
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium/Xu-Phos-catalyzed asymmetric carboamination towards isoxazolidines and pyrrolidines†
Yuzhuo
Wang
a,
Lei
Wang
c,
Mingjie
Chen
c,
Youshao
Tu
a,
Yu
Liu
 *a and
Junliang
Zhang
*b
*a and
Junliang
Zhang
*b
aCollege of Chemistry and Life Science, Jilin Province Key Laboratory of Carbon Fiber Development and Application, Changchun University of Technology, Changchun 130012, China. E-mail: yuliu@ccut.edu.cn
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, China. E-mail: junliangzhang@fudan.edu.cn
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, 3663 N. Zhongshan Road, Shanghai 200062, China
First published on 5th May 2021
Abstract
An efficient palladium-catalyzed enantioselective carboamination reaction of N-Boc-O-homoallyl-hydroxylamines and N-Boc-pent-4-enylamines with aryl or alkenyl bromides was developed, delivering various substituted isoxazolidines and pyrrolidines in good yields with up to 97% ee. The reaction features mild conditions, general substrate scope and scalability. The obtained products can be transformed into chiral 1,3-aminoalcohol derivatives without erosion of chirality. The newly identified Xu-Phos ligand bearing an ortho-OiPr group is responsible for the good yield and high enantioselectivity.
Introduction
Five-membered N-heterocyclic skeletons are commonly found in biological and pharmaceutical molecules (Fig. 1).1 For example, pyrrolidine-based compounds can act as receptor antagonists, and their diverse activities are determined by the different configurations (eg 2 and 3).2 Isoxazolidine is a versatile precursor for the synthesis of 1,3-amino alcohols, taking sitagliptin as a typical case (eg 5).3 Therefore, it is highly desirable to develop synthetic methods towards isoxazolidine and pyrrolidine compounds, especially for efficient construction of enantioenriched molecules.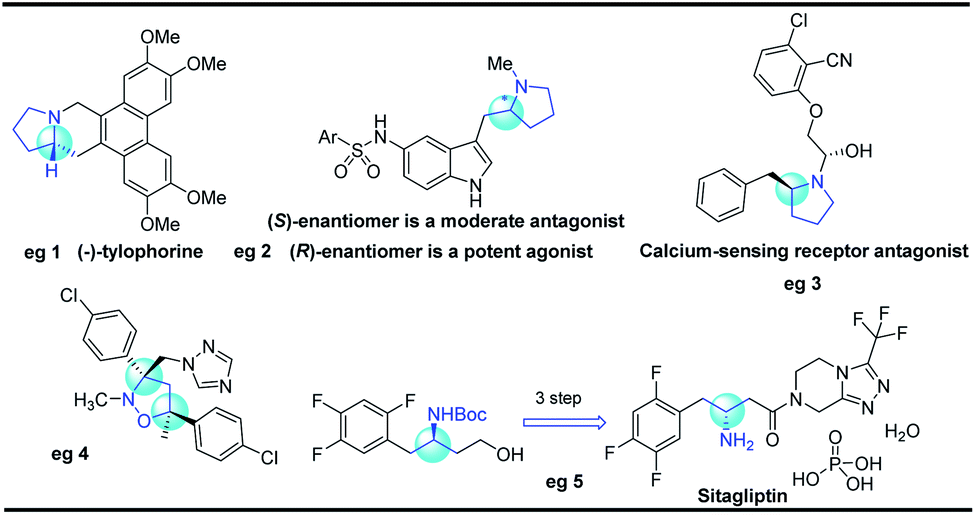 | ||
| Fig. 1 Biologically active molecules with pyrrolidine and isoxazolidine motifs. | ||
Isoxazolidines could be furnished by 1,3-dipolar cycloaddition reactions or cyclization of unsaturated hydroxylamines proceeding through electrophilic or free radical pathways, Michael addition reactions etc.4 Transition metal catalyzed cyclization strategies have also been showcased in recent years using palladium or gold as common catalysts.5 In 2010, Toste6a reported the Au(I)-catalyzed enantioselective synthesis of isoxazolidines from allenic hydroxylamines, which could be also extended for the preparation of pyrazolidines and tetrahydroxazines with high enantioselectivity (Scheme 1a). Recently, Gao and co-workers reported a tandem aza-Michael/hemiacetal reaction between (E)-4-phenylbut-2-enal and N-Boc-hydroxyl-amine for the synthesis of 2-hydroxyl-isoxazolidines (Scheme 1b).3 The Wolfe group reported an elegant palladium-catalyzed carboamination of alkenes, which provides facile access to enantioenriched pyrrolidines (Scheme 1c).7 Recently, Zhang and co-workers8 also implemented an enantioselective radical cyclization approach by metalloradical C–H alkylation reactions (Scheme 1d).
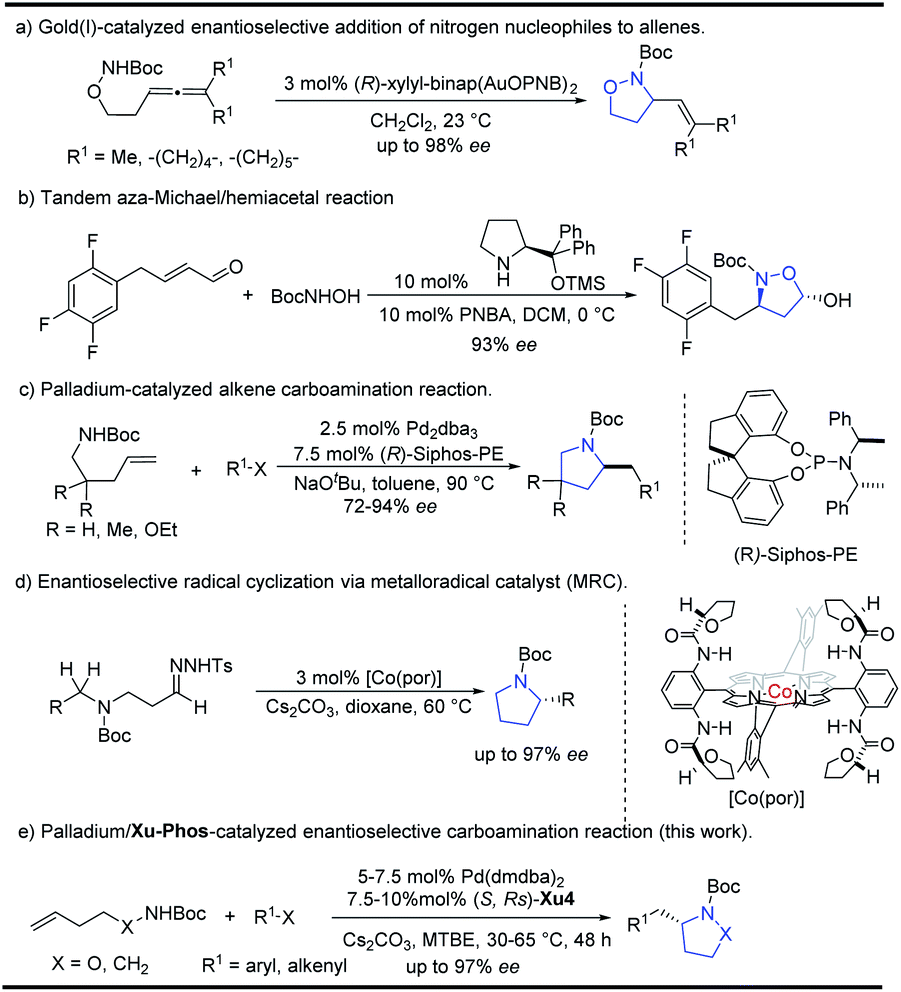 | ||
| Scheme 1 Enantioselective synthesis of isoxazolidines and pyrrolidines. | ||
Despite the above advances, the development of an efficient methodology for N-heterocyclic skeletons with high enantioselectivity is of great importance and still challenging, especially for the introduction of an alkenyl group. Based on our interests in the asymmetric synthesis of heterocyclic compounds,9 herein we wish to report our efforts in the development of palladium-catalyzed intermolecular carboamination of unsaturated hydroxylamines with aryl or alkenyl halides, in which the newly identified (S,Rs)-Xu4 bearing an ortho-OiPr group ligand showed a unique effect, leading to substituted isoxazolidines in relatively high yield and selectivity. Moreover, pyrrolidines could also be synthesized efficiently starting from the corresponding carbamates.
Results and discussion
In our initial study, N-Boc-O-homoallyl-hydroxylamine 1a and 4-bromobiphenyl were selected as the model substrates. A series of commercially available chiral ligands were investigated at first (Fig. 2). Although L1 afforded the desired product 3a with moderate enantioselectivity, other bisphosphine ligands L2–L5 suppressed the reaction, with no or only a small amount of 3a detected. The desired product could be obtained in low yield with poor enantioselectivity when using chiral phosphoramidite ligand L6, but L7 is not effective at all. Josiphos L8 delivered the product 3a with moderate yield but as a close to racemic mixture. The phoxphos derivative L9 is not effective for this reaction.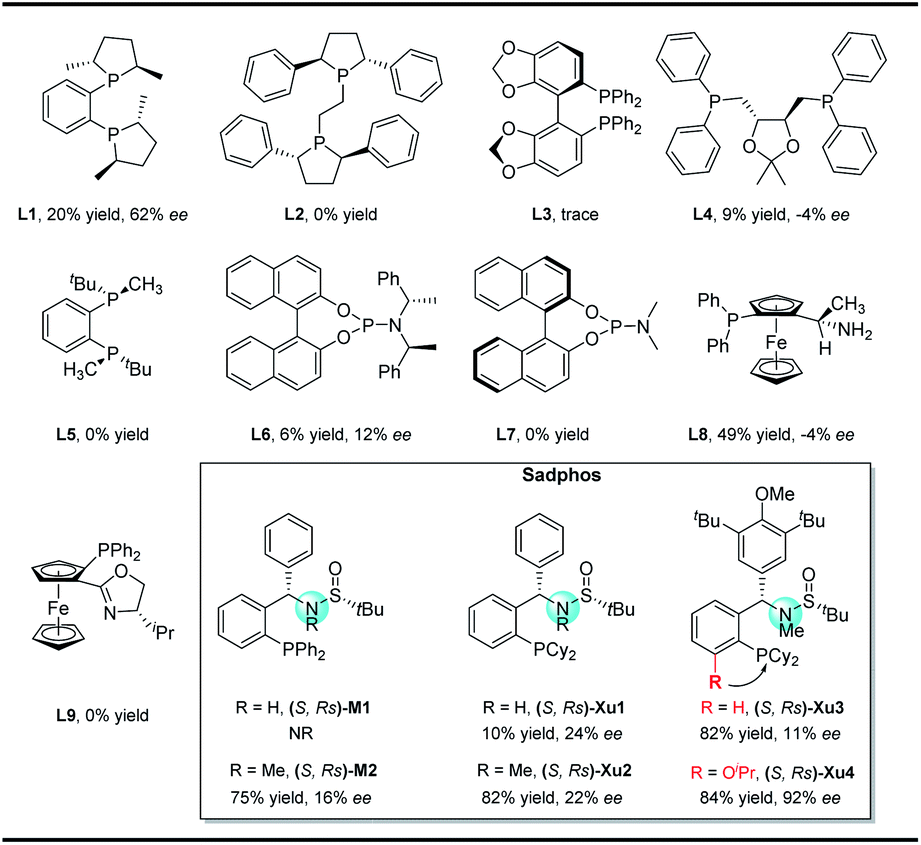 | ||
| Fig. 2 Representative chiral ligands tested in this work. | ||
Inspired by the success of our developed Sadphos ligands in asymmetric catalysis, we turned our attention to evaluate their performance in the present carboamination reaction. Of note, the Sadphos kits are commercially available from Strem Inc. now. As an initial trial, Ming-Phos9h,10 (S,Rs)-M1 with free NH was inactive, leaving the starting materials untouched. Surprisingly, when the amine moiety was protected by a methyl group ((S,Rs)-M2), 3a was obtained in 75% yield, albeit the enantioselectivity was unsatisfactory, indicating that the NH moiety of Ming-Phos might inhibit carboamination. The dicyclohexyl phosphine ligand Xu-Phos11 showed a consistent trend, and (S,Rs)-Xu2 bearing a N-methyl group resulted in higher yield and enantioselectivity. Inspired by these notable results, we investigated the modification of Xu-Phos ligand. (S,Rs)-Xu3 bearing a 3,5-di-tert-butyl-4-methoxybenzyl group did not provide a better result. Amazingly, the introduction of OiPr at the ortho-position of the phosphine moiety dramatically improved the enantioselectivity, delivering 3a in 84% yield and 92% ee.13 These results unambiguously prove the subtleness and unique efficacy of the Xu-Phos ligands. We attribute this ortho-effect to the repulsion of the iso-propyl group with the cyclohexyl group on the P-atom, which would push the cyclohexyl group close to the catalytic center and affect the enantioselectivity.
Other factors were also systematically examined consequently. Among the palladium sources tested (Table 1), PdCl2, Pd(OAc)2, Pd(MeCN)2Cl2 and [Pd(allyl)Cl]2 were obviously less active, generating 3a in lower yields (Table 1, entries 1–4). Pd2(dba)3 performed similarly to Pd(dmdba)2, with slightly reduced yield (Table 1, entry 5). Other bases including NaOtBu, KOtBu and K2CO3 led to lower yields (Table 1, entries 7–9). Various solvents were also screened, and MTBE was demonstrated to be the best choice (entries 10–14). To our delight, the enantioselectivity was further improved by lowering the temperature, and the yield could also be promoted with prolonged reaction time (Table 1, entries 15 and 16). Finally, using Pd(dmdba)2/(S,Rs)-Xu4 as the catalyst and Cs2CO3 as the base, the reaction proceeds smoothly in MTBE at 30 °C to afford the desired product in 88% yield and 95% ee (Table 1, entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd sources | Base | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.2 mmol), 4-bromobiphenyl (0.4 mmol), Cs2CO3 (2 equiv.), 5 mol% Pd, and 7.5 mol% ligand in 2.0 mL MTBE at 75 °C under Ar for 24 h. b Isolated yield. c ee was determined by HPLC analysis. d 50 °C, 24 h. e 30 °C, 48 h. f 2.5 mol% Pd. | |||||
| 1 | PdCl2 | Cs2CO3 | MTBE | 55 | 85 |
| 2 | Pd(OAc)2 | Cs2CO3 | MTBE | 64 | 80 |
| 3 | Pd(MeCN)2Cl2 | Cs2CO3 | MTBE | 24 | 91 |
| 4f | [Pd(allyl)Cl]2 | Cs2CO3 | MTBE | 70 | 92 |
| 5f | Pd2(dba)3 | Cs2CO3 | MTBE | 82 | 92 |
| 6 | Pd(dmdba)2 | Cs2CO3 | MTBE | 84 | 92 |
| 7 | Pd(dmdba)2 | NaOtBu | MTBE | 49 | 90 |
| 8 | Pd(dmdba)2 | KOtBu | MTBE | 38 | 91 |
| 9 | Pd(dmdba)2 | K2CO3 | MTBE | 40 | 91 |
| 10 | Pd(dmdba)2 | Cs2CO3 | Toluene | 59 | 89 |
| 11 | Pd(dmdba)2 | Cs2CO3 | THF | 78 | 90 |
| 12 | Pd(dmdba)2 | Cs2CO3 | DCM | 79 | 88 |
| 13 | Pd(dmdba)2 | Cs2CO3 | MeCN | 63 | 86 |
| 14 | Pd(dmdba)2 | Cs2CO3 | DMF | 79 | 89 |
| 15d | Pd(dmdba)2 | Cs2CO3 | MTBE | 82 | 93 |
| 16e | Pd(dmdba)2 | Cs2CO3 | MTBE | 88 | 95 |
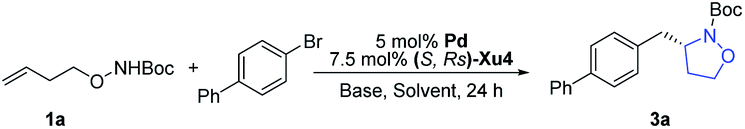
Enantioselective synthesis of substituted isoxazolidines
With the optimized reaction conditions in hand, a variety of aryl bromides were reacted with N-Boc-O-homoallyl-hydroxylamine 1a to verify the generality of the reaction system (Scheme 2). Both electron-donating and electron-withdrawing groups at the para-position of the aryl bromides are compatible, providing isoxazolidines 3a–3h with 91–96% ee. When 4-biphenylyl trifluoromethanesulfonate is used instead of p-bromobiphenyl at 75 °C, 3a can be prepared with 81% yield and 91% ee. In addition to halogens, electron-withdrawing groups at the para-position of the benzene ring such as an aldehyde group, ester group, cyano group, and trifluoromethyl group are all tolerated, delivering products 3i–3l in good yields with 91–95% ee. Aryl bromides bearing different substituents such as OMe, F, Cl, CHO, or COCH3 at the meta-position could also efficiently give the desired products 3m, 3o, 3r, 3s and 3u with up to 97% ee. The absolute configuration of 3r was confirmed by X-ray crystallography analysis.13 Moreover, when a methoxy or fluorine substituent was located at the ortho-position of the phenyl bromide, the products 3n and 3p were obtained with 93% and 88% ee, respectively in moderate yields. Disubstituted phenyl bromides could also be transformed smoothly, and the corresponding products 3t, 3v and 3w were furnished in 78–91% yields with up to 97% ee. The trisubstituted compound 3q on the phenyl ring can also be obtained in 64% yield and 84% ee. Naphthyl and phenanthryl isoxazolidines were furnished in notably high yields and enantioselectivity (3x–3aa). Moreover, heteroaromatic rings including quinolinyl and thienyl could also be well tolerated, affording 3ab and 3ac with no inferior effects. The introduction of an alkenyl group to the molecule could increase the diversity of the compounds due to the alkenyl group having abundant functional group transformations. To our delight, the alkenyl group could also be transferred into the final products from the corresponding alkenyl bromides by slight adjustment of the catalyst loading. Both cyclic and linear precursors performed gratifyingly, delivering the desired products 4a–4c in good yields with up to 91% ee. A preliminary and promising result showed that the present method is also promising for the synthesis of isoxazolidine 4d with an aza-quaternary carbon stereocenter, albeit the efficiency and enantioselectivity are not satisfactory yet and further modification of the chiral ligand is necessary.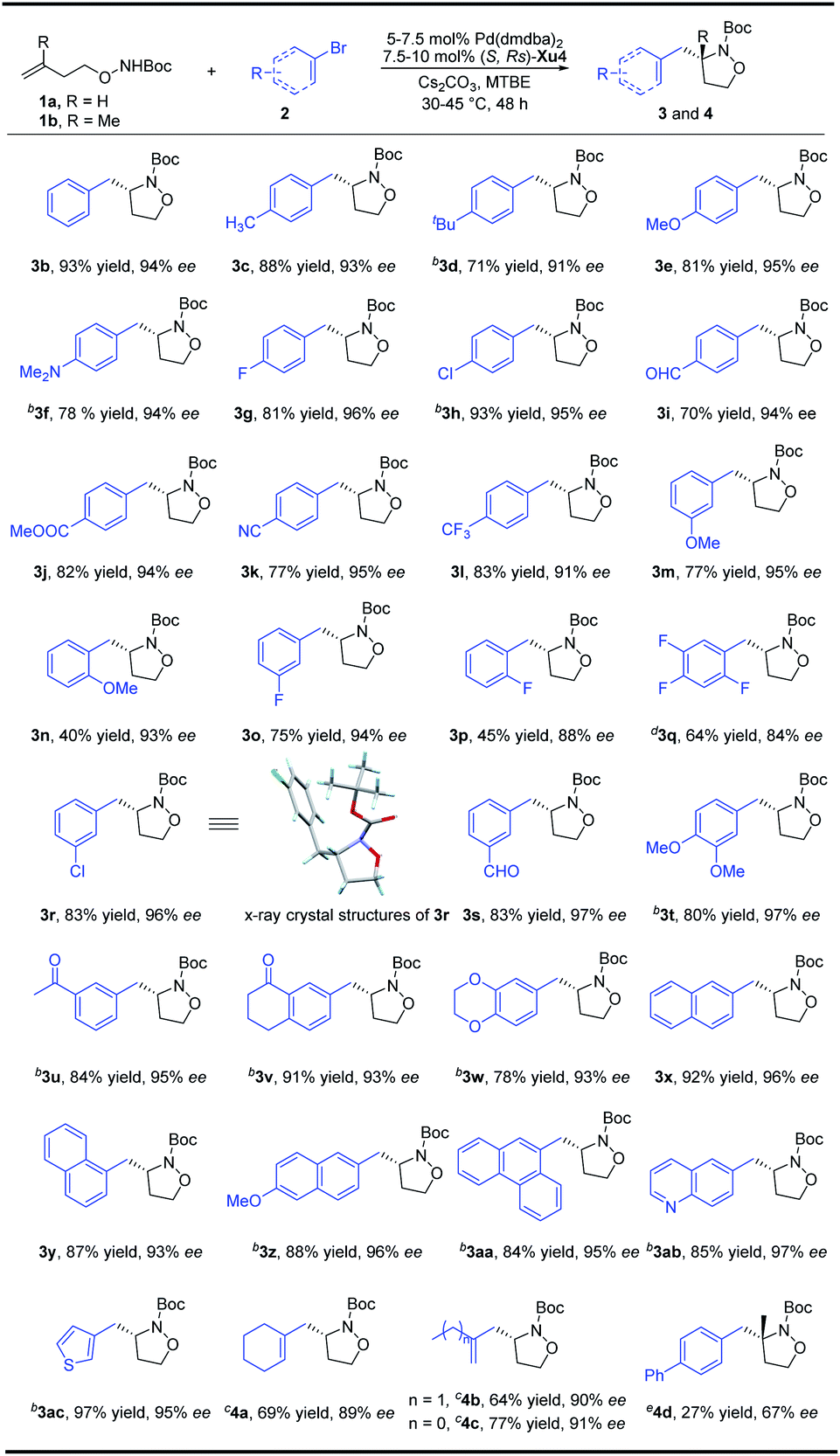 | ||
| Scheme 2 Synthesis of substituted isoxazolidines. aUnless otherwise noted, all reactions were carried out with 1a (0.2 mmol), aryl bromides (0.4 mmol), Cs2CO3 (2 equiv.), 5 mol% Pd(dmdba)2, and 7.5 mol% ligand in 2.0 mL MTBE at 30 °C under Ar for 48 h. b1a (0.4 mmol), aryl bromides (0.8 mmol), 4.0 mL MTBE. c1a (0.4 mmol), alkenyl bromides (0.8 mmol), Cs2CO3 (2 equiv.), 7.5 mol% Pd(dmdba)2, and 10 mol% ligand in 4 mL MTBE at 45 °C under Ar for 48 h. d1a (0.4 mmol), aryl bromides (0.8 mmol), 4.0 mL MTBE, 75 °C, 40 h. e1b (0.4 mmol), 65 °C. | ||
Enantioselective synthesis of aryl substituted pyrrolidines
With regard to the importance of pyrrolidine derivatives, we next turned to investigate this catalyst system in the asymmetric carboamination reaction of C-linked alkenyl carbamates (Fig. 3). The desired product 5a was obtained with similar enantioselectivity with the use of (S,Rs)-Xu4, (S,Rs)-Xu5 and (S,Rs)-Xu6 with different ortho-substituents, among which (S,Rs)-Xu4 gives the highest yield. Various substituted pyrrolidines 5a–5d were delivered in moderate to good yields with high enantioselectivity (Scheme 3). The benzofuranyl group (5e) could also be introduced into the final product easily. We next investigated the substituent effect on the alkyl chain and the corresponding product 5f was produced in low yield (15%) and relatively lower enantioselectivity (81% ee). The amide moiety also affects the reaction significantly, for instance, the Cbz-derived 5g was delivered in only 48% yield with 60% ee. The tosylated substrate produced the corresponding N-arylation product rather than the carboamination product, indicating that these two reaction pathways are competitive.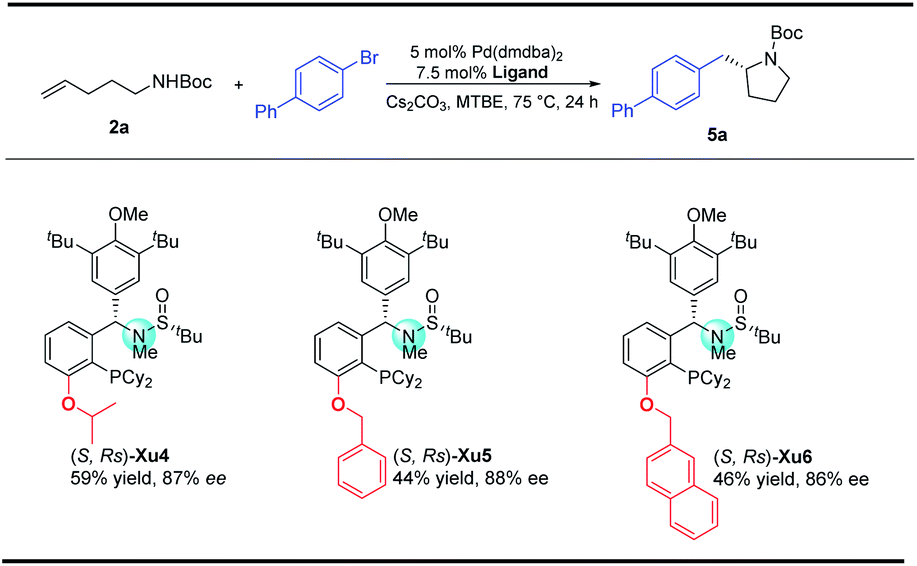 | ||
| Fig. 3 Screening ligands in the synthesis of pyrrolidines. | ||
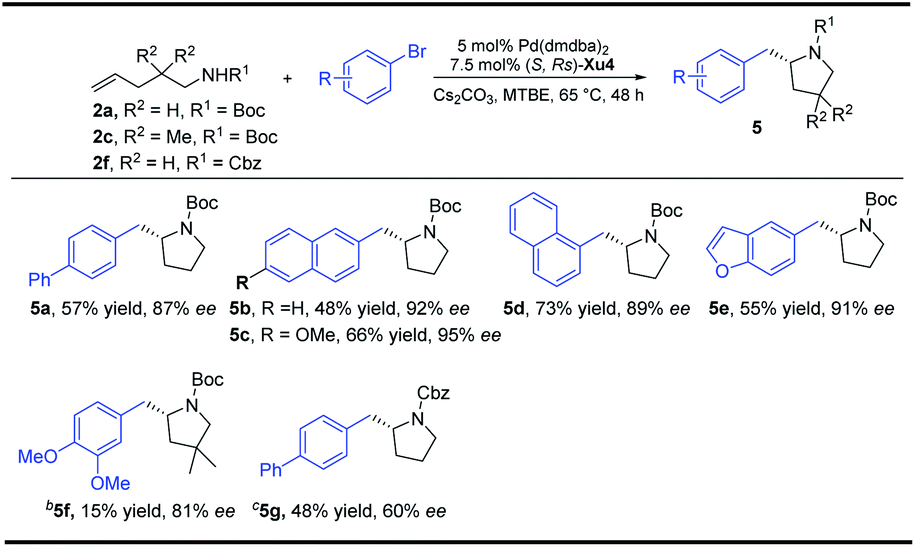 | ||
| Scheme 3 Synthesis of substituted pyrrolidines. aUnless otherwise noted, all reactions were carried out with 2a (0.4 mmol), aryl bromides (0.8 mmol), Cs2CO3 (2 equiv.), 5 mol% Pd(dmdba)2 and 7.5 mol% ligand in 4.0 mL MTBE at 65 °C under Ar for 48 h. b2c (0.2 mmol), 20 h. c2f (0.2 mmol), 24 h. | ||
To demonstrate the practicability of our protocol, a 6 mmol scale reaction was carried out under standard conditions, delivering 1.7 g of naphthyl isoxazolidine 3x in 90% yield with 96% ee. Further treatment with NaBH4 furnished chiral aminoalcohol 9a in 88% yield with 94% ee (Scheme 4).12
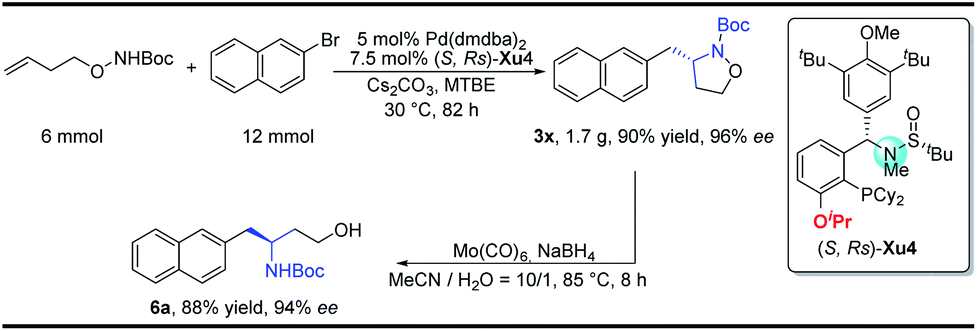 | ||
| Scheme 4 Gram-scale synthesis and synthetic applications. | ||
A catalytic cycle and asymmetric induction model were proposed in Scheme 5. The oxidative addition of aryl bromides to the (S,Rs)-Xu4/Pd(0) complex would generate Pd(II) species II. In the presence of a base, species II would form a Pd–N bond to deliver the intermediate III, which undergoes insertion or aminopalladation of the alkene to produce the intermediate IV. The final product was obtained via reductive elimination, and the catalytic species were also regenerated. Xu-Phos and Pd coordinate through P on the ligand and O on the sulfinamide to form the corresponding catalytic center. The cyclohexyl group is in the sensitive area of metal active species. The introduction of OiPr as a side arm group may push the cyclohexyl group closer to the catalytic center and produce a key dynamic steric hindrance effect.
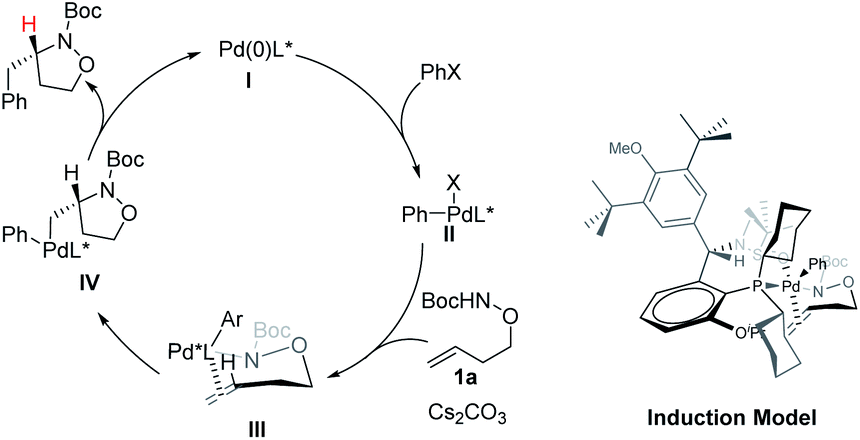 | ||
| Scheme 5 Catalytic cycle and chirality induction model. | ||
Conclusions
In summary, we have successfully developed a palladium-catalyzed asymmetric carboamination reaction of N-Boc-O-homoallyl-hydroxylamine and N-Boc-pent-4-enylamine with either aryl or alkenyl bromides under mild reaction conditions, furnishing various substituted isoxazolidines and pyrrolidines in moderate to high yields with high enantioselectivity. The newly identified ligand of (S,Rs)-Xu4 with OiPr at the ortho-position, which is easily prepared from commercially available starting materials, is responsible for the general substrate scope, good yield and high enantioselectivity. The application of this chiral ligand in other transition metal asymmetric reactions is ongoing in our lab.Author contributions
Y. Wang, L. Wang, M. Chen and Tu did the experiments and collected the data. Y. Liu and J. Zhang directed the research. Y. Wang, Y. Liu and J. Zhang wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the funding support from the NSFC (22031004 and 21921003) and Shanghai Municipal Education Commission (20212308).Notes and references
- (a) A. R. Minter, B. B. Brennan and A. K. Mapp, J. Am. Chem. Soc., 2004, 126, 10504–10505 CrossRef CAS PubMed; (b) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed; (c) J. R. Lewis, Nat. Prod. Rep., 2001, 18, 95–128 RSC; (d) T. R. Govindachari and N. Viswanathan, Heterocycles, 1978, 11, 587–613 CrossRef CAS; (e) M. Ali, S. H. Ansari and J. S. Qadry, J. Nat. Prod., 1991, 54, 1271–1278 CrossRef CAS; (f) F. Abe, Y. Iwase, T. Yamauchi, K. Honda and N. Hayashi, Phytochemistry, 1995, 39, 695–699 CrossRef CAS; (g) G. C. Palmer, M. J. Ordy, R. D. Simmons, J. C. Strand, L. A. Radov, G. B. Mullen, C. Richard-Kinsolving, V. S. Georgiev, J. T. Mitchell and S. D. Allen, Antimicrob. Agents Chemother., 1989, 33, 895–905 CrossRef CAS PubMed.
- (a) D. C. Cole, W. J. Lennox, S. Lombardi, J. W. Ellingboe, R. C. Bernotas, G. J. Tawa, H. Mazandarani, D. L. Smith, G. Zhang, J. Coupet and L. E. Schechter, J. Med. Chem., 2005, 48, 353–356 CrossRef CAS PubMed; (b) W. Yang, Y. Wang, J. Y. Roberge, Z. Ma, Y. Liu, R. Michael-Lawrence, D. P. Rotella, R. Seethala, J. H. M. Feyen and J. K. Dickson, Bioorg. Med. Chem. Lett., 2005, 15, 1225–1228 CrossRef CAS PubMed.
- H. Gao, J. Yu, C. Ge and Q. Jiang, Molecules, 2018, 23, 1440 CrossRef PubMed.
- (a) J. J. Tufariello, Acc. Chem. Res., 1979, 12, 396–403 CrossRef CAS; (b) M. Frederickson, Tetrahedron, 1997, 53, 403–425 CrossRef CAS; (c) Y. Xiang, J. Chen, R. F. Schinazi and K. Zhao, Tetrahedron Lett., 1995, 36, 7193–7196 CrossRef CAS; (d) Y. K. Chen, M. Yoshida and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 9328–9329 CrossRef CAS PubMed; (e) M. Lombardo, G. Rispoli, S. Licciulli, C. Trombini and D. D. Dhavale, Tetrahedron Lett., 2005, 46, 3789–3792 CrossRef CAS; (f) K. Moriyama, Y. Izumisawa and H. Togo, J. Org. Chem., 2011, 76, 7249–7255 CrossRef CAS PubMed; (g) B. Janza and A. Studer, J. Org. Chem., 2005, 70, 6991–6994 CrossRef CAS PubMed.
- (a) K. G. Dongol and B. Y. Tay, Tetrahedron Lett., 2006, 47, 927–930 CrossRef CAS; (b) J. Peng, W. Lin, S. Yuan and Y. Chen, J. Org. Chem., 2007, 72, 3145–3148 CrossRef CAS PubMed; (c) L. J. Peterson and J. P. Wolfe, Adv. Synth. Catal., 2015, 357, 2339–2344 CrossRef CAS PubMed; (d) B. R. Rosen, J. E. Ney and J. P. Wolfe, J. Org. Chem., 2010, 75, 2756–2759 CrossRef CAS PubMed; (e) R. W. Bates, J. A. Nemeth and R. H. Snell, Synthesis, 2008, 1033 CrossRef CAS; (f) J. Cornil, A. Guérinot, S. Reymond and J. Cossy, J. Org. Chem., 2013, 78, 10273–10287 CrossRef CAS PubMed; (g) M. Rigoulet, O. T. Boullay, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2020, 59, 16625–16630 CrossRef CAS PubMed; (h) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS.
- (a) R. L. LaLonde, Z. J. Wang, M. Mba, A. D. Lackner and F. Dean Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 ( Angew. Chem. , 2010 , 122 , 608–611 ) CrossRef CAS PubMed; (b) O. Kanno, W. Kuriyama, Z. Jane Wang and F. Dean Toste, Angew. Chem., Int. Ed., 2011, 50, 9919–9922 CrossRef CAS PubMed; (c) E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard and F. Dean Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS PubMed; (d) W. E. Brenzovich Jr, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard III and F. Dean Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5522 ( Angew. Chem. , 2010 , 122 , 5651–5654 ) CrossRef PubMed; (e) R. L. LaLonde, B. D. Sherry, E. Joo Kang and F. Dean Toste, J. Am. Chem. Soc., 2007, 129, 2452–2453 CrossRef CAS PubMed.
- D. N. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157–12159 CrossRef CAS PubMed.
- (a) Y. Wang, X. Wen, X. Cui and X. P. Zhang, J. Am. Chem. Soc., 2018, 140, 4792–4796 CrossRef CAS PubMed; (b) X. Wen, Y. Wang and X. P. Zhang, Chem. Sci., 2018, 9, 5082–5086 RSC; (c) J. V. Ruppel, R. M. Kamble and X. P. Zhang, Org. Lett., 2007, 9, 4889–4892 CrossRef CAS PubMed; (d) H. Lu, J. Tao, J. E. Jones, L. Wojtas and X. Peter Zhang, Org. Lett., 2010, 12, 1248–1251 CrossRef CAS PubMed; (e) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS PubMed; (f) X. Cui, X. Xu, L.-M. Jin, L. Wojtas and X. P. Zhang, Chem. Sci., 2015, 6, 1219–1224 RSC; (g) Y. Hu, K. Lang, C. Li, J. B. Gill, I. Kim, H. Lu, K. B. Fields, M. Marshall, Q. Cheng, X. Cui, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2019, 141, 18160–18169 CrossRef CAS PubMed; (h) K. Lang, C. Li, I. Kim and X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20902–20911 CrossRef CAS PubMed.
- (a) L. Wang, K. Zhang, Y. Wang, W. Li, M. Chen and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4421–4427 ( Angew. Chem. , 2020 , 132 , 4451–4457 ) CrossRef CAS PubMed; (b) B. Liu, W. Li, H. Wu and J. Zhang, Org. Chem. Front., 2019, 6, 694–698 RSC; (c) S. Xu, Z.-M. Zhang, B. Xu, B. Liu, Y. Liu and J. Zhang, J. Am. Chem. Soc., 2018, 140, 2272–2283 CrossRef CAS PubMed; (d) T.-Y. Lin, H.-H. Wu, J.-J. Feng and J. Zhang, Org. Lett., 2018, 20, 3587–3590 CrossRef CAS PubMed; (e) B. Liu, Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhang, Adv. Synth. Catal., 2018, 360, 2144–2150 CrossRef CAS; (f) Y. Wang, P. Zhang, X. Di, Q. Dai, Z.-M. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 15905–15909 ( Angew. Chem. , 2017 , 129 , 16121–16125 ) CrossRef CAS PubMed; (g) C.-Z. Zhu, J.-J. Feng and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 1351–1355 ( Angew. Chem. , 2017 , 129 , 1371–1375 ) CrossRef CAS PubMed; (h) Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhang, Angew.Chem. Int. Ed., 2016, 55, 6324–6328 ( Angew. Chem. , 2016 , 128 , 6432–6436 ) CrossRef CAS PubMed; (i) M. Chen, Z.-M. Zhang, Z. Yu, H. Qiu, B. Ma, H.-H. Wu and J. Zhang, ACS Catal., 2015, 5, 7488–7492 CrossRef CAS; (j) J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2015, 137, 3787–3790 CrossRef CAS PubMed.
- Z.-M. Zhang, P. Chen, W. Li, Y. Niu, X. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350–4354 ( Angew. Chem. , 2014 , 126 , 4439–4443 ) CrossRef CAS PubMed.
- (a) Z.-M. Zhang, B. Xu, Y. Qian, L. Wu, Y. Wu, L. Zhou, Y. Liu and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10373–10377 ( Angew. Chem. , 2018 , 130 , 10530–10534 ) CrossRef CAS PubMed; (b) Z.-M. Zhang, B. Xu, L. Wu, L. Zhou, D. Ji, Y. Liu, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 8110–8115 CrossRef CAS PubMed; (c) Z.-M. Zhang, B. Xu, L. Wu, Y. Wu, Y. Qian, L. Zhou, Y. Liu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14653–14659 ( Angew. Chem. , 2019 , 131 , 14795–14801 ) CrossRef CAS PubMed.
- (a) S. Cicchi, A. Goti, A. Brandi, A. Guarna and F. D. Sarlo, Tetrahedron Lett., 1990, 31, 3351–3354 CrossRef CAS; (b) R. W. Bates and C. J. Lim, Synlett, 2010, 866–868 CrossRef CAS.
- CCDC: 2053552 (3r) and 2053557 ((S,Rs)-Xu4). Please find the detailed crystal data in the supporting information.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2053552 (3r) and 2053557 ((S,Rs)-Xu4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01337h |
| This journal is © The Royal Society of Chemistry 2021 |