
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric addition of thiols to silyl glyoxylates for synthesis of multi-hetero-atom substituted carbon stereocenters†
Mingming
Guan
 ,
Shiyu
Wang
,
Yao
Luo
,
Weidi
Cao
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
,
Shiyu
Wang
,
Yao
Luo
,
Weidi
Cao
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: wdcao@scu.edu.cn; xmfeng@scu.edu.cn; Web: http://www.scu.edu.cn/chem_asl/
First published on 24th April 2021
Abstract
A chiral Lewis acid-catalyzed enantioselective addition of thiols to silyl glyoxylates was developed. The reaction proceeds well with a broad range of thiols and acylsilanes, affording the target tertiary chiral α-silyl–α-sulfydryl alcohols with multi-hetero-atom carbon stereocenters in excellent yields (up to 99%) and enantioselectivities (up to 98% ee). A series of control experiments were conducted to elucidate the reaction mechanism.
Introduction
Heteroatoms are prevalent in numerous organic compounds. As a significant subset of heteroatom-containing compounds, organosilanes are important skeletons of many known drugs and provide an opportunity to control pharmacokinetic properties.1 They have been also widely applied in modern organic synthesis due to their unique properties. Particularly, optically active α-hydroxysilanes, containing a carbon stereocenter linked with a silicon atom and an oxygen atom simultaneously, are highly useful reagents in stereoselective C–C bond formation and rearrangement reactions for delivering various chiral molecules.2 As a result, catalytic asymmetric preparation of chiral α-hydroxysilanes attracts much attention.Enantioselective access to α-silyl alcohols could be obtained from nucleophilic addition of organosilanes to carbonyl compounds,3 or from reduction4 and addition reactions5–9 of acylsilanes, which represent a class of unusual and fascinating carbonyl compounds.10 The addition of highly reactive (pro)nucleophilic organometallic reagents to acylsilanes has emerged for the construction of α-hydroxysilanes bearing two saturated or unsaturated hydrocarbyl groups. For example, Marek's5 and Chan's6 group reported zinc-catalyzed enantioselective alkynylations of acylsilanes to give alkyl substituted α-silyl alcohols (Scheme 1a, path i). Harutyunyan7 later extended the nucleophiles to a series of alkyl Grignard reagents in the presence of a copper ferrocenyl diphosphine complex catalyst (path ii), and the competing Meerwein–Ponndorf–Verley reduction could be largely suppressed by using an equivalent mixture of CeCl3 and BF3·Et2O. Recently, Oestreich developed the stereoselective addition of chiral allylic copper intermediates (in situ generated from boron nucleophile and 1,3-dienes) to acylsilanes (path iii).8 Alternatively, Wang, Han and co-workers realized an organocatalytic (vinylogous) aldol reaction with carbonyl compounds as nucleophiles (path iv).9 The advance in synthesis of chiral α-silyl alcohols by using “carbon” nucleophiles intrigued us to investigate enantioselective construction of multi-hetero-atom-bearing carbon stereogenic centers by using hetero-atom nucleophiles.11
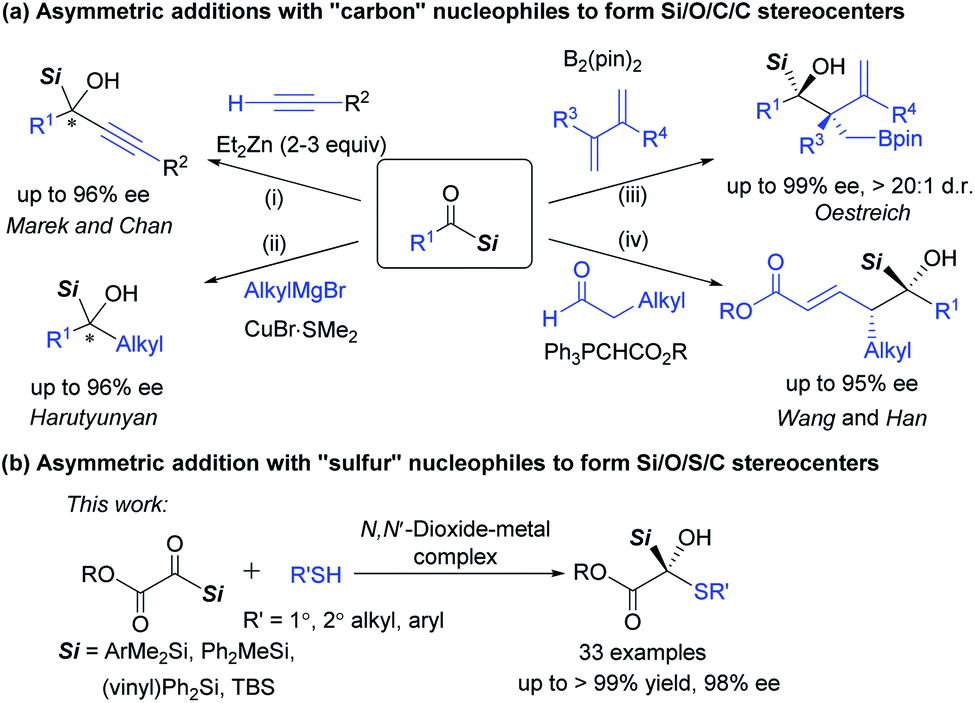 | ||
| Scheme 1 Enantioselective nucleophilic addition reactions of acylsilanes for the construction of chiral tertiary α-silyl alcohols. | ||
The asymmetric construction of a carbon–sulfur (C–S) bond plays a prominent role for the synthesis of valuable chiral organosulfur compounds.12 Significant progress has been achieved through the addition of “sulfur” nucleophiles to carbon–carbon or carbon–nitrogen double bonds, as well as thiolysis of epoxides or aziridines, and so on. However, the enantioselective addition of thiols to carbonyl compounds is hard to realize, although the corresponding thiohemiacetal and thiohemiketal products are common skeletons in many pharmaceuticals,13 which are generally considered as unstable structures and prone to racemization.
As a continuation of our interest in asymmetric sulfur-chemistry,14 we envisaged that chiral N,N′-dioxide–metal complex catalysts15 would be potentially useful in asymmetric addition of thiols to silyl glyoxylates,16 although the reaction was found to be highly reactive in some solvents including water without catalysts.11a Herein we report an efficient enantioselective addition of silyl glyoxylates with various thiols as nucleophiles for the formation of chiral tertiary α-silyl alcohols containing a thiohemiketal skeleton (Scheme 1b). The adducts with multi-hetero-atom carbon stereocenters were afforded in excellent yields, and the competing Brook rearrangement2 products were suppressed.11
Results and discussion
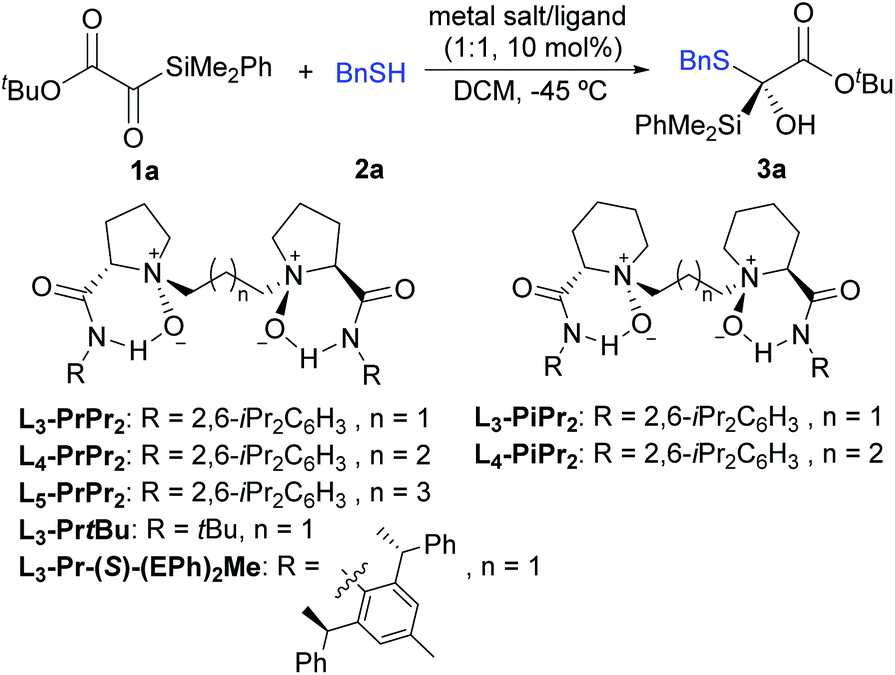
Our study commenced with silyl glyoxylate 1a and benzyl mercaptan 2a as the model substrates to optimize the reaction conditions (Table 1). Considering the strong background reaction response,11 the initial attempts focused on exploring various chiral N,N′-dioxide ligands with Er(OTf)3 (10 mol%) in DCM at a low temperature (−45 °C). To our delight, the Er(OTf)3/L3-PrPr2 complex could promote this reaction to afford the desired product 3a in 85% yield with 49% ee (entry 1). It was found that the chiral backbones, the amide substituents, and the linker of the ligands displayed obvious effects on the enantioselectivity of the reaction (entries 1–6, see Table S1 in the ESI† for details). The amide substituents with the tert-butyl group exhibited poor enantioselectivity compared to that with the aryl group (entry 3 vs. 1). The investigation of the linker of the ligands revealed that L5-PrPr2 provided higher enantioselectivity (87% ee) than L4-PrPr2 (81% ee) and L3-PrPr2 (49% ee) (entry 6 vs. entries 4 and 1). Comparatively, when other representative chiral ligands, such as bis(oxazoline) (BOX), pyridine-2,6-bis(oxazoline) (Pybox), BINAP or chiral phosphoric acid (CPA), were used instead of the N,N′-dioxide ligand, moderate yields with low ee values were achieved (see Table S9 in the ESI† for details). In view of the high reactivity, lowering the reaction temperature to −60 °C led to an increased ee value slightly to 89% (entry 11). Other reaction parameters including solvent (entries 7–10), additives and the concentration were also explored; unfortunately, no better results were obtained (see the ESI† for details). Notably, when protonic additives were used, the racemic version of the reaction process was increased through hydrogen bonding catalysis,11a leading to reduced enantiomeric excess. We next chose L5-PrPr2 as a chiral ligand to screen the metal salts. La(OTf)3 could provide 88% yield with moderate enantioselectivity (51% ee, entry 12). Nevertheless, Y(OTf)3 led to a slight improvement of enantioselectivity with quantitative yield (99% yield, 91% ee, entry 13). In addition, excellent yield (96%) and enantioselectivity (90% ee) could still be achieved when the catalyst loading was reduced to 2 mol% (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Metal salt | Solvent | Yieldb (%) | eec (%) |
a Unless otherwise noted, all reactions were performed with a metal salt/ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%), 1a (0.10 mmol), 2a (0.10 mmol) in DCM (1.0 mL) at −45 °C.
b Yield of isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d Run at −60 °C.
e The reaction was performed with 2 mol% of Y(OTf)3/L5-PrPr2 for 12 hours. :1, 10 mol%), 1a (0.10 mmol), 2a (0.10 mmol) in DCM (1.0 mL) at −45 °C.
b Yield of isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d Run at −60 °C.
e The reaction was performed with 2 mol% of Y(OTf)3/L5-PrPr2 for 12 hours.
|
|||||
| 1 | L3-PrPr2 | Er(OTf)3 | DCM | 85 | 49 |
| 2 | L3-PiPr2 | Er(OTf)3 | DCM | 88 | 55 |
| 3 | L3-PrtBu | Er(OTf)3 | DCM | 74 | 12 |
| 4 | L4-PrPr2 | Er(OTf)3 | DCM | 86 | 81 |
| 5 | L4-PiPr2 | Er(OTf)3 | DCM | 99 | 77 |
| 6 | L5-PrPr2 | Er(OTf)3 | DCM | 87 | 87 |
| 7 | L5-PrPr2 | Er(OTf)3 | CH3CN | 86 | 0 |
| 8 | L5-PrPr2 | Er(OTf)3 | THF | 85 | 10 |
| 9 | L5-PrPr2 | Er(OTf)3 | Et2O | NR | — |
| 10 | L5-PrPr2 | Er(OTf)3 | Toluene | NR | — |
| 11d | L5-PrPr2 | Er(OTf)3 | DCM | 95 | 89 |
| 12d | L5-PrPr2 | La(OTf)3 | DCM | 88 | 51 |
| 13d | L5-PrPr2 | Y(OTf)3 | DCM | 99 | 91 |
| 14d,e | L5-PrPr2 | Y(OTf)3 | DCM | 96 | 90 |
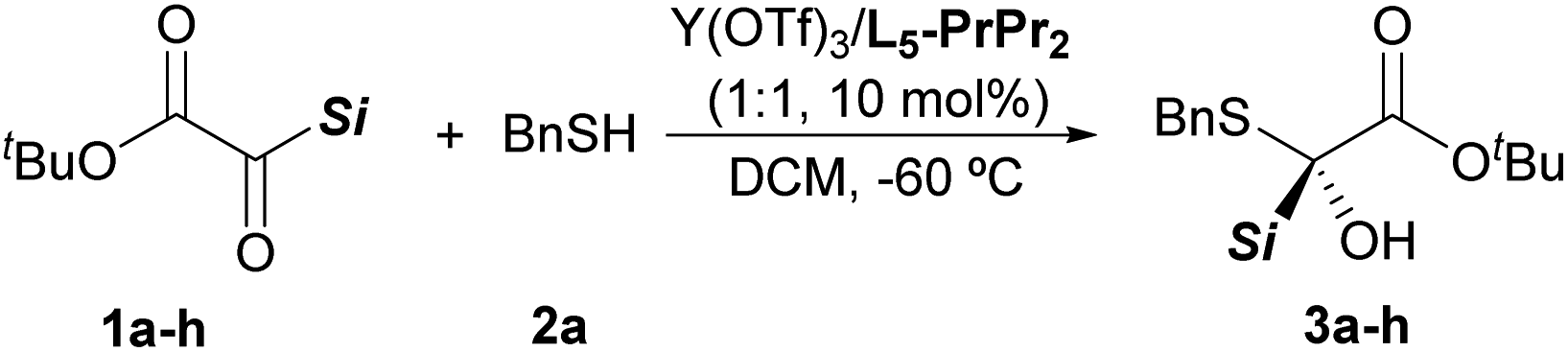
With the optimized reaction conditions in hand (Table 1, entry 13), the silyl glyoxylate scope was next investigated (Table 2). Good substrate compatibility was observed with this Lewis acid catalysis system, and PhMe2Si, Ph2MeSi and Ph2(vinyl)Si substituted silyl glyoxylate (entries 1–3) could be transformed into the corresponding products smoothly with excellent yields and enantioselectivities (3a–3c, 97–99% yields, 91–96% ee). The substituents of the phenyl group have little influence on the activity and enantioselectivity (entries 4–6). The aromatic silyl glyoxylate bearing methyl or fluoro groups at the para-position provided the desired α-silyl–α-sulfydryl alcohols with decreased yields and enantioselectivities (3d, 88% yield with 85% ee; 3e, 76% yield with 80% ee). Delightedly, the silyl glyoxylate with the 4-chloro group attached to the phenyl ring could be converted into 3f with 84% yield and 91% ee (entry 6). The alkyl silyl group (TBS) substituted silyl glyoxylate was also tolerated in this reaction and produced the product 3g in 87% yield with 80% ee by switching the ligand L5-PrPr2 to L3-Pr-(S)-EPh2Me (entry 7). However, when less sterically hindered groups, such as the TMS and TES group, were introduced into the silyl glyoxylate, poor results were obtained (see Table S10 in the ESI† for details).
|
|
|||
|---|---|---|---|
| Entry | Si | Yieldb (%) | eec (%) |
|
a Unless otherwise noted, all reactions were performed with Y(OTf)3/L5-PrPr2 (1:1, 10 mol%), 1 (0.10 mmol), 2a (0.10 mmol) in DCM (1.0 mL) at −60 °C.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d
L3-Pr-(S)-EPh2Me was used instead of L5-PrPr2.
e Run at −45 °C.
|
|||
| 1 | PhMe2Si | 99 (3a) | 91 |
| 2 | Ph2MeSi | 99 (3b) | 95 |
| 3 | Ph2(vinyl)Si | 97 (3c) | 96 |
| 4 | (p-Tolyl)-Me2Si | 88 (3d) | 85 |
| 5 | (4-Fluorophenyl)-Me2Si | 76 (3e) | 80 |
| 6 | (4-Chlorophenyl)-Me2Si | 84 (3f) | 91 |
| 7d,e | t BuMe2Si | 87 (3g) | 80 |
Subsequently, we turned our attention to the scope of mercaptans 2 (Scheme 2). Regardless of the electronic effect or steric hindrance of the substituents on the phenyl ring of benzyl thiols, this asymmetric reaction proceeded smoothly to afford 3h–3m with efficient yields (93–99%) and ee values (92–94%). For the primary alkyl thiols, the increase of the alkyl chain length did not affect the activity and enantioselectivity of the reaction; the corresponding products (3n–3r) were obtained in 96–99% yields with 91–98% ee. Interestingly, functionalized mercaptan reagents, containing silyl, alkenyl or ester groups at the terminal chain of thiols, could be also tolerated, readily yielding the desired products (3u–3w) with good results (95–99% yield, 86–93% ee). The absolute configuration of the product 3w was determined to be (R) by X-ray crystallography analysis.17 It was found that the nucleophilic addition with branched alkyl thiols, such as 2-propanethiol and cyclopentanethiol, proceeded successfully to give 3x and 3y in 90% yield with 94% ee and 98% yield with 96% ee, respectively. However, no product was observed when the sterically hindered t-butyl mercaptan was used. Apart from the above alkyl mercaptans, aryl mercaptans could also react with silyl glyoxylate 1b to produce the desired products with high yields and moderate to high enantioselectivities (3aa–3ah, 88–99% yields, 81–95% ee).
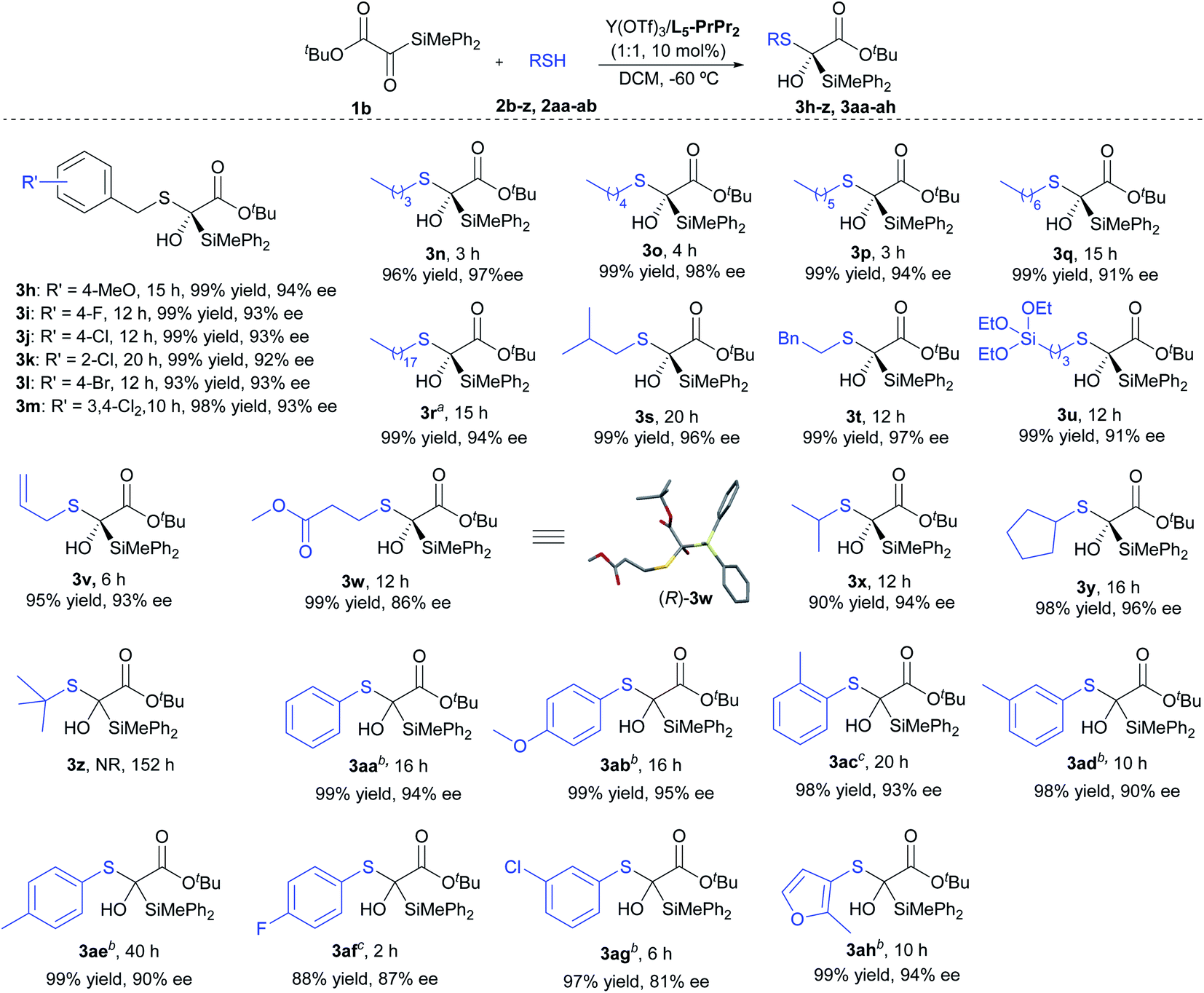 | ||
| Scheme 2 Substrate scope of mercaptans. Unless otherwise noted, all reactions were performed with Y(OTf)3/L5-PrPr2 (1:1, 10 mol%), 1b (0.10 mmol), and 2 (0.10 mmol) in DCM (1.0 mL) at −60 °C. aThe reaction was performed at −45 °C. bEu(OTf)3/L3-PrPr2 was used instead of Y(OTf)3/L5-PrPr2. cEr(OTf)3/L3-PrPr2 was used instead of Y(OTf)3/L5-PrPr2. | ||
To evaluate the synthetic value of the catalytic system, a gram-scale synthesis of three-heteroatomic substituted chiral α-silyl–α-sulfydryl alcohol was carried out. 1b (1.8 mmol) reacted with 2l (1.8 mmol) under the standard reaction conditions, providing the corresponding product 3r in 99% yield with 94% ee (Scheme 3).
 | ||
| Scheme 3 Gram-scale reaction of 3r. | ||
To explore the fact that Brook rearrangement products are rarely observed during the reaction, we conducted a series of control experiments. Firstly, the reaction between 1a and 2a was performed in the presence of the L5-PrPr2/Y(OTf)3 complex at a higher temperature (0 °C); the product of Brook rearrangement was still not detected (see the ESI† for details). When the benzyl 2-(dimethyl(phenyl)silyl)-2-oxoacetate 1h reacted with 2a at −60 °C, only addition product 3ai was observed in 94% yield with 61% ee (Scheme 4a). However, increasing the reaction temperature to 0 °C, a mixture of 3ai and Brook rearrangement product 4ai was obtained with a ratio of 10:1 (Scheme 4b), suggesting that the Brook rearrangement could be significantly suppressed under low temperature conditions.11b Moreover, the result that 45% ee of 3ai and racemate of 4ai were observed suggests that oxygen anion Int-A generated from 1h and 2a facilitated Brook rearrangement to afford the carbon anion Int-B, which rapidly underwent racemization before proton transfer due to the existence of the α-ester group (Scheme 4b, see the ESI† for details). Subsequently, the influence of the base was explored. For the tert-butyl 2-silyl-2-oxoacetate 1a, only racemic addition product 3a was obtained when Et3N (30 mol%) was added (see the ESI† for details). In contrast, when benzyl 2-silyl-2-oxoacetate 1h reacted with 2a under the same conditions, a mixture of addition product and Brook rearrangement product was observed with a ratio of 10:1 (Scheme 4c). These experiments suggests that the ester group of substrates and base (Et3N) have an important influence on the Brook rearrangement reaction. In addition, the conversion experiment of addition product 3ai into Brook rearrangement product 4ai was also performed with or without addition of Et3N. It was found that no 4ai was observed (Scheme 4d), which indicates that the formations of the addition product and Brook rearrangement product were competing processes from the oxygen anion Int-A (see the ESI† for details).
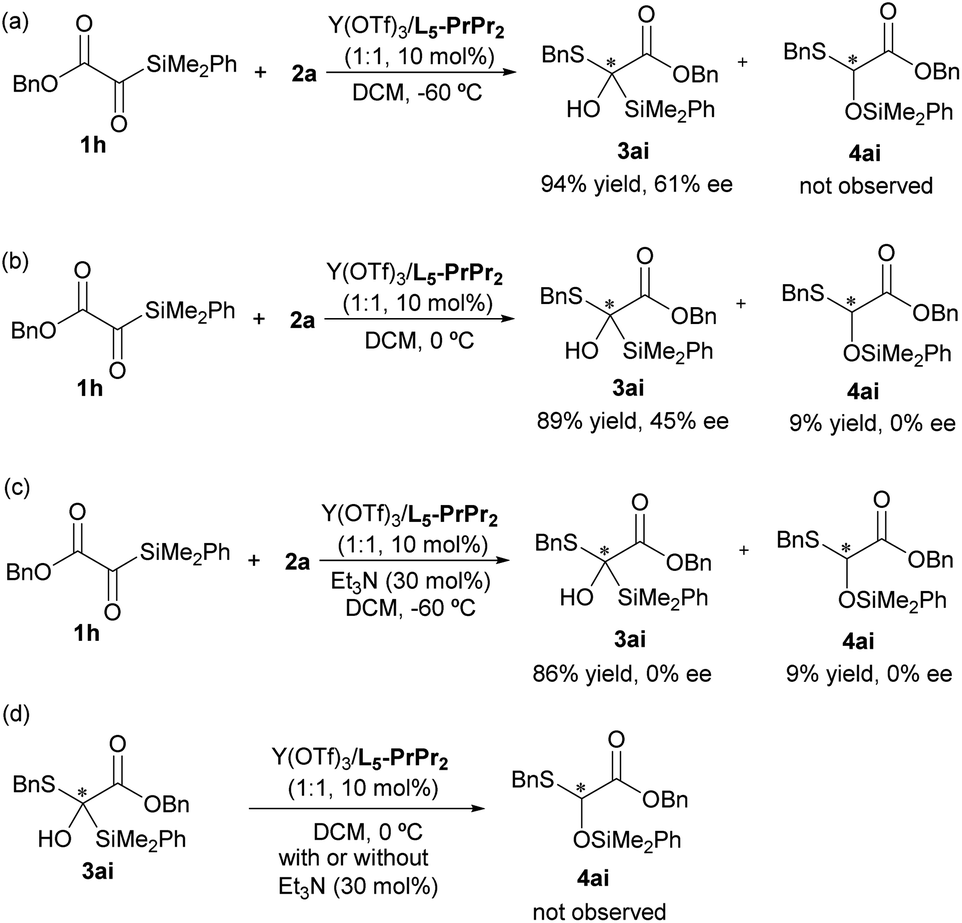 | ||
| Scheme 4 Control experiments. | ||
To further gain insight into the mechanism, the relationship between the ee values of the product 3a and ligand L5-PrPr2 was explored. A self-evident linear effect was observed, which indicates that the monomeric catalyst of L5-PrPr2 and Y(OTf)3 may be the main catalytically active species (see the ESI† for details). Based on the absolute configuration of 3w and X-ray single-crystal structure of the N,N′-dioxide/YIII complex,18 a proposed transition-state model was described in Scheme 5. The four oxygen atoms of ligand N,N′-dioxide, two oxygen atoms of silyl glyoxylate 1b and two molecules of the OTf− anion or a trace amount of H2O in solvent coordinate to YIII in an eight-coordinated manner. In combination with the above experimental results and our previous report,19 we conjectured that a π–π interaction between the aryl silyl group of the substrate and the aryl group of the ligand exists and is beneficial to the reactivities and enantioselectivities. The Si-face of the carbonyl group is blocked by the 2,6-diisopropylphenyl group of the ligand (Scheme 5b), and thus the “sulfur” nucleophile 2q preferably attacks from the Re-face to generate the (R)-configured product 3w (Scheme 5a).
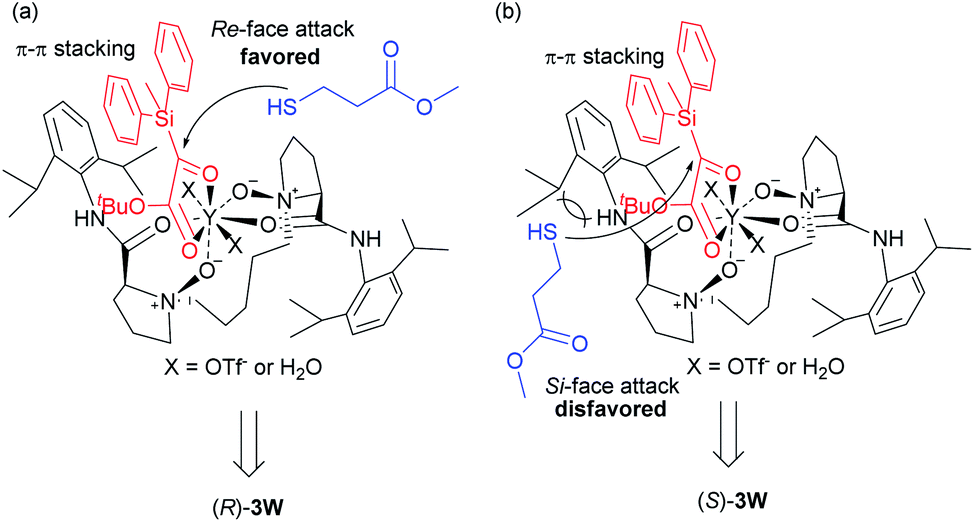 | ||
| Scheme 5 The proposed working mode. | ||
Conclusions
In summary, we have developed the first catalytic asymmetric addition of various thiol nucleophiles to silyl glyoxylates. The corresponding tertiary chiral α-silyl–α-sulfydryl alcohols containing multi-hetero-atom carbon stereocenters were obtained with high yields and good to excellent enantioselectivities (76–99% yields, 80–98% ee). A series of control experiments were conducted to elucidate the reaction mechanism. The application of this methodology to the modification of protein and pharmaceutical structures needs further research.Author contributions
M. G. performed experiments and prepared the ESI† and paper. S. W. repeated some experiments. Y. L. helped with crystal growth. W. C. and X. L. helped with modifying the paper and ESI.† X. F. conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (21801174 and 21890723) for financial support. We thank Dr Yuqiao Zhou for the assistance in X-ray analysis.Notes and references
- (a) K. Miura and A. Hosomi, in Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley-VCH, Weinheim, 2005, p. 409; See also Search PubMed; (b) G. K. Min, D. Hernandez and T. Skrydstrup, Acc. Chem. Res., 2013, 46, 457 CrossRef CAS PubMed; (c) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- (a) A. G. Brook, Acc. Chem. Res., 1974, 7, 77 CrossRef CAS; (b) W. H. Moser, Tetrahedron, 2001, 57, 2065 CrossRef CAS; (c) G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175 CrossRef CAS PubMed; (d) N. Lee, C.-H. Tan and D. Leow, Asian J. Org. Chem., 2019, 8, 25 CrossRef CAS; (e) L. Gao, W. Yang, Y. Wu and Z. Song, in Organic Reactions, ed. P. A. Evans, et al., Wiley-VCH, Weinheim, 2020, p. 5 Search PubMed.
- V. Cirriez, C. Rasson, T. Hermant, J. Petrignet, J. D. Alvarez, K. Robeyn and O. Riant, Angew. Chem., Int. Ed., 2013, 52, 1785 CrossRef CAS PubMed.
- (a) L. Panella, B. L. Feringa, J. G. de Vries and A. J. Minnaard, Org. Lett., 2005, 7, 4177 CrossRef CAS PubMed; (b) N. Arai, K. Suzuki, S. Sugizaki, H. Sorimachi and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 1770 CrossRef CAS PubMed; (c) J. Matsuo, Y. Hattori and H. Ishibashi, Org. Lett., 2010, 12, 2294 CrossRef CAS PubMed.
- (a) R. Unger, F. Weisser, N. Chinkov, A. Stanger, T. Cohen and I. Marek, Org. Lett., 2009, 11, 1853 CrossRef CAS PubMed; (b) P. Smirnov, J. Mathew, A. Nijs, E. Katan, M. Karni, C. Bolm, Y. Apeloig and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 13717 CrossRef CAS PubMed; (c) P. Smirnov, E. Katan, J. Mathew, A. Kostenko, M. Karni, A. Nijs, C. Bolm, Y. Apeloig and I. Marek, J. Org. Chem., 2014, 79, 12122 CrossRef CAS PubMed.
- F.-Q. Li, S. Zhong, G. Lu and A. S. C. Chan, Adv. Synth. Catal., 2009, 351, 1955 CrossRef CAS.
- J. Rong, R. Oost, A. Desmarchelier, A. J. Minnaard and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2015, 54, 3038 CrossRef CAS PubMed.
- J.-J. Feng and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 8211 CrossRef CAS PubMed.
- (a) M.-Y. Han, X. Xie, D. Zhou, P. Li and L. Wang, Org. Lett., 2017, 19, 2282 CrossRef CAS PubMed; (b) M.-Y. Han, W.-Y. Luan, P.-L. Mai, P. Li and L. Wang, J. Org. Chem., 2018, 83, 1518 CrossRef CAS PubMed.
- For reviews on acylsilane chemistry, see: (a) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540 RSC; (b) G. R. Boyce, S. N. Greszler, J. S. Johnson, X. Linghu, J. T. Malinowski, D. A. Nicewicz, A. D. Satterfield, D. C. Schmitt and K. M. Steward, J. Org. Chem., 2012, 77, 4503 CrossRef CAS PubMed; (c) D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927 CrossRef CAS; (d) X. Wang, F. Liu, Y. Li, Z. Yan, Q. Qiang and Q.-Q. Rong, ChemCatChem, 2020, 12, 5022 CrossRef CAS.
- The only example for the formation of racemic tertiary α-silyl alcohol with a three-heteroatomic substituted chiral carbon center, see: (a) M.-Y. Han, J. Lin, W. Li, W.-Y. Luan, P.-L. Mai and Y. Zhang, Green Chem., 2018, 20, 1228 RSC; (b) M.-Y. Han, F.-Y. Yang, D. Zhou and Z. Xu, Org. Biomol. Chem., 2017, 15, 1418 RSC.
- For the reviews on carbon–sulfur bond-forming reactions, see: (a) T. Kondo and T.-A. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (b) P. Bichler and J. A. Love, Top. Organomet. Chem., 2010, 31, 39 CrossRef CAS; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (d) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed; (e) H. Chen, W. Jiang and Q. Zeng, Chem. Rec., 2020, 20, 1269 CrossRef CAS PubMed.
- Selected examples: (a) K. Brocklehurst and J. P. G. Malthouse, Biochem. J., 1978, 175, 761 CrossRef CAS PubMed; (b) A. N. Matralis, M. G. Katselou, A. Nikitakis and A. P. Kourounakis, J. Med. Chem., 2011, 54, 5583 CrossRef CAS PubMed; (c) A. N. Matralis and A. P. Kourounakis, J. Med. Chem., 2014, 57, 2568 CrossRef CAS PubMed.
- (a) Y. H. Hui, J. Jiang, W. T. Wang, W. L. Chen, Y. F. Cai, L. L. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 4290 CrossRef CAS PubMed; (b) G. Wang, Y. Tang, Y. Zhang, X. H. Liu, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2017, 23, 554 CrossRef CAS PubMed; (c) Y. Zhang, F. C. Zhang, L. Chen, J. Xu, X. H. Liu and X. M. Feng, ACS Catal., 2019, 9, 4834 CrossRef CAS; (d) F. C. Zhang, Y. Zhang, Q. Tan, L. L. Lin, X. H. Liu and X. M. Feng, Org. Lett., 2019, 21, 5928 CrossRef CAS PubMed; (e) T. F. Kang, L. Z. Hou, S. Ruan, W. D. Cao, X. H. Liu and X. M. Feng, Nat. Commun., 2020, 11, 3869 CrossRef CAS PubMed; (f) F. Z. Chang, B. Shen, S. J. Wang, L. L. Lin and X. M. Feng, Chem. Commun., 2020, 56, 13429 RSC.
- For selected reviews on N,N′-dioxide ligands, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (e) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 Search PubMed ; For recently selected examples: ; (f) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 142, 15975 CrossRef PubMed; (g) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423 Search PubMed; (h) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1894 CrossRef; (i) F. Tan, M. Pu, J. He, J. Z. Li, J. Yang, S. X. Dong, X. H. Liu, Y.-D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 2394 CrossRef CAS PubMed.
- Selected examples: (a) Y. Matsuya, K. Wada, D. Minato and K. Sugimoto, Angew. Chem., Int. Ed., 2016, 55, 10079 CrossRef CAS PubMed; (b) M. Bouzrati-Zerelli, J. Kirschner, C. P. Fik, M. Maier, C. Dietlin, F. Morlet-Savary, J. P. Fouassier, J.-M. Becht, J. E. Klee and J. Lalevée, Macromolecules, 2017, 50, 6911 CrossRef CAS; (c) F.-G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364 CrossRef CAS PubMed; (d) C. Zhu, M.-Y. Han, X.-X. Liang, B. Guan, P. Li and L. Wang, Org. Lett., 2021, 23, 54 CrossRef CAS PubMed.
- The absolute configuration of 3w was determined to be R by X-ray crystal analysis and the others were also determined by comparing the circular dichroism spectra with those of 3w. CCDC 2056433 (3w) contain sthe supplementary crystallographic data for this paper.†.
- CCDC 2064772.†.
- S. J. Zou, W. W. Li, K. Fu, W. D. Cao, L. L. Lin and X. M. Feng, Adv. Synth. Catal., 2019, 361, 2295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, and HPLC spectra. X-ray crystallographic data for 3w (CIF). CCDC 2056433 and 2064772. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01096d |
| This journal is © The Royal Society of Chemistry 2021 |