
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-covalent assembly of proton donors and p-benzoquinone anions for co-electrocatalytic reduction of dioxygen†
Shelby L.
Hooe
 ,
Emma N.
Cook
,
Amelia G.
Reid
and
Charles W.
Machan
*
,
Emma N.
Cook
,
Amelia G.
Reid
and
Charles W.
Machan
*
Department of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 17th June 2021
Abstract
The two-electron and two-proton p-hydroquinone/p-benzoquinone (H2Q/BQ) redox couple has mechanistic parallels to the function of ubiquinone in the electron transport chain. This proton-dependent redox behavior has shown applicability in catalytic aerobic oxidation reactions, redox flow batteries, and co-electrocatalytic oxygen reduction. Under nominally aprotic conditions in non-aqueous solvents, BQ can be reduced by up to two electrons in separate electrochemically reversible reactions. With weak acids (AH) at high concentrations, potential inversion can occur due to favorable hydrogen-bonding interactions with the intermediate monoanion [BQ(AH)m]˙−. The solvation shell created by these interactions can mediate a second one-electron reduction coupled to proton transfer at more positive potentials ([BQ(AH)m]˙− + nAH + e− ⇌ [HQ(AH)(m+n)−1(A)]2−), resulting in an overall two electron reduction at a single potential at intermediate acid concentrations. Here we show that hydrogen-bonded adducts of reduced quinones and the proton donor 2,2,2-trifluoroethanol (TFEOH) can mediate the transfer of electrons to a Mn-based complex during the electrocatalytic reduction of dioxygen (O2). The Mn electrocatalyst is selective for H2O2 with only TFEOH and O2 present, however, with BQ present under sufficient concentrations of TFEOH, an electrogenerated [H2Q(AH)3(A)2]2− adduct (where AH = TFEOH) alters product selectivity to 96(±0.5)% H2O in a co-electrocatalytic fashion. These results suggest that hydrogen-bonded quinone anions can function in an analogous co-electrocatalytic manner to H2Q.
Introduction
The systematic optimization of molecular electrocatalysts requires an in-depth mechanistic understanding of the movement of electrons and protons to facilitate a reaction of interest.1 Biological models, particularly the water splitting reaction of photosystem II2 and complex IV in the electron transport chain,3 are common inspiration for the development of molecular electrocatalytic systems for artificial photosynthesis, where proton (H+) and electron (e−) equivalents need to be efficiently directed.4 Given the known role of tyrosine as a mediator of protons and electrons in the Mn-containing oxygen evolving complex of photosystem II,2a there is considerable incentive to identify co-catalytic phenol/quinone derivatives to improve the activity and/or alter the selectivity of Mn-based electrocatalytic processes related to the interconversion of H2O, H2O2, and O2.5 To the best of our knowledge, no co-electrocatalytic systems have been reported with Mn and quinone derivatives,6 although prior studies have noted that hydroquinones and aldehydes can drive partial O2 reduction during chemical oxidations mediated by Mn complexes.7It has been previously demonstrated that p-hydroquinone (H2Q) can function as an electron–proton transfer mediator (EPTM) in the co-electrocatalytic reduction of O2 to H2O by a Co(salophen) compound.8 In the co-electrocatalytic system, formal reduction and protonation of BQ to generate H2Q is proposed to occur in N,N-dimethylformamide (DMF) solution with acetic acid (AcOH; pKa(DMF) = 13 (ref. 9)) as a proton donor. In this system, H2Q is a discrete intermediate, which engages in the formal transfer of proton and electron equivalents to intermediate Co–O2[H]+/0 species. Importantly, the presence of the EPTM shifted product selectivity from H2O2 (91% efficiency under otherwise identical conditions with Co(salophen) and decamethylferrocene as the electron source) to H2O (overall quantitative, thanks to thermal reactivity between H2Q, Co(salophen), and O2).8a
The reduction chemistry of quinones in aprotic solvents is known to be highly dependent on solvent, added proton donor activity, and electrolyte.10 Under aprotic conditions, sequential one-electron reduction events are generally observed. Depending on proton donor activity and hydrogen-bonding strength, potential inversion can occur, enabling a two-electron reduction at a single potential. Under these conditions beginning from the neutral quinone ([Q]0), the second one-electron reduction (eqn (2)) is more favorable than the first (eqn (1)). Even when pKa comparisons are available, they can nonetheless fail to address the complex solvent mixtures which result at high concentrations of added weak proton donors,10f,11 which directly impact the stability of the BQ-based mono- and di-anions.10f,12
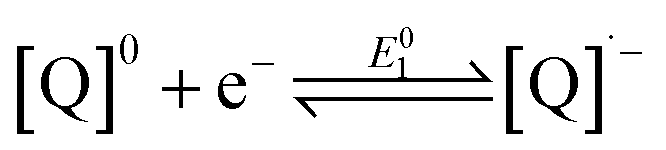 | (1) |
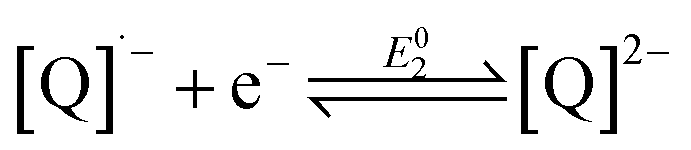 | (2) |
A pioneering study by Gupta and Linschitz10b noted that at certain hydrogen-bonding interaction strengths between the proton donor and para-quinone radical anions [Q]˙− in acetonitrile (MeCN) two-electron reduction waves could be observed. This type of effective potential inversion was possible with weak acids (e.g. 2,2,2-trifluoroethanol, TFEOH), when the second reduction was accompanied by proton transfer in a solvated shell of proton donors ([Q(AH)m]˙− + nAH + e− ⇌ [HQ(AH)(m+n)−1(A)]2−; where AH is a proton donor and A− is the conjugate base form of AH). The stabilization of the [Q]˙− and [HQ]− species, as well as the observation of electrochemical irreversibility at sufficient acid concentrations, were ascribed to the strong hydrogen-bonds formed between the anions and proton donors in solution facilitating at least one proton transfer to achieve potential inversion.10b Notably, this effect was also observed by Evans and co-workers using an ortho-quinone derivative in MeCN with high concentrations of added TFEOH.11e
We were interested in the possibility that the stabilizing effect of hydrogen-bonds on BQ anions could enable them to perform a similar function to H2Q for co-electrocatalytic O2 reduction. One advantage would be that these intermediates would be comprised of highly reversible interactions, minimizing the height of any new kinetic barriers in the co-electrocatalytic mechanism. In MeCN, the pKa(1) and pKa(2) of H2Q have been estimated computationally to be 26.20 and 40.96, respectively.10i This means that there is a range of approximately 14 pKa units where speciation should favor only partial protonation of [BQ]2−, if an appropriate acid is used. However, such species are known to be unstable and reactive: it has been demonstrated that without added proton donor two equivalents of [HQ]− disproportionate to a dianionic quinhydrone species [H2Q·BQ]2−.10h To the best of our knowledge, the use of hydrogen-bonded quinone anion adducts in an analogous role to their hydroquinone counterparts has only been applied to the study of quinone-based energy storage systems.13
Given the profound interest in Mn complexes as electrocatalysts for O2 reduction5c,14 and the single report on the use of the BQ/H2Q redox couple in tandem with a molecular electrocatalyst to facilitate O2 reduction,8b we sought to carry out a study on the co-electrocatalytic competency of hydrogen-bonded qunione anions using a Mn-based catalyst developed in our lab.15 This molecular Mn complex, Mn(tbudhbpy)Cl 1, where 6,6′-di(3,5-di-tert-butyl-2-phenolate)-2,2′-bipyridine = [tbudhbpy]2−, is a competent catalyst for the selective reduction of O2 to H2O2 (ca. 80% selectivity) in the presence of weak Brønsted acids (phenol derivatives and TFEOH). Herein, we report the selective electrocatalytic reduction of O2 to H2O (96 ± 0.5%) by a catalyst system comprised of Mn(tbudhbpy)Cl 1, p-benzoquinone (BQ), and TFEOH as a sacrificial proton donor (Fig. 1). We note that the computationally estimated pKa of TFEOH in MeCN is 35.4.16
 | ||
| Fig. 1 Comparison of product selectivity in previous versus current work with Mn(tbudhbpy)Cl 1 to summarize the overall co-electrocatalytic effect of BQ; L = [tbudhbpy]2−, TFEOH = 2,2,2-trifluoroethanol and [MnIII(L)Cl]0 = Mn(tbudhbpy)Cl 1. | ||
Mechanistic experiments suggest that a key component is the generation of hydrogen-bonded [H2Q(AH)3(A)2]2− adducts in situ at elevated concentrations of TFEOH, which we propose serve an analogous function to H2Q in transferring proton donors and electrons to Mn–O2[H]+/0 intermediates which are generated at more positive potentials. The number of TFEOH equivalents in the adducts shows a dependence on the concentration of BQ: 5.6 at 0.5 mM of BQ to 4.7 at 2.5 mM of BQ. Given that the solvation shell of these anion adducts in MeCN solution with elevated TFEOH concentrations is by all definitions a non-ideal solvent, it is important to note that the continuum of proton placement is not trivial to articulate. Should formal proton transfer from TFEOH occur, the anionic bases generated in solution are likely to remain strongly associated to any protonated quinone product. Based on NMR data obtained from coulometric experiments, we consider that these data suggest the average structure in solution is best described as [H2Q(TFEOH)3(TFEO)2]2− at sufficient excess concentration of TFEOH. While the known thermochemistry from previous DFT studies suggests that formation of [HQ(TFEOH)4(TFEO)]2− is possible at intermediate and low concentrations, this species is known to eventually dimerize to H2Q10h and therefore likely to be the thermodynamically favored product.
Results
CV analysis of BQ reduction with TFEOH
Cyclic voltammetry (CV) experiments were carried out with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte in acetonitrile (MeCN) solution. Under argon (Ar) saturation conditions, BQ displays two reversible redox features with E1/2 values of −0.89 V and −1.69 V versus the ferrocenium/ferrocene (Fc+/Fc) redox couple (Fig. 2A, black), consistent with previous reports.10b These reduction features are assigned to BQ/[BQ]˙− and [BQ]˙−/[BQ]2−, respectively. As has been observed previously, the second reduction feature shows a slightly diminished current response, which is consistent with previously proposed reactivity between the monoanion [BQ]− and the dianion [BQ]2−.10b,11c Coulometry performed at −2.1 V versus Fc+/Fc confirmed that these two redox features correspond to an overall two-electron reduction process under aprotic conditions (Fig. S1–S3†). These data are consistent with previous proposals of sequential one-electron BQ/[BQ]˙− and [BQ]˙−/[BQ]2− processes.10b | ||
| Fig. 2 (A) CVs of TFEOH titration with 0.5 mM BQ obtained under Ar saturation conditions. (B) Linear fit of −E1/2 (V vs. Fc+/Fc) versus log[TFEOH (M)] for the two-electron BQ/[BQ]2− reduction feature obtained from CV titration data in (A), using only reversible two-electron responses observed in the region from −0.85 to −0.70 V. Conditions: 0.1 M TBAPF6/MeCN; glassy carbon working electrode, glassy carbon counter electrode, Ag/AgCl pseudoreference electrode; 100 mV s−1 scan rate; referenced to internal ferrocene standard. | ||
Titrating increasing quantities of TFEOH into an Ar-saturated MeCN solution of 0.5 mM BQ resulted in the convergence of these two redox processes into a single irreversible redox feature at 1.37 M TFEOH (voltage at half-peak current −0.66 V vs. Fc+/Fc), which displayed an increased current response relative to the one-electron redox features under aprotic conditions (Table S1† and Fig. 2A, blue and black traces). The titration data at low [TFEOH] were consistent with the response observed for analogous experiments with 2.5 mM BQ, however the same degree of irreversibility was not observed at the higher BQ concentration (Fig. S4 and Table S2†). Comparing CV titration data with BQ and TFEOH under Ar and O2 did not reveal significant differences in the observed electrochemical response, indicating that the anionic species generated under these conditions does not interact with O2 in a catalytic fashion on the CV timescale (Fig. S5†).
Coulometric analysis of BQ reduction with TFEOH
Coulometry at −1.1 V versus Fc+/Fc with 2.5 mM BQ and 1.37 M TFEOH suggests that with an added proton donor this single redox feature corresponds to a two-electron reduction (Fig. S6†). NMR analysis of the product of this reduction shows excellent agreement with authentic samples of H2Q under analogous conditions (Fig. S7–S11†). Indeed, replicating these coulometry and control experiments with acetic acid (AcOH; pKa in MeCN = 23.5),6 which is expected to be sufficiently acidic to directly generate H2Q produced identical results (Fig. S12–S14†). This observation is inconsistent with the reported pKa of TFEOH in MeCN (35.4), which should thermodynamically preclude it from protonating [HQ]− (pKa H2Q in MeCN = 26.20) and suggests that additional thermal reactions are occurring. Previous studies have shown that the disproportionation of [HQ]− to a dianionic quinhydrone species [H2Q·BQ]2− occurs following mono-deprotonation (eqn (3)).10h This assignment was based on a distinct NMR spectrum obtained for the quinhydrone dimer observed experimentally relative to H2Q, which we do not observe. However, the previously reported conditions10h were aprotic, conducted with stoichiometric amounts of added base. Therefore, under our electrolysis conditions where 1.37 M TFEOH is present, we propose that the non-ideality of the solvation shell facilitates the transfer of a second proton, producing the anion-stabilized H2Q molecule observed in the coulometric studies, vide infra.| 2[HQ]− ⇌ [H2Q·BQ]2− | (3) |
Based on these observations, we next conducted CV experiments using BQ, H2Q, AcOH and TFEOH to establish whether evidence of eqn (3) was apparent. If eqn (3) was relevant on the electrochemical timescale under these conditions, there should be a quantifiable difference in the reduction of BQ, with and without a stoichiometric amount of H2Q present, consistent with a favorable equilibrium interaction (K > 1). With and without 1.37 M TFEOH present, we observed no evidence of an analogous strong interaction during the reduction of BQ when H2Q was present (Fig. S15 and S16†), suggesting minimal speciation of the dianionic quinhydrone species [H2Q·BQ]2− with an excess of proton donor present. Others have noted previously that much greater concentrations than those used here are required to observe this interaction, which resulted in diagnostic electrode adsorption features in the observed CV response.17 The reduction features of BQ with 1.37 M TFEOH present are approximately 0.36 V more negative than those with 1.37 M AcOH present, a trend which holds from 0.5 mM to 2.5 mM BQ (Fig. S17 and S18†). These observations are consistent with the expected difference in proton donor activity and do not exclude the formation of monoprotonated [HQ]−, particularly given the predicted pKa values. However, these experimental conditions represent a non-ideal solvent system not reflected in the theoretical approach and therefore the only direct structural evidence shows H2Q generation.
Quantification of TFEOH binding during BQ reduction
To better understand the speciation under electrochemical conditions, the relationship between the overall two-electron BQ redox response and TFEOH was analyzed by CV through TFEOH titration data under Ar saturation conditions according to the framework of Gupta and Linschitz (Fig. 2, S4 and S19;† Methods).10b At TFEOH concentrations where the two-electron BQ redox feature remains reversible, plotting the −E1/2 (V vs. Fc+/Fc) of the two-electron BQ redox feature against log[TFEOH (M)] gives a slope consistent with a 2e−/5.6TFEOH redox process ([BQ] = 0.5 mM; Fig. 2B). Based on prior reports,10b,11e this suggests that the two-electron reduced BQ species is the result of a stabilized monoanionic H-bonded intermediate eqn (4) undergoing a second reduction, which is concerted with proton transfer in the non-covalent assembly eqn (5).10b Our observation of a two-electron redox response is consistent with the standard potential of eqn (4) occurring at more negative potentials than that of eqn (5).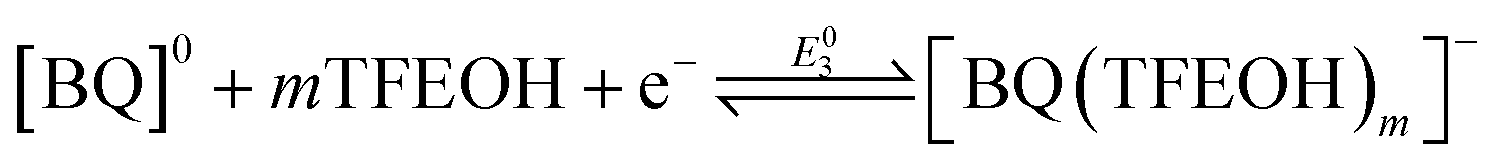 | (4) |
 | (5) |
At higher concentrations of BQ (2.5 mM), comparable analysis of the two-electron BQ reduction feature shows that the average number of TFEOH molecules engaged in hydrogen-bonding interactions diminishes slightly to 4.7 (Fig. S4†). Analysis of overall Keq for the equilibrium binding events depicted in eqn (4) and (5), produced values of 4.31 × 107 at 0.5 mM BQ and 2.31 × 106 at 2.5 mM BQ, respectively, comparable to those determined for other weak acids previously.10b Note that as the reaction in eqn (5) shifts to increasingly positive potentials in comparison to eqn (4), a disproportionation reaction to produce the same two-electron reduction product also becomes increasingly favorable (see Methods). Based on the variable concentration data in Fig. 2 and S4,† we assign an average value of 5 to m + n.
The experimental observation of irreversibility at higher TFEOH concentrations, which cannot be analyzed by the method above (Fig. 2), coupled with the multiple equivalents solvating the anionic species suggest that additional formal proton transfer can occur.10b,11e It is worth noting that the non-ideality of the solvent system should support additional proton transfer reactions, where high concentrations of TFEOH are expected to favorably solvate mono- and dianionic species.10f,11 As discussed above, previous computational studies estimate that TFEOH has sufficient proton activity to monoprotonate [BQ]2− ([HQ]− ⇌ H+ + [BQ]2−; pKa(MeCN) = 40.96), but a second protonation is thermodynamically disfavored ([H2Q] ⇌ H+ + [HQ]−; pKa(MeCN) = 26.20).10i However, our coulometric data shows that H2Q is present at longer timescales and we cannot exclude direct production or a relevant reaction timescale for the dimerization reaction eqn (3) under protic conditions. Therefore, under elevated concentrations of TFEOH, it is likely that a second proton transfer can occur, as evidenced in the NMR data obtained from coulometry, eqn (6).
| [HQ(TFEOH)4(TFEO)]2− ⇌ [H2Q(TFEOH)3(TFEO)2]2− | (6) |
Under these conditions, it is likely that the 2,2,2-trifluoroethoxide anions generated following proton transfer will associate strongly to H2Q,10b making definitive assignment of the protonation continuum non-trivial.
Co-electrocatalytic studies with 1, BQ and TFEOH
As we reported in our initial study on 1,15a the addition of TFEOH under O2 saturation conditions causes a catalytic increase in current (Fig. 3A, red). Upon the addition of 0.5 mM BQ to a solution of 1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1:BQ) and TFEOH (1.37 M) under O2 saturation conditions, a shift towards more positive potentials and a multielectron irreversible wave consistent with catalysis is observed (Fig. 3B, blue). Comparative electrochemical reaction conditions where Mn(tbudhbpy)Cl 1, O2, TFEOH, or BQ are omitted do not show analogous activity, indicating that an alternate electrochemical process is occurring when all four components are present. Importantly, these observations are consistent when greater concentrations of BQ (1.25 mM or 2.5 mM) are present (Fig. S20 and S21†).
:1 ratio of 1:BQ) and TFEOH (1.37 M) under O2 saturation conditions, a shift towards more positive potentials and a multielectron irreversible wave consistent with catalysis is observed (Fig. 3B, blue). Comparative electrochemical reaction conditions where Mn(tbudhbpy)Cl 1, O2, TFEOH, or BQ are omitted do not show analogous activity, indicating that an alternate electrochemical process is occurring when all four components are present. Importantly, these observations are consistent when greater concentrations of BQ (1.25 mM or 2.5 mM) are present (Fig. S20 and S21†).
 | ||
| Fig. 3 (A) CVs comparing 0.5 mM of Mn(tbudhbpy)Cl 1, with 1.37 M TFEOH both with (blue) and without (red) 0.5 mM BQ under O2 saturation conditions. (B) CVs comparing 0.5 mM of Mn(tbudhbpy)Cl 1, with 0.5 mM BQ both with (blue) and without (red) 1.37 M TFEOH under O2 saturation conditions. (C) CVs comparing 0.5 mM Mn(tbudhbpy)Cl 1, with 1.37 M TFEOH and 0.5 mM BQ under Ar (red) and O2 (blue) saturation conditions compared to a control CV in the absence of Mn(tbudhbpy)Cl 1 (black). Conditions: 0.1 M TBAPF6/MeCN; glassy carbon working electrode, glassy carbon counter electrode, Ag/AgCl pseudoreference electrode; 100 mV s−1 scan rate; referenced to internal ferrocene standard. | ||
Notably, with 1, 0.5 mM BQ, and 1.37 M TFEOH under Ar saturation conditions, a shift in the BQ reduction potential is observed (Fig. 3C, comparing red and black). Specifically, the BQ redox feature (0.5 mM) in the presence of 1.37 M TFEOH shifts to more positive potentials by 0.046 V when 1 (0.5 mM) is added. Based on this, we qualitatively interpret the positive shifts in the BQ reduction potential to be the result of an interaction between the hydrogen-bonded [H2Q(AH)3(A)2]2− and the one-electron reduced and monoprotonated product of 1. However, due to the closeness of the of BQ and Mn reduction potentials (Ep = −0.60 V and −0.77 V vs. Fc+/Fc with 0.2 M TFEOH, respectively) and their respective dependences on added TFEOH concentration, more rigorous quantification is not possible. We note that our previous studies have shown that the ligand framework of 1 is sensitive to protonation upon Mn(III)/(II) reduction in the presence of an acid.15 No shift is observed for the Mn(III)/(II) reduction because the reduced BQ species are not generated until more negative potentials.
We have established in our previous studies that the Mn catalyst binds O2 to generate a Mn(III)-superoxide, which is reduced to a hydroperoxide with proton transfer at more negative potentials.15 Since the potential for the reduction of this Mn(III)-superoxide intermediate overlaps with the reduction of BQ under these conditions, we propose that the observed co-electrocatalysis arises from an intermolecular reaction as summarized in eqn (7).
| 2[HQ(TFEOH)3(TFEO)2]2− + [MnIII(L(H))O2˙−]+ ⇌ 2[BQ]0 + [MnII(L(H))]+ + 4[TFEO]− + 2H2O + 6TFEOH | (7) |
We expect that [H2Q(AH)3(A)2]2− has comparable reducing power to H2Q and note that H2Q functions as a competent reductant of O2 and H2O2 under experimental conditions with 1 present in control studies (Fig. S22–S27†).7
Kinetic analysis of co-electrocatalytic conditions
To understand the relative kinetic relationships of the reaction components, variable concentration studies were carried out via CV (Fig. S28–S33†). Although the complexity of the reaction mixture precludes assigning concentration dependencies to the observed catalytic current, these data are consistent with the proposed equilibrium interactions described above, as well as the dependence of co-electrocatalytic activity on the presence of 1, BQ, TFEOH, and O2. In these data, a pronounced anodic wave is observed in the CV response near the [BQ]0/[H2Q(AH)3(A)2]2− feature on the return sweep under co-catalytic conditions when BQ is in excess of complex 1 (Fig. S29†). This waveform suggests a mismatch between the generation of [H2Q(AH)3(A)2]2− and its rate of reaction with the intermediate [Mn–(L(H))O2˙−]+ species, where L = [tbudhbpy]2− and (L(H)) denotes formal protonation of the O atom of the ligand framework.15a The resulting accumulation of unreacted [H2Q(AH)3(A)2]2− in the reaction–diffusion layer results in appreciable re-oxidation of [H2Q(AH)3(A)2]2− to [BQ]0 (reverse of eqn (6), eqn (5) and (4) as written). Control CV data with BQ, TFEOH and urea·H2O2 present in MeCN show minimal differences, suggesting that [H2Q(AH)3(A)2]2− is relatively stable in the presence of H2O2 on the CV timescale (Fig. S34†).Determining co-electrocatalytic product selectivity
Rotating ring-disk electrode (RRDE) experiments were carried out to quantify the electrocatalytic production of H2O2 (see Materials and methods for description). Control experiments with BQ and added TFEOH showed that the electrogenerated [H2Q(AH)3(A)2]2− decomposes to produce a small amount of H2O2 (10(±23)% (Fig. S35–S40 and Table S3†)). In CVs of BQ with TFEOH present under O2 saturation there is no multielectron catalytic response at reducing potentials (e.g., Fig. 3). We postulate that this discounts appreciable H2O generation during the catalytic response, which would otherwise explain the less-than-quantitative ring current corresponding to H2O2 oxidation. The proposed instability is consistent with the observations of others.13With 1 and 1.37 M TFEOH under O2 saturation conditions, the system exhibited comparable selectivity for H2O2 compared to our previous report, 68(±13)% (Fig. S41 and Table S3†). Here, the use of greater concentrations of TFEOH than previously reported15a resulted in a slight shift of the average product distribution, but was within error of the original report. Upon the addition of one equivalent of BQ relative to 1 under these conditions, the system showed selectivity for H2O2 within error of the BQ-free system (69(±0.3)%). An increased cathodic current response was observed, suggesting reaction rate enhancement under 1:1 co-electrocatalytic conditions (Fig. S42–S47†). At 2.5 equivalents of BQ (1.25 mM) relative to 1 (0.5 mM), the selectivity of the system shifted to H2O as the major product with 55(±4)% efficiency (Fig. S48–S53 and Table S3†). Lastly, with five equivalents of BQ (2.5 mM) relative to 1 (0.5 mM), the selectivity of the system became 96(±0.5)% selective for H2O (Fig. S54–S59 and Table S3†). Consistent with the role of added BQ in shifting product selectivity, the observed decreases in efficiency for H2O2 showed a linear relationship with respect to the concentration of added BQ (Fig. 4).
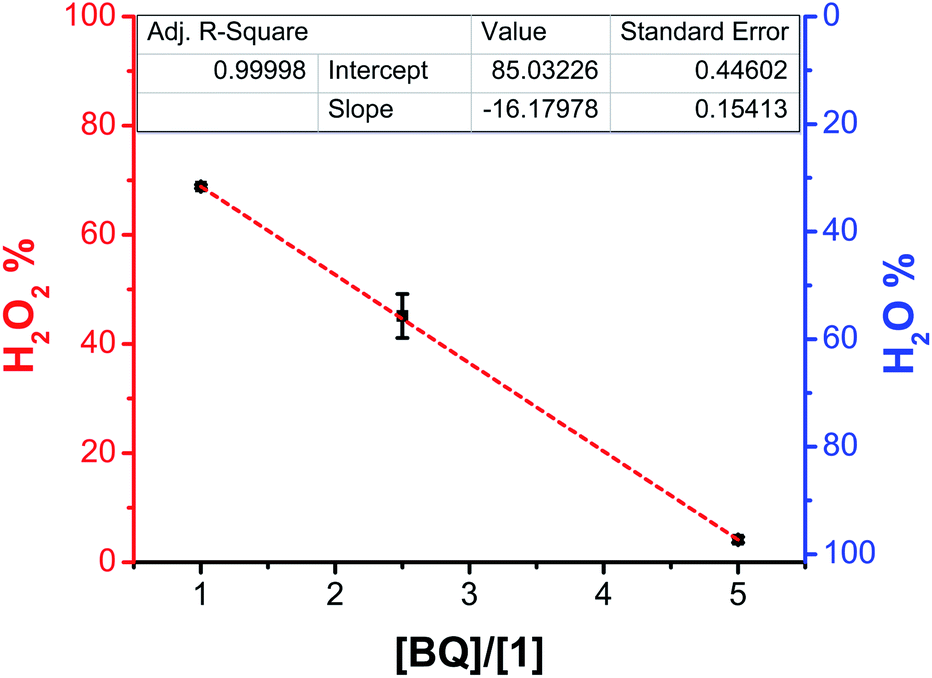 | ||
| Fig. 4 The observed relationship between the concentration of added BQ and the reaction product as characterized by RRDE at a rotation rate of 400 rpm. Conditions: 0.5 mM Mn(tbudhbpy)Cl 1; 1.37 M TFEOH; 0.5, 1.25, or 5 mM BQ; 0.1 M TBAPF6/MeCN. | ||
Discussion
We have demonstrated that the selectivity for electrocatalytic O2 reduction by Mn(tbudhbpy)Cl 1 with TFEOH as a proton donor can be altered from the 2H+/2e− product H2O2 to the 4H+/4e− product H2O when BQ is added as a co-catalyst. Furthermore, current increases occur relative to the intrinsic performance of 1, consistent with an enhanced rate of catalysis. Unlike the only previous report we are aware of,8b this co-electrocatalytic effect leverages a non-covalent assembly between the added proton donor and redox mediator to achieve these improvements (Fig. 5).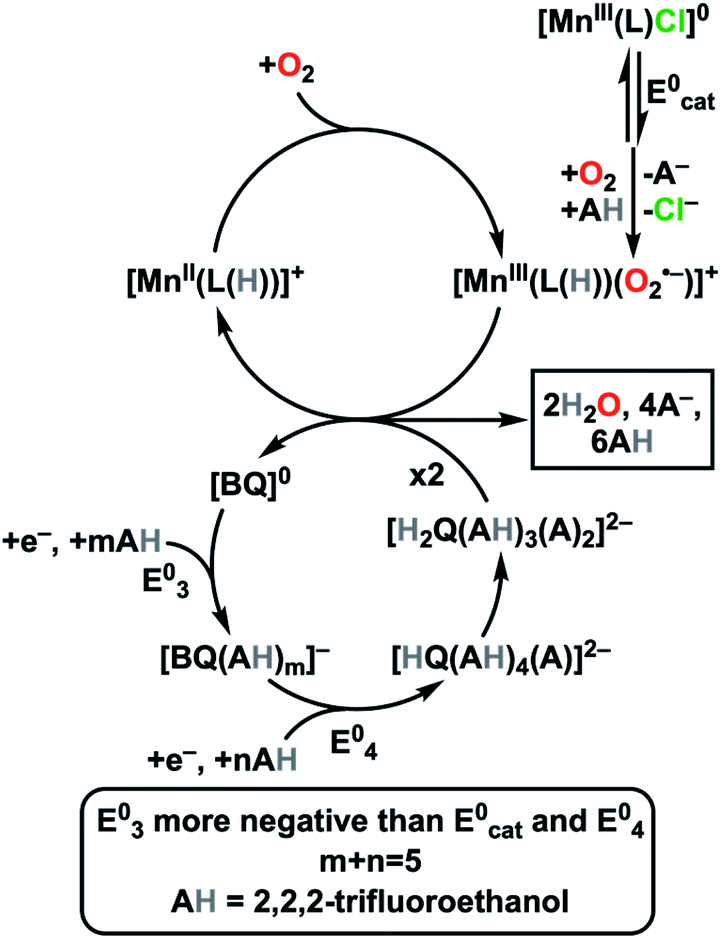 | ||
| Fig. 5 Proposed potential inversion mechanism for co-electrocatalytic O2 reduction to water by Mn(tbudhbpy)Cl 1 and BQ, where AH = TFEOH, m + n = 5, L = [tbudhbpy]2−, and (L(H)) denotes formal protonation of the O atom of the ligand framework. | ||
At high concentrations in MeCN, TFEOH acts to stabilize and solvate anionic quinone-derived species.11e These solvent clusters assist thermodynamically favored monoprotonation of the intermediate [BQ]− species as a part of a proton-coupled electron transfer reaction to generate a stabilized [HQ]− species. The concerted nature of this proton and electron transfer results in a standard potential which is more positive than the initial reduction, which is observed experimentally as an overall two-electron reduction feature.10f,18 At increased TFEOH concentrations, a second proton transfer is likely to occur: our electrolysis studies accompanied by NMR characterization show that an H2Q species is produced. The reduction corresponding to the overall formation of the expected hydrogen-bond stabilized [H2Q(AH)3(A)2]2− species is observed to shift to more positive potentials when 1 is present, suggesting an equilibrium interaction between the two species.
Based on our results, we propose that the hydrogen bond-stabilized [H2Q(AH)3(A)2]2− adduct serves as a redox mediator in these co-electrocatalytic reactions, delivering electrons and a single proton to Mn–O2[H]+ intermediate (Fig. 5).15 In doing so, BQ also facilitates the net transfer of TFEOH to a Mn catalyst during the reaction. As [H2Q(AH)3(A)2]2− is oxidized to BQ, several equivalents of TFEOH are released from strong hydrogen-bonding interactions, allowing formal proton and electron transfer to the Mn catalyst to occur. In the previously reported use of a BQ/H2Q couple to achieve a co-electrocatalytic effect, separate hydrogen atom transfer and proton-coupled electron transfer steps from H2Q were proposed to occur.8 We note that in control experiments, significant reactivity was not observed between H2O2 and the hydrogen-bond stabilized [H2Q(AH)3(A)2]2−; however, RRDE experiments suggest that it can generate small amounts of H2O2 from O2, demonstrating greater intrinsic activity than H2Q alone.
Conclusions
These results suggest that energy transduction reactions analogous to those mediated by EPTMs are kinetically feasible by leveraging potential inversion phenomena from non-covalent effects, greatly expanding hypothetical reaction conditions. This suggests that an expanded range of weak proton donors can be used to achieve co-electrocatalytic changes in product distribution, enabling alternative strategies to optimize kinetic parameters of a co-electrocatalytic reaction without causing the large increases in the overpotential of the reaction that result from using strong acids.Materials and methods
General
All chemicals and solvents (ACS or HPLC grade) were commercially available and used as received unless otherwise indicated. Mn(tbudhbpy)Cl 1 was prepared according to our previous report.15a For all air-sensitive reactions and electrochemical experiments, HPLC-grade solvents were obtained as anhydrous and air-free from a PPT Glass Contour Solvent Purification System. Gas cylinders were obtained from Praxair (Ar as 5.0; O2 as 4.0) and passed through the electrochemical working solvent with added molecular sieves prior to use. Gas mixing for variable concentration experiments was accomplished using a gas proportioning rotameter from Omega Engineering. UV-vis absorbance spectra were obtained on a Cary 60 from Agilent. An Anton-Parr Multiwave Pro SOLV, NXF-8 microwave reactor was used for microwave syntheses.Electrochemistry
All electroanalytical experiments were performed using a Metrohm Autolab PGSTAT302N potentiostat. Glassy carbon working (ϕ = 3 mm) and non-aqueous silver/silver chloride pseudoreference electrodes behind PTFE tips were obtained from CH Instruments. The pseudoreference electrodes were obtained by depositing chloride on bare silver wire in 10% HCl at oxidizing potentials and stored in a 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) acetonitrile solution in the dark prior to use. The counter electrode was a glassy carbon rod (ϕ = 3 mm). All CV experiments were performed in a modified scintillation vial (20 mL volume) as a single-chamber cell with a cap modified with ports for all electrodes and a sparging needle. Tetrabutylammonium hexafluorophosphate was purified by recrystallization from ethanol and dried in a vacuum oven before being stored in a desiccator. All data were referenced to an internal ferrocene standard (ferrocenium/ferrocene Fc+/Fc reduction potential under stated conditions) unless otherwise specified; ferrocene was purified by sublimation prior to use. All voltammograms were corrected for internal resistance.Rotating ring-disk electrode (RRDE) experiments were conducted using a Metrohm rotator with a Metrohm electrode consisting of a glassy carbon disk (ϕ = 5 mm) and a Pt ring. The average collection efficiency of the RRDE electrode was experimentally determined to be 25.5% using 0.5 mM ferrocene in 0.1 M TBAPF6/MeCN. The counter electrode in the RRDE experiments was a glassy carbon rod (Type 2, ϕ = 3 mm; Alfa Aesar) and the reference electrode was a non-aqueous Ag/AgCl reference electrode with a double-junction system from Metrohm. RRDE experiments were performed in a 50 mL graduated glass vessel from Metrohm as a single-chamber cell with adapted ports for all electrodes. All RRDE data was referenced to an internal ferrocene standard.
Quantifying TFEOH binding (adapted10b)
To determine the average number of TFEOH equivalents involved in the overall two-electron BQ redox response from the CV titration data (Fig. 2 and S4†), the slope of the −E1/2 (V vs. Fc+/Fc) versus log[TFEOH (M)] was used in the concentration region where the feature remained reversible (−0.85 V to −0.70 V). The slope of this plot can be used to determine the number of TFEOH molecules x which associate to the [BQ]2− dianion, where n is the number of electrons involved (2), F is Faradays' constant (96485 C mol−1), R is the ideal gas constant (J K−1 mol−1), T is temperature (K), eqn (8).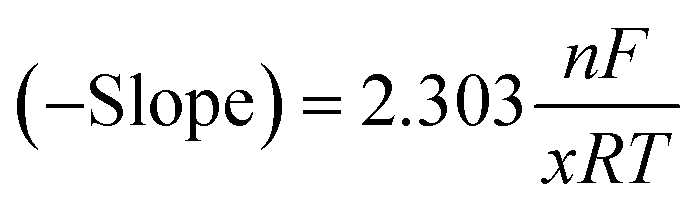 | (8) |
Using the linear fit equations obtained from the relevant regions of Fig. 2 and S4,† averaged E1/2 values (A and B) were determined for the respective representative concentration ranges and used to determine Keq according to eqn (9) using Δ[TFEOH].
 | (9) |
Description of RRDE quantification
The collection efficiency was determined in the same manner as our previous reports15a using 0.5 mM ferrocene (Fig. S60 and S61†). Under conditions for which Levich behavior was observed, the difference between the amount of current produced at the disk under O2 saturation conditions and the amount of current produced at the disk under Ar saturation conditions was taken as the corrected disk current for O2 saturation conditions (Idisk corrected) at each rotation rate. The difference between the amount of current produced at the ring under O2 saturation conditions and the amount of current produced at the ring under Ar saturation conditions was taken as the corrected ring current for O2 saturation conditions (Iring corrected) at each rotation rate. To calculate the H2O2%, the Idisk corrected was multiplied by the corresponding Nempirical value for the specific rotation rate to determine the maximum amount of ring current for H2O2 production (Iring max). The ratio of Iring corrected to Iring max was multiplied by a factor of 100 to determine the % H2O2 generated at the ring across all measured rotation rates (eqn (10)). | (10) |
To ensure that ring current used above corresponded only to H2O2 oxidation, multi-segmented CV sweeps were obtained with 0.5 mM 1, 1.37 M TFEOH, and 0.5 mM BQ under Ar and O2 saturation conditions (Fig. S62 and S63†). Beginning from the resting potential, the voltage was swept to a switching potential of +1.1 vs. Fc+/Fc, then to a switching potential at −0.87 V vs. Fc+/Fc, followed by final switching potential of +1.05 V vs. Fc+/Fc before completing the sweep at the resting potential. Data taken under Ar (Fig. S62,† black) do not display the expected oxidation features associated with H2Q or quinhydrone oxidation in control experiments (Fig. S64 and S65†). When catalytic potentials are swept, the return sweep shows anodic current increases at ∼+0.4 V vs. Fc+/Fc, before the expected oxidation response of H2O2, suggestive of the re-oxidation of other reaction intermediates. To account for the background current unique to catalytic conditions, two sets of RRDE experiments were conducted with ring potentials of +0.85 V and +0.4 V vs. Fc+/Fc for the systems which exhibited Levich behavior: BQ and TFEOH (Fig. S35 and S38†), 1:1 Mn to BQ with TFEOH (Fig. S42 and S45†), 1:2.5 Mn to BQ with TFEOH (Fig. S48 and S51†), and 1:5 Mn to BQ with TFEOH (Fig. S54 and S57†). The raw disk current responses for both ring potentials were averaged at identical rotation rates to produce the required values for the analysis described in the preceding paragraph. The difference between the ring currents at these two potentials under catalytic conditions was used for the H2O2 efficiency analysis to remove the current response from other reaction intermediates in the determination of Iring corrected. CV traces obtained under co-electrocatalytic conditions using the working electrode of the RRDE electrode confirmed that minimal reversibility was present at a 2.5 mM concentration of BQ (Fig. S66†) with a larger working electrode. Data were compared at a single rotation rate (400 rpm; Fig. 4) from the RRDE experiments with different ratios of 1:BQ to minimize variability from the multiple equilibria involved in the overall reaction. To obtain an average and standard deviation at a rotation rate of 400 rpm, the current densities at 0.8 V, 0.85 V, and 0.9 V vs. Fc+/Fc were each used to calculate the % H2O2.
Under conditions for which non-Levich behavior was observed, with 0.5 mM 1 and 1.37 M TFEOH (Fig. S41†), the % H2O2 was calculated using the method which has been previously established for similar systems which also display non-Levich behavior (eqn (11)).14k,14m
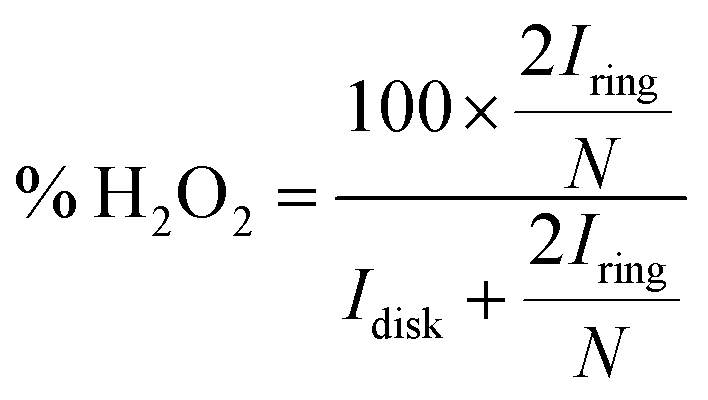 | (11) |
Both the ring and disk currents used to calculate % H2O2 in eqn (11) were the raw O2 currents determined by averaging the data from all the rotation rates at the catalyst E1/2 (−0.63 V vs. Fc+/Fc).
Disproportionation mechanism
As discussed in the main text, at high TFEOH concentrations, a two-electron reduction feature is observed. Since sequential one-electron reduction features are observed under aprotic conditions, this observation requires potential inversion. Under such conditions, it is worth noting that a disproportionation reaction eqn (12) becomes increasingly favorable as eqn (5) shifts increasingly positive of eqn (4).19 In this reaction, two equivalents of [BQ(TFEOH)m]− disproportionate into one equivalent of [BQ]0 and one equivalent of [HQ(TFEOH)4(TFEO)]2−, which is structurally equivalent to the product of eqn (5).11e| 2[BQ(TFEOH)m]− ⇌ [BQ]0 + [HQ(TFEOH)4(TFEO)]2− + (2m − 5)TFEOH | (12) |
Data availability
Additional spectroscopic, coulometric, and electrochemical data (including electroanalytical plots) are provided in the ESI.Author contributions
C. W. M. conceived and supervised the experiments; the manuscript was written by C. W. M. and S. L. H.; all experiments were carried out by S. L. H.; E. N. C. and A. G. R. assisted in developing experimental procedures specific to this manuscript. All authors have given approval to the final version of the manuscript and declare no competing financial interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Michael L. Pegis for helpful discussion concerning the RRDE quantification of non-Levich behavior by molecular electrocatalysts and Spenser Simpson for assistance and advice with NMR methods. The authors also thank ACS PRF 61430-ND3 and NSF CHE-2102156 for funding.References
- (a) J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef; (b) J.-M. Savéant, Angew. Chem., Int. Ed., 2019, 58, 2125–2128 CrossRef PubMed; (c) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- (a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed; (b) B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- R. J. Mailloux, Redox Biol., 2015, 4, 381–398 CrossRef CAS PubMed.
- L. Hammarström and S. Styring, Philos. Trans. R. Soc., B, 2008, 363, 1283–1291 CrossRef PubMed.
- (a) C. W. Machan, ACS Catal., 2020, 10, 2640–2655 CrossRef CAS; (b) L. E. Lieske, S. L. Hooe, A. W. Nichols and C. W. Machan, Dalton Trans., 2019, 48, 8633–8641 RSC; (c) G. Passard, D. K. Dogutan, M. Qiu, C. Costentin and D. G. Nocera, ACS Catal., 2018, 8, 8671–8679 CrossRef CAS.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS.
- (a) M. Fontecave and D. Mansuy, Tetrahedron, 1984, 40, 4297–4311 CrossRef CAS; (b) S.-I. Murahashi, T. Naota and N. Komiya, Tetrahedron Lett., 1995, 36, 8059–8062 CrossRef CAS; (c) M. S. Seo, J. Y. Kim, J. Annaraj, Y. Kim, Y.-M. Lee, S.-J. Kim, J. Kim and W. Nam, Angew. Chem., Int. Ed., 2007, 46, 377–380 CrossRef CAS PubMed; (d) J. Haber, T. Mlodnicka and J. Poltowicz, J. Mol. Catal., 1989, 54, 451–461 CrossRef CAS; (e) Y. Tohru, I. Kiyomi, N. Takushi and M. Teruaki, Bull. Chem. Soc. Jpn., 1994, 67, 2248–2256 CrossRef.
- (a) C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 4186–4193 CrossRef CAS PubMed; (b) C. W. Anson and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 18472–18475 CrossRef CAS PubMed.
- K. Izutsu, Commission on Electroanalytical Chemistry International Union of Pure Applied Chemistry: Acid-Base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell Scientific Publications, Oxford, England, 1990, vol. 35 Search PubMed.
- (a) M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed; (b) N. Gupta and H. Linschitz, J. Am. Chem. Soc., 1997, 119, 6384–6391 CrossRef CAS; (c) C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed; (d) M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed; (e) P. A. Staley, E. M. Lopez, L. A. Clare and D. K. Smith, J. Phys. Chem. C, 2015, 119, 20319–20327 CrossRef CAS; (f) D. H. Evans, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS PubMed; (g) P. D. Astudillo, J. Tiburcio and F. J. González, J. Electroanal. Chem., 2007, 604, 57–64 CrossRef CAS; (h) P. D. Astudillo, D. P. Valencia, M. A. González-Fuentes, B. R. Díaz-Sánchez, C. Frontana and F. J. González, Electrochim. Acta, 2012, 81, 197–204 CrossRef CAS; (i) T. M. Alligrant, J. C. Hackett and J. C. Alvarez, Electrochim. Acta, 2010, 55, 6507–6516 CrossRef CAS.
- (a) D. H. Evans and A. René, Phys. Chem. Chem. Phys., 2012, 14, 4844–4848 RSC; (b) N. A. Macías-Ruvalcaba and D. H. Evans, J. Phys. Chem. B, 2006, 110, 5155–5160 CrossRef PubMed; (c) A. René and D. H. Evans, J. Phys. Chem. C, 2012, 116, 14454–14460 CrossRef; (d) B. Uno, N. Okumura, M. Goto and K. Kano, J. Org. Chem., 2000, 65, 1448–1455 CrossRef CAS PubMed; (e) N. A. Macías-Ruvalcaba, N. Okumura and D. H. Evans, J. Phys. Chem. B, 2006, 110, 22043–22047 CrossRef PubMed.
- H. Shalev and D. H. Evans, J. Am. Chem. Soc., 1989, 111, 2667–2674 CrossRef CAS.
- R. R. S. Shi, M. E. Tessensohn, S. J. L. Lauw, N. A. B. Y. Foo and R. D. Webster, Chem. Commun., 2019, 55, 2277–2280 RSC.
- (a) R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS PubMed; (b) M. Balamurugan, N. Saravanan, H. Ha, Y. H. Lee and K. T. Nam, Nano Convergence, 2018, 5, 18 CrossRef; (c) J. Masa, K. Ozoemena, W. Schuhmann and J. H. Zagal, J. Porphyrins Phthalocyanines, 2012, 16, 761–784 CrossRef CAS; (d) J. Zagal, M. Páez, A. A. Tanaka, J. R. dos Santos and C. A. Linkous, J. Electroanal. Chem., 1992, 339, 13–30 CrossRef CAS; (e) J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS; (f) J. H. Zagal, F. Javier Recio, C. A. Gutierrez, C. Zuñiga, M. A. Páez and C. A. Caro, Electrochem. Commun., 2014, 41, 24–26 CrossRef CAS; (g) I. S. Flyagina, K. J. Hughes, M. Pourkashanian and D. B. Ingham, Int. J. Hydrogen Energy, 2014, 39, 21538–21546 CrossRef CAS; (h) J. Masa and W. Schuhmann, Chem.–Eur. J., 2013, 19, 9644–9654 CrossRef CAS PubMed; (i) W. Schöfberger, F. Faschinger, S. Chattopadhyay, S. Bhakta, B. Mondal, J. A. A. W. Elemans, S. Müllegger, S. Tebi, R. Koch, F. Klappenberger, M. Paszkiewicz, J. V. Barth, E. Rauls, H. Aldahhak, W. G. Schmidt and A. Dey, Angew. Chem., Int. Ed., 2016, 55, 2350–2355 CrossRef PubMed; (j) R. McGuire Jr, D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC; (k) M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed; (l) G. Passard, A. M. Ullman, C. N. Brodsky and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 2925–2928 CrossRef CAS PubMed; (m) D. J. Wasylenko, C. Rodríguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 CrossRef CAS PubMed; (n) K. Nagao, S. Hiroshi and O. Tetsuo, Chem. Lett., 1985, 14, 1917–1920 CrossRef.
- (a) S. L. Hooe, A. L. Rheingold and C. W. Machan, J. Am. Chem. Soc., 2018, 140, 3232–3241 CrossRef CAS; (b) S. L. Hooe and C. W. Machan, J. Am. Chem. Soc., 2019, 141, 4379–4387 CrossRef CAS PubMed.
- Y. C. Lam, R. J. Nielsen, H. B. Gray and W. A. Goddard, ACS Catal., 2015, 5, 2521–2528 CrossRef CAS.
- N. Gamboa-Valero, P. D. Astudillo, M. A. González-Fuentes, M. A. Leyva, M. d. J. Rosales-Hoz and F. J. González, Electrochim. Acta, 2016, 188, 602–610 CrossRef CAS.
- M. Aguilar-Martinez, N. A. Macias-Ruvalcaba, J. A. Bautista-Martinez, M. Gomez, F. J. Gonzalez and I. Gonzalez, Curr. Org. Chem., 2004, 8, 1721–1738 CrossRef CAS.
- J.-M. Savéant and C. Costentin, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, Wiley, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrolyses, NMR, CV, RDE, and RRDE data. See DOI: 10.1039/d1sc01271a |
| This journal is © The Royal Society of Chemistry 2021 |