
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic inhibition of SARS-CoV-2 cell entry by otamixaban and covalent protease inhibitors: pre-clinical assessment of pharmacological and molecular properties†
Tim
Hempel‡
 ab,
Katarina
Elez‡
a,
Nadine
Krüger‡
c,
Lluís
Raich
a,
Jonathan H.
Shrimp
d,
Olga
Danov
e,
Danny
Jonigk
ef,
Armin
Braun
e,
Min
Shen
d,
Matthew D.
Hall
d,
Stefan
Pöhlmann
cg,
Markus
Hoffmann
cg and
Frank
Noé
*abh
ab,
Katarina
Elez‡
a,
Nadine
Krüger‡
c,
Lluís
Raich
a,
Jonathan H.
Shrimp
d,
Olga
Danov
e,
Danny
Jonigk
ef,
Armin
Braun
e,
Min
Shen
d,
Matthew D.
Hall
d,
Stefan
Pöhlmann
cg,
Markus
Hoffmann
cg and
Frank
Noé
*abh
aDepartment of Mathematics and Computer Science, Freie Universität Berlin, Berlin, Germany. E-mail: frank.noe@fu-berlin.de
bDepartment of Physics, Freie Universität Berlin, Berlin, Germany
cInfection Biology Unit, German Primate Center – Leibniz Institute for Primate Research, Göttingen, Germany
dNational Center for Advancing Translational Sciences, National Institutes of Health, Rockville, MD, USA
eFraunhofer Institute for Toxicology and Experimental Medicine (ITEM), Biomedical Research in Endstage and Obstructive Lung Disease Hannover (BREATH), Member of the German Center for Lung Research (DZL), Member of Fraunhofer International Consortium for Anti-Infective Research (iCAIR), Hannover, Germany
fInstitute of Pathology, Hannover Medical School, Biomedical Research in Endstage and Obstructive Lung Disease Hannover (BREATH), Hannover, Germany
gFaculty of Biology and Psychology, University Göttingen, Göttingen, Germany
hDepartment of Chemistry, Rice University, Houston, TX, USA
First published on 26th August 2021
Abstract
SARS-CoV-2, the cause of the COVID-19 pandemic, exploits host cell proteins for viral entry into human lung cells. One of them, the protease TMPRSS2, is required to activate the viral spike protein (S). Even though two inhibitors, camostat and nafamostat, are known to inhibit TMPRSS2 and block cell entry of SARS-CoV-2, finding further potent therapeutic options is still an important task. In this study, we report that a late-stage drug candidate, otamixaban, inhibits SARS-CoV-2 cell entry. We show that otamixaban suppresses TMPRSS2 activity and SARS-CoV-2 infection of a human lung cell line, although with lower potency than camostat or nafamostat. In contrast, otamixaban inhibits SARS-CoV-2 infection of precision cut lung slices with the same potency as camostat. Furthermore, we report that otamixaban's potency can be significantly enhanced by (sub-) nanomolar nafamostat or camostat supplementation. Dominant molecular TMPRSS2-otamixaban interactions are assessed by extensive 109 μs of atomistic molecular dynamics simulations. Our findings suggest that combinations of otamixaban with supplemental camostat or nafamostat are a promising option for the treatment of COVID-19.
1 Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection causes coronavirus disease 2019 (COVID-19), a potentially fatal respiratory condition. The first cases of this illness were reported in late December 2019 in Wuhan, China. Due to the high human-to-human transmission rate of SARS-CoV-2, the World Health Organization declared the outbreak a pandemic.1 By early March 2021, the virus has infected more than 116.1 million and killed more than 2.5 million people worldwide.2 With no robust therapeutic options available at present, the development of new antivirals against SARS-CoV-2 is of imminent importance.SARS-CoV-2 exploits two membrane-bound host proteins for cell entry – angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2)4 (Fig. 1). Specifically, the spike (S) protein of SARS-CoV-2 binds to ACE2 and is subsequently cleaved by the transmembrane serine protease TMPRSS2, allowing membrane fusion and cell entry. The low risk of SARS-CoV-2 infection in groups with decreased levels of TMPRSS2, such as infants/children5 and androgen-deprived prostate cancer patients,6 is consistent with the suggested SARS-CoV-2 dependence on TMPRSS2. The S protein depends on TMPRSS2 for priming (proteolytic separation of its subunits) in human airway cells (primary targets of the virus),4,7 even though it also relies on the endosomal cysteine protease cathepsin L for entry into some cell lines.4,8 Additionally, recent evidence suggests that coagulation factors such as factor Xa (fXa) can directly cleave S and facilitate SARS-CoV-2 cell entry.9
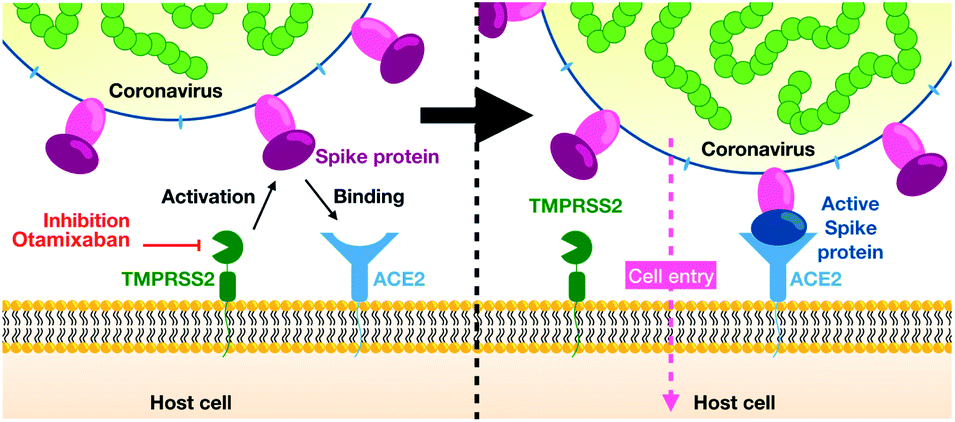 | ||
| Fig. 1 Overview of SARS-CoV-2 cell entry (reproduced from ref. 3 with permission from the Royal Society of Chemistry.). | ||
TMPRSS2 also activates other coronaviruses,10–16 as well as several strains of the influenza A virus.17–19 TMPRSS2 is dispensable for normal development and homeostasis in mice,20 suggesting that blockade of this protease might not be associated with substantial unwanted side effects.
Two synthetic serine protease inhibitors, camostat and nafamostat, approved in Japan for treatment of pancreatitis, inhibit TMPRSS2 and block SARS-CoV-2 infection.4,21,22 Besides these small molecules, protein-based inhibitors such as alpha-1-antitrypsin23 and aprotinin (BPTI),24 have been proposed as potential treatment options. However, demand for potent and easily administrable TMPRSS2 inhibitors still exists.
Otamixaban is a potent non-covalent synthetic inhibitor of factor Xa,25 investigated as an anticoagulant for managing acute coronary syndrome. It entered phase III clinical trials but was deemed inferior to unfractionated heparin combined with eptifibatide for reducing ischaemic events.26
Rensi et al.27 first identified otamixaban as a potential TMPRSS2 inhibitor in their virtual screening. We corroborated this hit computationally by docking the compound against several protein conformations extracted from extensive molecular dynamics simulations.3 Otamixaban was experimentally identified by the screening pipelines of both, the NIH/NCATS Cheminformatics group led by Shen28 and the Noé lab, using the NIH/NCATS TMPRSS2 high throughput screening (HTS) assay.29 It was most recently found to have an inhibitory effect in ref. 9.
Here we present TMPRSS2 enzyme activity assay results, SARS-CoV-2 S-driven cell entry in a human lung cell line (Calu-3), and precision cut human lung slices (PCLS). Based on extensive 109 μs in silico molecular dynamics simulations, we study the structural basis of TMPRSS2 inhibition via otamixaban and highlight interactions that could be exploited for increasing drug potency.
Under in vitro conditions, we find otamixaban to be a weak inhibitor of TMPRSS2 with an IC50 value 2–3 orders of magnitude above that of more established TMPRSS2 inhibitors – camostat and nafamostat. However, our PCLS experiment suggests that in lung tissue, the otamixaban treatment is as potent as the camostat standard.
Otamixaban is additionally known to act on a SARS-CoV-2 entry-related protease that is orthogonal to TMPRSS2, otamixaban's primary target fXa.29 This renders combination preparations with camostat or nafamostat a promising option.30 Furthermore, we observe strong synergistic effects when otamixaban is combined with these drugs in Calu-3 cells, which indicates that there is an additional, yet unknown, target involved in SARS-CoV-2 cell entry. We, therefore, suggest that otamixaban, most likely in a combination preparation with camostat or nafamostat, should be considered as a therapeutic strategy against COVID-19.
2 Results
Otamixaban inhibits TMPRSS2 and suppresses SARS-CoV-2 entry
We identify otamixaban as a potential TMPRSS2 inhibitor in the NIH/NCATS TMPRSS2 high throughput screening (HTS) assay.29 This is a single replicate experiment conducted at room temperature which is easily scalable to large amounts of different drug candidates (1536-well plates). The HTS assay indicates that otamixaban shows inhibitory activity against TMPRSS2 (Fig. 2a). We subsequently perform activity confirmation in a different facility (cf. Materials and methods) at physiological temperature with multiple replicates, using a similar, although low throughput, TMPRSS2 activity assay. We find that, as expected, both assays yield comparable results (Fig. 2a).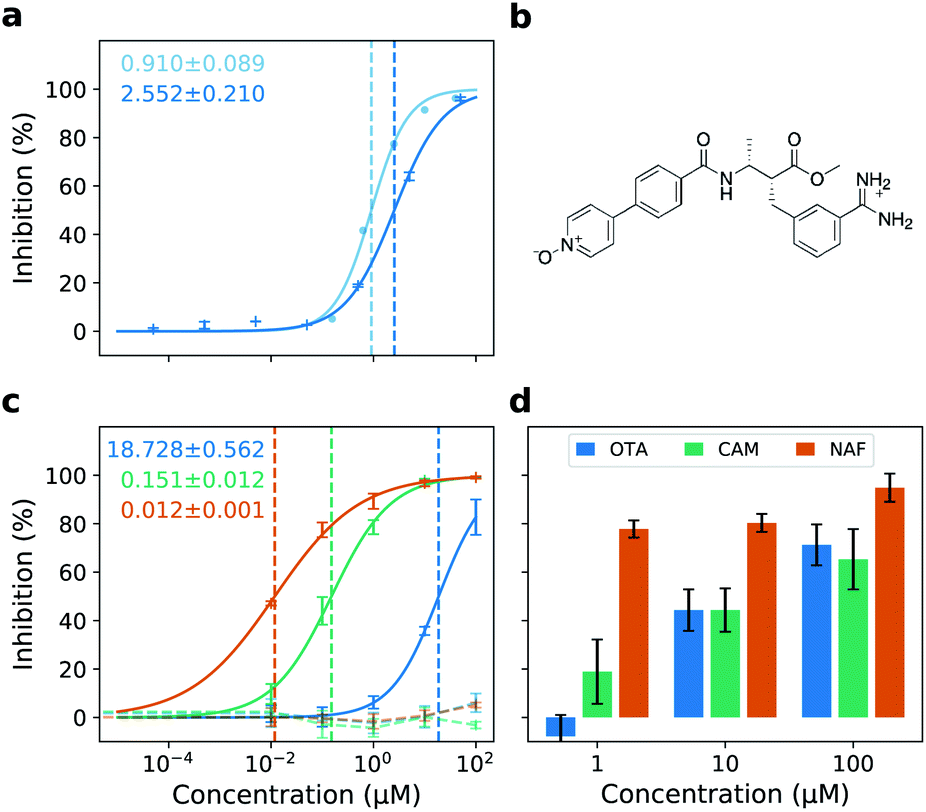 | ||
| Fig. 2 Otamixaban chemical structure and antiviral activity. (a) TMPRSS2 enzyme activity assays. NIH/NCATS high throughput screening assay (light blue, room temperature, one replicate); Hit confirmation assay (dark blue, physiological temperature, multiple replicates). IC50 estimates for either assay are printed using the same color code. (b) Chemical structure of otamixaban at pH = 7.4 (blood). (c) Dose–response curves in Calu-3 cells with their respective IC50 estimates. Inhibition of entry driven by SARS-2-S (solid lines) or VSV-G (control, dashed lines) was analyzed using VSV pseudotyped particles. The average of three biological replicates ± SD is shown. (d) Inhibition of SARS-CoV-2 infection by otamixaban (blue), camostat (green) and nafamostat (orange) in precision cut human lung slices (PCLS). For clarity, PCLS from a single donor are shown; confirmatory results from an independent second donor are presented in Fig. S2.† The results (mean) of three technical replicates are shown. Error bars indicate SD. | ||
We next test otamixaban in the Calu-3 cell line where we find it to inhibit SARS-CoV-2 S-mediated viral entry, but not entry driven by vesicular stomatitis virus (VSV) glycoprotein (VSV-G), which does not require TMPRSS2 activity to mediate cell entry. Inhibition of SARS-2-S-driven cell entry by otamixaban is much less potent compared to camostat and nafamostat (Fig. 2c). Being in the lower μm range, otamixaban's IC50 is 2 or 3 orders of magnitude larger than the IC50 of camostat or nafamostat, respectively (cf.Table 1). However, in human PCLS, inhibitory effects of otamixaban and camostat are comparable (Fig. 2d and S2†).
Otamixaban shows a strong synergistic effect when combined with camostat or nafamostat
In a subsequent step, we test otamixaban in the Calu-3 cell line in a combination preparation with constant small doses of camostat (1 nm) or nafamostat (0.1 nm), which are two orders of magnitude below their IC50 values (Table 1). We find that the dose–response behavior of otamixaban in combination with either drug is significantly enhanced, e.g. at a drug concentration of 10 μm, SARS-CoV-2 cell entry is inhibited by 80% using otamixaban with a camostat or nafamostat supplement compared to only 36% inhibition without the supplement.To quantify potential synergistic effects between the drugs, we use the combination index (CI) by Chou and Talalay,31 which classifies drug interactions into additive (CI = 1), synergistic (CI < 1), and antagonistic (CI > 1). We find that, at 50% inhibition, the CI is significantly smaller than 0.1 for both tested drug combinations (Table 1), indicating that otamixaban and camostat (or nafamostat) show very strong synergism.32
A simulation of the CI in the range between 10 and 95% inhibition (Fig. 3b) shows that the majority of predicted CI values can be considered strongly synergistic (<0.3), following the classification defined by Chou.32 Please note that the CI simulation depends on the learned parameters from the dose–response data, i.e. we expect prediction quality to be high around 50% inhibition. Peripheral deviations between camostat and nafamostat supplementation might therefore be an artifact of sparse sampling in that area.
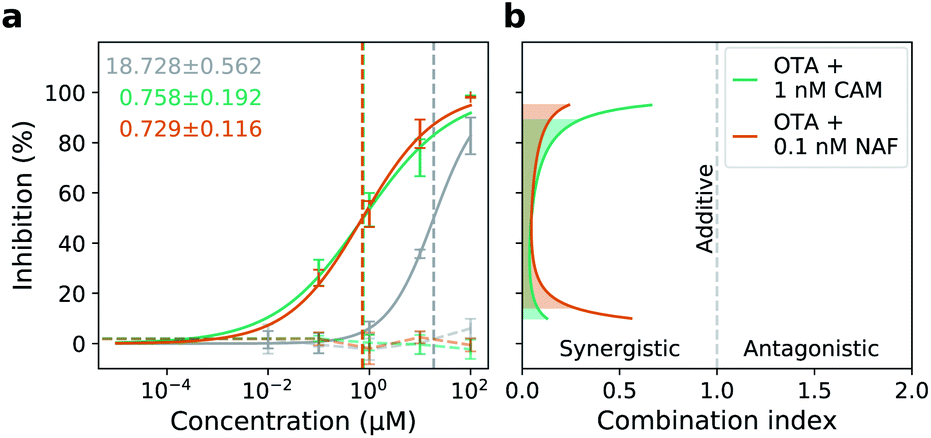 | ||
| Fig. 3 Synergistic antiviral activity of otamixaban combined with camostat and nafamostat. (a) Antiviral activity of otamixaban in combination with 1 nm camostat (green) or 0.1 nm nafamostat (orange) compared with otamixaban alone (grey) was analyzed using Calu-3 cells and VSV pseudotypes harboring either SARS-2-S (solid lines) or VSV-G (dashed lines) as described for Fig. 2b. Corresponding IC50 values are highlighted with dotted lines and annotated on the left. The average of three biological replicates ± SD is shown. (b) Combination index as a function of inhibition strength. The dashed line at CI = 1.0 indicates additivity (neither synergism nor antagonism); combination indices below the dotted line indicate synergism (CI < 1). Inhibition range with strong synergism (IC < 0.3) is marked by shaded areas. | ||
Due to this unprecedented synergistic effect, the dose of otamixaban can be significantly reduced when used in combination with camostat or nafamostat. Specifically, the dose reduction index (DRI) of otamixaban at 50% inhibition is 24.7 and 25.7, for the two combination preparations, respectively. In other words, to achieve 50% inhibition, only ∼1/25 (∼1/26) of the pure otamixaban concentrations are necessary when supplemented with nanomolar (sub-nanomolar) concentrations of camostat (nafamostat).
To shed light on the origin of the observed synergistic effect, we perform additional experiments in the HTS assay with supplemental drug concentrations, two orders of magnitude below their IC50 values (0.01 nm of camostat or 0.001 nm of nafamostat). We find that the dose–response curves of the combination preparations behave identically to otamixaban alone (Fig. S1,† CIs ≈ 0.9 at 50% inhibition). In other words, no synergistic effect is observed in the HTS assay.
Structural basis of TMPRSS2 inhibition by otamixaban
Analysis of our MD simulations, a set of 109 trajectories of 1 μs each, reveals that otamixaban binds TMPRSS2 with multiple metastable binding modes. We find three dominant metastable binding poses that make up for 60% of the observed simulation ensemble (Fig. S5†). Briefly, they are (a) a TMPRSS2-specific pose (42% of the total observed population), (b) similar to the crystal configuration observed with otamixaban's original target (18%), and (c) a pre-bound state with catalytic triad interactions (13%).In the most populated state, otamixaban is bound to the S1 pocket (Fig. S3a†) with the charged benzamidine moiety interacting with Asp435 (Fig. 4b). This interaction is fundamental for the recognition of natural substrates and is typically exploited by several serine protease inhibitors, among which we find camostat and nafamostat, both with a charged guanidinobenzoyl group instead of the benzamidine group of otamixaban.3 Hence, the binding site is shared among the two drugs in the tested combination preparations, indicating that the synergistic effects described above are not likely to arise from simultaneous binding. At the opposite side of the benzamidine group, otamixaban has a long, hydrophobic pyridine-N-oxide moiety, with a polar head that is neutral at physiological pH (cf.Fig. 2b). We find this moiety mainly interacting with the hydrophobic CH2 chains of Glu299 and Lys300, as well as the catalytic His296.
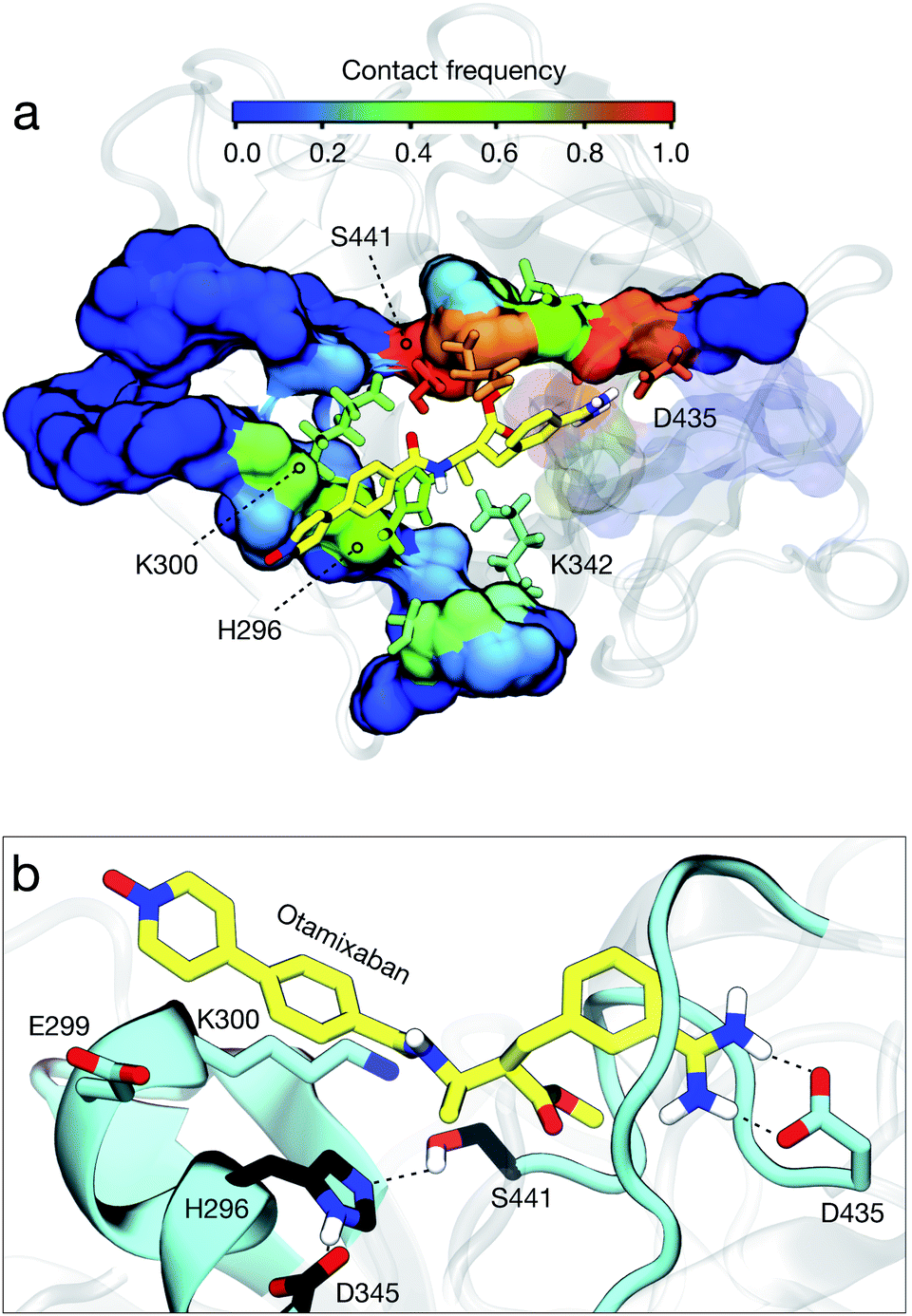 | ||
| Fig. 4 Otamixaban binding modes; the drug is depicted in yellow licorice representation. (a) Contact frequency analysis of the full drug to TMPRSS2. Residue contact frequency is color-coded as shown in the color bar. (b) Highest populated state observed in our MD simulations. Catalytic triad residues are depicted in black. | ||
The second state is comparable to the crystal structure of otamixaban with its original target, the coagulation factor Xa (fXa)25,33 (Fig. S3b†). We find that the S1 interactions with the benzamidine moiety (in particular with Asp435) are equivalent, with a subtle difference in the amidinium ring conformation (Fig. S4a†). Furthermore, the interactions of the opposite head differ as this moiety is designed to bind the fXa S4 pocket, a hydrophobic cavity formed by residues Tyr99, Phe174, and Trp215. In TMPRSS2, however, this cavity is less defined since the residue equivalent to Tyr99 is charged (Lys342), and the loop containing the residue equivalent to Phe174 is two amino acids longer and highly flexible (Tyr416–Asp417–Asn418; see alignment in Fig. S4b†). These differences may explain why small modifications in the pyridine-N-oxide moiety can exert a large impact on drug's selectivity across different serine proteases.34 We further analyzed the contacts that otamixaban forms with TMPRSS2, finding that it mainly blocks the catalytic region, impeding the binding of any substrate that could be processed (Fig. 4a).
In the pre-bound metastable state (Fig. S5b†), the drug is within the active site but outside the S1 pocket. It interacts with catalytic residues His296 and Asp345, mainly mediated by Coulomb interactions between its amidinium head and the aspartate.
Finally, it is interesting to note that otamixaban has two potential hydrolyzable centers, an ester and an amide, that could be in principle digested by TMPRSS2. Nonetheless, otamixaban is known to be a reversible non-covalent inhibitor, with the two chemical groups being stable in blood and urine.34,35 This prompts the question of why these two chemical groups are stable within the active site of the enzyme. To address this question, we searched for conformations fulfilling certain structural criteria that are suitable for catalysis, including the integrity of the catalytic machinery (Ser441-His296 h-bond), the nucleophilic attack (Ser441-reactive center), and the position of part of the substrate inside the oxyanion hole (formed by Gly439 and Ser441 NH groups). For the amide group, we found that it is never near the catalytic serine at the same time that it places its carboxylic group inside the oxyanion hole, two factors that are crucial to enhance the enzymatic activity.36 Even if these two criteria were met, we note that the N end of the amide is placed within the S1 pocket, inverted with respect to the orientation of natural substrates, which may further slow down a potential reaction. For the ester group, instead, we find reactive conformations, but just a very small fraction (0.13%) of the total, suggesting that hydrolysis could be possible, but it will be minor. These factors may explain why these two chemical groups of otamixaban are preserved within the enzymatic cavity, rendering a stable, non-covalent reversible inhibitor.
3 Discussion
The ongoing COVID-19 pandemic requires the development of potent drugs against SARS-CoV-2. A possible treatment strategy is to target the host cell protease TMPRSS2, an essential compound for viral host cell entry. In this work we identify otamixaban, a drug candidate admitted to phase III clinical trials, as a potential TMPRSS2 inhibitor.We show that even though otamixaban is only a weak TMPRSS2 inhibitor in cell culture, it is as potent as camostat in human lung tissue. One could argue that this intriguing result may arise from the fact that camostat is rapidly converted to a metabolite (GBPA) in the human body, which is slightly less potent.21 However, the difference in potency between camostat and its metabolic product is likely not large enough to explain the leveling out of otamixaban and camostat observed in lung tissue. Thus, it seems more plausible to hypothesize that the presence of otamixaban's primary target fXa, in PCLS but not in Calu-3 cells, might be at the root of the observed differences.
As otamixaban was originally developed for inhibiting fXa, a protease that was also reported to cleave the S protein,9 it is promising to combine otamixaban with known potent TMPRSS2 inhibitors.30 Furthermore, our experiments in Calu-3 cells reveal that otamixaban shows strong synergistic effects with camostat or nafamostat. This implies that there is another, yet unknown, protease that might contribute to SARS-CoV-2 cell entry. As a result, otamixaban doses can be significantly lowered by adding (sub-) nanomolar concentrations of known TMPRSS2 inhibitors camostat or nafamostat.
Given that these three drugs bind to the S1 pocket of TMPRSS2 (as shown above and in ref. 3), it seems unlikely that they have a cooperative mechanism towards the same target.
We did not observe synergy between drugs when we combined otamixaban with supplemental doses of camostat/nafamostat in our HTS assay (Fig. S1†). These observations suggest that the synergistic effect between otamixaban and camostat/nafamostat in Calu-3 cells is likely not due to drug–drug interactions or TMPRSS2 allosteric effects. Instead, it could be explained by a secondary target. Additional investigation is necessary to shed light upon the precise mechanism of synergy between the drugs.
Otamixaban's potency in combination preparations is also encouraging from a therapeutic point of view: COVID-19 patients often suffer from hypercoagulation37 and otamixaban was originally optimized for inhibiting coagulation factor Xa.25 It might thus inhibit a suitable target orthogonal to TMPRSS2, possibly mitigating severe symptoms of COVID-19.
Having arrived at the phase III of clinical trials, the safety and pharmacokinetics of otamixaban have already been characterized. Across a 100-fold dose range, otamixaban has proven to be well tolerated in a cohort of healthy subjects.38 It also showed rapid plasma distribution and elimination, with low variability of plasma exposure across subjects.35
The availability of clinical data alongside the synergistic effects described in this manuscript suggests that otamixaban in combination with nafamostat or camostat is a promising candidate for COVID-19 clinical trials.
4 Materials and methods
High throughput TMPRSS2 activity assay
A high throughput activity assay has been developed to test potential inhibitors of TMPRSS2.29 The experiment was performed in a 1536-well black plate according to the published protocol: Boc–Gln–Ala–Arg–AMC substrate (20 nl) and test compound (20 nl in DMSO) were added using an ECHO 655 acoustic dispenser (LabCyte). TMPRSS2 (150 nl) diluted in assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) was dispensed to that, using a BioRAPTR (Beckman Coulter), for a total assay volume of 5 μl. After incubation at room temperature for 1 h, fluorescence was measured.To detect inhibitors causing fluorescence quenching (those having a concentration-dependent decrease on AMC fluorescence), a counter-assay was performed as previously described.29 7-Amino-4-methylcoumarin (20 nl) and inhibitor or DMSO (20 nl) were added using an ECHO 655 acoustic dispenser (LabCyte). Assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) was added to that, for a total reaction volume of 5 μl.
PHERAstar with excitation at 340 nm and emission at 440 nm was used for detection and fluorescence was normalized relative to substrate alone (negative control) and substrate with 1 μm of nafamostat (positive control).
Hit confirmation TMPRSS2 activity assay
Compound dilutions ranging from 100 pm to 100 μm were prepared in assay buffer (50 mM Tris–HCl pH 8.0, 154 mM NaCl) containing a final DMSO concentration of 1%. In addition, recombinant TMPRSS2 (LifeSpan Biosciences, cat. no. LS-G57269) and the peptide substrate Boc–GlnAla–Arg–MCA (Peptide Institute, Inc., cat. no. 3135-v) were reconstituted in assay buffer, yielding concentrations of 2 μg ml−1 and 100 mM, respectively. Compound dilutions (25 μl per well, final concentration ranging from 50 pm to 50 μm) and recombinant TMPRSS2 (25 μl per well, final concentration of 1 μg ml−1) were mixed in black 96-well plates and incubated for 10 min. Next, assay buffer containing 20 μM Boc–Gln–Ala–Arg–MCA peptide substrate (50 μl per well, final concentration 10 μM) was added and samples were incubated for 3 h at 37 °C before fluorescence intensity was recorded using the Tecan Genios plate reader and the Magellan V6.4 Software (Tecan; excitation: 360 nm, emission: 465 nm).To define the upper and lower limits of the assay, the following controls were used: (i) to determine maximum TMPRSS2 enzyme activity, recombinant TMPRSS2 (25 μl per well, final concentration of 1 μg ml−1) was preincubated with assay buffer containing 1% DMSO but no compound (25 μl per well) before the peptide substrate (50 μl per well, final concentration of 10 μM) was added; (ii) to determine the unspecific background of the assay, peptide substrate (50 μl per well, final concentration of 10 μM) was incubated with assay buffer containing 0.5% DMSO (50 μl per well) in the absence of both recombinant TMPRSS2 and compound.
Cell culture
All cell lines were incubated at 37 °C in a humidified atmosphere with 5% CO2. Vero E6 (African green Monkey kidney; ATCC no. CRL-1586; kindly provided by Marcel A. Müller and Christan Drosten, Institute of Virology, Charité – Universitätsmedizin Berlin, Germany) and HEK-293T (human embryonic kidney; DSMZ no. ACC 635) cells were cultivated in Dulbecco's modified Eagle medium containing 10% fetal bovine serum (FCS, Biochrom), 100 U ml−1 of penicillin and 0.1 mg ml−1 of streptomycin (PAN-Biotech), while Calu-3 cells (human lung adenocarcinoma; kindly provided by Stephan Ludwig, Institute of Virology, University of Münster, Germany) were cultivated in minimum essential medium supplemented with 10% FCS, 100 U ml−1 of penicillin and 0.1 mg ml−1 of streptomycin (PAN-Biotech), 1× non-essential amino acid solution (from 100× stock, PAA) and 1 mM sodium pyruvate (Thermo Fisher Scientific). Transfection of HEK-293T was carried out by calcium-phosphate precipitation.Plasmids
The expression plasmids for severe acute respiratory syndrome coronavirus 2 spike glycoprotein (SARS-2-S) and vesicular stomatitis virus glycoprotein (VSV-G) that were used to generate VSV pseudotype particles harboring the respective viral proteins have been described elsewhere.4,39 Further, an empty pCG1 expression plasmid (kindly provided by Roberto Cattaneo, Mayo Clinic College of Medicine, Rochester, MN, USA) was used for the production of VSV particles bearing no viral protein.Reconstitution of compounds and storage
Camostat mesylate (Tocris, cat. no. 3193), nafamostat mesylate (Tocris, cat. no. 3081) and otamixaban (AdooQ BioScience, cat. no. A11517) were reconstituted in dimethyl sulfoxide (DMSO), yielding stock concentrations of 10 mM. For each compound stock, multiple small volume aliquots were prepared and stored at −20 °C. Of note, in order to avoid changes in compound activity due to multiple freeze–thaw cycles, residual material from thawed stocks was discarded. Please note that compounds for the high throughput activity assay were treated in a separate facility (cf. high throughput TMPRSS2 activity assay).Analysis of SARS-2-S-driven cell entry using VSV pseudotypes
VSV pseudotype particles bearing SARS-2-S, VSV-G or no viral protein (negative control) were produced according to an established protocol.40 In brief, HEK-293T cells transfected to express the respective viral protein or no viral protein were inoculated with a replication-deficient VSV vector that lacks the genetic information for VSV-G and instead possesses individual open reading frames for an enhanced green fluorescent protein and firefly luciferase (FLuc), VSVΔG-FLuc (kindly provided by Gert Zimmer, Institute of Virology and Immunology, Mittelhäusern, Switzerland).41 Following an incubation period of 1 h at 37 °C, the cells were washed and received a fresh culture medium. In the case of cells transfected with SARS-2-S expression vector or empty plasmid, the medium was further supplemented with anti-VSV-G antibody (culture supernatant from I1-hybridoma cells; ATCC no. CRL-2700). At 16–18 h post-inoculation, the culture supernatant containing the VSV pseudotype particles was collected, clarified from cellular debris by centrifugation (2000×g, 10 min, room temperature) and stored at −80 °C until further use. In order to analyze the effects of the tested compounds on SARS-2-S-driven VSV pseudotype entry into target cells, Calu-3 cells, which were grown in 96-well plates, were incubated for 1 h at 37 °C in the presence of different concentrations of the respective compounds or solvent (DMSO) diluted in culture medium (50 μl per well). Next, VSV pseudotypes were added (50 μl per well) and cells were further incubated for 16–18 h before the efficiency of pseudotype entry was assessed. For this, the culture supernatant was aspirated and cells were lysed by incubation with 1× cell culture lysis reagent (produced from 5-fold concentrated stock, Promega) for 30 min at room temperature. Thereafter, lysates were transferred into white 96-well plates. Finally, FLuc activity was recorded using a Hidex Sense plate luminometer (Hidex) following the addition of a commercial substrate (Beetle-Juice, PJK).Infection of ex vivo human lung cultures with authentic SARS-CoV-2 and quantification of viral titer
Experiments with human lung tissue were approved by the Ethics Committee of the Hannover Medical School (MHH, Hannover, Germany) and are in compliance with The Code of Ethics of the World Medical Association (number 2701-2015). All patients or their next of kin gave written informed consent for the use of lung tissue for research. Ex vivo human lung cultures (precision cut lung slices, PCLS) were prepared according to a published protocol.42 Only macroscopically disease-free parts of lung tissue obtained from lung cancer patients that underwent lobe resection at the Hannover Medical School (MHH, Hannover, Germany) were used for experiments. First, the lung lobe was inflated with DMEM/F-12 medium containing 2% agarose. Following agarose polymerization, the tissue was cut into slabs and tissue cores of 8 mm in diameter were cut into 300 μm thin slices. Cultivation of PCLS was carried out at 37 °C and 5% CO2 using Ham's F-12 medium containing 0.1 mg ml−1 streptomycin, 10 U ml−1 penicillin, 0.05 mg ml−1 gentamicin and 0.01 mg ml−1 enrofloxacin. For infection experiments, only PCLS with a ciliary activity of 75% or higher were used. The PCLS were placed into 48-well plates and incubated for 1 h at 37 °C and 5% CO2 in the presence of different concentrations of the respective compounds (PCLS treated with DMSO served as a control). Next, the culture medium was removed and PCLS were inoculated with SARS-CoV-2 isolate hCoV-19/Germany/FI1103201/2020 (kindly provided by Stephan Ludwig, Institute of Virology, University of Münster, Germany) using an infectious dose of 4 × 105 pfu ml−1 (250 μl per well). At 1 h post-inoculation, the inoculum was removed and PCLS were washed three times with phosphate-buffered saline (PBS) before culture medium (500 μl per well) containing the respective inhibitor or DMSO was added. Samples (100 μl) were collected at 1 h (washing control) and 24 h (end point) post-infection and stored at −80 °C for virus titration. Viral titers were determined by plaque titration. For this, Vero E6 cells that were grown in 12-well plates were inoculated with serial dilutions of supernatant (inoculum = 850 μl) and incubated for 1 h at 37 °C and 5% CO2. Thereafter, the inoculum was removed and cells were washed with PBS. Next, cells were overlaid with Eagle's minimal essential medium without phenol red (Lonza) containing 1% plaque agarose (Biozym) and incubated for 72 h at 37 °C and 5% CO2, before plaques were counted to determine viral titers (given as plaque-forming units per ml, pfu ml−1).Docking
Due to the lack of a crystal structure for TMPRSS2, we manually extracted 10 representative frames from a precursive MD dataset3 which was originally seeded with a homology model.27 Our MD construct includes only the protease domain (amino acids 256 to 491). We note that even though TMPRSS2 has a transmembrane domain, the catalytic domain (i.e. our MD setup) itself is not embedded or directly linked to the membrane, nor is there evidence of lipids playing a role in catalysis. We, therefore, assume that a simulation in the solvent is a realistic model.For the ligand structure, we downloaded the three-dimensional representation of the reference molecule from the ZINC database (ZINC ID: ZINC000001908051).43 We prepared both the receptor and the ligand structures with MGLTools.44
We performed protein–ligand docking using smina,45 a fork of AutoDock Vina.46 The search space was defined as a box of 30 Å3 centered on the Cα atom of the catalytic serine (Ser441). We identified five important residues whose rigidity could negatively influence ligand binding into the S1 pocket, namely: Glu299, Lys300, Asp435, Gln438 and Trp461. The side-chains of these residues were kept flexible throughout the run to explore their different conformations. We used the Vinardo scoring function47 and the default exhaustiveness of 10.
For each receptor structure docked against the ligand, we took the single highest-scored binding pose. Finally, we used the SMILES to determine the positions of missing hydrogens (omitted by smina) and added them back to the docking poses.
Molecular dynamics simulations
MD simulations were run with OpenMM 7.4.0 (ref. 48) with the amber14SB forcefield49 for protein and openff-1.1.0 (ref. 50) for the ligand parameters. We initiate a simulation box of side length 7.2 nm, use the TIP3P water model51 with a NaCl ion concentration of 0.1 mol l−1 at a neutral charge. The setups contain between 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 354 and 10429 water molecules, depending on the docking pose and TMPRSS2 conformation.
354 and 10429 water molecules, depending on the docking pose and TMPRSS2 conformation.
We run simulations in the NPT ensemble. The temperature is kept at 310 K (physiological temperature) and the pressure at 1 bar. Rigid water molecules, PME electrostatics, and a 1 nm cutoff for nonbonded interactions are used. We further applied a Langevin integrator with 4 fs integration step using hydrogen mass repartitioning (HMR, 4 amu hydrogen mass). HMR slows down covalent hydrogen bond vibrations such that a larger integration step is possible while conserving the thermodynamics and kinetics.52,53 Our trajectories accumulate to 109 μs, each containing a simulation time of 1 μs. VMD54 was used to visualize structures.
MD simulations were seeded from the docked structures, as described above. More specifically, we selected as starting complexes the four docking poses where the ligand had identical chirality to the molecule in the ZINC database (ZINC ID: ZINC000001908051). Please note, again, that our protein structures are based on a homology model27 as detailed in ref. 3.
We analyzed MD simulations by using inverse minimal distances between the different drug groups (benzamidine-group, methyl-ester, pyridine-N-oxid, methyl-group) to each protein residue with PyEMMA 2.5.7.55 We further performed a linear VAMP56 dimension reduction operation with a lag time of 5 ns, used 10 dimensions with the highest kinetic variance, and performed a regular spatial clustering with a minimal distance of 0.9. The discrete state with the most population was subsequently estimated by the argmax of a histogram.
Based on the same VAMP projection, we used a different discretization (k-means algorithm with 300 cluster centers) to estimate a 16 state hidden Markov model57 (HMM) at a lag time of 2 ns. The HMM model is used to assign metastable states (binding modes) and to extract metastable trajectories (Viterbi algorithm58). We use the metastable trajectories to verify that the model captures the metastable dynamics (Fig. S6†) and to assess empirical state probabilities through histogram counting.
Dose–response curve fitting
We normalized the inhibition efficiency against the respective control(s) for each of the experiments. We then used the generalized four-parameter log-logistic model to fit the dose–response curves on the normalized data. The model is defined as: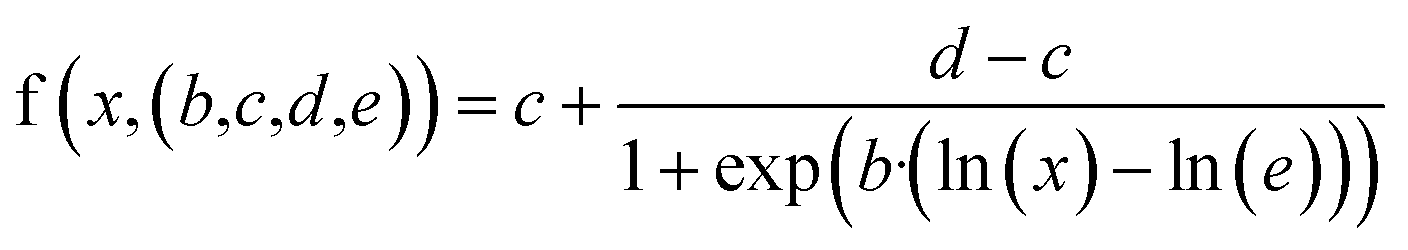 | (1) |
Drug combination analysis
The combination index (CI) for two mutually exclusive drugs is computed using the following equation31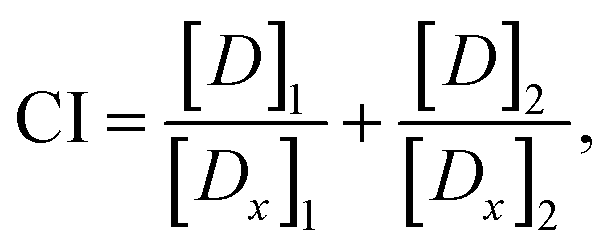 | (2) |
We further note that, since it is a priori unclear whether the drugs in the combination preparations are mutually exclusive or not, we have computed the correction terms for mutually non-exclusive drugs, 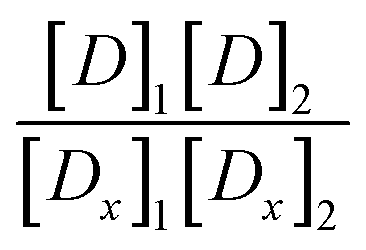 , and found them to be negligibly small for both combination preparations.
, and found them to be negligibly small for both combination preparations.
To estimate the CI for different inhibition values, we have analytically inverted the dose–response function (eqn (1)). It is evaluated from 10 to 95% inhibition, i.e. in the range in which the experimental measurements allow a reasonable curve fit.
The dose reduction index (DRI) for drug i is calculated as:32
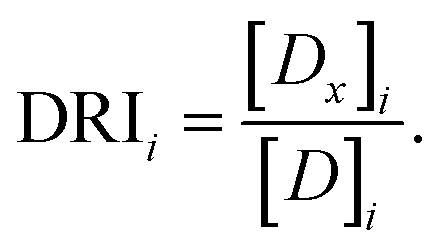 | (3) |
Data availability
Structures of otamixaban in complex with TMPRSS2 are published online at https://github.com/noegroup/tmprss2_structures.Author contributions
Frank Noé, Stefan Pöhlmann, Markus Hoffmann, Matt Hall designed research. Jon Shrimp, Matt Hall, Nadine Krüger, Stefan Pöhlmann, Markus Hoffmann conducted single-drug/TMPRSS2 inhibition assays. Katarina Elez, Nadine Krüger, Min Shen, Matt Hall, Stefan Pöhlmann, Markus Hoffmann, Frank Noé analyzed single-drug/TMPRSS2 inhibition assays. Tim Hempel, Katarina Elez, Lluís Raich, Frank Noé proposed drug combination experiments and analyzed drug combination data. Markus Hoffmann, Nadine Krüger, Stephan Pöhlmann conducted drug combination experiments. Tim Hempel, Katarina Elez, Lluís Raich, Frank Noé conducted & analyzed computational experiments (docking, MD simulations). Olga Danov, Danny Jonigk, Armin Braun prepared and provided human PCLS. Tim Hempel, Katarina Elez, Lluís Raich, Stefan Pöhlmann, Markus Hoffmann, Frank Noé wrote the manuscript.Conflicts of interest
T. H., K. E., L. R., F. N., N. K., M. H. and S. P. are inventors of a patent application (EP21187449) submitted by Freie Universität Berlin based on the findings presented in this manuscript. The remaining authors declare no conflicts of interest.Acknowledgements
We acknowledge financial support from Deutsche Forschungsgemeinschaft DFG (SFB/TRR 186, Project A12 and SFB 1114, Project C03), the European Commission (ERC CoG 772230 “ScaleCell”), the Berlin Mathematics center MATH+ (AA1-6 and AA1-10), the federal ministry of education and research BMBF (BIFOLD and RAPID Consortium) and the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 897414. S. P. acknowledges funding by BMBF (RAPID Consortium, 01KI1723D and 01KI2006D; RENACO, 01KI20328A; SARS1S2 01KI20396; COVIM consortium, 01KX2021), the county of Lower Saxony and the German Research Foundation (DFG) (PO 716/11-1, PO 716/14-1). N. K. acknowledges funding from BMBF (ANI-CoV, 01KI2074A). This work was supported by the National Center for Advancing Translational Sciences, Division of Preclinical Innovation. The work in the Braun lab was funded by Bundesministerium für Bildung und Forschung (RENACO) and the Fraunhofer Internal Programs under Grant No. Anti-Corona 840260 DRECOR (Drug Repurposing for Corona). We further acknowledge European Research Council Consolidator Grant (XHale, 771883) to Danny Jonigk. We thank Simon Olsson (Chalmers University of Technology, Göteborg) for his ample advice in the initial phase of the project.References
- B. Hu, H. Guo, P. Zhou and Z.-L. Shi, Characteristics of SARS-CoV-2 and COVID-19, Nat. Rev. Microbiol., 2020, 19, 141–154 CrossRef PubMed.
- The World Health Organization, Weekly Epidemiological Update, Coronavirus disease 2019 (COVID-19), 9 March 2021 Search PubMed.
- T. Hempel, et al., Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat, Chem. Sci., 2021, 12, 983–992 RSC.
- M. Hoffmann, et al., SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor, Cell, 2020, 181(2), 271–280.e8 CrossRef CAS.
- B. A. Schuler, et al., Age-determined expression of priming protease TMPRSS2 and localization of SARS-CoV-2 in lung epithelium, J. Clin. Invest., 2021, 131(1), e140766 CrossRef CAS PubMed.
- M. Montopoli, et al., Androgen-Deprivation Therapies for Prostate Cancer and Risk of Infection by SARS-CoV-2: A Population-Based Study (N = 4532), Ann. Oncol., 2020 Search PubMed.
- M. Hoffmann, et al., Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2, Nature, 2020, 585(7826), 588–590 CrossRef CAS.
- T. Ou, et al., Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2, PLoS Pathog., 2021, 17(1), e1009212 CrossRef CAS PubMed.
- E. R. Kastenhuber, et al., Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry, bioRxiv., 2021 DOI:10.1101/2021.03.31.437960.
- S. Matsuyama, et al., Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2, J. Virol., 2010, 84(24), 12658–12664 CrossRef CAS PubMed.
- A. Shulla, et al., A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry, J. Virol., 2010, 85(2), 873–882 CrossRef.
- I. Glowacka, et al., Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response, J. Virol., 2011, 85(9), 4122–4134 CrossRef.
- S. Bertram, et al., Cleavage and Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by Human Airway Trypsin-Like Protease, J. Virol., 2011, 85(24), 13363–13372 CrossRef CAS PubMed.
- S. Gierer, et al., The Spike Protein of the Emerging Betacoronavirus EMC Uses a Novel Coronavirus Receptor for Entry, Can Be Activated by TMPRSS2, and Is Targeted by Neutralizing Antibodies, J. Virol., 2013, 87(10), 5502–5511 CrossRef CAS.
- S. Bertram, et al., TMPRSS2 Activates the Human Coronavirus 229E for Cathepsin-Independent Host Cell Entry and Is Expressed in Viral Target Cells in the Respiratory Epithelium, J. Virol., 2013, 87(11), 6150–6160 CrossRef CAS.
- K. Shirato, M. Kawase and S. Matsuyama, Middle East Respiratory Syndrome Coronavirus Infection Mediated by the Transmembrane Serine Protease TMPRSS2, J. Virol., 2013, 87(23), 12552–12561 CrossRef CAS PubMed.
- E. Böttcher, et al., Proteolytic Activation of Influenza Viruses by Serine Proteases TMPRSS2 and HAT from Human Airway Epithelium, J. Virol., 2006, 80(19), 9896–9898 CrossRef.
- C. Chaipan, et al., Proteolytic Activation of the 1918 Influenza Virus Hemagglutinin, J. Virol., 2009, 83(7), 3200–3211 CrossRef CAS PubMed.
- K. Sakai, et al., The Host Protease TMPRSS2 Plays a Major Role in in Vivo Replication of Emerging H7N9 and Seasonal Influenza Viruses, J. Virol., 2014, 88(10), 5608–5616 CrossRef PubMed.
- T. S. Kim, C. Heinlein, R. C. Hackman and P. S. Nelson, Phenotypic Analysis of Mice Lacking the Tmprss2-Encoded Protease, Mol. Cell. Biol., 2006, 26(3), 965–975 CrossRef CAS PubMed.
- M. Hoffmann, Camostat Mesylate Inhibits SARS-CoV-2 Activation by TMPRSS2-Related Proteases and Its Metabolite GBPA Exerts Antiviral Activity, EBioMedicine, 2021, 65, 103255 CrossRef CAS PubMed.
- M. Hoffmann, et al., Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19, Antimicrob. Agents Chemother., 2020, 64(6), e00754–20 CrossRef.
- N. P. Azouz, et al., Alpha 1 Antitrypsin is an Inhibitor of the SARS-CoV-2–Priming Protease TMPRSS2, Pathog. Immun., 2021, 6(1), 55–74 Search PubMed.
- D. Bestle, et al., TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells, Life Sci. Alliance, 2020, 3(9), e202000786 CrossRef PubMed.
- K. R. Guertin, et al., Optimization of the _-Aminoester class of factor Xa inhibitors. part 2: Identification of FXV673 as a potent and selective inhibitor with excellent In vivo anticoagulant activity, Bioorg. Med. Chem. Lett, 2002, 12(12), 1671–1674 CrossRef CAS.
- P. G. Steg, Anticoagulation With Otamixaban and Ischemic Events in Non–ST-Segment Elevation Acute Coronary Syndromes, JAMA, J. Am. Med. Assoc., 2013, 310(11), 1145 CrossRef CAS PubMed.
- S. Rensi, et al., Homology Modeling of TMPRSS2 Yields Candidate Drugs That May Inhibit Entry of SARS-CoV-2 into Human Cells, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12009582.
- X. Hu, et al., Discovery of TMPRSS2 Inhibitors from Virtual Screening as a Potential Treatment of COVID-19, ACS Pharmacol. Transl. Sci., 2021, 4(3), 1124–1135 CrossRef CAS PubMed.
- J. H. Shrimp, et al., An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19, ACS Pharmacol. Transl. Sci., 2020, 3(5), 997–1007 CrossRef CAS.
- W. Sun, P. E. Sanderson and W. Zheng, Drug combination therapy increases successful drug repositioning, Drug Discovery Today, 2016, 21(7), 1189–1195 CrossRef CAS PubMed.
- T.-C. Chou and P. Talalay, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS.
- T.-C. Chou, Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies, Pharmacol. Rev., 2006, 58(3), 621–681 CrossRef CAS.
- V. Chu, et al., Pharmacological Characterization of a Novel Factor Xa Inhibitor, FXV673, Thromb. Res., 2001, 103(4), 309–324 CrossRef CAS.
- K. Guertin and Y.-M. Choi, The Discovery of the Factor Xa Inhibitor Otamixaban: From Lead Identification to Clinical Development, Curr. Med. Chem., 2007, 14(23), 2471–2481 CrossRef CAS PubMed.
- A. Paccaly, et al., Pharmacokinetics of Otamixaban, a Direct Factor Xa Inhibitor, in Healthy Male Subjects: Pharmacokinetic Model Development for Phase 2/3 Simulation of Exposure, J. Clin. Pharmacol., 2006, 46(1), 37–44 CrossRef CAS PubMed.
- L. Hedstrom, Serine Protease Mechanism and Specificity, Chem. Rev., 2002, 102(12), 4501–4524 CrossRef CAS.
- M. Y. Abou-Ismail, A. Diamond, S. Kapoor, Y. Arafah and L. Nayak, The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management, Thromb. Res., 2020, 194, 101–115 CrossRef CAS.
- A. Paccaly, et al., Pharmacodynamic markers in the early clinical assessment of otamixaban, a direct factor Xa inhibitor, Thromb. Haemostasis, 2005, 94(12), 1156–1163 CrossRef CAS PubMed.
- C. Brinkmann, et al., The Glycoprotein of Vesicular Stomatitis Virus Promotes Release of Virus-like Particles from Tetherin-Positive Cells, PLoS One, 2017, 12(12), e0189073 CrossRef.
- H. Kleine-Weber, et al., Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization, J. Virol., 2019, 93(2) CrossRef CAS.
- M. Berger Rentsch and G. Zimmer, A Vesicular Stomatitis Virus Replicon-Based Bioassay for the Rapid and Sensitive Determination of Multi-Species Type I Interferon, PLoS One, 2011, 6(10), e25858 CrossRef.
- V. Neuhaus, et al., Assessment of the Cytotoxic and Immunomodulatory Effects of Substances in Human Precision-Cut Lung Slices, J. Visualized Exp., 2018, 135, e57042 Search PubMed.
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337 CrossRef CAS.
- G. M. Morris, et al., AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed.
- D. R. Koes, M. P. Baumgartner and C. J. Camacho, Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise, J. Chem. Inf. Model., 2013, 53(8), 1893–1904 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- R. Quiroga and M. A. Villarreal, Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening, PloS One, 2016, 11(5), e0155183 CrossRef.
- P. Eastman, et al., OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics, PLoS Comput. Biol., 2017, 13(7), e1005659 CrossRef PubMed.
- J. A. Maier, et al., ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- Y. Qiu, et al., Development and Benchmarking of Open Force Field v1.0.0, the Parsley Small Molecule Force Field, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-l070l-v3.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J. Chem. Theory Comput., 2015, 11(4), 1864–1874 CrossRef CAS.
- F. Rao and M. Spichty, Thermodynamics and Kinetics of Large-Time-Step Molecular Dynamics, J. Comput. Chem., 2012, 33(5), 475–483 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graph., 1996, 14(1), 33–38 CrossRef CAS.
- M. K. Scherer, et al., PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models, J. Chem. Theory Comput., 2015, 11(11), 5525–5542 CrossRef CAS.
- H. Wu and F. Noé, Variational Approach for Learning Markov Processes from Time Series Data, J. Nonlinear Sci., 2019, 30(1), 23–66 CrossRef CAS.
- F. Noé, H. Wu, J.-H. Prinz and N. Plattner, Projected and Hidden Markov Models for Calculating Kinetics and Metastable States of Complex Molecules, J. Chem. Phys., 2013, 139(18), 184114 CrossRef.
- L. R. Rabiner, A Tutorial on Hidden Markov Models and Selected Applications in Speech Recognition, Proc. IEEE, 1989, 77(2), 257–286 CrossRef.
- P. Virtanen, et al., SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01494c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |