelength emission (λem = 748 nm in CH3CN), its ΦF value was low (0.07). Notably, all these derivatives, except for 1e, retained high quantum yields even in CH3CN, despite their long-wavelength emission.
Evaluation of the photostability
The introduction of additional heteroatoms can be expected to affect the photostability of the present compounds.35 Therefore, the photostability of 1a, 3, and reference compound 10 (Fig. 5a), which does not contain a P(O)Ph group, was evaluated in degassed CH3CN under irradiation from a 449 nm high-power LED lamp equipped with a 450/10 nm hard-coated bandpass filter. We have previously demonstrated that the introduction of a bulky substituent on the B atom results in increased photostability.36 Accordingly, we designed 1f, which should exhibit improved photostability relative to 1a, on account of the bulky substituent at the boron atom, and evaluated its photophysical properties. To quantitatively evaluate the photostability of these compounds, we attempted to determine their total quantum yield of photodecomposition (Φdec), which is defined as the sum of the quantum yields of all photoreactions producing products under light irradiation.37–39 The Φdec values estimated were considered to be the minimum possible values, as Φdec is underestimated when the decomposition products absorb light at the monitored wavelength.
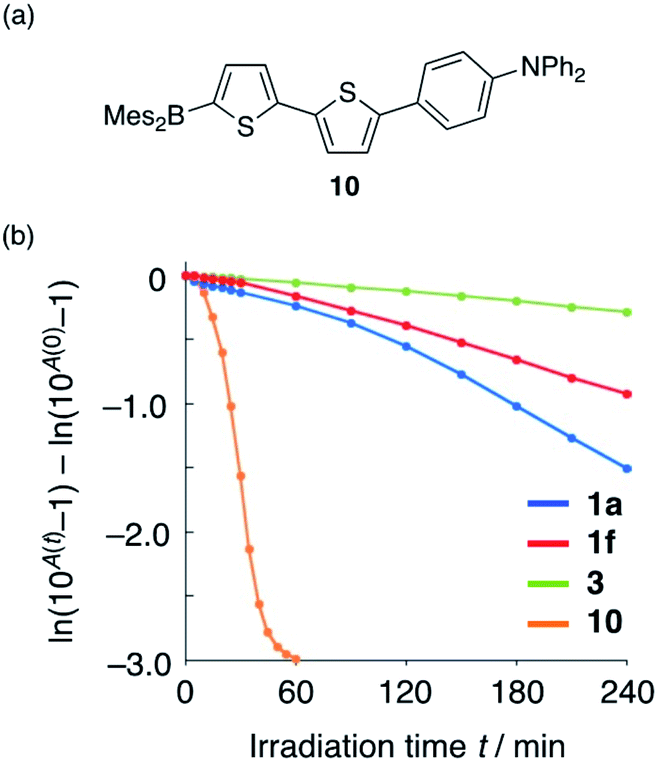 | ||
| Fig. 5 (a) Chemical structure of 10 and (b) monitoring the absorption decay upon exposure to irradiation from LED light (λem = 449 nm). | ||
The results of the irradiation experiments are shown in Fig. 5b. While the change in absorbance over time was almost linear for 3 and 1f, 1a and 10 showed non-linear behaviour, probably due to the influence of the absorption of the photo-decomposed product(s). Although the Φdec values for 1a and 10 cannot be determined quantitatively, given that a linear slope value is required for the calculation, a qualitative comparison demonstrates that 1a is substantially more photostable than 10, confirming that the introduction of the P(O)Ph group improves the photostability of the fluorophore. Furthermore, bulky aryl-substituted 1f showed improved photostability (Φdec = 5.8 × 10−6). This value approaches that of 3 (2.7 × 10−6) and is two orders of magnitude smaller than that of Alexa Fluor 488 (2.3 × 10−4 in DMSO/buffer = 7/3), which is widely used as a representative photostable dye in bioimaging. However, it should be noted here that these values cannot be compared directly as different solvents were used in the measurements. The improved photostability of 1f suggests that the steric congestion around the boron atom greatly enhances its photostability.
Application to bioimaging
The bright far-red to NIR emission of the D–A–A dyes 1, even in polar solvents, suggest promising potential for fluorescence imaging of biological samples. To examine the utility of these dyes in such applications, 1c was employed as a representative example. Prior to conducting the imaging experiments, we confirmed the solubility of 1c in phosphate-buffered saline (PBS; pH = 7.4) containing 2% bovine serum albumin (BSA), which is the most abundant protein in the blood plasma, by dynamic light scattering (DLS) measurements (Fig. S17†). The solution of 1c in the presence of BSA only showed a peak comparable to that of a solution of BSA without 1c, indicating that 1c was solved under these conditions. Moreover, the solution exhibited red fluorescence with a λem of 632 nm (Fig. S18†), which is slightly shorter than that in CHCl3. This fact implies that 1c is bound to the hydrophobic pocket of BSA.40With these results in hand, we tested 1c in two kinds of imaging. First, we used 1c for the in vivo imaging of blood vessels in Japanese medaka (Oryzias latipes) larvae one week after hatching. For that purpose, a solution of 1c in DMSO (<1 μL, 1 mM) was directly injected into the peritoneal cavity of the fish using a microinjection system. The fish was then placed into water with 0.3% salinity and cultured for 1 h. We conducted a whole-body imaging analysis of the larvae using a confocal microscopy system (λex = 488 nm; emission collection: 570–620 nm). Fig. 6a shows a 3D image of the fish that was reconstructed by combining five 1272.8 μm × 1272.8 μm × 918 μm images; as shown, the blood vessels are clearly visible in the living fish. This result suggests that 1c is rapidly absorbed into the bloodstream, where it most likely binds to proteins such as albumin.
 | ||
| Fig. 6 (a) Confocal image of the entire body of a Japanese medaka larvae and (b) two-photon excitation image of blood vessels in mouse brain. | ||
Then, we performed deep imaging of the blood vessels in mouse brain using two-photon excitation microscopy. Immediately after administering 100 μL of 1c (0.9 mM in PBS containing 18% DMSO and 1.6% BSA) to a mouse via intravenous injection, images were recorded through an open-skull window at a two-photon excitation wavelength of 880 nm (Fig. 6b). Notably, in addition to emission of the dye in the bloodstream in the red region (λem = 601–657 nm), some blood vessels also exhibited green fluorescence in the 500–550 nm region, which indicates that the microenvironment of the dye is considerably hydrophobic. Although the identity of the biological component that is stained with 1c to produce this green fluorescence has not yet been clarified, its observation suggests that the hydrophobic dye may have detached from the albumin and then adsorbed on the hydrophobic region of the blood vessel wall.
Conclusions
We have developed a series of diarylboryl and dithienophosphole P-oxide-containing D–A–A-type fluorophores, which exhibit far-red to NIR emissions. Thus, the tuning of the acceptor–spacer is a complementary useful strategy for gaining such red-shifted emissions with the conventional modification of the terminal acceptor boryl group in the D–π–A scaffolds.20 Unlike analogues that bear formyl or cyano groups in place of the boryl group, the boryl-substituted D–A–A-type fluorophores retain a high fluorescence quantum yield even in polar solvents such as CH3CN. An analysis of the excited-state dynamics revealed that the higher kr and the lower knr values of the developed dyes are responsible for the high quantum yields. TD-DFT calculations demonstrated that the higher kr values can be attributed to higher oscillator strengths for the electronic transition from S1 to S0. In terms of nonradiative decay, the internal conversion from S1 to S0 is likely a major pathway, in which the boryl group contributes to decreasing the non-adiabatic coupling associated with the quinoidal stretching mode of the dithienophosphole oxide moiety. This result should provide an important insight into the effect of the boryl groups in the widely studied boron-based D–π–A fluorophores. The introduction of the P(O)Ph group also greatly influences the photostability of the dyes; the presence of bulky substituents on the boron atom improves the photostability. Moreover, one of the synthesized dyes, 1c, was successfully applied for whole-body imaging of blood vessels and two-photon bioimaging.
Author contributions
S. Y. and Y. S. conceived the idea. Y. S. synthesized all the compounds and evaluated their properties. N. I. and T. Y. conducted the theoretical calculations. M. T., R. K., T. S., and T. I. conducted imaging experiments. S. Y. and T. B. discussed the π-electron systems studied. Y. S., N. I., M. T., T. Y., and S. Y. wrote the manuscript, and all authors discussed and commented on the manuscript. S. Y. directed the project.Ethical statement
All animal experiments were approved by the Ethics Committee for Animal Experiments of Ehime University (#05-RE-4-16). The experimental procedures we employed were conducted in accordance with the approved guidelines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MEXT/JSPS KAKENHI Grant Numbers JP18H03909 and JP18H05261 (to S. Y.), JP16H06280 (ABiS), JP15H05952 (to T. I. and T. S.), JP19K12218 and JP20H05038 (to T. S.) and by AMED Grant Number JP20gm1210001 (to T. I.). This work was also funded by Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering, and Kato Memorial Bioscience Foundation (to T. S.). ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan. T. B. thanks the Canada Research Chairs program for support.Notes and references
- (a) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482–2506 CrossRef CAS PubMed; (b) F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- (a) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed; (b) S. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994–2006 RSC.
- (a) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC; (b) J.-M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 RSC; (c) L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Org. Biomol. Chem., 2020, 18, 5747–7563 RSC.
- (a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (c) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS.
- (a) F. Jäkle, Chem. Rev., 2010, 110(7), 3985–4022 CrossRef PubMed; (b) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- (a) Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef; (b) D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS; (c) H. Ye, D. H. Kim, X. Chen, A. S. D. Sandanayaka, J. U. Kim, E. Zaborova, G. Canard, Y. Tsuchiya, E. Y. Choi, J. W. Wu, F. Fages, J.-L. Brédas, A. D'Aléo, J.-C. Ribierre and C. Adachi, Chem. Mater., 2018, 30, 6702–6710 CrossRef CAS; (d) D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef CAS PubMed; (e) J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242 CrossRef PubMed; (f) Q. Liang, J. Xu, J. Xue and J. Qiao, Chem. Commun., 2020, 56, 8988–8991 RSC.
- T. Yamanaka, H. Nakanotani, S. Hara, T. Hirohata and C. Adachi, Appl. Phys. Express, 2017, 10, 074101 CrossRef.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, 1900321 CrossRef PubMed.
- (a) Q. Yang, Z. Ma, H. Wang, B. Zhou, S. Zhu, Y. Zhong, J. Wang, H. Wan, A. Antaris, R. Ma, X. Zhang, J. Yang, X. Zhang, H. Sun, W. Liu, Y. Liang and H. Dai, Adv. Mater., 2017, 29, 1605497 CrossRef PubMed; (b) Q. Yang, Z. Hu, S. Zhu, R. Ma, H. Ma, Z. Ma, H. Wan, T. Zhu, Z. Jiang, W. Liu, L. Jiao, H. Sun, Y. Liang and H. Dai, J. Am. Chem. Soc., 2018, 140, 1715–1724 CrossRef CAS PubMed.
- (a) J. C. Doty, B. Babb, P. J. Grisdale, M. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1972, 38, 229–236 CrossRef CAS; (b) S. M. Berger, M. Ferger and T. B. Marder, Chem.–Eur. J., 2021 DOI:10.1002/chem.202005302.
- (a) Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, J. Chem. Soc., Chem. Commun., 1990, 1489–1492 RSC; (b) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem., 2000, 154, 5–12 CrossRef CAS; (c) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- (a) M. Lequan, R. M. Lequan and K. C. Ching, J. Mater. Chem., 1991, 1, 997–999 RSC; (b) M. Lequan, R. M. Lequan, K. C. Ching, M. Barzoukas, A. Fort, H. Lahoucine, G. Bravic, D. Chasseau and J. Gaultier, J. Mater. Chem., 1992, 2, 719–725 RSC.
- (a) Z.-q. Liu, Q. Fang, D. Wang, G. Xue, W.-t. Yu, Z.-s. Shao and M.-h. Jiang, Chem. Commun., 2002, 2900–2901 RSC; (b) Z. Liu, Q. Fang, D. Wang, D. Cao, G. Xue, W. Yu and H. Lei, Chem.–Eur. J., 2003, 9, 5074–5084 CrossRef CAS PubMed; (c) J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed.
- (a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed; (b) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed; (c) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- (a) S. Griesbeck, E. Michail, F. Rauch, H. Ogasawara, C. Wang, Y. Sato, R. M. Edkins, Z. Zhang, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 13164–13175 CrossRef CAS PubMed; (b) S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem. Sci., 2019, 10, 5405–5422 RSC; (c) S. Griesbeck, M. Ferger, C. Czernetzi, C. Wang, R. Bertermann, A. Friedrich, M. Haehnel, D. Sieh, M. Taki, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 7679–7688 CrossRef CAS PubMed; (d) S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed; (e) J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- B. Wang, H. Pan, J. Jia, Y.-Q. Ge, W.-Q. Cai, J.-W. Wang and C.-H. Zhao, Tetrahedron, 2014, 70, 5488–5493 CrossRef CAS.
- P. Gautam, R. Maragani and R. Misra, RSC Adv., 2015, 5, 18288–18294 RSC.
- S. Yamaguchi and K. Tamao, Bull. Chem. Soc. Jpn., 1996, 69, 2327–2334 CrossRef CAS.
- (a) T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed; (b) M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1–44 CrossRef CAS; (c) M. Hissler, C. Lescop and R. Réau, Pure Appl. Chem., 2007, 79, 201–212 CAS; (d) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4691–4727 CrossRef PubMed.
- (a) A. Fukazawa, S. Suda, M. Taki, E. Yamaguchi, M. Grzybowski, Y. Sato, T. Higashiyama and S. Yamaguchi, Chem. Commun., 2016, 52, 1120–1123 RSC; (b) A. Fukazawa, J. Usuba, R. A. Adler and S. Yamaguchi, Chem. Commun., 2017, 53, 8565–8568 RSC.
- T. Baumgartner, T. Neumann and B. Wirges, Angew. Chem., Int. Ed., 2004, 43, 6197–6201 CrossRef CAS PubMed.
- C. Romero-Nieto and T. Baumgartner, Synlett, 2013, 24, 920–937 CrossRef CAS.
- (a) Y. Dienes, S. Durben, T. Kárpáti, T. Neumann, U. Englert, L. Nyulászi and T. Baumgartner, Chem.–Eur. J., 2007, 13, 7487–7500 CrossRef CAS PubMed; (b) C. Romero-Nieto, K. Kamada, D. T. Cramb, S. Merino, J. Rodriguez-Lopez and T. Baumgartner, Eur. J. Org. Chem., 2010, 5225–5231 CrossRef CAS.
- H. V. Huynh, X. He and T. Baumgartner, Chem. Commun., 2013, 49, 4899–4901 RSC.
- Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Würthner, C. M. Marian, L. Ji and T. B. Marder, Angew. Chem., Int. Ed., 2020, 59, 17137–17144 CrossRef CAS PubMed.
- Y. Niu, W. Li, Q. Peng, H. Geng, Y. Yi, L. Wang, G. Nan, D. Wang and Z. Shuai, Mol. Phys., 2018, 116, 1078–1090 CrossRef CAS.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 6121–6133 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16, Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- (a) C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374–10381 CrossRef CAS PubMed; (b) M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS PubMed; (c) C. Wang, M. Taki, Y. Sato, Y. Tamura, H. Yaginuma, Y. Okada and S. Yamaguchi, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15817–15822 CrossRef CAS PubMed.
- M. Ito, E. Ito, M. Hirai and S. Yamaguchi, J. Org. Chem., 2018, 83, 8449–8456 CrossRef CAS PubMed.
- M. Hanazawa, R. Sumiya, Y. Horikawa and M. Irie, J. Chem. Soc., Chem. Commun., 1992, 206–207 RSC.
- T. Sumi, Y. Takagi, A. Yagi, M. Morimoto and M. Irie, Chem. Commun., 2014, 50, 3928–3930 RSC.
- H. Sotome, T. Nagasaka, K. Une, C. Okui, Y. Ishibashi, K. Kamada, S. Kobatake, M. Irie and H. Miyasaka, J. Phys. Chem. Lett., 2017, 8, 3272–3276 CrossRef CAS PubMed.
- (a) Ž. Ban, S. Griesbeck, S. Tomić, J. Nitsch, T. B. Marder and I. Piantanida, Chem.–Eur. J., 2020, 26, 2195–2203 CrossRef PubMed; (b) H. Amini, Ž. Ban, M. Ferger, S. Lorenzen, F. Rauch, A. Friedrich, I. Crnolatac, A. Kenđel, S. Miljanić, I. Piantanida and T. B. Marder, Chem.–Eur. J., 2020, 26, 6017–6028 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, photophysical properties, evaluation of the photostability, theoretical calculations, in vivo imaging, and NMR spectra for all new compounds. See DOI: 10.1039/d1sc00827g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |