DOI:
10.1039/D1SC01550H
(Edge Article)
Chem. Sci., 2021,
12, 8252-8259
Alkylsulfenyl thiocarbonates: precursors to hydropersulfides potently attenuate oxidative stress†
Received
17th March 2021
, Accepted 14th May 2021
First published on 14th May 2021
Abstract
The recent discovery of the prevalence of hydropersulfides (RSSH) species in biological systems suggests their potential roles in cell regulatory processes. However, the reactive and transient nature of RSSH makes their study difficult, and dependent on the use of donor molecules. Herein, we report alkylsulfenyl thiocarbonates as a new class of RSSH precursors that efficiently release RSSH under physiologically relevant conditions. RSSH release kinetics from these precursors are tunable through electronic modification of the thiocarbonate carbonyl group's electrophilicity. In addition, these precursors also react with thiols to release RSSH with a minor amount of carbonyl sulfide (COS). Importantly, RSSH generation by these precursors protects against oxidative stress in H9c2 cardiac myoblasts. Furthermore, we demonstrate the ability of these precursors to increase intracellular RSSH levels.
Introduction
Recently, hydrogen sulfide (H2S) has been reported to exert several favorable physiological effects including vasorelaxation, inhibition of myocardial ischemia-reperfusion injury, and slowing the development of atherosclerosis.1–3 Many of the biological effects of H2S have been attributed to the formation of hydropersulfide (RSSH) species.4–6 Indeed, recent studies have demonstrated that RSSH species such as cysteine hydropersulfide (Cys-SSH) and glutathione hydropersulfide (GSSH) are endogenously produced at significant levels in various organisms, including mammals.7,8 Furthermore, cysteine residues in a variety of proteins and enzymes have been reported to be modified to the corresponding hydropersulfides.9,10 The chemical properties and abundance of these species suggest that RSSH play important roles in various physiological processes.11 CysSSH can be generated via the enzymes cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS), both of which utilize cystine (CysSSCys) as a substrate.7,12 In addition, recently it has been reported that CysSSH can be synthesized by cysteinyl-tRNA synthetases (CARSs) utilizing CysSH as a substrate.13 The reaction of H2S with oxidized thiols such as disulfides (RSSR) and sulfenic acids (RSOH) can also generate RSSH.14 Despite these findings, the physiological functions of RSSH remain poorly understood.
RSSH species possess unique chemical properties that are not found in other biologically relevant sulfur species. For instance, RSSH are significantly more nucleophilic than their thiol (RSH) counterparts, likely due to the alpha effect.14–16 Additionally, RSSH have been proposed to behave as potent antioxidants and may play an important role in maintaining the cellular redox balance.17 The high nucleophilicity of RSSH can result in the efficient scavenging of reactive electrophiles. Recently, Fukuto and co-workers have reported that increased levels of intracellular RSSH exhibit enhanced resistance to electrophilic stress in HEK293T cells.18 Furthermore, Akaike and co-workers demonstrated that CSE overexpression leads to increased intracellular levels of RSSH species in A549 cells, which confers resistance toward hydrogen peroxide (H2O2)-mediated oxidative stress.7
Interestingly, unlike RSH, RSSH in the protonated state are electrophilic and can react with nucleophiles, including RSH. The electrophilic properties of RSSH may result in nucleophilic attack by protein thiols (PSH) giving rise to generation of protein hydropersulfides (PSSH). In fact, this transformation has been proposed to protect protein thiols from irreversible modification during oxidative and/or electrophilic insult.11 Consistent with this hypothesis, Greiner and co-workers demonstrated that the phosphatase and tensin homolog (PTEN) is protected from irreversible H2O2-mediated inhibition when the protein is in the PSSH form.19 Accumulating evidence suggests that the electrophilic nature of RSSH serves to regulate redox signaling and contribute to various cellular functions.20 For example, RSSH can activate the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) by reacting with the thiol group on Kelch-like ECH-associated protein 1 (KEAP1), which leads to cellular protection by expressing antioxidant response element (ARE)-dependent genes.21
Thus, the chemical properties of RSSH are distinct and far more diverse than thiols. However, due to the dichotomy of their nucleophilic and electrophilic character, RSSH are intrinsically unstable and subject to rapid disproportionation reactions to produce multiple species including polysulfides, H2S, and sulfur (S8). The metastable nature of RSSH remains a bottleneck in developing RSSH-based therapeutics.
One approach employed to address the intrinsic instability of RSSH relies on donor molecules capable of releasing RSSH in situ. To date, various donors have been developed in which the terminal sulfhydryl group is masked with a suitable protecting group. In the presence of specific stimuli such as cellular thiols,22 reactive oxygen species (ROS),23–25 pH,26–29 light,30 or esterase,31,32 these donors release RSSH. Notably, many of these precursors exhibit cytoprotective and cardioprotective effects, suggesting their potential application as reactive sulfur species-based therapeutics. Although several RSSH donor molecules have been developed, a significant limitation of current precursors is the lack of controllable RSSH release rates. In addition, some RSSH donors produce electrophilic byproducts that can complicate biological studies. Accessing RSSH donors with varying release kinetics under physiological conditions is necessary to evaluate the impact on potential therapeutic applications.
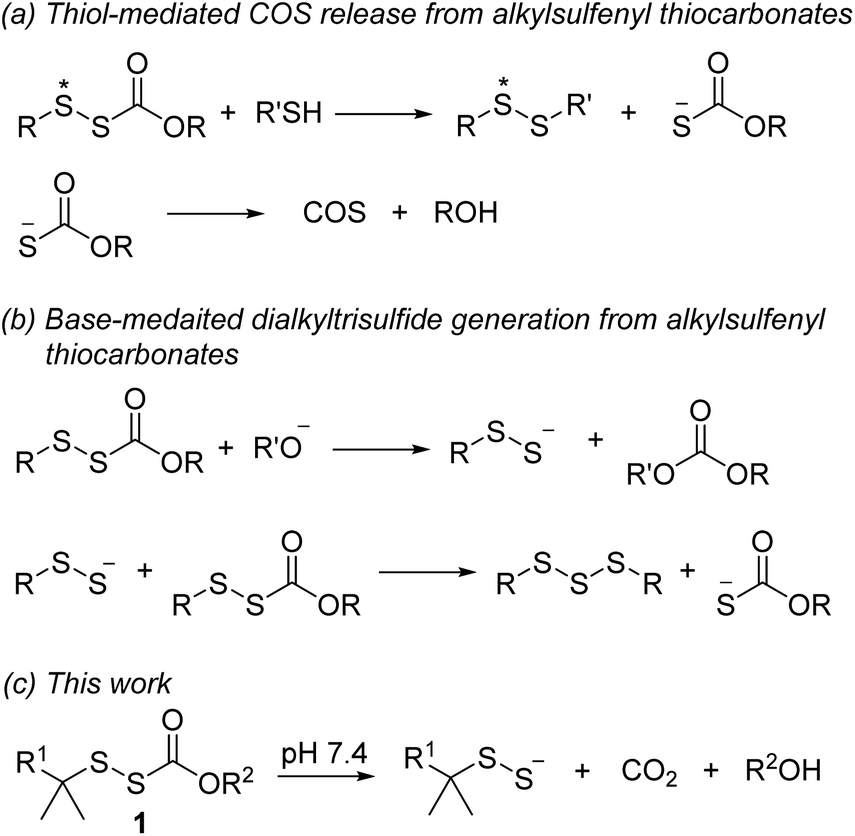
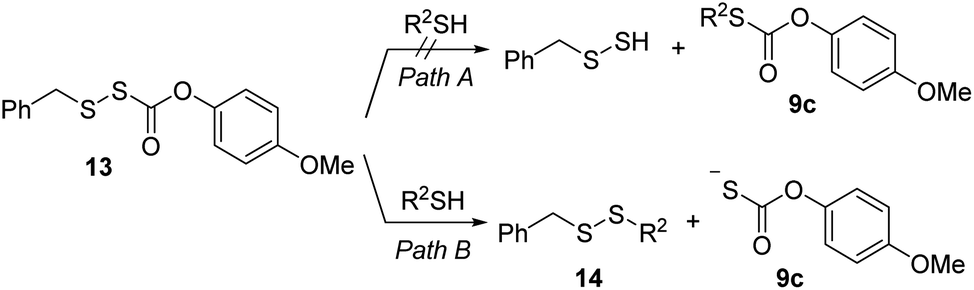
Alkylsulfenyl thiocarbonates (RSSC(O)OR) are an interesting class of sulfur compounds that have received relatively limited attention. In the presence of thiols, these compounds produce carbonyl sulfide (COS), likely via thiol attack at the internal sulfur atom (marked with an asterisk, Scheme 1a) of the disulfide to produce an unsymmetrical disulfide and a thiocarbonate intermediate, which spontaneously decomposes to release COS.33,34 In the presence of a strong base like potassium tert-butoxide, RSSC(O)OR can react to form dialkyl trisulfides, presumably via the intermediacy of RSSH (Scheme 1b).35,36 However, to the best of our knowledge, hydrolysis of these compounds under physiologically relevant conditions to release RSSH has not been reported. We envisioned enhancing the electrophilicity of the thiocarbonate carbonyl carbon in compound 1 by varying the electronic properties of the R2 group to promote hydrolysis under physiological conditions (Scheme 1c). Additionally, modification of the R2 group in compound 1 should allow the rate of hydrolysis to be tuned, thereby varying the RSSH release kinetics. We also anticipated increasing the steric hindrance at the internal sulfur atom of the precursor would be important to minimize its reaction with thiol, which precludes RSSH generation. Notably, sterically hindered RSSH are also expected to be superior substrates for trans-persulfidation reactions due to the preference of nucleophilic attack of thiols on the terminal sulfur atom.
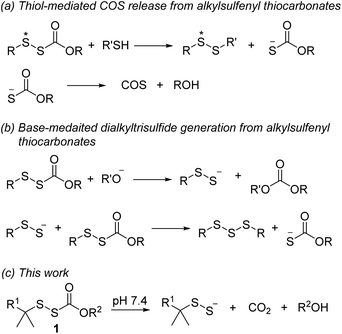 |
| | Scheme 1 RSSH and COS generation from alkylsulfenyl thiocarbonates. | |
Results and discussion
Synthesis of alkylsulfenyl thiocarbonates
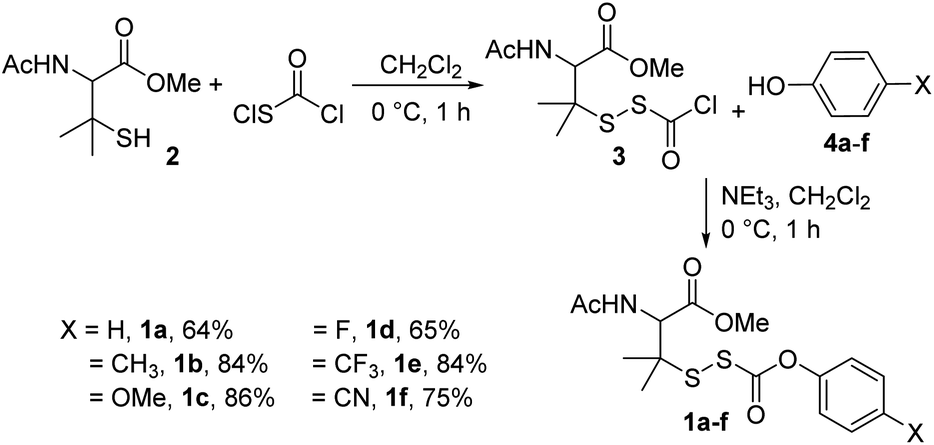
To test our hypothesis, we synthesized precursors 1a–fvia treatment of N-acetyl-penicillamine methyl ester (2) with chlorocarbonylsulfenyl chloride to obtain intermediate 3, which was then immediately treated with various para-substituted phenols 4a–f (Scheme 2; see ESI† for synthetic and analytical details).
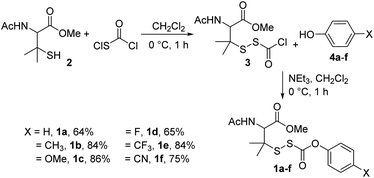 |
| | Scheme 2 Synthesis of alkylsulfenyl thiocarbonates 1a–f with overall yields. | |
RSSH generation studies
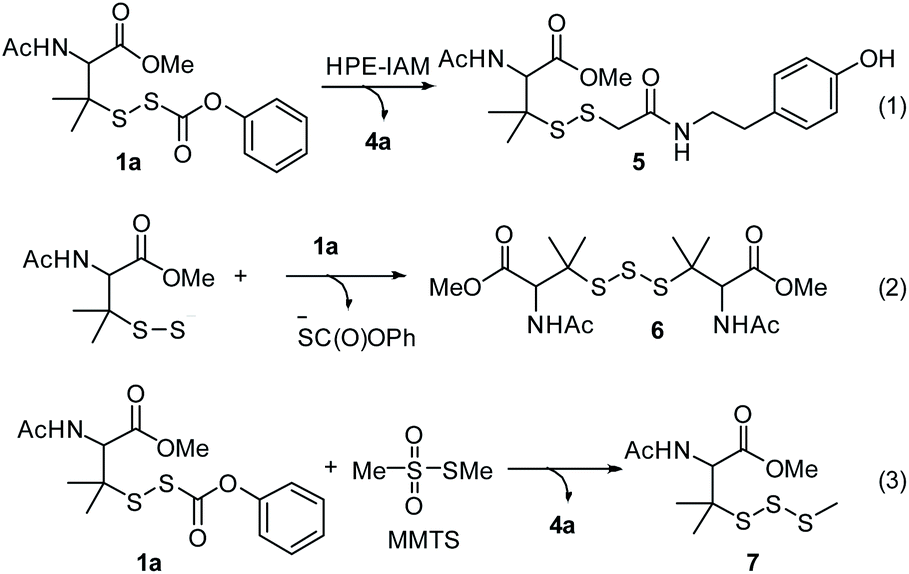
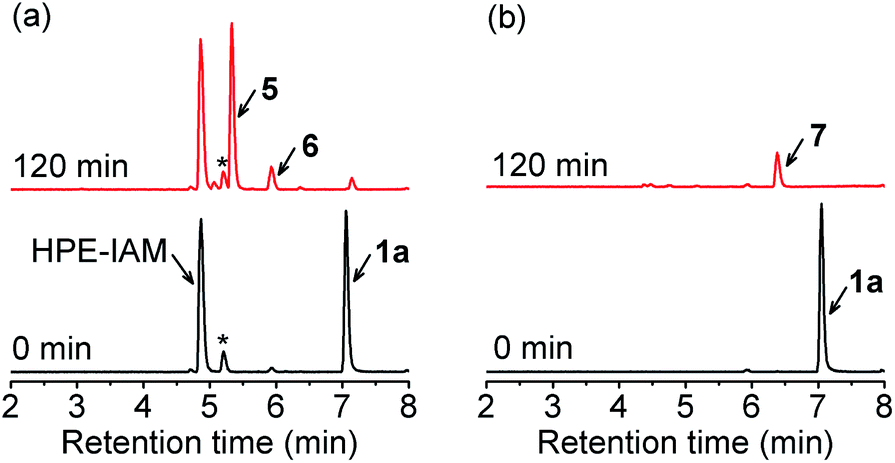
With these precursors in hand, we first examined RSSH generation from 1a by trapping with β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) under physiological conditions. UPLC-MS analysis following incubation of 1a with 10 equiv. of HPE-IAM in pH 7.4 ammonium bicarbonate buffer shows RSS-HPE-AM 5 formation (Scheme 3, eqn (1), and Fig. 1a), confirming RSSH generation. A small amount of dialkyl trisulfide 6 formation is also observed (Fig. 1a), suggesting that precursor 1a is a competitive trap for the initially released RSSH (Scheme 3, eqn (2)). To minimize the reaction of released RSSH with precursor 1a, we examined RSSH generation in the presence of the more electrophilic trap, S-methyl methanethiosulfonate (MMTS). MMTS has been reported as an alkylating reagent for identification of small molecule and protein hydropersulfides.31,37 Incubation of 1a with 10 equiv. of MMTS shows RSS-S-Me 7 formation with no evidence of dialkyl trisulfide 6 (Fig. 1b and S4†), supporting that MMTS is a more efficient trap than HPE-IAM.
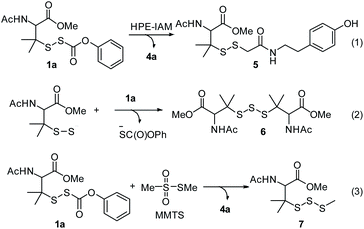 |
| | Scheme 3 RSSH generation from precursor 1a. | |
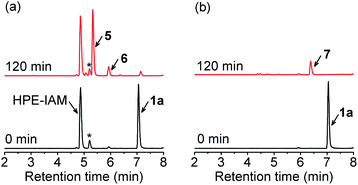 |
| | Fig. 1 UPLC-MS traces showing RSSH generation from 1a (100 μM) in the presence of (a) HPE-IAM (1 mM), and (b) MMTS (1 mM) in ammonium bicarbonate buffer (pH 7.4, 50 mM) containing the metal chelator diethylenetriaminepentaacetic acid (DTPA) (100 μM) at 37 °C. Asterisk indicates the presence of an impurity in the commercial HPE-IAM sample. | |
After confirming RSSH generation from 1a, we analyzed the kinetics of RSSH release from alkylsulfenyl thiocarbonates in the presence of MMTS by high-performance liquid chromatography (HPLC). Due to the lack of a chromophore in MMTS adduct 7, we monitored the decay of the RSSH precursor and formation of the corresponding phenol byproduct. First-order rate constants were obtained by plotting the precursor decay and phenol byproduct formation as a function of time. HPLC analysis of 1a incubation with 10 equiv. of MMTS in phosphate buffered saline (PBS, pH 7.4, 100 mM) shows disappearance of 1a (k = 5.39 × 10−3 min−1; t1/2 = 129 ± 2 min) with concomitant formation of phenol (k = 5.33 × 10−3 min−1; t1/2 = 130 ± 1 min) (Fig. 2). The observation of similar rate constants for both 1a decay and phenol (4a) formation indicates a rapid reaction of relased RSSH with MMTS, and that these observed rate constants indicate the RSSH release rate. Importantly, 92% of the byproduct phenol 4a is observed. In addition, we also examined the effect of pH on the kinetics of RSSH release. As expected, the rate of 1a hydrolysis is enhanced at pH 8.0 (t1/2 = 37 ± 3 min) and is slowed significantly at pH 6.0 (t1/2 = 1599 ± 48 min), supporting the proposed base catalyzed hydrolysis to release RSSH.
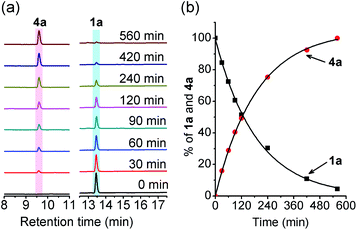 |
| | Fig. 2 (a) HPLC traces showing hydrolysis of 1a (100 μM) in the presence of MMTS (1 mM) in PBS (pH 7.4, 100 mM) containing the metal chelator DTPA (100 μM) at 37 °C. An aliquot of the reaction mixture was withdrawn at the specified time and quenched with 0.1% formic acid. The decay of 1a was monitored at 240 nm and formation of phenol 4a at 275 nm. (b) Kinetics of 1a decomposition and phenol 4a formation. Data represent the average of three experiments. The curve is the calculated best fit to a single-exponential function. | |
We then compared RSSH release kinetics for precursors containing a variety of para-substituted phenols. We hoped to decrease the rate of RSSH release through the introduction of electron-donating groups. Indeed, we observe a small decrease in the RSSH release rate from 1b, equipped with a 4-Me substituent (137 ± 4 min) (Table 1). Similarly, precursor 1c, equipped with a 4-OMe substituent, shows a half-life 147 ± 2 min, suggesting a minor effect of electron donating groups on RSSH release rate. As also expected, we find that electron withdrawing groups enhance the rate of RSSH release. For example, 4-F substituted precursor 1d releases RSSH with a half-life of 96 ± 3 min. Similarly, 4-CF3 substituted precursor 1e, and 4-CN substituted precursor 1f generate RSSH with a half-life of 56 ± 3 min and 28 ± 1 min, respectively. Importantly, UPLC-MS analysis of 1a–f following incubation with MMTS shows adduct 7 formation as the exclusive product (Fig. S5–S9†), and HPLC analysis confirms phenol byproduct 4a–f formation in excellent yields (Table 1). Hammett analysis of the kinetic data of RSSH release from precursors 1a–f indicates a ρ value of 0.71 (Fig. S47†), consistent with a mild substituents effect on the hydrolysis of these precursors. Taken together, these results indicate that the RSSH release kinetics of alkylsulfenyl thiocarbonates can be tuned through electronic modulation of the thiocarbonate carbonyl carbon in precursor 1.
Table 1 Half-lives for precursors 1a–f and phenol 4a–f yields
| Precursor |
X |
t
1/2 (min)a |
ArOH |
4a–f yield (%) |
|
RSSH precursors (100 μM) were incubated in the presence of MMTS (1 mM) in pH 7.4 PBS containing DTPA at 37 °C.
Reduced aqueous solubility required this experiment to be carried out in PBS with 50% acetonitrile. Reported data represent averages ± SD (n = 3).
|
|
1a
|
4-H |
129 ± 2 |
4a
|
92 ± 2 |
|
1b
|
4-CH3 |
137 ± 4 |
4b
|
93 ± 4 |
|
1c
|
4-OMe |
147 ± 2 |
4c
|
92 ± 1 |
|
1d
|
4-F |
96 ± 3 |
4d
|
96 ± 6 |
|
1e
|
4-CF3 |
56 ± 3b |
4e
|
97 ± 1 |
|
1f
|
4-CN |
28 ± 1 |
4f
|
97 ± 2 |
To further explore the substrate scope, we synthesized O-benzyl substituted RSSH precursor 8 (Scheme 4). Here, we observe significantly slower RSSH release (t1/2 = 2235 ± 94 min), indicating slow hydrolysis which is likely due to reduced electrophilicity of the thiocarbonate carbonyl carbon (O-benzyl substituent in 8vs. O-phenyl in precursors 1a–f).
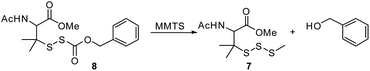 |
| | Scheme 4 RSSH generation from O-benzyl substituted RSSH precursor 8. | |
RSSH generation in the presence of thiol
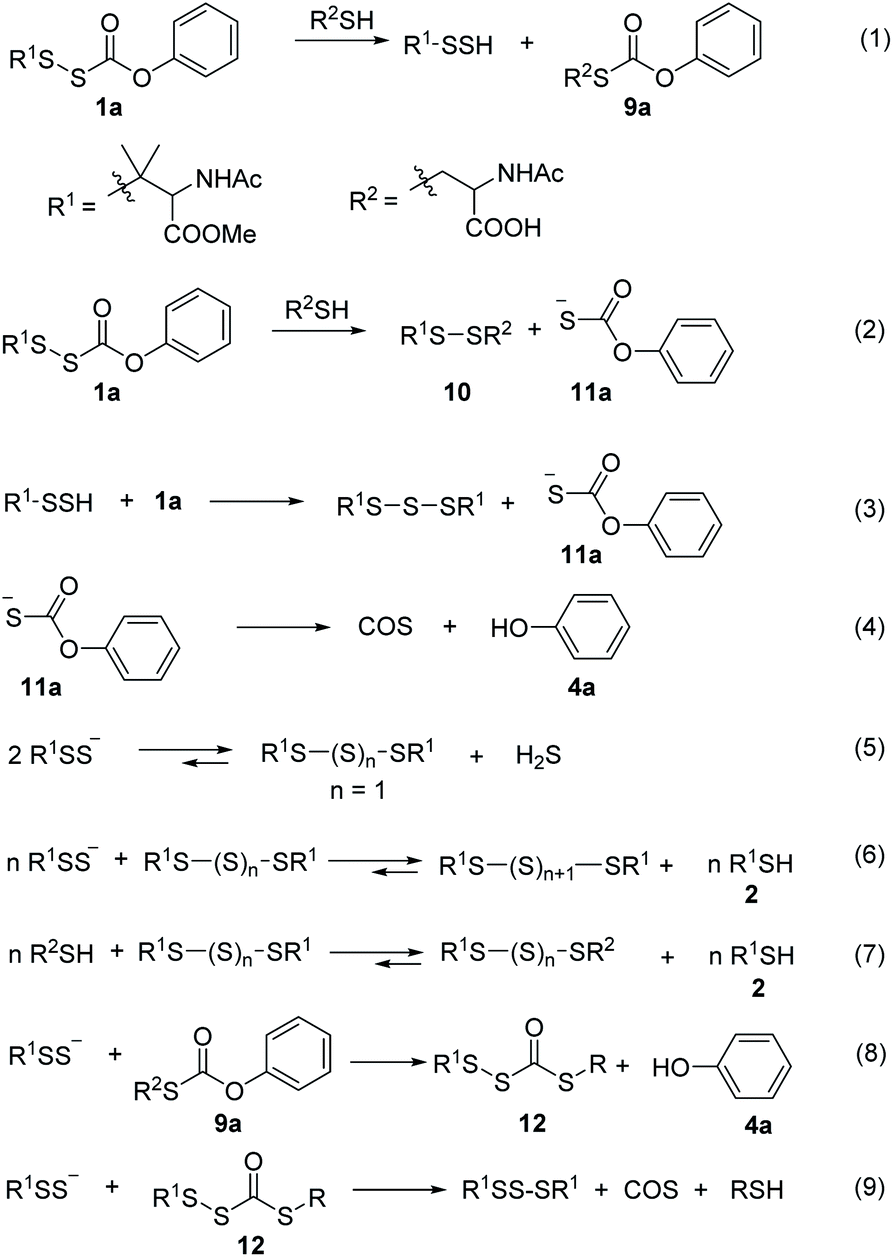
Next, we examined the ability of alkylsulfenyl thiocarbonate 1a to release RSSH in the presence of thiols. We anticipated that if thiol attacks at the carbonyl carbon of the thiocarbonate moiety, we should observe RSSH and/or RSSH-derived polysulfides, along with S-alkyl-thiocarbonate 9a (Scheme 5, eqn (1)). Alternatively, thiol can attack at the internal sulfur atom of the disulfide to produce unsymmetrical disulfide 10 and thiocarbonate intermediate 11a, which can rapidly decompose to release COS and phenol (Scheme 5, eqn (2) and (4)). If formed, COS will be hydrolyzed to H2S in biological systems by the ubiquitous enzyme carbonic anhydrase (CA).38
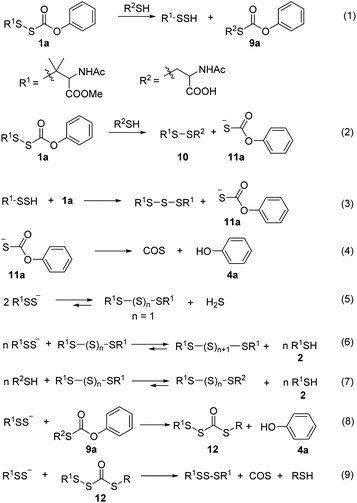 |
| | Scheme 5 Proposed mechanism of RSSH and COS generation from 1a in the presence of thiol. | |
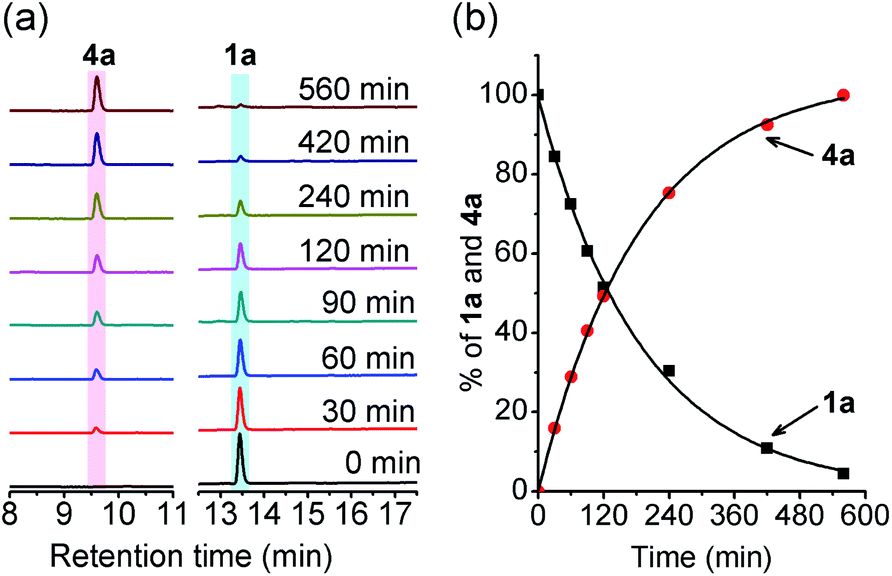
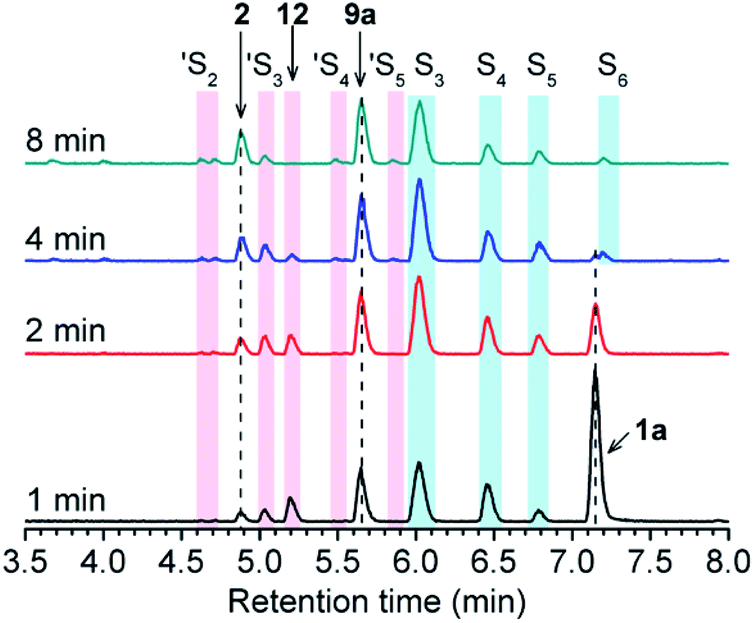
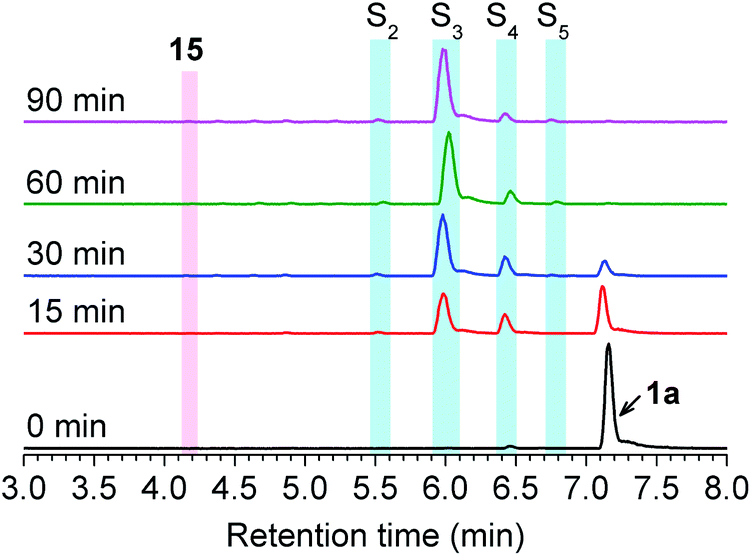
Initially, the decomposition of 1a was analyzed in the presence of 5 equiv. of N-acetylcysteine (NAC) in PBS (pH 7.4) by HPLC. Rapid decomposition of 1a is observed in the presence of NAC (t1/2 = 0.64 ± 0.11 min) (ESI,† Table 3), indicating a rapid reaction between NAC and 1a. To investigate the mechanism, we monitored this reaction by UPLC-MS. As shown in Fig. 3, disappearance of the peak attributed to 1a is observed with concomitant formation of symmetrical dialkyl polysulfides (cyan highlight, R1SSnSR1, n = 1–4), presumably formed by the decomposition of RSSH through disproportionation (Scheme 5, eqn (5) and (6)). Additionally, a small amount of unsymmetrical dialkyl polysulfide (pink highlight, R1SSnSR2, n = 0–3) is observed, likely formed via NAC reaction with symmetrical dialkyl polysulfides (Scheme 5, eqn (7)). As expected, a new peak at 5.65 min corresponding to N-acetyl cysteine-thiocarbonate 9a is also observed, confirming thiol attack at the perthiocarbonate carbonyl carbon of 1a to release RSSH. Due to the unstable nature of RSSH under these experimental conditions, we measured the yield of the byproduct 9a as an indication of RSSH yield. The observation of RSSH-derived polysulfides formation as major products led us to anticipate the efficient formation of byproduct 9a. However, incubation of 1a with 5 equiv. of N-acetylcysteine methyl ester shows only 53% formation of 9a. The lower observed yield of 9a is likely due to its further reaction with RSSH under these conditions. This hypothesis is supported by UPLC-MS data, where we observe appearance of a new peak at 5.2 min with m/z = 427.0659 (Fig. 3 and S12†). This peak is assigned to byproduct 12, potentially formed by RSSH reaction with 9a as shown in Scheme 5, eqn (8). Furthermore, disappearance of 12 is also observed, likely due to further reaction with RSSH producing dialkyl trisulfide and COS (Scheme 5, eqn (9)).
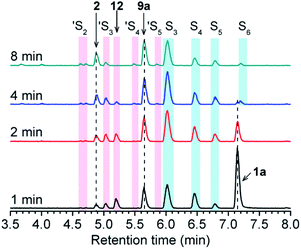 |
| | Fig. 3 UPLC-MS chromatograms of RSSH generation from 1a (100 μM) in the presence of NAC (500 μM) in pH 7.4 ammonium bicarbonate (50 mM) containing the metal chelator DTPA (100 μM) at 37 °C. Aliquots taken at various times were quenched with 1% formic acid, and analyzed by UPLC-MS. RSSH-derived symmetrical dialkyl polysulfide, labeled as S3 to S6 (R1SSnSR1, n = 1–4, cyan highlight), and unsymmetrical dialkyl polysulfides labeled as ′S2 to ′S5 (R1SSnSR2, n = 0–3, pink highlight) formation is evident. A peak at 5.65 min attributed to thiocarbonate 9a is also observed. | |
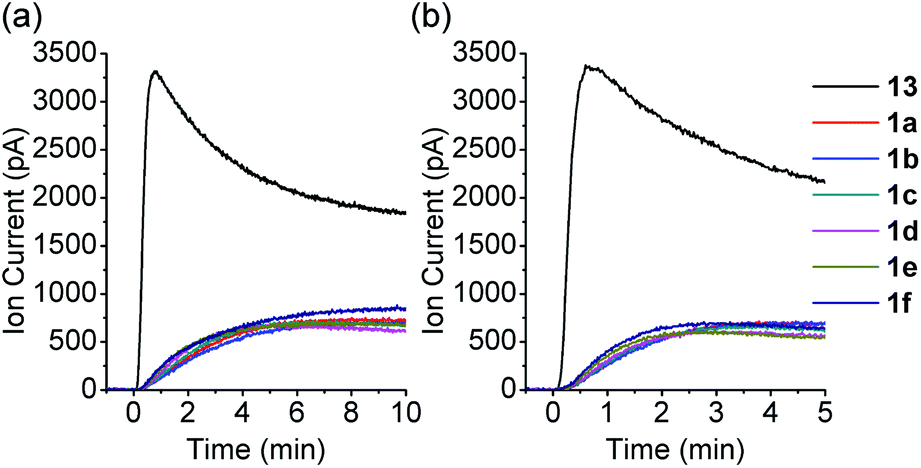
COS generation from 1a was analyzed in the presence of NAC by membrane inlet mass-spectrometry (MIMS), a technique used for detection of dissolved hydrophobic gases in aqueous solutions.39,40 For comparison, analogous experiments were conducted with the primary alkyl RSSH precursor 13 (Scheme 6). Thiol is expected to react with 13 selectively at the disulfide's internal sulfur atom due to its steric accessibility to produce unsymmetrical disulfide and thiocarbonate intermediate 9c, which subsequently decomposes to release COS (Scheme 6 and Fig. S34†). Comparison of COS release from 1a in the presence of NAC shows only approximately 20% COS production with respect to that observed for precursor 13 (Fig. 4a). Under these conditions, the observed COS signal is likely a result of both NAC and initially released RSSH reaction with 1a to produce thiocarbonate intermediate 11a (Scheme 5, eqn (2) and (3)). Taken together, these results demonstrate that 1a predominantly produces RSSH, even in the presence of thiols. We also examined COS release from precursors 1b–f in the presence of NAC. As shown in Fig. 4a, similar COS generation is observed for all precursors indicating electron donating/withdrawing groups do not have a major effect on COS release rates. The reactivity of these precursors toward GSH was also measured. As expected, a slightly faster COS release is observed in the presence of GSH (pKa = 8.83) compared with NAC (pKa = 9.52) (Fig. 4b), presumably due to a higher concentration of the glutathione thiolate under these conditions.
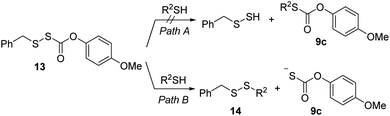 |
| | Scheme 6 Proposed Mechanism of 13 reaction with thiol. | |
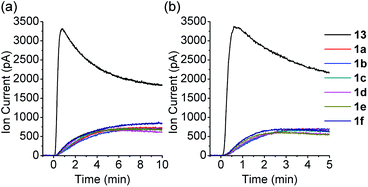 |
| | Fig. 4 COS MIMS signals at m/z 60 observed following the reaction of control compound 9 (50 μM) and RSSH precursors 1a–f (50 μM) with (a) NAC, and (b) GSH (0.25 mM, 5 equiv.) in PBS (pH 7.4, 10 mM) with DTPA (100 μM) at 37 °C. | |
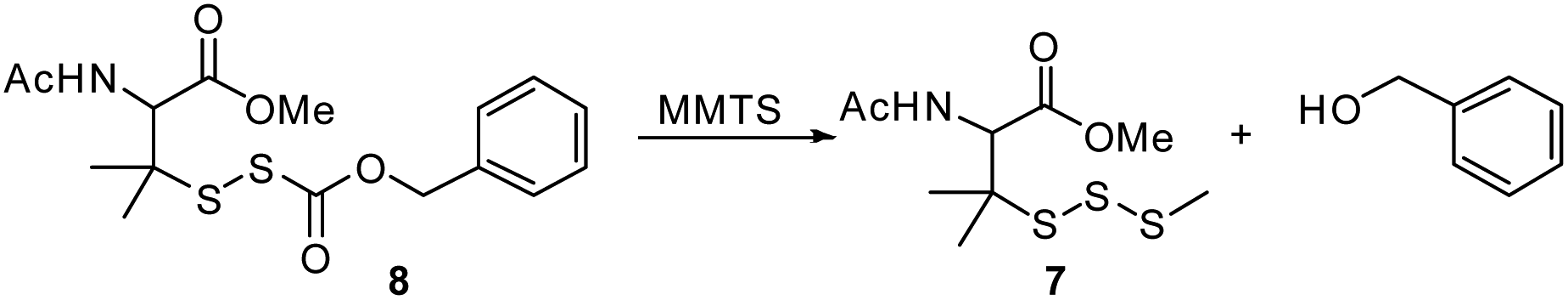
Reaction of O-benzyl substituted precursor 8 with NAC was also examined. As expected, a rapid decomposition of 8 is observed in the presence of thiol (t1/2 = 4.0 ± 0.7 min vs. 2235 min in the absence of NAC). With respect to 1a, 8 decomposes about six times slower in the presence of thiol. Similarly, slower COS generation is observed from precursor 8 in the presence of thiol compared to 1a (Fig. S36†). We also examined whether the reduced electrophilicity of the carbonyl carbon of precursor 8 favors thiol attack at the disulfide, which would preclude RSSH generation. However, UPLC-MS analysis shows R1SSnSR1 (n = 1–4) and R1SSnSR2 (n = 0–3) formation (Fig. S23†), indicating that 8 releases RSSH even in the presence of thiol. Thus, the results presented here confirm that alkylsulfenyl thiocarbonate thiolysis can be achieved under physiological conditions to release RSSH.
RSSH generation in the presence of amine
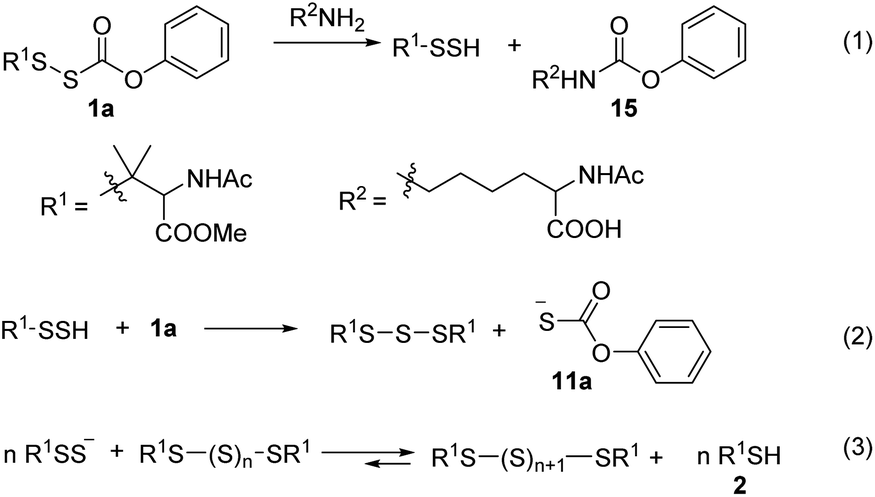
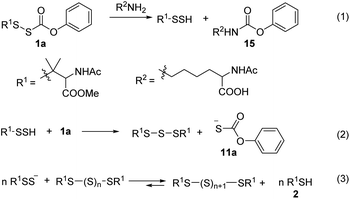
We tested the ability 1a to release RSSH in the presence of amines. We anticipated that if amine attacks at the carbonyl carbon of the thiocarbonate moiety, we should observe RSSH and/or RSSH-derived polysulfides, along with alkyl-carbamate 15 (Scheme 7, eqn (1)). Incubation of 1a with L-lysine (5 equiv.) shows disappearance of the peak attributed to 1a with concomitant formation of symmetrical dialkyl polysulfides (Fig. 5). However, only a very minor peak corresponding to expected byproduct 15 is observed, suggesting that RSSH release from 1a is mainly via hydrolysis even in the presence of amine.
 |
| | Scheme 7 Proposed Mechanism of 1a reaction with amine. | |
 |
| | Fig. 5 UPLC-MS chromatograms of RSSH generation from 1a (100 μM) in the presence of lysine (500 μM) in pH 7.4 ammonium bicarbonate (50 mM) containing the metal chelator DTPA (100 μM) at 37 °C. Aliquots taken at various times were quenched with 1% formic acid and analyzed by UPLC-MS. RSSH-derived symmetrical dialkyl polysulfide, labeled as S2 to S5 (R1SSnSR1, n = 0–3, cyan highlight), formation is evident. The very minor peak at 4.17 min corresponds to carbamate 15. | |
RSSH protection against oxidative stress
Oxidative stress has been implicated in the etiologies of many cardiovascular diseases.41,42 Oxidative stress usually arises from overproduction of reactive oxygen species (ROS). ROS constitutes both oxygen free radicals, such as superoxide, hydroxyl radicals, and peroxyl radicals, and non-radicals, such as hydrogen peroxide, and hypochlorous acid. It has been proposed that RSSH generation can be an endogenous cellular protectant in response to oxidative stress.17 Furthermore, it appears likely that the proposed mechanism of protection can be augmented pharmacologically with the addition of RSSH donor species.
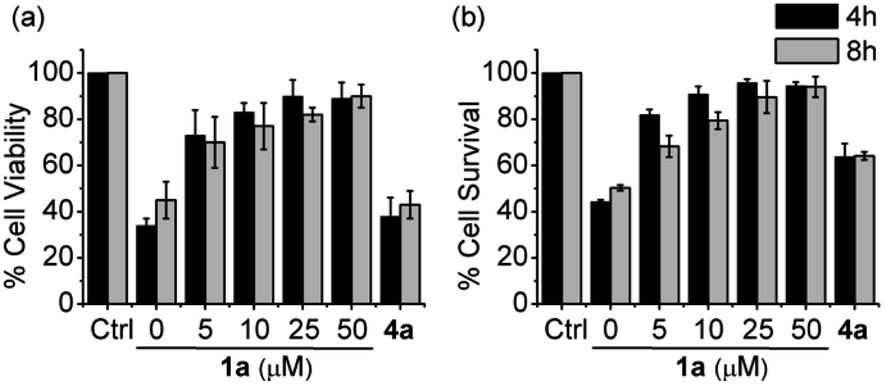
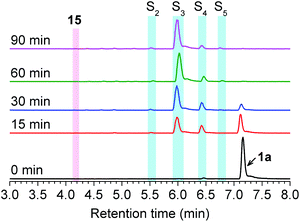
To test this hypothesis, we evaluated RSSH-mediated cellular protection against oxidative-stress. First, we examined the cytotoxicity of 1a on H9c2 cardiac myoblasts using the Sytox viability assay, which measures cell membrane integrity.43 Both precursor 1a and its byproduct phenol show no toxicity toward H9c2 cells up to 100 μM (Fig. S43†). We then measured the cytoprotective effects of 1a against oxidative stress. As expected, treatment of H9c2 cells with hydrogen peroxide (200 μM) results in reduced cell viability (Fig. 6a), measured using the CCK-8 viability assay, which measures cellular reducing capacity. Interestingly, pre-treating myoblasts with precursor 1a for 4 h results in a dose-dependent decrease of H2O2-induced toxicity (Fig. 6a). Remarkably, pre-treatment with only 5 μM 1a for 8 h also shows protection against 200 μM of H2O2, demonstrating the potency of these newly developed alkylsulfenyl thiocarbonate RSSH precursors against oxidative stress. Under similar conditions, the phenol byproduct shows no protective effect against H2O2-mediated toxicity, indicating that the protection is due to RSSH. In addition, we have measured the protective effects of thiol 2 in H9c2 cells. This thiol does indeed show some protection against H2O2-mediated toxicity, but to a much lesser extent compared with 1a (Fig. S44†), indicating important role of RSSH in cytoprotection against oxidative stress. The cytoprotective effect of 1a was also independently measured using the Sytox viability assay due to the potential background reduction of CCK-8 by reactive sulfur species leading to artifactual viability measurements.44 As shown in Fig. 6b, 1a consistently shows protective effects against H2O2-mediated toxicity. The protective effects against oxidative stress warrant future investigation as the potency of 1a suggests cytoprotective properties beyond direct scavenging of free-radical products derived from H2O2. Taken together, these results demonstrate the potential therapeutic benefit of RSSH donors in the context of oxidative stress and suggest that precursors that increase intracellular RSSH levels are potentially useful agents against oxidative stress-induced diseases.
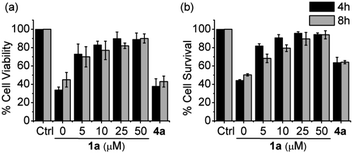 |
| | Fig. 6 Cell viability of H9c2 cardiac myoblasts pretreated with RSSH precursor 1a at 0, 5, 10, 25, and 50 μM and the byproduct phenol 4a at 50 μM for 4 and 8 h followed by exposure to H2O2 (200 μM) for 2 h. (a) Quantification of viability was carried out using Cell Counting Kit-8 (CCK-8). Results are expressed as the mean ± SEM (n = 5 for each treatment group) with three independent experiments. (b) Quantification of cytotoxicity was carried out using Sytox green nucleic acid stain. Results are expressed as the mean ± SEM (n = 5 for each treatment group) with five independent experiments. | |
Intracellular RSSH release
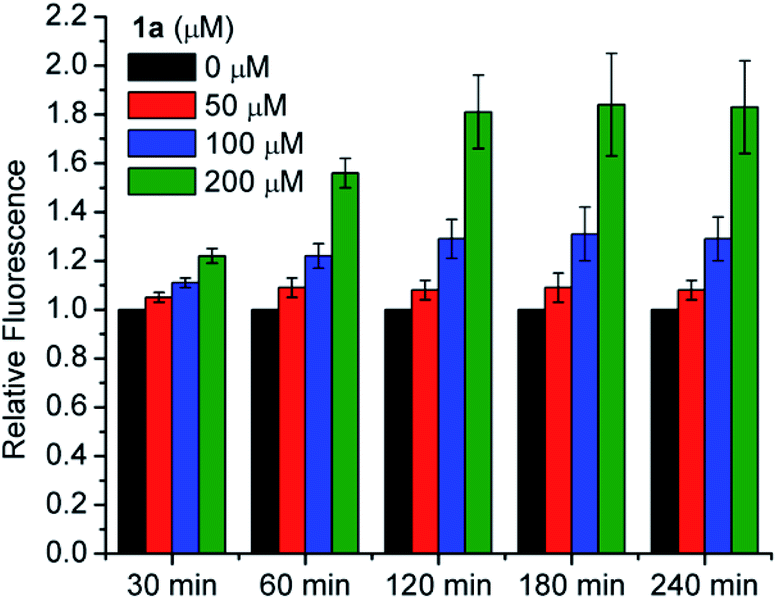
We also tested for increased intracellular RSSH levels resulting from incubation with 1a utilizing SSP4, a fluorescent probe that has been used to detect of intracellular sulfane sulfur.45,46 Sulfane sulfur refers to a sulfur atom with six valence electrons and no charge. Biologically important sulfane sulfur compounds include RSSH, polysulfides (RSnSH or RSSnSR, n ≥ 1), inorganic polysulfides (HSSnH, n ≥ 1), and elemental sulfur (S8). Briefly, cells were incubated with SSP4 in the presence of cetrimonium bromide (CTAB) for 20 min and then washed with serum-free media before 1a is introduced. We did not observe a significant increase of the SSP4 fluorescence signal following treatment with 50 μM 1a due to the low amount of RSSH released at this concentration falling below the detection limit of the probe (Fig. 7). However, dose-dependent increases in fluorescence intensity are observed for 100 and 200 μM 1a over a period of 3 h. Since the SSP4 probe is known to react with a variety of sulfane sulfur species, the fluorescence signal observed upon treatment of 1a is likely due to RSSH and/or other sulfane sulfur species that might be present in the equilibrium with RSSH (e.g., Scheme 5). We measured the sulfane sulfur in H9c2 cells treated with N-acetyl O-methyl cysteine trisulfide (N-Ac-OMe-Cys-(S)3) using the SSP4 probe and find that a higher fluorescence signal for N-Ac-OMe-Cys-(S)3 compared with 1a (Fig. S45†). However, sulfane sulfur detection under cell-free conditions also shows a much higher fluorescence signal intensity for Na2S3 and N-Ac-OMe-Cys-(S)3 compared with 1a (Fig. S46†). Based on these data we believe that 1a efficiently generates RSSH intracellularly, but reduced reactivity of the 1a-derived sterically hindered RSSH and/or dialkyl polysulfides towards SSP4 probe likely contributes to the observed lower fluorescence intensity. Thus, we conclude that the cytoprotective effects of 1a are attributed to increasing sulfane sulfur pools within the cultured cells.
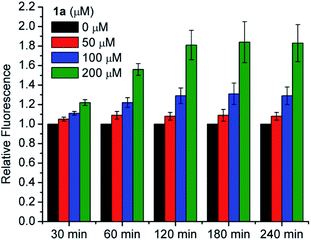 |
| | Fig. 7 Intracellular RSSH release in H9c2 cardiac myoblasts. H9c2 cells were pretreated with SSP4 (20 μM) and CTAB (500 μM) for 20 min, followed by incubation with RSSH precursor 1a at 0, 50, 100, 25, and 200 μM. Fluorescence intensity was measured at the indicated times using a plate reader at the excitation and emission wavelengths of 482 nm and 515 nm, respectively. Results are normalized to the 0 μM value at each time point and expressed as the mean ± SEM (n = 3 for each treatment group) with three independent experiments. | |
Conclusions
This study reports a new strategy for efficient RSSH generation based on the hydrolysis of alkylsulfenyl thiocarbonates under physiologically relevant conditions. We demonstrate that RSSH generation from these precursors can be tuned by modifying the thiocarbonate carbonyl group's electrophilicity. These alkylsulfenyl thiocarbonates also generate RSSH in the presence of thiol. As a proof-of-principle of possible protective effects associated with RSSH, we have demonstrated the potential benefit of these donors in the context of oxidative stress in H9c2 cardiac myoblasts. Furthermore, these precursors also increase intracellular sulfane sulfur levels in H9c2 cells. We hope that this series of RSSH precursors may be used as a research tool for delineating RSSH cell signaling mechanisms.
Author contributions
V. S. K carried out the synthesis, collected the spectral data, and performed UPLC-MS-based assays. S. C. A. performed the HPLC analysis to measure the kinetics of RSSH release. B. M. P performed the cell toxicity experiments, and intracellular sulfane sulfur analysis. J. P. T. and N. P. contributed to the conceptualization, discussion, manuscript writing, review, and editing.
Conflicts of interest
The authors declare that there is no conflict of interests.
Acknowledgements
We gratefully acknowledge the National Science Foundation (CHE-1900285 to J.P.T.) and the National Institutes of Health (T32 GM080189 for support of B.M.P. and R01 HL136918 to N.P.) for generous support for this research. We also thank Drs Christopher McGinity (National Cancer Institute) and Jon Fukuto (Sonoma State University) for advice on intracellular RSSH detection.
Notes and references
- L. Zhang, Y. Wang, Y. Li, L. Li, S. Xu, X. Feng and S. Liu, Front. Pharmacol., 2018, 9, 1066 CrossRef PubMed.
- H. Kimura, Molecules, 2014, 19, 16146–16157 CrossRef PubMed.
- L. A. Barr and J. W. Calvert, Circ. J., 2014, 78, 2111–2118 CrossRef CAS PubMed.
- J. M. Fukuto, L. J. Ignarro, P. Nagy, D. A. Wink, C. G. Kevil, M. Feelisch, M. M. Cortese-Krott, C. L. Bianco, Y. Kumagai, A. J. Hobbs, J. Lin, T. Ida and T. Akaike, FEBS Lett., 2018, 592, 2140–2152 CrossRef CAS PubMed.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72 Search PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- H. Kunikata, T. Ida, K. Sato, N. Aizawa, T. Sawa, H. Tawarayama, N. Murayama, S. Fujii, T. Akaike and T. Nakazawa, Sci. Rep., 2017, 7, 41984 CrossRef CAS PubMed.
- É. Dóka, I. Pader, A. Bíró, K. Johansson, Q. Cheng, K. Ballagó, J. R. Prigge, D. Pastor-Flores, T. P. Dick, E. E. Schmidt, E. S. J. Arnér and P. Nagy, Sci. Adv., 2016, 2, e1500968 CrossRef PubMed.
- S. Longen, F. Richter, Y. Köhler, I. Wittig, K.-F. Beck and J. Pfeilschifter, Sci. Rep., 2016, 6, 29808 CrossRef CAS PubMed.
- J. M. Fukuto, J. Lin, V. S. Khodade and J. P. Toscano, Antioxid. Redox Signaling, 2020, 33, 1295–1307 CrossRef CAS PubMed.
- P. K. Yadav, M. Martinov, V. Vitvitsky, J. Seravalli, R. Wedmann, M. R. Filipovic and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 289–299 CrossRef CAS PubMed.
- T. Akaike, T. Ida, F.-Y. Wei, M. Nishida, Y. Kumagai, M. M. Alam, H. Ihara, T. Sawa, T. Matsunaga, S. Kasamatsu, A. Nishimura, M. Morita, K. Tomizawa, A. Nishimura, S. Watanabe, K. Inaba, H. Shima, N. Tanuma, M. Jung, S. Fujii, Y. Watanabe, M. Ohmuraya, P. Nagy, M. Feelisch, J. M. Fukuto and H. Motohashi, Nat. Commun., 2017, 8, 1177 CrossRef PubMed.
- E. Cuevasanta, M. Lange, J. Bonanata, E. L. Coitiño, G. Ferrer-Sueta, M. R. Filipovic and B. Alvarez, J. Biol. Chem., 2015, 290, 26866–26880 CrossRef CAS PubMed.
- D. Benchoam, J. A. Semelak, E. Cuevasanta, M. Mastrogiovanni, J. S. Grassano, G. Ferrer-Sueta, A. Zeida, M. Trujillo, M. N. Möller, D. A. Estrin and B. Alvarez, J. Biol. Chem., 2020, 295, 15466–15481 CrossRef CAS PubMed.
- J. Zarenkiewicz, V. S. Khodade and J. P. Toscano, J. Org. Chem., 2021, 86, 868–877 CrossRef CAS PubMed.
- L. Álvarez, C. L. Bianco, J. P. Toscano, J. Lin, T. Akaike and J. M. Fukuto, Antioxid. Redox Signaling, 2017, 27, 622–633 CrossRef PubMed.
- C. L. Bianco, T. Akaike, T. Ida, P. Nagy, V. Bogdandi, J. P. Toscano, Y. Kumagai, C. F. Henderson, R. N. Goddu, J. Lin and J. M. Fukuto, Br. J. Pharmacol., 2019, 176, 671–683 CrossRef CAS PubMed.
- R. Greiner, Z. Pálinkás, K. Bäsell, D. Becher, H. Antelmann, P. Nagy and T. P. Dick, Antioxid. Redox Signaling, 2013, 19, 1749–1765 CrossRef CAS PubMed.
- S. Kasamatsu, A. Nishimura, M. Morita, T. Matsunaga, H. Abdul Hamid and T. Akaike, Molecules, 2016, 21 Search PubMed.
- S. Koike, S. Nishimoto and Y. Ogasawara, Redox Biol., 2017, 12, 530–539 CrossRef CAS PubMed.
- C.-t. Yang, N. O. Devarie-Baez, A. Hamsath, X.-d. Fu and M. Xian, Antioxid. Redox Signaling, 2019, 33, 1092–1114 CrossRef PubMed.
- P. Bora, P. Chauhan, S. Manna and H. Chakrapani, Org. Lett., 2018, 20, 7916–7920 CrossRef CAS.
- Y. Wang, K. M. Dillon, Z. Li, E. W. Winckler and J. B. Matson, Angew. Chem., Int. Ed., 2020, 59, 16698–16704 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon, Y. Wang, R. J. Carrazzone and J. B. Matson, Angew. Chem., Int. Ed., 2018, 57, 6324–6328 CrossRef CAS PubMed.
- J. Kang, S. Xu, M. N. Radford, W. Zhang, S. S. Kelly, J. J. Day and M. Xian, Angew. Chem., Int. Ed., 2018, 57, 5893–5897 CrossRef CAS PubMed.
- I. Artaud and E. Galardon, ChemBioChem, 2014, 15, 2361–2364 CrossRef CAS PubMed.
- V. S. Khodade and J. P. Toscano, J. Am. Chem. Soc., 2018, 140, 17333–17337 CrossRef CAS PubMed.
- V. S. Khodade, B. M. Pharoah, N. Paolocci and J. P. Toscano, J. Am. Chem. Soc., 2020, 142, 4309–4316 CrossRef CAS PubMed.
- A. Chaudhuri, Y. Venkatesh, J. Das, M. Gangopadhyay, T. K. Maiti and N. D. P. Singh, J. Org. Chem., 2019, 84, 11441–11449 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, Z. Li, Z. Yuan, C. L. Organ, R. K. Trivedi, S. Wang, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 11749–11753 CrossRef CAS PubMed.
- Z. Yuan, Y. Zheng, B. Yu, S. Wang, X. Yang and B. Wang, Org. Lett., 2018, 20, 6364–6367 CrossRef CAS PubMed.
- S. J. Brois, J. F. Pilot and H. W. Barnum, J. Am. Chem. Soc., 1970, 92, 7629–7631 CrossRef CAS.
- Y. Zhao, M. M. Cerda and M. D. Pluth, Chem. Sci., 2019, 10, 1873–1878 RSC.
- D. N. Harpp and A. Granata, Tetrahedron Lett., 1976, 17, 3001–3004 CrossRef.
- D. N. Harpp and A. Granata, J. Org. Chem., 1979, 44, 4144–4148 CrossRef CAS.
- J. Pan and K. S. Carroll, ACS Chem. Biol., 2013, 8, 1110–1116 CrossRef CAS PubMed.
- C. P. Chengelis and R. A. Neal, Toxicol. Appl. Pharmacol., 1980, 55, 198–202 CrossRef CAS PubMed.
- G. Hoch and B. Kok, Arch. Biochem. Biophys., 1963, 101, 160–170 CrossRef CAS PubMed.
- M. R. Cline, C. Tu, D. N. Silverman and J. P. Toscano, Free Radical Biol. Med., 2011, 50, 1274–1279 CrossRef CAS PubMed.
- N. S. Dhalla, R. M. Temsah and T. Netticadan, J. Hypertens., 2000, 18, 655–673 CrossRef CAS PubMed.
- R. D'Oria, R. Schipani, A. Leonardini, A. Natalicchio, S. Perrini, A. Cignarelli, L. Laviola and F. Giorgino, Oxid. Med. Cell. Longevity, 2020, 2020, 5732956 Search PubMed.
- L. J. Jones and V. L. Singer, Anal. Biochem., 2001, 293, 8–15 CrossRef CAS PubMed.
- H. Tominaga, M. Ishiyama, F. Ohseto, K. Sasamoto, T. Hamamoto, K. Suzuki and M. Watanabe, Anal. Commun., 1999, 36, 47–50 RSC.
- W. Chen, C. Liu, B. Peng, Y. Zhao, A. Pacheco and M. Xian, Chem. Sci., 2013, 4, 2892–2896 RSC.
- E. Marutani, M. Sakaguchi, W. Chen, K. Sasakura, J. Liu, M. Xian, K. Hanaoka, T. Nagano and F. Ichinose, MedChemComm, 2014, 5, 1577–1583 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01550h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a