
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective formation of Pt12L24 nanospheres by ligand design†
Eduard O.
Bobylev
,
David A.
Poole III
,
Bas
de Bruin
 and
Joost N. H.
Reek
*
and
Joost N. H.
Reek
*
Van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
First published on 28th April 2021
Abstract
Supramolecular self-assemblies are used across various fields for different applications including their use as containers for catalysts, drugs and fluorophores. M12L24 spheres are among the most studied, as they offer plenty of space for functionalization, yielding systems with unique properties in comparison to their single components. Detailed studies on the formation of M12L24 structures using palladium cornerstones (that have generally dynamic coordination chemistry) aided in the development of synthetic protocols. The more robust platinum-based systems received thus far much less attention. The general use of platinum-based assemblies remains elusive as parameters and design principles of the ligand building blocks are not fully established. As platinum-based nanospheres are more robust due to the kinetically more stable nitrogen–platinum bond, we studied the sphere formation process in detail in order to develop descriptors for the formation of platinum-based nanospheres. In a systematic study, using time-dependent mass spectrometry, 1H-NMR and DOSY NMR, we identified new kinetically trapped intermediates during the formation of Pt12L24 spheres and we developed key parameters for selective formation of Pt12L24 spheres. Molecular mechanics calculations and experimental result support the importance of charge and steric bulk placed at the endo-site of the ditopic linker for selective sphere formation. Applicability of these principles is demonstrated by employing various ditopic ligands with different bend-angles for the synthesis of a range of Pt2L4, Pt3L6, Pt4L8 and Pt12L24 polyhedra with platinum cornerstones in excellent yields, thus paving the way for future applications of well-defined robust platinum nanospheres of different shapes and sizes with the general composition PtnL2n.
Introduction
Inspired by the importance of self-assembly processes found in natural systems, supramolecular chemistry has been developed as a strategy to generate functional large molecular objects. In this context, chemists have succeeded in synthesizing a wide variety of polyhedral coordination cages with the general formula MnL2n, n being 2, 3, 4, 6, 8, 9, 10, 12, 24 and 30.1–10 These symmetrical structures are formed from the square-planar complexation of metal ions with ditopic pyridine linkers. Whereas for small structures of the composition M2L4 various metals have been utilized (e.g. M = Pd, Cu, Pt, Zn, Ni),4,10–12 only palladium and platinum based structures have been reported for larger assemblies based on ditopic pyridine ligands.13–15 Depending on the size of the self-assembled nanospheres, different applications are envisioned. Small M2L4 assemblies can be used as catalysts16,17 or as hosts to bind small guests.18–20 Larger structures, such as M6L12 or M12L24 assemblies, are able to incorporate bigger guests or even multiple guest molecules.21–23 The large inner (endo) volume of M12L24 spheres allows preorganization of up to 24 molecules. As such, it resembles a unique system to study transition metal complexes at high local concentration, which is beneficial when reactions proceed via dinuclear pathways or when catalyst and substrates are pre-organized.21,22,24 The effect of the second coordination sphere on catalytic reactions,22 tandem catalysis25 and spectroscopic cooperativity effects can also be studied.26–29For several applications it is important to control the size of the sphere that is formed during the assembly process, but this can be challenging. Key parameters for selective palladium-based systems have been identified previously by the group of Fujita. One main design principle is the dihedral angle (bend angle) between the two pyridines of the applied linker (Fig. 1).2 This bend angle is locked at a certain degree by employing a rigid spacer between the central aromatic system and the pyridines (Fig. 1). The outcome of sphere formation is well-described by the bend angle strategy because of geometric constraints, with only few exceptions.1,30 Other parameters such as solvent, concentration and counterion of the palladium source are typically adjusted to the system requirements and generally do not play a major role in the selectivity of sphere formation.15
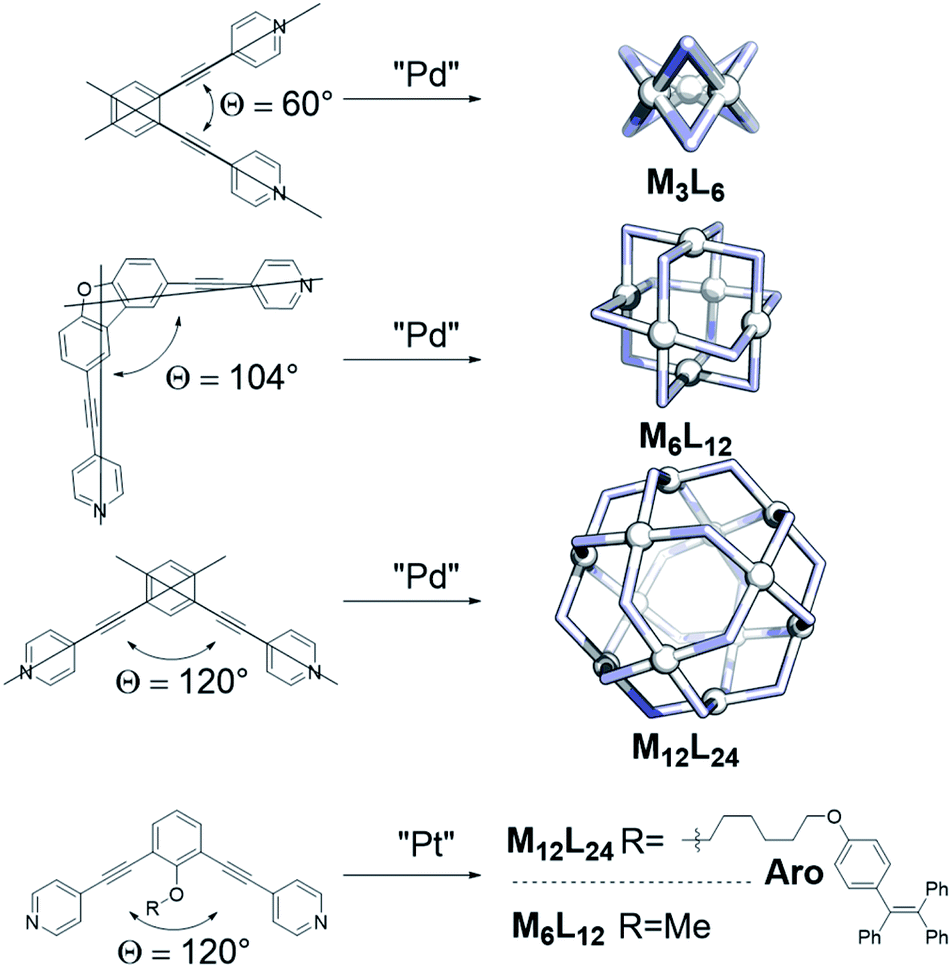 | ||
| Fig. 1 Selected examples demonstrating the bend angle approach for palladium-spheres; two literature known building blocks which show that the bend angle is not sufficient to predict formation of platinum spheres. | ||
The dynamic pyridyl–palladium bond leads to dynamic cage behaviour, which make palladium-based spheres excellent candidates for studying cooperativity effects and structural features. However, palladium-spheres are not sufficiently stable for all applications.23,24,31 The analogous platinum systems display superior stability under a variety of conditions including the presence of coordinating substrates, due to the stronger and kinetically more stable Pt–N(pyridine) bonds.24 Moreover, platinum-spheres are less prone to degradation by reducing agents23 and display good fluorescent properties,26–29 making them promising candidates for biological applications.32,33 Research has been focused mainly on the formation of palladium-based systems34–37 and their application across the fields.3,9,21,22,25,30,38–42 Only a relatively small number of reports describe platinum-based MnL2n analogues.22–24,29,31,32,43,44
The strong bond between Pt and pyridine allows limited conditions for product formation as sufficient dynamicity is required. Two examples, from the group of Fujita and the group of Stang, are depicted in Fig. 1 using a combination of high temperature and agents that destabilize the Pt–N bond.29,31 Interestingly, even though most structural features of the linkers such as the bend angle and electronic properties as well as similar synthetic protocols were employed, one of the ditopic ligands formed a Pt6L12 (R = Me) sphere31 and the other a Pt12L24 (R = Aro, Fig. 1) sphere.29 These examples demonstrate that the bend angle as practical parameter for the prediction of palladium based nanospheres is a poor or incomplete descriptor for platinum nanosphere formation. We therefore decided to investigate the factors determining platinum nanosphere formation in detail to guide their synthesis. The results are reported in this paper. In order to obtain a clear picture, the formation nanospheres was carefully monitored using MS-analysis and NMR techniques providing insights into sphere formation pathways. The metastable structures formed along the pathway to the formation of M12L24 assemblies were next used as a starting point to develop design principles for platinum spheres (combined with known principles guiding palladium sphere formation). With these descriptors, the outcome of sphere formations with platinum cornerstones can be predicted and ligands can be designed to yield desired structures in various shapes and sizes, as was confirmed in this study. Keeping in mind the numerous advantages of platinum-based systems, this work should stimulate further development of these more robust architectures in different applications.
Results and discussion
Model studies
The goal of our investigation was to selectively form Pt12L24 assemblies with broadly functionalizable building blocks. A relatively simple building block with acetylene linkers LOMe (Fig. 2) was chosen for initial studies. This building block has a bend angle of 120° geometrically required for M12L24 assemblies.2,36 Furthermore, it has no particular electronic or steric demands and can act as a starting point for exo or endo functionalization. Because spherical systems are typically prepared by thermodynamic control, molecular modelling was performed on the desired Pt12LOMe24 assembly and other possible spherical structures. These possible intermediate structures for platinum based M12L24 systems are based on observations made for the analogues palladium spheres. Five possible intermediates Pd6L12, Pd8L16, Pd9L18, Pd10L20 and Pd11L22, were predicted in silico.36 Two of these palladium-based structures (Pd8L16, Pd9L18) were trapped by ligand design and characterized in detail.35 We included all known and predicted intermediates in our molecular modelling. According to the minimized structures for all spherical objects of the general formula PtnL2n for n = 6–12 (Fig. 2, SI7† for details, description of the method can be found in ref. 37), the Pt12LOMe24 sphere represents the thermodynamic minimum. Because the Pt12LOMe24 assembly is thermodynamically even more favourable over other structures than was the case for the palladium analogue (Fig. 2), selective formation was expected when structures could form under thermodynamic control.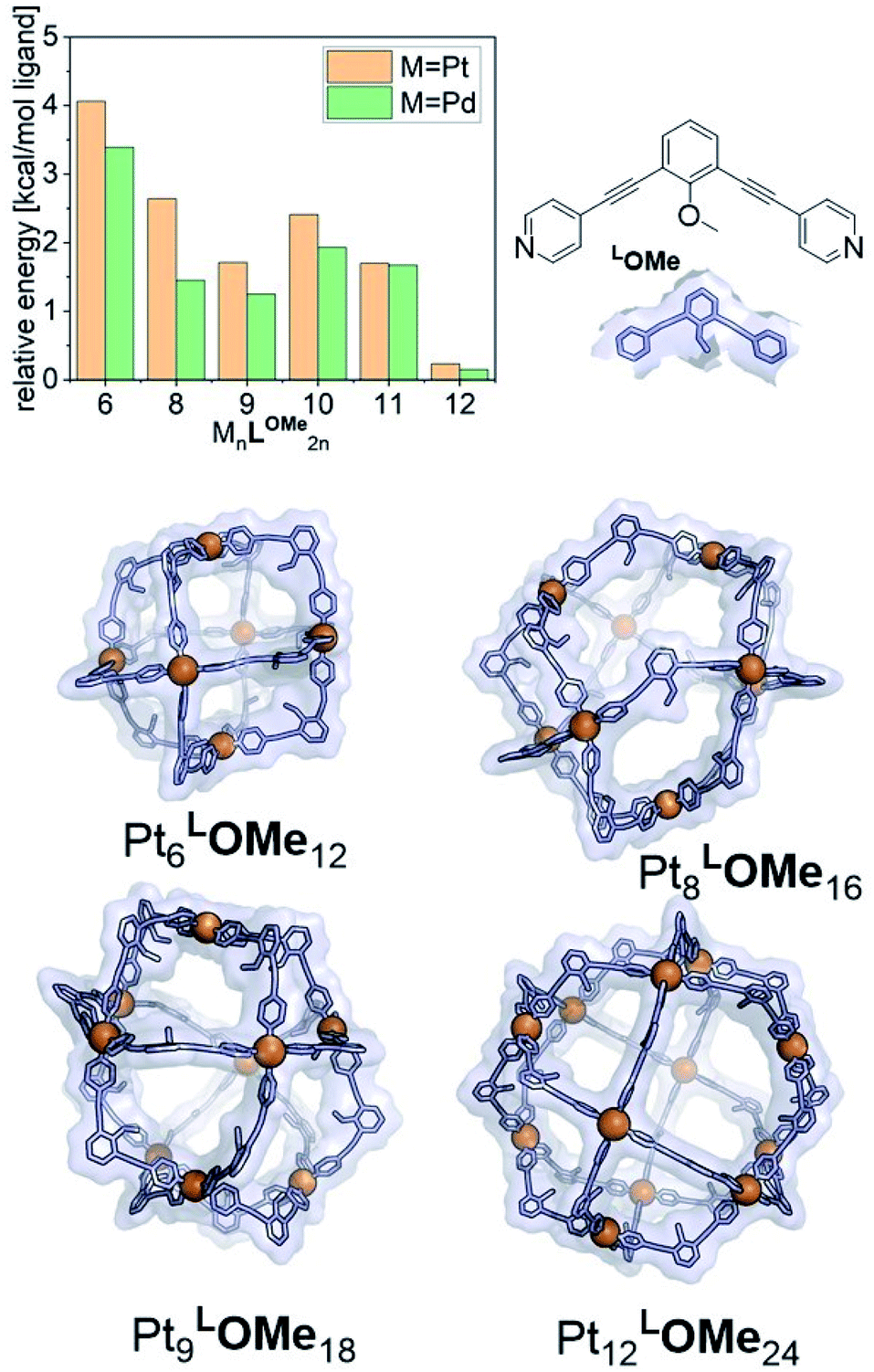 | ||
| Fig. 2 Examples of minimized structures for PtnLOMe2n spheres and the per-ligand energy of differently sized spheres relative to the minimum energy structure, cuboctohedral Pt12LOMe24. These energies include the Boltzmann weighted contributions of less favourable configurations such as the triangular bicopula form of the Pt12LOMe24. | ||
Experiments were set up to form the Pt12L24 assembly with LOMe under thermodynamic control by varying the reaction temperature. Sphere synthesis was performed by mixing 1 eq. of building block LOMe with 0.55 eq. [Pt(BF4)2(MeCN)4] in acetonitrile in closed high pressure tubes for 3 days at the desired temperature (Fig. 3A). Progress was monitored using 1H-NMR, DOSY and MS-analysis. At 70 °C, a downfield shift of the pyridine protons accompanied by broadening of the signals upon coordination to platinum was observed. The surprisingly broad signals are in contrast to the sharp signals obtained for the well-defined palladium-based sphere. A lower diffusion coefficient observed by DOSY indicates formation of structures in the size range of the M12L24 spheres (S65–S67†). However, no well-defined sphere structures were detected by MS analysis. The kinetically inert Pt–N bond may prevent formation of the thermodynamically favoured spheres due to kinetic trapping of less stable intermediates. Therefore, we increased the reaction temperature gradually. At 85 °C, the 1H-NMR and DOSY spectra remained similar to the spectra obtained at 70 °C (Fig. 3B, S74–S75†). MS analysis showed multiple signals corresponding to different charged states of the sphere with isotopic patterns corresponding to [Pt12(LOMe)24(BF4−)24−x]x+ (x = 7–11). Because these self-assembled spheres show a linear response with the concentration in the MS spectra15 (S113–S115†), provided that the parameters such as the ion strength and the solvent remain the same, the quantity of the desired sphere in solution can be monitored by the characteristic peaks of the M12L24 sphere, i.e. [Pt12(LOMe)24(BF4−)13]11+ (m/z = 992 Da) (Fig. 3C). Increasing the reaction temperature further to 110 °C, 130 °C and 150 °C led to relatively sharp signals in the 1H-NMR spectra (Fig. 3B). All signals are split into multiple sets, indicative for the presence of multiple species. The intensity of the identified 11+ species increased with increasing the reaction temperature (Fig. 3C, S69†). However, after prolonged heating at 150 °C, the signal became less intense (after 5 days). The decrease of counts is accompanied with a colour change of the solution (from colourless to orange), observation of new species in 1H-NMR (around 5 ppm, S73†) and appearance of free building block LOMe. Presumably the sphere disassembles by decomposition of both the platinum precursor to nanoparticles and the ligand LOMe to other adducts (S73†). Besides the desired Pt12(LOMe)24 also other structures can exist in solution as indicated by the multiple sets of signals in the 1H-NMR spectra. A detailed analysis of the MS data confirmed the presence of multiple spherical objects. Besides analogues to intermediates which have been identified/predicted for palladium as Pt8LOMe16, Pt9LOMe18, Pt10LOMe20 and Pt11LOMe22, also a non-Archimedean-solid type Pt7LOMe14 was observed. MSMS analysis of the M12L24 assembly show that these structures were not formed by fragmentation during the MS experiments (S116†). As such, it is important to analyse all multiple charged species, as otherwise some of the formed assemblies could be missed. Analysis of the even charged species may be complicated, because they can show an overlap of differently charged states of different assemblies (e.g., overlap of [Pt12(LOMe)24(BF4−)12]12+ and [Pt8(LOMe)16(BF4−)8]8+). All structures indicated here show signals corresponding to [Ptn(LOMe)2n(BF4−)2n−x]x+ for multiple charged states with a matching isotope pattern (S34†). Calibration of the MS signals using of a mixture of nanospheres prepared at 150 °C for 3d (which gave overall the most counts, Table S19†) and measured at a range of concentrations (diluted with a Pd12LOMe24 sphere to maintain the ion concentration constant) show a linear correlation between the concentration of the different sized spheres and their intensity in the MS spectra (S113–S115†). With this correlation in hand, the relative amount of spheres formed with different methods can be compared. By following unique signals assigned to each of those assemblies over a range of different temperature, their relative stability was estimated (Fig. 4). Though this method is prone to quantitative errors (∼10% as described in S70–S71† from experiments in triplo), qualitative analysis showed excellent reproducibility (S70–S71†). At 85 °C, the dominant (highest intensity) species was assigned to Pt6LOMe12, similar to what has been found before in literature.31 After heating the sample for 3d at 110 °C the Pt7LOMe14 assembly disappears (least stable) and the M6L12 structure becomes less pronounced (moderately stable). Heating the sample at 150 °C led to disappearance of the M10L20 assembly. Spheres with 8, 9 and 12 platinum cornerstones increased constantly over the period of the study. The slope of increase was most dominant for the M12L24 assembly (most stable). After heating the sample for prolonged time at 150 °C or addition of 2-chloropyridine as a destabilizing agent (S72†), the M8L16 structure decreased more than the M9L18 assembly (more stable). The experimentally obtained relative stability is in good agreement to our computational results (see Fig. 4 and 2).
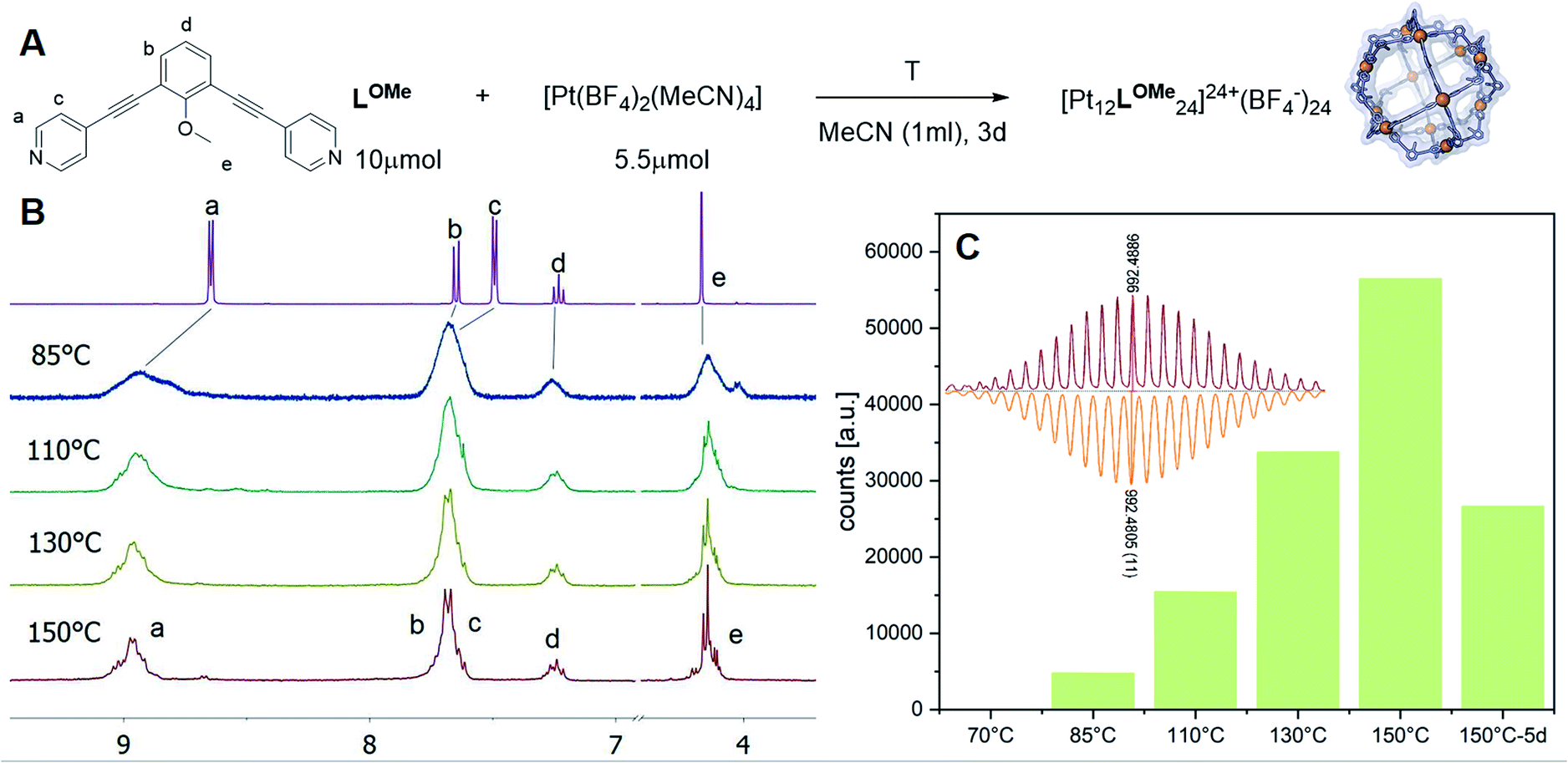 | ||
| Fig. 3 (A) Reaction scheme of the formation of [Pt12LOMe24]; (B) 1H-NMR spectra of the complexation of ligand LOMe at different temperatures; (C) Development of the assigned 11 + species of the [Pt12LOMe24] assembly at different temperatures (inset: measured spectra = red; calculated spectra = orange). | ||
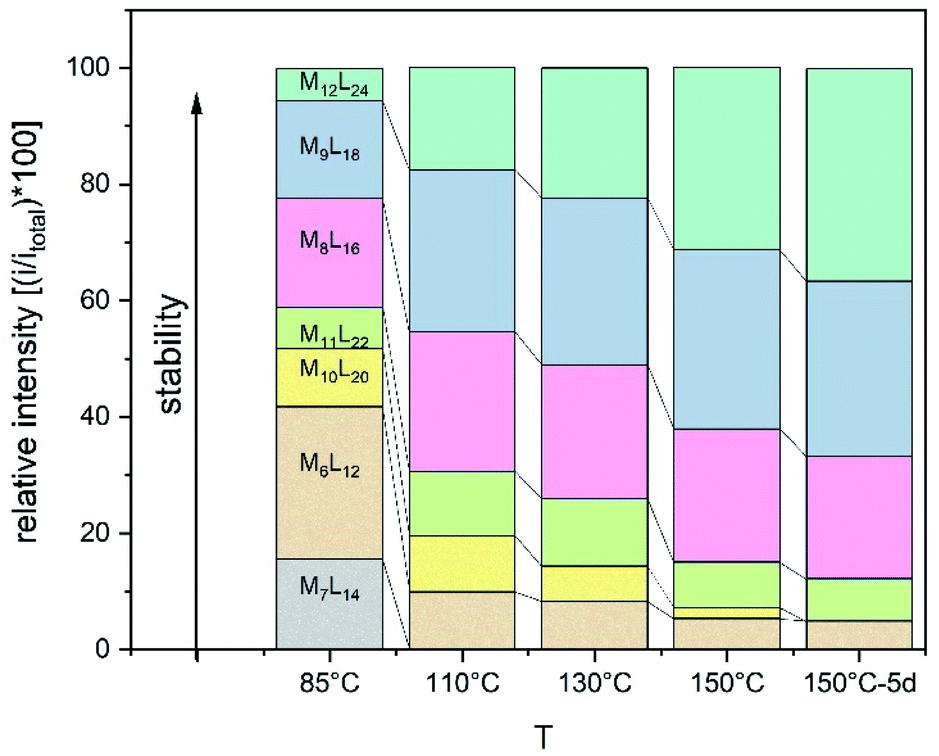 | ||
| Fig. 4 Development of unique signals assigned to each of PtnLOMe2n assemblies for n = 6–12 at different temperatures. | ||
Because pure spheres could not be obtained, we attempted to purify the sphere from the mixtures by crystallization and column chromatography. Crystallization did not turn out fruitful using a number of different anti solvents (Et2O, THF, diisopropylether, chloroform). Interestingly, a mixture of nanospheres prepared at 150 °C, could be separated by column chromatography. Eluting with pure MeOH, using silica as the stationary phase afforded pure Pt12LOMe24 assembly according to MS analysis in small quantities (<0.5 mg, <1% yield; S119†). While optimizing the separation conditions (such as stationary phase, solvent, etc.) is not within the scope of this work, the experiment shows that the kinetic stability of platinum spheres sufficiently high for purification by column chromatography.
The formation of an Pt12L24 assembly with a simple acetylene bridged ligand has been achieved by adjusting the temperature. However, despite using destabilizing agents and elevated temperature, selective formation of the thermodynamically favoured Pt12L24 assembly was not possible using LOMe. Two initially kinetically trapped assemblies Pt7L14 and Pt10L20 disappeared by increasing the reaction temperature. Other spheres such as the Pt9L18 and Pt8L16 remained in solution regardless of the temperature applied. The relative energy gap between the M9L18, M8L16 and the M12L24 assembly is similar for both, platinum-based systems and the palladium-based analogues. It is possible to form Pd12L24 self-assemblies selectively with the relatively small thermodynamic gap between the desired Pd12L24 and the smaller sized structures (PdnL2n for n = 6–11). This shows that the thermodynamic difference (which is even bigger for Pt) should also allow the selective formation of Pt12L24 assemblies. Because the desired Pt12L24 assembly could not be formed by adjusting the temperature, intermediates of the general formula PtnL2n for n = 8, 9, 11 are kinetically trapped due to the stronger Pt–N bond, as they do not disappear upon increasing the reaction temperature (Fig. 4). In contrast to these observations made for platinum, palladium-based systems have a weaker Pd–N bond and can thus escape from a kinetic trapped mixture of intermediates, thus forming the thermodynamically most favoured Pd12L24 assembly selectively.
Formation pathway and strategy
In order to develop a strategy to selectively form M12L24 assemblies, the formation pathway was studied further. The proposed pathway of palladium-based systems proceeds via an initially formed coordination oligomer and subsequently through a set of kinetically trapped small sized intermediates of the general formula MnL2n (n = 6, 8 and 9). These Pd intermediates were found relatively stable by computation, but could only be analysed as short-lived intermediates with 1H-NMR spectroscopy and HR ESI-MS studies.34 It is not clear whether the thermodynamically controlled Pd12L24 assemblies form by a linear pathway from small sized spheres and intermediates or alternatively that the intermediates re-enter an oligomeric state and reform spheres.We observed after mixing of LOMe with the corresponding platinum salt at 70 °C (or lower) no higher charged species in the HR CSI-MS spectra, DOSY shows a low diffusion coefficient (S74–S77†) and broad signals were observed in the 1H-NMR spectra. The species formed in the initial period is likely oligomeric in nature (O) (Fig. 5). When this oligomeric material (O) is heated to 85 °C (or higher), a sharper 1H-NMR spectrum appears and also the HR CSI-MS spectra display signals attributed to various types of spheres with the general formula PtnL2n. In a cage formation experiment carried out at 85 °C similar amounts of different spherical objects (e.g., Pt9L18 and Pt6L12) are formed (Fig. 4). This implies that the initial oligomeric material (O) can fold in different ways forming randomly different sized spheres. Increasing the temperature to 150 °C causes the disappearance of the Pt7L14 and a decrease of the signals attributed to the Pt6L12 and Pt10L20 assemblies. These different PtnL2n assemblies thus represent kinetically unstable structures. Their decrease is accompanied with an increase of concentrations of the larger assemblies Pt8L16, Pt9L18 and Pt12L24 assemblies. This transition to the bigger assemblies can proceed via two slightly different pathways. (1) the initially formed intermediates (I) are further converted to the larger spheres. Herein, we combine all non- or over-saturated spherical structures which are derived from PtnL2n to these intermediates (I) (e.g., Ptn+1L2n, PtnL2n+1, PtnL2n−1 and PtnL2nMeCN1, Fig. 5). (2) the initial oligomer (O) can be re-formed from the intermediates, from which all types of spheres are formed again. In this scenario the intermediates and cages are in equilibrium via the oligomer state.
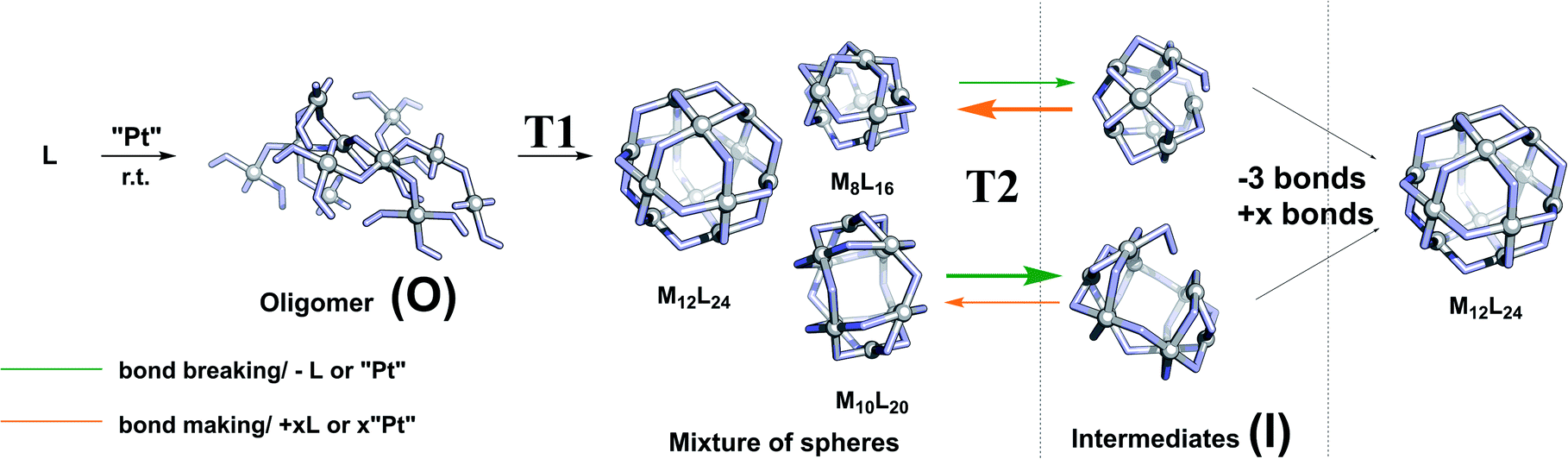 | ||
| Fig. 5 Proposed formation mechanism of platinum-based spheres (T1 = 85 °C; T2 = 150 °C); middle: example of a few spheres formed during this process (M8L16, M12L24 and M10L20); arrows indicate formation and deformation of most unstable spheres; productive pathways are depicted with bold arrows. | ||
To arrive at a strategy that leads to selective formation of Pt12L24 assemblies, it is important to know how the observed small sized assemblies convert to the desired structure. The mechanism of conversion from intermediates to desired structure (route 1 or 2) has implications for the design rules of building blocks and conditions for the preparation of pure Pt12L24 assemblies. As such, additional exchange experiments under various conditions were carried out and analysed by NMR and HR CSI-MS. In these experiments we generated self-assembled structures based on slightly different building blocks LOMe and LOBn and we mixed the solutions at various stages of sphere formation and monitored the degree of exchange, resulting PtnLOMe2n−xLOBnx assemblies.
In a first experiment (Fig. 6, Case I), two oligomers OOMe and OOBn containing either LOMe or LOBn are formed in separate solutions by mixing the ligands with platinum precursor at room temperature. Coordination of platinum is confirmed by a downfield shift of the pyridine protons and DOSY shows that the formed structure has a lower diffusion coefficient in line with oligomer formation (S79–S81†). MS analysis of the individual oligomer mixtures showed no occurrence of spherical structures as multiple charged species were absent. The oligomers solutions were mixed in a 1 to 1 ratio and heated at 150 °C for one day. Analysis of the solution revealed a mixture of different sized spheres with a statistical distribution of the two ligands LOMe and LOBn as expected from building blocks with no self-sorting function (Fig. 6, S88–S91†). This experiment shows that building block exchange from the oligomer state occurs without kinetic barriers.
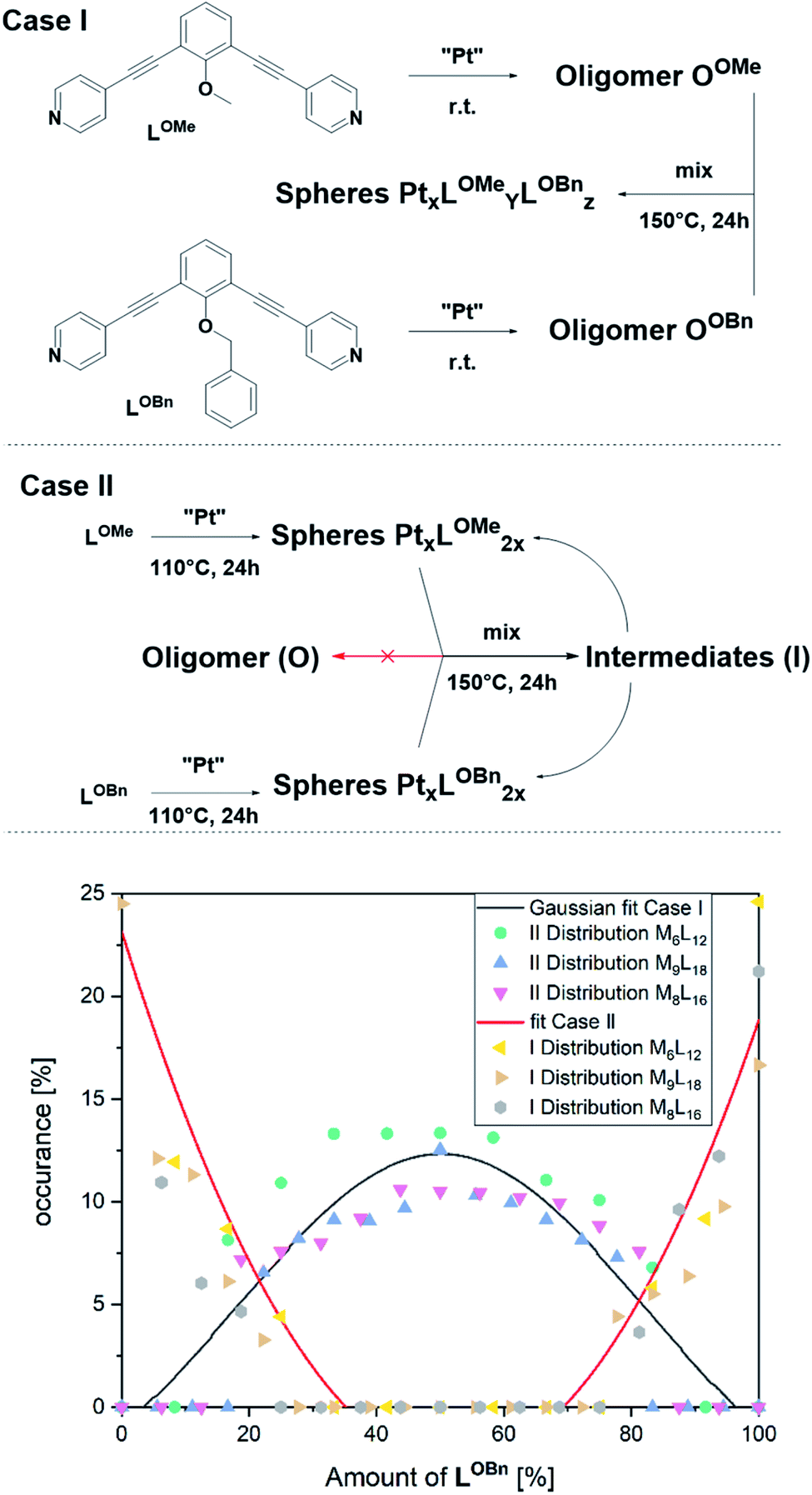 | ||
| Fig. 6 (Case I) Formation of two individual oligomers and the resulting statistical distribution of the ligands in the spherical objects obtained after heating a mix of oligomers; (Case II) Formation of a small number of individual spheres and the resulting non-statistical distribution of the ligands in the spherical objects obtained after heating; MS Distribution of the two ligands obtained by averaging the relative intensities of the respective M6L12, M8L16 and M9L18 assemblies (Case I gives a Gaussian distribution of the two applied ligands; Case II was fitted quadratically to give a line to guide the eye). | ||
In the second experiment, the spheres of LOMe and LOBn are prepared in separate solutions at 110 °C for one day (Fig. 6, Case II), combined and further heated at 110 °C. Importantly, no changes are observed by MS analysis (S94†) indicating that building blocks do not exchange when the assemblies are in well-defined sphere states, in line with kinetic inertness of platinum assemblies up to 110 °C. However, if the mixture is heated at 150 °C more spherical structures (S95†) are formed showing building block exchange under these conditions. The formation of more spherical compounds at 150 °C is accompanied with slow exchange of single ligands (Fig. 6, S96–S99†). The exchange of single ligands is occurring for all type of assemblies (MnL2n for n = 6–12) in a similar fashion. As the first experiment shows that in the oligomer state there is a full exchange of building blocks, and the second experiments shows only slow exchange of single building blocks from the spheres (similar result was also obtained mixing spherical sample of LOMe with oligomeric sample of LOBn, S102†), we can conclude that formation of platinum-based spheres from the oligomer state is irreversible. Indeed, such transition from sphere to oligomer requires the breaking of multiple Pt–pyridyl bonds.
At 150 °C, the exchange of single ligands is kinetically allowed. Importantly, this implies that formation of pure Pt12L24 spheres requires the direct conversion of formed intermediates to the desired Pt12L24.
As the conversion of lower to higher spheres is crucial, we studied it in more detail. Analysis of the structures shows that it is required to break (at least) 4 Pt–N bonds to obtain the Pt12L24 sphere from any of the smaller intermediates (section SI6†). The intermediates Pt8L16 and Pt9L18 spheres are kinetically inert as they remain in solution even at elevated temperatures. In the formation of Pt12L24 assemblies these therefore represent trapped states. Experiments show that at 150 °C exchange of single ligands from both the Pt8L16 and Pt9L18 spheres is possible, however, the conversion of these M8L16 and M9L18 sphere intermediates to the desired Pt12L24 is not possible (e.g., Fig. 5 and section SI6†). As the palladium–pyridyl bond is weaker, these M8L16 and M9L18 intermediates do not represent trapped states for the formation of Pd12L24 spheres.
Ligand design by using ditopic ligands with functional groups
As some of the intermediates are kinetically trapped states we set-out a strategy for selective formation of Pt12L24 assemblies that relies on destabilization of these intermediate-sized structures. A look at the modelled spheres shows a clear growth of the interior space available per ligand with increasing sphere size (Fig. 7). For example, the volume available for the M12L24 sphere is 25% larger than that of the M9L18 intermediate state. By endo functionalization of the ditopic ligand building blocks, either with sterically bulk or charged functional groups, the smaller spheres are destabilized by steric hindrance (or charge repulsion) to a larger extend than the M12L24 spheres. These intermediates therefore might not form, or are destabilized such that they no longer form kinetic traps.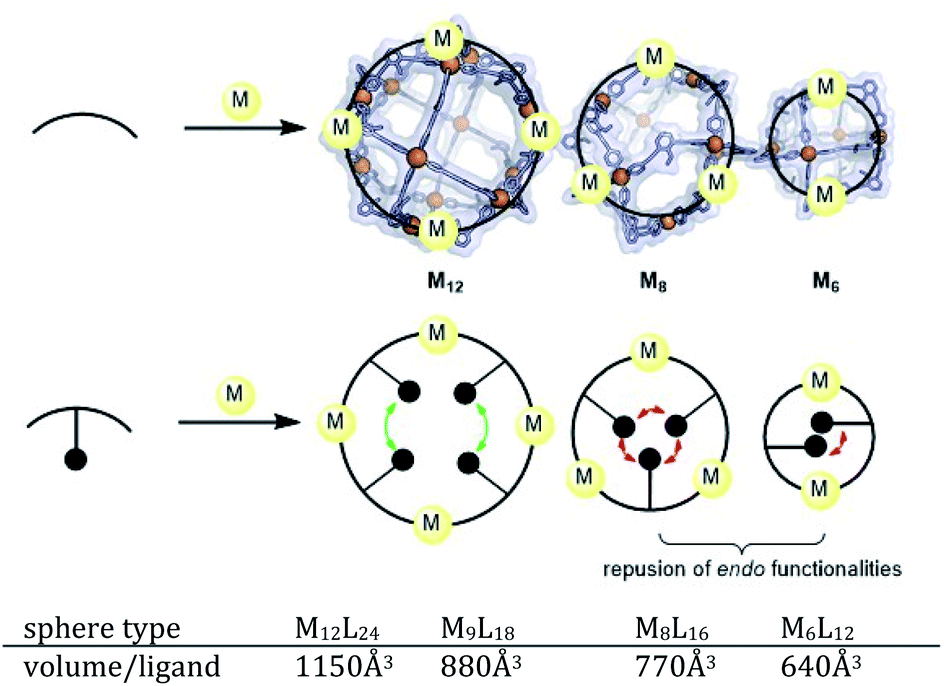 | ||
| Fig. 7 Schematic representation of our strategy for destabilization of small size spheres. Representation of M6L12, M8L16 and M12L24 nanospheres. The sphere frames are optimized at molecular mechanics level (MMFF) and shown in space-filling-style. | ||
A set of ditopic ligands with additional functional groups was synthesized to test our hypothesis of selective formation of Pt12L24 spheres by a combination of electrostatic/steric repulsion, using the typically bend angle of 120°. The set (Fig. 8) contains two building blocks with charged units at the endo site (LPy and LImi), and building block with a charged unit at the exo site LexoPy to carefully study the effect of electrostatic repulsion. Two ligand building blocks, LPO and LCou, with bulky functional groups at the endo site were also used. All novel ligands were synthesized by a modular approach (section SI1†).
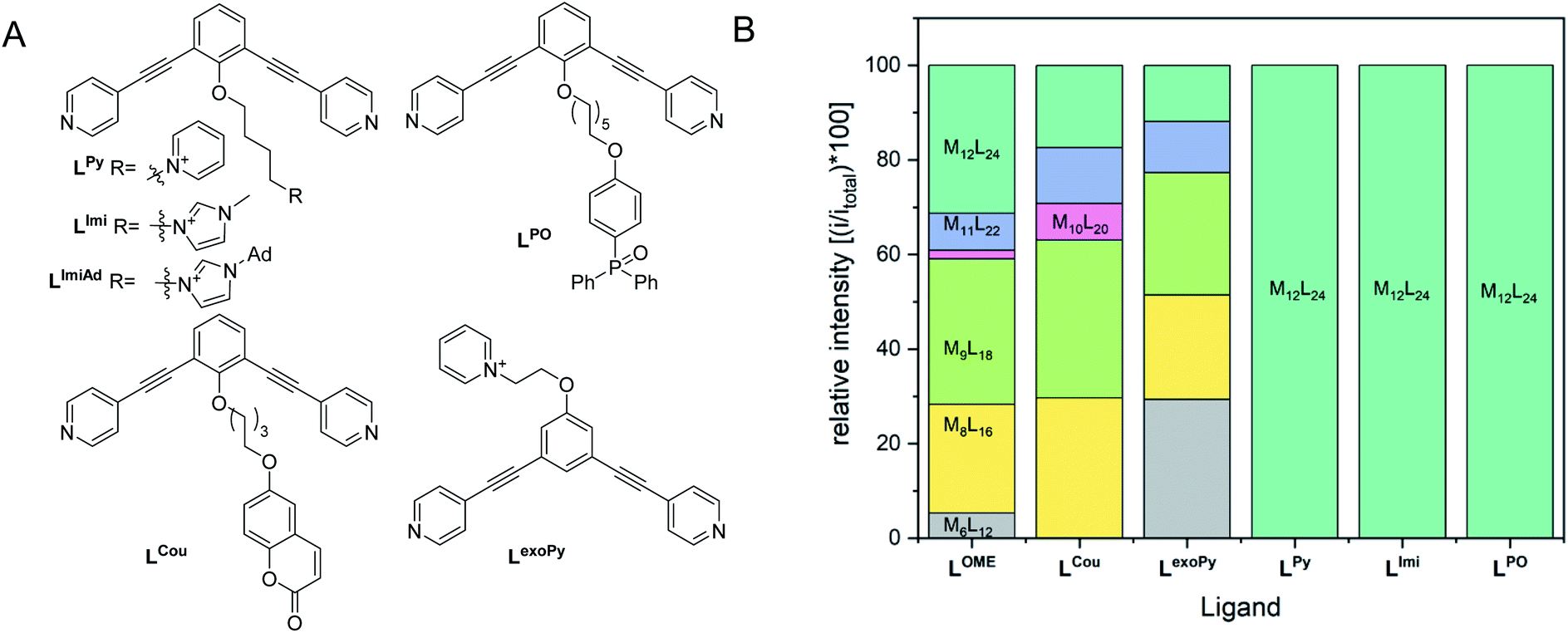 | ||
| Fig. 8 (Left) Structures of applied ligands in our investigation for selective formation of Pt12L24 spheres by steric or electrostatic repulsion; (Right) distribution of spherical structures obtained after complexation of the applied ligands with platinum precursor. | ||
First, two sets of spherical structures (M6–M12) were modelled based on charged ligand LPy and the bulky ligand LPO. In agreement with our previous observations, also for these assemblies the M12L24 sphere represents the thermodynamic minimum. Assembly formation with LPO was performed with 0.55 eq. platinum precursor. After heating the sample for 2d at 150 °C, a single set of sharp signals was observed in the 1H-NMR spectra (S43–S46†). HR CSI-MS analysis of the sphere solution showed selective formation of the desired Pt12LPO24 structure (Fig. 8B, S47†). Signals of any other sphere (MnL2n) were absent in the MS spectrum. These experiments show that indeed the presence of steric groups at the endo site prevents the formation of kinetically trapped states. For the building block LAro, also discussed in the introduction (Fig. 1) the formation of the Pt12L24 species was reported, suggesting that also for this sphere the selective sphere formation is possible because of the presence of the bulky functional group present at the endo site. The coumarin functionalized building block LCou, which is sterically less demanding, was also used for the formation of spheres under identical conditions, and this yielded a mixture of spheres (Fig. 8, S39–S42†). This shows that the introduction of steric groups is a successful strategy to obtain Pt12L24 spheres in a selective fashion, provided that the groups introduced at the endo side are sufficiently large. As a small note, the presence of 24 units of LPO occupy the complete interior space of the Pt12L24 sphere, complicating applications in which space is required, for example the binding of guest molecules or utilize the system in catalytic transformations.
As a second strategy, we attempted to selectively form Pt12L24 spheres using charge repulsion. After heating a sample containing LImi and 0.55 eq. platinum precursor to 150 °C for 2d, only the desired Pt12L24 sphere was formed as shown by a single set of protons in 1H-NMR and HR CSI-MS analysis (Fig. 9 and 8). When sphere formation was attempted at 85 °C a small amount of the corresponding Pt8LImi16 and Pt9LImi18 assemblies were detected using MS analysis (S78†). These structures disappear upon heating, indicating an escape from the otherwise kinetically trapped assemblies (S52†). In contrast to this selective formation of a Pt12LImi24 assembly, utilizing the exo functionalized building block LexoPy, yielded a mixture of spheres when the same conditions for sphere formation were applied (Fig. 8, see S58–S60† for details). This experiment demonstrates the importance of endo functionalization for the successful selective sphere formation with platinum cornerstones, as exo functionalization does not take advantage of differences in the available inner-volume between the kinetically trapped smaller assemblies and the desired larger Pt12L24 spheres.
Having demonstrated that building blocks with the proper bend angle and with sufficient steric bulk or charges at the endo site of the ligand leads to selective formation of Pt12L24 assemblies at 150 °C, we decided to perform additional experiments (Fig. 8B). The general applicability with different positively charged groups was confirmed using a ligand bearing a different functional group LPy (Fig. 8, details S53–S57†). Mixing LPy with the platinum pre-cursor for 2d at 150 °C selectively yields the Pt12L24 sphere in pure form, as shown by MS analysis (S57†). Further-more, the 1H-NMR spectrum of the formed complex is well resolved with a downfield shift of the pyridine protons (S53–S56†). From our previous experiments using a similarly sized ligand LCou, the steric repulsion introduced by a single aromatic group is not sufficient for selective formation (Fig. 8). Because selective sphere formation using charge repulsion does not require bulky substituents, these building blocks render themselves suitable for further functionalization (available space in self-assembly, Fig. 9). This was previously demonstrated by the binding of 24 gold complexes in the guanidine based nanosphere.22 In addition, both, the imidazolium-based building block LImi as well as LPy can be further derivatized with functional groups of interest for guest uptake or catalytic investigations within well-defined Pt12L24 assemblies. Exemplary, an adamantane group was placed on the imidazolium linker (LImiAd) (Fig. 8), which yields together with platinum also Pt12LImiAd24 self-assemblies with good selectivity (S61–S64†), also indicating that there is sufficient space available for further chemistry.
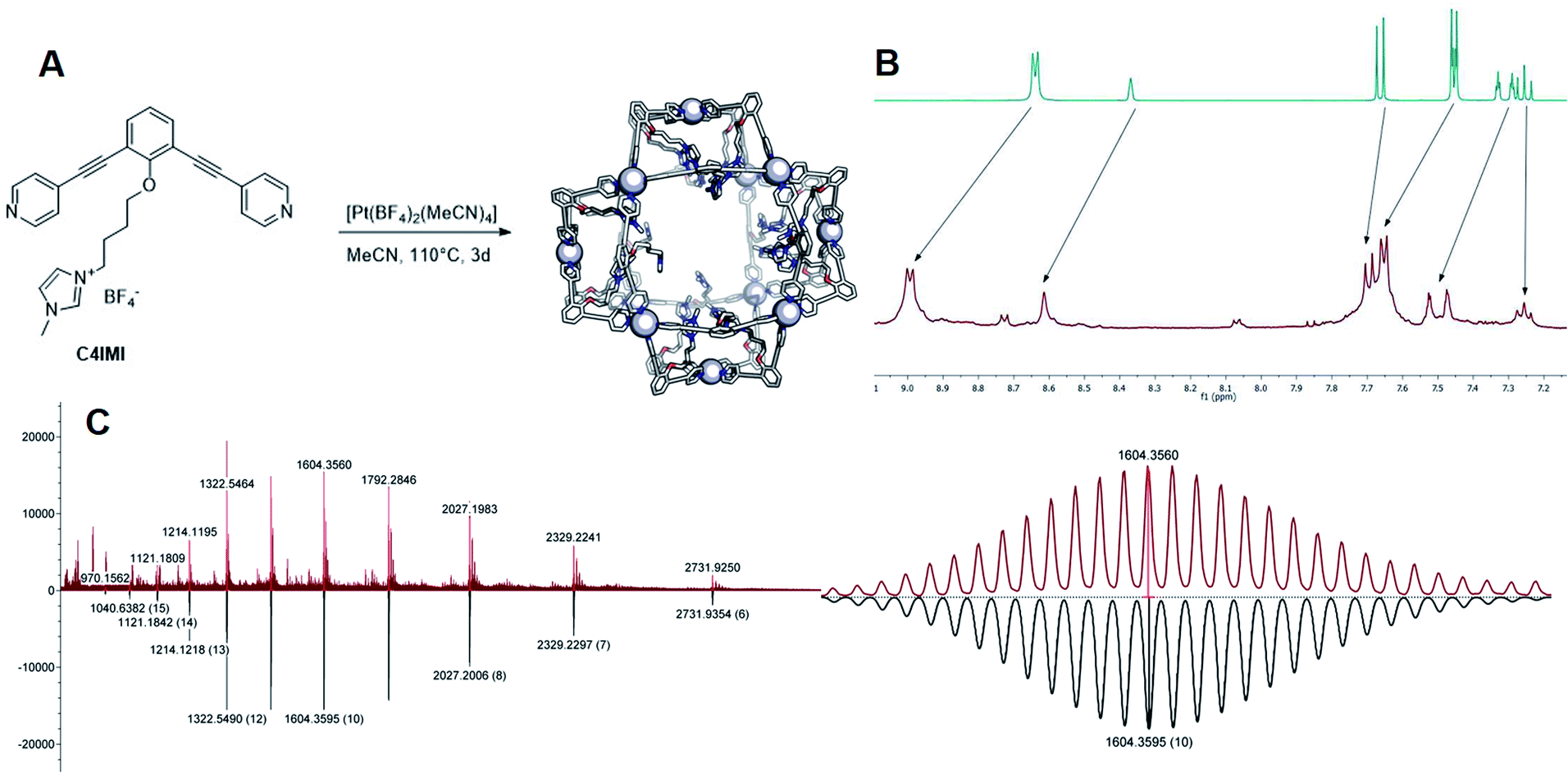 | ||
| Fig. 9 (A) Synthesis of the [Pt12(LImi)24] assembly, the sphere frames are optimized at molecular mechanics level (MMFF), nitrogen = blue, oxygen = red, carbon = white and the metal is depicted as white sphere; (B) 1H-NMR spectra of the ligand (top) and the self-assembly (bottom); (C) MS data of the self-assembly recorded at 60 °C in MeCN (top: measured; bottom: calculated) and zoom in into the 10+ species. | ||
Application for other structures
After exploring the formation of Pt12L24 spheres, the methodology was expanded to other systems. As described before, the bend angle between the pyridine bonds allows selective formation of palladium based spherical objects of different size. We choose building blocks known for successful sphere formation of the palladium analogue with bend-angles of Θ = 0° and 60° (Lmeta and Lpara, Fig. 10).1,13 Because platinum spheres form through smaller size kinetically trapped structures at elevated temperatures, these two bend angles are ideal to check sphere formation as both form the smallest possible spherical assemblies, being the M2L4, M3L6 and M4L8 structures. Sphere formation was performed by mixing 1 eq. of building block Lmeta or Lpara with 0.55 eq. [Pt(BF4)2(MeCN)4] in acetonitrile at 150 °C for 24 h (Fig. 10, section SI5†). After this period sharp 1H-NMR spectra with a downfield shift of all pyridine protons upon coordination of platinum were observed. Furthermore, HR CSI-MS analysis and DOSY NMR supported the formation of the desired assemblies. In line with the experiments for Pt12L24 sphere formation, when reaction temperatures were applied up to 110 °C a mixture of assemblies was obtained (Fig. S106†), suggesting that also for these smaller spheres the presence of kinetically trapped states may prevent the formation of pure assemblies. The elevated temperature of 150 °C allowed formation of the small spherical objects in excellent yields (94% based on 1H-NMR, see section SI5†).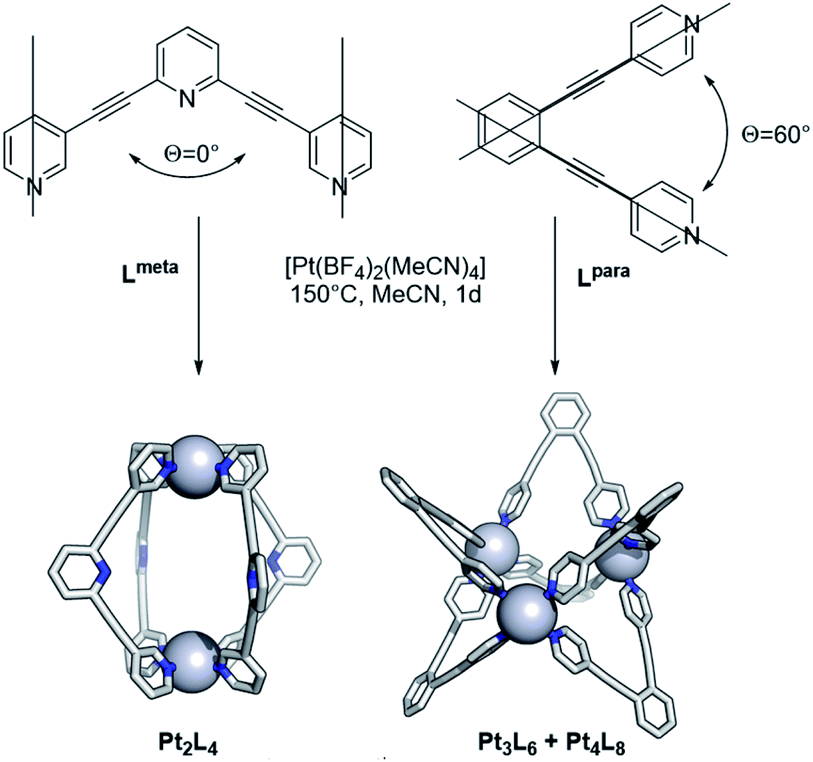 | ||
| Fig. 10 Complexation of Lmeta and Lpara at high temperature (meta and para refer to the connection of the pyridine ring with respect to the nitrogen atom). | ||
Conclusions
Whereas it has previously been demonstrated that using the bend angle of ditopic pyridyl building blocks is an effective descriptor to predict the formation of the size of PdnL2n, this descriptor alone is not sufficient for platinum analogues. A systematic study on the selective formation of platinum-based spheres using a combination of experimental and theoretical approaches is described. Due to the stronger and less dynamic platinum–nitrogen bond (compared to palladium–nitrogen), a high temperature is required to thermodynamically drive the equilibrium towards the most stable assemblies. Mixing ditopic ligands with the platinum precursor at 70 °C leads to mostly oligomer formation. Upon increasing the reaction temperature during the sphere formation, mixtures of various spheres are formed. Upon raising the temperature, some of the kinetically trapped intermediate PtnL2n species disappear from the reaction mixture, but a mixture of PtnL2n assemblies is still observed with simple (unfunctionalized) ligands. Sphere decomposition is observed before the thermodynamic Pt12L24 species is formed as the only nanosphere. Introducing the proper amount of steric bulk or an endohedral charge on the ligand gives rise to selective formation of well-defined Pt12L24 assemblies in excellent yield. These groups destabilize the smaller spheres that are the kinetic traps, such as the Pt8L16 and Pt9L18 and therefore an escape from these trapped states is possible. The herein presented charged building blocks can be easily modified to yield a variety of robust Pt12L24 assemblies in excellent yields. The synthetic strategy has been extended to other building blocks, including those with other bend angles that lead to other structures (Pt2L4–Pt4L6). MS was demonstrated to be an ideal tool to study complex sphere mixtures, helping to identify kinetically trapped intermediates pathing the way for strategies to escape those.Within the field of supramolecular self-assemblies, features of platinum-based assemblies such as electronic and kinetic inertness, fluorescent properties and biological relevance are already well recognized. By providing a strategy for selective formation of PtnL2n assemblies with easily modifiable ligands, we hope to promote further research on platinum based self-assemblies and their applications across fields.
Author contributions
Conceptualization: EOB, JNHR; formal analysis: EOB, DAP; funding acquisition: JNHR, BdB; investigation: EOB, DAP; supervision: JNHR; validation: EOB, DAP, BdB, JNHR; visualization: EOB, DAP; writing – original draft: EOB; writing – review & editing: EOB, DAP, BdB, JNHR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge the University of Amsterdam for financial support to RPA sustainable chemistry. We would like to thank Eline M. Meijer for discussions.Notes and references
- D. K. Chand, K. Biradha, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Chem.–Asian J., 2006, 1, 82–90 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, H. Yokoyama, N. Mizuno, T. Kumasaka and M. Fujita, Chem, 2016, 1, 91–101 CAS.
- K. Harris, Q. F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC.
- L. M. Mesquita, J. Anhauser, D. Bellaire, S. Becker, A. Lutzen and S. Kubik, Org. Lett., 2019, 21, 6442–6446 CrossRef CAS PubMed.
- K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 13, 1638–1640 RSC.
- Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed.
- S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064–6065 CrossRef CAS PubMed.
- T. Tsutsui, L. Catti, K. Yoza and M. Yoshizawa, Chem. Sci., 2020, 11, 8145–8150 RSC.
- Z. Li, N. Kishi, K. Hasegawa, M. Akita and M. Yoshizawa, Chem. Commun., 2011, 47, 8605–8607 RSC.
- Z. Li, N. Kishi, K. Yoza, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2012, 18, 8358–8365 CrossRef CAS PubMed.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- V. Marti-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef CAS PubMed.
- R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- R. A. S. Vasdev, L. F. Gaudin, D. Preston, J. P. Jogy, G. I. Giles and J. D. Crowley, Front. Chem., 2018, 6, 563 CrossRef CAS PubMed.
- A. Schmidt, V. Molano, M. Hollering, A. Pothig, A. Casini and F. E. Kuhn, Chem.–Eur. J., 2016, 22, 2253–2256 CrossRef CAS PubMed.
- S. Gonell, X. Caumes, N. Orth, I. Ivanovic-Burmazovic and J. N. H. Reek, Chem. Sci., 2019, 10, 1316–1321 RSC.
- Q. Q. Wang, S. Gonell, S. H. Leenders, M. Durr, I. Ivanovic-Burmazovic and J. N. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS.
- R. Zaffaroni, E. O. Bobylev, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2020, 142, 8837–8847 CrossRef PubMed.
- S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Adv. Synth. Catal., 2016, 358, 1509–1518 CrossRef CAS.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed.
- K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed.
- J. B. Pollock, T. R. Cook, G. L. Schneider, D. A. Lutterman, A. S. Davies and P. J. Stang, Inorg. Chem., 2013, 52, 9254–9265 CrossRef CAS PubMed.
- J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2012, 134, 10607–10620 CrossRef CAS.
- X. Yan, P. Wei, Y. Liu, M. Wang, C. Chen, J. Zhao, G. Li, M. L. Saha, Z. Zhou, Z. An, X. Li and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 9673–9679 CrossRef CAS.
- C. L. Liu, E. O. Bobylev, Y. Fu, D. A. Poole 3rd, K. Robeyns, C. A. Fustin, Y. Garcia, J. N. H. Reek and M. L. Singleton, Chem.–Eur. J., 2020, 26, 11960–11965 CrossRef CAS.
- D. Fujita, A. Takahashi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2011, 133, 13317–13319 CrossRef CAS PubMed.
- F. Kaiser, A. Schmidt, W. Heydenreuter, P. J. Altmann, A. Casini, S. A. Sieber and F. E. Kühn, Eur. J. Inorg. Chem., 2016, 2016, 5189–5196 CrossRef CAS.
- A. Pothig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef PubMed.
- S. Kai, T. Shigeta, T. Kojima and S. Hiraoka, Chem.–Asian J., 2017, 12, 3203–3207 CrossRef CAS PubMed.
- D. Fujita, H. Yokoyama, Y. Ueda, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 155–158 CrossRef CAS PubMed.
- M. Yoneya, S. Tsuzuki, T. Yamaguchi, S. Sato and M. Fujita, ACS Nano, 2014, 8, 1290–1296 CrossRef CAS.
- D. A. Poole, E. O. Bobylev, S. Mathew and J. N. H. Reek, Chem. Sci., 2020, 11, 12350–12357 RSC.
- H. Li, J. Luo and T. Liu, Chem.–Eur. J., 2016, 22, 17949–17952 CrossRef CAS PubMed.
- L. Zeng, Y. Xiao, J. Jiang, H. Fang, Z. Ke, L. Chen and J. Zhang, Inorg. Chem., 2019, 58, 10019–10027 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. Sun, M. J. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Nat. Chem., 2016, 8, 33–41 CrossRef CAS PubMed.
- M. Wang, J. Liu, T. Luo, Y. Xue, L. Mao and P. J. Stang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef.
- W.-L. Jiang, J.-C. Shen, Z. Peng, G.-Y. Wu, G.-Q. Yin, X. Shi and H.-B. Yang, J. Mater. Chem. A, 2020, 8, 12097–12105 RSC.
- E. Puig, C. Desmarets, G. Gontard, M. N. Rager, A. L. Cooksy and H. Amouri, Inorg. Chem., 2019, 58, 3189–3195 CrossRef CAS PubMed.
- D. K. Chand, G. Balaji, R. Manivannan and J. Athilakshmi, Tetrahedron Lett., 2006, 47, 2867–2869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01295a |
| This journal is © The Royal Society of Chemistry 2021 |