
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing steric hindrance to manipulate and stabilize metal halide perovskites for optoelectronics
Yanfeng
Miao
a,
Yuetian
Chen
 a,
Haoran
Chen
a,
Xingtao
Wang
a and
Yixin
Zhao
*ab
a,
Haoran
Chen
a,
Xingtao
Wang
a and
Yixin
Zhao
*ab
aSchool of Environmental Science and Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: yixin.zhao@sjtu.edu.cn
bShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
First published on 18th May 2021
Abstract
The chemical instability of metal halide perovskite materials can be ascribed to their unique properties of softness, in which the chemical bonding between metal halide octahedral frameworks and cations is the weak ionic and hydrogen bonding as in most perovskite structures. Therefore, various strategies have been developed to stabilize the cations and metal halide frameworks, which include incorporating additives, developing two-dimensional perovskites and perovskite nanocrystals, etc. Recently, the important role of utilizing steric hindrance for stabilizing and passivating perovskites has been demonstrated. In this perspective, we summarize the applications of steric hindrance in manipulating and stabilizing perovskites. We will also discuss how steric hindrance influences the fundamental kinetics of perovskite crystallization and film formation processes. The similarities and differences of the steric hindrance between perovskite solar cells and perovskite light emission diodes are also discussed. In all, utilizing steric hindrance is a promising strategy to manipulate and stabilize metal halide perovskites for optoelectronics.
 Yanfeng Miao | Yanfeng Miao received his Ph.D. degree at the Institute of Advanced Materials (IAM), Nanjing Tech University, in 2019. He is currently a postdoctoral fellow at Shanghai Jiao Tong University. His current research interests focus on efficient and stable perovskite light-emitting diodes and perovskite solar cells. |
 Yixin Zhao | Yixin Zhao is a professor at Shanghai Jiao Tong University. He graduated from Shanghai Jiao Tong University with B.S. and M.S. degrees in chemistry and received his Ph.D. degree from Case Western Reserve University in 2010, followed by working as a postdoctoral fellow at Penn State University and National Renewable Energy Laboratory. His current research interests focus on perovskite solar cells and perovskite-based functional materials for solar energy conversion and environmental remediation application. |
Introduction
Metal halide perovskites have become one of the most promising semiconductor materials during the past decade because of their excellent performances in charge carrier mobility, absorption coefficient, and photoluminescence quantum efficiency.1–4 The efficiencies of perovskite solar cells (PSCs) and perovskite light-emitting diodes (PeLEDs) have reached impressive values of 25% (power conversion efficiency, PCE) and 23% (external quantum efficiency, EQE), respectively.5–10 However, the inferior operational stability caused by the weak ionic and hydrogen bonding in perovskite structures is one of the main obstacles hindering their further applications and potential commercialization.11,12 The most widely studied three-dimensional (3D) perovskites consist of frameworks organized with corner-sharing octahedra (e.g. [PbI6]4−) and small-sized cations localized in cages. Besides bonding effects, non-covalent steric interactions also influence the structure of perovskite crystals by repulsive forces between overlapping electron clouds. In chemical reactions, steric hindrance influences the active sites and reaction rate.13–17 Steric sizes of A, B and X in metal halide perovskites should also be suitable to meet an appropriate tolerance factor and octahedral factor to maintain the perovskite structure. Therefore, steric hindrance could help to tune and stabilize the crystal structure of perovskites.Over the past few years, there have been some noticeable studies based on steric hindrance using anions and cations with different sizes, rigidity and functional groups to modify the crystal structure, film quality and device performance.18–22 Steric hindrance has been shown to have direct effects on the optical bandgap, tolerance factor, phase stability, trap passivation, hydrophobicity and conductivity of perovskite materials (Fig. 1).22–28 Several general strategies have been developed to obtain stable perovskite structures such as utilizing additives, and developing two-dimensional (2D) perovskites and perovskite nanocrystals (NCs). In these methods, some extra components such as pseudohalides, bulky organic cations and long chain ligands can determine the properties of perovskite devices as steric hindrance plays an important role in these processes.6,29–33 There is also some burgeoning research on rational design based on steric hindrance, which could improve both efficiency and stability.22,34–36 These features have made the perspective of steric hindrance very attractive to researchers to understand the fundamental kinetics of perovskite crystallization and film formation processes.
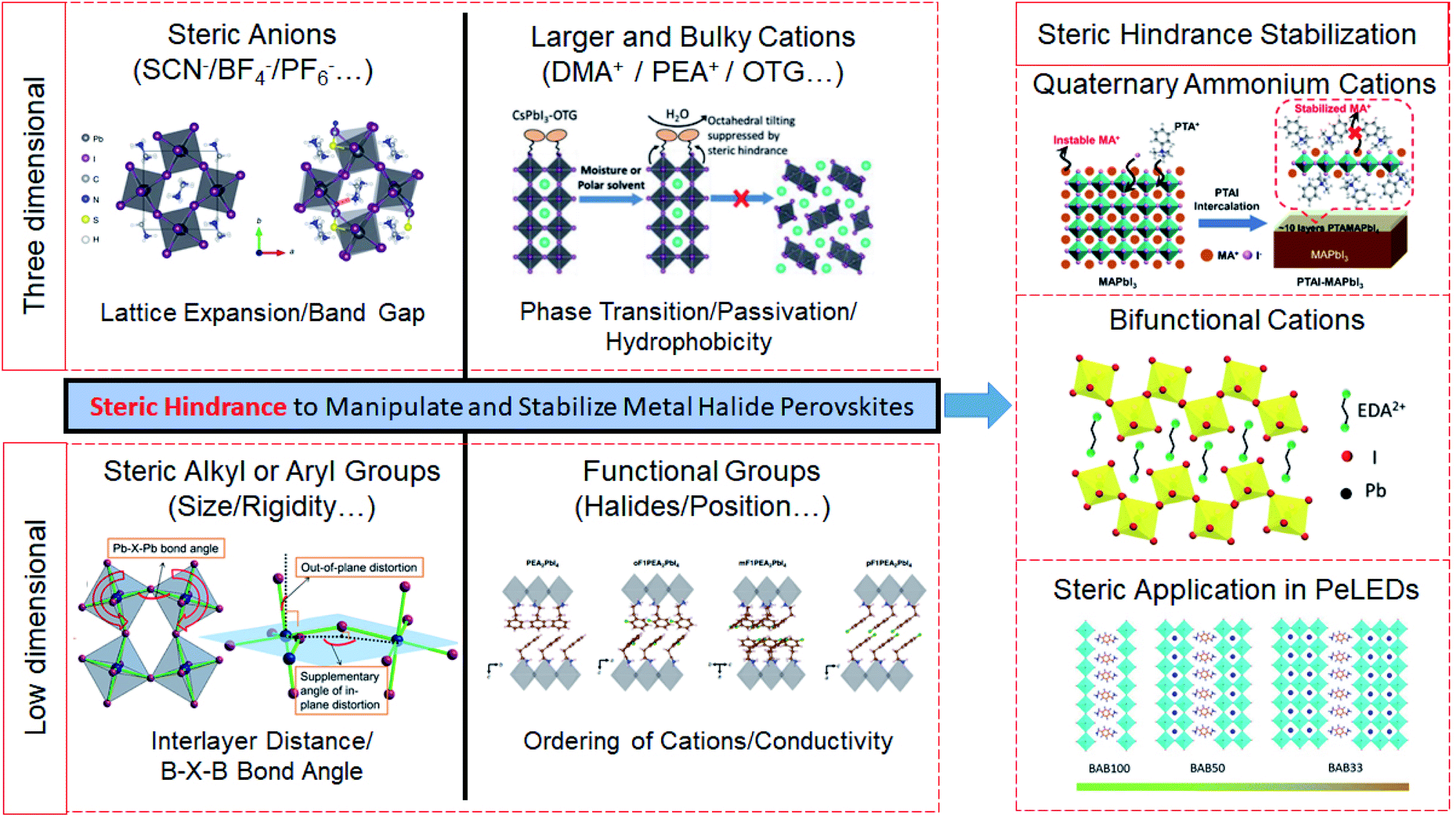 | ||
| Fig. 1 Schematic illustration of the steric hindrance in three-dimensional perovskites and low-dimensional perovskites, and the steric hindrance induced stabilization of perovskites. Reproduced with permission from ref. 23. Copyright 2016 Springer Nature. Reproduced with permission from ref. 24. Copyright 2019 John Wiley and Sons. Reproduced with permission from ref. 25. Copyright 2017 American Chemical Society. Reproduced with permission from ref. 26. Copyright 2019 Springer Nature. Reproduced with permission from ref. 22. Copyright 2020 John Wiley and Sons. Reproduced with permission from ref. 27. Copyright 2018 American Association for the Advancement of Science. Reproduced with permission from ref. 28. Copyright 2019 American Association for the Advancement of Science. | ||
In this perspective, we mainly focus on the role of steric hindrance in manipulating and stabilizing perovskites. We will discuss steric hindrance related work, reveal the wide applicability of steric hindrance and promote the realization of commercialized stable perovskite devices. We also note that the properties of perovskite structures are not simply influenced by one factor but determined by the combination of several interactions between components such as ionic bonds, hydrogen bonds and steric hindrance. Therefore, this perspective will mainly discuss the role of steric hindrance in perovskites and illustrate how steric hindrance and other interactions co-determine the properties of perovskites.
Steric hindrance in 3D perovskites
3D perovskites have a general formula of ABX3 where A is a small-sized cation such as CH3NH3+ (MA+), CH(NH2)2+ (FA+), and Cs+, B is a divalent metal cation such as Pb2+ or Sn2+ and X is a halide. The Goldschmidt tolerance factor (t) within the limit of 0.813–1.107 is usually used to determine whether a 3D perovskite structure can be formed by using the following equation:rA, rB and rX are the ionic radii of A, B and X, respectively.37 Theoretical calculations and experiments have shown that the steric size of A, B and X will influence not only the tolerance factor, but also structural parameters such as metal–halide–metal (B–X–B) bond angles and B–X bond lengths which will determine the optical properties and stability of lattice structures. In this section, we will focus on the steric hindrance in 3D perovskites and review how the steric size of anions and cations influences the properties, especially the stability, of perovskite materials.
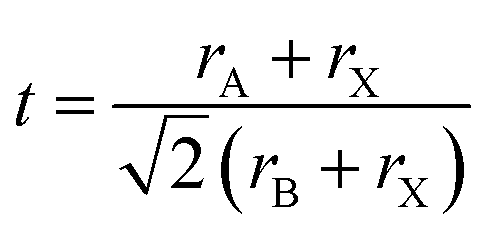
Anions
The 3D lead halide perovskite lattice structure is based on corner-sharing [BX6]4− frames consisting of metal cations and halide anions. The bandgap of perovskite materials is mainly determined by the p and s orbitals of metal cations and p orbitals of halide anions.38–43 Halides such as I−, Br− and Cl− are the most commonly used anions. However, the oxidation of halides is one of the main challenges for realizing stable metal halide perovskites. Halides in a perovskite lattice will form elementary X2 and further accelerate the degradation of perovskites.44–47 Replacing some halide ions with other anions of appropriate sizes is an effective way to stabilize perovskites.48 Pseudohalides such as SCN− (217 pm), BF4− (218 pm) and PF6− (255 pm) with a similar ionic radius to I− (220 pm) are potential candidates for improving the structural stability of APbI3 perovskites.49–51The lone pair of electrons in SCN− ions not only has a stronger interaction with Pb2+ than with I−, but also forms hydrogen bonds with MA.23 The calculated formation constant for PbI42− and Pb(SCN)42− are 3.5 and 7, respectively, which means SCN− can improve the stability of perovskite frame structures. Jiang et al. replaced two thirds of I− with SCN− and the humidity stability was significantly improved.52 The PCE of MAPbSCN2I based PSCs only dropped from 8.3% to 7.4% after 14 days of storage. However, further studies reported that MAPbSCN2I is in fact a layered perovskite MA2PbSCN2I2 with a bandgap of 1.53 eV (Fig. 2a).53,54 The stronger interaction between Pb2+ and SCN− can also be applied to stabilize the FAPbI3 perovskite by phase transition. Recent work by Lu et al. revealed that SCN− from MASCN or FASCN vapor treatment can replace I− on face-sharing δ-FAPbI3 and induce the formation of corner-sharing α-FAPbI3 with much reduced trap density and improved stability (Fig. 2b).55 Our work reported that SCN− incorporated into CsPbBr3 nanocrystals will cause an abnormal blue PL shift (Fig. 2c). We revealed that the steric shape of rod-like SCN− will induce the expansion of the CsPbBr3 crystal lattice and increase the energy of the conduction band.51
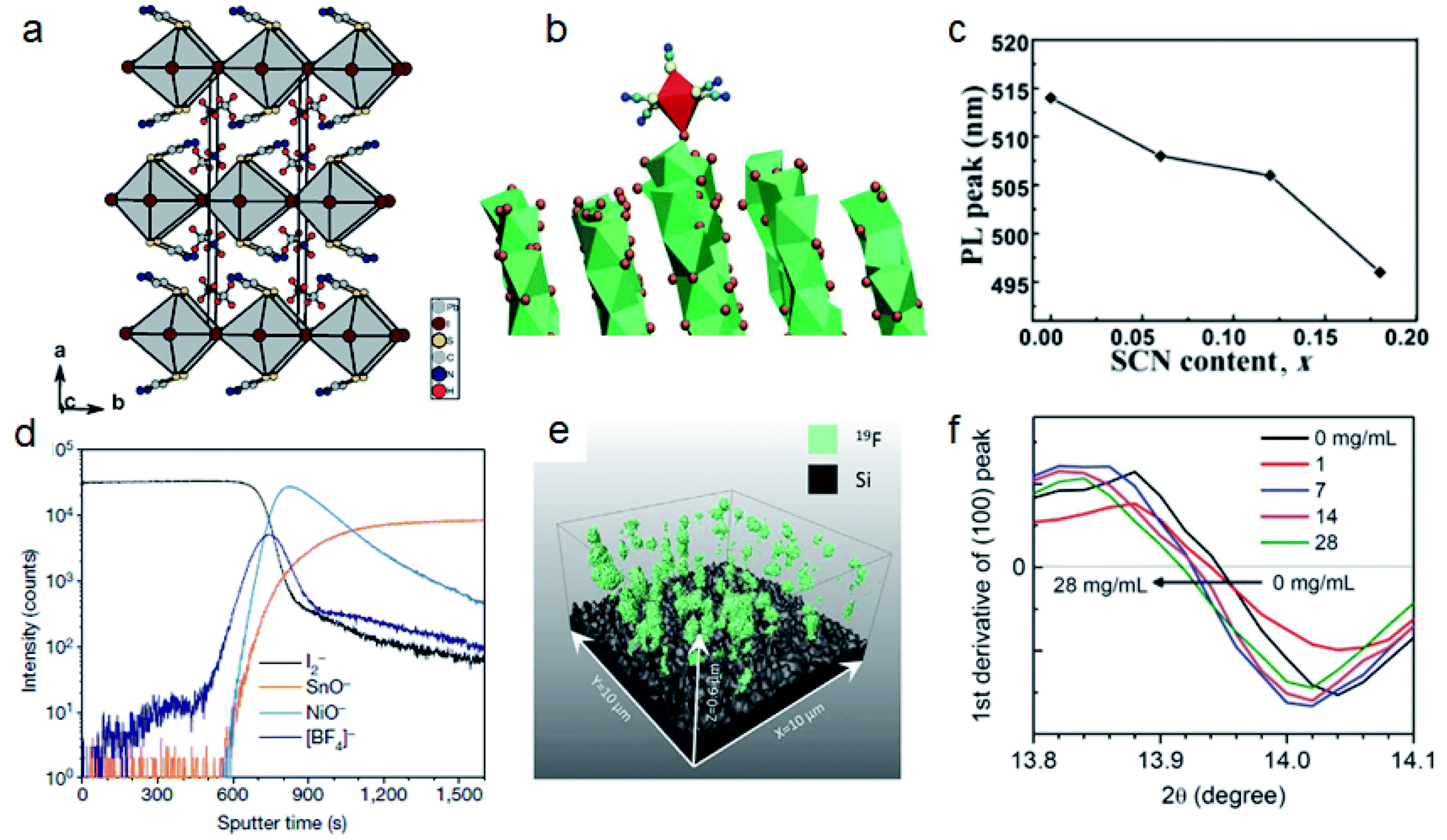 | ||
| Fig. 2 (a) Crystal structure of MA2PbSCN2I2. Reproduced with permission from ref. 53. Copyright 2015 John Wiley and Sons. (b) Molecular dynamics simulation of SCN− induced face-sharing octahedra to the corner-sharing octahedra of the FAPbI3 crystal structure. Reproduced with permission from ref. 55. Copyright 2020 American Association for the Advancement of Science. (c) PL peak positions of CsPbBr3−x(SCN)x (x = 0.06, 0.12, 0.18) nanocrystals with different SCN contents. Reproduced with permission from ref. 51. Copyright 2018 Springer Nature. (d) ToF-SIMS depth profile of [BF4]− in perovskite films. Reproduced with permission from ref. 56. Copyright 2019 Springer Nature. (e) 3D ToF-SIMS map of 19F in perovskite films. Reproduced with permission from ref. 44. Copyright 2020 American Association for the Advancement of Science. (f) (100) peak positions in XRD patterns of FA0.88Cs0.12PbI3 films post-treated with FAPF6 with different concentrations. Reproduced with permission from ref. 58. Copyright 2018 John Wiley and Sons. | ||
Different from SCN−, BF4− has weak interaction with Pb2+ but can also improve the stability of perovskites by steric hindrance induced lattice strain relaxation. Zhang et al. added a small amount (0.5–2% molar ratio) of BF4− into (FAPbI3)0.83(MAPbBr3)0.17 precursor solutions.49 They found that the XRD peak of (001) shifted from 14.20° to 14.14°, which means that BF4 had been incorporated into the perovskite structure and the elongated Pb–BF4 bond can cause lattice relaxation. The PCE increased from 17.55% to 20.16%, and the device could retain 86% of the initial efficiency after 300 h under AM 1.5G illumination. Snaith and co-authors applied two kinds of BF4− based ionic liquids: 1-butyl-3-methylimidazolium tetrafluoroborate (BMIMBF4) and 1-butyl-1-methylpiperidinium tetrafluoroborate (BMPBF4) to modify (FA0.83MA0.17)0.95Cs0.05Pb(I0.9Br0.1)3 and Cs0.17FA0.83Pb(I0.77Br0.23)3 perovskite films, respectively.44,56 In contrast, they found that BF4− in these two salts cannot be incorporated into the perovskite lattice and pairing with different cations will influence the distribution of BF4−. In the case of BMIMBF4, BF4− is located mainly at the buried interface (Fig. 2d), while BF4− of BMPBF4 is distributed over the entire film (Fig. 2e). These two salts can improve both efficiency and stability of PSCs. BMIMBF4 treated PSCs exhibited an increase of PCE from 18.5% to 19.8% and can maintain 95% efficiency under AM 1.5G at ∼75 °C for more than 1800 h. BMPBF4 based PSCs reached a PCE of 20.1%, and the unencapsulated device can maintain 80% efficiency for 1010 hours at 60 °C.
PF6− is another noncoordinating anion similar to BF4−.50,57 Chen et al. post treated a FA0.88Cs0.12PbI3 film with FAPF6. The resulting XRD patterns show that the lattice constant increased, which means that some I− ions were replaced by PF6− (Fig. 2f).58 The larger steric size of PF6− leads to the expansion of the lattice and reduces the bandgap. The efficiency of these PSCs increased from 17.79% to 19.25% and the PF6− treated device retained more than 80% PCE after 528 h under 50–70% humidity.
Cations
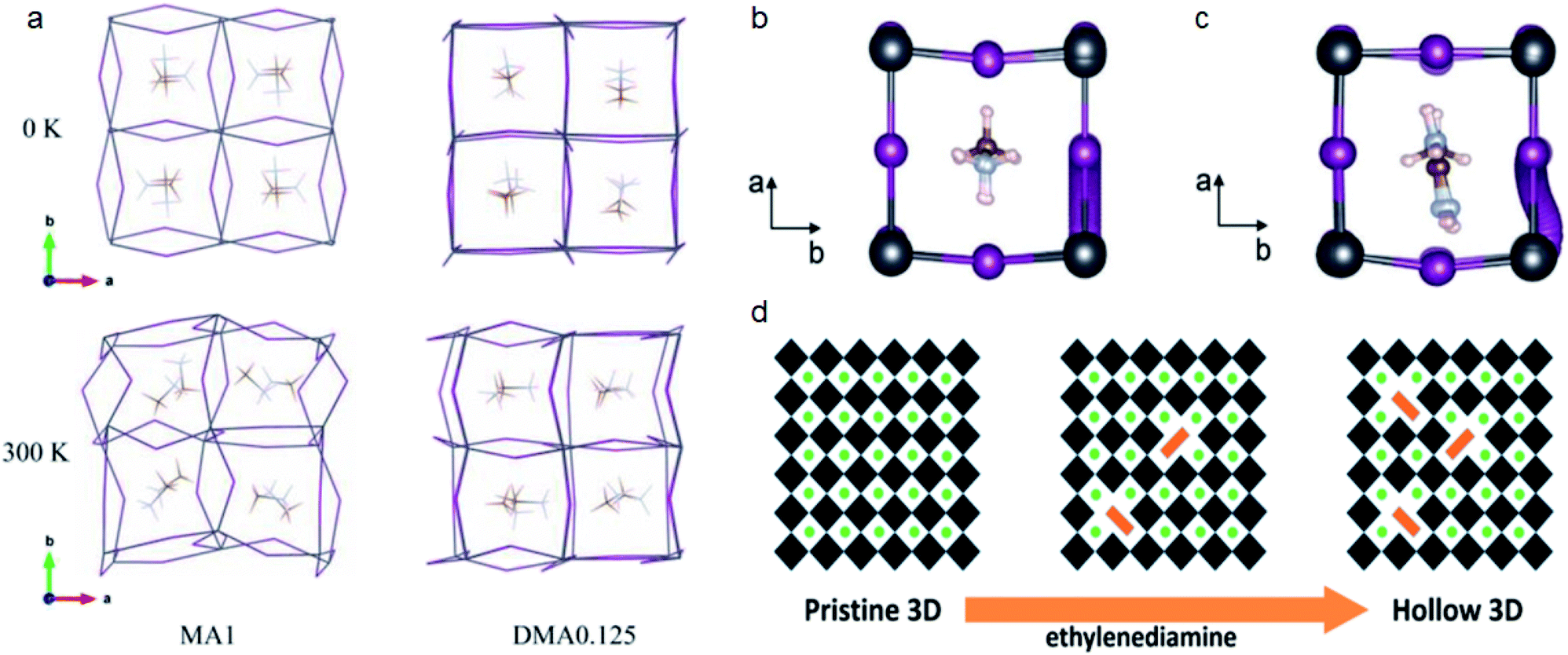 | ||
| Fig. 3 (a) Rigidity changes for MAPbI3 and DMA0.125MA0.875PbI3 structures at different temperatures. Reproduced with permission from ref. 61. Copyright 2019 John Wiley and Sons. (b and c) Iodide migration pathway for MAPbI3 and Ace0.25MA0.75PbI3, respectively. Reproduced with permission from ref. 21. Copyright 2020 John Wiley and Sons. (d) Ethylenediamine induced formation of hollow 3D perovskites. Reproduced with permission from ref. 71. Copyright 2018 American Chemical Society. | ||
The incorporation of GA can stabilize the perovskite structure by reducing the degree of octahedral tilting. The lattice rigidity is enhanced and the defect formation energy is increased.62,66,67 The amount of larger cations incorporated into the lattice can be controlled by adjusting the sizes of the mixed A and X ions.67 Zhou et al. found that the ratio of GA incorporation into a Cs0.15FA0.85Pb(I0.85Br0.15)3 based perovskite can be improved by increasing the amount of Cs and Br. This is because their smaller size can provide more space for the larger GA. GA introduced into the lattice can then interact with I and improve the stability of the perovskite and the PSC can maintain 80% efficiency under AM 1.5G for 8 h.
Ion-migration in perovskites can be inhibited by the steric hindrance of larger cations.21,66 Tan et al. compared the activation energy for iodide migration in MAPbI3 and CH3C(NH2)2+ (Ace) incorporated perovskite Ace0.25MA0.75PbI3.21 In the case of MA, I migrates directly from the lattice. When MA is partially replaced by Ace, the I migrated along an S-shaped curve because of the larger steric size of Ace (Fig. 3b and c). The inhibited I migration can help improve the stability of PSCs and the Ace based device can retain 55% of the initial efficiency after 2100 h under AM 1.5G.
Besides the all corner-sharing 3D structure, some larger cations can replace the position of Pb/I and form a deficient perovskite (d-perovskite).68–71 Leblanc et al. reported d-MAPbI3 prepared by mixing MA+ and hydroxyethyl ammonium (HEA+) cations. These d-MAPbI3 perovskites have similar absorption profiles to that of MAPbI3 but with reduced Pb2+ concentration (Pb0.83I2.83), as well as improved flexibility and stability. Spanopoulos et al. also found that hollow 3D perovskite structures can be realized by introducing ethylenediammonium (EDA) cations (Fig. 3d).71 Addition of EDA can help improve the air stability of Sn based perovskites. The XRD pattern of MA0.6EDA0.4Sn0.72I2.84 remains unchanged for 9 days which indicates its greater stability than MASnI3.
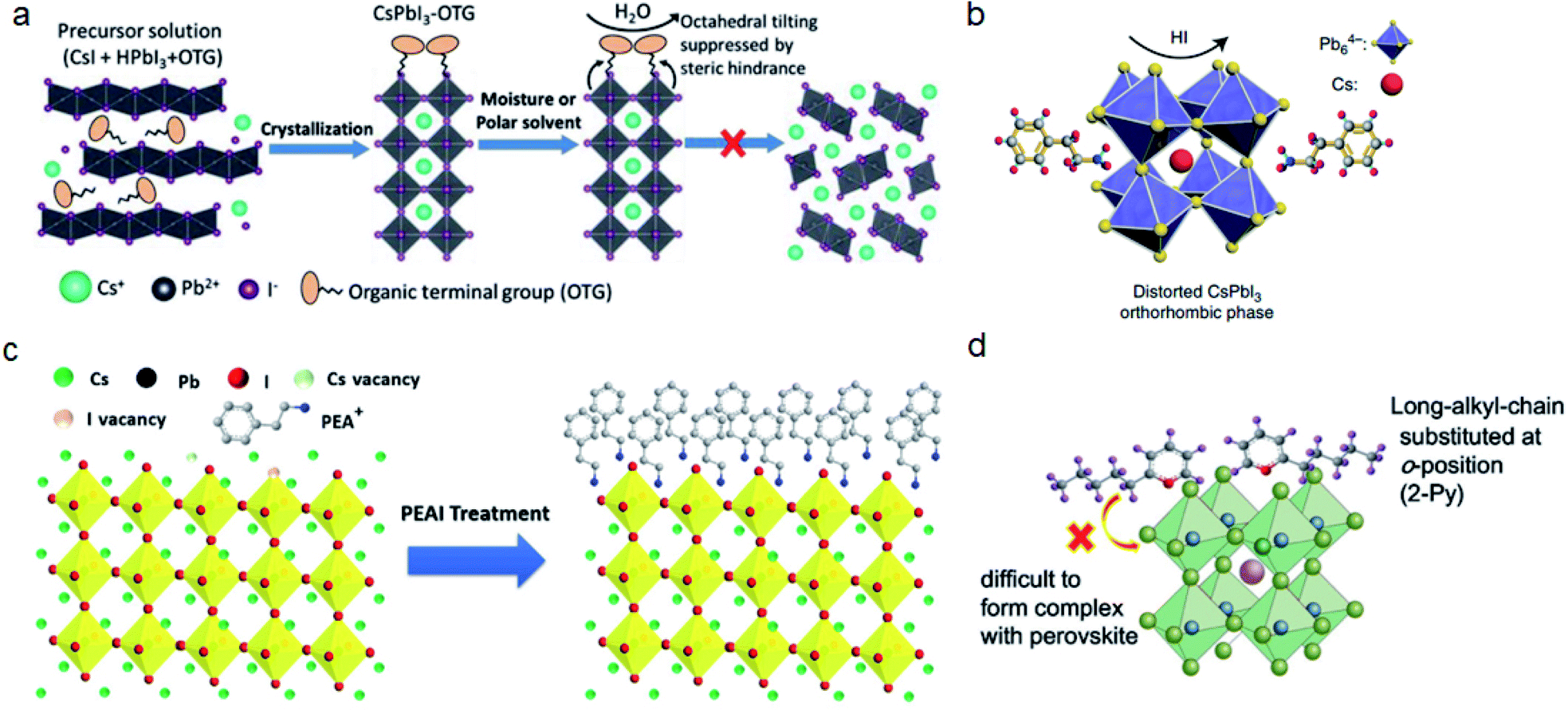 | ||
| Fig. 4 (a) Octahedral tilting of CsPbI3 is suppressed by the steric hindrance of OTG. Reproduced with permission from ref. 24. Copyright 2019 John Wiley and Sons. (b) PEAI additive induced formation of distorted CsPbI3. Reproduced with permission from ref. 20. Copyright 2018 Springer Nature. (c) Surface termination of CsPbI3 by PEAI post-treatment. Reproduced with permission from ref. 76. Copyright 2018 Elsevier. (d) The reaction of pyridine with perovskite is inhibited by using 2-Py with long alkyl chains. Reproduced with permission from ref. 79. Copyright 2016 John Wiley and Sons. | ||
The alignment of bulky cations influences the water-resistivity of perovskite films. Wang et al. compared three phenylalkylamines: aniline, benzylamine, and phenethylamine. They found that benzylamine molecules are aligned perpendicularly on the film, therefore increasing the water penetration distance.77 Post-treatment may also leave a crystalline layer of salt on the surface of the perovskite film, which could inhibit the charge transfer in the device. This unwanted aggregation can be prevented by applying cations with larger steric repulsion. For example, 4-tert-butyl-benzylammonium iodide (tBBAI) has a tert-butyl substitution, which could increase the steric hindrance and form an amorphous morphology on the perovskite film.78
Some post-treatment cations, such as pyridine, not only passivate trap states but also may further react with perovskite films which could reduce the stability of PSCs. By rational molecular design, 2-Py with a long alkyl chain substituted at the o-position can effectively inhibit the further reaction between the perovskite and pyridine (Fig. 4d).79 Since halogen migration and its reaction with a metallic electrode is one of the main reason for device degradation, using a hole-transporting layer (HTL) with high steric hindrance can restrain halogen migration and improve the stability of perovskite solar cells.80 Li et al. compared two HTLs: HL-1 and HL-2, where HL-2 has a longer chain alkyl group compared with HL-1. They found that PSCs based on HL-2 show reduced iodide migration and improved stability.73
Bulky cations are also commonly used in the synthesis of perovskite NCs. Perovskite NCs will undergo aggregation after long term storage; thus rational design of ligands with steric hindrance can help to maintain the colloidal and optical stability. Luo et al. reported a comparison between two kinds of branched ligands: (3-aminopropyl)triethoxysilane (APTES) and polyhedral oligomeric silsesquioxane PSS-[3-(2-aminoethyl)amino]propylheptaisobutyl substituted (NH2-POSS).81 The steric hindrance of APTES can inhibit the dissolution of NCs back into DMF, therefore preventing the penetration of protic molecules into the surface of perovskite NCs. As a result, NCs with uniform crystal size and improved long-term stability can be achieved and well-maintained.
Steric hindrances in low-dimensional perovskites
When larger or bulky cations are the main cations in the perovskite component, the perovskite structure cannot maintain a 3D framework and a low-dimensional perovskite may be formed.82 2D perovskites with a corner-sharing inorganic layer structure, especially quasi-2D perovskites, have similar optical and electrical properties but much higher stability than 3D perovskites. This is because long chain cations can lock the unstable octahedral frame and inhibit the penetration of water molecules owing to the steric effect. As we mainly discuss the steric effect at the interface between organic layers and inorganic layers, we will combine the discussion on 2D perovskite, quasi-2D perovskite and 2D capping layers in this section.Most commonly reported 2D perovskites are (100)-oriented, and they can be further classified into Ruddlesden–Popper (RP), Dion–Jacobson (DJ) and alternating cations in the interlayer space (ACI) phases because of the different stacking modes and displacements of inorganic layers.83,84 RP phases have two spacer layers of large organic cations (Fig. 5a).85 ACI phases have one layer of large size cations and one layer of small size cations. DJ phases have one spacer layer of organic cations between the inorganic layers.86 Different structures are mainly affected by the chemical and spatial structure of cations. We will focus on the influence of steric hindrance on the structure and the optical and electrical-properties of 2D perovskites and perovskite based devices.
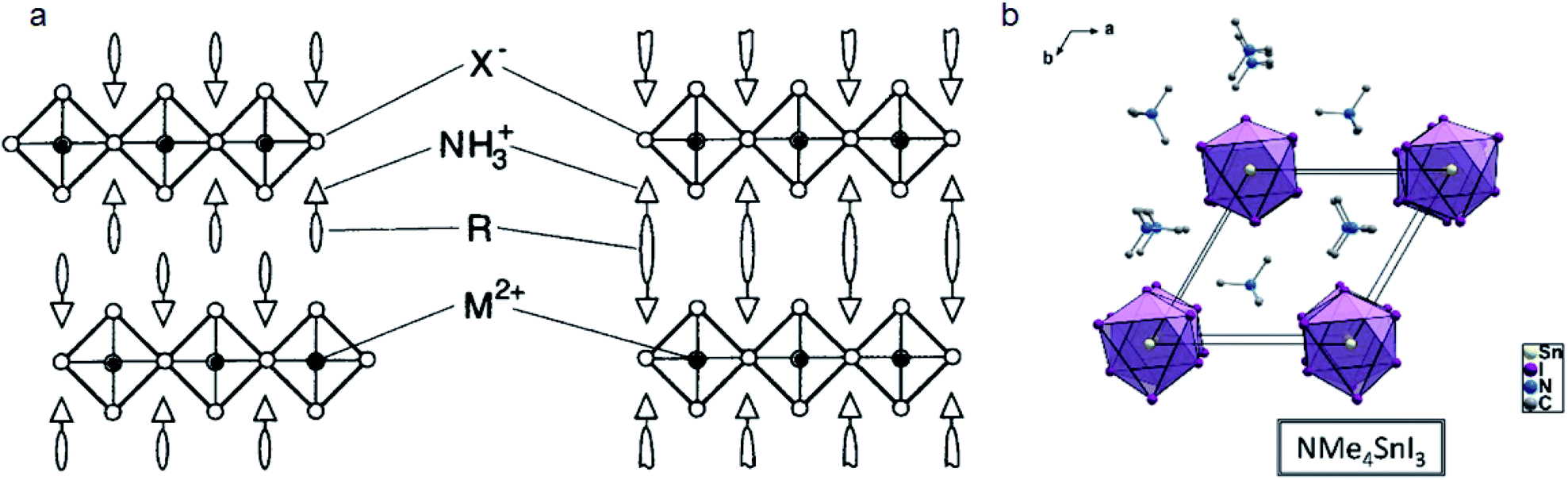 | ||
| Fig. 5 (a) Crystal structure of the RP phase (left) and DJ phase (right) 2D perovskites. Reproduced with permission from ref. 85. Copyright 1999 John Wiley and Sons. (b) Crystal structures of NMe4 based 1D perovskites. Reproduced with permission from ref. 87. Copyright 2017 American Chemical Society. | ||
A large-sized spacer cation often consists of an ammonium and a terminal component such as an alkyl or aromatic group. Inorganic layers are bridged by cations by hydrogen bonding between the ammonium groups and octahedra, as well as van der Waals interactions between terminal groups. The steric effect caused by organic spacers will influence the configuration of inorganic layers and thereby the optical and electrical properties of 2D perovskite materials and optoelectronic devices. We will therefore discuss the steric effect on 2D perovskites with respect to the ammonium group, alkyl/aryl group and functional group.
Ammonium groups
Ammonium groups that can immobilize on the surface of inorganic layers by interacting with metal atoms have a direct influence on the properties of 2D perovskites. Stoumpos et al. studied different kinds of ammonium cations based on tin iodide.87 They found that single ammonium (CH3)mNH4−m+ cations have very different structures. The number of methyl groups (m) on the N atom will influence the connectivity of the octahedra and face-sharing 1D perovskites will be formed when m ≥ 2, such as in DMA+, (CH3)3NH+ (TMA+) and (CH3)4N+ (NMe4+) (Fig. 5b). Very recently, our group reported that tetra-butyl ammonium (TBA+) can intercalate into the CsPbI3 film and form a 1D TBAPbI3 layer which is quite hydrophobic.88 TBAPbI3 single crystals can remain unchanged even when immersed in water for 12 hours. In the case of (NH2)nCH3−n+, the number of ammonium groups will also influence the structure of 2D perovskites. These 2D perovskites will have a combination of corner-sharing and face-sharing structures which is defined as hexagonal perovskite polytypes such as GA+. When GA is mixed with MA, a (GA)(MA)nPbnI3n+1 (n = 1, 2, and 3) perovskite will form an ACI phase perovskite.89,90The position of the N atom will also influence the steric hindrance. Ruben and co-workers compared the substitution position of a N atom on pyridinium based cations.91 For example, 3BrPyH can form a 2D (3BrPyH)2PbI4 perovskite, while 2BrPyH and 4BrPyH will form 1D (2BrPyH)PbI3 and (4BrPyH)4Pb3I10 respectively. The steric size of the unconnected part will influence the face sharing or corner sharing mode and thereby define the structure of perovskites. Li et al. reported that 2-(aminomethyl)pyridine (2AMPY), 3-(aminomethyl)pyridine (3AMPY) and 4-(aminomethyl)pyridine (4AMPY) which have different N positions will form 2D, 0D and 1D perovskites, respectively.92 Mao et al. also studied DJ phase 2D perovskites based on 3-(aminomethyl)piperidinium (3AMP) and 4-(aminomethyl)piperidinium (4AMP) which have different N positions.86 The position of N in the piperidinium organic backbone yields different hydrogen-bonding modes. The H-bonds in 3AMP and 4AMP are formed with terminal I− and bridging I− respectively. Therefore, the steric hindrance of the penetrated ring will induce different hydrogen-bonding modes and thereby influence Pb–I–Pb angles and optical band gaps.
Alkyl or aryl groups
Besides the ammonium groups that directly interact with inorganic layers, the steric hindrance of the alkyl or aryl groups connected with ammonium groups will also influence the perovskite properties.The interlayer distance can also directly influence the conductivity of 2D perovskites. Yu et al. compared the interlayer distances of cations with different sizes. The reduced distance can obviously improve the charge carrier mobility of perovskite films (Fig. 6a).95 Polymer cations such as polyethylenimine (PEI) can provide a more compact organic layer between inorganic layers which can facilitate the charge transport of 2D films.96 The stronger steric hindrance of polymer cations can also make the 2D films more hydrophobic, thereby improving the moisture stability of perovskite solar cells. The length of the organic spacer will further influence the stability of perovskites.94,97 Koh et al. reported that CH3(CH2)3NH3+ (C4) and CH3(CH2)7NH3+ (C8) can both form 2D perovskites by post-treatment to the Cs0.05(MA0.17FA0.83)0.95Pb(I0.83Br0.17)3 surface and C8 exhibits a better passivation effect and water repelling effect.98 Spanopoulos et al. found that the higher tilting angles of CH3(CH2)4NH3+ (C5) yield a more distorted structure. The resulting heat stability and light stability are both improved for C5 than for C4 and CH3(CH2)5NH3+ (C6).99 The length of spacer cations will influence cation penetration which influences the Pb–X–Pb bond angle. Du et al. synthesized several acene alkylamine cation based 2D perovskites.25 They found that the number of methyl unit plays a more important role than the number of acene groups. 2-(2-Naphthyl)ethanammonium (NEA) and PEA have larger NH3 penetration and Pb–X–Pb bond angles than phenylmethylammonium (PMA) and 1-(2-naphthyl)methanammonium (NMA). Very recently, Xue et al. compared three conjugated organic cations, pyrene-based ammonium (PRA), pyrene-based methylammonium (PRMA) and pyrene-based ethylammonium (PREA).36 They found that the A-site cations can modify the band-edge state of perovskites. PRA without alkyl chains has shallow interaction depth because of the steric hindrance from the pyrene group. PREA with long alkyl chains can relieve the steric hindrance, thus leading to deeper vertical intercalation with inorganic layers (Fig. 6b).
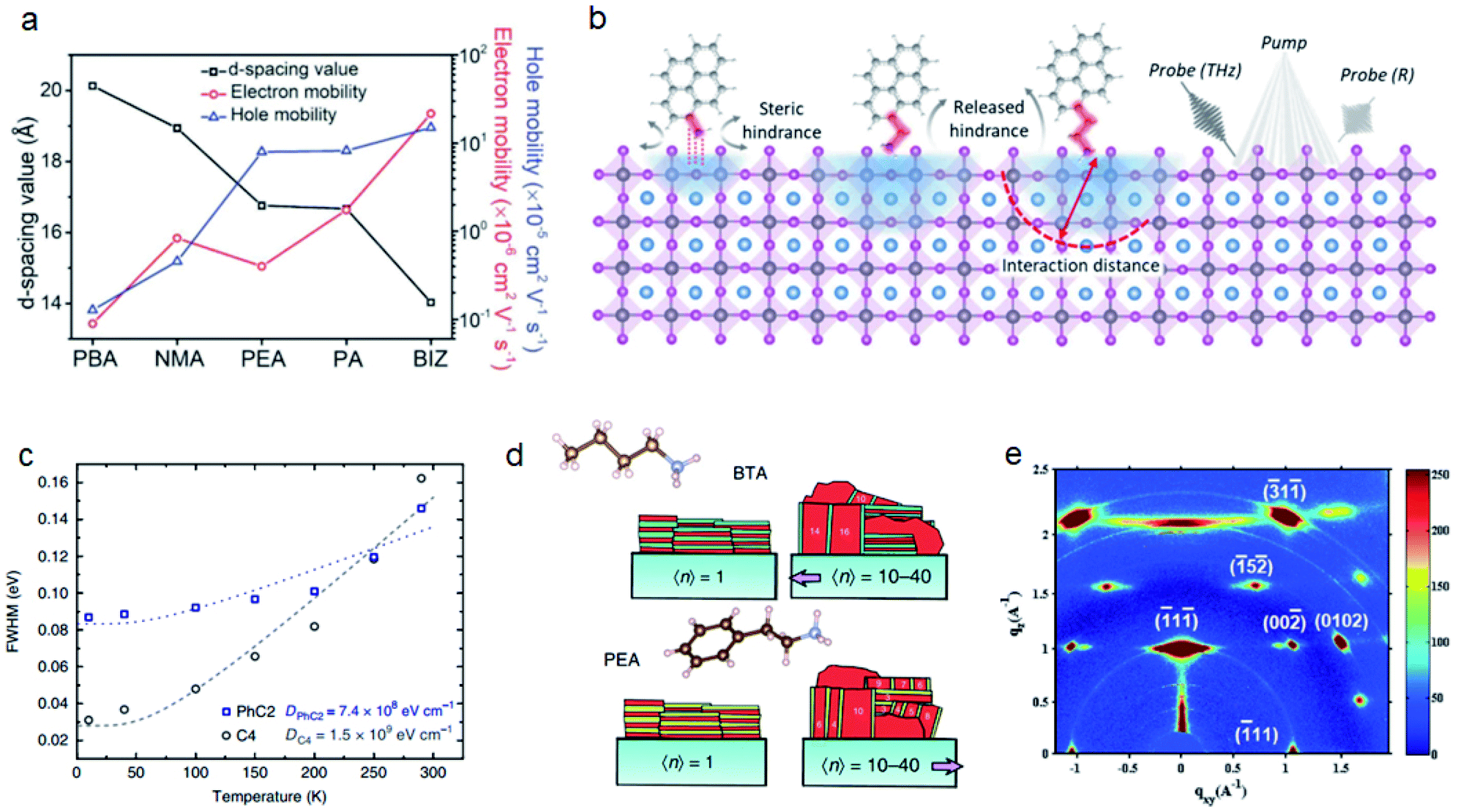 | ||
| Fig. 6 (a) Mobilities of different 2D perovskites with different interlayer distances. Reproduced with permission from ref. 95. Copyright 2018 John Wiley and Sons. (b) Schematic illustration on steric hindrance of PRA, PRMA and PREA, their interaction distance and measurements by transient reflection spectroscopy. Reproduced with permission from ref. 36. Copyright 2021 American Association for the Advancement of Science. (c) The FWHM of PL spectra for PhC2 and C4 based 2D perovskites under different temperatures. Reproduced with permission from ref. 100. Copyright 2018 Springer Nature. (d) The orientations of BTA and PEA based 2D perovskites. Reproduced with permission from ref. 102. Copyright 2018 Springer Nature. (e) GIWAXS data of iso-C4 based 2D perovskites. Reproduced with permission from ref. 104. Copyright 2017 John Wiley and Sons. | ||
Functional groups
Large size spacer cations can be further modified by other elements or groups such as halides (F/Cl/Br/I), –OH, MeO–, unsaturated bonds, and so on. These functional groups will also have steric interactions with the inorganic framework or between the organic layers.25 Mitzi and co-workers compared the crystal structure of (2-XC6H4C2H4NH3)2SnI4 (X: F, Cl, Br) with halides of different sizes.105 They found that (2-ClC6H4C2H4NH3)2SnI4 and (2-FC6H4C2H4NH3)2SnI4 have a similar gauche conformation while (2-BrC6H4C2H4NH3)2SnI4 exhibits anti conformation. The larger Br will cause structural distortion effects such as reduced Sn–I–Sn angles and blue shift on the absorption spectra (Fig. 7a). Their recent work revealed that functional group induced steric effects will influence the formation energies and thermal properties.106 Knutson et al. also reported that the steric impact of organic cations can tune the bandgap of perovskites by more than 1 eV.107 Steric bulk near the ammonium head such as 2-bromophenethylammonium (2-BrPEA), 2-trifluoromethylphenethylamine (2-CF3PEA) and 1-pyreneethylamine (1-PYREA) will inhibit the penetration of ammonium into the perovskite sheet, thereby reducing in-plane distortion.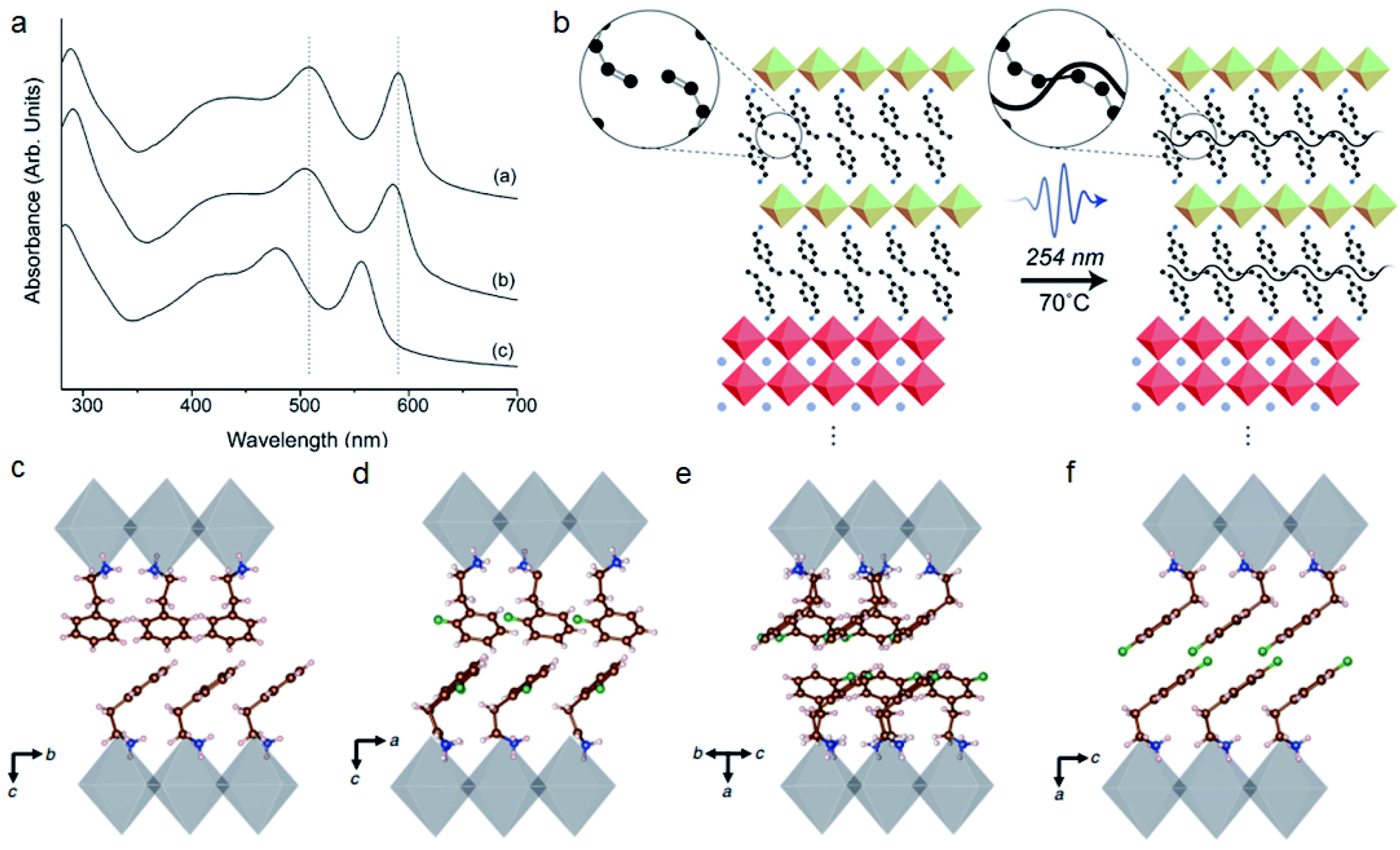 | ||
| Fig. 7 (a) Absorption spectra of the (2-XC6H4C2H4NH3)2SnI4 (X: F, Cl, Br) perovskite. Curve a, F; curve b, Cl and curve c, Br. Reproduced with permission from ref. 105. Copyright 2003 American Chemical Society. (b) Formation of new covalent bonds under UV light for BzA with a vinyl group at the para position. Reproduced with permission from ref. 108. Copyright 2019 American Chemical Society. (c–f) Organic cation packing arrangements of PEA2PbI4, 2FPEA2PbI4, 3FPEA2PbI4 and 4FPEA2PbI4, respectively. Reproduced with permission from ref. 26. Copyright 2019 Springer Nature. | ||
Functional groups with unsaturated bonds may form a cross-linked structure and improve the connectivity of organic layers, which can efficiently improve the conductivity and stability of 2D perovskites. For example, double bonds and triple bonds can lead to the formation of a cross-linked structure upon UV light or thermal annealing.108,109 The vinyl group at the para position of benzylammonium (BzA) can form new covalent bonds under UV light, which can improve the operational stability and reduce the hysteresis of PSCs (Fig. 7b).108 Cross-linking combined with oxygen or iodine doping will also change the energy bandgap of organic layers from 3.0 to 1.4 eV and induce a thousand-fold conductivity increase.109 In some conditions, in order to obtain tunable perovskite quantum wells, functionalized groups with methyl groups impart steric effects, which can inhibit intermolecular π–π interactions.110
Similar to the position of ammonium groups, the position of functional groups will also influence the steric effects. Recently 4-fluorophenethylammonium (4FPEA) was shown to have potential for high efficiency 2D perovskite solar cells because of its enhanced π-orbital overlap and decreased interlayer distance.26,111–114 Several groups have found that the position of monofluorination on PEA will influence the organic cation packing arrangements (Fig. 7c–f).26,115 The formation energy of 4FPEA is lower than that of other positions and will promote the formation of phase pure, flat and oriented 2D perovskites.
Steric hindrance stabilization
Quaternary cations
Compared with the commonly used ammonium cations, quaternary ammonium cations with larger steric hindrance were shown to have a significant effect on passivating the MA+ vacancy and improving the ambient stability of PSCs.35,116 Zheng et al.116 found that quaternary ammonium halides such as choline iodide (CHI) and choline chloride (CHCl) can passivate both positive traps and negative traps on the surface of perovskites. This passivation can improve Voc significantly for more than 100 mV. Our group also found that CHI can passivate the cracks and pinholes in β-CsPbI3 films.65 The PCE of all inorganic PSCs can be improved to 18.4%. The PSC can be retained at 92% of the initial PCE under 1 sun for 500 hours.Several works have shown that light or temperature induced lattice expansion will improve the efficiency and stability of perovskite solar cells.117,118 As we have mentioned before, the steric size of ammonium will influence the tilting angles of Pb–I–Pb. This steric effect on the perovskite lattice can inhibit ion migration and improve the structural stabilities of perovskites. Recently, we reported that the passivation of phenyltrimethylammonium iodide (PTAI) to MAPbI3 can form a mixed-2D PTAMAPbI4 perovskite, which can construct a dynamic barrier for MA+ migration and thereby inhibit the degradation of perovskite films (Fig. 8a). The MAPbI3 film exhibits exceptional stability with unchanged properties even when soaked in PTAI/IPA solution.22 The work by Yangi et al. also reported the formation of mixed-2D perovskites with cations of phenylformamidinium (PFA).119 We also discovered that bifunctional PTABr post-treatment to CsPbI3 can further improve the phase stability by gradient Br doping and the PCE can reach 17.06%.120 Furthermore, PTACl treatment to CsPbI3 can passivate the surface traps effectively and boost the PCE to more than 19%.121
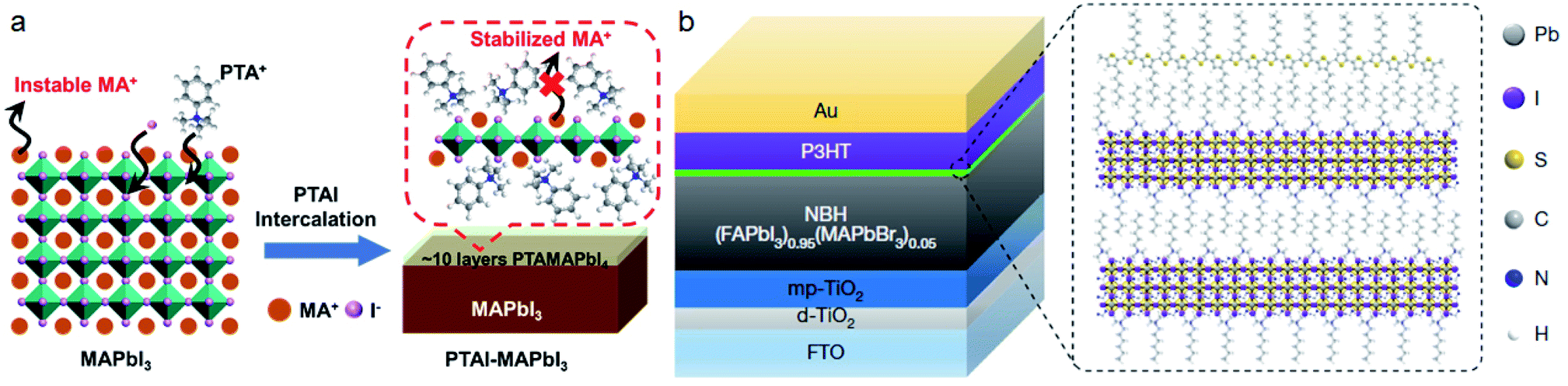 | ||
| Fig. 8 (a) Schematic mechanism for the stabilization of MA+ by PTAI intercalation. Reproduced with permission from ref. 22. Copyright 2020 John Wiley and Sons. (b) PSC based on a P3HT hole transport layer with an n-hexyl trimethyl ammonium bromide (HTAB) based wide-bandgap perovskite as the interfacial layer. Reproduced with permission from ref. 35. Copyright 2019 Springer Nature. | ||
The steric hindrance provided by bulky quaternary cations can also prevent the interaction between the lone pair of electrons of water molecules and the perovskite surface. Yang et al. compared five bulky cations and classified them into three types: MA has one methyl group, tetra-methyl ammonium (TMA) and hexadecyl trimethyl ammonium (CTA) have three or four methyl groups, tetra-ethyl ammonium (TEA), TBA and tetra-hexyl ammonium (THA) have four longer alkyl groups. Among them, TEA with stronger steric hindrance can induce larger Pb–I tilt angles and reduce the tendency of water adsorption.72 The steric effect of quaternary cations is also certified by the fabricated PSCs based on the P3HT hole transport layer. Jung et al. prepared a wide-bandgap layer on a perovskite film by n-hexyl trimethyl ammonium bromide post-treatment (Fig. 8b).35 The stability is significantly improved and the PSC can maintain 80% efficiency after 1008 h under a humidity of 85%.
Bifunctional cations
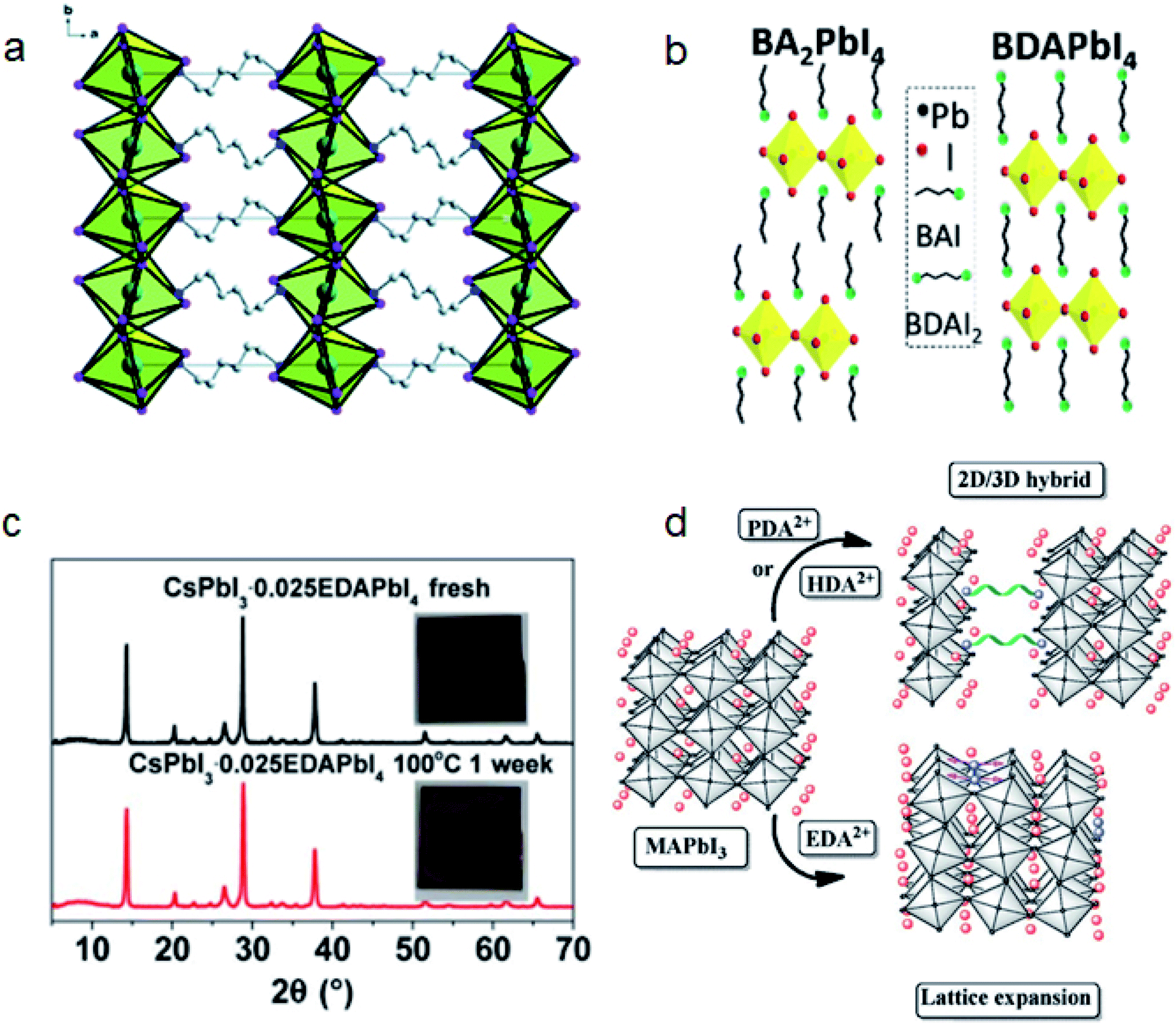 | ||
| Fig. 9 (a) Schematic structure of HDAPbI4. Reproduced with permission from ref. 122. Copyright 2016 Royal Society of Chemistry. (b) Schematic structures of BA2PbI4 and BDAPbI4. Reproduced with permission from ref. 126. Copyright 2018 IOP Publishing Ltd. (c) XRD patterns of fresh and aged CsPbI3–0.025EDAPbI4 perovskite films at 100 °C for 1 week. Reproduced with permission from ref. 27. Copyright 2017 American Association for the Advancement of Science. (d) Effect of diammonium cations with different lengths on the formation of 2D perovskites or lattice expansion. Reproduced with permission from ref. 137. Copyright 2017 John Wiley and Sons. | ||
Black phase CsPbI3 perovskites are believed to be unstable at room temperature as they tend to transform to the yellow phase perovskite that is unsuitable for photovoltaics.129 In 2017, our group reported that EDA based 2D EDAPbI4 can cross link CsPbI3 crystals and the extrusion by the steric effect of EDA can inhibit the phase transition.27 The film condition can keep unchanged upon annealing at 100 °C for a week (Fig. 9c). The DJ phase EDAPbI4 can also maintain the conductivity of CsPbI3 films and CsPbI3·0.025EDAPbI4 based PSC achieved a record PCE of 11.8% at that time.
Tin-based perovskites are attractive as nontoxic, efficient PSCs because of their lead-free composition and broader absorption properties. However, Sn(II) in tin based perovskite can be easily oxidized to Sn(IV) and induce the degradation of perovskites. The strong binding energy and rigid structure of DJ phase perovskites show potential for realizing stable tin based perovskites. Chen et al. reported that (4AMP)(FA)3Sn4I13 based PSCs can obtain a PCE of 4.22% and retain 90% PCE for 100 h under AM 1.5G.130 They suspected that the synergistic effect of reduced vacancy formation, improved binding energy and charge transport together improved the efficiency and stability of tin based PSCs. Li et al. reported that adding 1,4-butanediamine (BDA) ligands into a tin based perovskite can inhibit the oxidation of Sn2+.131 BDA has a large penetration depth between NH3+ and terminal iodides. This steric effect can reduce the electronic interaction and hydrogen bonding, thereby reducing the octahedral distortion, resulting in the DJ phase 2D perovskite having a similar band gap to 3D perovskites.124,131
The energy quantum well (QW) distribution and orientation will influence the optical and electrical properties of DJ phase perovskites.132,133 Zheng et al. compared 1,3-propanediamine (PDA), BDA, 1,5-pentamethylenediamine (PeDA) and 1,6-hexamethylenediamine (HDA) with 3, 4, 5 and 6 carbon chains respectively.134,135 They found that PDA and BDA that have shorter chain lengths can yield oriented and shallow distributed QWs. The chain length of diammonium cations will also influence QW barrier thickness, charge carrier mobility and the 3D-to-2D transformation activation energy.122,136 Lu et al. compared the perovskite structures induced by PDA, HDA and EDA. They found that PDA and HDA will induce 3D to 2D transformation more easily than EDA because of their longer chain lengths (Fig. 9d).137 Zhao et al. also compared a series of diammonium iodides, BDA, 1,8-octamethylenediamine (ODA) and 2,2′-(ethylenedioxy)bis(ethylamine) (EDBE).136 They found that BDA and EDBE can induce the transformation of 3D MAPbI3 to 2D perovskite by post treatment, while ODA only passivates the grain boundary without phase transformation. They suspected that the unique anti–gauche isomerization of ODA elevated the 3D to 2D activation energy.
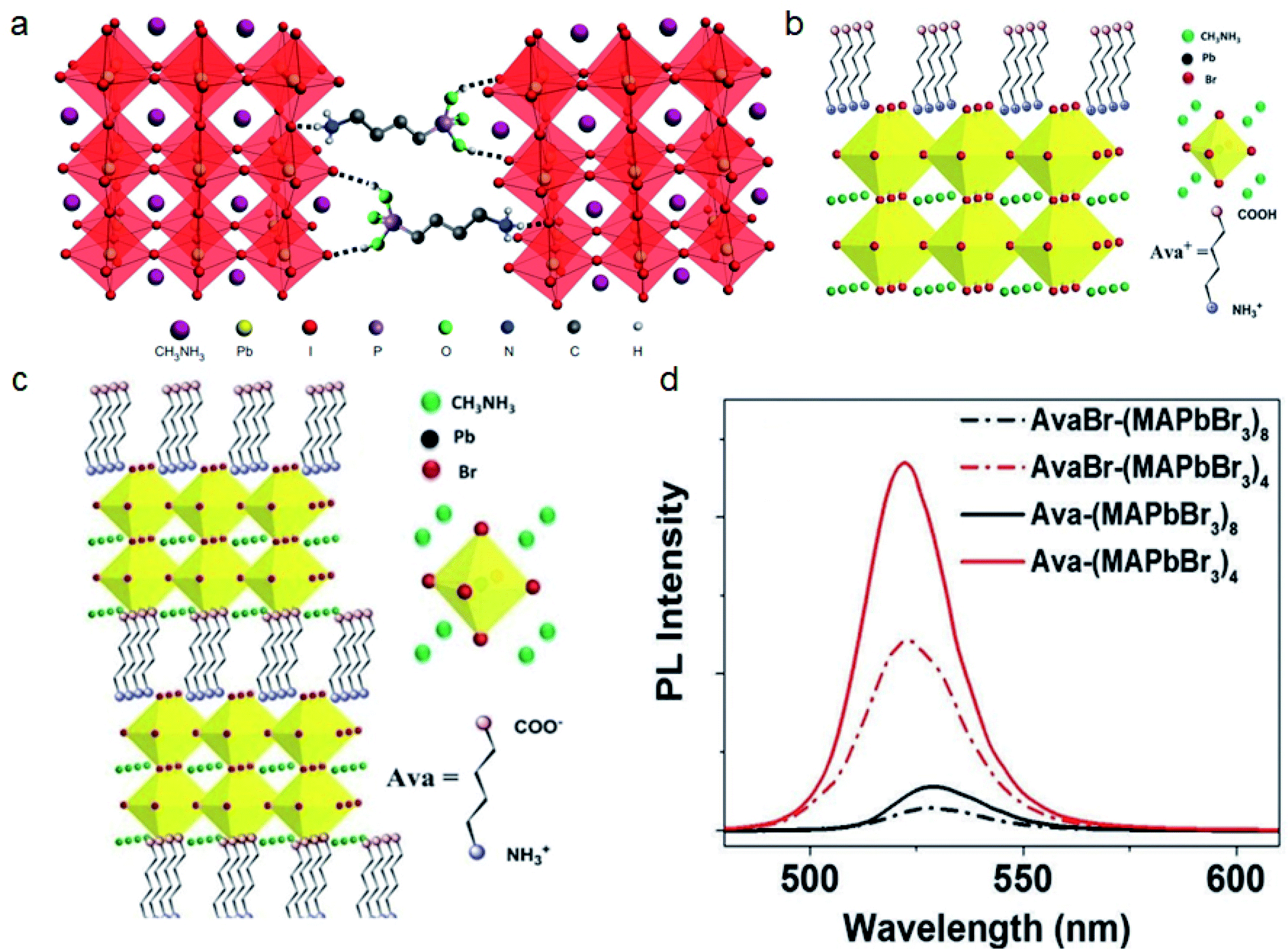 | ||
| Fig. 10 (a) 3D perovskites cross-linked by 4-ABPACl. Reproduced with permission from ref. 138. Copyright 2015 Springer Nature. (b) 5-Ava perovskites cross-linked by Pb–COO bonds and interactions between NH3+ and [PbBr6]4− units. (c) Single linked 5-AvaBr on 3D perovskites. (d) PL spectra of 5-Ava and 5-AvaBr treated perovskite films. Reproduced with permission from ref. 139. Copyright 2017 John Wiley and Sons. | ||
Han and co-workers applied 5-ammoniumvaleric acid iodide (5-AvaI) in MAPbI3 based mesoscopic perovskite solar cells and achieved a PCE of 12.8%.140 The effect of 5-AvaI was considered to be the result of passivating traps and improving the contact with mesoporous TiO2. Our group applied 5-Ava in the MAPbBr3 system and further revealed that the COO− and NH3+ groups on 5-Ava can cross-link perovskites via Pb–COO bonds and the interaction between NH3+ and [PbBr6]4− units. We found that 5-AvaBr will be incorporated into the perovskite lattice and forms the RP phase AvaxMA1−xPbBr3 structure as reported in previous work.140–142 In contrast, 5-Ava will form the crosslinked DJ phase Ava(MAPbBr3)n structure because the COOH on 5-AvaBr will not form such Pb–COO bonds (Fig. 10b and c). This cross-linked structure will sterically reduce the interlayer distance and improve the PLQE of the in situ formed MAPbBr3 QD films up to 80% (Fig. 10d).139 The 5-Ava also shows potential for crosslinking and stabilizing the FASnI3 based perovskite by reducing the Sn4+ content.143
Steric hindrance design for PeLEDs
Perovskite materials are also attractive for light-emitting diodes benefiting from the high PLQE and high emission purity. PSCs and PeLEDs have a similar layered device structure, i.e., anode/HTL/perovskite layer/ETL/cathode. The generation of the excited state is the key step for them. They all require low trap density and reduced nonradiative recombination. That is to say, efficient PSCs are usually efficient PeLEDs. There are also some different requirements between PSCs and PeLEDs because the excited states are separated by an electrical field in PSCs, but are recombined to emit photons in PeLEDs. Therefore, tuning of steric hindrance in perovskite materials can enable us to design specific films for different optoelectronic devices.In order to separate charge carriers effectively, PSCs usually have a type-II band alignment which can drive electrons and holes to different electrodes.144 In the case of LEDs, a type-I band alignment can accumulate electrons and holes to increase the charge carrier density and to improve the radiative recombination rate and PLQE.145–147 Yuan et al. changed the n value in PEA2(CH3NH3)n−1PbnI3n+1 and found that a film prepared with a certain n value will consist of a variety of quasi-2D perovskites with different n values.148 This type-I structure can act as an energy funnel and significantly improve PLQE (Fig. 11a). Besides the addition of different kinds of cations, the steric hindrance induced band alignment can also be modified by changing the concentration of cations. Quintero-Bermudez et al. found that the ligand density at the surface influences the work function of the material and shows tunable type-I or type-II properties.146
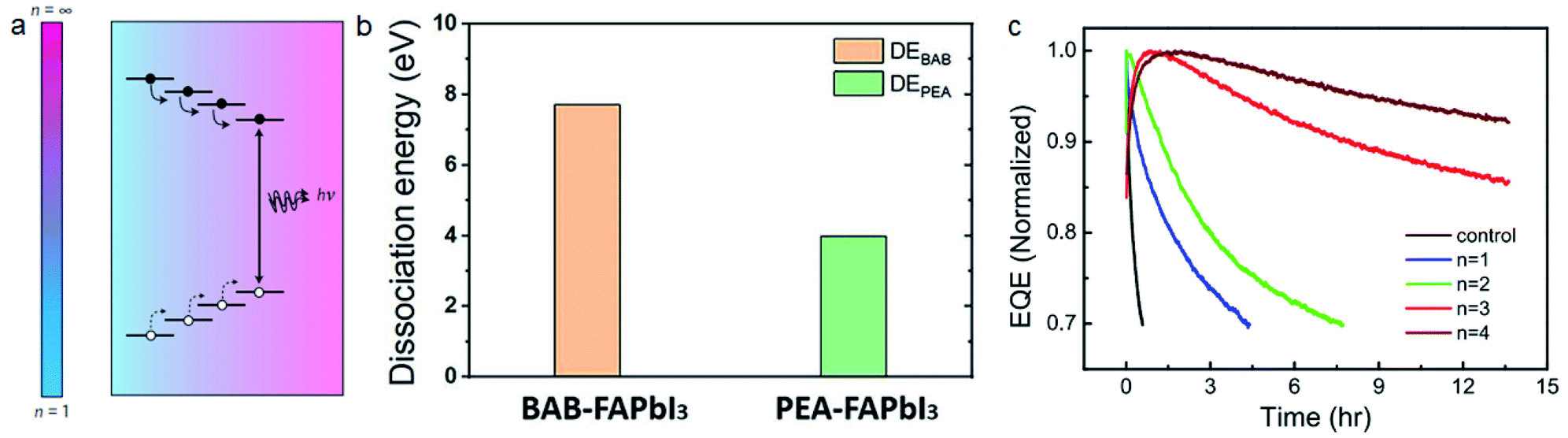 | ||
| Fig. 11 (a) Charge transport in MQW based films. Reproduced with permission from ref. 148. Copyright 2016 Springer Nature. (b) Molecule dissociation energy of BAB and PEA based perovskites. Reproduced with permission from ref. 28. Copyright 2019 American Association for the Advancement of Science. (c) Stability of PeLEDs with different phenylalkylammonium iodide molecules. Reproduced with permission from ref. 34. Copyright 2021 Springer Nature. | ||
The bifunctional effect of 5-Ava is also applied in the fabrication of high efficiency PeLEDs. Cao et al. found that 5-Ava not only reduces the trap density of perovskite films but also undergoes a dehydration reaction with ZnO/polyethylenimine ethoxylated (PEIE) to form an organic layer which reduces the leakage current, thus boosting the EQE up to 20.7%.8 They reported that 6-aminocaproic acid (6-ACA) and 7-aminoheptanoic acid (7-AHA) with longer chain lengths can also improve the efficiency of PeLEDs. Wang et al. found that γ-aminobutyric acid (3-ABA) which has a similar structure to 5-Ava can control the distribution of n values in the PEA2Csn−1PbnBr3n+1 perovskite and suppress the formation of thicker quantum wells.149 Bifunctional 3-ABA can also passivate trap states of perovskite films and achieve a high EQE of 6.3% for blue PeLEDs.
It's more difficult to realize stable PeLEDs because of the higher current density and internal electrical field under working conditions. 2D perovskite materials with bulky cations can significantly improve the stability of PeLEDs. Wang et al. reported that adding 1-naphthylmethylamine iodide (NMAI) into FAPbI3 precursor solution can also realize quasi-2D perovskite films with multiple quantum wells.150 The PeLED reaches a T50 lifetime (time to 50% of the initial brightness) of 2 h under a current density of 10 mA cm−2, while the control device without NMAI can only work for 1 min. The steric hindrance from large cations contribute to both type-I energy transport and improved stability. DJ-phase 2D perovskites with cross-linked inorganic layers also show potential for stable PeLEDs. Shang et al. fabricated 2D PeLEDs with 1,4-bis(aminomethyl)benzene (BAB) and PEA.28 BAB based DJ phase perovskites exhibit higher molecule dissociation energy than PEA based RP phase perovskites (Fig. 11b). BAB based PeLEDs have a T50 lifetime of 100 h under a current density of 25 mA cm−2. Surface passivation is another way for improving the stability of PeLEDs while keeping the high conductivity of 3D perovskite materials.151 Guo et al. passivated FAPbI3 films with four kinds of phenylalkylammonium iodide molecules of different alkyl chain lengths (phenylmethylammonium iodide (PMAI, n = 1), phenethylammonium iodide (PEAI, n = 2), phenpropylammonium iodide (PPAI, n = 3), and phenylbutanammonium iodide (PBAI, n = 4)).34 They found that the lifetime of PeLEDs is significantly improved with longer alkyl chain lengths (Fig. 11c). Density functional theory (DFT) calculations show that phenylalkylammonium with longer chain lengths exhibits larger steric hindrance and inhibits the iodide migration. The PPAI treated PeLED reached a peak EQE of 17.5% and a record T50 lifetime of 130 h under a current density of 100 mA cm−2.
Conclusion and outlook
In conclusion, the past decade has witnessed the rapid development of metal halide perovskite materials and perovskite-based optoelectronic devices. The key structure for metal halide perovskites is the [BX]64− octahedron. This structure endows perovskites with “flexible” properties and can be easily formed. However, this “flexibility” also makes the perovskite structure more prone to degradation. Among various strategies to stabilize the perovskite structure, steric hindrance mainly stabilizes the cations and halide anions by inhibiting their migration. Other than the well-known tolerance factor tuning, steric hindrance can relieve the lattice strain of the crystal structures of 3D perovskites, which effectively improves the intrinsic phase stability of perovskites. Surface termination and incorporation of additives in 3D perovskites by large-sized cations can also passivate surface defects and suppress the degradation of perovskites caused by humidity. In low-dimensional perovskites, the steric hindrance of spacer cations can significantly change the spatial distance, orientation, conductivity and optoelectronic properties of layered perovskites. There are several specific cations that can significantly stabilize perovskites such as quaternary cations, diammonium cations and bifunctional cations. Steric hindrance has proved to be a promising strategy for both efficient and stable perovskite optoelectronic devices. However, the current development of steric hindrance in perovskites is still in the early stages without enough rational molecular design and detailed fundamental mechanism understanding.The concept of steric hindrance has been widely used in selective chemical reactions, especially asymmetric synthesis, and tremendous molecular design samples and theoretical rules have been developed in this field.152–154 Therefore, we believe that there is still enough potential for making good use of steric hindrance with a rational molecular design strategy to further improve the efficiency and stability of perovskite devices. Especially, the combination of previous knowledge on asymmetric syntheses based on steric hindrance and the latest advances in in situ material characterization and theoretical investigations would help us to further explore the mechanism of steric hindrance. With ongoing fundamental investigations and interdisciplinary studies, we believe that more and more molecular and crystal structure design strategies based on steric hindrance will be realized to help achieve the goal of high efficiency and high stability for perovskite materials and their optoelectronic applications.
Author contributions
Y. Z. conceived the idea of the perspective. Y. M., Y. C., H. C. and X. W. wrote and edited the initial manuscript. Y. M. and Y. Z. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (Grant 22025505, 51861145101, and 21777096), Program of Shanghai Academic/Technology Research Leader (Grant Number 20XD1422200), Cultivating fund of Frontiers Science Center for Transformative Molecules (2019PT02) and China Postdoctoral Science Foundation (2020M671110).References
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- S. D. Stranks, V. M. Burlakov, T. Leijtens, J. M. Ball, A. Goriely and H. J. Snaith, Phys. Rev. Appl., 2014, 2, 034007 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. I. Seok, Science, 2020, 370, 108–112 CrossRef CAS PubMed.
- Z. K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y. K. Kim, C. S. Moon, N. J. Jeon, J. P. Correa-Baena, V. Bulovic, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler, D.-H. Kim, S. H. Jo, L. Martínez-Sarti, P. Tan, A. Sadhanala, G.-S. Park, Y.-W. Kim, B. Hu, H. J. Bolink, S. Yoo, R. H. Friend, A. M. Rappe and T.-W. Lee, Nat. Photonics, 2021, 15, 148–155 CrossRef CAS.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- C. K. Ingold, Q. Rev., Chem. Soc., 1957, 11, 1–14 RSC.
- H. Choi, J. H. Ko, Y. H. Kim and S. Jeong, J. Am. Chem. Soc., 2013, 135, 5278–5281 CrossRef CAS PubMed.
- E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2020, 120, 4578–4611 CrossRef CAS PubMed.
- J. Sun, H. Yang and W. Tang, Chem. Soc. Rev., 2021, 50, 2320–2336 RSC.
- Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. Madron du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed.
- M. R. Filip, G. E. Eperon, H. J. Snaith and F. Giustino, Nat. Commun., 2014, 5, 5757 CrossRef CAS PubMed.
- J. Im, C. C. Stoumpos, H. Jin, A. J. Freeman and M. G. Kanatzidis, J. Phys. Chem. Lett., 2015, 6, 3503–3509 CrossRef CAS PubMed.
- K. Wang, Z. Jin, L. Liang, H. Bian, D. Bai, H. Wang, J. Zhang, Q. Wang and S. Liu, Nat. Commun., 2018, 9, 4544 CrossRef PubMed.
- S. Tan, I. Yavuz, N. De Marco, T. Huang, S. J. Lee, C. S. Choi, M. Wang, S. Nuryyeva, R. Wang, Y. Zhao, H. C. Wang, T. H. Han, B. Dunn, Y. Huang, J. W. Lee and Y. Yang, Adv. Mater., 2020, 32, e1906995 CrossRef PubMed.
- X. Wang, Y. Wang, T. Zhang, X. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 1469–1473 CrossRef CAS PubMed.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef CAS PubMed.
- T. Wu, Y. Wang, Z. Dai, D. Cui, T. Wang, X. Meng, E. Bi, X. Yang and L. Han, Adv. Mater., 2019, 31, e1900605 CrossRef PubMed.
- K. Z. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS PubMed.
- J. Hu, I. W. H. Oswald, S. J. Stuard, M. M. Nahid, N. Zhou, O. F. Williams, Z. Guo, L. Yan, H. Hu, Z. Chen, X. Xiao, Y. Lin, Z. Yang, J. Huang, A. M. Moran, H. Ade, J. R. Neilson and W. You, Nat. Commun., 2019, 10, 1276 CrossRef PubMed.
- T. Zhang, M. I. Dar, G. Li, F. Xu, N. Guo, M. Grätzel and Y. Zhao, Sci. Adv., 2017, 3, e1700841 CrossRef PubMed.
- Y. Shang, Y. Liao, Q. Wei, Z. Wang, B. Xiang, Y. Ke, W. Liu and Z. Ning, Sci. Adv., 2019, 5, eaaw8072 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J. C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk and M. G. Kanatzidis, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- C. Liang, H. Gu, Y. Xia, Z. Wang, X. Liu, J. Xia, S. Zuo, Y. Hu, X. Gao, W. Hui, L. Chao, T. Niu, M. Fang, H. Lu, H. Dong, H. Yu, S. Chen, X. Ran, L. Song, B. Li, J. Zhang, Y. Peng, G. Shao, J. Wang, Y. Chen, G. Xing and W. Huang, Nat. Energy, 2020, 6, 38–45 CrossRef.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- R. Wang, J. Xue, K.-L. Wang, Z.-K. Wang, Y. Luo, D. Fenning, G. Xu, S. Nuryyeva, T. Huang, Y. Zhao, J. L. Yang, J. Zhu, M. Wang, S. Tan, I. Yavuz, K. N. Houk and Y. Yang, Science, 2019, 366, 1509–1513 CrossRef CAS PubMed.
- Y. Guo, S. Apergi, N. Li, M. Chen, C. Yin, Z. Yuan, F. Gao, F. Xie, G. Brocks, S. Tao and N. Zhao, Nat. Commun., 2021, 12, 644 CrossRef CAS PubMed.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T. Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- J. Xue, R. Wang, X. Chen, C. Yao, X. Jin, K.-L. Wang, W. Huang, T. Huang, Y. Zhao, Y. Zhai, D. Meng, S. Tan, R. Liu, Z.-K. Wang, C. Zhu, K. Zhu, M. C. Beard, Y. Yan and Y. Yang, Science, 2021, 371, 636–640 CrossRef CAS PubMed.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS.
- J.-H. Lee, N. C. Bristowe, J. H. Lee, S.-H. Lee, P. D. Bristowe, A. K. Cheetham and H. M. Jang, Chem. Mater., 2016, 28, 4259–4266 CrossRef CAS.
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- Y. Zhao and K. Zhu, J. Am. Chem. Soc., 2014, 136, 12241–12244 CrossRef CAS PubMed.
- G. Li, T. Zhang, N. Guo, F. Xu, X. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2016, 55, 13460–13464 CrossRef CAS PubMed.
- Y.-H. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS PubMed.
- J. S. W. Godding, A. J. Ramadan, Y.-H. Lin, K. Schutt, H. J. Snaith and B. Wenger, Joule, 2019, 3, 2716–2731 CrossRef CAS.
- M. I. Saidaminov, J. Kim, A. Jain, R. Quintero-Bermudez, H. Tan, G. Long, F. Tan, A. Johnston, Y. Zhao, O. Voznyy and E. H. Sargent, Nat. Energy, 2018, 3, 648–654 CrossRef CAS.
- S. G. Motti, D. Meggiolaro, A. J. Barker, E. Mosconi, C. A. R. Perini, J. M. Ball, M. Gandini, M. Kim, F. De Angelis and A. Petrozza, Nat. Photonics, 2019, 13, 532–539 CrossRef CAS.
- D. Wang, W. Li, Z. Du, G. Li, W. Sun, J. Wu and Z. Lan, ACS Appl. Mater. Interfaces, 2020, 12, 10579–10587 CrossRef PubMed.
- J. Zhang, S. Wu, T. Liu, Z. Zhu and A. K. Y. Jen, Adv. Funct. Mater., 2019, 29, 1808833 CrossRef CAS.
- C. H. Hendon, R. X. Yang, L. A. Burton and A. Walsh, J. Mater. Chem. A, 2015, 3, 9067–9070 RSC.
- Y. Lou, Y. Niu, D. Yang, Q. Xu, Y. Hu, Y. Shen, J. Ming, J. Chen, L. Zhang and Y. Zhao, Nano Res., 2018, 11, 2715–2723 CrossRef CAS.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., Int. Ed., 2015, 54, 7617–7620 CrossRef CAS PubMed.
- M. Daub and H. Hillebrecht, Angew. Chem., Int. Ed., 2015, 54, 11016–11017 CrossRef CAS PubMed.
- A. M. Ganose, C. N. Savory and D. O. Scanlon, J. Phys. Chem. Lett., 2015, 6, 4594–4598 CrossRef CAS PubMed.
- H. Lu, Y. Liu, P. Ahlawat, A. Mishra, W. R. Tress, F. T. Eickemeyer, Y. Yang, F. Fu, Z. Wang, C. E. Avalos, B. I. Carlsen, A. Agarwalla, X. Zhang, X. Li, Y. Zhan, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, L. Zheng, A. Hagfeldt and M. Grätzel, Science, 2020, 370, eabb8985 CrossRef CAS PubMed.
- S. Bai, P. Da, C. Li, Z. Wang, Z. Yuan, F. Fu, M. Kawecki, X. Liu, N. Sakai, J. T. Wang, S. Huettner, S. Buecheler, M. Fahlman, F. Gao and H. J. Snaith, Nature, 2019, 571, 245–250 CrossRef CAS PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- J. Chen, S. G. Kim and N. G. Park, Adv. Mater., 2018, 30, e1801948 CrossRef PubMed.
- Z. Shi, Y. Zhang, C. Cui, B. Li, W. Zhou, Z. Ning and Q. Mi, Adv. Mater., 2017, 29, e1701656 CrossRef PubMed.
- W. Peng, X. Miao, V. Adinolfi, E. Alarousu, O. El Tall, A. H. Emwas, C. Zhao, G. Walters, J. Liu, O. Ouellette, J. Pan, B. Murali, E. H. Sargent, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 10686–10690 CrossRef CAS.
- H. Chen, Q. Wei, M. I. Saidaminov, F. Wang, A. Johnston, Y. Hou, Z. Peng, K. Xu, W. Zhou, Z. Liu, L. Qiao, X. Wang, S. Xu, J. Li, R. Long, Y. Ke, E. H. Sargent and Z. Ning, Adv. Mater., 2019, 31, e1903559 CrossRef.
- Y. Huang, L. Qiao, Y. Jiang, T. He, R. Long, F. Yang, L. Wang, X. Lei, M. Yuan and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 17834–17842 CrossRef CAS PubMed.
- F. Shao, P. Qin, D. Wang, G. Zhang, B. Wu, J. He, W. Peng, T. C. Sum, D. Wang and F. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 740–746 CrossRef CAS PubMed.
- W. M. J. Franssen, C. M. M. van Heumen and A. P. M. Kentgens, Inorg. Chem., 2020, 59, 3730–3739 CrossRef CAS PubMed.
- Y. Wang, M. I. Dar, L. K. Ono, T. Zhang, M. Kan, Y. Li, L. Zhang, X. Wang, Y. Yang, X. Gao, Y. Qi, M. Gratzel and Y. Zhao, Science, 2019, 365, 591–595 CrossRef CAS PubMed.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- Y. Zhou, H. Xue, Y. H. Jia, G. Brocks, S. Tao and N. Zhao, Adv. Funct. Mater., 2019, 29, 1905739 CrossRef CAS.
- A. Leblanc, N. Mercier, M. Allain, J. Dittmer, T. Pauporte, V. Fernandez, F. Boucher, M. Kepenekian and C. Katan, ACS Appl. Mater. Interfaces, 2019, 11, 20743–20751 CrossRef PubMed.
- A. Leblanc, N. Mercier, M. Allain, J. Dittmer, V. Fernandez and T. Pauporte, Angew. Chem., Int. Ed., 2017, 56, 16067–16072 CrossRef CAS PubMed.
- C.-M. Tsai, Y.-P. Lin, M. K. Pola, S. Narra, E. Jokar, Y.-W. Yang and E. W.-G. Diau, ACS Energy Lett., 2018, 3, 2077–2085 CrossRef CAS.
- I. Spanopoulos, W. Ke, C. C. Stoumpos, E. C. Schueller, O. Y. Kontsevoi, R. Seshadri and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 5728–5742 CrossRef CAS PubMed.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- X. Li, M. Cai, Z. Zhou, K. Yun, F. Xie, Z. Lan, J. Hua and L. Han, J. Mater. Chem. A, 2017, 5, 10480–10485 RSC.
- M. A. Stoeckel, M. Gobbi, S. Bonacchi, F. Liscio, L. Ferlauto, E. Orgiu and P. Samori, Adv. Mater., 2017, 29, 1702469 CrossRef PubMed.
- F. Cai, J. Cai, L. Yang, W. Li, R. S. Gurney, H. Yi, A. Iraqi, D. Liu and T. Wang, Nano Energy, 2018, 45, 28–36 CrossRef CAS.
- Y. Wang, T. Zhang, M. Kan, Y. Li, T. Wang and Y. Zhao, Joule, 2018, 2, 2065–2075 CrossRef CAS.
- F. Wang, W. Geng, Y. Zhou, H. H. Fang, C. J. Tong, M. A. Loi, L. M. Liu and N. Zhao, Adv. Mater., 2016, 28, 9986–9992 CrossRef CAS PubMed.
- H. Zhu, Y. Liu, F. T. Eickemeyer, L. Pan, D. Ren, M. A. Ruiz-Preciado, B. Carlsen, B. Yang, X. Dong, Z. Wang, H. Liu, S. Wang, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar, X. Li and M. Gratzel, Adv. Mater., 2020, 32, 1907757 CrossRef CAS PubMed.
- Y. Yue, N. Salim, Y. Wu, X. Yang, A. Islam, W. Chen, J. Liu, E. Bi, F. Xie, M. Cai and L. Han, Adv. Mater., 2016, 28, 10738–10743 CrossRef CAS PubMed.
- C. Besleaga, L. E. Abramiuc, V. Stancu, A. G. Tomulescu, M. Sima, L. Trinca, N. Plugaru, L. Pintilie, G. A. Nemnes, M. Iliescu, H. G. Svavarsson, A. Manolescu and I. Pintilie, J. Phys. Chem. Lett., 2016, 7, 5168–5175 CrossRef CAS PubMed.
- B. Luo, Y. C. Pu, S. A. Lindley, Y. Yang, L. Lu, Y. Li, X. Li and J. Z. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8864–8868 CrossRef CAS PubMed.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- J. C. Blancon, J. Even, C. C. Stoumpos, M. G. Kanatzidis and A. D. Mohite, Nat. Nanotechnol., 2020, 15, 969–985 CrossRef PubMed.
- D. B. Mitzi, Prog. Inorg. Chem., 1999, 48, 1–121 CAS.
- L. Mao, W. Ke, L. Pedesseau, Y. Wu, C. Katan, J. Even, M. R. Wasielewski, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 3775–3783 CrossRef CAS PubMed.
- C. C. Stoumpos, L. Mao, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2017, 56, 56–73 CrossRef CAS PubMed.
- X. Liu, X. Wang, T. Zhang, Y. Miao, Z. Qin, Y. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60 DOI:10.1002/anie.202102538.
- C. M. M. Soe, C. C. Stoumpos, M. Kepenekian, B. Traore, H. Tsai, W. Nie, B. Wang, C. Katan, R. Seshadri, A. D. Mohite, J. Even, T. J. Marks and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 16297–16309 CrossRef CAS PubMed.
- T. Luo, Y. Zhang, Z. Xu, T. Niu, J. Wen, J. Lu, S. Jin, S. F. Liu and K. Zhao, Adv. Mater., 2019, 31, e1903848 CrossRef.
- V. Gómez, O. Fuhr and M. Ruben, CrystEngComm, 2016, 18, 8207–8219 RSC.
- Y. Li, G. Zheng, C. Lin and J. Lin, Solid State Sci., 2007, 9, 855–861 CrossRef CAS.
- T. Ishihara, J. Takahashi and T. Goto, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 11099–11107 CrossRef CAS.
- W. Paritmongkol, N. S. Dahod, A. Stollmann, N. Mao, C. Settens, S.-L. Zheng and W. A. Tisdale, Chem. Mater., 2019, 31, 5592–5607 CrossRef CAS.
- M. Yu, C. Yi, N. Wang, L. Zhang, R. Zou, Y. Tong, H. Chen, Y. Cao, Y. He, Y. Wang, M. Xu, Y. Liu, Y. Jin, W. Huang and J. Wang, Adv. Opt. Mater., 2018, 7, 1801575 CrossRef.
- K. Yao, X. Wang, Y.-x. Xu, F. Li and L. Zhou, Chem. Mater., 2016, 28, 3131–3138 CrossRef CAS.
- A. Lemmerer and D. G. Billing, Dalton Trans., 2012, 41, 1146–1157 RSC.
- T. M. Koh, V. Shanmugam, X. Guo, S. S. Lim, O. Filonik, E. M. Herzig, P. Müller-Buschbaum, V. Swamy, S. T. Chien, S. G. Mhaisalkar and N. Mathews, J. Mater. Chem. A, 2018, 6, 2122–2128 RSC.
- I. Spanopoulos, I. Hadar, W. Ke, Q. Tu, M. Chen, H. Tsai, Y. He, G. Shekhawat, V. P. Dravid, M. R. Wasielewski, A. D. Mohite, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 5518–5534 CrossRef CAS PubMed.
- X. Gong, O. Voznyy, A. Jain, W. Liu, R. Sabatini, Z. Piontkowski, G. Walters, G. Bappi, S. Nokhrin, O. Bushuyev, M. Yuan, R. Comin, D. McCamant, S. O. Kelley and E. H. Sargent, Nat. Mater., 2018, 17, 550–556 CrossRef CAS PubMed.
- D. Ghosh, A. J. Neukirch and S. Tretiak, J. Phys. Chem. Lett., 2020, 11, 2955–2964 CrossRef CAS PubMed.
- R. Quintero-Bermudez, A. Gold-Parker, A. H. Proppe, R. Munir, Z. Yang, S. O. Kelley, A. Amassian, M. F. Toney and E. H. Sargent, Nat. Mater., 2018, 17, 900–907 CrossRef CAS PubMed.
- X. Li, W. Ke, B. Traore, P. Guo, I. Hadar, M. Kepenekian, J. Even, C. Katan, C. C. Stoumpos, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 12880–12890 CrossRef CAS PubMed.
- Y. Chen, Y. Sun, J. Peng, W. Zhang, X. Su, K. Zheng, T. Pullerits and Z. Liang, Adv. Energy Mater., 2017, 7, 1700162 CrossRef.
- Z. Xu, D. B. Mitzi, C. D. Dimitrakopoulos and K. R. Maxcy, Inorg. Chem., 2003, 42, 2031–2039 CrossRef CAS PubMed.
- T. Li, W. A. Dunlap-Shohl, Q. Han and D. B. Mitzi, Chem. Mater., 2017, 29, 6200–6204 CrossRef CAS.
- J. L. Knutson, J. D. Martin and D. B. Mitzi, Inorg. Chem., 2005, 44, 4699–4705 CrossRef CAS PubMed.
- A. H. Proppe, M. Wei, B. Chen, R. Quintero-Bermudez, S. O. Kelley and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 14180–14189 CrossRef CAS PubMed.
- C. Ortiz-Cervantes, P. I. Roman-Roman, J. Vazquez-Chavez, M. Hernandez-Rodriguez and D. Solis-Ibarra, Angew. Chem., Int. Ed., 2018, 57, 13882–13886 CrossRef CAS PubMed.
- Y. Gao, E. Shi, S. Deng, S. B. Shiring, J. M. Snaider, C. Liang, B. Yuan, R. Song, S. M. Janke, A. Liebman-Pelaez, P. Yoo, M. Zeller, B. W. Boudouris, P. Liao, C. Zhu, V. Blum, Y. Yu, B. M. Savoie, L. Huang and L. Dou, Nat. Chem., 2019, 11, 1151–1157 CrossRef CAS PubMed.
- F. Zhang, D. H. Kim, H. Lu, J. S. Park, B. W. Larson, J. Hu, L. Gao, C. Xiao, O. G. Reid, X. Chen, Q. Zhao, P. F. Ndione, J. J. Berry, W. You, A. Walsh, M. C. Beard and K. Zhu, J. Am. Chem. Soc., 2019, 141, 5972–5979 CrossRef CAS PubMed.
- J. Shi, Y. Gao, X. Gao, Y. Zhang, J. Zhang, X. Jing and M. Shao, Adv. Mater., 2019, 31, e1901673 CrossRef PubMed.
- H. Pan, X. Zhao, X. Gong, Y. Shen and M. Wang, J. Phys. Chem. Lett., 2019, 10, 1813–1819 CrossRef CAS PubMed.
- W. Fu, H. Liu, X. Shi, L. Zuo, X. Li and A. K. Y. Jen, Adv. Funct. Mater., 2019, 29, 1900221 CrossRef.
- D. B. Mitzi, C. D. Dimitrakopoulos and L. L. Kosbar, Chem. Mater., 2001, 13, 3728–3740 CrossRef CAS.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- L. Yangi, Y. Li, Y. Pei, J. Wang, H. Lin and X. Li, J. Mater. Chem. A, 2020, 8, 7808–7818 RSC.
- Y. Wang, T. Zhang, M. Kan and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 12345–12348 CrossRef CAS PubMed.
- Y. Wang, X. Liu, T. Zhang, X. Wang, M. Kan, J. Shi and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16691–16696 CrossRef CAS PubMed.
- M. Safdari, P. H. Svensson, M. T. Hoang, I. Oh, L. Kloo and J. M. Gardner, J. Mater. Chem. A, 2016, 4, 15638–15646 RSC.
- M. Safdari, D. Phuyal, B. Philippe, P. H. Svensson, S. M. Butorin, K. O. Kvashnina, H. Rensmo, L. Kloo and J. M. Gardner, J. Mater. Chem. A, 2017, 5, 11730–11738 RSC.
- H. Wang, C. C. S. Chan, M. Chu, J. Xie, S. Zhao, X. Guo, Q. Miao, K. S. Wong, K. Yan and J. Xu, Sol. RRL, 2020, 123, 1900578 CrossRef.
- Q. He, M. Worku, L. Xu, C. Zhou, H. Lin, A. J. Robb, K. Hanson, Y. Xin and B. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 1159–1168 CrossRef CAS PubMed.
- T. Zhang, Y. Hui, L. Chen, G. Li, B. Mao and Y. Zhao, J. Phys. D: Appl. Phys., 2018, 51, 404001 CrossRef.
- C. Ma, D. Shen, T. W. Ng, M. F. Lo and C. S. Lee, Adv. Mater., 2018, 30, e1800710 CrossRef.
- S. Ahmad, P. Fu, S. Yu, Q. Yang, X. Liu, X. Wang, X. Wang, X. Guo and C. Li, Joule, 2019, 3, 794–806 CrossRef CAS.
- J. A. Steele, H. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. Yuan, C. Martin, E. Solano, K. Lejaeghere, S. M. J. Rogge, C. Notebaert, W. Vandezande, K. P. F. Janssen, B. Goderis, E. Debroye, Y.-K. Wang, Y. Dong, D. Ma, M. Saidaminov, H. Tan, Z. Lu, V. Dyadkin, D. Chernyshov, V. Van Speybroeck, E. H. Sargent, J. Hofkens and M. B. J. Roeffaers, Science, 2019, 365, 679–684 CrossRef CAS PubMed.
- M. Chen, M.-G. Ju, M. Hu, Z. Dai, Y. Hu, Y. Rong, H. Han, X. C. Zeng, Y. Zhou and N. P. Padture, ACS Energy Lett., 2018, 4, 276–277 CrossRef.
- P. Li, X. Liu, Y. Zhang, C. Liang, G. Chen, F. Li, M. Su, G. Xing, X. Tao and Y. Song, Angew. Chem., Int. Ed., 2020, 59, 6909–6914 CrossRef CAS PubMed.
- B. E. Cohen, Y. Li, Q. Meng and L. Etgar, Nano Lett., 2019, 19, 2588–2597 CrossRef CAS PubMed.
- Y. Li, J. V. Milic, A. Ummadisingu, J. Y. Seo, J. H. Im, H. S. Kim, Y. Liu, M. I. Dar, S. M. Zakeeruddin, P. Wang, A. Hagfeldt and M. Gratzel, Nano Lett., 2019, 19, 150–157 CrossRef CAS PubMed.
- Y. Zheng, T. Niu, J. Qiu, L. Chao, B. Li, Y. Yang, Q. Li, C. Lin, X. Gao, C. Zhang, Y. Xia, Y. Chen and W. Huang, Sol. RRL, 2019, 3, 1900090 CrossRef.
- T. Niu, H. Ren, B. Wu, Y. Xia, X. Xie, Y. Yang, X. Gao, Y. Chen and W. Huang, J. Phys. Chem. Lett., 2019, 10, 2349–2356 CrossRef CAS PubMed.
- T. Zhao, C.-C. Chueh, Q. Chen, A. Rajagopal and A. K. Y. Jen, ACS Energy Lett., 2016, 1, 757–763 CrossRef CAS.
- J. Lu, L. Jiang, W. Li, F. Li, N. K. Pai, A. D. Scully, C.-M. Tsai, U. Bach, A. N. Simonov, Y.-B. Cheng and L. Spiccia, Adv. Energy Mater., 2017, 7, 1700444 CrossRef.
- X. Li, M. I. Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Gratzel, Nat. Chem., 2015, 7, 703–711 CrossRef CAS PubMed.
- T. Zhang, L. Xie, L. Chen, N. Guo, G. Li, Z. Tian, B. Mao and Y. Zhao, Adv. Funct. Mater., 2017, 27, 1603568 CrossRef.
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Gratzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- G. Grancini, C. Roldan-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- N. Mercier, CrystEngComm, 2005, 7, 429–432 RSC.
- M. E. Kayesh, K. Matsuishi, R. Kaneko, S. Kazaoui, J.-J. Lee, T. Noda and A. Islam, ACS Energy Lett., 2019, 4, 278–284 CrossRef CAS.
- J. Qing, X.-K. Liu, M. Li, F. Liu, Z. Yuan, E. Tiukalova, Z. Yan, M. Duchamp, S. Chen, Y. Wang, S. Bai, J.-M. Liu, H. J. Snaith, C.-S. Lee, T. C. Sum and F. Gao, Adv. Energy Mater., 2018, 8, 1800185 CrossRef.
- C. Qin, T. Matsushima, W. J. Potscavage, A. S. D. Sandanayaka, M. R. Leyden, F. Bencheikh, K. Goushi, F. Mathevet, B. Heinrich, G. Yumoto, Y. Kanemitsu and C. Adachi, Nat. Photonics, 2019, 14, 70–75 CrossRef.
- R. Quintero-Bermudez, A. H. Proppe, A. Mahata, P. Todorovic, S. O. Kelley, F. De Angelis and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 13459–13467 CrossRef CAS.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
- M. Yuan, L. N. Quan, R. Comin, G. Walters, R. Sabatini, O. Voznyy, S. Hoogland, Y. Zhao, E. M. Beauregard, P. Kanjanaboos, Z. Lu, D. H. Kim and E. H. Sargent, Nat. Nanotechnol., 2016, 11, 872–877 CrossRef CAS PubMed.
- Y. K. Wang, D. Ma, F. Yuan, K. Singh, J. M. Pina, A. Johnston, Y. Dong, C. Zhou, B. Chen, B. Sun, H. Ebe, J. Fan, M. J. Sun, Y. Gao, Z. H. Lu, O. Voznyy, L. S. Liao and E. H. Sargent, Nat. Commun., 2020, 11, 3674 CrossRef CAS PubMed.
- N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao, W. Zou, C. Yi, Y. Sun, Y. Cao, R. Yang, Y. Wei, Q. Guo, Y. Ke, M. Yu, Y. Jin, Y. Liu, Q. Ding, D. Di, L. Yang, G. Xing, H. Tian, C. Jin, F. Gao, R. H. Friend, J. Wang and W. Huang, Nat. Photonics, 2016, 10, 699–704 CrossRef CAS.
- Y. Miao, Y. Ke, N. Wang, W. Zou, M. Xu, Y. Cao, Y. Sun, R. Yang, Y. Wang, Y. Tong, W. Xu, L. Zhang, R. Li, J. Li, H. He, Y. Jin, F. Gao, W. Huang and J. Wang, Nat. Commun., 2019, 10, 3624 CrossRef PubMed.
- Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS.
- A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Chem. Rev., 2020, 120, 11479–11615 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |