
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of polycyclic structures with vicinal all-carbon quaternary stereocenters via an enantioselective photoenolization/Diels–Alder reaction†
Min
Hou‡
a,
Mengmeng
Xu‡
a,
Baochao
Yang
a,
Haibing
He
b and
Shuanhu
Gao
 *ab
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering East China Normal University 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: shgao@chem.ecnu.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University 3663 North Zhongshan Road, Shanghai 200062, China
First published on 27th April 2021
Abstract
All-carbon quaternary stereocenters are ubiquitous in natural products and significant in drug molecules. However, construction of all-carbon stereocenters is a challenging project due to their congested chemical environment. And, when vicinal all-carbon quaternary stereocenters are present in one molecule, they will dramatically increase its synthetic challenge. A chiral titanium promoted enantioselective photoenolization/Diels–Alder (PEDA) reaction allows largely stereohindered tetra-substituted dienophiles to interact with highly active photoenolized hydroxy-o-quinodimethanes, delivering fused or spiro polycyclic rings bearing vicinal all-carbon quaternary centers in excellent enantiomeric excess through one-step operation. This newly developed enantioselective PEDA reaction will inspire other advances in asymmetric excited-state reactions, and could be used in the total synthesis of structurally related complex natural products or drug-like molecules for drug discovery.
Introduction
Chemical synthesis of complex molecules with chiral stereogenic centers and three-dimensional skeletons has been a significant topic in organic synthesis. Tuning the reactivities and controlling the stereoselectivities are central to bond-forming reactions. For small-molecule drug discovery, scientists from both academia and the pharmaceutical industry are pursuing the innovation of chemical structures from traditional achiral molecules to privileged three-dimensional structures. The great potential of lead compounds bearing sp3-hybridized stereogenic centers relies on (1) more structural diversity; (2) suitable pharmacophores to selectively target the desired proteins; and (3) easily tunable physical properties to meet the in vivo metabolic stability. Therefore, they will significantly expand the chemical space of drug-like molecules and facilitate pharmaceutical discovery.All-carbon quaternary stereocenters exist in many natural products (Fig. 1A). Such stereocenters are frequently located at the bridgeheads of fused rings, such as the highlighted structures (blue) in daphniglaucin C. Spiro quaternary carbon stereocenters are also found in polycyclic structures, such as the red scaffolds in scopadulcic acid A. Due to their congested chemical environment, construction of all-carbon stereogenic centers is a challenging project.1 And, when vicinal all-carbon quaternary stereocenters are present in one molecule, they will dramatically increase its synthetic challenge. Multiple chemical transformations are normally required to build this structural motif. A few methodologies have been disclosed to stereoselectively construct vicinal quaternary centers in a single-step transformation to date.2 This mainly resulted from the low reactivities of the substrate because of the stereogenic effects, and harsh reaction conditions are needed to overcome the energy barrier separating the reactants and products. And, it's even harder to control the diastereo- and enantioselectivity during this process.
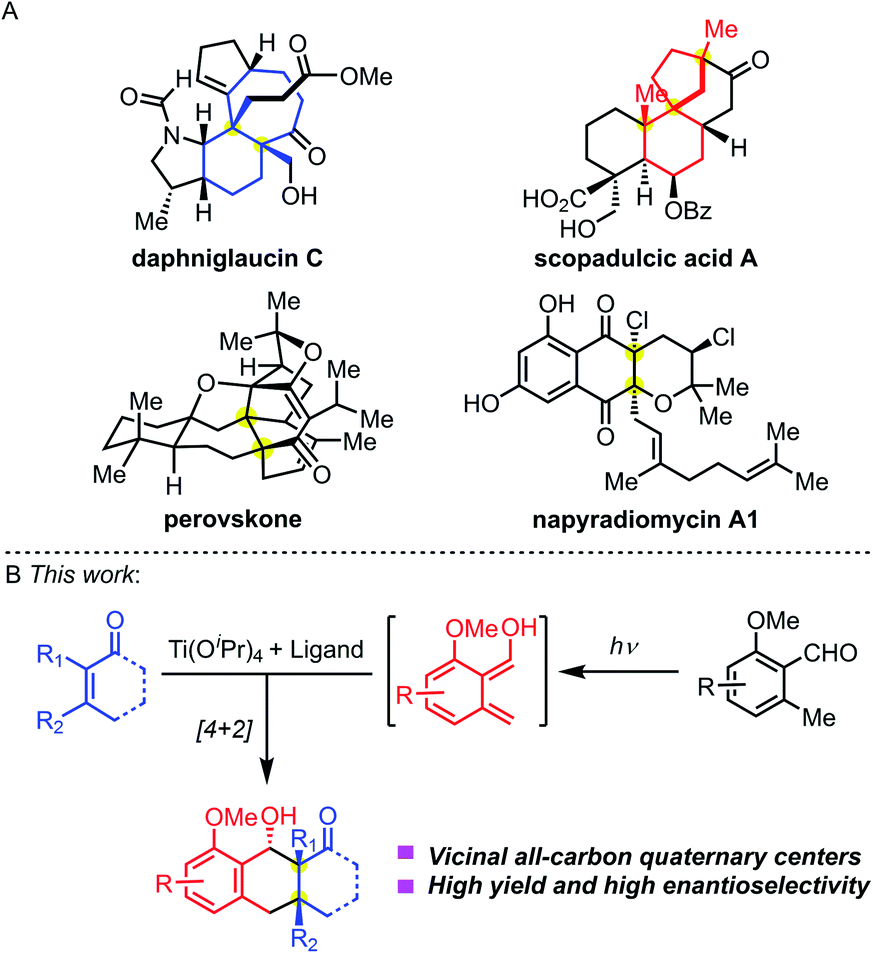 | ||
| Fig. 1 (A) Bioactive nature products bearing vicinal quaternary stereocenters. (B) Enantioselective PEDA reaction. | ||
The photoenolization/Diels–Alder (PEDA) reaction was discovered by Yang and coworkers in 1961,3 which relies on the Norrish type I/II photo reaction.4 Through UV light-induced enolization of 2-alkyl benzaldehyde, the reaction generates a highly active diene hydroxy-o-quinodimethane via an excited-state species, which can be trapped by a dienophile through Diels–Alder cycloaddition to form an adduct. The PEDA reaction has emerged as an effective, atom-economical method to construct highly functionalized polycyclic molecules in organic synthesis,5 materials chemistry,6 and biology,7 providing new ideas for retrosynthetic disconnection. Historically, Nicolaou and co-workers studied the asymmetric PEDA reaction using the Narasaka–Mikami catalyst [(R)-BINOL]TiCl2 which didn't give satisfactory results.5b In 2003, Bach and co-workers achieved an asymmetric PEDA reaction by using a unique hydrogen-binding chiral agent which controlled the enantioselectivity of the photo-cycloaddition process.8 Recently, Melchiorre and co-workers creatively used a cinchona–thiourea catalyst to achieve an enantioselective PEDA reaction of highly reactive maleimides.9 However, the asymmetric PEDA reaction involving sterically hindered multi-substituted olefins to construct vicinal all-carbon quaternary stereocenters has been elusive.
In contrast to the wealth of asymmetric catalytic systems of ground-state reactions, few effective methods exist for excited-state reactions.10 As for the PEDA reaction, we envisioned that two challenges need to be overcome: (1) maintaining the reactivity of the diene and dienophile during cycloaddition in the chiral environment and (2) suppressing the background and side reactions. We aimed to meet both challenges by combining the Lewis acid Ti(Oi-Pr)4 with a chiral ligand (Fig. 1B).11 We reasoned that the chiral Ti-complex would chelate both the diene and dienophile, temporarily converting an intermolecular PEDA reaction into an intramolecular one. We envisioned that this investigation would not only provide solutions to achieve the synthesis of these natural products containing an anthracenol core bearing vicinal quaternary centers (Fig. 1A), but also offer opportunities to explore the fundamentals of the enantioselectivity of UV-light mediated cycloadditions.
Results and discussion
Our initial screening of reaction conditions showed this PEDA reaction to be sensitive to different titanium(IV) species, and even to an equivalent of Ti(Oi-Pr)4.11 Photolysis using Ti(Oi-Pr)4 dosages >2.0 equivalents gave stable and comparable yield; therefore, we initially conducted our experiment using 3.0 equivalents of Ti(Oi-Pr)4 with 1.0 equivalent of a chiral ligand, employing the electron-rich 3,6-dimethoxy-2-methylbenzaldehyde 1 and electron-deficient 1-cyclohexenyl-ethanone 2 as model substrates. Four types of commonly used bidentate chiral ligands (Table S1 in the ESI†) were examined and it was found that chiral TADDOL L1 gave a more promising result, generating the tricyclic product 3 in 70% yield with 67% ee (Table 1A). Then we tested the reaction with variants of TADDOL L2–12 and observed that the stereochemistry of the ketal motif strongly affected the chirality of the chelated Ti–TADDOL complex. Replacing the ligand from the methyl group (L1) to an iso-propyl one (L3) improved the ee from 67% to 75%. Meta-substituted groups on the aromatic rings also strongly influenced the enantioselectivity of the photoreaction: ligands L9–L11, respectively, containing bis-(trifluoromethyl), di-tert-butyl or dimethoxy groups at the meta position gave products with ee values from 82% to 89%. Ligands L9A–D bearing a spiro skeleton in the backbone gave 3 in a similar yield but a lower ee than L10.
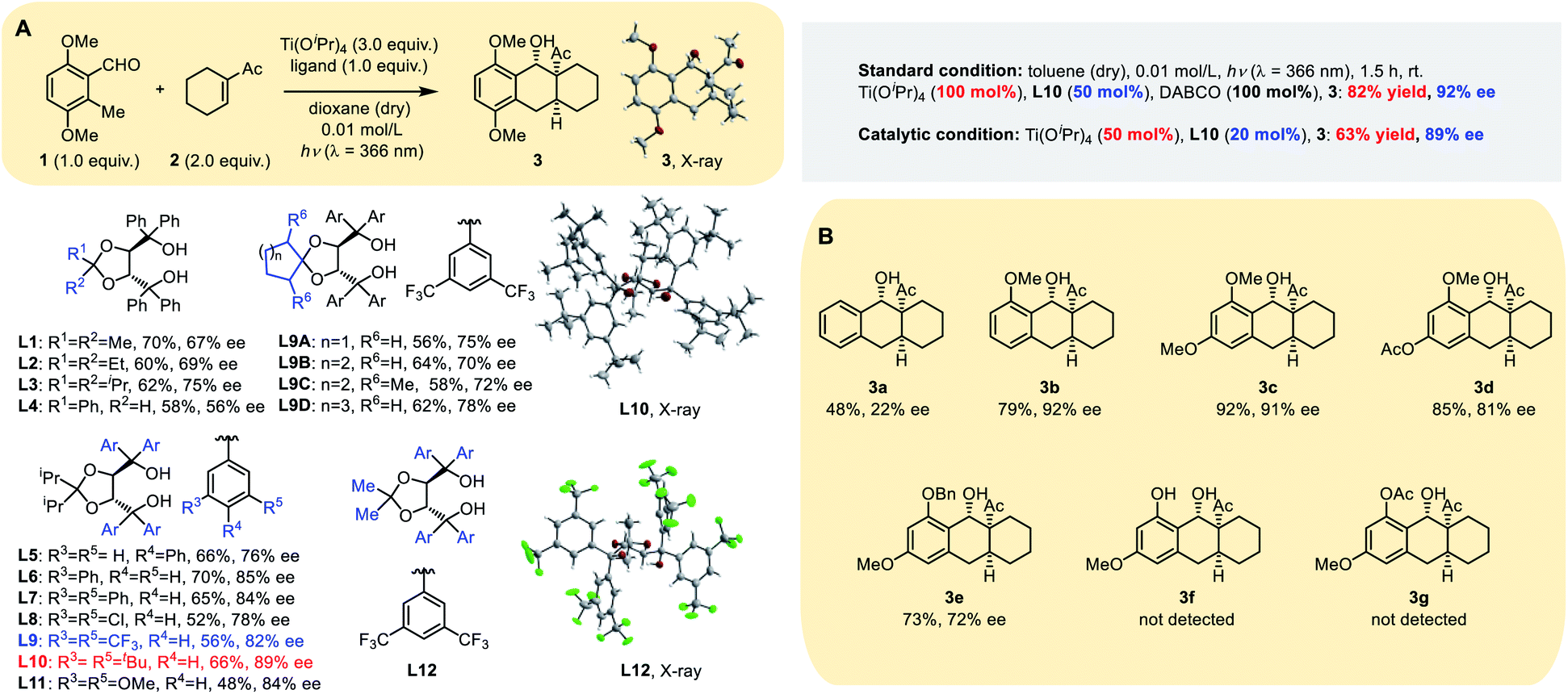
|
Extensive screening showed that most aprotic solvents worked, including tetrahydrofuran, dichloromethane, 1,2-dichloroethane and toluene. Anhydrous toluene gave the best photocycloaddition yield (70%) and ee (91%). To our delight, reducing the amount of the dienophile (1.5 equiv.) and using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Ti(Oi-Pr)4 (100 mol%) to chiral ligand L10 (50 mol%) gave a comparable ee (90%) and better yield (78%) (Table S2, ESI†). In order to realize the catalytic asymmetric PEDA reaction, we explored three strategies: (1) using the pre-prepared catalyst or additives; (2) changing the wavelength of the excitation light source (for the absorption spectra of the substrate see Fig. S1 in the ESI†); (3) changing the ratio of titanium to ligand (Table S5, ESI†). Finally, we successfully obtained catalytic asymmetric reaction conditions (50 mol% Ti(Oi-Pr)4 with 20 mol% equivalent L10), which gave a good yield and ee value when using the template substrate. However, we found that these conditions were not applicable to some other substrates during the reaction scope investigation. We speculated that this might be due to the reactivity disparity of different substrates. When the circulation rate of the catalyst does not match the cycloaddition rate, it might increase the rate of the racemic background reaction and side reactions, such as the oxa-D-A reaction, resulting in a lower ee value and yield.
:1 ratio of Ti(Oi-Pr)4 (100 mol%) to chiral ligand L10 (50 mol%) gave a comparable ee (90%) and better yield (78%) (Table S2, ESI†). In order to realize the catalytic asymmetric PEDA reaction, we explored three strategies: (1) using the pre-prepared catalyst or additives; (2) changing the wavelength of the excitation light source (for the absorption spectra of the substrate see Fig. S1 in the ESI†); (3) changing the ratio of titanium to ligand (Table S5, ESI†). Finally, we successfully obtained catalytic asymmetric reaction conditions (50 mol% Ti(Oi-Pr)4 with 20 mol% equivalent L10), which gave a good yield and ee value when using the template substrate. However, we found that these conditions were not applicable to some other substrates during the reaction scope investigation. We speculated that this might be due to the reactivity disparity of different substrates. When the circulation rate of the catalyst does not match the cycloaddition rate, it might increase the rate of the racemic background reaction and side reactions, such as the oxa-D-A reaction, resulting in a lower ee value and yield.
Inspired by the pioneering discoveries of Melchiorre and MacMillan groups that tertiary amine quinuclidine and 1,4-diazabi-cyclo[2.2.2]octane (DABCO) could act as an electrontransfer agent or base in photoreactions,9,12 we intended to examine whether the background racemic reaction would further decrease by adding an organo-base. We tested a variety of amines, including triethylamine (Et3N), imidazole (IMD), hexamethylenetetramine (HMTA), DABCO and quinuclidine (Quin) (Tables S3 and S4, ESI†). All five amines led to similar enantioselectivity, but the constrained tertiary amines HMTA, DABCO and Quin gave better yields. Dynamics studies showed that DABCO and Quin inhibited the background reaction more than other tertiary amines and the additive can increase the yield and ee value in small but steady increments. Moreover, dynamics studies also demonstrated that the addition of a chiral ligand could evidently accelerate the reaction (Fig. S2, ESI†) with nearly a double increase in the yield from 46% to 82%. The experimental results indicated that ligand acceleration plays a key role in suppressing background reactions. Ligand acceleration is often observed in titanium catalyzed systems13,14 and could effectively suppress the racemic background reaction, which has been attributed to the fast chelation rate for substrate binding and the fast dissociation rate for product release, as a result of steric hindrance.14
In order to investigate the function of the methoxy group at the ortho position of the aldehyde group, we carried out the following substrate studies (Table 1B). When the methoxy group was absent, the reaction yield and ee value were extremely low (3a and 3f); when –OMe was replaced by a benzyloxy group, the reaction yield was comparable but the ee value was significantly reduced (3e). We speculate that the ortho-methoxy group of the aldehyde group plays a key role by coordinating with the catalyst. In addition, when the aromatic ring contained an electron withdrawing group, it also influenced the reactivity and enantioselectivity of the reaction (3d and 3g).
We then investigated the scope of the reaction with electron rich benzaldehydes (1, 1a, 4–6) and cyclic tetrasubstituted olefins (7–10) as sterically hindered dienophiles using the standard reaction conditions (Table 2). Adding L10 improved substrate reactivity greatly and accelerated the reaction. For example, photolysis of 1 with 2,3-dimethylcyclohexenone in the presence of Ti(Oi-Pr)4 gave the desired product 11 in 38% yield. Adding the chiral ligand L10 led to 7 as a single diastereomer in 77% yield with 91% ee. Under the optimized conditions, we found that this photocycloaddition occurred smoothly to form fused tricyclic products containing vicinal all-carbon quaternary stereocenters at the bridgeheads, and the chiral ligand can be easily recycled (>95% yield). Alkyl substituents on cyclic enones (methyl, n-propyl, n-heptyl and benzyl groups) provided the corresponding adducts 11–17 in good yields with excellent diastereo- and enantioselectivities of 85–99% ee. For product 18, the reaction was dominated by the endo selectivity based on the NOE spectrum, and the dr of the reaction was 2.3:1. However, when we used 3.0 equivalents of titanium as the Lewis acid and no ligand was added for the racemic reaction, it was found that the reaction mainly underwent the exo-cycloaddition, and the dr was 1.8:1 (Table S6, ESI†). This result shows that the chiral ligands can not only control the enantioselectivity of the reaction, but also influence the diastereoselectivity. For most of the substrates, adding ligands could improve the yield and diastereoselectivity of the reaction. The reaction also tolerated a change in ring size from cyclohexenone to cyclopentenone, affording 19–28 with comparable reaction yields and enantioselectivities of 89–98% ee. The absolute configuration of 22 was determined by X-ray crystallographic analysis, and the configuration of the other adducts was assigned by analogy.
| a Ligand L10 (0.8 equiv.) was used. |
|---|
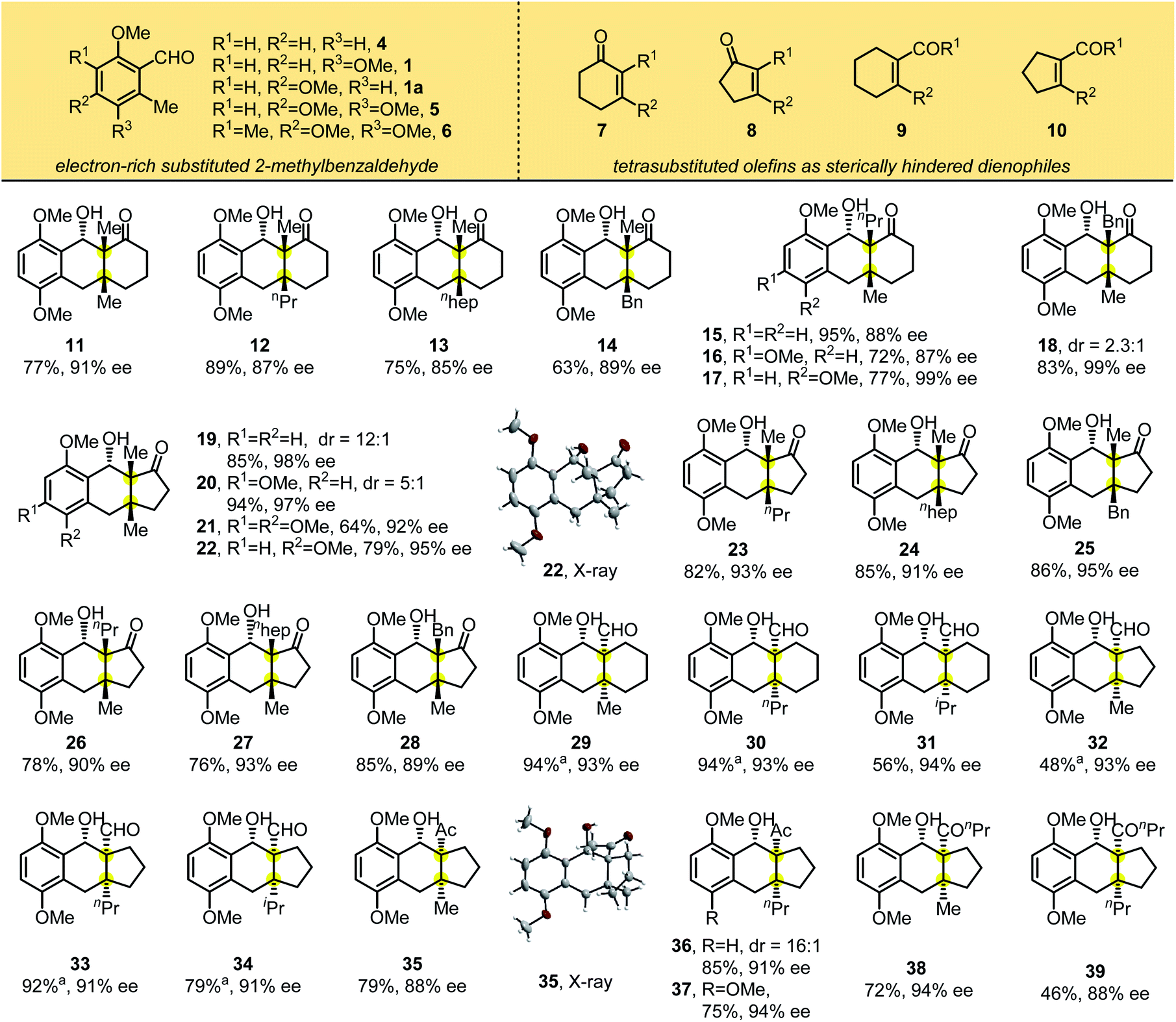
|
We further examined substrates bearing electron-withdrawing exocyclic groups such as aldehydes and ketones. In the presence of L10, the substrate 3,6-dimethoxy-2-methylbenzaldehyde 1 reacted smoothly with cyclohexenones and cyclopentenones substituted with aldehydes (to yield products 29–34) or ketones (to yield 35–39). These tricyclic adducts were generated in good to excellent yields with excellent diastereo- and enantioselectivities. X-ray crystallographic analysis of 35 showed that photolytic products 29–39 had the same absolute configuration.
We then designed tetrasubstituted olefins 40–44, bearing exo-unsaturated aldehyde and carbonyl groups as dienophiles, which were used to forge the tricyclic adducts with spiro all-carbon quaternary stereocenters (Table 3). These substrates were well tolerated, giving the desired products 46–52 with unique spiro-skeletons ranging from cyclobutane to cyclohexane and even pyran rings in synthetically useful yields with good enantioselectivities. The acyclic tetrasubstituted olefin 44 was also tested in the PEDA reaction, and the corresponding bicyclic adducts 53 and 54 were achieved in good enantioselectivities and moderate yield, which might be caused by its flexible structure. When we performed the reaction with beta-cyclocitral (45), which exerts unusual steric effects because it bears gemi-dimethyl groups adjacent to a tetrasubstituted unsaturated aldehyde, we were surprised to obtain the corresponding tricyclic adducts 55–59 bearing three vicinal all-carbon quaternary centers in good yields with excellent diastereo- and enantioselectivities. The yields of 55–57 were even better than those of products with two vicinal all-carbon quaternary centers. Our ability to generate the photoadduct 59, bearing a fully substituted aromatic ring and an icetexane-based diterpene skeleton, lays the foundation for the total synthesis of these natural products.15
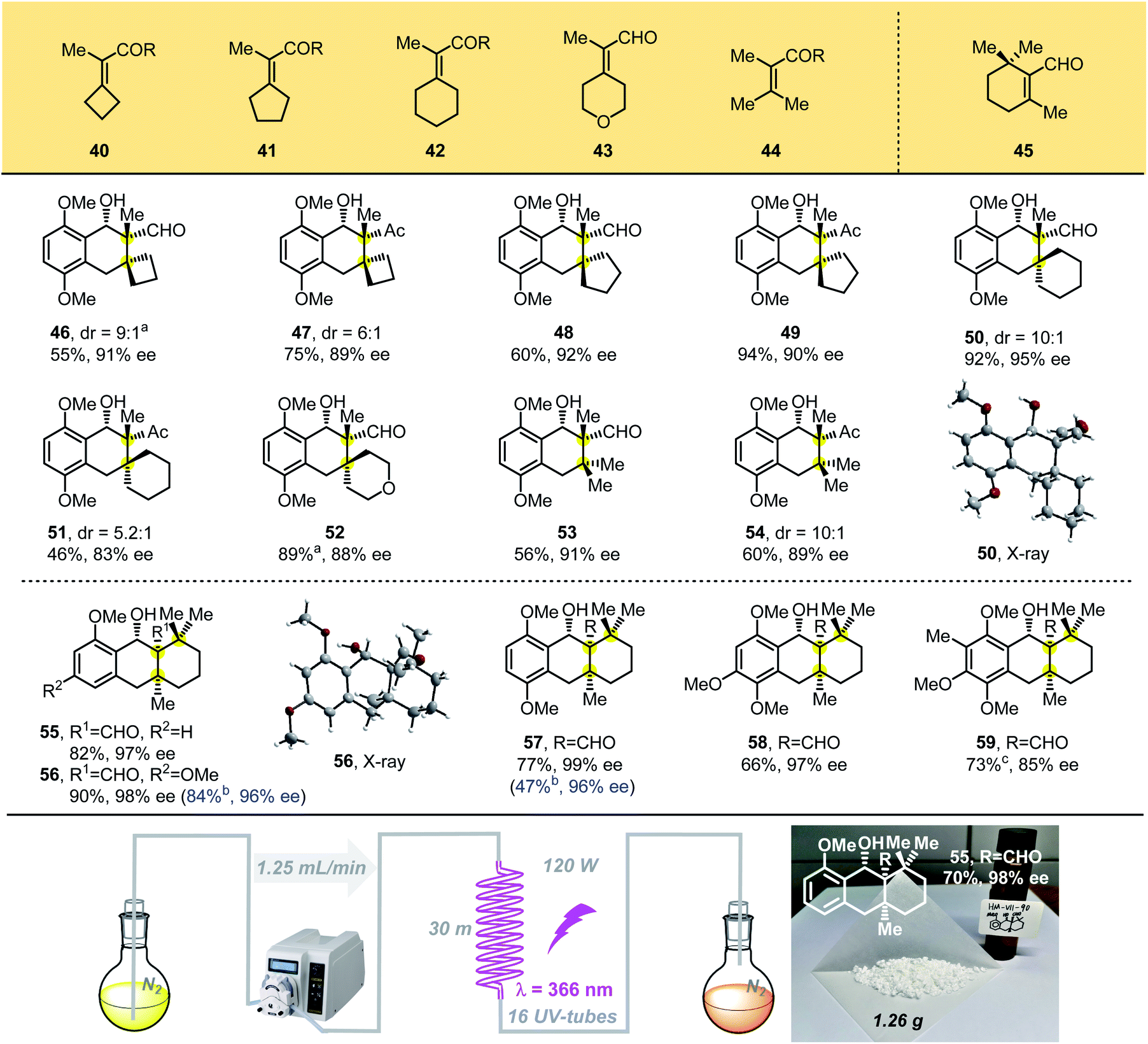
To scale-up this enantioselective PEDA reaction, a continuous-flow reactor was assembled using crocheted fluorinated ethylene propylene (FEP) tubing placed inside a Rayonet chamber.16 With this system, the enantioselective PEDA reaction could be performed on a large scale easily, which afforded 1.26 g of cycloadduct 55 (70% yield, 98% ee) under a flow rate of 1.25 mL min−1. This technology proves to be more efficient than the conventional batch photochemistry and extends the utility of the PEDA reaction.
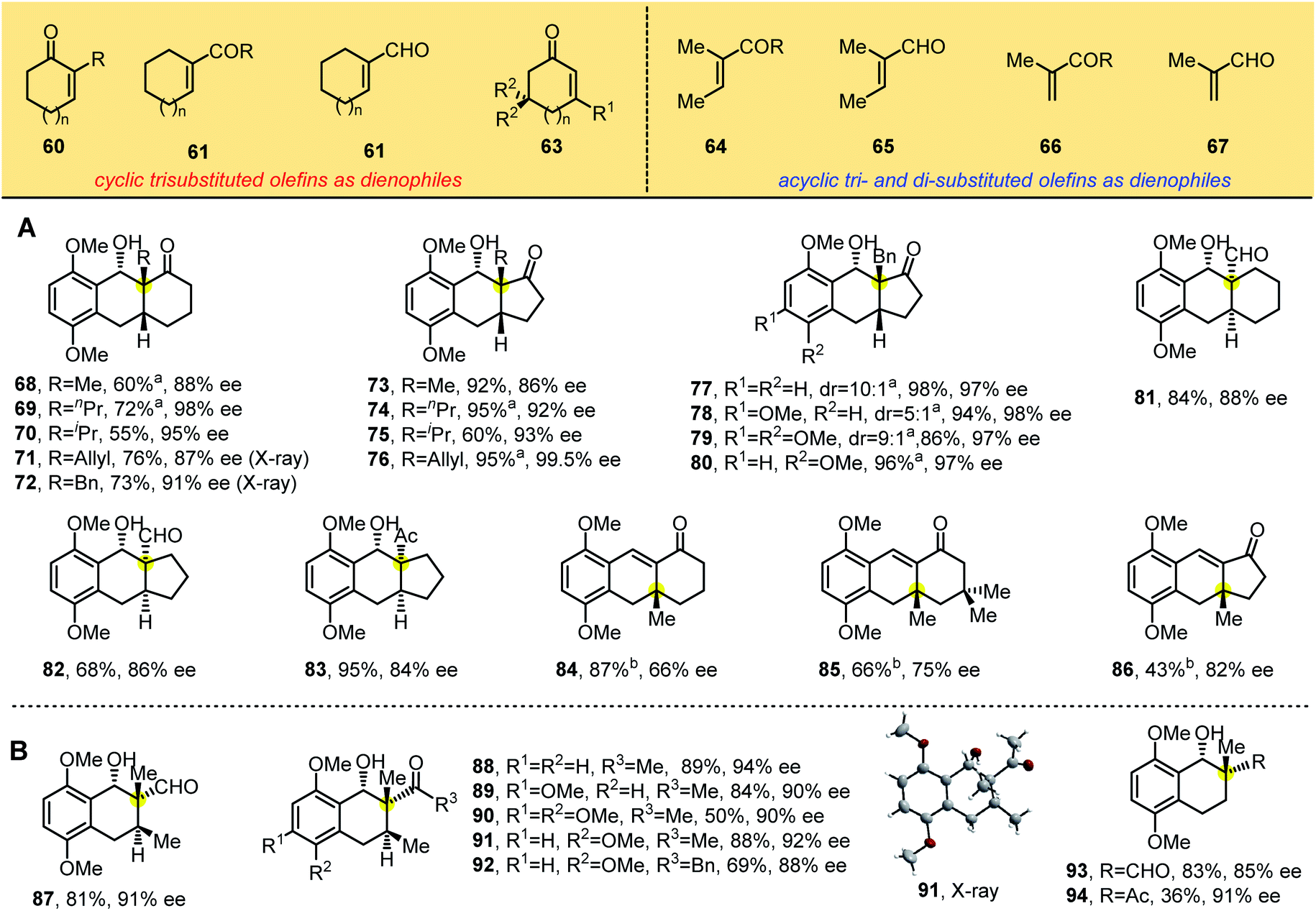
To build the chiral tricyclic anthracenol containing one all-carbon quaternary stereocenter, we explored trisubstituted olefins (60–67) as dienophiles in the reaction (Table 4), starting with 1,1-disubstituted cyclic dienophiles bearing electron-withdrawing groups such as ketones and aldehydes, which we reacted with substituted 2-methylbenzaldehydes 1, 1a and 4–5. We found that the PEDA reaction occurred smoothly and produced the desired products 68–83 in good yields and enantioselectivities ranging from 84% to 99%. X-ray crystallographic analyses of 71 and 72 showed the same structure (containing one all-carbon quaternary center) as photoadducts in Tables 2 and 3, in which the hydroxyl group at the benzylic position is cis to the electron-withdrawing groups. These results suggest that alkyl substitutions adjacent to the electron-withdrawing group in dienophiles promote enantioselectivity. Consistent with this idea, 3-methyl cyclohexenone and cyclopentenone generated enones 84–86 with 66–82% ee through photocycloaddition and benzylic hydroxyl group elimination. These lower enantioselectivities indicate that the position of the substitution strongly influences stereoselectivity: when the ortho substituent of the carbonyl group is a hydrogen atom, the smaller steric hindrance is unfavorable to control enantioselectivity. When we switched the dienophiles to flexible di- and trisubstituted acyclic olefins (Table 4B), we found that α,β-unsaturated enones and aldehydes gave products 87–94 as single diastereomers in synthetically useful yields with 85–94% ee. X-ray crystallographic analysis of 91 revealed the absolute configuration of these bicyclic products.
Mechanistic studies
To gain insights into the transition state and mechanism of this enantioselective PEDA reaction, we analyzed nonlinear effects (Fig. S3, ESI†). Complexes of titanium with bidentate ligands such as TADDOL are thought to exist as oligomers because of the strong Ti–O bond and high coordination number.17 We envisioned that the Ti–TADDOLate complex in our enantioselective photocycloaddition might exist as a dimeric or dinuclear species, based on our observation that a 2:1 ratio of Ti(Oi-Pr)4 to the chiral ligand gave the highest enantioselectivity. Using a 1:1 ratio of L10 to Ti(Oi-Pr)4 (1.0 equiv. of each) showed a slight but real negative non-linear effects. Upon expanding the ratio of Ti(Oi-Pr)4 to L10 to 6:1 (3.0:0.5 equiv.) the nonlinear effects were eliminated. This nonlinear behavior is similar to that of Ti–BINOLate systems reported by Walsh.18 These results suggest that the (TADDOLate)Ti(Oi-Pr)2 complex preferentially associates with excess Ti(Oi-Pr)4 to form a dinuclear species containing a single TADDOL ligand instead of the oligomeric [(TADDOLate)Ti(Oi-Pr)2]2 complex. According to this model, the presence of a sufficient amount of the chiral ligand may generate some oligomeric [(TADDOLate)Ti(Oi-Pr)2]n complexes, which would explain the observed nonlinear behavior. Analysis of the nonlinear effects with spiro-cycloadduct 46 also supports this model (Fig. S4, ESI†).
Given these results, we propose the mechanism and reaction pathway of the asymmetric PEDA reaction in Scheme 1. UV-irradiation of 3,6-dimethoxy-2-methylbenzaldehyde 1 generates triplet state-1 and triplet state-2via intersystem crossing under excited-state conditions (excitation from a singlet to a triplet state). The oxygen radical induces an intramolecular hydrogen shift from the benzylic methyl group, forming 1,4-biradical-3. Tautomerization produces two transient hydroxy-o-quinodimethane species, Z- and E-dienol. The structural features of these intermediates and their chemical environment strongly influence the equilibrium in this photo-reversible reaction.19 Triplet state-1 and E-dienol quickly return to the ground-state benzaldehyde 1. Since the triplet intermediate has a short life,20 we speculate that DABCO may serve as a proton shuttle to return the dienols to the starting material.12 Meanwhile, the ligand acceleration effect can inhibit the racemic background reaction. This makes the reaction conducive to achieving a higher ee value. Then the Z-dienol is chelated by the cage-like chiral dinuclear species Ti–TADDOLate-I to form a relatively stable complex TS-1.17,21 The ortho methoxy may serve as a key neighboring group that helps to stabilize the short-lived photoenolized hydroxy-o-quinodimethane diene.11 Due to its strong Lewis acidity, the Ti-complex TS-1 preferentially associates with the electron-withdrawing groups in the dienophile, such as enones 2, forming the transient TS-2, which in turn facilitates a Diels–Alder reaction. The chiral cage-like environment accelerates and controls the diastereo- and enantioselectivity of the cycloaddition. The [4 + 2] reaction occurs from the endo direction, viaTS-2, to give the cycloadduct 3. In this figure, ligand acceleration is crucial to suppress the racemic background reaction, while the chiral dinuclear species Ti–TADDOLate-I serves as a bifunctional catalyst by chelating both the diene and dienophile. This not only stabilizes and maintains the reactivity of the photoenolized hydroxy-o-quinodimethane species, but also activates weakly reactive dienophiles.
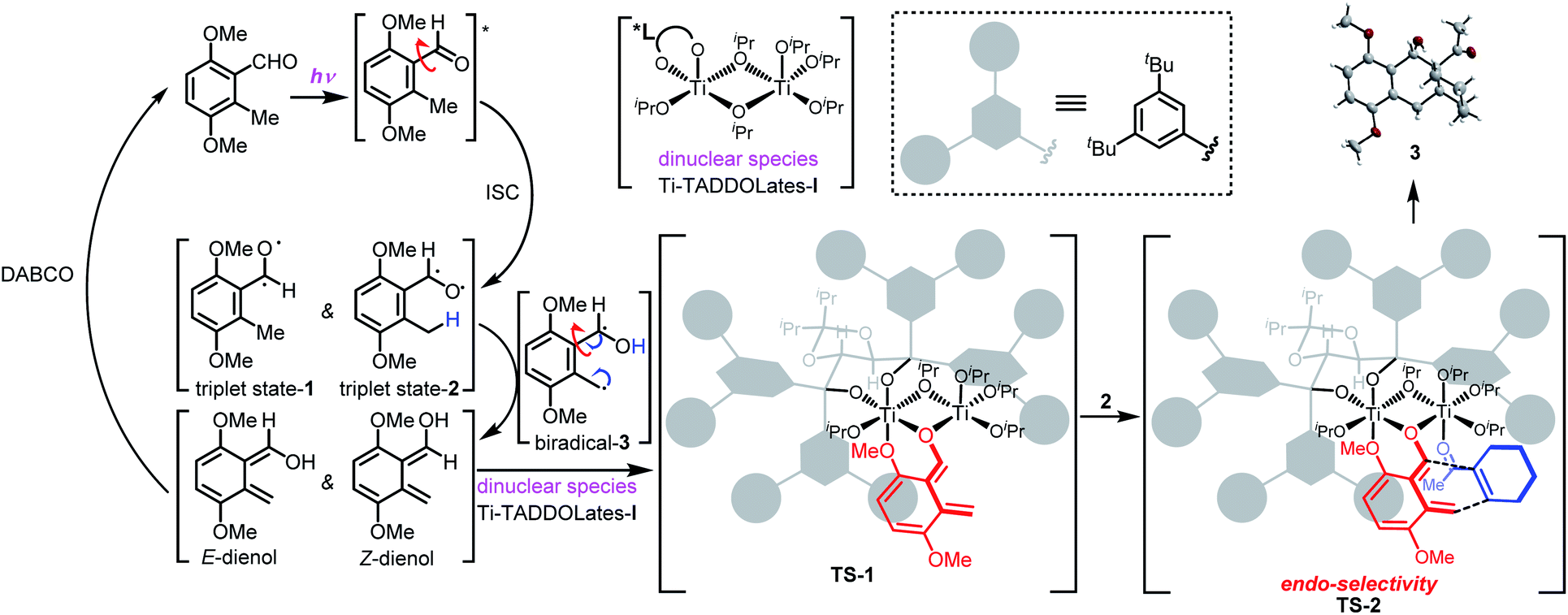 | ||
| Scheme 1 Plausible reaction process of the enantioselective PEDA reaction. | ||
Conclusions
In summary, we have developed an asymmetric PEDA reaction promoted by Ti(Oi-Pr)4 and a substoichiometric amount of a TADDOL-type ligand. The chiral dinuclear Ti–TADDOLate species formed in situ acts as a bifunctional catalyst. Within the chiral cage-like environment, largely stereohindered dienophiles can interact with the transient photoenolized hydroxy-o-quinodimethane, delivering fused or spiro polycyclic rings bearing 2–3 vicinal all-carbon quaternary centers in good yield and excellent enantiomeric excess. This method may create new possibilities for enantioselective organic reactions under excited-state conditions. And, it's also the first example of using direct UV-light mediated cycloaddition to build chiral adjacent quaternary centers at bridgeheads, which demonstrates its potential in the synthesis of complex natural polyketides and icetexane-based diterpenes for medicinal studies and drug discovery.Author contributions
M. Hou and M. Xu performed the synthetic experiments. S. Gao, B. Yang and H. He supervised the research and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21971068, 21772044), Program of Shanghai Academic Technology Research Leader (18XD1401500), Program of Shanghai Science and Technology Committee (18JC1411303), “Shuguang Program” (19SG21), and “the Fundamental Research Funds for the Central Universities” for generous financial support.Notes and references
- (a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (b) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (c) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (d) R. Long, J. Huang, J. Gong and Z. Yang, Nat. Prod. Rep., 2015, 32, 1584–1601 RSC; (e) Y. Wei, L.-Q. Lu, T.-R. Li, B. Feng, Q. Wang, W.-J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2016, 55, 2200–2204 CrossRef CAS PubMed; (f) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 133–137 CrossRef CAS PubMed; (g) M.-M. Li, Q. Xiong, B.-L. Qu, Y.-Q. Xiao, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 17429–17434 CrossRef CAS PubMed.
- (a) M. Buschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed; (b) B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176–9181 CrossRef CAS PubMed; (c) A. Jolit, P. M. Walleser, G. P. A. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180–6183 CrossRef CAS PubMed; (d) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed; (e) H. Zhang, L. Hong, H. Kang and R. Wang, J. Am. Chem. Soc., 2013, 135, 14098–14101 CrossRef CAS PubMed; (f) Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197–8200 CrossRef CAS PubMed; (g) K. Ohmatsu, N. Imagawa and T. Ooi, Nat. Chem., 2014, 6, 47–51 CrossRef CAS PubMed; (h) F. Zhou, L. Zhu, B.-W. Pan, Y. Shi, Y.-L. Liu and J. Zhou, Chem. Sci., 2020, 11, 9341–9365 RSC.
- (a) N. C. Yang and D.-D. H. Yang, J. Am. Chem. Soc., 1958, 80, 2913–2914 CrossRef CAS; (b) N. C. Yang and C. Rivas, J. Am. Chem. Soc., 1961, 83, 2213 CrossRef CAS; (c) C. Chen, Org. Biomol. Chem., 2016, 14, 8641–8647 RSC.
- R. G. W. Norrish and C. H. Bamford, Nature, 1937, 140, 195–196 CrossRef CAS.
- (a) K. C. Nicolaou and D. Gray, Angew. Chem., Int. Ed., 2001, 40, 761–763 CrossRef CAS; (b) K. C. Nicolaou, D. Gray and J. Tae, J. Am. Chem. Soc., 2004, 126, 607–612 CrossRef CAS PubMed; (c) K. C. Nicolaou, D. Gray and J. Tae, J. Am. Chem. Soc., 2004, 126, 613–627 CrossRef CAS PubMed; (d) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC.
- O. A. Mukhina, N. N. B. Kumar, T. M. Arisco, R. A. Valiulin, G. A. Metzel and A. G. Kutateladze, Angew. Chem., Int. Ed., 2011, 50, 9423–9428 CrossRef CAS PubMed.
- Q. Li, T. Dong, X. Liu and X. Lei, J. Am. Chem. Soc., 2013, 135, 4996–4999 CrossRef CAS PubMed.
- B. Grosch, C. N. Orlebar, E. Herdtweck, W. Massa and T. Bach, Angew. Chem., Int. Ed., 2003, 42, 3693–3696 CrossRef CAS PubMed.
- L. Dell'Amico, A. Vega-Penaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3317 CrossRef PubMed.
- (a) C. Müller, A. Bauer, M. M. Maturi, M. C. Cuquerella, M. A. Miranda and T. Bach, J. Am. Chem. Soc., 2011, 133, 16689–16697 CrossRef PubMed; (b) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1046 CrossRef CAS PubMed; (c) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (d) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed; (e) S. Poplata and T. Bach, J. Am. Chem. Soc., 2018, 140, 3228–3231 CrossRef CAS PubMed; (f) M. Leverenz, C. Merten, A. Dreuw and T. Bach, J. Am. Chem. Soc., 2019, 141, 20053–20057 CrossRef CAS PubMed; (g) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (h) S. Cuadros and P. Melchiorre, Eur. J. Org. Chem., 2018, 2884–2891 CrossRef CAS; (i) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed; (j) C. M. Holden and P. Melchiorre in Photochemistry, Royal Society of Chemistry, 2019, vol. 47, pp. 344−378 Search PubMed; (k) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed; (l) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS PubMed.
- (a) B. Yang, K. Lin, Y. Shi and S. Gao, Nat. Commun., 2017, 8, 622–631 CrossRef PubMed; (b) D. Xue, M. Xu, C. Zheng, B. Yang, M. Hou, H. He and S. Gao, Chin. J. Chem., 2019, 37, 135–139 CrossRef CAS; (c) D. Jiang, K. Xin, B. Yang, Y. Chen, Q. Zhang, H. He and S. Gao, CCS Chem., 2020, 2, 800–812 CrossRef CAS.
- (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed; (b) F. R. Petronijević, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323–18326 CrossRef PubMed; (c) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- D. J. Berrisford, C. Bolm and K. B. Sharpless, Angew. Chem., Int. Ed., 1995, 34, 1059–1070 CrossRef CAS.
- (a) D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS; (b) D. Seebach, R. Dahinden, R. E. Marti, A. K. Beck, D. A. Plattner and F. N. M. Kuehnle, J. Org. Chem., 1995, 60, 1788–1799 CrossRef CAS.
- E. M. Simmons and R. Sarpong, Nat. Prod. Rep., 2009, 26, 1195–1217 RSC.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- D. J. Ramon and M. Yus, Chem. Rev., 2006, 106, 2126–2208 CrossRef CAS PubMed.
- (a) J. Balsells, T. J. Davis, P. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2002, 124, 10336–10348 CrossRef CAS PubMed; (b) K. M. Waltz, P. Carroll and P. J. Walsh, Organometallics, 2004, 23, 127–134 CrossRef CAS.
- R. Haag, J. Wirz and P. J. Wagner, Helv. Chim. Acta, 1977, 60, 2595–2607 CrossRef CAS.
- P. K. Das, M. V. Encinas, R. D. Small Jr and J. C. Scaiano, J. Am. Chem. Soc., 1979, 101, 6965–6970 CrossRef CAS.
- K. V. Gothelf, R. G. Hazell and K. A. Jorgensen, J. Am. Chem. Soc., 1995, 117, 4435–4436 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1947390, 1947392, 1947396, 1947384, 1947385, 1947395, 1947393, 1947386, 1947388 and 1947387. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00883h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |