
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphite mediated asymmetric N to C migration for the synthesis of chiral heterocycles from primary amines†
Soniya
Rani
ab,
Soumya Ranjan
Dash
 bc,
Asish
Bera
ab,
Md Nirshad
Alam
ab,
Kumar
Vanka
bc and
Pradip
Maity
*a
bc,
Asish
Bera
ab,
Md Nirshad
Alam
ab,
Kumar
Vanka
bc and
Pradip
Maity
*a
aOrganic Chemistry Division, CSIR-National Chemical Laboratory, Pune-411008, India. E-mail: p.maity@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cPhysical and Material Chemistry Division, CSIR-National Chemical Laboratory, Pune 411008, India
First published on 28th May 2021
Abstract
A phosphite mediated stereoretentive C–H alkylation of N-alkylpyridinium salts derived from chiral primary amines was achieved. The reaction proceeds through the activation of the N-alkylpyridinium salt substrate with a nucleophilic phosphite catalyst, followed by a base mediated [1,2] aza-Wittig rearrangement and subsequent catalyst dissociation for an overall N to C-2 alkyl migration. The scope and degree of stereoretention were studied, and both experimental and theoretical investigations were performed to support an unprecedented aza-Wittig rearrangement–rearomatization sequence. A catalytic enantioselective version starting with racemic starting material and chiral phosphite catalyst was also established following our understanding of the stereoretentive process. This method provides efficient access to tertiary and quaternary stereogenic centers in pyridine systems, which are prevalent in drugs, bioactive natural products, chiral ligands, and catalysts.
Introduction
The recent focus on sustainable chemistry has encouraged chemists to develop the direct and diverse functionalization of abundant and renewable chemicals.1 In this context, primary alkyl amines have emerged as an inexpensive, stable, and abundant source for the generation of reactive intermediates. In a recent application of its use as an alkylating agent,2 the Watson group in 2017 developed an elegant nickel catalysed method to generate alkyl radicals from stable Katritzky pyridinium salts of primary amines for their coupling with arylboronic acids.3 In the same year, the Glorius group reported an iridium based photoredox catalysed protocol for alkyl radical generation and its coupling with heteroaromatic compounds.4 Since then, this approach has become a focus of intensive study via new and elegant deaminative protocols with diverse reaction partners under photoredox and dual catalysis or with redox-active coupling partners (Fig. 1a).5,6 These deaminative processes to alkyl radicals lead to racemic products from both achiral and enantioenriched amines. Additionally, the C–N bond cleavage processes generate triphenylpyridine as a part of the waste stream.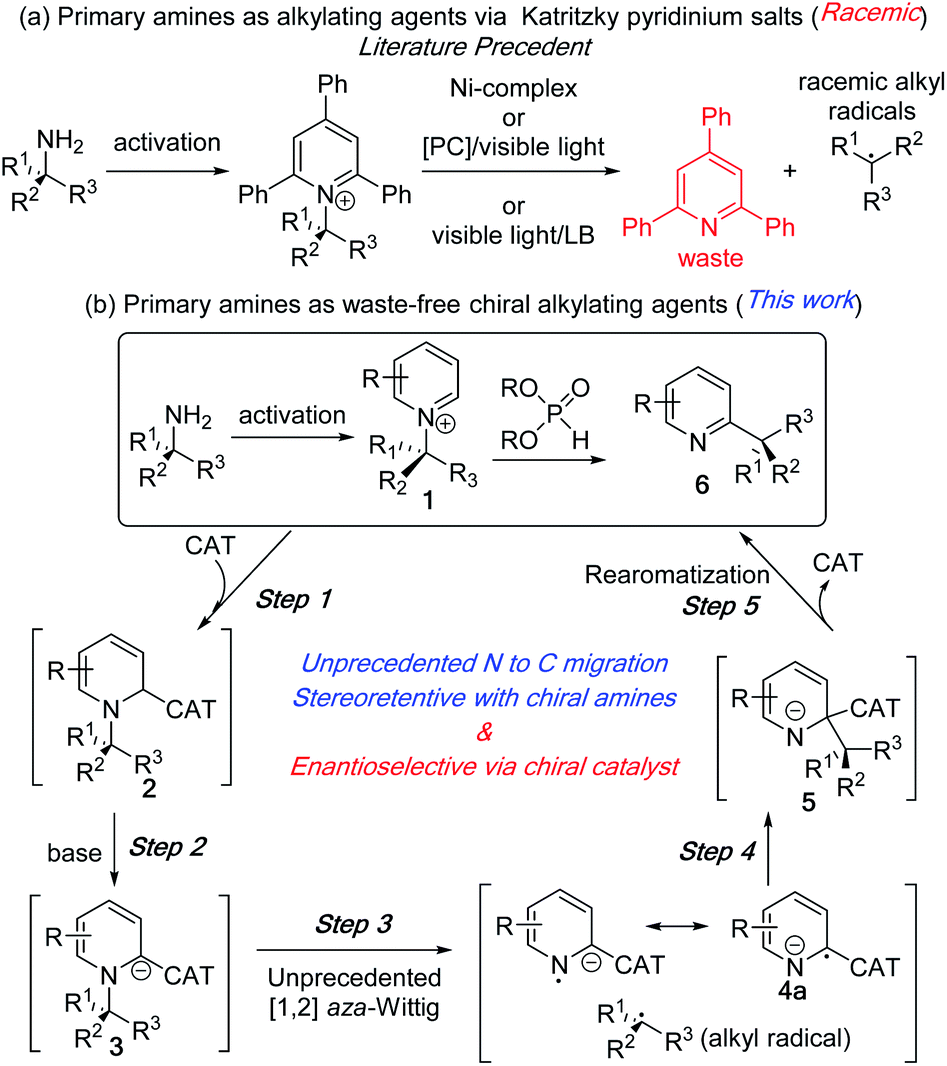 | ||
| Fig. 1 Primary amines as alkylating reagent. (a) Literature precedents. (b) Proposed N to C dissociative alkyl migration. | ||
We reasoned that the unique reactivity of pyridinium salts could be exploited to generate enantioenriched products that retain the pyridine ring system. The synthetic utility of our approach lies in the various simple methods for generating alkylpyridinium salts other than Katritzky salts (1) from amines.7 Despite the prevalence of chiral alkyl pyridines (6) and the corresponding saturated heterocycles in numerous pharmaceutical drugs and biologically active compounds,8 their enantioselective synthesis via catalytic methods remains a challenge.9–12 In this communication, we report an intramolecular alkylation process with chirality transfer to synthesize chiral pyridines from enantioenriched pyridinium salts without the loss of pyridines as waste (Fig. 1b). We also report a catalytic enantioselective version of this unprecedented N to C alkyl migration from racemic N-alkylpyridinium salt.
The reaction development strategy is based on our experience in various selective molecular rearrangements and radical reactions.11e,13 We envisioned that a suitable catalyst adduct (2), upon deprotonation could form a reactive carbanion (3) and subsequent stable radical anion (4a) to reduce the activation energy to effect an [1,2] aza-Wittig rearrangement (3 to 5, Fig. 1b). To the best of our knowledge, the anionic [1,2] aza-rearrangement is not known, presumably due to its high activation energy requirement.14 The [1,2] alkyl migration (5) intermediate would undergo a facile catalyst dissociation to waste-free intramolecular C–H alkylation product (6). This subsequent rearomatization exothermic step after the aza-Wittig should favour the overall thermodynamics. Although the C–N bond breaks during the rearrangement, the stereochemical outcome of this intramolecular process would be intriguing.15 A highly stereoretentive migration or a chiral catalyst controlled stereoselective recombination of radical pair could provide us with multiple pathways to control the stereoselectivity.
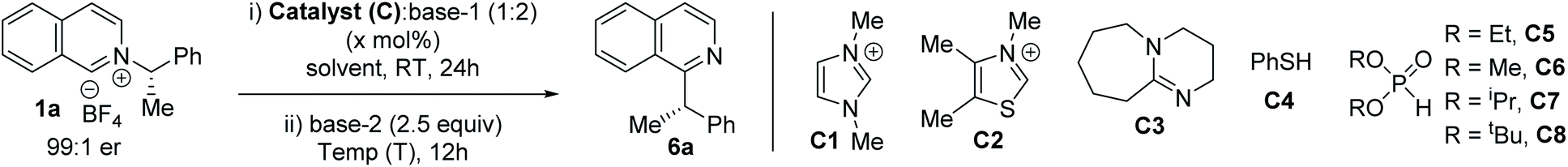
To explore the feasibility of alkyl migration, racemic (S)-1-phenylethylamine salt of isoquinoline (1a) was selected as the substrate. A variety of catalysts were screened based on their nucleophilicity, and subsequent anion and radical stabilization ability (Table 1).13,16 NHC catalysts, DBU, and thiophenol were unsuccessful with different bases (entries 1–4). But, to our delight, the addition of stoichiometric diethylphosphite anion at room temperature, followed by treatment with 2.5 equivalents of LiHMDS at 0 °C led to 58% of the migration product 6a (Table 1, entry 5).17 With a suitable catalyst in hand, we performed the reaction with enantioenriched 1a to explore the stereochemical outcome. The chiral pyridinium salt ((S)-1a) was synthesized from commercially available chiral (S)-1-phenylethylamine.7b The migration in THF at 0 °C with (S)-1a led to an encouraging 86% enantiospecificity (es) with retention of the configuration (entry 6).9j Next, we optimize our stereoretentive method, starting with screening the role of solvent. MTBE instead of THF led to 88% es, while toluene led to 91% es (entries 7 and 8). Cyclohexane as solvent did not improve the stereoretention (90% es, entry 9), but DCM as solvent led to 96% es (entry 10). The counter cation of the HMDS base also plays a significant role, with sodium being more effective than lithium (entry 11).18 Other phosphites were tested next, which reveals better results with smaller alkoxy groups (entries 12–14). Finally, the optimal reaction temperature was determined to be −20 °C (entries 15–17). Although the racemic diethylphosphite can be used in catalytic amounts under strict deoxygenated conditions at lower temperature (entry 18), we utilized stoichiometric amounts of the phosphites to simplify the operation to obtain the product at a shorter reaction time, since diethylphosphite is non-toxic and cheaper than the reaction solvents. Since THF is detrimental to the stereoretention (entries 6 vs. 9), we performed the reaction with 2 M THF solution of NaHMDS instead of 1 M, which improved the es to 97% (entry 19), leading to substituted isoquinoline (S)-6a in 72% yield and 92% ee from enantioenriched (98% ee) (S)-1-phenylethylamine salt (1a).19 The optimized reaction works well on 1 mmol scale, with no change in stereoretention and comparable yield (entry 20).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | C (mol%) | T (°C) | Base-1 | Base-2 | Solvent | 6a, yield (%) | 6a, % ee (es) |
| a Reactions were carried out with 0.3 mmol 1a, 4 ml solvent, and 1 M THF solution of MHMDS base. b With catalytic (20 mol%) dimethylphosphite diethylphosphite catalyst (see ESI for procedure). c With 2 M NaHMDS in THF. d 1 mmol scale. | |||||||
| 1 | C1 (50) | 60 | Cs2CO3 | Cs2CO3 | THF | ND | — |
| 2 | C2 (50) | 60 | Cs2CO3 | Cs2CO3 | THF | ND | — |
| 3 | C3 (50) | 60 | Cs2CO3 | Cs2CO3 | THF | ND | — |
| 4 | C4 (100) | 25 | K2CO3 | LiHMDS | THF | ND | — |
| 5 | C5 (100) | 0 | K2CO3 | LiHMDS | THF | 58 | — |
| 6 | C5 (100) | 0 | K2CO3 | LiHMDS | THF | 63 | 69 (86) |
| 7 | C5 (100) | 0 | K2CO3 | LiHMDS | MTBE | 61 | 73 (88) |
| 8 | C5 (100) | 0 | K2CO3 | LiHMDS | Toluene | 64 | 80 (91) |
| 9 | C5 (100) | 0 | K2CO3 | LiHMDS | Cyclohexane | 47 | 78 (90) |
| 10 | C5 (100) | 0 | K2CO3 | LiHMDS | DCM | 67 | 90 (96) |
| 11 | C5 (100) | 0 | K2CO3 | NaHMDS | DCM | 70 | 90 (96) |
| 12 | C6 (100) | 0 | K2CO3 | NaHMDS | DCM | 66 | 88 (95) |
| 13 | C7 (100) | 0 | K2CO3 | NaHMDS | DCM | 55 | 85 (93) |
| 14 | C8 (100) | 0 | K2CO3 | NaHMDS | DCM | 26 | 59 (80) |
| 15 | C5 (100) | −20 | K2CO3 | NaHMDS | DCM | 72 | 91 (96) |
| 16 | C5 (100) | −40 | K2CO3 | NaHMDS | DCM | 69 | 91 (96) |
| 17 | C5 (100) | −60 | K2CO3 | NaHMDS | DCM | 70 | 91 (96) |
| 18b | C5 (20) | −60 | K2CO3 | NaHMDS | DCM | 67 | 91 (96) |
| 19c | C5 (100) | −20 | K2CO3 | NaHMDS | DCM | 72 | 92 (97) |
| 20d | C5 (100) | −20 | K2CO3 | NaHMDS | DCM | 70 | 92 (97) |
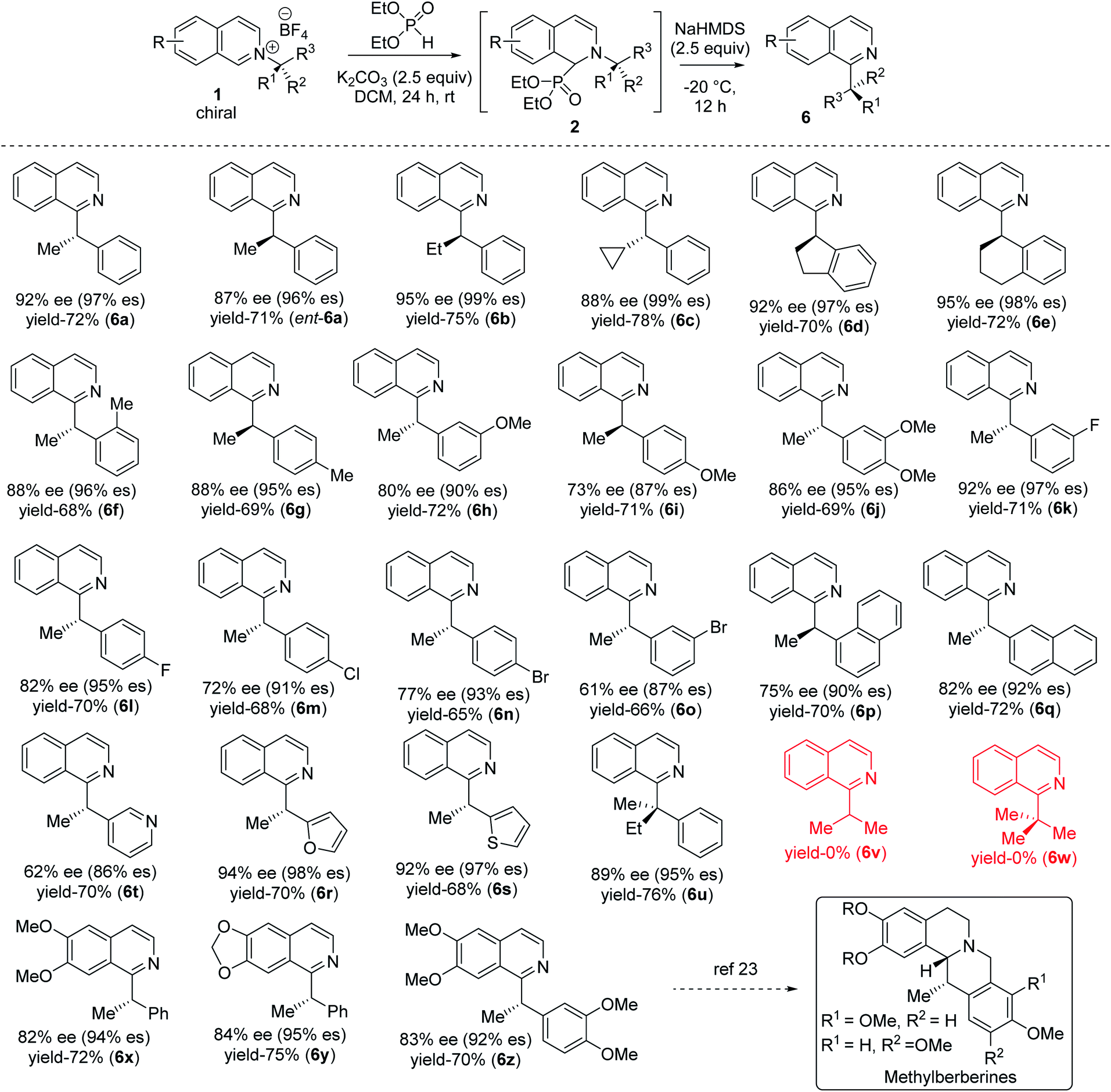
With optimized reaction conditions in hand, we tested the generality of our stereoretentive method for both chiral amines20 and pyridine derivatives. First, we explored the scope and efficiencies of different chiral amines with isoquinoline (Table 2). Gratifyingly, the enantiomer of 1a (ent-1a) led to an efficient synthesis of ent-6a. Alkyl groups other than methyl at the migrating carbon such as ethyl (6b), cyclopropyl (6c), fused cyclopentyl (6d), and cyclohexyl (6e) were all resulted in good yields with almost quantitative chirality transfer. These examples showed that the alkyl group at migrating carbon has very little influence on the reaction outcome. The α-cyclopropyl group at migrating carbon is significant with no ring-opening product was observed.21 Next, we varied the aryl groups at the migrating carbon. Methyl at ortho- (6f) and para-position (6g) of the phenyl ring led to similar results, indicating a minimal steric effect on yield and enantiospecificity. Methoxy substitution at both C-3 (6h) and C-4 (6i) led to lower stereoretention (90 and 87%), whereas 3,4-dimethoxy substitution (6j) lead to 95% es. The yields are comparable to that of parent substrate for all methoxyaryl substrates. Halo substituents at C-3 and C-4 positions were well tolerated. Fluoro substituent at C-3 led to 97% es (6k), while C-4 fluro resulted in 95% es (6l). Both chloro and bromo substitution resulted in slightly lower chirality transfer (6m,n,o). α-Napthyl instead of phenyl formed product with 90% es (6p), and β-naphthyl yielded the corresponding products with 92% es (6q). Heteroaryl groups at the migrating center such as electron-rich 2-furan (6r) and 2-thiophene (6s) both migrated with excellent enantiospecificity (98 & 97% es), while electron-deficient 3-pyridine (6t) led to a moderate 86% es. Finally, we synthesized an isoquinolinium salt of quaternary chiral amine (1u). Under optimized reaction conditions, it rearranged successfully to the corresponding all carbon quaternary alkyl chiral products with better yield (76%) and very good 95% es (6u). Isoquinolinium salts of isopropyl (6v) and tert-butyl (6w) amine did not lead to the product which indicates (hetero) aryl stabilization on migrating alkyl radical is necessary.
| a Conditions: 1 (0.3 mmol) was dissolved in 3 ml DCM and added to diethylphosphite (0.3 mmol) and K2CO3 (0.75 mmol) in 1 ml DCM at 25 °C and stirred for 24 h. Then the reaction mixture was cooled to −20 °C and NaHMDS (2.5 equiv., 2 M in THF) was added and stirred for 12 h. |
|---|
|
A diverse range of substituted pyridines was subjected to the reaction condition to find the generality of our method with azaarenes. Electron rich bis-alkoxy groups on isoquinolines were tested first since the corresponding products could be easily converted to the numerous natural products of biological importance.22 4,5-Bis-alkoxyisoquinoline derivatives of (S)-1-phenylethylamine (6x, 6y) resulted in 94 and 95% es with 72 and 75% yields respectively. With 4,5-dimethoxyisoquinoline and 3,4-dimethoxyphenylethylamine, the product 6z is obtained in 70% yield with 92% es. 6z and its reduced forms were utilized for the synthesis of various methylberberine alkaloids.22a,c
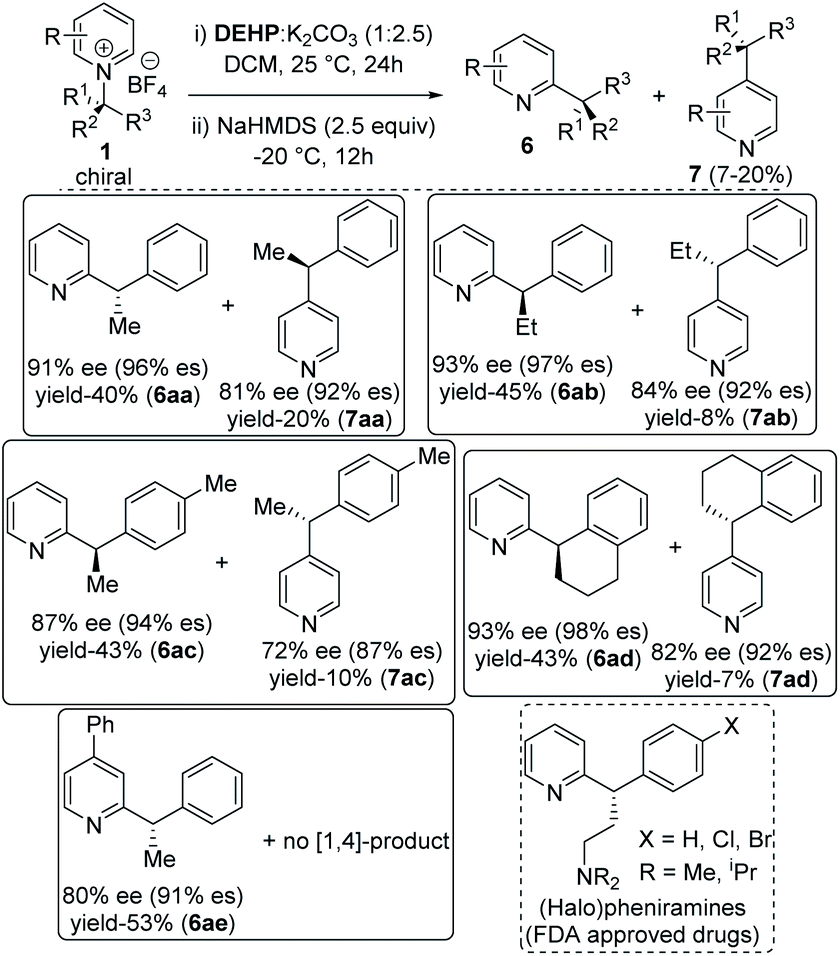
Finally, the alkyl migration protocol was tested on unsubstituted pyridine substrates, which are common pharmacophores in many approved pharmaceutical drugs.7,23 Under standard reaction condition, the C-2 alkyl migrated products formed in moderate yields but with excellent enantiospecificity (Table 3). The stereoretention trend in alkyl migration runs parallel to that in isoquinoline substrates. For 4-unsubstituted pyridines, a minor [1,4] migrated side products 7 also formed with 4–7% lower enantiospecificities. Competitive [1,4] Wittig rearrangements from common radical pair intermediates are known in literature, with a single report by Maleczka Jr group describes stereoretention pattern similar to our findings.24 With pyridinium substrates, either the same C-2 added catalyst or a C-4 added catalyst intermediate can undergo the [1,4] migration.
|
Quantum chemical calculations (DFT, details in ESI†) were performed to gain insight into the crucial C–N bond breaking (step 3, Fig. 1b) and C–C bond forming (step 4, Fig. 1b) steps of our proposed phosphite catalysed aza-Wittig, and subsequent rearomatization protocol.25 First, the C–N bond dissociation energy (BDE) was calculated at the PBE/def-TZVP level of theory. The ΔG for the homolytic C–N bond cleavage was calculated to be exothermic by 0.8 kcal mol−1, supporting the stability of radical 4avia the catalyst mediated captodative effect and extended conjugation.13,16 An alternative heterolytic bond cleavage path with 2.4 kcal mol−1 higher energy via an E2 could also be identified during the calculations. To avoid any bias from the chosen DFT method, and since the difference between homolytic and heterolytic BDE was close, we used different levels of theory to compare the two possibilities.26 In all cases, the homolytic dissociation was found to be favourable (Table 4). We also calculated homolytic and heterolytic dissociation for substrate 1u with a quaternary migrating group, in which the homolytic cleavage is favoured by 5.7 kcal mol−1 (at the PBE/def-TZVP level of theory).
| Substrate | DFT methods | Homo | Hetero | Homo–hetero |
|---|---|---|---|---|
| 1a | PBE/def-TZVP | −0.8 | +1.6 | −2.4 |
| B3LYP/def2-TZVPP | −5.0 | −1.4 | −3.6 | |
| B3LYP/6-31+G** | −16.9 | −10.5 | −6.4 | |
| M06-2X/6-31+G** | +1.8 | +7.2 | −5.4 | |
| M06-2X/6-311+G** | +4.1 | +8.0 | −3.9 | |
| 1u | PBE/def-TZVP | −6.9 | −1.2 | −5.7 |
| B3LYP/def2-TZVPP | −11.1 | −3.9 | −7.2 |
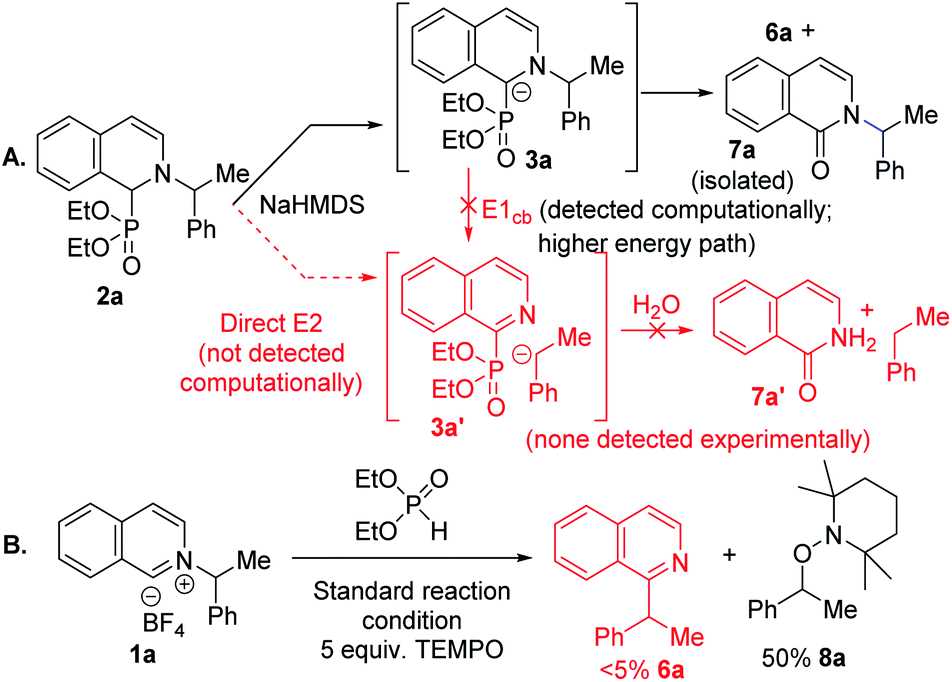
Experimental mechanistic studies were carried out to support or discredit the alternative paths identified in the computational studies (Scheme 1). We noticed that in the presence of traces of oxygen, the corresponding pyridone side product formed with the C–N bond intact under the N to C migration protocol (Scheme 1a). This result is suggestive of a common carbanionic intermediate 3a for its competing Nef oxidation versus alkyl migration.21a An E1cb pathway would lead to C–N bond cleaved intermediate 3a′, or its hydrolyzed product 7a′, which were not detected. A direct base mediated E2 pathway to 3a′ could also be a possibility, but was not detected computationally. To further distinguish between heterolytic E1cb/E2 and homolytic radical pair paths, a radical trapping experiment was conducted with TEMPO. The addition of five equivalents of TEMPO led to a complete shut down of the alkyl migration with 50% TEMPO trapped alkyl product formation (Scheme 1b). No alkyl migration product under excess radical trap reagent along with trapped alkyl product supports a radical pair mechanism.27
 | ||
| Scheme 1 Mechanistic studies. (A) Homolytic vs. heterolytic paths via intermediate trapping. (B) Radical trap with TEMPO. | ||
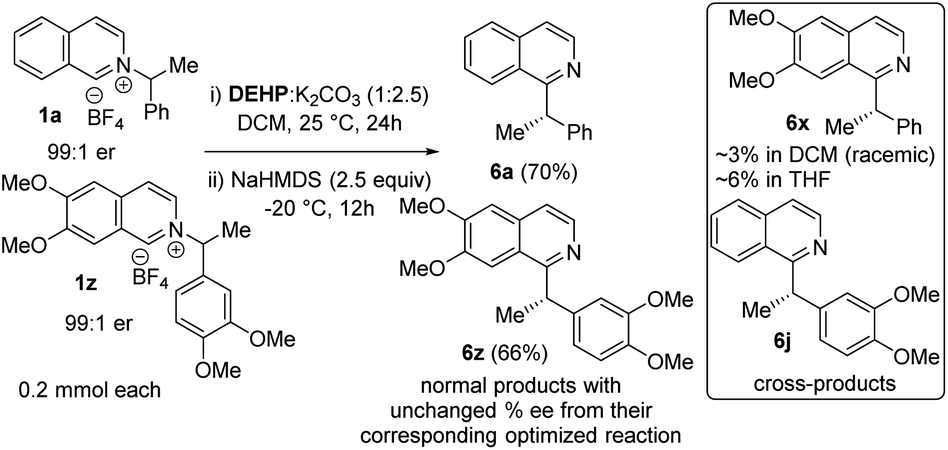
Next, we examined the source of high stereoretention for this dissociative alkyl migration (Scheme 2). For [1,2] oxa-Wittig rearrangements, the commonly accepted explanation for stereoretention at the migrating centre is that the radical pair formation and recombination occurs within the solvent cage.15 This solvent-caged radical pair recombination is faster than racemization through solvent-cage escape and recapture. On the other hand, out-of-plane rotation within the solvent cage can also lead to racemization.28 To gain insight, we conducted a crossover experiment with 1a and 1z in DCM and THF. The cross-products formations in DCM are ∼2–3%, which supports predominantly solvent-caged radical recombination. In THF, the cross-products are ∼4–6%, which is consistent with less stereoretention in that solvent. The chiral HPLC analysis of cross-products showed that those were rendered almost racemic, supporting solvent cage escape as one possible pathway for racemization. The normal products, which now have less ‘solvent-cage escape and recapture’ possibility in the crossover experiment, did not result in significantly higher stereoretention. Therefore an out-of-plane rotation within the solvent cage cannot be ruled out.
 | ||
| Scheme 2 Crossover experiment & stereoretention. | ||
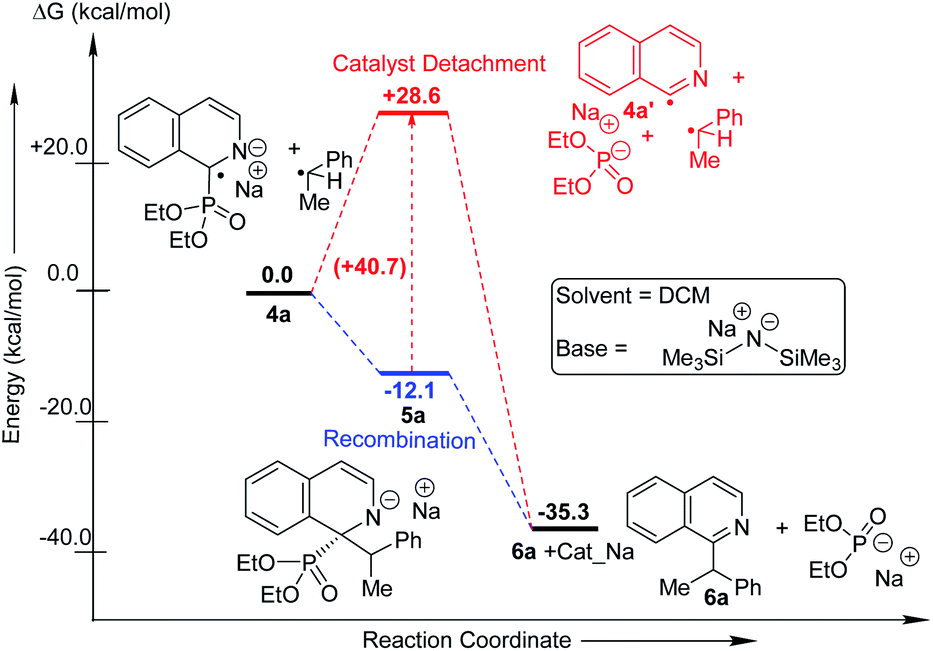
We performed computational studies for radical pair recombination in order to determine the exact juncture of catalyst detachment (step 4, Fig. 1b; Scheme 3). The catalyst detachment from radical 4a to 4a′, prior to its recombination with alkyl radical, signifies no catalyst control on the subsequent C–C bond formation. Although we predict radical 4a formation from anionic intermediate 3a would be stabilized by captodative effect and extended delocalization, the formation of 4a′ might be feasible owing to the rearomatization of the heterocycle. The calculations reveal that this is highly unlikely, since the ΔG for the corresponding imine radical (4a′) formation is endothermic by 28.6 kcal mol−1. The loss of conjugation and captodative stability seems to outweigh the rearomatization in 4a′. The recombination of the catalyst attached 4a at C-2 with the alkyl radical could, therefore, be influenced by the catalyst. The subsequent catalyst release from 5a leading to the rearomatized final product (6a) is found to be highly exothermic (23.1 kcal mol−1), which makes the overall transformation favourable.
 | ||
| Scheme 3 Computed C–C bond formation and subsequent rearomatization energy profile. | ||
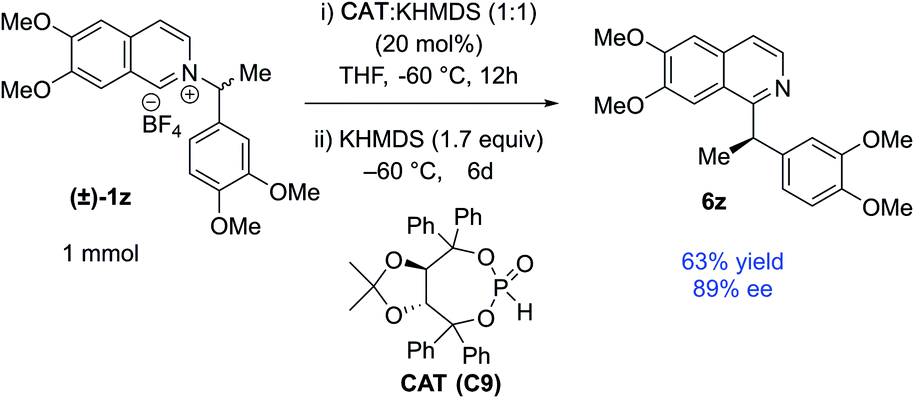
The catalyst involvement in the C–C bond formation and the possibility of racemization via dissociative path led us to speculate if a chiral phosphite catalyst could induce enantioselective transformation starting with racemic pyridinium salts. During reaction optimization, we observed variable degrees of racemization with different catalysts (Table 1). The significant drop in stereoretention with a bulky tert-butyl phosphite (80 vs. 96% es, Table 1, entries 12 & 14) could be due to its slower recombination, with racemization paths in play. Therefore, a bigger chiral phosphite catalyst could also slow the recombination for the racemic alkyl radical to allow its rotation to catalyst controlled enantioselection. Since the reaction medium influences the stereoretention (Table 1, entries 7–10), solvents with high racemization might also provide time for the alkyl radical to rotate for the enantioselection. We chose racemic 1z as a test substrate, since it can be converted to natural products, and its stereoretentive migration generated the product in a modest 83% ee (92% es, Table 2). To our delight, 20 mol% TADDOL-phosphite catalyst (C9) generates chiral 6z with 89% ee in THF (Scheme 4, see ESI† for details). This result represents a proof-of-concept for the chiral phosphite catalyzed enantioselective N to C migration of racemic pyridinium salts.
 | ||
| Scheme 4 Chiral phosphite catalyzed enantioselective N to C migration. | ||
In conclusion, a phosphite mediated stereoretentive C–H alkylation of N-alkylpyridinium salt was developed. Chiral alkylpyridines were obtained from the corresponding chiral amines with a broad range of functional groups. An all-carbon quaternary alkyl group also migrated efficiently. The mechanistic experiments suggest a phosphite catalyzed bis-radical [1,2] aza-Wittig followed by rearomatization via catalyst release for product formation. Exploring the source of stereoretention and a variable degree of racemization led us to establish a chiral phosphite catalyzed enantioselective version with racemic alkylpyridinium salt. The dissociative reaction path presents us with the possibility of [1,n] alkyl migrations other than the present [1,2] in and around pyridines, which we are exploring currently. Synthesis of natural and bioactive compounds from the chiral alkylpyridines is also under development.
Data availability
The complete data supporting this article have been uploaded as part of the supplementary material.Author contributions
S. R. and A. B. performed the experiments and developed the method. S. R. D. performed the DFT calculation in consultation with K. V. and P. M.; Md N. A. finalized the data. P. M. directed the project and wrote the manuscript with the feedback from other authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the SERB (EMR/2015/000711 and CRG/2019/001065). S. R. and A. B. thanks CSIR, and Md. N. A. thanks UGC for a fellowship. The support from central analytical facility, NCL is greatly acknowledged. We thank Dr S. H. Chikkali (NCL, Pune) for experimental help and Prof. U. K. Tambar (UTSW Medical Center, Dallas) for useful discussions and help in writing the manuscript. The support and the resources provided by ‘PARAM Brahma Facility’ under the National Supercomputing Mission, Government of India at the Indian Institute of Science Education and Research (IISER) Pune are gratefully acknowledged.Notes and references
- (a) V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181 CrossRef CAS PubMed; (b) N. Burn, P. Hesemann and D. Esposito, Chem. Sci., 2017, 8, 4724 RSC; (c) M. Pelckmans, T. Renders, S. V. Vyver and B. F. Sels, Green Chem., 2017, 19, 5303 RSC.
- (a) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790 CrossRef CAS PubMed; (b) S. L. Rössler, B. J. Jelier, E. Magnier, G. Fagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264 CrossRef; (c) D. Kong, P. J. Moon and R. J. Lundgren, Nat. Catal., 2019, 2, 473 CrossRef CAS; (d) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS; (e) J. T. M. Correia, V. A. Fernandes, B. T. Matsuo, J. A. Delgado, W. C. de Souza and M. W. Paixao, Chem. Commun., 2020, 56, 503 RSC; (f) K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045 CrossRef CAS PubMed.
- C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313 CrossRef CAS PubMed.
- F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336 CrossRef CAS PubMed.
- (a) J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227 CrossRef CAS PubMed; (b) M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362 CrossRef CAS; (c) J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030 CrossRef CAS PubMed; (d) M.-M. Zhang and F. Liu, Org. Chem. Front., 2018, 5, 3443 RSC; (e) F. J. R. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2019, 9, 236 CrossRef CAS; (f) X. Jiang, M. M. Zhang, W. Xiong, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 2402 CrossRef CAS PubMed; (g) S. Plunkett, C. H. Basch, S. O. Santana and M. P. Watson, J. Am. Chem. Soc., 2019, 141, 2257 CrossRef CAS; (h) H. Yue, C. Zhu, L. Shen, Q. Geng, K. J. Hock, T. Yuan, L. Cavallo and M. Rueping, Chem. Sci., 2019, 10, 4430 RSC; (i) S.-Z. Sun, C. Romano and R. Martin, J. Am. Chem. Soc., 2019, 141, 16197 CrossRef CAS PubMed; (j) J. Yi, S. O. Badir, L. M. Kammer, M. Ribagorda and G. A. Molander, Org. Lett., 2019, 21, 3346 CrossRef CAS PubMed; (k) S. Ni, C.-X. Li, Y. Mao, J. Han, Y. Wang, H. Yan and Y. Pan, Sci. Adv., 2019, 5, eaaw9516 CrossRef CAS PubMed; (l) M. E. Hoerner, K. M. Baker, C. H. Basch, E. M. Bampo and M. P. Watson, Org. Lett., 2019, 21, 7356 CrossRef; (m) Z.-K. Yang, N.-X. Xu, C. Wang and M. Uchiyama, Chem.–Eur. J., 2019, 25, 5433 CrossRef CAS PubMed; (n) Z.-F. Zhu, J.-L. Tu and F. Liu, Chem. Commun., 2019, 55, 11478 RSC; (o) F. T. Pulikottil, R. Pilli, R. V. Suku and R. Rasappan, Org. Lett., 2020, 22, 2902 CrossRef CAS; (p) C.-G. Yu and Y. Matsuo, Org. Lett., 2020, 22, 950 CrossRef CAS; (q) J. Wang, M. E. Hoerner, M. P. Watson and D. J. Weix, Angew. Chem., Int. Ed., 2020, 59, 13484 CrossRef CAS.
- (a) J. Wu, L. He and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700 CrossRef CAS PubMed; (b) F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210 CrossRef CAS PubMed; (c) J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697 CrossRef CAS PubMed; (d) M. Yang, T. Cao, T. Xu and S. Liao, Org. Lett., 2019, 21, 8673 CrossRef CAS PubMed; (e) J. Hu, B. Cheng, X. Yang and T.-P. Loh, Adv. Synth. Catal., 2019, 361, 4902 CrossRef CAS; (f) M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem.–Eur. J., 2019, 25, 8240 CrossRef CAS PubMed; (g) Q. Xia, Y. Li, X. Wang, P. Dai, H. Deng and W.-H. Zhang, Org. Lett., 2020, 22, 7290 CrossRef CAS PubMed.
- (a) C. D. Vanderwal, J. Org. Chem., 2011, 76, 9555 CrossRef CAS PubMed; (b) R. S. Paton, S. E. Steinhardt, C. D. Vanderwal and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 3895 CrossRef CAS PubMed; (c) T. M. Nguyen, M. Salvatori, R. S. del, J.-C. Wypych and C. Marazano, J. Org. Chem., 2007, 72, 5916 CrossRef CAS PubMed; (d) Y. Genisson, C. Marazano, M. Mehmandoust, D. Gnecco and B. C. Das, Synlett, 1992, 5, 431 CrossRef.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) H. Yang, E. Wang, P. Yang, H. Lv and X. Zhang, Org. Lett., 2017, 19, 5062 CrossRef CAS PubMed; (c) K.-S. Zhou, P. Yi, T. Yang, F.-M. Yanf, K.-H. Lee, B.-Y. Zhao, Y.-H. Wang and C.-J. Tan, Org. Lett., 2019, 21, 5051 CrossRef CAS PubMed; (d) J. Li, Y. Liu, X. Song, T. Wu, J. Meng, Y. Zheng, Q. Qin, D. Zhao and M. Cheng, Org. Lett., 2019, 21, 7149 CrossRef CAS PubMed; (e) S. Varga, P. Angyal, G. Martin, O. Egyed, T. Holczbauer and T. Soos, Angew. Chem., Int. Ed., 2020, 59, 13547 CrossRef CAS; (f) K. M. Lambert, J. B. Cox, L. Liu, A. C. Jackson, S. Yruegas, K. B. Wiberg and J. L. Wood, Angew. Chem., Int. Ed., 2020, 59, 9757 CrossRef CAS.
- For stereospecific cross-coupling of C–X with optically pure organometallic reagents: (a) L. Li, C.-Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607 CrossRef CAS; (b) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef CAS PubMed; (c) G. A. Molander and S. R. Wisniewski, J. Am. Chem. Soc., 2012, 134, 16856 CrossRef CAS; (d) Q. Zhou, H. D. Srinivas, S. Dasgupta and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 3307 CrossRef CAS; (e) G. A. Molander, S. R. Wisniewski and M. Hosseini-Sarvari, Adv. Synth. Catal., 2013, 355, 3037 CrossRef CAS PubMed; (f) L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027 CrossRef CAS PubMed; (g) Q. Zhou, K. M. Cobb, T. Tan and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 12057 CrossRef CAS PubMed; (h) K. R. Campos, A. Klapars, J. H. Waldman, P. G. Dormer and C. Chen, J. Am. Chem. Soc., 2006, 128, 3538 CrossRef CAS PubMed; (i) H. M. Wisniewska, E. C. Swift and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 9083 CrossRef CAS PubMed; (j) J. Llveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958 CrossRef PubMed.
- For asymmetric C–X alkylation: (a) S. Ge and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 16330 CrossRef CAS PubMed; (b) N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2015, 137, 10480 CrossRef CAS; (c) D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed; (d) G. J. Lovinger, M. D. Aparece and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 3153 CrossRef CAS.
- For asymmetric C–H alkylation: (a) G. Song, W. N. O. Wylie and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed; (b) G. A. Molander and S. R. Wisniewski, J. Am. Chem. Soc., 2012, 134, 16856 CrossRef CAS PubMed; (c) S. Yu, H. L. Sang and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 15896 CrossRef CAS PubMed; (d) G. Bertuzzi, D. Pecorari, L. Bernardi and M. Fochi, Chem. Commun., 2018, 54, 3977 RSC; (e) A. Motaleb, S. Rani, T. Das, R. G. Gonnade and P. Maity, Angew. Chem., Int. Ed., 2019, 58, 14104 CrossRef CAS PubMed.
- For racemic and asymmetric C−H aminoalkylation: (a) A. Kundu, M. Inoue, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2018, 140, 7332 CrossRef CAS PubMed; (b) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS; (c) X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Org. Lett., 2018, 20, 6298 CrossRef CAS PubMed.
- (a) Md N. Alam, S. R. Dash, A. Mukherjee, S. Pandole, U. K. Marelli, K. Vanka and P. Maity, Org. Lett., 2021, 23, 890 CrossRef CAS PubMed; (b) A. Motaleb, A. Bera and P. Maity, Org. Biomol. Chem., 2018, 16, 5081 RSC; (c) Md N. Alam, K. M. Lakshmi and P. Maity, Org. Biomol. Chem., 2018, 16, 8922 RSC.
- Although these reports by Wolfe group are reported as [1,2] aza-Wittig, excess Lewis acid along with base is required and the authors recognize a possible Stevens pathway via zwitterionic ammonium intermediate: (a) R. K. Everett and J. P. Wolfe, J. Org. Chem., 2015, 80, 9041 CrossRef CAS PubMed; (b) R. K. Everett and J. P. Wolfe, Tetrahedron Lett., 2015, 56, 3393 CrossRef CAS.
- The corresponding oxa-[1,2] Wittig is a well-known rearrangement, proceeds via a widely accepted dissociative radical pair mechanism, and exhibit substrate and reaction condition dependent variable degrees of chirality transfer: (a) K. Tomooka, H. Yamamoto and T. Nakai, Liebigs Ann./Recl., 1997, 1275 CrossRef CAS; (b) S. L. Schreiber and M. T. Goulet, Tetrahedron Lett., 1987, 28, 1043 CrossRef CAS; (c) K. Tomoka, M. Kikuchi, K. Igawa, P.-H. Keong and T. Nakai, Tetrahedron Lett., 1999, 40, 1917 CrossRef; (d) K. Tomoka, H. Yamamoto and T. Nakai, Angew. Chem., Int. Ed., 2000, 39, 4500 CrossRef; (e) K. Tomoka, M. Kikuchi, K. Igawa, M. Suzuki, P.-H. Keong and T. Nakai, Angew. Chem., Int. Ed., 2000, 39, 4502 CrossRef; (f) K. Tomooka, H. Yamamoto and T. Nakai, J. Am. Chem. Soc., 1996, 118, 3317 CrossRef CAS; (g) R. E. Maleczka Jr and F. Geng, J. Am. Chem. Soc., 1998, 120, 8551 CrossRef.
- For captodative stabilization of radical intermediates, see: (a) H. Viehe, Z. D. Anousek and R. Merenyi, Acc. Chem. Res., 1985, 18, 148 CrossRef CAS; (b) J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901 CrossRef CAS.
- Stoichiometric amount of catalysts were taken for strong MHMDS base at 0 °C to avoid starting material decomposition.
- (a) J. Barluenga, F. J. Fañanás, R. Sanz, C. Marcos and M. Trabada, Org. Lett., 2002, 9, 1587 CrossRef PubMed; (b) F. Gao, B.-S. Kim and P. J. Walsh, Chem. Sci., 2016, 7, 976 RSC.
- The enantiomeric excess of salts 1 was determined by chiral HPLC after converting those to the corresponding pyridones.
- Few chiral amines are commercially available and others were synthesized following a recent literature method: (a) M. Xiao, X. Yue, R. Xu, W. Tang, D. Xue, C. Li, M. Lei, J. Xiao and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 10528 CrossRef CAS PubMed; (b) V. Bagutski, T. G. Elford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 1080 CrossRef CAS PubMed.
- Cyclopropanes are known to remain intact in a radical reaction; either via reversible ring opening, or faster radical recombination before ring opening: (a) S. Umemiya, K. Nishino, I. Sato and Y. Hayashi, Chem.–Eur. J., 2014, 20, 15753 CrossRef CAS PubMed; (b) X. Gou and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469 CrossRef PubMed; (c) J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Nat. Commun., 2018, 9, 2445 CrossRef PubMed.
- (a) L. Haiyan, L. Gaoping and J. Liu, WO2010075469A1, 2010; (b) S. Zhou and R. Tong, Chem.–Eur. J., 2016, 22, 7084 CrossRef CAS PubMed; (c) L. Mengozzi, A. Gualandi and P. G. Cozzi, Chem. Sci., 2014, 5, 3915 RSC; (d) S. Zhou and R. Tong, Org. Lett., 2017, 19, 1594 CrossRef CAS PubMed; (e) J. Yu, Z. Zhang, S. Zhou, W. Zhang and R. Tong, Org. Chem. Front., 2018, 5, 242 RSC.
- We synthesized pyridinium salts following a modified literature protocol: G. H. R. Viana, I. C. Santos, R. B. Alves, L. Gil, C. M. Marazano and R. P. F. Gil, Tetrahedron Lett., 2005, 46, 7773 CrossRef CAS.
- For competitive [1,2] & [1,4] oxa-[1,4] Wittig rearrangements: F. Wang, J. Wang, Y. Zhang and J. Yang, Tetrahedron, 2020, 76, 130857 CrossRef CAS . For stereoretentive oxa-[1,2] & [1,4]: L. M. Mori-Quiroz and R. E. Maleczka Jr, J. Org. Chem., 2015, 80, 1163 CrossRef PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 Search PubMed; (c) R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- (a) H.-Y. Yoo, K. N. Houk, J. K. Lee, M. A. Scialdone and A. I. Meyers, J. Am. Chem. Soc., 1998, 120, 205 CrossRef CAS; (b) F. Haeffner, K. N. Houk, S. M. Schulze and J. K. Lee, J. Org. Chem., 2003, 68, 92310 CrossRef PubMed.
- (a) W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654 RSC; (b) X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620 CrossRef CAS PubMed; (c) Z. Feng and D. J. Tantillo, J. Am. Chem. Soc., 2021, 143, 088 Search PubMed.
- (a) F. D. Greene, M. A. Berwick and J. C. Stowell, J. Am. Chem. Soc., 1970, 92, 867 CrossRef CAS; (b) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. Shenvi, Chem. Sci., 2020, 11, 12401 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01217g |
| This journal is © The Royal Society of Chemistry 2021 |