
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-lived lanthanide emission via a pH-sensitive and switchable LRET complex†
Tamara
Boltersdorf
a,
Felicity N. E.
Gavins
b and
Nicholas J.
Long
 *a
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, London, W12 0BZ, UK. E-mail: n.long@imperial.ac.uk
bDepartment of Life Sciences, Centre for Inflammation Research and Translational Medicine (CIRTM), Brunel University London, Uxbridge, Middlesex UB8 3PH, UK
First published on 17th May 2021
Abstract
Lanthanide-based luminescence resonance energy transfer (LRET) can be used as a tool to enhance lanthanide emission for time-resolved cellular imaging applications. By shortening lanthanide emission lifetimes whilst providing an alternative radiative pathway to the formally forbidden, weak lanthanide-only emission, the photon flux of such systems is increased. With this aim in mind, we investigated energy transfer in differently spaced donor–acceptor terbium–rhodamine pairs with the LRET “on” (low pH) and LRET “off” (high pH). Results informed the design, preparation and characterisation of a compound containing terbium, a spectrally-matched pH-responsive fluorophore and a receptor-targeting group. By combining these elements, we observed switchable LRET, where the targeting group sensitises lanthanide emission, resulting in an energy transfer to the rhodamine dye with an efficiency of E = 0.53. This strategy can be used to increase lanthanide emission rates for brighter optical probes.
Introduction
Fluorescence microscopy has been widely used in biomedical research settings for cellular imaging due to its high sensitivity and spatial resolution.1,2 The technique is often combined with carefully designed fluorescent molecular probes to gain insights into physiological and pathophysiological mechanisms.3 Although a variety of organic fluorophores (such as derivatives of cyanine, rhodamine, coumarin etc.) have been developed previously, their utility for imaging is limited by low photostability, small Stokes shifts and spectral overlap with biological autofluorescence.Signals from emissive lanthanide probes have the potential to overcome these limitations. Specifically, lanthanide species have sharp emission profiles, high photostability, large Stokes shifts, and long lifetimes, which can be temporally separated from autofluorescent background signals.4,5 Therefore, these compounds should enable more sensitive detection of biological targets that are otherwise swamped with short-lived background signals.6 However, the ability to use lanthanide probes for sensitive imaging is compromised by (1) inefficient excitation pathways at cell-compatible excitation and (2) a low photon flux resulting from long metal-based lifetimes with low emission rates. As a result, either high concentrations of the probe or longer acquisition times become necessary7,8 and there is little or no improvement in sensitivity when comparing lanthanide-based cellular microscopy with conventional fluorophores.9–11
One way to address point (2), is to employ a resonance energy transfer in order to increase lanthanide photon flux. Resonance energy transfer can occur between two luminescent molecules in sufficient proximity when there is a spectral overlap between donor emission and acceptor absorption.12 The interaction often proceeds via nonradiative dipole–dipole interactions (Förster mechanism).13 The energy transfer leads to a reduction in donor lifetime as well as a simultaneous increase in the acceptor's fluorescence intensity and drops off with distance.14 As a result, such donor–acceptor pairs have been mainly used in the context of biological assays to gain information on nanometre scale distances.15–17 In the context of lanthanide photophysics, energy transfer where an organic chromophore donor can sensitise lanthanide-acceptor emission are commonly used.18,19 In these cases, the short-lived organic moiety acts as the donor and long-lived emission from the excited lanthanide state is observed. By constructing an energy transfer from a lanthanide to an organic dye, the donor luminescence is shortened and an alternative radiative deactivation pathway to metal-based emission becomes available, both factors that should increase emission.
Lanthanide-based luminescence resonance energy transfer (LRET), where the long-lived metal acts as the donor and an organic dye as the acceptor, has been previously reported, for example, to measure voltage-induced changes occurring in potassium ion channels,17,20 elucidate the geometry of DNA double-helix formation,21 study protein–protein interactions22 and detect ligands binding to cellular receptors.23 Similarly, lanthanide complexes have been used as donors for quantum dots or dyes in specialised assays, where the proximity to an acceptor indicates a binding event.24–27 In self-assembled transition metal-lanthanide constructs, energy transfer from a lanthanide to a d-block metal has given rise to interesting optical properties, for example, where Cr(III) acts as an acceptor to a lanthanide donor.28–31
However, in these examples the energy transfer between two separate donor and acceptor molecules is used to assess proximity or binding events but does not look at combining donor and acceptor into the same molecule to enhance long-lived emission properties. To the best of our knowledge, the idea to improve lanthanide photon flux using LRET has only recently been demonstrated for the first time by Cho et al.8 who showed that co-injecting an energetically-matched organic fluorophore with a separate Eu(III)-based probe can greatly enhance emissive properties in vivo. However, in their system, the metal donor and fluorophore acceptor are not incorporated into the same molecule but interact only when they diffuse near each other. To build upon and improve the concept, we have combined a long-lived donor and a short-lived acceptor lumiphore into the same molecule to create a “switchable” intramolecular LRET.
Further to the donor and acceptor unit, we incorporated Quin C1,32 a Formyl Peptide Receptor 2 (FPR2/ALX, also known as FPR-like 1 [FPRL1]) ligand into the same molecule. FPR2/ALX is expressed on the cell surface of immune cells33 and is known to produce ligand-dependent responses that can be both pro- or anti-inflammatory,34,35 and thus is an interesting target for preclinical studies. Assembled together, these units create a targeted, responsive cellular probe.
Results and discussion
As the acceptor unit, we chose a rhodamine B derivative with pH-dependent emission and absorption bands,36,37 to enable the same system to be studied with the LRET “on”, when the acceptor can absorb in the relevant region or “off”, when the fluorophore is non-emissive. The Tb(III) donor emission is energetically matched with the absorbance of the chosen rhodamine acceptor (Fig. 1A). When combined and excited, our system should result in long-lived donor-only signals, short-lived acceptor-only emission and donor-excited acceptor emission at low pH (Fig. 1B). We further incorporated Quin C1, a FPR2/ALX ligand into the same molecule. Apart from binding to a cellular receptor, the targeting group can sensitise lanthanide-donor emission, which in turn enhances the energy transfer between Tb and rhodamine (Fig. 1C and D).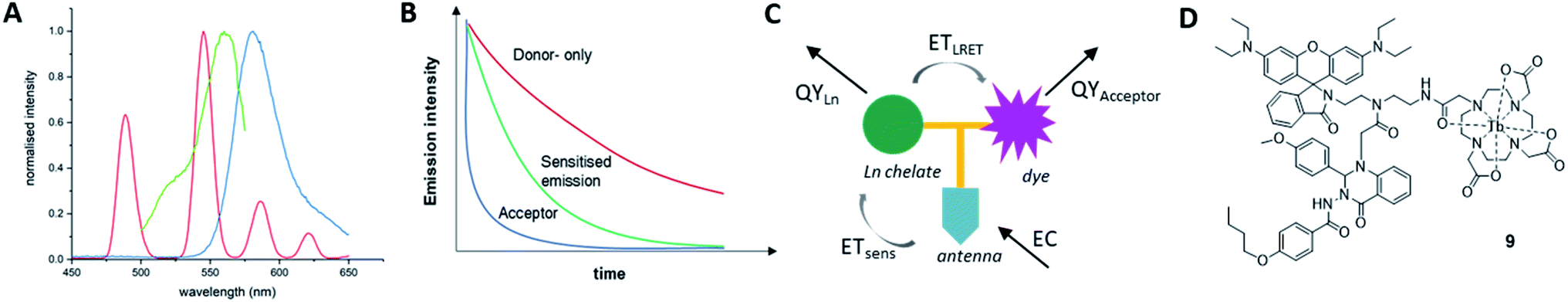 | ||
| Fig. 1 (A) Emission profile of a Tb(III)-complex (red, λexc = 350 nm, time-gate: 0. ms) and steady state excitation (green, λem = 580 nm) and emission profiles (blue, λexc = 350 nm) of rhodamine-B. The spectra demonstrate good overlap between donor emission and acceptor excitation. (B) Schematic representation of donor-only (red), acceptor-only (blue) and sensitised emission (green) lifetimes in an LRET system. (C) Schematic of processes determining emission in LRET systems. Total emission consists of EC = extinction coefficient, ETsens = efficiency of lanthanide sensitisation energy transfer, ETLRET = efficiency of LRET, QY = quantum yield. (D) Molecular structure of final compound 9. | ||
Varying intramolecular donor–acceptor distances
To investigate how the relative magnitude of energy transfer varies with distance in our system, we prepared three rhodamine–terbium compounds with varying linker lengths: an ethylenediamine bridge (1),38 a diethylenetriamine bridge (2), and a combination of the two (3, Fig. 2A, synthetic details and characterisation given in the ESI†). All three compounds contained a spiro-cyclisable, rhodamine B-derived motif as the acceptor. The chosen rhodamine dye is strongly fluorescent in its ring-opened analogue (at low pH) but neither absorbs nor emits at relevant wavelengths in its cyclic form (at neutral and high pH), effectively enabling the acceptor to be turned “on” or “off”, depending on pH conditions.39 Time-gated emission measurements, that exclude short-lived, acceptor-only signals were recorded at pH 7, when the acceptor is “off” and at pH 3, when the acceptor unit is “on” (Fig. 3). The energy transfer was quantified by comparing percentage increase in luminescence intensity ratios at 580 nm (maximum of acceptor-based emission) and 490 nm (local maximum of terbium-based emission) at pH 3 (containing donor-only and sensitised acceptor emission) compared to that at pH 7 (containing donor-only signals), as outlined in eqn (1).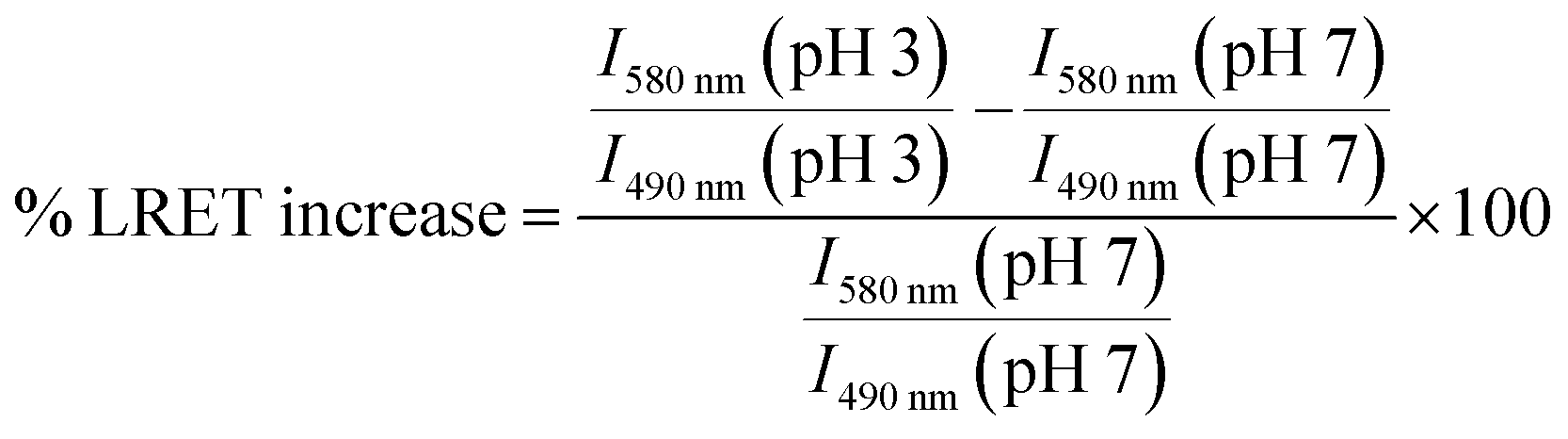 | (1) |
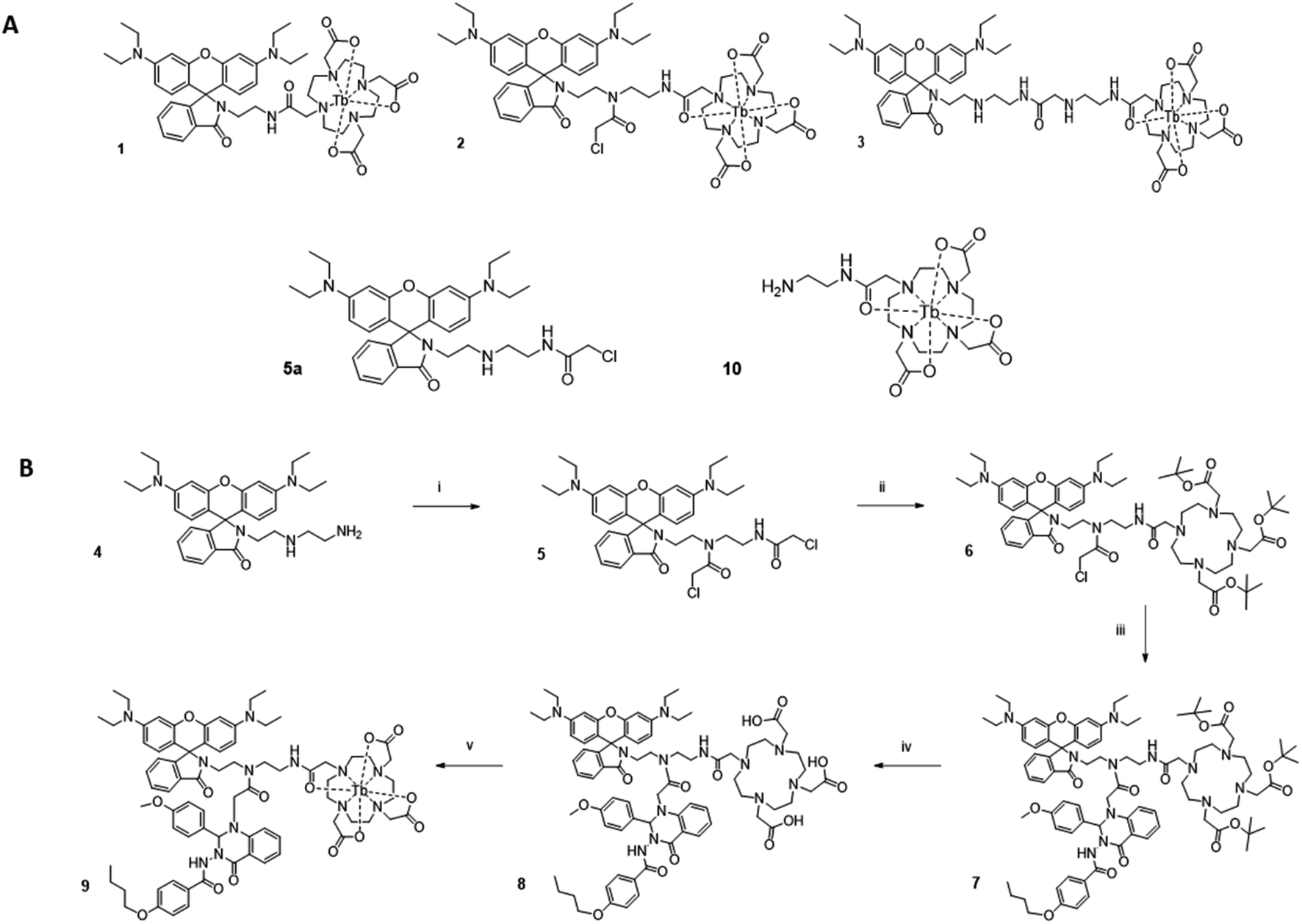 | ||
| Fig. 2 (A) Molecular structures of rhodamine–terbium constructs 1, 2 and 3 with varying chain lengths between donor and acceptor unit. The rhodamine fragment is depicted in its cyclic, non-emissive form. (B) Overview of synthetic pathway. Reagents: (i) chloroacetyl chloride, triethylamine, dichloromethane, 24% yield, (ii) tert-butyl DO3A, triethylamine, acetonitrile, 79% yield, (iii) Quin C1, N,N-diisopropylethylamine, acetonitrile, 54% yield, (iv) conc. hydrochloric acid, 60% yield, (v) Tb(III) trichloride hexahydrate, water, pH 5.5, 51% yield. | ||
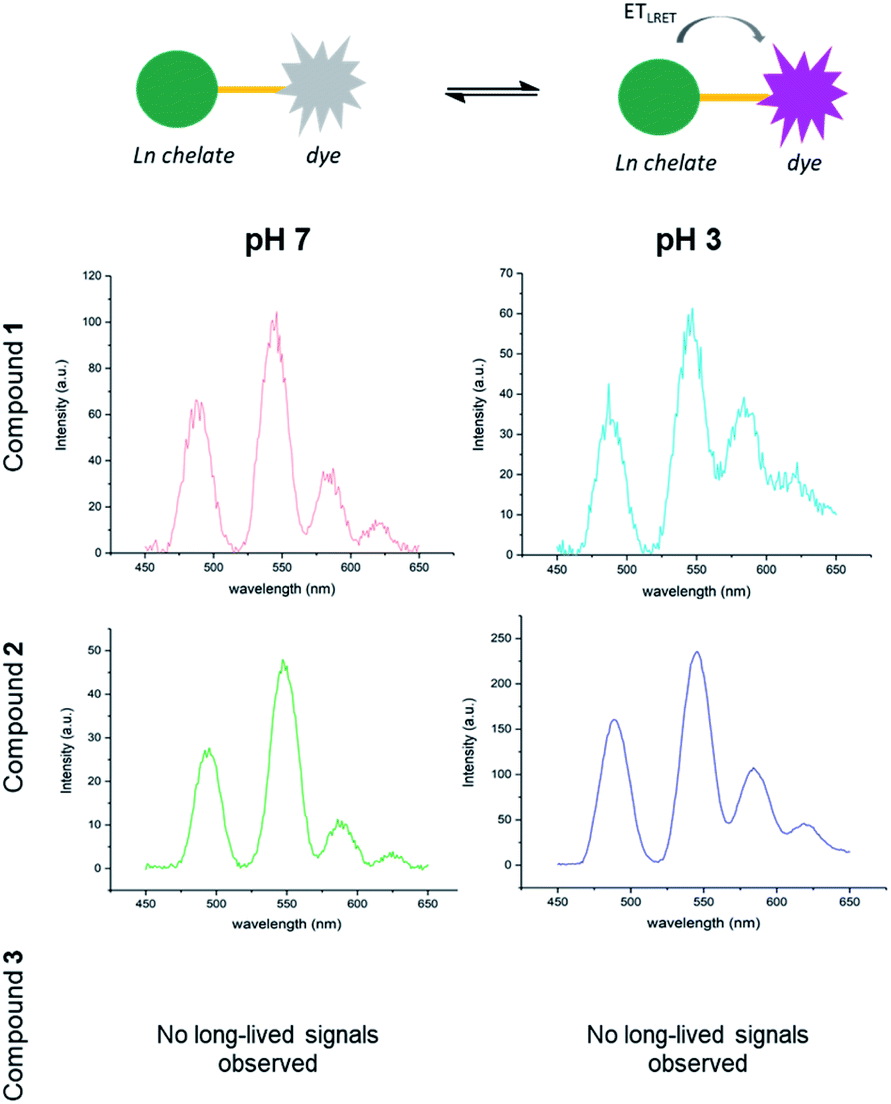 | ||
| Fig. 3 Switchable LRET between terbium donor and rhodamine dye acceptor with three different linker lengths in compounds 1, 2 and 3 at pH 7 (left) and pH 3 (right). Emission recorded in 0. mM solutions with 0.2 ms delay (λexc = 350 nm). | ||
No communication between the two units was found in compound 3, indicating that the linker length was too long for efficient energy transfer. In fact, hardly any terbium-based long-lived signals were observed. The absence of terbium signals may be due to a combination of inefficient lanthanide excitation, with little or no sensitisation occurring and increased solvent quenching effects when the ion is less shielded by a proximal hydrophobic dye. For compounds 1 and 2, the percentage increase in intensity ratios observed at pH 3 compared to pH 7 was calculated as 74% and 26% respectively using the procedures described above. The observed trend is consistent with a drop-off in energy transfer as the donor–acceptor chain length is increased and demonstrates that in both compounds 1 and 2 the donor–acceptor pair can still communicate, whereas the distance in compound 3 is too large. Notably, terbium-based emission intensity (e.g. at 490 nm) decreased in compound 1 as the pH was decreased, but increased in compound 2 with decreasing pH. This may be a result of varying steric constraints around the metal centre between the two compounds, which has been shown to be related to DOTA conformational populations.40 Non-radiative deactivation pathways will differ depending on the metal chelator conformation as well as proximity of the hydrophobic dye.
Preparation of a receptor-targeted, LRET complex
With these results in hand, we concluded that the intermediate chain length compound 2 could simultaneously enable coupling of the FPR targeting group Quin C1, while still holding the energy matched lumiphores in sufficient proximity for an energy transfer to occur. We have previously shown that Quin C1 acts not only as a targeting group, but can further function as an antenna, providing a more efficient route to populate the lanthanide emissive state.41 Accordingly, the combined compound should result in an increase in LRET. To test this hypothesis, Quin C1 was reacted with the chloroacetone moiety on compound 2 before complexation with terbium to obtain LRET complex 9 (synthetic route detailed in Fig. 2B, synthetic details in ESI†).The choice of a switchable rhodamine analogue within compound 9 has two key advantages. Firstly, the same system can be studied with the energy transfer “on” (low pH) or “off” (high pH), enabling separate detection of donor and acceptor signals and, as a result, direct quantification of the energy transfer. Secondly, inflammation and associated disease states are often accompanied by a local extracellular environment with lower pH ranges.42 We have previously shown that the rhodamine fragment in combination with Quin C1 binds to neutrophil FPR2/ALX and switches “on” in vitro under stimulated conditions that mimic acute inflammation.43 In an extension, this complex could find future application as a switchable LRET sensor for inflammatory environments.
Photophysical properties of compound 9
Excitation of compound 9 at 350 nm in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/methanol solution at pH 7 produced one emission maximum at 454 nm that was assigned to the organic targeting group component (Fig. 4A). The excitation spectrum of 9 (λem = 450 nm) resembles that of Quin C1. As the pH is decreased, the rhodamine spirocyclisation equilibrium becomes increasingly shifted to the ring-opened form, resulting in characteristic absorption, excitation and emission bands of both the rhodamine unit and the dihydroquinazolinone-derivative at pH 3 (Fig. 4B). Upon application of a 0.2 ms delay to ensure that short-lived acceptor-only signals had decayed, time-gated emission spectra at pH 7 and pH 3 were collected. At 0.1 mM concentrations, changes in emission signal resulting from diffusion-related, intermolecular energy transfer should be minimal.44 At pH 7, the spectrum features a characteristic terbium emission profile (Fig. 4C). At pH 3, the lanthanide emission appears to be superimposed on donor-excited long-lived rhodamine luminescence with a maximum at 580 nm, representing the LRET-based dye signals (Fig. 4D). The percentage increase in intensity ratio (580 nm to 490 nm) between the compound at pH 7 and the compound at pH 3 was determined as 113% using eqn (1). In line with expectations, the value has increased compared to that found for compound 2, which has the same donor–acceptor linker length but does not contain the Quin C1 moiety. This was attributed to the fact that the 2,3-dihydroquinazolinone derivative acts as an antenna and enhances ETsens (Fig. 1), resulting in increased donor emission and a larger LRET.
:1 water/methanol solution at pH 7 produced one emission maximum at 454 nm that was assigned to the organic targeting group component (Fig. 4A). The excitation spectrum of 9 (λem = 450 nm) resembles that of Quin C1. As the pH is decreased, the rhodamine spirocyclisation equilibrium becomes increasingly shifted to the ring-opened form, resulting in characteristic absorption, excitation and emission bands of both the rhodamine unit and the dihydroquinazolinone-derivative at pH 3 (Fig. 4B). Upon application of a 0.2 ms delay to ensure that short-lived acceptor-only signals had decayed, time-gated emission spectra at pH 7 and pH 3 were collected. At 0.1 mM concentrations, changes in emission signal resulting from diffusion-related, intermolecular energy transfer should be minimal.44 At pH 7, the spectrum features a characteristic terbium emission profile (Fig. 4C). At pH 3, the lanthanide emission appears to be superimposed on donor-excited long-lived rhodamine luminescence with a maximum at 580 nm, representing the LRET-based dye signals (Fig. 4D). The percentage increase in intensity ratio (580 nm to 490 nm) between the compound at pH 7 and the compound at pH 3 was determined as 113% using eqn (1). In line with expectations, the value has increased compared to that found for compound 2, which has the same donor–acceptor linker length but does not contain the Quin C1 moiety. This was attributed to the fact that the 2,3-dihydroquinazolinone derivative acts as an antenna and enhances ETsens (Fig. 1), resulting in increased donor emission and a larger LRET.
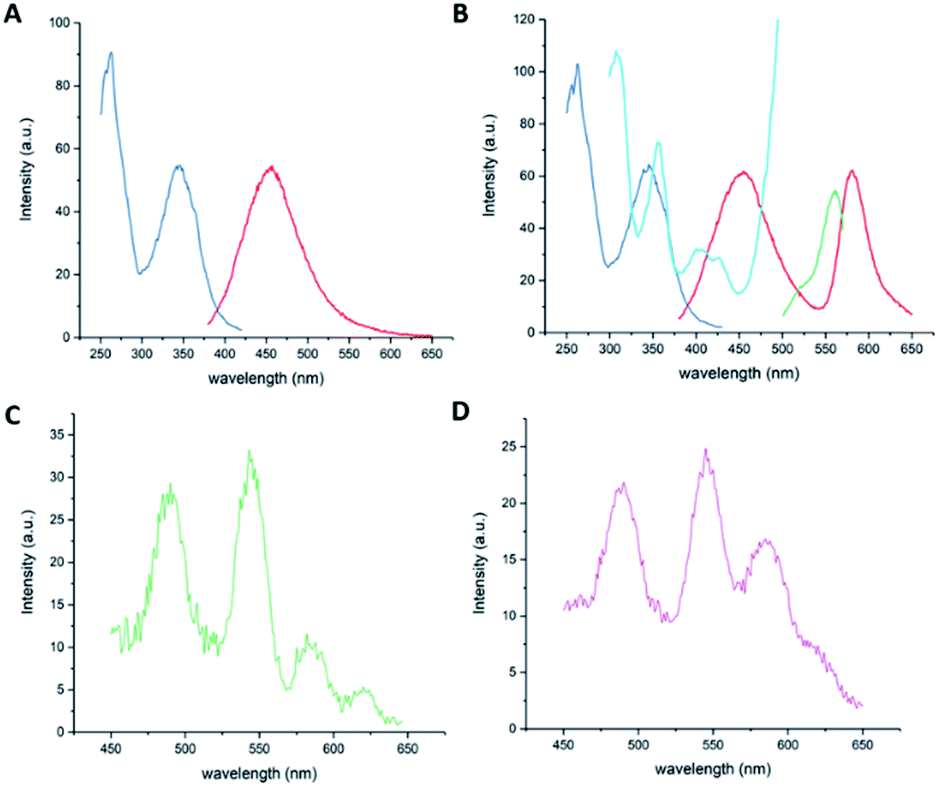 | ||
| Fig. 4 (A) Steady state emission (red, λexc = 350 nm) and excitation (blue, λem = 450) spectra of compound 9 (0. mM) in 1:1 water/methanol (v/v) mixtures at pH 7. (B) Steady state emission (red, λexc = 350 nm) and excitation spectra (λem = 450 in blue and λem = 580 nm in pale blue (5 nm slit width) and green (2.5 nm slit width)) of compound 9 (0. mM) in 1:1 water/methanol (v/v) mixtures at pH 3. (C) Time-gated emission spectra (λexc = 350 nm, delay = 0.2 ms) of 9 (0. mM) in 1:1 methanol/water solutions at pH 7 and (D) pH 3. | ||
Determination luminescence lifetimes
Long-lived luminescence lifetimes of compound 9 at pH 3 and pH 7 were acquired. The short-lived acceptor-only signals were excluded by the 0.1 ms time gate. Using the in-built OriginPro 8 curve fitting function, we found that the data obtained for long-lived signals were not in agreement with a single exponential decay, implying the presence of two long-lived components. A bi-exponential fit was achieved (eqn (S1), ESI†) with lifetime components of τ1 = 100 ± 4 μs (21.8% fractional intensity) and τ2 = 1544 ± 8 μs (78.2% fractional intensity) at pH 7 (Fig. 5A, χ2 = 0.9998) and τ1 = 107 ± 3 μs (45.0% fractional intensity) and τ2 = 721 ± 8 μs (55.0% fractional intensity) at pH 3 (Fig. 5B, χ2 = 0.9997). τ2 was assigned to terbium-based signals, whereas the assignment of τ1 proved more complex. One possible assignment is donor-excited acceptor emission from a minor population of the donor with a high rate of resonance energy transfer, as has been previously described in intermolecular LRET systems where biexponential decay was observed.45 Another option is that a solvated species of the lanthanide could result in a terbium lifetime decrease as reflected by τ1.4 At pH 7, the rhodamine is predominantly in its non-emissive, cyclic form and the contribution of τ1 to the overall decay is small (as reflected by the A1 constant, eqn (S1) (ESI)†). At pH 3, donor-based lifetimes (τ2) are substantially decreased (to less than half the original value) and the contribution of τ1 to the overall decay is raised (as reflected by the A1 constant, consistent with lanthanide-based LRET at pH 3).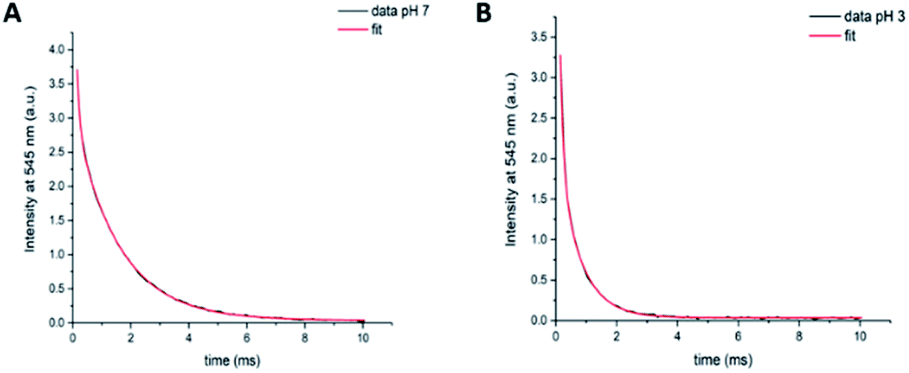 | ||
| Fig. 5 Time-gated emission lifetime decay components of 9 ( mM) at 545 nm (λexc = 350 nm, 0. ms delay) in 1:1 water/methanol solutions. Data and biexponential fit using eqn (S1) (ESI†) obtained at pH 7 (A) and pH 3 (B). | ||
Determination of LRET efficiency
Finally, the efficiency of an energy transfer can be calculated based on experimentally derived lifetimes of the donor-only (τD, which was approximated as τ2 at pH 7) and the donor in presence of the acceptor (τDA, which was approximated as τ2 at pH 3) according to the following relationship –eqn (2):12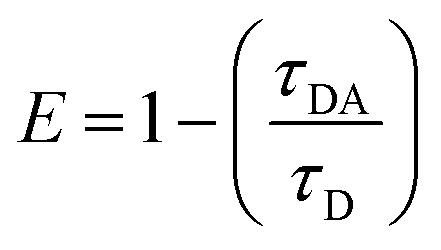 | (2) |
By this method, we were able to determine the LRET efficiency as E = 0.53 for compound 9. The energy transfer rate was calculated as 740 s−1 (eqn (S2), ESI†). It should be noted that shortening the molecular distance between donor and acceptor can lead to substantially higher energy transfer rates (see eqn (S3), ESI†).
Conclusions
Here we describe the design, preparation and characterisation of a compound with switchable LRET. First, donor–acceptor distances within the same molecule were varied and the relative energy transfer was quantified. Results informed design choices of a more complicated molecule balancing synthetic accessibility with proximity of the lumiphore pair. A terbium chelate as the long-lived donor was combined with a pH-responsive organic dye (LRET acceptor) and a receptor-targeting group (Quin C1). Quin C1 acts as a lanthanide sensitiser and enhances LRET donor emission via energy transfer to the emissive terbium state. By using a pH-switchable fluorophore as the LRET acceptor, we were able to compare the same compound with and without acceptor-based emission as a convenient strategy to examine photophysical properties. In the final compound 9, we observed the desired lifetime decrease in long-lived emission when the acceptor is “on” (from 1544 μs to 721 μs), and showed that we have an efficient energy transfer system with an LRET efficiency value of E = 0.53 (113% increase in intensity ratios from pH 7 (no LRET) to pH 3 (LRET)). These results suggest that LRET can be used as a novel strategy to develop brighter complexes for lanthanide-based cellular imaging applications.Author contributions
NJL and TB conceived the idea and designed experiments. NJL and FNEG supervised the project. TB conducted synthesis and spectroscopic studies. All the authors contributed to writing, reviewing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC Centre for Doctoral Training in Neurotechnology for funding (EP/L016737/1). FNEG acknowledges funding from the Royal Society Wolfson Foundation (RSWF\R3\183001) and NJL is grateful for a Royal Society Wolfson Research Merit Award.Notes and references
- N. J. Long and W. T. Wong, The Chemistry of Molecular Imaging, Wiley, 2015 Search PubMed.
- S. W. Hell, Nat. Methods, 2009, 6, 24–32 CrossRef CAS.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC.
- J.-C. G. Bünzli and S. V. Eliseeva, Springer Ser. Fluoresc., 2011, 13, 311–337 Search PubMed.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2014, 50, 384–399 RSC.
- M. Rajendran and L. W. Miller, Biophys. J., 2015, 109, 240–248 CrossRef CAS PubMed.
- U. Cho, D. P. Riordan, P. Ciepla, K. S. Kocherlakota, J. K. Chen and P. B. Harbury, Nat. Chem. Biol., 2018, 14, 15–21 CrossRef CAS PubMed.
- J. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- R. C. Leif, L. M. Vallarino, M. C. Becker and S. Yang, Cytometry, Part A, 2006, 69, 767–778 CrossRef PubMed.
- J. M. Zwier and N. Hildebrandt, in Reviews in Fluorescence, ed. C. G. Geddes, 2016, pp. 17–44 Search PubMed.
- P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275–302 CrossRef CAS PubMed.
- L. Stryer, Annu. Rev. Biochem., 1978, 47, 819–846 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed.
- D. M. Dolino, S. S. Ramaswamy and V. Jayaraman, J. Visualized Exp., 2014, 91, e51895 Search PubMed.
- P. R. Selvin, Nat. Struct. Mol. Biol., 2000, 7, 730–734 CrossRef CAS PubMed.
- D. J. Posson, P. Ge, C. Miller, F. Bezanilla and P. R. Selvin, Nature, 2005, 436, 848–851 CrossRef CAS PubMed.
- T. Zhang, C. Chan, R. Lan, W. Wong and K. Wong, Chem.–Eur. J., 2014, 20, 970–973 CrossRef CAS PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- A. Cha, G. E. Snyder, P. R. Selvin and F. Bezanilla, Nature, 1999, 946, 809–813 CrossRef PubMed.
- P. R. Selvin and J. E. Hearst, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10024–10028 CrossRef CAS PubMed.
- H. E. Rajapakse, N. Gahlaut, S. Mohandessi, D. Yu, J. R. Turner and L. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13582–13587 CrossRef CAS PubMed.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef CAS PubMed.
- L. J. Charbonnière and N. Hildebrandt, Eur. J. Inorg. Chem., 2008, 3241–3251 CrossRef.
- D. Geißler, S. Linden, K. Liermann, K. D. Wegner, L. J. Charbonniere and N. Hildebrandt, Inorg. Chem., 2014, 53, 1824–1838 CrossRef PubMed.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef CAS PubMed.
- S. Comby and J.-C. G. Bünzli, Lanthanide near-Infrared Luminescence in Molecular Probes and Devices, Elsevier Science, Amsterdam, 2007 Search PubMed.
- M. Cantuel, G. Bernardinelli, D. Imbert, J.-C. G. Bünzli, G. Hopfgartner and C. Piguet, J. Chem. Soc., Dalton Trans., 2002, 1929–1940 RSC.
- M. Cantuel, G. Bernardinelli, G. Muller, J. P. Riehl and C. Piguet, Inorg. Chem., 2004, 43, 1840–1849 CrossRef CAS PubMed.
- S. Torelli, D. Imbert, M. Cantuel, G. Bernardinelli, S. Delahaye, A. Hauser, J.-C. G. Bünzli and C. Piguet, Chem.–Eur. J., 2005, 11, 3228–3242 CrossRef CAS PubMed.
- M. Cantuel, F. Gumy, J.-C. G. Bünzli and C. Piguet, Dalton Trans., 2006, 2647–2660 RSC.
- M. Nanamori, X. Cheng, J. Mei, H. Sang, Y. Xuan, C. Zhou, M. Wang and R. D. Ye, Mol. Pharmacol., 2004, 66, 1213–1222 CrossRef CAS PubMed.
- C. N. Serhan and P. M. Murphy, Pharmacol. Rev., 2009, 61, 119–161 CrossRef PubMed.
- D. A. Dorward, C. D. Lucas, G. B. Chapman, C. Haslett, K. Dhaliwal and A. G. Rossi, Am. J. Pathol., 2015, 185, 1172–1184 CrossRef CAS PubMed.
- J. G. Filep, M. Sekheri and D. El, Eur. J. Pharmacol., 2018, 833, 339–348 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- W. L. Czaplyski, G. E. Purnell, C. A. Roberts, R. M. Allred and E. J. Harbron, Org. Biomol. Chem., 2014, 12, 526–533 RSC.
- C. Rivas, G. J. Stasiuk, J. Gallo, F. Minuzzi, G. A. Rutter and N. J. Long, Inorg. Chem., 2013, 52, 14284–14293 CrossRef CAS PubMed.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS.
- M. Meyer, V. Dahaoui-Gindrey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 178–180, 1313–1405 CrossRef CAS.
- T. Boltersdorf, J. Ansari, E. Y. Senchenkova, L. Jiang, A. J. P. White, M. Coogan, N. E. Gavins and N. J. Long, Dalton Trans., 2019, 48, 16764–16775 RSC.
- A. Lardner, J. Leukocyte Biol., 2001, 69, 522–530 CAS.
- T. Boltersdorf, J. Ansari, E. Y. Senchenkova, J. Groeper, D. Pajonczyk, A. Vital, G. Kaur, J. S. Alexander, T. Vogl, U. Rescher, N. J. Long and F. N. E. Gavins, Theranostics, 2020, 10, 6599–6614 CrossRef CAS.
- D. D. Thomas, W. F. Carlsen and L. Stryer, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 5746–5750 CrossRef CAS PubMed.
- T. Heyduk and E. Heyduk, Anal. Biochem., 2001, 289, 60–67 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01503f |
| This journal is © The Royal Society of Chemistry 2021 |