
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing a three-center single-electron metal–metal bond in a fullerene cage†
Fei
Jin‡
a,
Jinpeng
Xin‡
a,
Runnan
Guan
a,
Xiao-Ming
Xie
b,
Muqing
Chen
 *a,
Qianyan
Zhang
*b,
Alexey A.
Popov
*c,
Su-Yuan
Xie
b and
Shangfeng
Yang
*a
*a,
Qianyan
Zhang
*b,
Alexey A.
Popov
*c,
Su-Yuan
Xie
b and
Shangfeng
Yang
*a
aHefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Materials for Energy Conversion, Department of Materials Science and Engineering, Synergetic Innovation Center of Quantum Information & Quantum Physics, University of Science and Technology of China, Hefei 230026, China. E-mail: sfyang@ustc.edu.cn
bState Key Lab for Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Mate-rials, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
cLeibniz Institute for Solid State and Materials Research (IFW Dresden), Helmholtzstrasse 20, Dresden, 01069, Germany
First published on 2nd April 2021
Abstract
Trimetallic carbide clusterfullerenes (TCCFs) encapsulating a quinary M3C2 cluster represent a special family of endohedral fullerenes with an open-shell electronic configuration. Herein, a novel TCCF based on a medium-sized rare earth metal, dysprosium (Dy), is synthesized for the first time. The molecular structure of Dy3C2@Ih(7)-C80 determined by single crystal X-ray diffraction shows that the encapsulated Dy3C2 cluster adopts a bat ray configuration, in which the acetylide unit C2 is elevated above the Dy3 plane by ∼1.66 Å, while Dy–Dy distances are ∼3.4 Å. DFT computational analysis of the electronic structure reveals that the endohedral cluster has an unusual formal charge distribution of (Dy3)8+(C2)2−@C806− and features an unprecedented three-center single-electron Dy–Dy–Dy bond, which has never been reported for lanthanide compounds. Moreover, this electronic structure is different from that of the analogous Sc3C2@Ih(7)-C80 with a (Sc3)9+(C2)3−@C806− charge distribution and no metal–metal bonding.
Introduction
Endohedral clusterfullerenes featuring encapsulation of an unstable metallic cluster within a fullerene cage have been attracting considerable interest over the past two decades due to the versatility of the types of encapsulated cluster and its charge transfer to the outer fullerene cage.1–8 Soon after the discovery of Sc3N@C80 as the first endohedral clusterfullerene encapsulating a trimetallic nitride cluster in 1999,9 the first metal carbide clusterfullerene (CCF), Sc2C2@C84, was identified by Shinohara and coworkers in 2001.10 The identification that a C2 moiety can be encapsulated in the form of metal carbide revealed a competition between CCFs M2C2@C2n−2 and genuine dimetallofullerenes (di-EMFs) M2@C2n in the hot carbon vapor during the fullerene formation. Importantly, CCFs and di-EMFs have different electronic structures and metal valence states. For Sc, Y, and heavy lanthanides, di-EMFs feature a (M2)4+@C2n4− distribution with M–M bonding and formally divalent metal M2+,11–14 while the charge distribution in dimetallic CCFs is (M2)6+(C2)2−@C2n4− with the more traditional M3+ state of rare-earth metals.8,15,16 Thus, by accumulating a certain negative charge, the C2 group has a paramount influence on the valence state of endohedral metal atoms.The C2 group in CCFs has a variable formal charge state adapting to the metal cluster composition, from (C2)2− in M2C2@C2n to (C2)6− in Sc4C2@C80.17,18 In Sc3C2@C80, the first trimetallic CCF (TCCF) and the only TCCF characterized by single-crystal X-ray diffraction (SC-XRD),19 the acetylide unit has a formal (C2)3− charge state.20 A similar charge distribution is also proposed for Lu3C2@C88.21 In mixed-metal M2TiC2@C80 TCCFs (M = Sc, Lu, Dy), the acetylide unit adopts a (C2)4− state to balance the tetravalent Ti4+.22,23 In these five TCCFs, rare-earth metals are present in their 3+ oxidation state and thus do not form metal–metal bonding interactions. Interestingly, three-center M–M bonding was predicted in Y3@C80 and Er3@C74 trimetallofullerenes (tri-EMFs) based on computational analysis, but structural characterization of such tri-EMFs is still lacking.24,25
In this work, we aim at filling the gap in the knowledge about rare-earth TCCFs and synthesized a novel TCCF based on a medium-sized rare-earth metal, dysprosium (Dy) – Dy3C2@C80. Astonishingly, SC-XRD and computational analysis of Dy3C2@C80 revealed that, dramatically different from the analogous Sc3C2@C80 based on the small-sized metal Sc, Dy3C2@C80 exhibits an unusual (Dy3)8+(C2)2−@C806− formal charge distribution and features a unique three-center single-electron Dy–Dy–Dy bond, which has never been reported in molecular lanthanide chemistry.26–28
Results and discussion
Synthesis, isolation and molecular structure of Dy3C2@Ih(7)-C80
Dy3C2@C80 was synthesized by a modified Krätschmer–Huffman DC arc discharge method using a mixture of Dy2O3 and graphite (molar ratio of Dy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ti:C = 1:1:15) as the raw material under 200 mbar He and 10 mbar N2 gas.29–32 Isolation of Dy3C2@C80 was fulfilled by three-step HPLC (see ESI S1† for details). The purity of the isolated Dy3C2@C80 was confirmed by laser desorption time-of-flight (LD-TOF) mass spectroscopic (MS) and HPLC analyses (Fig. 1a and b). The LD-TOF MS spectrum shows a dominant mass peak at m/z = 1471.5 with the isotopic distribution resembling the calculated one for Dy3C82. The HPLC profile with a single peak at a retention time of 53.5 min confirms the high purity of the isolated Dy3C82. Based on the integrated area of the corresponding HPLC peaks, the yield of Dy3C82 relative to Dy3N@Ih(7)-C80 is estimated to be 1:320 (see ESI Table S1†).
:Ti:C = 1:1:15) as the raw material under 200 mbar He and 10 mbar N2 gas.29–32 Isolation of Dy3C2@C80 was fulfilled by three-step HPLC (see ESI S1† for details). The purity of the isolated Dy3C2@C80 was confirmed by laser desorption time-of-flight (LD-TOF) mass spectroscopic (MS) and HPLC analyses (Fig. 1a and b). The LD-TOF MS spectrum shows a dominant mass peak at m/z = 1471.5 with the isotopic distribution resembling the calculated one for Dy3C82. The HPLC profile with a single peak at a retention time of 53.5 min confirms the high purity of the isolated Dy3C82. Based on the integrated area of the corresponding HPLC peaks, the yield of Dy3C82 relative to Dy3N@Ih(7)-C80 is estimated to be 1:320 (see ESI Table S1†).
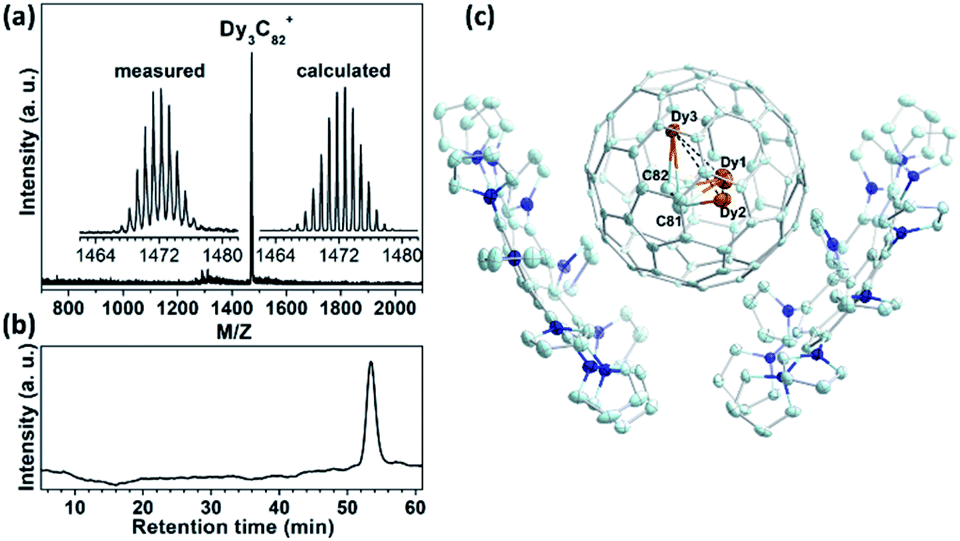 | ||
| Fig. 1 Confirmation of the purity and determination of the molecular structure of Dy3C2@Ih-C80. (a) LD-TOF mass spectrum of purified Dy3C2@Ih-C80; the insets show the measured and calculated isotopic distributions of Dy3C82. (b) HPLC profile of fraction A-3-3 (4.6 × 250 mm Buckyprep column; toluene as an eluent; flow rate 1.0 ml min−1; injection volume 1 ml; 25 °C). (c) The thermal ellipsoid drawing of Dy3C2@Ih(7)-C80·2DPC. Only the major Dy3C2 site is shown. Solvent molecules and H atoms are omitted for clarity. | ||
The molecular structure of Dy3C82 was established by SC-XRD using co-crystallization with a recently developed decapyrrylcorannulene (DPC) host.33 Mixing a CS2 solution of fullerene with a toluene solution of DPC afforded cocrystal Dy3C2@Ih(7)-C80·2(DPC)·3(C7H8), and its SC-XRD analysis confirmed unambiguously that the isolated Dy3C82 is Dy3C2@Ih(7)-C80 TCCF. The cocrystal falls into the monoclinic P21/c space group, its asymmetric unit cell is composed of four complete Dy3C2@Ih-C80 fullerene molecules and four pairs of 2DPC. Fig. 1c illustrates the relative orientations of the Dy3C2@Ih(7)-C80 and the DPC molecules in the cocrystal, in which only one orientation of the fullerene cage together with the major site of the Dy3C2 cluster is shown for clarity. The nearest DPC-cage distances are 3.322(11) and 3.341(12) Å, respectively, which are comparable to those of the cocrystals of DPC with other endohedral fullerenes.33 It is noteworthy that so far only two TCCFs with a homometallic M3C2 cluster have been reported, Sc3C2@Ih(7)-C80 (ref. 19 and 34–38) and Lu3C2@D2(35)-C88,21 both with rare-earth metals of relatively small ionic radius (R3+ is 0.75 Å for Sc and 0.86 Å for Lu39), and only Sc3C2@Ih(7)-C80 has been structurally determined by SC-XRD.19 Therefore, Dy3C2@Ih(7)-C80 represents the first TCCF based on a medium-sized rare-earth metal (R(Dy3+) = 0.91 Å) with the molecular structure determined unambiguously by SC-XRD.
While the Ih(7)-C80 cage is fully ordered in the crystal, Dy atoms are disordered over 14 positions, whose site occupancies range from 0.051(2) to 0.501(2) (see ESI Fig. S2b†). The further structural analysis is based on the major Dy1–Dy3 sites with the largest occupancies of 0.482(2), 0.501(2) and 0.429(3), respectively. Dy atoms are located near the junctions of one pentagon and two hexagons as shown in Fig. 2a, with the shortest Dy–C(cage) distances of 2.202(16), 2.253(12), and 2.254(9) Å, suggesting a strong metal–cage interaction. The Dy⋯Dy distances are 3.381(1), 3.361(2), and 3.389(1) Å, respectively, which are all shorter than Sc⋯Sc distances in Sc3C2@Ih(7)-C80 (3.407(3), 3.712(3), and 3.989(3) Å)19 or Dy⋯Dy distances in Dy3N@Ih(7)-C80 (3.476(3), 3.541(3), and 3.564(3) Å).40
 | ||
| Fig. 2 Configuration of the Dy3C2 cluster within Dy3C2@Ih(7)-C80. (a) Position of the Dy3C2 cluster within Dy3C2@Ih(7)-C80 (major Dy sites) with the closest fullerene fragments. (b) The geometric structure of the Dy3C2 cluster with bond lengths determined by single-crystal X-ray diffraction. The numbers above the carbon atoms indicate the vertical displacements of carbon atoms above the Dy3 planes. Distances are in Å. C and Dy atoms are shown in cyan and orange, respectively. | ||
In view of a considerable disorder of heavy Dy atoms, positions of endohedral carbons are not very well defined. The best refinement results were obtained for a bat ray configuration of the Dy3C2 cluster shown in Fig. 2. The center of the C2 unit is elevated above the Dy1–Dy2–Dy3 plane by ∼1.66 Å. The bat ray configuration was also found in the SC-XRD study of Sc3C2@Ih(7)-C80, but the elevation of the C2 unit above the Sc3 plane is considerably smaller (0.44 Å).19 Besides, the C–C distance within the Dy3C2 cluster is determined to be 1.134(22) Å, which is smaller than that within Sc3C2 (1.29(3) Å (ref. 19)). Overall, these results reveal the strong influence of the metal size on the geometry of the encapsulated M3C2 cluster within the TCCF. The rare earth metals with larger size push the C2 unit further above the M3 plane and at the same time afford shorter M⋯M distances.
DFT computational studies of M3C2@Ih(7)-C80 (M = Dy, Sc)
The different shapes of the Dy3C2 and Sc3C2 clusters motivated us to conduct a DFT computational study (Priroda and Orca codes41,42) of the molecular and electronic structure of M3C2@C80 (M = Dy, Sc) molecules. In Sc3C2@C80, earlier computations and SC-XRD studies revealed two quasi-isoenergetic structures of the Sc3C2 unit.19,20 In the lower-energy bat ray configuration, the C2 unit is almost parallel to the Sc3 plane and is elevated above it (Fig. 3a). The trifoliate structure has a trigonal bipyramidal shape with the Sc3 triangle in the base and two C atoms in the apexes. Using these structures as starting configurations for optimization of Dy3C2@C80 showed that Dy3C2 also has a minimum for a trifoliate configuration albeit with high relative energy, but the bat ray configuration changed considerably during optimization by elevating the C2 unit by 1.620 Å above the Dy3 plane and giving a structure quite similar to that found by SC-XRD. Analogous results were obtained in computations of Y3C2@C80 (see the ESI†). Thus, the larger ionic radii of Dy3+ and Y3+ do not afford enough place for the C2 unit when it is too close to the M3 plane. Importantly, this bat ray structure of the Dy3C2 cluster is 89 kJ mol−1 more stable than the trifoliate configuration. Different positions of the C2 group also result in substantial differences in the DFT-optimized Dy⋯Dy distances. For the bat ray structure, the predicted distances of 3.440, 3.382, and 3.408 Å agree well with the SC-XRD results, while the trifoliate configuration has longer Dy⋯Dy distances of 3.637, 3.651, and 3.939 Å. The transition state connecting these two cluster configurations is found at a relative energy of 140 kJ mol−1. The bat ray structure with an elevated C2 unit similar to the Dy3C2 cluster is also found to be an energy minimum for Sc3C2@C80, but this configuration is 65 kJ mol−1 less stable than the trifoliate and flattened bat-ray configurations. Thus, in agreement with the experimental results, our calculations confirm that the lowest energy configuration of the Dy3C2 cluster has relatively short Dy⋯Dy distances and a significantly elevated position of the C2 unit, which is different from the optimal geometry of the Sc3C2 cluster. | ||
| Fig. 3 DFT computational study on the electronic structure of M3C2@C80 (M = Dy, Sc). (a) DFT-optimized configurations of the Dy3C2 (top) and Sc3C2 (bottom) clusters in M3C2@C80 and their relative energies. Dy is depicted in green, Sc is shown in magenta, and “TS” means transition state. (b) DFT-computed valence spin density distribution in Dy3C2@C80. (c) Singly occupied MO (SOMO) in Dy3C2@C80. (d) Kohn–Sham MO energy levels in the lowest energy configurations of Dy3C2@C80 and Sc3C2@C80 (red and pale blue lines denote the occupied and unoccupied components of the singly occupied MO, and dashed arrows highlight the gap between the SOMO components). Computations are performed with the PBE functional, def2-TZVPP basis set for C and Sc, and 4f-in-core ECP55MWB-II basis set of Dolg and coworkers for Dy.43–46 | ||
Confinement of the Dy3C2 cluster within the Ih-C80 cage not only changes the molecular structure in comparison to the Sc congener, but also results in a completely new bonding situation. Previous studies showed that the electronic configuration of Sc3C2@C80 is best described as (Sc3)9+(C2)3−@C806− with the spin density mainly localized on the C23− unit and Sc atoms featuring their typical Sc3+ oxidation state.20 Surprisingly, in the Dy analog the charge distribution changes to (Dy3)8+(C2)2−@C806− (see ESI S5† for the analysis of the canonical and Pipek–Mezey localized orbitals of the cluster). Acetylide unit C2 in Dy3C2@C80 has negligible spin population, while the main part of spin density is shared between three Dy ions (Fig. 3b). This spin distribution reflects the shape of the SOMO, which has Dy–Dy–Dy bonding character and corresponds to the three-center single-electron bonding of all three Dy atoms (Fig. 3c) with main contributions from the 6s, 6p, and 5d atomic orbitals of Dy. The presence of the M–M bonding in medium-size rare-earth M3C2@C80 TCCFs is further corroborated by topological analysis of the electron density as described in the ESI.† Dimetallofullerenes were already shown to stabilize unique single-electron lanthanide–lanthanide bonds.47–50 Dy3C2@C80 described in this work is the first example of the new lanthanide bonding motif, i.e., three-center single-electron Dy–Dy–Dy bond. Three-center metal–metal bonding was earlier predicted in trimetallofullerene Y3@C80, but in this molecule the Y–Y–Y bonding MO is occupied by two electrons.22,23 Interestingly, Dy3C2@C80 with a trifoliate cluster configuration has a more traditional (Dy3)9+(C2)3−@C806− electron distribution without metal–metal bonding (see the ESI† for further analysis). Thus, variation of the C2 unit position within the M3C2 cluster has a strong influence on the valence state of metal atoms.
To clarify whether the shape of the Dy3C2 cluster in Dy3C2@C80 results from the templating effect of the fullerene cage or has a universal character for rare-earth TCCFs, we performed optimization of the molecule with a larger fullerene cage, Y3C2@C88 (Y3+ has a comparable ionic radius (0.90 Å) to Dy3+). Three different shapes of the cluster were used in the starting configurations: trifoliate, planar bat ray, and bat ray with an elevated C2 unit like in Dy3C2@C80. In all cases, optimization converged to the structure with a flattened bat ray configuration, and re-optimization of the latter with Dy instead of Y also left the shape intact (see ESI Fig. S4†). This result agrees with the earlier study of Lu3C2@C88, which predicted a planar shape of the Lu3C2 cluster, with the C2 unit lying in the triangle plane of three metals.21 Finally, an earlier computational study of Y3C2@C96 also favoured the flattened shape of the cluster.24 Based on these results, we conclude that the unique structure of the Dy3C2 cluster supporting the Dy–Dy–Dy bonding in Dy3C2@C80 is a result of the favourable matching between the fullerene cage size and metal size. When the metal is smaller like Sc or the fullerene cage has a larger inner space (say, C88 or C96), the M3C2 cluster tends to adopt a flattened bat ray shape, which does not lead to direct metal–metal bonding.
Electronic properties of Dy3C2@Ih(7)-C80
We next characterized the electronic properties of Dy3C2@Ih(7)-C80 by UV-vis-NIR spectroscopy and electrochemistry (Fig. 4). The electronic absorption spectrum of yellow Dy3C2@Ih(7)-C80 solution in toluene looks featureless without discernible absorption peaks and is similar to the spectrum of Sc3C2@Ih(7)-C80. Both compounds have their absorption onset at around 850–900 nm (Fig. 4a). Electrochemical characterization by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) gives more distinct characteristics of the compound (Fig. 4b). Dy3C2@Ih(7)-C80 exhibits one reversible oxidation step at +0.19 V, one reversible reduction step at E1/2 = −0.99 V, and one irreversible reduction with a peak potential of −1.65 V. The electrochemical gap ΔEEC of Dy3C2@Ih(7)-C80 is thus 1.18 V. It is remarkable that the ΔEEC gap of Sc3C2@Ih(7)-C80 is only 0.47 V, as its first oxidation and reduction potentials are found at −0.03 V and −0.50 V, respectively (see ESI Fig. S10 and Table S6†).34 Both TCCFs have their SOMO localized on the M3C2 cluster, but their different electronic configurations result in a considerable difference of the frontier MO energies (Fig. 3d). | ||
| Fig. 4 Electronic properties of Dy3C2@Ih(7)-C80. (a) UV-vis-NIR absorption spectra of Dy3C2@Ih(7)-C80 and Sc3C2@Ih(7)-C80 dissolved in toluene. Inset: the photograph of Dy3C2@Ih(7)-C80 solution in toluene. (b) Cyclic voltammogram (top) and differential pulse voltammogram (bottom) of Dy3C2@Ih(7)-C80 measured in o-DCB solution with TBAPF6 as the supporting electrolyte (scan rate: 100 mV s−1); ferrocene Fe(Cp)2 was added as the internal standard. Each redox step of Dy3C2@C80 is marked with a solid dot; the asterisk denotes the oxidation peak of ferrocene. Variation of the formal charge of the Dy3 fragment in different potential ranges is indicated. | ||
The Dy–Dy–Dy-bonding nature of the SOMO in (Dy3)8+ indicates that reduction and oxidation should substantially change the Dy–Dy bonding situation in Dy3C2@Ih(7)-C80. Indeed, DFT calculations show that Dy–Dy bond lengths in the anion shorten to 3.363/3.366/3.320 Å as a result of the stronger Dy–Dy bonding provided by the population of the Dy3-bonding MO by the second electron in the (Dy3)7+ fragment. In the Dy3C2@C80+ cation, the Dy–Dy distances increase to 3.668/3.473/3.542 Å as the metal–metal bonding is diminished when the Dy3-bonding MO is completely depopulated in (Dy3)9+. The electrochemical gap of 1.10 V estimated by DFT with the polarized continuum model of the solvent agrees well with the experimental value, confirming the reliable interpretation of the redox process by theory.
Conclusions
In summary, for the first time we synthesized and isolated a novel TCCF based on a medium-sized rare-earth metal, dysprosium (Dy) – Dy3C2@Ih(7)-C80. Its molecular structure elucidated by SC-XRD reveals Dy3C2 with a bat ray configuration similar to Sc3C2@C80. But the larger size of Dy pushes the acetylide unit above the Dy3 plane and at the same time results in shorter Dy–Dy distances than Sc–Sc distances in Sc3C2@C80. The electronic configuration of Dy3C2@C80 is described as (Dy3)8+(C2)2−@C806− and features a unique three-center single-electron Dy–Dy–Dy bond, which represents the first example of such bonding motif in molecular lanthanide chemistry. Furthermore, the population of the Dy–Dy–Dy-bonding SOMO can be changed electrochemically, providing access to ionic species with either strengthened or weakened Dy–Dy bonds. Our finding on the unprecedented three-center single-electron metal–metal bond offers new insight into the bonding theory of lanthanide coordination compounds.Author contributions
S. F. Y. conceived and designed this research. F. J. and J. P. X. synthesized, isolated the samples and conducted characterizations. R. N. G. helped with sample separation and characterizations. M. Q. C. helped with X-ray crystallographic measurements. A. A. P. did the DFT calculations. X. M. X., Q. Y. Z. and S. Y. X. afforded decapyrrylcorannulene. F. J., J. P. X., A. A. P. and S. F. Y. co-wrote the paper, and all the authors commented on it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Key Research and Development Program of China (2017YFA0402800), National Natural Science Foundation of China (51925206, U1932214, 21721001 and 91961113), and Deutsche Forschungsgemeinschaft (PO 1602/5 and PO 1602/7). We thank the staff of the BL17B beamline of the National Facility for Protein Science Shanghai (NFPS) at the Shanghai Synchrotron Radiation Facility for assistance during data collection and Ulrike Nitzsche for assistance with computational resources at IFW Dresden.Notes and references
- S. Yang, F. Liu, C. Chen, M. Jiao and T. Wei, Chem. Commun., 2011, 47, 11822–11839 RSC.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- R. Guan, M. Chen, F. Jin and S. Yang, Angew. Chem., Int. Ed., 2020, 59, 1048–1073 CrossRef CAS PubMed.
- P. Yu, W. Shen, L. Bao, C. Pan, Z. Slanina and X. Lu, Chem. Sci., 2019, 10, 10925–10930 RSC.
- D. Hao, L. Yang, B. Li, Q. Hou, L. Li and P. Jin, J. Phys. Chem. A, 2020, 124, 2694–2699 CrossRef CAS PubMed.
- W. Shen, S. Hu and X. Lu, Chem.–Eur. J., 2020, 26, 5748–5757 CrossRef CAS PubMed.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55–57 CrossRef CAS.
- C. R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS PubMed.
- A. A. Popov, S. M. Avdoshenko, A. M. Pendás and L. Dunsch, Chem. Commun., 2012, 48, 8031–8050 RSC.
- W. Shen, L. Bao, Y. Wu, C. Pan, S. Zhao, H. Fang, Y. Xie, P. Jin, P. Peng, F.-F. Li and X. Lu, J. Am. Chem. Soc., 2017, 139, 9979–9984 CrossRef CAS PubMed.
- C. Pan, W. Shen, L. Yang, L. Bao, Z. Wei, P. Jin, H. Fang, Y.-P. Xie, T. Akasaka and X. Lu, Chem. Sci., 2019, 10, 4707–4713 RSC.
- X. Lu, S. Hu, W. Shen, G. Duan, L. Yang, P. Jin, Y. Xie and T. Akasaka, Chem.–Eur. J., 2019, 25, 11538–11544 CrossRef PubMed.
- P. Jin, C. Tang and Z. Chen, Coord. Chem. Rev., 2014, 89, 270–271 Search PubMed.
- X. Lu, T. Akasaka and S. Nagase, Acc. Chem. Res., 2013, 46, 1627–1635 CrossRef CAS PubMed.
- K. Tan, X. Lu and C. R. Wang, J. Phys. Chem. B, 2006, 110, 11098–11102 CrossRef CAS PubMed.
- T.-S. Wang, N. Chen, J.-F. Xiang, B. Li, J.-Y. Wu, W. Xu, L. Jiang, K. Tan, C.-Y. Shu, X. Lu and C.-R. Wang, J. Am. Chem. Soc., 2009, 131, 16646–16647 CrossRef CAS PubMed.
- H. Fang, H. Cong, M. Suzuki, L. Bao, B. Yu, Y. Xie, N. Mizorogi, M. M. Olmstead, A. L. Balch, S. Nagase, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2014, 136, 10534–10540 CrossRef CAS PubMed.
- K. Tan and X. Lu, J. Phys. Chem. A, 2006, 110, 1171–1176 CrossRef CAS PubMed.
- W. Xu, T.-S. Wang, J.-Y. Wu, Y.-H. Ma, J.-P. Zheng, H. Li, B. Wang, L. Jiang, C.-Y. Shu and C.-R. Wang, J. Phys. Chem. C, 2011, 115, 402–405 CrossRef CAS.
- K. Junghans, C. Schlesier, A. Kostanyan, N. A. Samoylova, Q. Deng, M. Rosenkranz, S. Schiemenz, R. Westerström, T. Greber, B. Büchner and A. A. Popov, Angew. Chem., Int. Ed., 2015, 54, 13411–13415 CrossRef CAS PubMed.
- K. Junghans, K. B. Ghiassi, N. A. Samoylova, Q. Deng, M. Rosenkranz, M. M. Olmstead, A. L. Balch and A. A. Popov, Chem.–Eur. J., 2016, 22, 13098–13107 CrossRef CAS PubMed.
- A. A. Popov, L. Zhang and L. Dunsch, ACS Nano, 2010, 4, 795–802 CrossRef CAS PubMed.
- Y.-J. Guo, H. Zheng, T. Yang, S. Nagase and X. Zhao, Inorg. Chem., 2015, 54, 8066–8076 CrossRef CAS PubMed.
- S. T. Liddle, Molecular metal-metal bonds, Wiley-VCH Verlag GmbH & Co. KGaA, 2015 Search PubMed.
- C.-S. Cao, Y. Shi, H. Xu and B. Zhao, Coord. Chem. Rev., 2018, 365, 122–144 CrossRef CAS.
- M. V. Butovskii and R. Kempe, New J. Chem., 2015, 39, 7544–7558 RSC.
- S. Hu, P. Zhao, W. Shen, P. Yu, W. Huang, M. Ehara, Y. Xie, T. Akasaka and X. Lu, Nanoscale, 2019, 11, 13415–13422 RSC.
- S. Yang, C. Chen, A. Popov, W. Zhang, F. Liu and L. Dunsch, Chem. Commun., 2009, 6391–6393 RSC.
- C. Chen, F. Liu, S. Li, N. Wang, A. A. Popov, M. Jiao, T. Wei, Q. Li, L. Dunsch and S. Yang, Inorg. Chem., 2012, 51, 3039–3045 CrossRef CAS PubMed.
- F. Liu, T. Wei, S. Wang, J. Guan, X. Lu and S. Yang, Fullerenes, Nanotubes, Carbon Nanostruct., 2014, 22, 215–226 CrossRef CAS.
- Y.-Y. Xu, H.-R. Tian, S.-H. Li, Z.-C. Chen, Y.-R. Yao, S.-S. Wang, X. Zhang, Z.-Z. Zhu, S.-L. Deng, Q. Y. Zhang, S. F. Yang, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Nat. Commun., 2019, 10, 485 CrossRef CAS PubMed.
- Y. Iiduka, T. Wakahara, T. Nakahodo, T. Tsuchiya, A. Sakuraba, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, T. Kato, M. T. H. Liu, N. Mizorogi, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 12500–12501 CrossRef CAS PubMed.
- T. Wang, J. Wu, W. Xu, J. Xiang, X. Lu, B. Li, L. Jiang, C. Shu and C. Wang, Angew. Chem., Int. Ed., 2010, 49, 1786–1789 CrossRef CAS PubMed.
- B. Wu, T. Wang, Y. Feng, Z. Zhang, L. Jiang and C. Wang, Nat. Commun., 2015, 6, 6468 CrossRef CAS PubMed.
- E. Nishibori, I. Terauchi, M. Sakata, M. Takata, Y. Ito, T. Sugai and H. Shinohara, J. Phys. Chem. B, 2006, 110, 19215–19219 CrossRef CAS PubMed.
- S. Taubert, M. Straka, T. O. Pennanen, D. Sundholm and J. Vaara, Phys. Chem. Chem. Phys., 2008, 10, 7158–7168 RSC.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. F. Yang, S. I. Troyanov, A. A. Popov, M. Krause and L. Dunsch, J. Am. Chem. Soc., 2006, 128, 16733–16739 CrossRef CAS PubMed.
- D. N. Laikov and Y. A. Ustynuk, Russ. Chem. Bull., 2005, 54, 820–826 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- J. Yang and M. Dolg, Theor. Chem. Acc., 2005, 113, 212 Search PubMed.
- T. Zuo, L. Xu, C. M. Beavers, M. M. Olmstead, W. Fu, T. D. Crawford, A. L. Balch and H. C. Dorn, J. Am. Chem. Soc., 2008, 130, 12992–12997 CrossRef CAS PubMed.
- L. Bao, M. Chen, C. Pan, T. Yamaguchi, T. Kato, M. M. Olmstead, A. L. Balch, T. Akasaka and X. Lu, Angew. Chem., Int. Ed., 2016, 55, 4242–4246 CrossRef CAS PubMed.
- F. Liu, G. Velkos, D. S. Krylov, L. Spree, M. Zalibera, R. Ray, N. A. Samoylova, C.-H. Chen, M. Rosenkranz, S. Schiemenz, F. Ziegs, K. Nenkov, A. Kostanyan, T. Greber, A. U. B. Wolter, M. Richter, B. Büchner, S. M. Avdoshenko and A. A. Popov, Nat. Commun., 2019, 10, 571 CrossRef CAS PubMed.
- F. Liu, L. Spree, D. S. Krylov, G. Velkos, S. M. Avdoshenko and A. A. Popov, Acc. Chem. Res., 2019, 52, 2981–2993 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2057452. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00965f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |