
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent structural evolution of lactam- and imide-functionalized polymers applied in organic field-effect transistors and organic solar cells
Yankai
Zhou
ab,
Weifeng
Zhang
*a and
Gui
Yu
 *ab
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Research/Education Centre for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: yugui@iccas.ac.cn; zhangwf@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 7th May 2021
Abstract
Organic semiconductor materials, especially donor–acceptor (D–A) polymers, have been increasingly applied in organic optoelectronic devices, such as organic field-effect transistors (OFETs) and organic solar cells (OSCs). Plenty of high-performance OFETs and OSCs have been achieved based on varieties of structurally modified D–A polymers. As the basic building block of D–A polymers, acceptor moieties have drawn much attention. Among the numerous types, lactam- and imide-functionalized electron-deficient building blocks have been widely investigated. In this review, the structural evolution of lactam- or imide-containing acceptors (for instance, diketopyrrolopyrrole, isoindigo, naphthalene diimide, and perylene diimide) is covered and their representative polymers applied in OFETs and OSCs are also discussed, with a focus on the effect of varied structurally modified acceptor moieties on the physicochemical and photoelectrical properties of polymers. Additionally, this review discusses the current issues that need to be settled down and the further development of new types of acceptors. It is hoped that this review could help design new electron-deficient building blocks, find a more valid method to modify already reported acceptor units, and achieve high-performance semiconductor materials eventually.
 Yankai Zhou | Yankai Zhou received his M.S. degree from the University of Science and Technology, Beijing in 2019. He is presently a PhD student in the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). His current research is focused on pi-conjugated organic semiconductors. |
 Weifeng Zhang | Weifeng Zhang received his PhD from Hong Kong Baptist University in 2011 under the supervision of Professor M. S. Wong. Thereafter, he joined the key laboratory of organic solids at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) as a postdoctoral researcher. He was promoted to Associate Professor in 2015 at ICCAS. His research interests include the syntheses, characterization, and application of novel organic/polymeric semiconducting materials. |
 Gui Yu | Gui Yu is currently a professor at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). He graduated from Jilin University in 1988 and received his M.S. (1993) and PhD (1997) degrees from the Changchun Institute of Physics, Chinese Academy of Sciences. After obtaining his PhD, he worked as a postdoctoral fellow at ICCAS. Thereafter, he was appointed to the position of Associate Professor in 1999 and promoted to Professor in 2008. His current research mainly focuses on the syntheses and optoelectronic devices of organic/polymeric semiconducting materials, graphene, and covalent organic frameworks. |
1. Introduction
Since chlorine-doped aromatic carbohydrate films were firstly found to be conductive in the 1950s,1,2 organic semiconducting materials have attracted a large amount of interest and attention from researchers around the world. In 1986, the first organic field-effect transistor (OFET) based on polythiophene was fabricated, from then on a large number of organic semiconducting materials, possessing outstanding properties and novel chemical structures, have been designed and synthesized.3 In recent decades, the strategy of combining electron-rich (donor) and electron-deficient (acceptor) units alternately together has become increasingly popular and has been applied to obtain all kinds of D–A type polymeric semiconductors. As is well known, the structure of the acceptor unit has a great influence on its corresponding polymer, no matter physicochemical properties or optoelectronic performances. Therefore, the design of novel acceptors and their structural optimization have received considerable attention. In recent years, many structurally varied acceptors were synthesized and numerous high-performance semiconducting polymers were reported. Among the well-known electron-deficient units and the corresponding polymers lactam- and imide-based acceptors and corresponding polymers have been attracting the attention of scientists until now, due to their excellent optoelectronic properties and wide applications in high-performance OFETs and organic solar cells (OSCs).Lactam is a cyclic amide, which consists of a carbonyl group adjacent to an amino group. Imide has the same structure with one more carbonyl group connected to the amino group. These two groups were increasingly used to construct acceptors for high-performance semiconducting polymers, which can be explained by the following points: firstly, the nitrogen atom in the structure adopts sp2 hybridization, with its p-orbital conjugation with the π-orbital from the adjacent carbonyl group. The formed p–π conjugation facilitates evenly distributed electron density and better planarity, both of which could positively impact the polymer properties.4–7 Secondly, constructing acceptor units with strong electron-withdrawing lactam or imide groups provides the molecules and polymers with low-lying energy levels of the lowest unoccupied molecular orbital (LUMO), thus promoting electron injection efficiency and enhancing the device air stability.8–10 Thirdly, a variety of side chains can be substituted on the amide nitrogen atom to adjust the polymer solubility, film morphology, and the self-assembly of the side chain.11–13 Some functional side chains were also incorporated into polymers to fabricate devices with a specific function.14–16 In recent years, numerous lactam- and imide-functionalized polymers with high carrier mobilities have been reported. Considering the mobility of silicon FETs, which is around 1 cm2 V−1 s−1, the ultra-high hole mobility of over 14.0 cm2 V−1 s−1 and the balanced hole/electron mobilities around 6.5 cm2 V−1 s−1 achieved by polymer semiconductors have made considerable progress. Despite that some of these preferable performances were probably extracted from the saturation region, they are still valid tools to assess the application potential of different polymers. Besides, the PCE values of OSCs were improved to 14%, even higher. All the above impressive advances are made by those lactam or imide-containing polymers.
In this progress review, we first summarize the recent structural evolution of classic lactam-containing electron-deficient building blocks, mainly focus on diketopyrrolopyrrole (DPP) and isoindigo (IID) units, and investigate how such modification methods eventually lead to the different physicochemical properties and optoelectronic performances of their corresponding polymers. Then, various imide-functionalized acceptors (naphthalene diimide (NDI), perylene diimide (PDI), etc.) and polymers with modified molecular structures are also covered. Finally, we discuss the development strategy and feasible optimization methods for current acceptors and polymers, followed by the current issues we are meeting and the future development of analogous molecules.
2. Polymers with lactam moieties
As the commonly used structural building block, the lactam moiety is famous for its strong electron-withdrawing nature and changeable substituted sidechains. Owing to their outstanding features, lactam-functionalized acceptor building blocks have been widely used in obtaining high-performance small molecules and polymer semiconductors. Among the numerous building blocks, diketopyrrolopyrrole (DPP) and isoindigo (IID) units are the most representative lactam-containing acceptor units. It is convenient to regulate their molecular solubility and electron deficiency because of their adjustable molecular frameworks and less modification complexity.17–20 Therefore, those two building blocks have developed rapidly in recent years. A large number of derivatives and analogs of DPP and IID units are designed and numerous high-performance DPP- and IID-based polymers have been reported in recent two decades, which we introduce below.2.1. DPP-based polymers
As a popular molecule in pigment chemistry, DPP contains two lactam groups and has been increasingly applied in the field of organic electronics during the past several years.21–23 Multiple polymeric materials have been designed and synthesized based on the DPP unit, due to its proper frontier molecular orbital (FMO) energy levels and its adjustable molecular structure. To sum up, there are two main ways to construct a new type of electron-accepting DPP-based unit. One is the introduction of flanked aromatic rings on both sides of the DPP core according to the methodology proposed by Rochat and co-workers,24 such as thiophene, furan, benzene, and other aromatic units, for improved molecular planarity and electron deficiency. The other strategy is adjusting the molecular structure of the DPP core, no matter to increase the conjugation length or to change the way of N-substitution. Such methodologies are beneficial to obtain varieties of high-performance polymers containing DPP or its multiple derivatives. Accordingly, there are mainly three types of structures for DPP derivatives, (i) having symmetrical flanking units; (ii) having asymmetrical flanking units; (iii) having modified DPP core structures.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC61BM was 1:3. After optimization with an additional 9% 1-chloronaphthalene, the PCE of P4(P5)/PC71BM was improved and the max PCE reached 5.0% (Fig. 2a) due to its fiber-like interpenetrating morphologies (Fig. 2b). Moreover, another furan-DPP-based polymer P6 achieved balanced ambipolar hole and electron mobilities, with the PCE value of the OSC device still reaching 3.7%.30 By changing the donor building block to the dithienothiophene unit, polymer P7 gave an impressive enhancement in the hole mobility of 3.56 cm2 V−1 s−1, due to its good crystallinity and appropriate interlayer distance (Fig. 2c).31 Besides, polymer P8, containing furan-DPP and (E)-1,2-di-2-thienylethylene units, showed improved OSC performance in comparison with that of P6.32 It was found that the P8:PC71BM blend film afforded the max photovoltaic performance when the weight ratio was 1:4 (Fig. 2d), with a PCE of 4.56%. Besides the furan ring, the selenophene ring was also chosen as the bridge unit of the DPP core. The high polarizability of the selenophene atom is prone to form the Se⋯O intramolecular interaction in the DPP molecule, thus facilitating high molecular planarity. Shahid and co-workers prepared two polymers P9 and P10.33 Both the polymers showed narrower optical band gaps than that of thiophene-DPP-based polymers, facilitating the achievement of balanced electron and hole mobilities of 0.1 cm2 V−1 s−1. By changing the donor to a fluorinated bithiophene unit, Liu and co-workers synthesized the selenophene-DPP-based polymer (P11) with a lowered HOMO energy level.34 The easier hole carrier injection gave a much improved hole mobility of 0.95 cm2 V−1 s−1 compared with the hole mobilities of P9 and P10. Besides, the additional NMe4I contributed a higher hole mobility of up to 1.51 cm2 V−1 s−1 under ambient conditions, indicating its further potential in better FET performance. It is well known that most of the asymmetric five-membered bridges, such as furan, thiophene, and selenophene flanked in the above DPP units, are more electron-rich compared with six-membered aromatic rings. Therefore, synthesizing DPP bridged with six-membered aromatic rings could possibly enhance electron deficiency. The benzene ring, in this regard, has the potential to make a difference. As shown in Fig. 1, polymer P12 was prepared with phenyl-flanked DPP.35 In comparison with thiophene-flanked counter polymers, the electron deficiency of phenyl rings provided P12 with lower HOMO/LUMO energy levels and smaller band gaps, which led to a PCE of 1.51% based on the P12:PCBM blend film. Research on phenyl-DPP-based polymers was limited in the amount just because of its large dihedral angles between the benzene ring and DPP core. Such nonplanar conformation, to some extent, limited the application of such polymers. To relieve the above-mentioned steric effect, the nitrogen atom was embedded in the flanking units. Jung and co-workers reported polymer P13 based on pyridine-flanked DPP (PyDPP).36 Since the increased electron deficiency of PyDPP was induced by electron-withdrawing nitrogen atoms, P13 showed lower HOMO/LUMO energy levels than those of thiophene- and phenyl-bridged DPP units (Fig. 3a). A PCE of 4.9% with a high open-circuit voltage (Voc) up to 0.92 V was achieved by the P13:PC71BM blend film. The PCE value was much higher than that of P12. On account of the alleviated repulsive interaction and improved π-conjugation of the molecular backbone owing to the intramolecular N⋯H hydrogen bond, P13-based devices also exhibited excellent ambipolar transport performance with the max hole and electron mobilities of 2.78 and 6.30 cm2 V−1 s−1, respectively.37 Such preferable performance was attributed to the π-spacing distance of 3.60 for P13, which was much shorter than that of its thiophene-based counter polymer, thereby favoring the intermolecular hopping of the charge carrier and eventually achieving higher mobilities. More recently, Liu and co-workers synthesized another pyridine-DPP-based polymer P14, possessing deep-lying FMO energy levels.38 The resulting easier electron injection and increased hole carrier injection barrier of P14 successfully led to its electron dominant transport performance with a μe of 2.22 cm2 V−1 s−1. Compared with the pyridine unit, the pyrazine unit has two nitrogen atoms substituted on the para-position of the benzene ring, which could further draw down the FMO energy levels of the polymer, i.e., P15, which was constructed on the pyrazine-bridged DPP (PzDPP) unit.39 Apart from the much-lowered FMO energy levels, P15 featured a suppressed backbone distortion due to the formed multiple hydrogen bonds. Unlike other DPP derivatives, most of which showed hole dominant ambipolar transport performances, P15 exhibited unipolar n-type performance with its electron mobility reaching 1.67 cm2 V−1 s−1. Moreover, they tested the electrical conductivity of P15 and gave a high value of 8.4 S cm−1 with its power factor reaching 57.3 μW m−1 K−2. Considering that the electrical conductivities of most n-doped polymers were around 1 S cm−1 with their power factors of no more than 10 μW m−1 K−2, the pyrazine-based polymer P15 exhibited an impressive enhancement. The nitrogen embedding strategy was not only applied in the benzene ring, the thiophene unit could be similarly modified as well. The thiazole-based DPP in polymer P16 helped achieve electron-dominant performance by adjusting its FMO energy levels and interchain packing.40 Besides, the P16/PDPP5T blend film also afforded a PCE of 2.9%. Such a PCE value was improved by another thiazole-DPP-based polymer P17, which had a low photon energy loss, thus exhibiting a PCE up to 5.6%, with a low energy loss of less than 0.6 eV after blending with PC71BM.41 Shortly afterward, Chen and co-workers synthesized three polymers (P18, P19, and P20) with different side chains.42 The pyridine-flanked DPP could address the limitation of large dihedral angles and provided a more electron-deficient condition. Therefore, high and balanced ambipolar charge transport properties were achieved by P18-P20, whose highest electron and hole mobilities reached 1.14 and 1.46 cm2 V−1 s−1, respectively. As described above, all the bridged moieties of DPP units are aromatic single rings. Besides, fused rings with extended conjugation length provided the polymer rigid backbone with better planarity and delocalized FMO distribution along the polymer main chain. Bronstein and co-workers extended the thiophene flanking unit to thieno[3,2-b]thiophene to increase the intermolecular association and synthesized the P21.43 Such extension contributed to the evenly distributed HOMO energy level and facilitated intermolecular charge carrier hopping. Eventually, ambipolar performance was achieved, with hole and electron mobilities of 1.95 and 0.07 cm2 V−1 s−1, respectively. And the OSC device could also afford a PCE of 5.4%. To further increase the polymer electron affinity (EA) and reduce the steric hindrance between the flanking unit and DPP core, polymer P22 was synthesized with the thieno[2,3-b]pyridine-bridged DPP (TPDPP) unit.44 The single-crystal structure of the TPDPP molecule substituted with two short 2-ethylhexyl side chains showed a small dihedral angle of 7.1° between the core and bridges, which facilitated the appropriate planarity of P22 (Fig. 3b). Such a planar molecular backbone ensured good main-chain conjugation and the delocalization of π-electrons. The P22/PTB7-Th blend film showed a PCE value of 2.72% with the open-circuit voltage up to 1.04 V. With the replacement of thiophene by a benzene ring, P23 and P24 were synthesized based on the more electron-deficient quinoline-flanked DPP monomer (QDPP).45 The additional fused benzene ring provided P23 and P24 with extended π-conjugation lengths and shorter π–π stacking distances, which invited ambipolar transport performances for both the polymers. The flexible FET devices on PET substrates (Fig. 3c) of P23 exhibited balanced hole/electron mobilities of around 0.5–0.7 cm2 V−1 s−1. For polymer P24, the electron mobility reached 6.04 cm2 V−1 s−1 (Table 1). Moreover, the ambipolar mobilities of P23 and P24 were much higher than those of their pyridine-based counter polymers (Fig. 3d).
:PC61BM was 1:3. After optimization with an additional 9% 1-chloronaphthalene, the PCE of P4(P5)/PC71BM was improved and the max PCE reached 5.0% (Fig. 2a) due to its fiber-like interpenetrating morphologies (Fig. 2b). Moreover, another furan-DPP-based polymer P6 achieved balanced ambipolar hole and electron mobilities, with the PCE value of the OSC device still reaching 3.7%.30 By changing the donor building block to the dithienothiophene unit, polymer P7 gave an impressive enhancement in the hole mobility of 3.56 cm2 V−1 s−1, due to its good crystallinity and appropriate interlayer distance (Fig. 2c).31 Besides, polymer P8, containing furan-DPP and (E)-1,2-di-2-thienylethylene units, showed improved OSC performance in comparison with that of P6.32 It was found that the P8:PC71BM blend film afforded the max photovoltaic performance when the weight ratio was 1:4 (Fig. 2d), with a PCE of 4.56%. Besides the furan ring, the selenophene ring was also chosen as the bridge unit of the DPP core. The high polarizability of the selenophene atom is prone to form the Se⋯O intramolecular interaction in the DPP molecule, thus facilitating high molecular planarity. Shahid and co-workers prepared two polymers P9 and P10.33 Both the polymers showed narrower optical band gaps than that of thiophene-DPP-based polymers, facilitating the achievement of balanced electron and hole mobilities of 0.1 cm2 V−1 s−1. By changing the donor to a fluorinated bithiophene unit, Liu and co-workers synthesized the selenophene-DPP-based polymer (P11) with a lowered HOMO energy level.34 The easier hole carrier injection gave a much improved hole mobility of 0.95 cm2 V−1 s−1 compared with the hole mobilities of P9 and P10. Besides, the additional NMe4I contributed a higher hole mobility of up to 1.51 cm2 V−1 s−1 under ambient conditions, indicating its further potential in better FET performance. It is well known that most of the asymmetric five-membered bridges, such as furan, thiophene, and selenophene flanked in the above DPP units, are more electron-rich compared with six-membered aromatic rings. Therefore, synthesizing DPP bridged with six-membered aromatic rings could possibly enhance electron deficiency. The benzene ring, in this regard, has the potential to make a difference. As shown in Fig. 1, polymer P12 was prepared with phenyl-flanked DPP.35 In comparison with thiophene-flanked counter polymers, the electron deficiency of phenyl rings provided P12 with lower HOMO/LUMO energy levels and smaller band gaps, which led to a PCE of 1.51% based on the P12:PCBM blend film. Research on phenyl-DPP-based polymers was limited in the amount just because of its large dihedral angles between the benzene ring and DPP core. Such nonplanar conformation, to some extent, limited the application of such polymers. To relieve the above-mentioned steric effect, the nitrogen atom was embedded in the flanking units. Jung and co-workers reported polymer P13 based on pyridine-flanked DPP (PyDPP).36 Since the increased electron deficiency of PyDPP was induced by electron-withdrawing nitrogen atoms, P13 showed lower HOMO/LUMO energy levels than those of thiophene- and phenyl-bridged DPP units (Fig. 3a). A PCE of 4.9% with a high open-circuit voltage (Voc) up to 0.92 V was achieved by the P13:PC71BM blend film. The PCE value was much higher than that of P12. On account of the alleviated repulsive interaction and improved π-conjugation of the molecular backbone owing to the intramolecular N⋯H hydrogen bond, P13-based devices also exhibited excellent ambipolar transport performance with the max hole and electron mobilities of 2.78 and 6.30 cm2 V−1 s−1, respectively.37 Such preferable performance was attributed to the π-spacing distance of 3.60 for P13, which was much shorter than that of its thiophene-based counter polymer, thereby favoring the intermolecular hopping of the charge carrier and eventually achieving higher mobilities. More recently, Liu and co-workers synthesized another pyridine-DPP-based polymer P14, possessing deep-lying FMO energy levels.38 The resulting easier electron injection and increased hole carrier injection barrier of P14 successfully led to its electron dominant transport performance with a μe of 2.22 cm2 V−1 s−1. Compared with the pyridine unit, the pyrazine unit has two nitrogen atoms substituted on the para-position of the benzene ring, which could further draw down the FMO energy levels of the polymer, i.e., P15, which was constructed on the pyrazine-bridged DPP (PzDPP) unit.39 Apart from the much-lowered FMO energy levels, P15 featured a suppressed backbone distortion due to the formed multiple hydrogen bonds. Unlike other DPP derivatives, most of which showed hole dominant ambipolar transport performances, P15 exhibited unipolar n-type performance with its electron mobility reaching 1.67 cm2 V−1 s−1. Moreover, they tested the electrical conductivity of P15 and gave a high value of 8.4 S cm−1 with its power factor reaching 57.3 μW m−1 K−2. Considering that the electrical conductivities of most n-doped polymers were around 1 S cm−1 with their power factors of no more than 10 μW m−1 K−2, the pyrazine-based polymer P15 exhibited an impressive enhancement. The nitrogen embedding strategy was not only applied in the benzene ring, the thiophene unit could be similarly modified as well. The thiazole-based DPP in polymer P16 helped achieve electron-dominant performance by adjusting its FMO energy levels and interchain packing.40 Besides, the P16/PDPP5T blend film also afforded a PCE of 2.9%. Such a PCE value was improved by another thiazole-DPP-based polymer P17, which had a low photon energy loss, thus exhibiting a PCE up to 5.6%, with a low energy loss of less than 0.6 eV after blending with PC71BM.41 Shortly afterward, Chen and co-workers synthesized three polymers (P18, P19, and P20) with different side chains.42 The pyridine-flanked DPP could address the limitation of large dihedral angles and provided a more electron-deficient condition. Therefore, high and balanced ambipolar charge transport properties were achieved by P18-P20, whose highest electron and hole mobilities reached 1.14 and 1.46 cm2 V−1 s−1, respectively. As described above, all the bridged moieties of DPP units are aromatic single rings. Besides, fused rings with extended conjugation length provided the polymer rigid backbone with better planarity and delocalized FMO distribution along the polymer main chain. Bronstein and co-workers extended the thiophene flanking unit to thieno[3,2-b]thiophene to increase the intermolecular association and synthesized the P21.43 Such extension contributed to the evenly distributed HOMO energy level and facilitated intermolecular charge carrier hopping. Eventually, ambipolar performance was achieved, with hole and electron mobilities of 1.95 and 0.07 cm2 V−1 s−1, respectively. And the OSC device could also afford a PCE of 5.4%. To further increase the polymer electron affinity (EA) and reduce the steric hindrance between the flanking unit and DPP core, polymer P22 was synthesized with the thieno[2,3-b]pyridine-bridged DPP (TPDPP) unit.44 The single-crystal structure of the TPDPP molecule substituted with two short 2-ethylhexyl side chains showed a small dihedral angle of 7.1° between the core and bridges, which facilitated the appropriate planarity of P22 (Fig. 3b). Such a planar molecular backbone ensured good main-chain conjugation and the delocalization of π-electrons. The P22/PTB7-Th blend film showed a PCE value of 2.72% with the open-circuit voltage up to 1.04 V. With the replacement of thiophene by a benzene ring, P23 and P24 were synthesized based on the more electron-deficient quinoline-flanked DPP monomer (QDPP).45 The additional fused benzene ring provided P23 and P24 with extended π-conjugation lengths and shorter π–π stacking distances, which invited ambipolar transport performances for both the polymers. The flexible FET devices on PET substrates (Fig. 3c) of P23 exhibited balanced hole/electron mobilities of around 0.5–0.7 cm2 V−1 s−1. For polymer P24, the electron mobility reached 6.04 cm2 V−1 s−1 (Table 1). Moreover, the ambipolar mobilities of P23 and P24 were much higher than those of their pyridine-based counter polymers (Fig. 3d).
| Polymers | UV-vis λsolmax/λfilmmax (nm) | HOMO/LUMOa (eV) | Band Gap (eV) Eoptg/Eecg | μ h/μe (cm2 V−1 s−1) | Max PCE (%) | Device configuration OFET (OSCs) | Ref. |
|---|---|---|---|---|---|---|---|
| a The frontier energy levels calculated from the cyclic voltammetry or UPS. b The performance of the polymer having additives. c The mobilities extracted in the linear region. | |||||||
| P1 | — | −5.17/−3.61 | 1.40/1.56 | 0.04/0.01 | 4.70 | BGBC | 26 |
| P2 | 838/857 | −5.07/— | — | 3.10/— | — | TGBC | 27 |
| P3 | — | — | — | 2.1(26.2b)/— | — | BGBC | 28 |
| P4 | —/789 | −5.40/−3.80 | 1.41/1.60 | — | 3.80(5.00b) | (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 29 |
| P5 | —/767 | −5.50/−3.80 | 1.35/1.70 | — | 3.00(4.10b) | (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 29 |
| P6 | — | — | 1.59/1.66 | 3 × 10−3/7 × 10−3 | 3.70 | BGBC | 30 |
| P7 | 782/782 | −5.36/— | 1.37/— | 3.56/— | — | BGTC | 31 |
| P8 | —/796 | −5.19/−3.63 | 1.56/— | — | 4.56 | (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 32 |
| P9 | 827/882 | −5.20/−4.02 | 1.18/— | 0.10/0.10 | — | BGBC | 33 |
| P10 | 823/864 | −5.10/−3.92 | 1.21/— | 0.26/0.05 | — | BGBC | 33 |
| P11 | 790/836 | −5.30/−3.97 | 1.33/— | 0.95(1.51b)/— | — | BGTC | 34 |
| P12 | 539/563 | −5.47/−3.74 | 1.70/1.73 | — | 1.51 | (ITO/polymer:PCBM/Al) | 35 |
| P13 | 664/684 | −5.77/−3.86 | 1.71/1.91 | — | 4.90 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 36 |
| P14 | 654/672 | −5.75/−4.03 | 1.72/— | 0.58/2.22 | — | TGBC | 38 |
| P15 | 713/— | −5.89/−4.03 | 1.86/— | —/1.67 | — | TGBC | 39 |
| P16 | — | −5.63/−4.00 | 1.44/1.63 | 6 × 10−3/0.13 | 2.90 | TGBC (ITO/ZnO/PDPP5T:polymer/MoO3/Ag) | 40 |
| P17 | — | −4.78/−3.11 | 1.28/1.67 | — | 5.60 | (ITO/MoO3/active layer/LiF/Al) | 41 |
| P18 | 770/702 | −5.73/−3.66 | 1.42/2.07 | 0.77/0.497 | — | TGBC | 42 |
| P19 | 770/776 | −5.80/−3.72 | 1.39/2.08 | 1.46/1.14 | — | TGBC | 42 |
| P20 | 770/702 | −5.81/−3.72 | 1.41/2.09 | 0.489/0.202 | — | TGBC | 42 |
| P21 | 812/746 | −5.06/−3.68 | 1.37/— | 1.95/0.03 | 5.40 | TGBC (ITO/PEDOT/polymer:PCBM/LiF/Al) | 43 |
| P22 | 711/— | — | — | —/0.1 | 2.72 | TGBC | 44 |
| P23 | — | −5.42/−3.68 | 1.74/— | 0.5/0.72 | — | TGBC | 45 |
| P24 | — | −5.64/−3.84 | 1.80/— | 0.21/6.04 | — | TGBC | 45 |
| P25 | — | −4.80/−3.49 | 1.36/1.31 | 5.87/— | 6.10 | BGBC | 46 |
| P26 | — | −4.80/−3.49 | 1.34/1.31 | 12.5/— | 6.50 | BGBC | 46 |
| P27 | 780/785 | −5.48/−4.03 | 1.45/— | 3.05/— | 5.90 | BGBC (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 47 |
| P28 | 774/774 | −5.36/−3.93 | 1.43/— | 0.32/— | 5.70 | BGBC (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 47 |
| P29 | — | −5.28/−3.55 | 1.53/1.73 | 0.18/0.48 | 5.48 | TGBC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 48 |
| P30 | — | −5.14/−3.50 | 1.54/1.64 | 0.55/0.08 | 7.56 | TGBC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 48 |
| P31 | — | −4.73/−3.24 | 1.49/— | 1.20/0.40 | 5.59 | TGBC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 49 |
| P32 | — | −5.09/−3.63 | 1.46/— | 1.68/0.14 | 6.96 | TGBC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 49 |
| P33 | — | −5.29/−3.83 | 1.46/— | 1.50/0.35 | 5.62 | TGBC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 49 |
| P34 | 831/833 | −5.24/−3.91 | 1.17/1.33 | 0.074/0.053 | — | TGBC | 50 |
| P35 | 832/833 | −5.29/−3.85 | 1.18/1.44 | 0.48/0.12 | — | TGBC | 50 |
| P36 | 832/832 | −5.27/−3.74 | 1.19/1.53 | 1.09/0.25 | — | TGBC | 50 |
| P37 | 833/833 | −5.24/−3.73 | 1.16/1.51 | 0.76/0.13 | — | TGBC | 50 |
| P38 | 796/806 | −5.33/−3.79 | 1.20/1.54 | 0.86/0.44 | — | TGBC | 50 |
| P39 | 785/782 | −5.29/−4.21 | —/1.08 | 0.021/1.2 × 10−4 | — | TGBC | 50 |
| P40 | — | −5.06/−3.68 | 1.44/— | 0.12/— | — | BGBC | 51 |
| P41 | — | −5.16/−3.20 | 1.49/1.96 | — | 5.90 | (ITO/MoO3/polymer:PCBM/LiF/Al) | 59 |
| P42 | — | −5.19/−3.20 | 1.49/1.99 | — | 6.60 | (ITO/MoO3/polymer:PCBM/LiF/Al) | 59 |
| P43 | 569/642 | −5.51/−3.52 | 1.59/1.99 | 0.034/— | 5.10 | BGTC (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 62 |
| P44 | 785/825 | −5.19/−3.50 | 1.27/2.09 | 0.02/0.02 | — | BGBC | 64 |
| P45 | 524/538 | −5.39/−3.35 | 1.90/2.04 | —/1 × 10−3 | — | BGBC | 65 |
| P46 | 579/579 | −5.25/−3.50 | 1.68/1.75 | 1 × 10−3/1 × 10−3 | — | BGBC | 65 |
| P47 | 615/639 | −5.68/−3.96 | 1.69/1.72 | 0.21/0.18 | — | TGBC | 66 |
| P48 | 788/806 | −5.49/−4.17 | 1.32/— | 0.053/0.021 | — | BGBC | 67 |
| P49 | 615/678 | −5.37/−3.96 | 1.38/1.41 | — | — | — | 68 |
| P50 | 687/712 | −5.66/−4.20 | 1.33/1.46 | 0.069/0.667 | — | BGBC | 69 |
| P51 | 675/703 | −5.78/−4.31 | 1.39/1.47 | 0.021/0.227 | — | BGBC | 69 |
| P52 | —/665 | −5.56/−4.08 | 1.46/1.48 | —/0.01 | — | BGBC | 70 |
| P53 | 834/858 | −5.00/−3.70 | 1.10/1.30 | 1.24/0.82 | — | BGBC | 72 |
| P54 | 790/815 | −5.00/−3.60 | 1.20/1.40 | 1.37/— | — | BGBC | 72 |
| P55 | 789/798 | −5.28/−3.79 | 1.25/1.49 | 1.08/2.23 | — | TGBC | 76 |
| P56 | 628/624 | −5.58/−3.87 | 1.71/— | — | 7.30 | (ITO/ZnO/PEOz/PBFTAZ:polymer/MoO3/Ag) | 79 |
| P57 | —/645 | −5.82/−3.83 | 1.50/— | — | 6.30 | (ITO/PEDOT:PSS/polymer: PCBM/LiF/Al) | 80 |
| P58 | 703/709 | −5.69/−3.82 | 1.61/1.59 | — | 10.7 | (ITO/ZnO/PEIE/polymer:PCBM/MoO3/Ag) | 81 |
| P59 | 728/729 | −5.54/−3.70 | 1.53/1.84 | 3.49/2.77 | — | TGBC | 82 |
| P60 | 686/689 | −5.82/−3.83 | 1.62/1.99 | 3.94/3.50 | — | TGBC | 82 |
| P61 | 711/720 | −5.66/−3.46 | 1.54/2.20 | 5.33/2.06 | — | TGBC | 83 |
| P62 | 718/720 | −5.63/−3.55 | 1.57/2.08 | 6.41/6.76 | — | TGBC | 83 |
| P63 | 726/731 | −5.72/−3.64 | 1.40/2.08 | 2.75/9.70 | — | TGBC | 83 |
| P64 | 742/756 | −5.67/−3.64 | 1.50/2.03 | 7.28/6.43 | — | BGBC | 84 |
| P65 | 1050/1035 | −4.86/−3.73 | 0.92/1.13 | 0.16/0.14 | — | BGBC | 85 |
| P66 | 927/925 | −4.84/−3.46 | 1.04/1.38 | 0.39/— | — | BGTC | 86 |
| P67 | 880/868 | −4.98/−3.53 | 1.15/1.45 | 0.45/— | — | BGTC | 86 |
| P68 | 730/759 | −5.12/−3.49 | 1.36/1.63 | 14.4/— | — | TGBC | 87 |
| P69 | 848/839 | −5.13/−3.58 | 1.21/1.55 | 3.93/1.07 | — | TGBC | 88 |
| P70 | 914/926 | −4.90/−3.90 | 1.05/1.00 | 0.40/0.70 | — | TGBC | 89 |
| P71 | 897/909 | −4.80/−3.70 | 1.13/1.10 | 0.20/0.20 | — | TGBC | 89 |
| P72 | 864/875 | −4.80/−3.60 | 1.19/1.20 | 0.40/0.10 | — | TGBC | 89 |
| P73 | —/665 | −5.10/−3.50 | 1.60/— | 0.31/— | 9.1 | (ITO/ZnO/polymer: PCBM/MoO3/Ag) | 90 |
| P74 | 670/665 | −5.15/−3.67 | 1.48/— | — | 5.0 | (ITO/ZnO/polymer: PCBM/MoOx/Ag) | 91 |
| P75 | 642/640 | −5.39/−3.80 | 1.59/— | — | 5.6 | (ITO/ZnO/polymer: PCBM/MoOx/Ag) | 91 |
| P76 | 660/665 | −5.34/−3.86 | 1.58/— | — | 5.1 | (ITO/ZnO/polymer: PCBM/MoOx/Ag) | 91 |
| P77 | — | −5.07/−3.51 | 1.56/— | — | 6.2 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 92 |
| P78 | — | −5.10/−3.54 | 1.56/— | — | 6.92 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 92 |
| P79 | 729/729 | −5.20/−3.67 | 1.53/— | — | 9.63 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 92 |
| P80 | 505/501 | — | 2.32/— | — | 5.75 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 93 |
| P81 | 523/523 | — | 2.23/— | — | 6.32 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 93 |
| P82 | 551/575 | — | 2.16/— | 0.55/— | — | BGBC | 94 |
| P83 | 505/541 | — | 2.29/— | 6 × 10−3/— | — | BGBC | 94 |
| P84 | — | −5.42/−3.57 | —/1.85 | — | 11.0 | (ITO/PEDOT:PSS/polymer:m-ITIC/PDINO/Al) | 96 |
| P85 | 781/801 | −5.54/−3.84 | 1.37/1.70 | 8.8 × 10−2/— | 3.39 | BGTC (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 97 |
| P86 | 737/773 | −5.30/−3.72 | 1.39/1.58 | 0.18/— | 4.79 | BGTC (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 97 |
| P87 | 673/668 | −5.34/−3.62 | 1.55/1.71 | 8.95 × 10−4/— | 5.03 | BGBC (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 99 |
| P88 | 663/677 | −5.77/−3.66 | 2.03/2.11 | — | 3.12(5.26b) | (ITO/PFN/polymer:PCBM/MoO3/Ag) | 100 |
| P89 | 708/706 | −5.24/−3.56 | 1.44/1.68 | 4.82/1.11 | — | TGBC | 101 |
| P90 | 706/703 | −5.11/−3.55 | 1.45/1.56 | 8.09/0.74 | — | TGBC | 101 |
| P91 | 594/606 | −5.24/−3.72 | 1.71/1.52 | — | 3.49(6.41b) | (ITO/PEDOT:PSS/polymer:PCBM/LiF/Al) | 102 |
| P92 | 600/606 | −5.41/−3.34 | 1.72/2.07 | 0.35/— | — | BGBC | 103 |
| P93 | 600/626 | −5.40/−3.62 | 1.68/1.78 | 0.037/— | — | BGBC | 103 |
| P94 | 628/628 | −5.25/−3.36 | 1.63/1.89 | 0.33/— | — | BGBC | 104 |
| P95 | 624/630 | −5.35/−3.39 | 1.64/1.96 | 0.39/— | — | BGBC | 104 |
| P96 | 624/632 | −5.20/−3.36 | 1.60/1.84 | 0.30/— | — | BGBC | 104 |
| P97 | 616/628 | −5.27/−3.43 | 1.63/1.84 | 0.48/— | — | BGBC | 104 |
| P98 | 646/648 | −5.82/−3.89 | 1.58/1.93 | 0.47/— | — | BGBC | 104 |
| P99 | 632/642 | −5.99/−3.93 | 1.60/2.06 | 0.31/— | — | BGBC | 104 |
| P100 | 944/986 | −5.33/−3.86 | 0.95/1.47 | 0.16/0.14 | — | BGBC | 105 |
| P101 | — | −5.70/−3.88 | 1.42/1.82 | 1.70/1.37 | — | TGBC | 106 |
| P102 | 1107/1107 | −5.18/−3.94 | 0.71/1.24 | 0.45/0.22 | — | TGBC | 107 |
| P103 | — | −5.72/−4.15 | 1.31/1.67 | —/1.74 | — | TGBC | 108 |
| P104 | — | −5.80/−4.37 | 1.32/1.43 | —/3.22 | — | TGBC | 108 |
| P105 | 812/876 | −5.87/−3.92 | 1.26/1.95 | 5.97/7.07 | — | TGBC | 109 |
| P106 | 838/844 | −5.96/−4.32 | 1.31/1.64 | —/1.24 | — | TGBC | 110 |
| P107 | 806/794 | −5.60/−4.06 | 1.23/1.54 | 0.10/0.14 | — | BGBC | 111 |
| P108 | 787/777 | −5.76/−3.79 | 1.45/1.97 | 0.12/0.14 | — | BGBC | 112 |
| P109 | 816/806 | −5.65/−3.84 | 1.41/1.81 | 0.51/0.50 | — | BGBC | 112 |
| P110 | 844/845 | −5.68/−3.77 | 1.31/1.91 | 0.15/0.33 | — | BGBC | 113 |
| P111 | 699/699 | −5.60/−3.71 | 1.23/1.89 | 0.19/0.088 | — | BGBC | 114 |
| P112 | 711/698 | −5.71/−3.70 | 1.22/2.01 | 0.10/0.075 | — | BGBC | 114 |
| P113 | — | −5.32/−4.05 | 1.27/— | 1.92/— | — | BGBC | 115 |
| P114 | 787/787 | −5.16/−3.58 | 1.31/1.58 | 1.55/0.021 | — | BGTC | 116 |
| P115 | 788/781 | −5.24/−3.58 | 1.29/1.67 | 1.79/0.087 | — | BGTC | 116 |
| P116 | — | −5.47/−3.79 | 1.55/1.68 | 0.055/0.069 | — | TGBC | 117 |
| P117 | — | −5.39/−3.77 | 1.52/1.62 | 0.10/0.14 | — | TGBC | 117 |
| P118 | —/850 | −5.40/−4.40 | 1.01/— | —/10−5 | — | BGTC | 119 |
| P119 | —/927 | −5.20/−4.20 | 1.01/— | —/0.03 | — | BGTC | 119 |
| P120 | —/1128 | −5.00/−4.20 | 0.84/— | —/0.001 | — | BGTC | 119 |
| P121 | 690/— | −5.99/−4.49 | 0.89/1.50 | —/0.18 | — | — | 120 |
| P122 | 693/707 | −5.25/−3.77 | 1.48/— | —/0.85 | — | TGBC | 122 |
| P123 | 384/390 | −5.55/−3.91 | 1.63/1.64 | — | 9.42 | (ITO/PEDOT:PSS/PffBT4T-2OD:PCBM/polymer/Ag) | 125 |
| P124 | 385/391 | −5.47/−4.18 | 1.56/1.29 | — | 10.1 | (ITO/PEDOT:PSS/PffBT4T-2OD:PCBM/polymer/Ag) | 125 |
| P125 | 713/698 | −5.84/−3.81 | —/2.03 | 1.70/8.50 | — | TGBC | 127 |
| P126 | 666/664 | −6.20/−3.88 | —/2.32 | —/3.50 | — | TGBC | 127 |
| P127 | 682/683 | −5.90/−3.77 | —/2.13 | 0.70/3.10 | — | TGBC | 127 |
| P128 | 673/669 | −6.24/−3.85 | —/2.39 | —/2.20 | — | TGBC | 127 |
| P129 | —/676 | −5.71/−3.80 | 1.50/1.91 | 0.003/3.95 | — | TGBC | 128 |
| P130 | —/917 | −5.40/−3.87 | 1.08/1.53 | 0.008/7.37 | — | TGBC | 128 |
| P131 | — | −5.60/−4.40 | 1.20/— | 0.10/0.27 | — | BGTC | 129 |
| P132 | 833/814 | −5.60/−4.00 | —/1.60 | — | 5.57 | (ITO/PEDOT: PSS/BDDT:polymer/Ca/Al) | 130 |
| P133 | — | −6.00/−4.20 | —/1.80 | —/5.60 × 10−3 | — | BGTC | 131 |
| P134 | — | −6.05/−4.22 | —/1.83 | —/0.12 | — | BGTC | 131 |
| P135 | — | −6.05/−4.22 | —/1.83 | —/0.55 | — | BGTC | 131 |
| P136 | 500/515 | −5.41/−3.29 | 2.00/2.12 | — | 5.04 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 132 |
| P137 | 502/518 | −5.47/−3.46 | 1.98/2.01 | — | 6.35 | (ITO/PEDOT:PSS/polymer:PCBM/Ca/Al) | 132 |
| P138 | 607/610 | −6.05/−4.01 | 1.71/2.04 | —/0.11 | 7.52 | BGTC (ITO/ZnO/PTB7−Th:polymer/MoO3/Ag) | 133 |
| P139 | 638/640 | −5.76/−3.96 | 1.59/1.80 | 8.70 × 10−4/1.79 × 10−3 | — | BGTC | 133 |
| P140 | 440/444 | −5.40/−3.88 | 1.06/1.52 | 0.02/— | — | BGBC | 134 |
| P141 | 524/— | −5.99/−3.33 | 2.07/2.66 | —/0.039 | — | TGBC | 135 |
| P142 | 551/— | −5.76/−3.27 | 1.94/2.49 | —/0.029 | — | TGBC | 135 |
| P143 | 592/— | −5.85/−3.38 | 1.87/2.47 | —/0.19 | — | TGBC | 135 |
| P144 | —/562 | −5.90/−3.96 | 1.74/1.94 | — | 6.85 | (ITO/PEDOT:PSS/PBDT-TS1:polymer/Mg/Al) | 137 |
| P145 | 427/432 | −5.97/−3.97 | 1.74/2.00 | — | 3.52 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Ca/Al) | 138 |
| P146 | 407/410 | −6.18/−3.95 | 1.86/2.23 | — | 0.97 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Ca/Al) | 138 |
| P147 | 408/413 | −5.73/−4.04 | 1.56/1.69 | — | 1.43 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Ca/Al) | 138 |
| P148 | 444/625 | −5.56/−3.70 | 1.86/— | 0.04/0.30 | — | TGBC | 139 |
| P150 | 498/755 | −5.01/−3.70 | 1.31/— | 3 × 10−3/0.03 | — | TGBC | 139 |
| P151 | 380/— | −6.00/−3.80 | 1.79/2.20 | — | 4.70 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Ca/Al) | 140 |
| P152 | — | −5.82/−4.12 | 1.70/— | — | 6.39 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Au) | 141 |
| P153 | — | −5.94/−4.15 | 1.79/— | — | 5.35 | (ITO/PEDOT:PSS/PTB7-Th:polymer/Au) | 141 |
| P154 | 402/547 | −5.94/−4.03 | 1.91/— | — | 8.59 | (ITO/ZnO/PTB7-Th:polymer/V2O5/Al) | 142 |
| P155 | — | −5.45/−3.80 | 1.29/1.65 | —/0.30 | — | BGTC | 144 |
| P156 | — | −5.14/−3.74 | 0.94/1.40 | —/0.09 | — | BGTC | 144 |
| P157 | — | — | — | — | 0.03 | (ITO/PEDOT:PSS/polymer/Al) | 145 |
| P158 | — | — | — | — | 0.03 | (ITO/PEDOT:PSS/polymer/Al) | 145 |
| P159 | 491/507 | −5.39/−3.36 | 2.03/— | — | 9.48 | (ITO/PEDOT:PSS/polymer:IDIC/Au) | 147 |
| P160 | 510/516 | −5.25/−3.25 | 2.00/— | — | 8.31 | (ITO/PEDOT:PSS/polymer:IDIC/Au) | 147 |
| P161 | 575/583 | −5.55/−3.80 | 1.75/— | 0.63(0.21c)/— | 13.3 | TGBC (ITO/PEDOT:PSS/polymer:IT-4F/PDINO/Al) | 148 |
| P162 | 580/580 | −5.63/−3.86 | 1.77/— | 0.93(0.23c)/— | 12.7 | TGBC (ITO/PEDOT:PSS/polymer:IT-4F/PDINO/Al) | 148 |
| P163 | 551/587 | −5.34/−3.46 | 1.81/— | — | 8.63 | (ITO/PEDOT:PSS/polymer:PCBM/PFN-Br/Al) | 149 |
| P164 | 545/608 | −5.90/−3.60 | 1.80/2.30 | — | 10.2 | (ITO/PEDOT:PSS/polymer:ITIC/PDINO/Al) | 150 |
| P165 | 578/598 | −5.97/−3.63 | 1.81/2.34 | — | 12.1 | (ITO/PEDOT:PSS/polymer:ITIC/PDINO/Al) | 150 |
| P166 | — | −5.56/−3.75 | 1.80/1.81 | — | 9.30 | (ITO/PEDOT:PSS/polymer:PBDTTT-C-T:PCBM/Ca/Al) | 151 |
| P167 | 518/567 | −5.65/−3.90 | 1.75/— | — | 9.21 | (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 153 |
| P168 | 533/593 | −5.39/−3.59 | 1.80/— | 0.74/— | — | TGBC | 154 |
| P169 | 480/534 | −5.67−/3.74 | 1.93/— | 0.32/— | — | TGBC | 154 |
| P170 | 610/630 | −5.20/−3.45 | 1.78/1.75 | — | 14.2 | (ITO/PEDOT:PSS/polymer:Y6/PFN-Br/Ag) | 155 |
| P171 | —/578 | −6.18/−4.05 | —/2.13 | —/0.06 | — | BGTC | 156 |
| P172 | —/535 | −6.15/−4.00 | —/2.15 | —/2.55 | — | BGTC | 156 |
| P173 | —/641 | −5.48/−3.17 | 1.78/— | 0.06/— | 5.60 | BGTC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 158 |
| P174 | —/660 | −5.49/−3.25 | 1.71/— | 0.05/— | 8.00 | BGTC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 158 |
| P175 | —/654 | −5.62/−3.20 | 1.73/— | 0.03/0.02 | 2.40 | BGTC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 158 |
| P176 | —/602 | −6.02/−3.36 | 1.91/— | —/0.05 | 0.05 | BGTC (ITO/ZnO/polymer:PCBM/MoOx/Ag) | 158 |
| P177 | 587/588 | −5.43/−3.45 | 1.98/— | —/3.10 | 6.67 | TGBC (ITO/PEDOT:PSS/PTB7-Th:polymer/LiF/Al) | 159 |
| P178 | 606/616 | −5.43/−3.55 | 1.88/— | —/1.23 | 8.61 | TGBC (ITO/PEDOT:PSS/PTB7-Th:polymer/LiF/Al) | 159 |
| P179 | — | −5.27/−3.43 | 1.84/— | —/1.13 | 6.85 | TGBC (ITO/PEDOT:PSS/PTB7-Th:polymer/ETL/Al) | 160 |
| P180 | — | −5.28/−3.30 | 1.98/— | —/0.84 | 0.03 | TGBC (ITO/PEDOT:PSS/PTB7-Th:polymer/ETL/Al) | 160 |
| P181 | —/588 | −6.08/−3.50 | 1.96/— | —/2.73 | 6.50 | TGBC (ITO/PEDOT:PSS/PTB7-Th:polymer/LiF/Al) | 161 |
| P182 | — | −5.94/−3.46 | 1.74/2.48 | — | 8.10 | (ITO/PEDOT:PSS/active layer/LiF/Al) | 162 |
| P183 | — | −5.78/−3.77 | 2.01/— | —/1.61 | — | TGBC | 163 |
| P184 | — | −5.97/−4.09 | 1.88/— | — | 19.5 | PVSCs (ITO/PTAA/perovskite/polymer/BCP/Ag) | 164 |
| P185 | — | −5.79/−4.01 | 1.78/— | — | 20.8 | PVSCs (ITO/PTAA/perovskite/polymer/BCP/Ag) | 164 |
| P186 | 628/666 | −5.44/−3.05 | 1.65/2.39 | — | 8.61 | (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 165 |
| P187 | 618/658 | −5.41/−3.01 | 1.67/2.40 | — | 10.2 | (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 165 |
| P188 | 590/635 | −5.48/−2.99 | 1.72/2.49 | — | 8.47 | (ITO/ZnO/polymer:PCBM/MoO3/Ag) | 165 |
| P189 | 643/630 | −5.59/−3.66 | 1.60/1.93 | 1.17 × 10−2/2.78 × 10−3 | — | TGBC | 166 |
| P190 | 594/585 | −5.85/−3.75 | 1.62/2.10 | 1.38 × 10−1/7.39 × 10−2 | — | TGBC | 166 |
| P191 | 560/587 | −5.70/−3.68 | 1.67/2.02 | 7.42 × 10−3/8.04 × 10−4 | — | TGBC | 166 |
 | ||
| Fig. 1 The molecular structures of polymers P1–P55. | ||
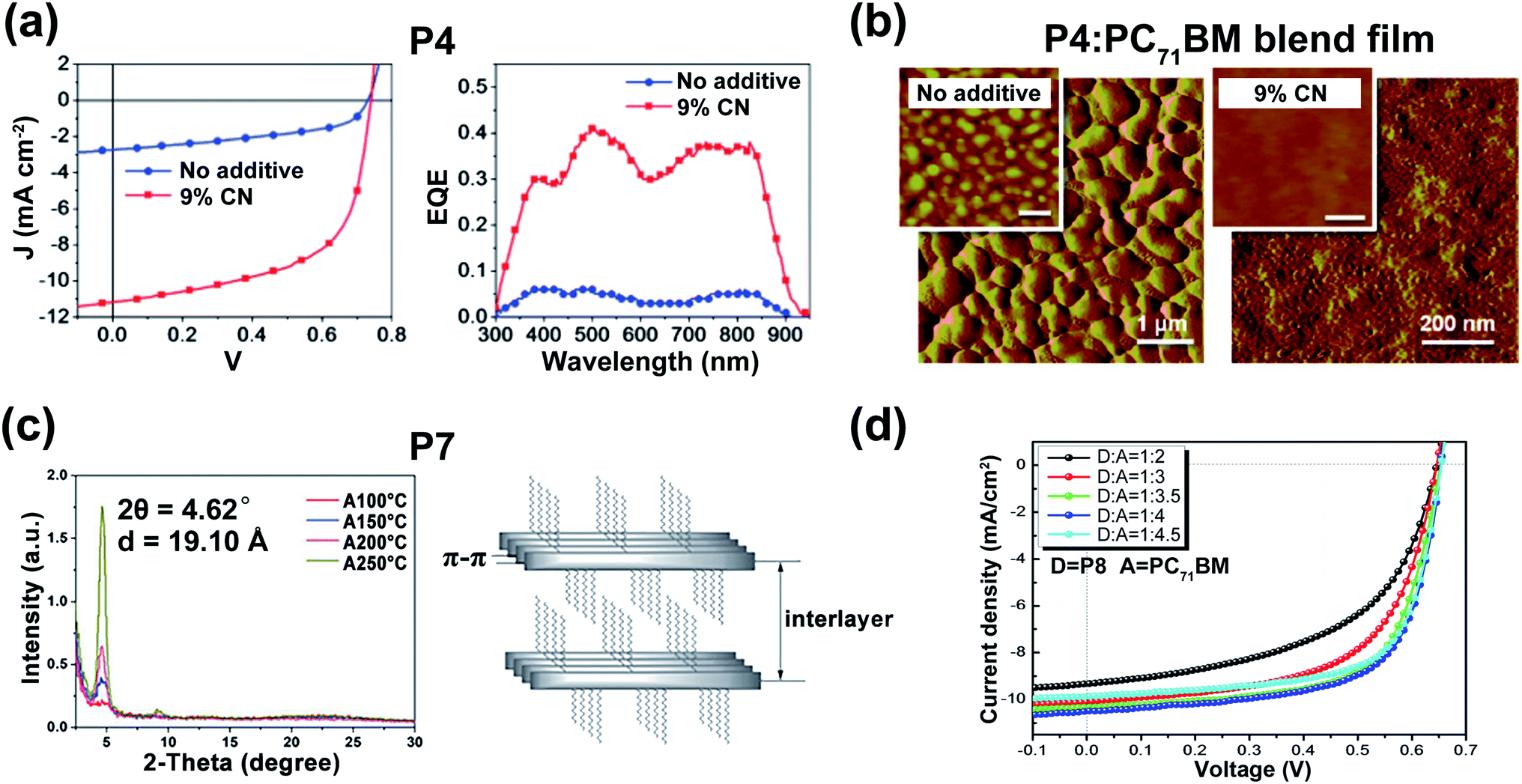 | ||
| Fig. 2 (a) The J–V curves (left) and the external quantum efficiency spectra (right) of the optimized devices based on P4:PC71BM blend films (weight ratio = 1:3). Reproduced from ref. 29 with permission from the American Chemical Society, copyright 2010. (b) The AFM phase images of P4:PC71BM blend films (weight ratio = 1:3). Reproduced from ref. 29 with permission from the American Chemical Society, copyright 2010. (c) XRD data (left) and the illustration of lamellar structure (right) of P7 film. Reproduced from ref. 31 with permission from the Royal Society of Chemistry, copyright 2015. (d) The OSC performances of P8:PC71BM with different ratios. Reproduced from ref. 32 with permission from the Royal Society of Chemistry, copyright 2017. | ||
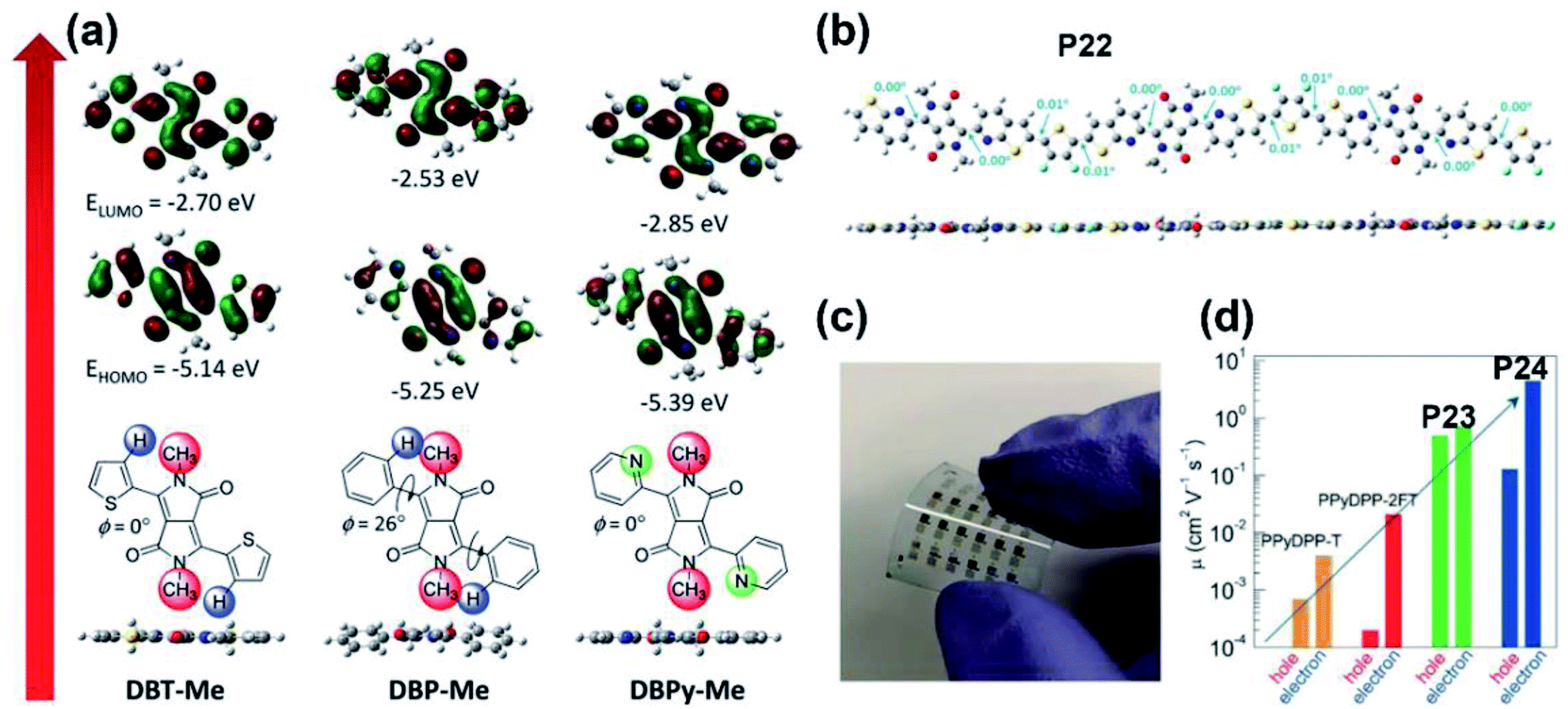 | ||
| Fig. 3 (a) The optimized geometries and FMO energy levels of the thiophene-, phenyl-, and pyridine-flanked DPP monomers with methyl substitution. Reproduced from ref. 37 with permission from Wiley-VCH, copyright 2014. (b) The DFT-simulated backbone geometry of the P22 trimer. Reproduced from ref. 44 with permission from the American Chemical Society, copyright 2017. (c) The flexible OFET array fabricated on the PET substrate. Reproduced from ref. 45 with permission from Wiley-VCH, copyright 2018. (d) The comparison of ambipolar mobilities between P23/P24 and their pyridine-based counter polymers. Reproduced from ref. 45 with permission from Wiley-VCH, copyright 2018. | ||
In addition to substituting the DPP core with dual electron donors, researchers also tried to replace one of the flanking rings with the electron-withdrawing unit, thus adjusting the electron deficiency of building blocks. For instance, the structures of P27 and P28 were built up with the thiophene/thiazole-flanked DPP units in asymmetric and random ways, respectively.47 In comparison with P28, the ordered structure of P27 afforded better planarity with smaller dihedral angles, thereby forming a larger domain size and adopting more percentage of favorable face-on orientation in its film. Those properties provided P27 with a relatively higher hole mobility of 3.05 cm2 V−1 s−1 and a higher PCE of 5.90% than those of P28. Just like the thiazole unit, pyridine is another available electron-deficient aromatic ring that can be used as the linker. Qiu and co-workers asymmetrically substituted DPP units with thiophene and pyridine rings in polymers P29 and P30.48 Owing to the more electron-deficient pyridine units, P29 and P30 showed deep-lying HOMO energy levels and narrow bandgaps, which led to the promising hole/electron mobilities of 0.18/0.48 and 0.08/0.55 cm2 V−1 s−1, respectively. In agreement with their wide absorption spectra, the P29:PC71BM and P30:PC71BM gave their respective PCE of 5.48% and 7.56%, both of which were higher than those of their pyridine-DPP-based analogs. To further substitute π-extended aromatic rings in DPP units, thiophene/benzothiophene flanked DPP was used in polymers P31-P33.49 Such changes not only widened their absorption bands but also simultaneously improved their Jsc and PCE values. P32 afforded the highest PCE of 7.0% among the three polymers. The PCEs of the other two polymers also achieved 5.6%. Moreover, the electron/hole mobilities of P31, P32, and P33 were 1.20/0.40 cm2 V−1 s−1, 1.68/0.14 cm2 V−1 s−1, and 1.50/0.35 cm2 V−1 s−1, respectively. Benefitting from the extended π-conjugation and delocalized orbitals, both hole and electron mobilities got improved, especially the electron mobility, which was almost four times higher than that of the PyTDPP-based counterpart. The DPP flanked by π-extended aromatic rings was proved to be preferable for application in both FETs and OSCs, which facilitated more studies on this strategy. Song and co-workers reported two novel DPP derivative units, DDPP-PhCO and DDPP-PhCN, in which two thiophene-based DPP units were combined with polycyclic aromatics.50 DDPP-PhCO-based P34-P38 and DDPP-PhCN-based P39 exhibited much-widened absorption spectra due to their extended π-conjugation. Despite the limited adjustment of LUMO energy levels, additional cyano and carbonyl groups induced the polymer films to have orderly packing, resulting in ambipolar transport characteristics with the max hole and electron mobilities reaching 1.09 and 0.44 cm2 V−1 s−1, respectively.
As mentioned in the previous section and other related studies, furan-DPP-containing polymers performed well in their solubilities, carrier transport behaviors, and OSC device performance. Therefore, the furan-containing asymmetrical DPP building block was inserted into the polymer backbone for better solution processability. The furan/thieno[3,2-b]thiophene-DPP (TTFDPP)-based polymer P40 combined the advantages of two flanking units together and achieved an improved solubility, self-assembly ability, and more conjugated molecular backbone (Fig. 4a).51 The DFT simulation revealed a planar backbone, with dihedral angles no more than 3.2° in the TTFDPP moiety (Fig. 4b), and delocalized HOMO/LUMO orbitals over the whole molecular backbone. Owing to the enhanced solubility, the P40 film was processed from mixed chloroform and toluene solvents, with its FET device exhibiting typical p-type performance.
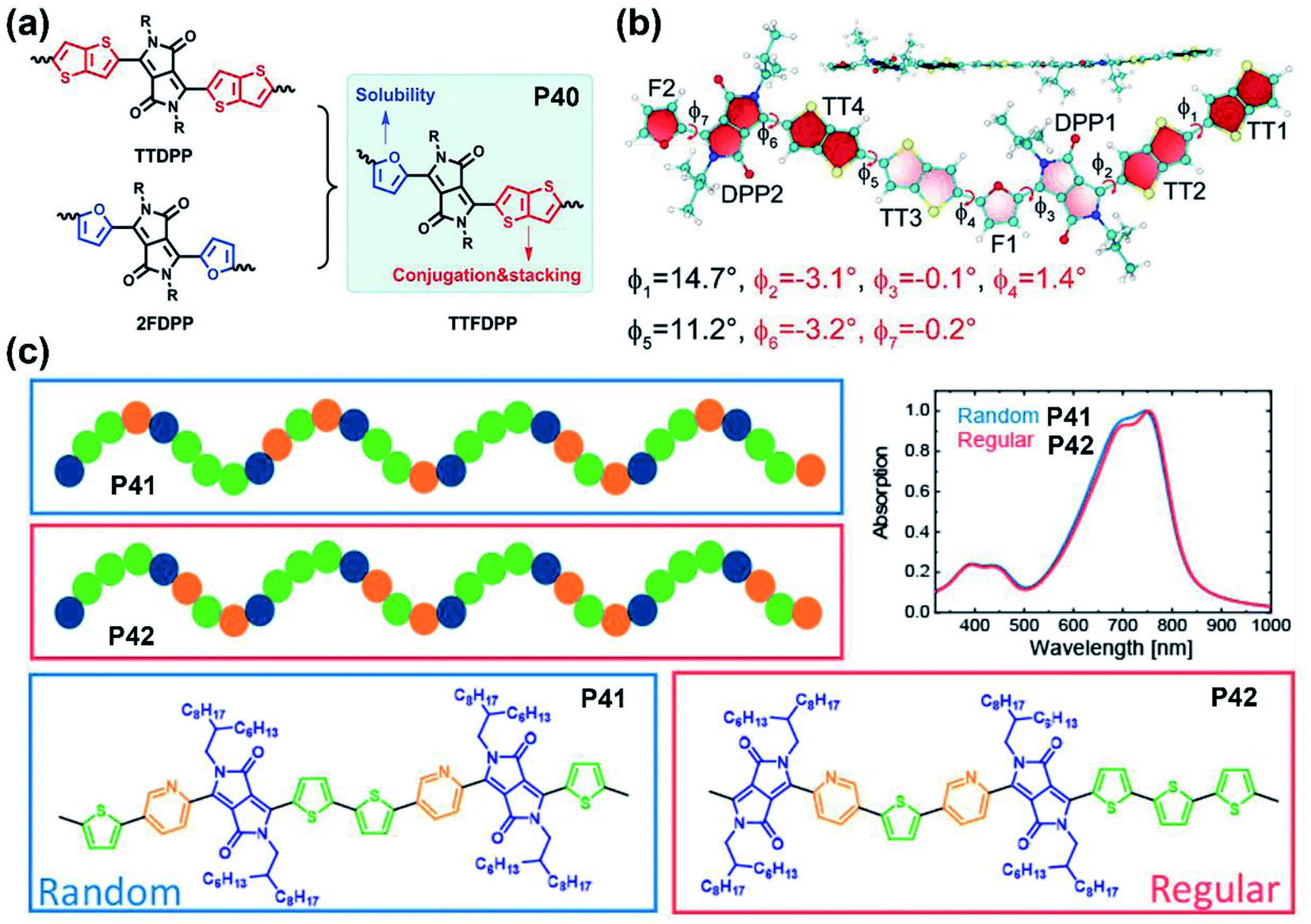 | ||
| Fig. 4 (a) The synthetic strategy of P40 to combine the advantages of two other polymers. Reproduced from ref. 51 with permission from Wiley-VCH, copyright 2018. (b) Optimized structure of the P40 dimer with the dihedral angles marked. Reproduced from ref. 51 with permission from Wiley-VCH, copyright 2018. (c) The illustration of backbone shapes and the UV-vis absorption spectra of P41 and P42. Reproduced from ref. 59 with permission from the American Chemical Society, copyright 2020. | ||
As the performance regulator of the corresponding polymers, the different DPP flanking units could firstly adjust the planarity and decrease the backbone distortion through the intramolecular interactions and hydrogen bonds in the lactam structure.52 Secondly, some flanking units like furan are another factor that could positively affect the processing solubility of polymers, besides the lactam nitrogen alkylation.53 This function directly leads to the feasibility of obtaining polymers with shorter sidechain substitution, which is conducive to the decreased lamellar d-spacing distance, increased crystallinity, and the ordered intermolecular packing state of polymer films.54,55 Finally, the incorporation of electron-deficient flanking units could tune FMOs to the lower level, which is pivotal to the carrier injection from the metal electrode to the semiconducting layer.56 Compared with polymers containing symmetrically flanked DPP units, both superiorities and defects existed in asymmetrically bridged DPP-based polymers. For one thing, the substitution of two different aromatic rings of the DPP unit could adjust the polymer in two or more aspects simultaneously, such as backbone planarity, conjugation length, solubility, etc.57 For another thing, the above properties could be fine-tuned by asymmetrical substitution. Nevertheless, asymmetrically flanked DPP units were combined with donors randomly, which results in incomparable symmetry and regularity of their molecular backbones to their symmetric counterparts. Such changes could make polymer films adopt different packing states, let alone their photoelectric performances.57,58 Besides, compared with DPPs with the same flanking rings, those asymmetrically bridged DPP units are relatively harder to obtain due to the multiple synthetic steps, low yield, and difficulties in the purification process. It is noted that the backbone randomness of asymmetrically flanked DPP-based polymers should not be neglected, which potentially affected the physicochemical properties or device performance. More recently, Leenaers and co-workers reported two polymers containing the thiophene/pyridine asymmetrically flanked DPP unit, P41 and P42, containing a regiorandom and regioregular molecular backbone, respectively (Fig. 4c).59 Despite their similar optical properties, the regioregular polymer P42 gave a relatively higher PCE of 6.6% than 5.9% of regioregular polymer P41 (Table 1). Overall, the asymmetrical substitution strategy can effectively tune the polymer properties if the reaction sites of the copolymerization are controllable, which is also a future challenge.
As the most structurally similar DPP-analog, pyrrolo[3,2-b]pyrrole-2,5-dione (isoDPP), with less synthetic complexity, has the position of the nitrogen atom and carbonyl group opposite to the DPP core. Song and co-workers synthesized a thiophene-bridged isoDPP with the amide nitrogen atoms substituted by butyl chain alkylated phenyl groups.61 Besides the p-type carrier transport performance, a PCE of 2.0% in photovoltaic cells without thermal annealing treatment was also achieved. Both performances were further improved by another isoDPP-based polymer P43, which showed n-channel carrier transport behavior and afforded a high PCE value of 5.1% (Table 1).62
The thiophene-isoDPP backbone was not comparable to that of thiophene-DPP, as indicated by their DFT-simulated dihedral angles which were 24.5° for thiophene-isoDPP and almost 0° for thiophene-DPP (Fig. 5a). Besides, the isoDPP-induced poor crystallinity was also revealed by another study.63 Those differences could be explained by their varied structures. Despite the simply exchanged positions of the carbonyl groups and nitrogen atoms in thiophene-DPP and thiophene-isoDPP, they had considerably different internal structures. Caused by the different conjugation ways, the length of the carbon–carbon double bonds in the isoDPP core was shorter than those in the DPP core and the adjacent single bonds elongated correspondingly (Fig. 5a and b), such changes directly lead to the more apparent steric effect between the flanking unit and the substituted sidechain, eventually resulting in the different backbone planarity and packing state. The structural disadvantage was inevitable but the adjustable structure and appropriate energy levels indicated the application potential of isoDPP nevertheless. For instance, in comparison with isoDPP- or DPP-based polymers, combining both the isoDPP and DPP moieties in one single polymer could potentially achieve a polymer film with better physicochemical properties and optoelectronic performance, i.e., P44. The combination of thiophene-isoDPP and thiophene-DPP isomers provided the P44 good film with aggregation and proper energy levels, leading to the balanced ambipolar transport behavior of P44 with around 0.02 cm2 V−1 s−1 for both hole and electron mobilities.64 This performance was higher than those of its reference polymers based on only the DPP or isoDPP unit.
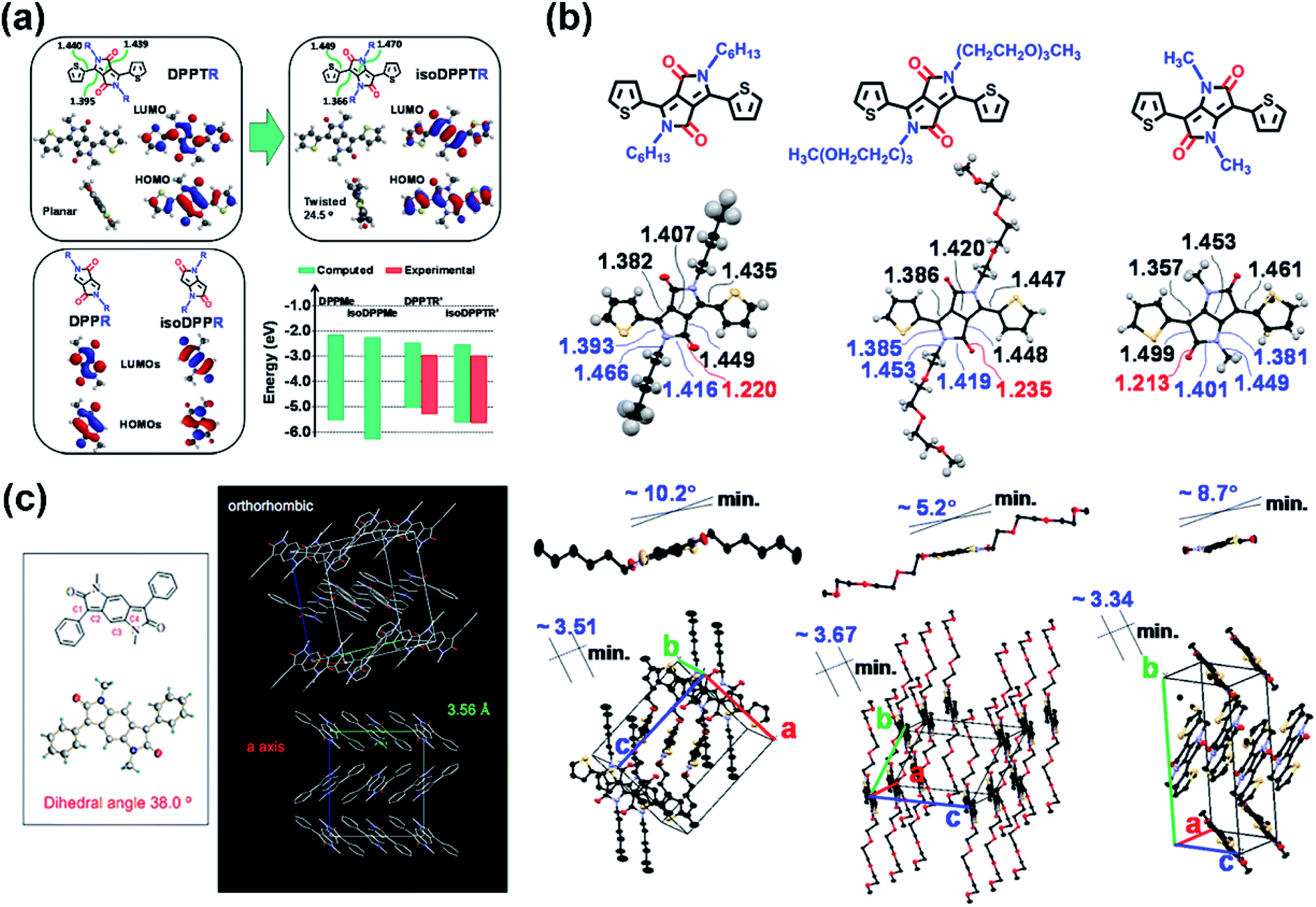 | ||
| Fig. 5 (a) The simulated structures of DPP and isoDPP monomers as well as the summary of their estimated and experimental HOMO/LUMO energy levels. Reproduced from ref. 62 with permission from the American Chemical Society, copyright 2013. (b) The chemical and crystal structures of two different chain substituted DPP units and the methyl-substituted isoDPP unit. Reproduced from ref. 62 with permission from the American Chemical Society, copyright 2013. (c) Single crystal structure of the building block of P45–P46. Reproduced from ref. 65 with permission from the American Chemical Society, copyright 2011. | ||
The easily changeable isoDPP structure could be modified by many methods. The extended isoDPP unit, benzodipyrrolidone (BDP), having two lactam structures fused with a benzene ring, was synthesized as early as 1980 as the dyestuff. Because of its DPP-similar structure and lactam moieties, the BDP moiety was deduced to be favorable for organic semiconductor materials. Two BDP-based polymers, P45 and P46, were reported by Cui and co-workers.65 The single-crystal analysis of the methyl-substituted phenyl-BDP (BDPDP) molecule discovered the high planarity of the BDP core despite a torsion angle of 38° between the adjacent benzene ring and BDP core. The orthorhombic packing structure revealed the π–π interaction of the middle benzene ring and the benzene ring adjacent to the lactam structure with a distance of 3.56 Å (Fig. 5c). P45 and P46 exhibited n-type and ambipolar type behaviors, respectively, with the mobilities of around the magnitude of 10−3 cm2 V−1 s−1. The reasons for such properties could be complex, such as molecular weight, device architecture, the solvent used for the spin-coating process, and some other reasons. The structural novelty of BDP and its polymers attracted scientists to further improve its performance in electronics. Polymer P47 was obtained and had the same structure as P46 but with a higher number-averaged molecular weight (Mn) of around 13.9 kDa (polydispersity: 5.20), which was higher than 8 kDa (polydispersity: 2.29) of P46.66 The higher molecular weight, together with device optimization, successfully enhanced its performance and gave it balanced electron and hole mobilities of 0.18 and 0.21 cm2 V−1 s−1, respectively, due to the reduced contact resistance. Additionally, the nitrogen embedded BDP building block (PzDP) was also designed and utilized in polymer P48, which had a deep-lying LUMO energy level of −4.17 eV.67 Besides, the existence of nitrogen atoms decreased the torsional angles between the benzene ring and the PzDP core, due to the N⋯H hydrogen bond. The spin-coated polymer P48 film also exhibited ambipolar FET performance. Despite the moderate mobilities, such effective adjustment to the narrower bandgap was still worthy of being borrowed. To further extend the BDP unit, the naphthodipyrrolidones (NDP) unit, with one more benzene ring fused in the BDP unit, was chosen as the building block of P49.68 The single-crystal analysis of the phenyl-flanked NDP unit (PhNDP) revealed its intermolecularly slipped π–π stacking state, with a shorter distance of 3.38 Å. Moreover, the planar backbone of PhNDP, with a dihedral angle of 25°, led to broad absorption bands and a narrow bandgap of the P49 film. The above characteristics indicated the easier carrier injection, ambipolar transport property, and excellent air stability of NDP-containing polymers. As Zhang and co-workers reported, two NDP-containing polymers P50 and P51, both of which have narrow band gaps and suitable FMO energy levels, showed electron-dominated ambipolar transport behaviors, with the max electron mobility of 0.667 cm2 V−1 s−1 achieved by P50.69 The relatively better performance of P50 was attributed to the lack of side-chain-induced backbone nonplanarity, which affected the chain orderly packing and the formation of large crystalline domains. Though ambipolar NDP polymers were obtained, unipolar n-type semiconductor materials based on the NDP unit were still unreported until the NDP-based polymer P52 was synthesized, whose NDP block had no alkyl sidechain substitutions.70 In comparison with its sidechain-substituted counter polymer, P52 showed more apparently bathochromic-shifted UV-vis absorption spectra and narrower bandgaps due to the enhanced intramolecular interaction between imide N and O atoms. Therefore, the P52 film afforded unipolar n-type transport performance, with the mobility 40 times higher than its counter polymer.
In addition to modifying the conjugation length of the DPP core and connecting two functional groups of the DPP unit with an aromatic ring, the double bond was creatively introduced to connect two lactam structures. The bipyrrolylidene-2,2′(1H,1′H)-dione (BPD) unit was firstly reported by Cai and co-workers, who achieved a hole mobility of 1.4 cm2 V−1 s−1 for BDP-based small molecules.71 Later, two thiophene-BPD-based polymers, P53 and P54, were also reported.72 Benefitting from the good planarity and delocalized FMO orbitals, two polymers showed appropriate HOMO/LUMO energy levels and narrow band gaps. Despite the comparable energy levels, P53 afforded ambipolar transport performance with hole/electron mobilities of 1.24/0.82 cm2 V−1 s−1, respectively, whereas P54 afforded a unipolar p-type transport performance with a hole mobility of 1.37 cm2 V−1 s−1. Such different transport types of P53 and P54 were attributed to their different sidechain orientations, in which bimodal and edge-on orientations were adopted by P53 and P54, respectively. In the last five years, some other studies have focused on BPD units, including siloxane-terminated side chain substitution,73 and copolymerization with varied donor units.74 Most of these polymers showed balanced ambipolar transport behaviors for their applications in FET due to the appropriate FMO energy levels. The apparently enhanced FET performance and the changed transfer type suggested the BPD framework a competitive candidate for promising building blocks to construct more applicable semiconductors. As the analog of BPD, the (E)-[4,4′-biimidazolylidene]-5,5′(1H,1′H)-dione (BID) unit with additional nitrogen atoms embedded in the lactam ring was also used as the building block, which induced stronger intermolecular interactions of the corresponding polymer mainchains, thereby achieving p-channel carrier transport behaviors for its corresponding polymers.75 Despite the moderate mobilities of those BID-based polymers, the effectively adjusted band gap and near-infrared absorption region could be helpful for OSC application, which still worthy of investigation in the future.
Most of the commonly investigated DPP structures had both their lactam nitrogen substituted by alkyl groups or other functional sidechains. Recently, P55 was reported based on a novel building block, half-fused DPP, with one of the lactam nitrogen atoms embedded in a six-membered ring fused with the adjacent thiophene flanking unit.76 Besides the much more red-shifted absorption relative to the DPP-counterpart, the more rigid backbone and decreased steric hindrance provided the P55 film with more ordered lamellar packing and denser π–π stacking, with the distances of 28.5 and 3.45 Å, respectively (Fig. 6a). The preferable polymer chain packing and the edge-on dominant bimodal orientation led to the max hole and electron mobilities of 1.08 and 2.23 cm2 V−1 s−1, respectively (Table 1). Efforts were also made to obtain DPP having both lactams fused to the adjacent units, with the promising solubility of alkylated thiophene flanking rings.77 Similar optical and chemical properties to the half-fused DPP were observed, which implied various applications in optoelectronic devices.
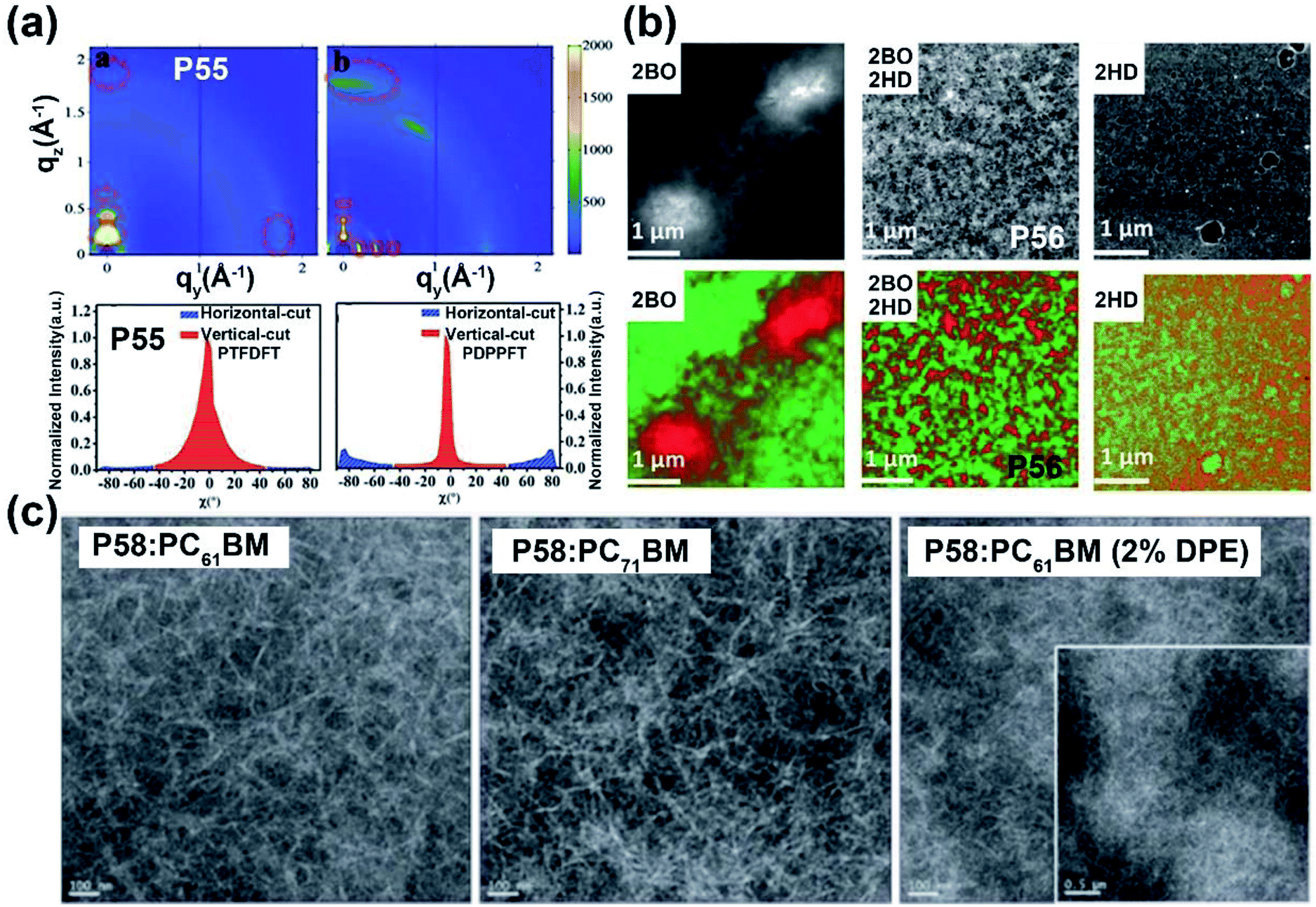 | ||
| Fig. 6 (a) 2D-GIWAXS patterns of P55 and reference polymer thin films (up) and the corresponding pole figures (bottom). Reproduced from ref. 76 with permission from Wiley-VCH, copyright 2020. (b) Dark field STEM images (up) of the active layers containing P56 and other two reference polymers, respectively, and the EELS maps of phase-separation patterns (bottom) between the electron-donor and electron-acceptor materials. Reproduced from ref. 79 with permission from Wiley-VCH, copyright 2018. (c) TEM images of different P58: PCBM thin films. Reproduced from ref. 81 with permission from the Royal Society of Chemistry, copyright 2019. | ||
2.2. Polymers with IID and its derivatives
In the previous part, we comprehensively introduced semiconductor polymers based on DPP and its derivatives. As the other most investigated lactam-containing building block of polymers applied in optoelectronic devices, the IID unit was originally used in the medical and pharmaceutical fields. Thereafter, a series of six IID-based polymers was firstly synthesized by Stalder and co-workers. The systematic investigation of their physicochemical properties demonstrated the wide application of the IID unit.78 From then on, the IID unit has been developed to numerous analogs and derivatives, which could be generally divided into two main types. One type is monomers with a modified IID core. The other one is monomers with various conjugated bridges between their indolinone units.:2HD was 2:1.79 The P56:PBFTAZ blend film exhibited a max PCE value of 7.3%, much higher than those of 2BO-IID- and 2HD-IID-based polymers (Table 1). This was caused by the high degree of donor and acceptor mixing, which had evenly distributed phase-separation as revealed by electron energy-loss spectroscopy (EELS) (Fig. 6b). The alkylation was not only used to modify the electron-deficient unit. As shown in the P57 structure, donor alkylation was also used to prepare polymers with its PCE value reaching 6.30% due to the improved film morphology.80 The further improvement of the PCE value of up to 10.7% was achieved by the polymer P58, which had both the sidechains of IID and thiophene ring elongated.81 Longer sidechains provided P58 with good solubility in halogenated solvents and thereby facilitated the formation of bi-continuous phase separation and finer nano-fibrils in the film (Fig. 6c), implying the good contact for P58 and PCBM. Likewise, synthesizing the more electron-deficient IID unit could have a significant influence on the device performance similarly to the alkylated IID units. The fluorine atom was incorporated into polymers P59 and P60, both of which showed ambipolar transport behaviors.82P59 exhibited the highest hole/electron mobilities of 3.49/2.77 cm2 V−1 s−1. The polymer P60 gave an improved and more balanced ambipolar transport behavior with the max hole/electron mobilities of 3.94/3.50 cm2 V−1 s−1, respectively. Such enhancement could be associated with its more planar backbone, which was attributed to the more fluorine-related intramolecular interactions, such as F⋯H and F⋯S interactions, in addition, P60 with a farther sidechain branching point exhibited an n-channel transport behavior with the max electron mobility of 4.97 cm2 V−1 s−1, which was caused by the decreased d-spacing and π–π stacking distance provided by multiple fluorinations and effective mainchain packing. To investigate the effect of different fluorination degrees on the polymers, P61-P63 were constructed with the units IID, 1FIID, and 2FIID substituted with 0, 1, and 2 fluorine atoms, respectively.83 The optical band gaps, FMO energy levels, and their π–π distances decreased in the order of P61, P62, and P63. Such regularly changed properties led to their different transport mobilities. P61 and P63 showed hole-dominated and electron-dominated ambipolar transport behaviors, with the μh/μe of 5.33/2.06 and 2.75/9.70 cm2 V−1 s−1, respectively. However, P62 showed a more balanced transport performance with high μh/μe values of 6.41/6.76 cm2 V−1 s−1. Their different FET performances indicated the adjustable transport types by varied backbone fluorination. Besides introducing fluorine atoms, nitrogen-embedded IID monomers also captured a lot of attention. As Huang and co-workers reported, the 7,7′-diazaisoindigo unit, in which 7-position carbon atoms were replaced by nitrogen atoms, possessed preferable backbone planarity.84 Besides, the corresponding polymer P64 showed small torsional angles between its donor and acceptor moieties due to the S⋯N intramolecular interaction. Such a planar polymer backbone of P64 agreed with its high hole mobility of 7.28 cm2 V−1 s−1.
The above electron-withdrawing atom substitution can effectively adjust the energy levels and relieve the backbone distortion of polymers. Apart from this, the aromatic rings fused with the lactam structure are also changeable for adjustment. Ashraf and co-workers replaced the benzene rings with thiophene rings in polymer P65, which afforded a balanced ambipolar transport behavior with mobility of around 0.1 cm2 V−1 s−1.85 Thereafter, P66 and P67 were synthesized based on thienoisoindigo units, both polymers showed improved mobilities of 0.39 and 0.45 cm2 V−1 s−1, respectively, compared with that of P65, due to their enhanced film crystallinity.86 As another thienoisoindigo-based polymer, P68 exhibited an impressive charge carrier mobility.87 Different from the thiophene-contained donors in P66 and P67, the naphthalene donor of P68 facilitated the well-ordered layer-by-layer lamellar packing state with both edge-on and face-on crystal domains, thus achieving a high μh up to 14.4 cm2 V−1 s−1 in the device with the high-dielectric-constant poly(vinylidenefluoride-trifluoroethylene) as the gate dielectric, which promoted accumulated positive charge carriers induced by the multiple fluorine atoms of the new dielectric (Fig. 8a). Similarly, further N-embedding modification of the thienoisoindigo core was completed by Li and co-workers, who synthesized the polymer P69.88 Owing to the smooth surface of the dominant edge-on orientation content of the film, the P69 film gave the max μh/μe of 3.93/1.07 cm2 V−1 s−1 in its TGBC FET devices, and unipolar p-type transport performance with mobility of 3.41 cm2 V−1 s−1 was also achieved in BGTC devices. As shown in Fig. 7, the polymers P70-P72 contained the extended thienoisoindigo, thieno[3,2-b] thiophene isoindigo (IIDTT), as the acceptor unit.89 The extended backbone facilitated an increased backbone coplanarity, which resulted in the more effective π-orbital overlap. Therefore, P70-P72 exhibited ambipolar transport performances and P70 showed the highest μh/μe of 0.4/0.7 cm2 V−1 s−1, respectively. Yue and co-workers further fused IIDTT with additional thiophene rings and synthesized a novel building block, thieno[3,2-b][1]benzothiophene isoindigo (TBTI).90 The corresponding polymer P73 showed strong XRD diffraction peak intensity with a large coherence length, demonstrating that the further extension of the conjugated length effectively improved the film order and the crystallinity. Hence, both the PCE of 9.1% and the hole mobility of 0.31 cm2 V−1 s−1 were achieved in its OSC and OFET devices, respectively. The TBTI-based polymers P74-P76 were also synthesized with high PCE values surpassing 5.0%, which could be attributed to the well-distributed small domain size of P74-P76 and the PC70BM in their respective blend films and their appropriate ionization potentials and electron affinities.91
 | ||
| Fig. 7 The molecular structures of polymers P56–P100. | ||
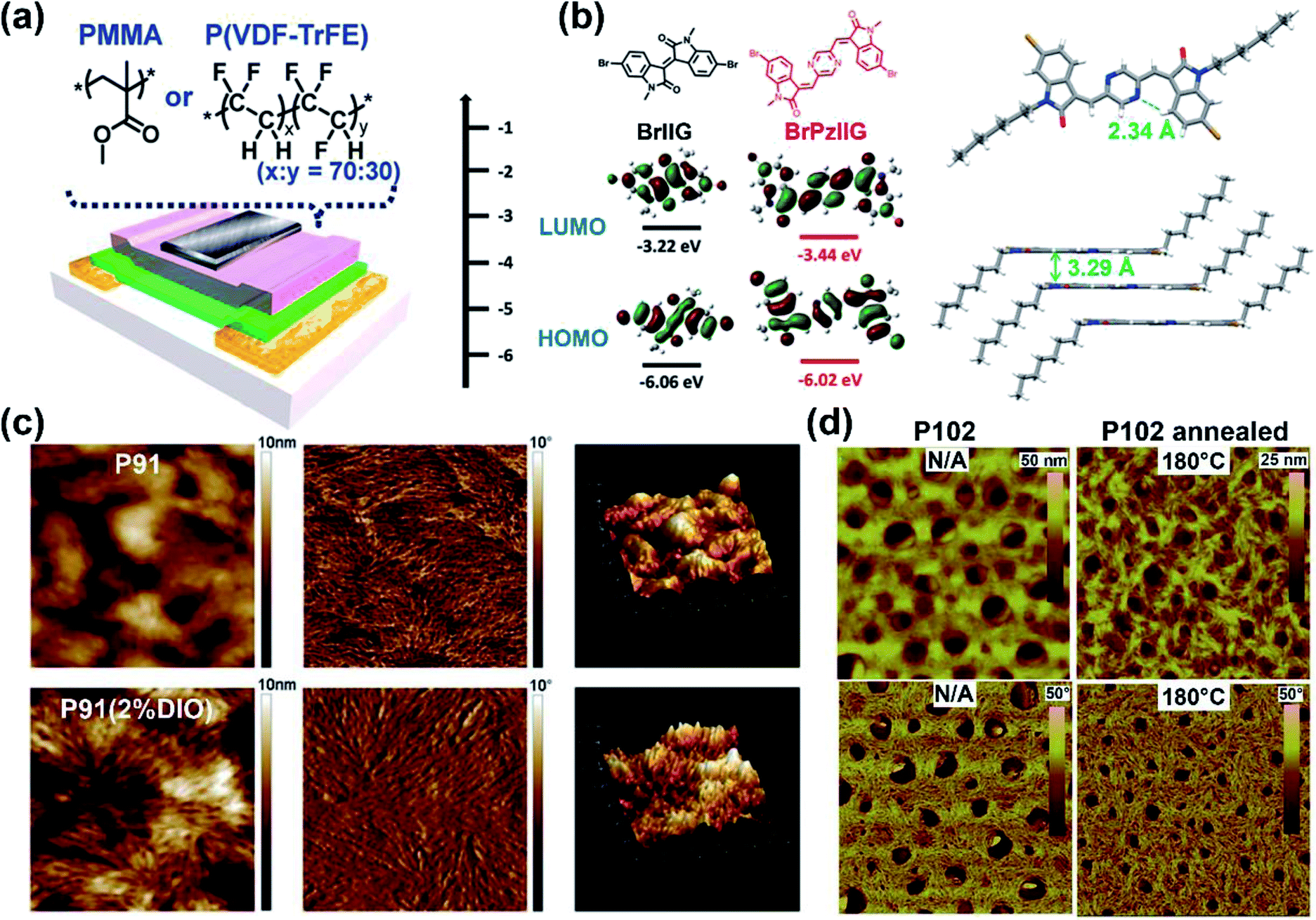 | ||
| Fig. 8 (a) The OFET device with the top-gate bottom-contact (TG-BC) configuration and the chemical structures of PMMA and P(VDF–TrFE) as the gate dielectric layer. Reproduced from ref. 87 with permission from the American Chemical Society, copyright 2014. (b) DFT estimated HOMO and LUMO energy levels of PzIIG and its counter monomer IIG (left) ant the single-crystal structures of the monomer PzIIG (right). Reproduced from ref. 100 with permission from the Royal Society of Chemistry, copyright 2017. (c) AFM images of the P91:PC61BM blend film (height images: left; phase images: middle) and the topography images (right). Reproduced from ref. 102 with permission from the Royal Society of Chemistry, copyright 2014. (d) AFM images of P102 thin films before and after thermal treatment: height images (up), phase images (bottom). Reproduced from ref. 107 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Besides the five-membered lactam structure, the six-membered bis-lactam monomer 1,4-naphthyridine-2,6-dione (NTD) was used as the acceptor in P77-P79.92 The intramolecular S⋯O interaction improved the molecular coplanarity and all three polymer films adopted face-on orientation with small π–π stacking distances. P79, with the smallest π-stacking distance of 3.61 Å, exhibited the highest PCE up to 9.63%. Owing to its stronger electron-accepting capacity and structural planarity, such a six-membered lactam moiety was widely modified and used. P80 and P81 featured novel acceptor dibenzo[c,h][2,6]naphthyridine-5,11(6H,12H)-dione (DBND), which had further benzene rings fused with the NTD unit.93 The only difference was that the DBND unit adopted O-alkylation rather than common N-alkylation. Despite the large dihedral angles of 30° existing between DBND and donor moieties, both P80 and P81 had short π–π distances of 3.74 and 3.38 Å, respectively, and the denser film packing gave a higher PCE value of 6.32% for P81 than the 5.75% of P80. Subsequently, they disclosed the other two DBND-based polymers P82 and P83 applied in FET transistors.94 The N-alkylated P82 exhibited a more red-shifted UV-vis absorption spectrum and had a larger I0−0/I0−1 value compared with the O-alkylated P83, implying a tighter aggregation of P82. Moreover, P82 had higher intermolecular self-assembly binding energy due to its stronger intermolecular interaction. The above results led to the smaller π–π distance and interconnected crystalline zones in the P82 film, which was consistent with its better unipolar p-type transport performance with a hole mobility of 0.55 cm2 V−1 s−1. Furthermore, P84 used thieno[2′,3′:5′,6′]pyrido[3,4-g]thieno[3,2-c]isoquinoline-5,11(4H,10H)-dione (TPTI) as the acceptor unit due to its strong electron-accepting ability and structural planarity.95 The terpolymer P84, with 70% TPTI-thiophene, afforded the highest PCE up to 11.02%, which was about 20% higher than those of other polymers with different ratios of donors. In comparison with all these blend polymer films (polymer: m-ITIC) having bimodal backbone orientation, the P84-based blend film exhibited more percentage of face-on orientation, which had vertical charge-transportation channels for better photovoltaic application.96
Just as the asymmetrically constructed DPP units, the IID unit and its derivatives could also have their indolinone moieties changed. To relieve the steric hindrance in the twisted indolinone moieties around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the isoindigo monomer, P85 and P86 were developed on thieno-benzo-isoindigo (TBIID).97 The optical bandgaps and FMO energy levels of P85 and P86 fell between those of IID- and thieno-IID-based counter polymers. The decreased torsional angles between two indolinones were around 0.11 and 0.19° for P85 and P86, respectively, favoring well-ordered polymer packing and the formation of large crystalline domains. Both P85 and P86 exhibited unipolar p-type transport properties and P86 afforded a max PCE of 4.79% in its OSC devices. The TBIID unit was still widely used in polymers nowadays for its good OSC performance guarantee.98 Besides the asymmetrical fused rings for isoindigo, there was also some other way of asymmetrical combination. The asymmetrical fluorine substitution was carried out in P87, in which one of the indolinone units had fluorine substitution on its 7th carbon atom.99 The P87:PCBM film gave a PCE value of 5.03%, slightly higher than those of the above-mentioned TBIID-based polymers (Table 1). This is because the fluorine-related inter- and intra-chain interactions could not only facilitate better backbone planarity but also form a well-mixed film with PCBM.
C bond of the isoindigo monomer, P85 and P86 were developed on thieno-benzo-isoindigo (TBIID).97 The optical bandgaps and FMO energy levels of P85 and P86 fell between those of IID- and thieno-IID-based counter polymers. The decreased torsional angles between two indolinones were around 0.11 and 0.19° for P85 and P86, respectively, favoring well-ordered polymer packing and the formation of large crystalline domains. Both P85 and P86 exhibited unipolar p-type transport properties and P86 afforded a max PCE of 4.79% in its OSC devices. The TBIID unit was still widely used in polymers nowadays for its good OSC performance guarantee.98 Besides the asymmetrical fused rings for isoindigo, there was also some other way of asymmetrical combination. The asymmetrical fluorine substitution was carried out in P87, in which one of the indolinone units had fluorine substitution on its 7th carbon atom.99 The P87:PCBM film gave a PCE value of 5.03%, slightly higher than those of the above-mentioned TBIID-based polymers (Table 1). This is because the fluorine-related inter- and intra-chain interactions could not only facilitate better backbone planarity but also form a well-mixed film with PCBM.
The polymer P88 contained an acceptor PzIIG, having the 1,4-pyrazine ring as the bridge between two indolinones.100 The PzIIG had little difference in the HOMO/LUMO energy levels relative to those of the IID counterpart but gave a dense and ordered packing state, whose π–π stacking distance was as small as 3.29 Å (Fig. 8b), due to the formed N⋯H hydrogen bond between 1,4-pyrazine and indolinone. The P88:PC61BM blend film exhibited a max PCE of 3.12%, which was further enhanced to 5.26% after adding 3% additive of 1,8-diiodooctane and changing PC61BM to PC71BM (Table 1). Similarly, quinoidal polymers P89 and P90 were obtained, with their IID indolinone units connected with a thiophene ring.101 The extended quinoidal acceptor provided the two polymers with narrow optical band gaps. Though there were torsional angles of around 26° between inserted thiophene and indolinone units for both P89 and P90, the annealing treatment facilitated two polymer films to adopt favorable edge-on orientations with strong π–π interactions. Therefore, their off-center spin-coating films afforded the max hole/electron mobilities of 4.82/1.11 cm2 V−1 s−1 for P89 and 8.09/0.74 cm2 V−1 s−1 for P90, respectively. Such outstanding performances could be attributed to the centrifugal force, which led to orderly aggregated polymer chains and improved crystallinities.
Likewise, the acceptor IBTI, with two indolinones connected with a bithiophene unit, was used in P91 × 102 The S⋯N intramolecular interactions relieved the molecular distortion and provided a well-delocalized HOMO over the entire backbone. It was found that the highest PCE value of 3.49% was achieved when the weight ratio of P91:PC61BM reached 1:2. With additional DIO, the P91:PC61BM blend film showed well-proportioned and interpenetrated morphology with good miscibility (Fig. 8c) and finally gave a considerably improved PCE of 6.41%. Considering the rotatable single bonds in bithiophene units, the vinyl groups were introduced into bithiophene to form a stable planar backbone in the obtained VDTOI units and two polymers P92 and P93.103 The increased planarity helped obtain smooth and uniform polymer films. Both P92 and P93 showed p-type transport performances and the highest hole mobility of 0.35 cm2 V−1 s−1 was achieved by the P92 film, due to its higher crystallinity and effective molecular packing state with the π–π stacking distance of 3.71 Å. More recently, the fluorinated TVT moiety and nitrogen-embedded indolinone were combined to afford four polymers P94-P97.104 With much-lowered FMO energy levels and ignorable torsional angles provided by multiple intrachain interactions, all the polymer films showed p-type transport performances. P97 and P98 exhibited the highest μh of 0.48 and 0.47 cm2 V−1 s−1, respectively, attributed to their relatively smaller π-stacking distances of around 3.50 Å. Nonetheless, all those polymers featured unipolar p-type transport performance, whatever the conjugated bridge was bithiophene or TVT unit. This is because their electron-donating nature could hardly tune the LUMO energy level to a preferable one for efficient electron injection. To settle down such a problem, the benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione unit was inserted between two indolinones, leading to a more electron-deficient building block (IBDT) in P100.105 The polymer P100 had much red-shifted absorption spectra with the optical band gap approaching 0.95 eV. Such a narrow energy gap gave the P100 film a highly balanced hole and electron mobility of 0.16 and 0.14 cm2 V−1 s−1, respectively. Higher mobilities were achieved by the other IBDT-based polymer P101.106 The more rigid thieno[3,2-b]thiophene donor contributed to the high crystallinity and ordered interchain packing of the P101 film, which adopted edge-on orientation with the π–π stacking distance of 3.45 Å. All these results resulted in its high μh/μe of 1.70/1.37 cm2 V−1 s−1, respectively. When the outer benzene rings in IBDT indolinone moieties were replaced by thiophene rings, the resulting monomer in P102 featured a more planar backbone due to the S⋯O interaction.107 In comparison with its reference polymer and analog polymer P100, the thiophene replacement provided the P102 with a lowered LUMO energy level and enhanced HOMO energy level, leading to its ultra-narrow band gap of 0.71 eV. Besides, the annealed P102 film showed a large and continuous nano-fibrillar structure, with an interconnected network, rather than clustered crystals in its as-cast film (Fig. 8d). For the above reasons, P102 afforded higher hole and electron mobilities of 0.45 and 0.22 cm2 V−1 s−1, respectively, relative to the mobilities of P100. It has been proved that azaisoindigo was preferable for improving the polymer backbone planarity and charge carrier transport performance.108 Likewise, nitrogen-embedded IBDT (AzaBDOPV)-based polymer P104 was prepared, with a much lowered LUMO energy level and smaller dihedral angles than those of its IBDT-based counter polymer P103 (Fig. 9a). The typical n-type transport performance was achieved for P104, with the highest mobility of 3.22 cm2 V−1 s−1, much higher than that of P103. Such better carrier transport performance was attributed to its shorter π–π distance of around 3.44 Å and its strong crystallinity with fiber-like crystallized zones. Later, when the AzaBDOPV and dithienylbenzothiadiazole units were copolymerized, the resulting polymer P105 afforded balanced FET performance with greatly enhanced mobilities compared to that of P104.109 Using 1-octanethiol self-assembled monolayer modified source and drain electrodes, the spin-coated P105 film displayed the highest hole/electron mobilities of 5.97/7.07 cm2 V−1 s−1, respectively, in its TGBC FET device under ambient conditions. Moreover, P105-based flexible FET devices gave balanced hole/electron mobilities of 4.68/4.72 cm2 V−1 s−1. The denser film stacking state and increased crystallinity of the P105 film, with a short π–π stacking distance of 3.46 Å and clear grains with fiber-like polycrystalline networks, could be helpful to realize its favorable carrier mobilities. At present, incorporating multiple electron-withdrawing atoms in the polymer backbone was proved to be effective for achieving satisfactory optoelectronic properties. In polymer P106, F4BDOPV with multiple fluorine substitutions was chosen as the acceptor.110 The numerous fluorine atoms in P106 formed both the F⋯H hydrogen bond and F⋯S intramolecular interaction, which led to less backbone distortion than its unfluorinated counter polymer (Fig. 9b), and synchronously increased the interchain packing with a small π–π stacking distance of 3.52 Å. The good crystallinity provided the P106 film with an electron mobility of 1.24 cm2 V−1 s−1 (Fig. 9c), which could still maintain 60% after 30 days of exposure under ambient conditions. Such promising good air-stability could be illustrated by its deep-lying energy level.
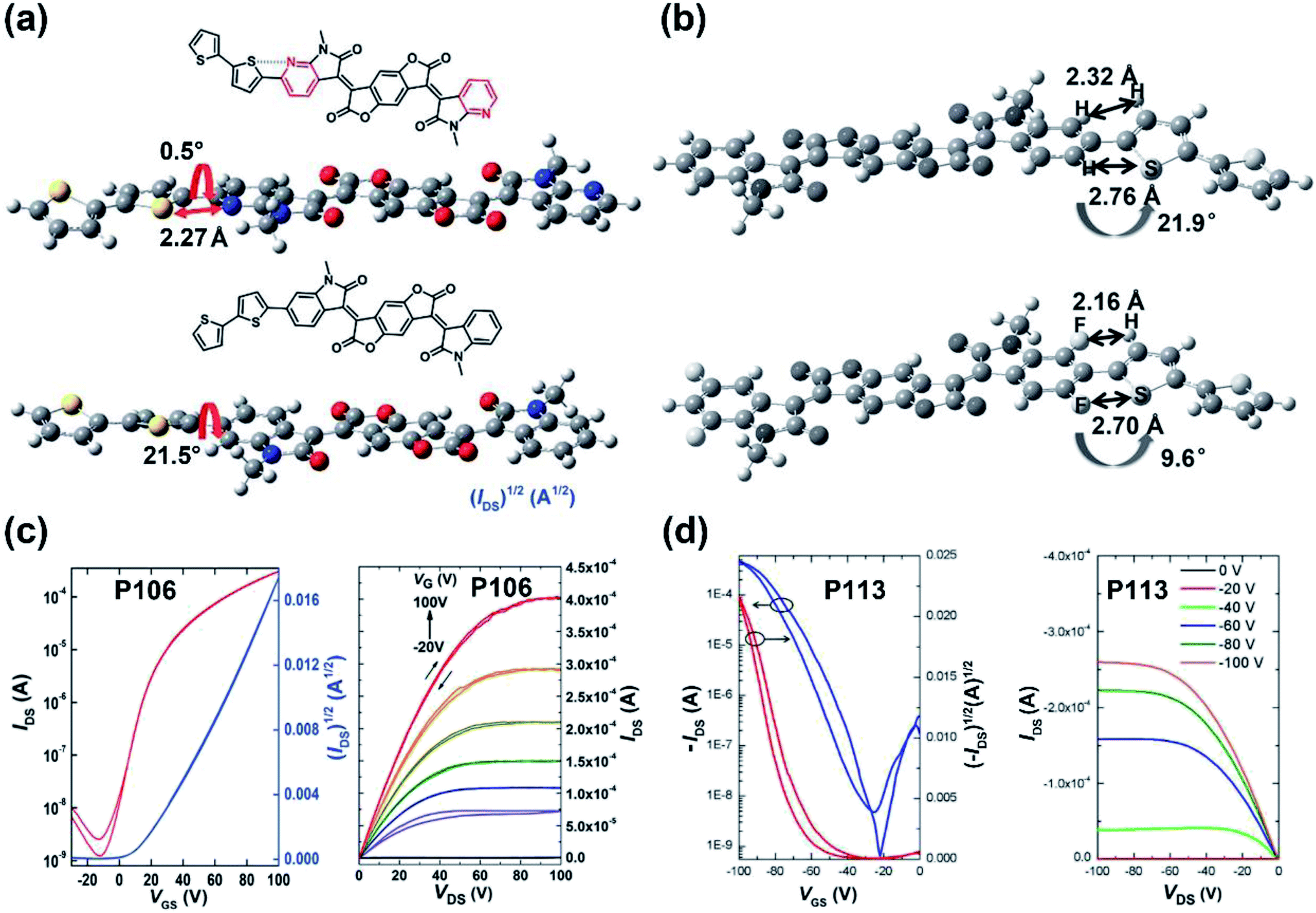 | ||
| Fig. 9 DFT-optimized structures with marked dihedral angles and the intramolecular interaction distances of the AzaBDOPV-2T fragment (a, up), Reproduced from ref. 108 with permission from the Royal Society of Chemistry, copyright 2016, Jian Pei, et al., licensed under CC BY 3.0, https://creativecommons.org/licenses/by/3.0/. BDOPV-2T fragment (a, bottom/b, up), and F4BDOPV-2T fragment (b, bottom). Reproduced from ref. 110 with permission from Wiley-VCH, copyright 2016. (c) The transfer (left) and output (right) characteristics of P106-based FET devices. Reproduced from ref. 110 with permission from Wiley-VCH, copyright 2016. (d) The transfer (left) and output (right) characteristics of P113-based FET devices. Reproduced from ref. 115 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Several modifications of indolinone units were developed, including the incorporation of electronegative atoms, the extension of conjugation, and the replacement of structural building blocks. Meanwhile, researchers endeavored to develop novel inserting core units like structural analogs to IBDF, possessing a planar backbone, extended π-system, and stronger electron with-drawing capacity. Then the novel building block IBDT, in which the benzodifurandione moiety of the IBDF unit was replaced with the benzodithiophenedione moiety, came into the sight of scientists. P107, as an IBDT-based polymer, had a comparable HOMO energy level to the IBDF-based counter polymer, but with a lower-lying LUMO energy level due to its smaller exciton binding energy.111 Finally, the P107 film gave a highly balanced ambipolar carrier transport property with the highest μe/μh of 0.14/0.10 cm2 V−1 s−1, respectively. To achieve ambipolar transport behavior as P107 did but with higher mobilities, π-extended IBDF (INDF) was used in P108 and P109.112 Compared with IBDF, the newly introduced naphthalene ring provided the INDF with an extended conjugation length and a weaker electron-accepting ability. Such changes favored stronger intermolecular interaction and relatively higher FMO energy levels hence achieving more balanced ambipolar FET performances. Though the dihedral angles between the indolinone and central core were slightly increased 3–5° relative to that of IBDF, smooth films with edge-on orientation motif were observed for both the polymers, thus exhibiting highly balanced ambipolar transport performances, with the max μe/μh of 0.14/0.12 cm2 V−1 s−1 for P108 and 0.50/0.51 cm2 V−1 s−1 for P109. Later, the different sidechains were substituted in both donor and acceptor units of another INDF-based polymer, P110.113 The increased solubility of the whole polymer backbone gave a much apparent red-shifted absorption compared to that of P109, indicating a stronger tendency of mainchain aggregation in solution and denser film packing in the solid state. Finally, good FET performance of P110 film was obtained, with the max μe/μh of 0.15 and 0.33 cm2 V−1 s−1, respectively, which were comparable to those of most polymers with INDF.
The improved solubility was proved to be capable of enhancing polymer device performance by adjusting their molecular packing states, and therefore, more available alkylation positions can provide sufficient sidechain modification. For instance, P111 and P112 contained the lactam-modified building block IBDP.114 The two more lactam structures in the acceptor allowed the application of shorter substituted side chains to enhance solubility and the polymer solution processability, without sacrificing the coplanarity. The promising sidechain self-assembly enabled the fully stretched sidechain and produced long d-spacing distances, which reached 27.1 Å for the P111 film and 25.5 Å for the P112 film, respectively. Both P111 and P112 showed ambipolar transport performances with their respective μe/μh of 0.088/0.19 and 0.075/0.10 cm2 V−1 s−1. The four lactam moieties of the IBDP monomer ensured its modifiable solubility and mainchain packing state by diverse sidechain substitutions. After moving the branching point of the sidechain in indolinone away from the backbone of P113, it showed a better lamellar edge-on packing state than P111 and P112 did, thereby facilitating stronger interchain packing.115P113 eventually exhibited typical p-type transport performance and exhibited higher mobility up to 1.92 cm2 V−1 s−1 owing to the fiber-like networks of its film morphology (Fig. 9d). Just like the extension of IBDF to obtain the INDF monomer, the same strategies were applied in the polymers P114 and P115, in which the phenyl-extended IBDP served as the electron-deficient building block.116 Such change had their HOMO energy levels raised to around −5.20 eV, which was preferable for hole carrier injection, thereby leading to their hole-dominated ambipolar performances with the max μh/μe of 1.55/0.021 cm2 V−1 s−1 for P114 and 1.79/0.087 cm2 V−1 s−1 for P115, respectively (Fig. 10).
 | ||
| Fig. 10 The molecular structures of polymers P101–P121. | ||
Generally speaking, the above building blocks, including IBDF, INDF, and other analogs, could be mainly regarded as two types of connection between IID units. One is that two IID units were fused by sharing one benzene ring (e.g., P111-P113) and the other one was the direct fusion of two IID units (e.g., P114-P115). Moreover, the third type of such kind of acceptor was incorporated in the polymers P116 and P117, in which two IID units were fused with the 2,3,6,7-tetrahydro-s-indacene-1,5-dione moiety by intramolecular Friedel–Crafts acylation of carboxylic acids.117 The extended backbone and additional carbonyl groups facilitated the delocalization of both the HOMO/LUMO orbitals and lower energy levels. P116 and P117 showed balanced ambipolar transport performances with both the highest hole and electron mobilities of around 0.10 cm2 V−1 s−1, achieved by P117 due to its more percentage of edge-on orientation crystal domains.
Owing to the current polymerization mechanism, like Stille, Suzuki–Miyaura, and Kumada coupling, the traditional bonding way between aromatic rings is always associated with the single bond, which potentially leads to the backbone distortion and causes the energetic disorder.118 To settle down such a problem, Onwubiko and co-workers utilized a new synthetic approach and disclosed a series of polymers, P118-P120.119 The way of aldol polymerization enabled the building blocks to connect via the double bond. The resulting rigid backbone and planar backbone resulted in apparently broadened absorption spectra and more red-shifted absorption peaks in the region from 850 to 1200 nm. All the polymers exhibited unipolar n-type transport performances and the highest air-stable electron mobility of 0.03 cm2 V−1 s−1 was achieved by P119. Later, another ladder polymer P121 was also reported, whose two building blocks were combined only with a double bond.120 Simultaneously, good backbone planarity was achieved due to the multiple double bonds and the intramolecular hydrogen bonds between two adjacent units. The P121 film eventually displayed unipolar n-type transport performance with the highest electron mobility of 0.18 cm2 V−1 s−1 (Table 1). Though all those polymers did not show good performance in FET devices as expected, which could be possibly attributed to the unideal sidechain self-assembly in their films caused by the rigid-backbone-induced quick interchain packing process, such a double-bond strategy can be used in those block copolymers for adjustment in the film morphology and the device performance.
3. Polymers with imide moieties
Besides DPP and IID monomers, there are some other novel electron-deficient building blocks that featured imide functional groups. Compared with the lactam moiety, additional carbonyl groups of the imide moiety make the molecule more electron-deficient. Hence naphthalene diimide (NDI), perylene diimide (PDI), and some other imide-functionalized acceptors were increasingly used to construct polymer semiconducting materials. Though studies on imide-functionalized polymers were not as early as their corresponding small molecules, their photoelectric performances were rapidly improved and exceeded those of small molecules as their chemical structures were continuously modified.3.1. Polymers based on NDI and its derivatives
Featuring multiple aromatic ring fusion and dual electron-withdrawing imide functional groups, the NDI unit is the ideal electron-deficient building block for semiconducting polymer materials and has been widely investigated. Especially in recent several years, many corresponding analogs and derivatives have been designed and reported to fabricate optoelectronic-device-favorable polymers.The first NDI-based polymer, reported by Guo and co-workers,121 showed promising physical and chemical properties, which attracted the interest of scientists around the world. Later, Yan and co-workers synthesized the polymer P122, which afforded the electron mobility up to 0.85 cm2 V−1 s−1 under ambient conditions (Table 1).122 Thereafter, wide applications in organic solar cells made it well known as N2200. Until today, N2200 is still the most common material used for fabricating high-performance OSC devices. More recently, N2200-containing all-polymer solar cells (all-PSCs) successfully afforded a considerably impressive PCE value of over 10% on 1 cm2 device area.123 Owing to the excellent properties and various applications of N2200, there were numerous attempts to synthesize a better polymer through structural modification. One of the successful attempts was F-N2200, which was obtained by substituting two fluorine atoms onto the bithiophene unit of N2200.124 Owing to the strengthened intramolecular stacking and more effective Si-polymer contact provided by the additional F–H interaction, Si-PEDOT:PSS-based solar cells with F-N2200 as the interlayer showed higher PCE up to 14.5% than 12.6% of its N2200 counterpart. When additional well-soluble donors were introduced, P123 and P124 were obtained.125 Benefitting from their amine or ammonium bromide group substitution, both the polymers showed good solubility in water and alcohol, which enable different films to be obtained with varied thicknesses. 5 nm-thick P123 and P124 films eventually exhibited the best OSC performances with the max PCE of 9.42% and 10.11%, respectively. In fact, shortly after the report of the first NDI-based polymers, a high electron transport mobility of up to 6.0 cm2 V−1 s−1 was achieved by the NDI-based small molecule.126 Nevertheless, the NDI-based polymer with high FET mobilities was not reported until 2017, when Zhao and co-workers reported four NDI-based conjugated polymers, P125-P128.127 All polymers had broad UV-vis absorption bands, indicating an interactive polymer backbone, and P125/P126 showed broader ones due to the strong electronic coupling effect of selenophene. All polymer films adopted edge-on-dominated bimodal orientations with small π–π stacking distances (Fig. 12a). P125 and P127 exhibited ambipolar transport properties with the highest μe/μh of 8.5/1.7 and 3.1/0.7 cm2 V−1 s−1, respectively, whereas their fluorinated counter polymers P126 and P128 afforded unipolar n-type transport performances with the max μe of 3.5 and 2.2 cm2 V−1 s−1, respectively. Such differences were caused by the increased hole injection barrier of fluorinated P126 and P128, thereby curtailing the injection of the hole carrier and facilitating n-channel carrier transport performances. For well optimizing the backbone conformation, vinylene spacers were introduced into P129 and P130 to form multiple hydrogen bonds.128 The improved planarities resulted in short π–π stacking distances for P129 and P130, thus facilitating electron-dominant ambipolar transport behaviors, with the respective high electron mobilities of 3.95 and 7.37 cm2 V−1 s−1 being obtained from the transfer and output characteristics of P129 and P130 (Fig. 12b). The much higher electron mobility of the P130 film could be explained by its dense π–π stacking in both out-of-plane and in-plane two directions with the small distances of 3.45 and 3.40 Å (Fig. 12c).
The NDI core was constituted by several fused rings. Further heteroaromatic-fusion on it achieved a planar and rigid backbone for good interchain π–π stacking. The thiophene-fused NDI (NDTI) unit in P131 showed a highly planar backbone, having a one-dimensional columnar structure with the π–π stacking distance of around 3.43 Å in its single-crystal structure.129 This preferable packing state provided the P131 film with ambipolar performance with μh and μe of 0.10 and 0.27 cm2 V−1 s−1, respectively, indicating the comparable application potential of the NDTI unit to the NDI unit. Moreover, P132, in which NDTI was combined with dithienothiophene, featured a much red-shifted absorption and a PCE of 5.57%, which was over twice as high as that of the NDI-based counter polymer.130 Using the same NDTI unit in the synthesis of the polymer P133-P135, Wang and Takimiya incorporated bithiopheneimide and thiazole units to achieve different polymer backbone geometries.131 Different from the wave-line backbone of P133, thiazole incorporation changed the backbones of P134-P135 to a straight-line geometry (Fig. 13a), which induced improved intermolecular packing (Fig. 13b) and hence the FET performance. Despite the ambipolar transport behaviors of all the polymers, P134 and P135 had much-improved electron mobilities. P135 afforded an electron mobility of 0.55 cm2 V−1 s−1, which was nearly a hundred-fold higher than that of P134 and also suppressed most of the other NDTI-containing polymers. Such results for P134-P135 were also associated with their different backbone orientations, in which the farther branching point provided P135 with bimodal orientation rather than the face-on and edge-on orientation of P133 and P134, respectively (Fig. 13c).
The extended NDI backbone contributed to a more extended conjugation and low-lying LUMO energy level of polymers, which were preferable for FET application. However, low LUMO levels of NDI-based polymers led to low Voc (<0.6 V) for OSC devices. Therefore, the 1,2,5,6-naphthalenediimide (1,2,5,6-NDI) unit, with less aromatic fused rings, was built up in P136 and P137.132 The decreased backbone electron deficiency enabled both polymers to have raised LUMO energy levels of around −3.30 eV, which successfully enhanced their Voc to above 0.9 V, with the highest PCEs of 5.04% for P136 and 6.35% for P137. These results showed a big influence of the different unit extensions on the final performance of their corresponding polymers. Besides, the controllable copolymerization site was also proved to be efficacious, e.g., P138 and P139 based on the novel electron-deficient building block dithieno[3,2-a:3′,2′-j][5,6,11,12]chrysene diimides (DTCDI) had more extended π-conjugation compared with 1,2,5,6-NDI.133 The electron-deficient nature of DTCDI resulted in much lower LUMO energy levels of P138 and P139, and more importantly, its unique structure ensured different reaction sites. The DTCDI-3,9-connected P138 film displayed uniform fibrous nanostructures with a larger grain size than that of the DTCDI-thiophene α,α′-connected P139 film. Such differences explained their huge different device performances. P138 showed a PCE of 7.52% in its OSCs and a FET electron mobility reaching 0.11 cm2 V−1 s−1, which was greatly higher than that of P138. The dual imide structures in the above-mentioned electron-deficient building blocks were symmetrically constructed in the skeleton, thus facilitating even distribution of FMO orbitals. With only one imide moiety located at one side of the main chain, the polymer P140 was developed based on a single-imide-featured TCNDIO unit.134 Despite one less imide structure of TCNDIO, P140 also had a wide absorption band range with a narrow optical band gap of 1.06 eV and comparable energy levels to other dual-imides-functionalized polymers. Because of a small torsional angle of 10.6° in the TCNDIO monomer, the P140 film adopted face-on orientation with the π–π stacking distance of around 3.79 Å. Finally, P140 showed a p-channel charge transport behavior with a hole mobility of 0.02 cm2 V−1 s−1. From another perspective, such a strategy could be further optimized to achieve a decreased density of side-chain substitution thus facilitating more effective intermolecular mainchain stacking.
Different from the original extension position of the NDI unit, in which the aromatic rings were fused next to the phenyl, the thienopyridine-fused naphthalene amide (TPNA) unit was obtained by fusing at the diagonal position of the NDI unit.135 Three types of polymers (homopolymer, D–A polymer, and all-acceptor polymer) were synthesized, namely P141, P142, and P143, with deep-lying HOMO energy levels ranging from −5.99 to −5.76 eV. The promising backbone planarity and π-conjugation conferred unipolar electron transport properties to all three polymers. Compared with P141 and P142, P143 had better planarity, lower-lying HOMO energy levels, and a closer π–π stacking distance of 3.49 Å, hence exhibiting better FET performance with the highest μe of 0.19 cm2 V−1 s−1 (Table 1).
Obviously, NDI-based polymers have both advantages and disadvantages. The brominated NDI monomer has less synthetic complexity, high yield, and a simple purification process. These positive aspects promise convenient access to varieties of polymers containing NDI or its structural analogs. NDI-based polymers feature good air-stable electron transport behaviors due to their deep-lying LUMO energy levels. Although NDI and its derivatives are widely applicable, there are still some drawbacks in them. The molecular structures of NDIs curtail the good coplanarity of the polymer main chain backbone, which makes most NDI-containing polymers have their LUMO located only at the NDI units, thus leading to much lower charge carrier mobilities than those of DPP- or IID-based counter polymers. And such drawbacks might be the main thing to surmount in the nearest future.
3.2. Polymers based on PDI and its derivatives
The PDI unit is also another popular imide-functionalized electron-deficient building block for application in semiconductor polymers. Compared with the NDI unit, the PDI unit is structurally similar but has more fused benzene rings and has been investigated earlier. In 2007, Zhan and co-workers firstly reported a PDI-based polymer, exhibiting good solubility and thermal stability.136 Such polymers afforded n-type FET performance (1.3 × 10−2 cm2 V−1 s−1) and also gave a max PCE over 1% in its OSC devices, both of which were at high levels at that time. Since then, numerous semiconducting polymers have been developed based on PDI and its derivatives, especially in recent several years. As shown in Fig. 11, polymer P144 showed good solubility in green solvents due to its long side chain in the NDI unit.137 The much-increased solubility enabled the anisole-processed PBDT-TS1:P144 film to afford a max PCE of 5.43% and an improved PCE of 6.58% in its inverted all-PSCs (Fig. 14a and Table 1), both of which were comparable to the highest value achieved by the film treated with halogenated solvent. Polymers with various D–A pairs, A–A type polymers, P145-P147, were also reported based on three available stannylated units (TPTQ, FTPTQ, and TPTI), containing lactam moieties.138 In comparison with the FMO energy levels of other PDI-based polymers, the additional acceptor units in P145-P147 showed limited adjustment to their LUMO energy levels but greatly dragged down their HOMO energy levels. The different internal polarization resulted in their different PCE values and P145-based devices afforded the highest PCE of 3.52% among the three polymers, which could be explained by its stronger face-on orientation after modification and the better binding affinity with the electron-donating material PTB7-Th. | ||
| Fig. 11 The molecular structures of polymers P122–P158. | ||
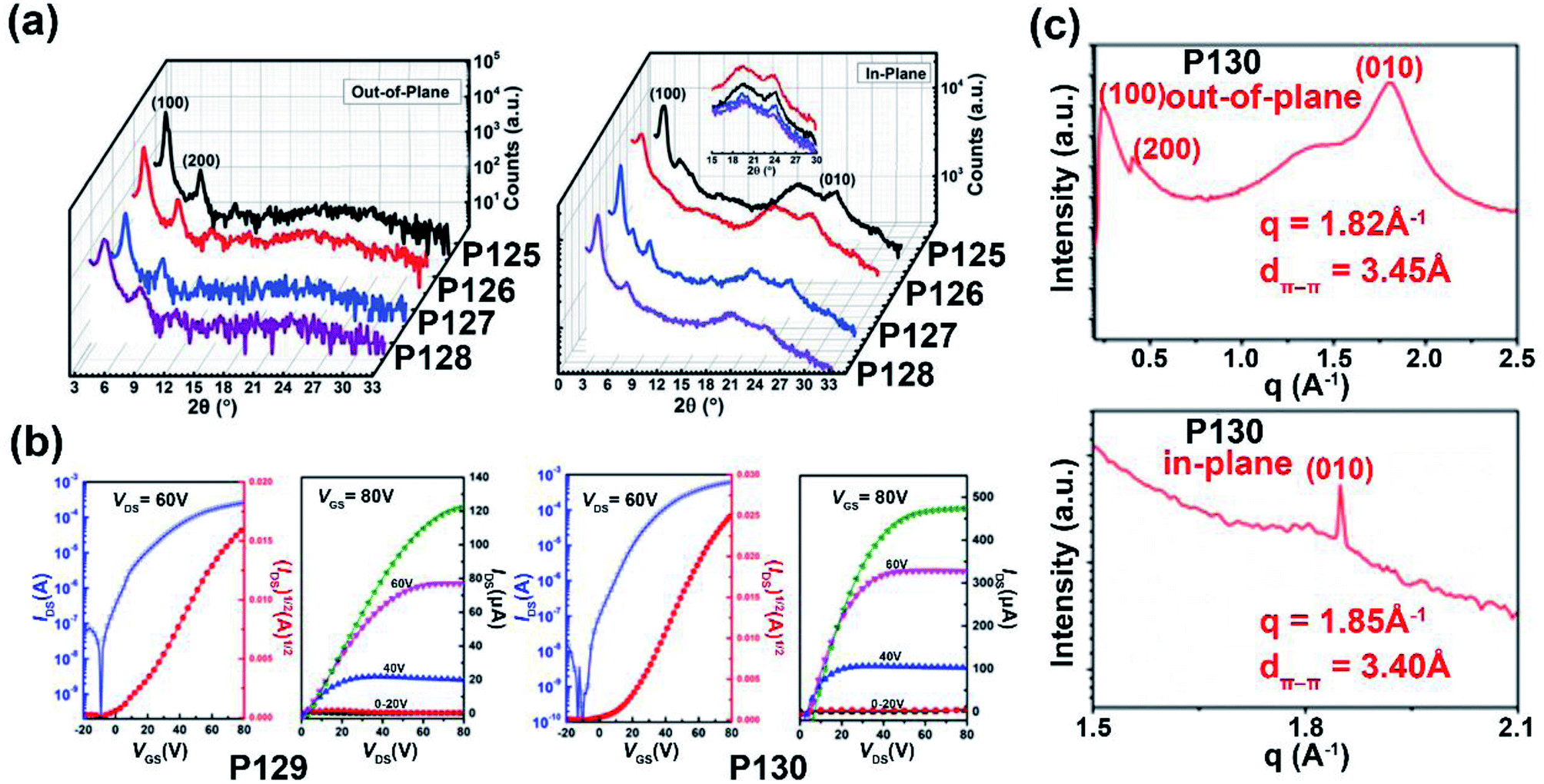 | ||
| Fig. 12 (a) The GIXRD profiles of P125-P128 films (annealed at 200 °C) in the out-of-plane (left) and in-plane direction (right). Reproduced from ref. 127 with permission from Wiley-VCH, copyright 2017. (b) The transfer and output characteristics of P129- and P130-based OFET devices. Reproduced from ref. 128 with permission from the American Chemical Society, copyright 2019. (c) 1D profile of P130 in the out-of-plane (up) direction and in the in-plane direction (bottom). Reproduced from ref. 128 with permission from the American Chemical Society, copyright 2019. | ||
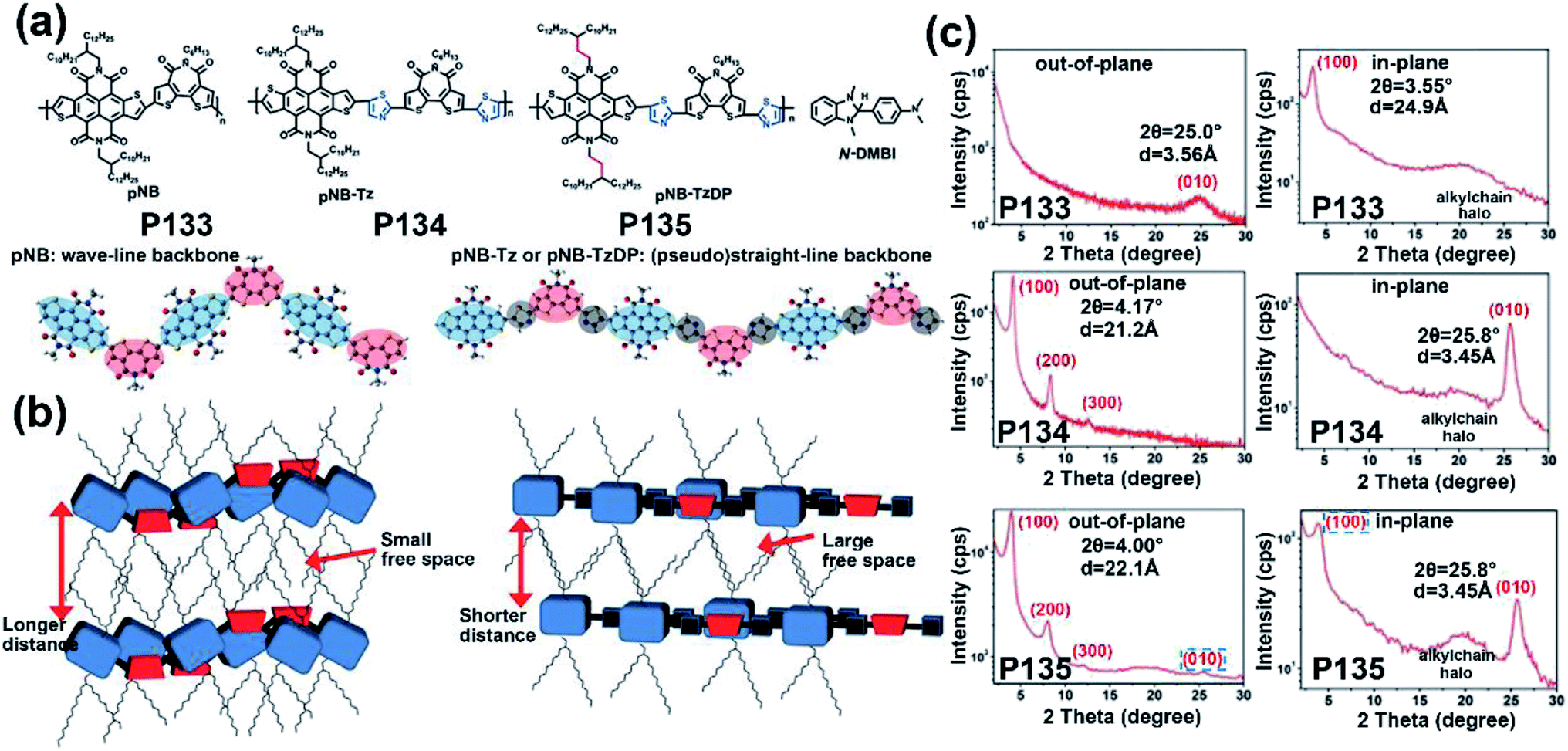 | ||
| Fig. 13 (a) Molecular structures of P133-P135 and n-doped N-DMBI (up) and their optimized backbone geometries (bottom). Reproduced from ref. 131 with permission from Wiley-VCH, copyright 2020. (b) The illustration of interpolymer the docking models of P133 (left) and P134 (right). Reproduced from ref. 131 with permission from Wiley-VCH, copyright 2020. (c) 1D GIXRD patterns of P133-P135 in the out-of-plane direction (left) and in the in-plane direction (right). Reproduced from ref. 131 with permission from Wiley-VCH, copyright 2020. | ||
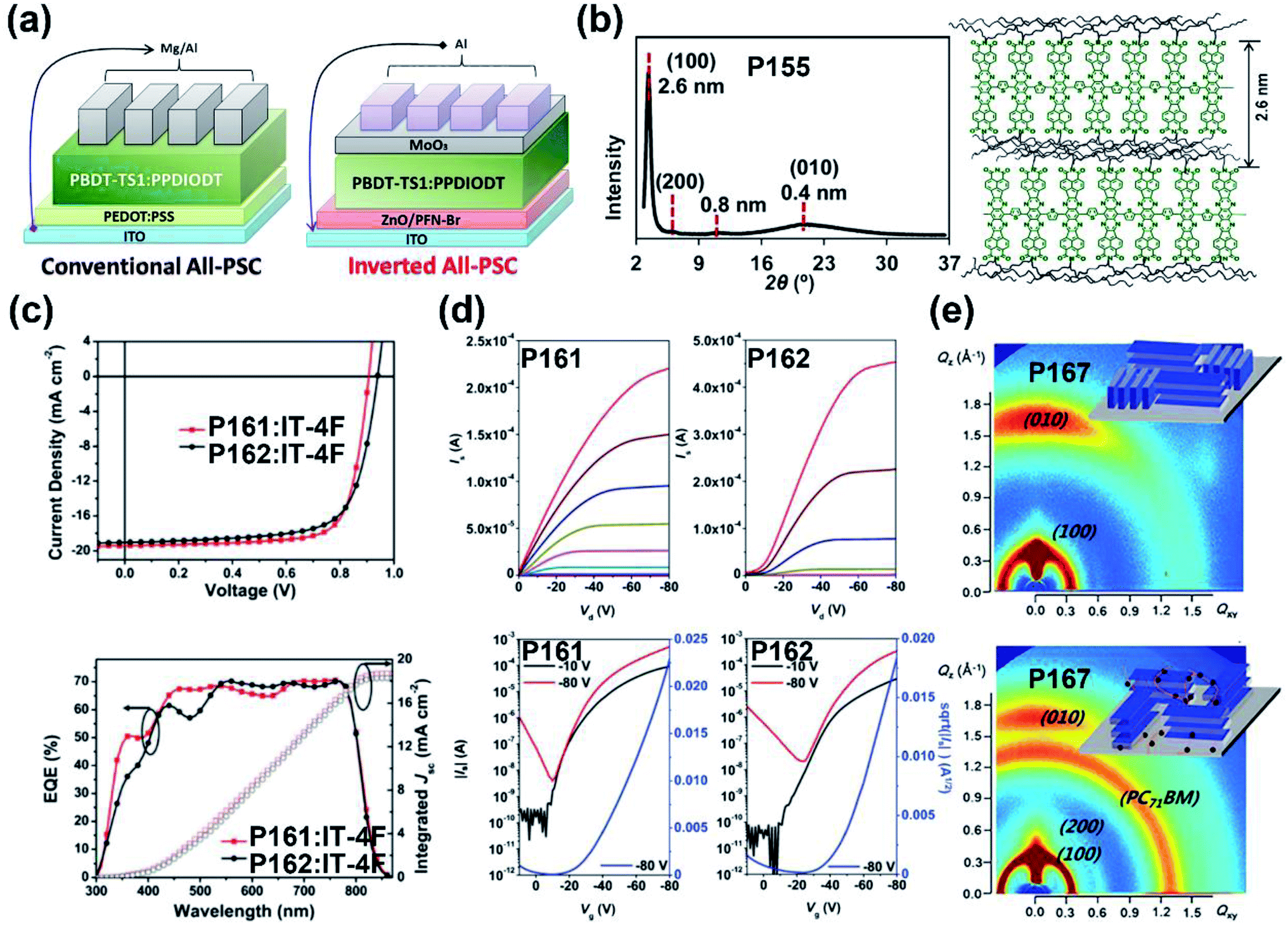 | ||
| Fig. 14 (a) The device architectures of the conventional (left) and inverted (right) all-PSCs. Reproduced from ref. 137 with permission from Wiley-VCH, copyright 2016. (b) The XRD pattern of the annealed P155 film on the glass substrate (left) and the ordered lamellar packing state deduced from the diffraction patterns (right). Reproduced from ref. 144 with permission from the American Chemical Society, copyright 2013. (c) J–V characteristics (up) and EQE spectra (bottom) of the P161- and P162-based solar cell devices, with the IT-4F as the acceptor material. Reproduced from ref. 148 with permission from Wiley-VCH, copyright 2019. (d) The output (up) and transfer (bottom) characteristics of P161 and P162. Reproduced from ref. 148 with permission from Wiley-VCH, copyright 2019. (e) 2D-GIXRD patterns of the neat P167 film (up) and P167:PC71BM blend film (bottom). Reproduced from ref. 153 with permission from the Royal Society of Chemistry, copyright 2015. | ||
As is well known, the extended and electron-deficient acceptor unit of D–A type polymers can adjust the FMO energy levels and bandgaps, which are beneficial for their device performances. Thus, with the π-extended dithienocoronenediimide as the acceptor, P148 and P149 were synthesized to achieve a high degree of regioregularity.139 Because of the poor solubility of P149, the additional alkylation of the donor was carried out in polymer P150, which gave it more widened and red-shifted absorption bands compared with those of P148. The electron-rich nature of thiophene fused in dithienocoronenediimide led to the higher HOMO/LUMO energy levels of P148 and P150 than those of P144-P147. The planar molecular backbone, enhanced regioregularity of dithienocoronenediimide, and the narrow energy gaps eventually provided P148 and P150 with ambipolar transport behaviors with higher electron mobilities than those of their PDI-based counter polymers. To avoid the high-rigidity-caused poor solubility of polymer P149 and to modulate the miscibility with other polymers for high-performance OSCs, Yang and co-workers reported a polymer P151, comprising asymmetric thianaphthene-fused perylenediimide (TDI).140 The slightly relieved backbone rigidity provided the polymer with good solubility, thus forming uniform blend film morphology with a smooth surface, facilitating a higher PCE of 4.70% than its PDI- or dithienocoronenediimide-based reference polymers did. It should be pointed out that the advantages that the extended planar backbone could bring to the polymers were much more important than the possibly resulting poor solubility, which was easier to modify, for instance, P152 and P153, which were based on naphthodiperylenetetraimide (NDP) units with two fused PDI units.141 The super rigid and π-extended backbone led to improved backbone coplanarity and the delocalization of π-electrons, with their LUMO energy levels below −4.10 eV. Both the polymers were blended with PTB7-Th and behaved well in their OSC devices with the highest PCE of 6.39%. Such preferable performance was surpassed by another NDP-based polymer, P154, in which NDP monomers were connected with vinylene units.142 The larger polycyclic NDP units provided the corresponding polymers with tunable chain flexibility and an improved polymeric packing state. The AFM images revealed a smooth and uniform blend film of P154 with small root-mean-square (RMS), leading to better contact between the active layer and the interfacial electrode. The PTB7-Th:P154 blend film afforded a high PCE up to 8.59%, proving the efficiency of the fusion strategy to improve the PCE values. Thereafter, more fused derivatives were reported. Recently, Yu and co-workers developed several novel small molecules, in which two NDP units were fused with the benzene rings of IID, BDOPV, etc.143 All the extended building blocks had narrower band gaps than those of their respective fused inserting units, implying their potential usage in the polymer backbone. In addition to extending the NDI or PDI unit cores laterally, the longitudinally extended tetraazabenzodifluoranthene diimide (BFI) monomers in P155 and P156 could also evenly distribute the FMO but in the perpendicular direction to the mainchain.144 The vertical extension also provided the two polymers with wide and NIR absorption regions just as the lateral extension could achieve. Moreover, both polymers had orderly packed films, especially for the P155 film, which had ordered lamellar crystalline with the d-spacing (2.6 nm) roughly the same as the backbone length of the BFI unit (Fig. 14b). The n-type transport properties were achieved for P155 and P156 with the highest mobility of 0.30 cm2 V−1 s−1 being achieved by P155 due to its good solubility and high quality of the thin film.
The above NDI, PDI, and their electron-withdrawing derivatives were always used in the polymer main conjugated backbones to build up the connection of donor and acceptor units. Creatively, Drewniak and co-workers introduced NDI and PDI units in the sidechain of P157 and P158, respectively.145 Such attempts provided the two polymers with a moderate PCE of around 0.03% in their OSC devices (Table 1). However, introducing the donor unit at the farther position of the main chain backbone could potentially form the interaction between donors and acceptors from different polymer chains during the chain packing process and might be a feasible method to enhance the interchain charge transport in the near future.
As summarized above, a large number of polymers with favorable performances in OFETs and OSCs were reported, whereas there are still some problems that existed. Limited by the molecular structure, there are too many byproducts that appear during the synthesis of the brominated PDI monomer, which directly reduces the yield of the desired product and makes it hard to be purified. Besides, PDI and its derivatives have the same problem in steric hindrance as the NDI does when connected with other units. The resulting poor molecular backbone planarity ultimately led to moderate or even low device performances for those polymers.
3.3. Polymers based on other imide-functionalized units
In addition to the widely investigated NDI and PDI units, there are also some other imide-functionalized electron-deficient building blocks, such as phthalimide (PHI), thieno[3,4-c]pyrrole-4,6-dione (TPD), and bithiophene imide (BTI) units.Different from NDI and PDI units, the PHI unit is less electron-deficient and showed absorption in the short wavelength range, thus having the potential to obtain polymers with wide band gaps and achieve unipolar FET performance. In 2009, the PHI unit was firstly introduced into polymers for optoelectronic devices by Guo and co-workers and the corresponding polymer eventually afforded the highest μh of 0.28 cm2 V−1 s−1.146 Then the fluorinated PHI unit was utilized to synthesize P159, which had lower FMO energy levels than those of PHI-based reference polymer P160.147 Both the polymers exhibited good OSC performances with the highest PCE value of 8.31% for P160 and 9.48% for P159, with their Voc over 0.90 V (Table 1). Thereafter, fluorinated benzothiadiazole and alkylated thiophene units were introduced into the backbones of P161 and P162.148 In comparison with P159 and P160, the acceptor units in polymers P161 and P162 greatly lowered their HOMO/LUMO energy levels and achieved narrower band gaps. Moreover, the alkylated thiophene units also facilitated a better blend film miscibility with a highly interpenetrating and continuous network. Because of these advantages, both P161 and P162 showed favorable PCE values of 13.31% and 12.74%, respectively (Fig. 14c). Apart from the high PCE, two polymers afforded unipolar p-type transport performances with the hole mobilities of 0.63 and 0.93 cm2 V−1 s−1 for P161 and P162, respectively (Fig. 14d), due to their better crystallinity and delocalized π-conjugation. These results revealed the effectiveness of fluorine introduction and the dual acceptor strategy in improving the OSC performances.
A new member of the family of PHI-derived monomers, 4,8-di(thien-2-yl)-6-octyl-2-octyl-5H-pyrrolo[3,4-f]benzotriazole-5,7(6H)-dione (TZBI), for constructing high-performance polymers was reported by Lan and co-workers.149P163 featured a wide optical band gap of 1.81 eV and high HOMO/LUMO energy levels. After thermal annealing and the application of the water/alcohol interlayer in the OSC devices, P163:PC71BM gave a high PCE of 8.63%, which was surpassed by two of its analogs P164 and P165.150 Both the polymers showed apparently lowered FMO energy levels and almost the same optical band gaps as P163. Because of the smooth film surface of their blend films and relative balanced hole/electron mobilities, P164 and P165 afforded the max PCE values of 10.24% and 12.12%, respectively. These results also demonstrated that the branched side chain strategy and fluorination together could improve the performance of OSCs by facilitating ordered film morphology and more balanced transport performance. Similar to the PHI unit, thieno[3,4-c]pyrrole-4,6-dione (TPD) was structurally simple, symmetric, and planar, which was preferable for semiconducting polymers. The first TPD-based polymer, P166, was firstly synthesized in 2009 and afforded a PCE of 5.5%.151 Thereafter, the P166-based ternary blend film with PBDTTT-CT and PC71BM exhibited a much-enhanced PCE of 9.3%, attributed to the good miscibility and favorable morphology of the ternary blend film.152 When the thienothiophene-flanked TPD and thiophene units are combined, the polymer P167 was obtained.153 In comparison with the neat P167 film, the P167:PC71BM blend film, having an additional 1,8-diiodooctane (DIO), provided an enhanced crystallinity, a smaller π–π stacking distance of 3.69 Å, and more percentage of the face-on orientation component (Fig. 14e). The appearance of more face-on stacking was preferable for charge carrier transport, thereby achieving a higher PCE up of 9.21%. To achieve a larger planar backbone and stronger electron-withdrawing ability, the bithieno[3,4-c]pyrrole-4,6-dione (BiTPD) unit, with two adjacent TPD units, was incorporated into polymers P168, P169,154 and P170.155 The sulfur–oxygen interaction in BiTPD supported three polymers with good planarities. P168- and P169-based TGBC FET devices exhibited p-type transport performances with their highest hole mobilities of 0.74 and 0.32 cm2 V−1 s−1, respectively. The higher mobility of P168 could be associated with the evenly dispersed granular grains in its annealed film and higher crystallinity. Moreover, benefiting from the face-on packed blend film and the induced polymer vertical π-overlap, the P170-based OSC afforded the highest PCE of 14.2%, which is much higher than those of P166 and P167. Similar to the BiTPD structure, which directly combined two TPD units, the TPD and NDI units in P171 and P172 were also connected via the thiazole unit in different ways.156 Though P171 and P172 had comparable FMO energy levels, P172 gave a more torsional backbone (Fig. 16a). Interestingly, both polymers afforded unipolar n-type transport performances whereas in different orders of magnitude. The backbone-distorted P172 exhibited a much higher electron mobility of 2.55 cm2 V−1 s−1 than the 0.06 cm2 V−1 s−1 of P171. Such a disruptive phenomenon could be attributed to the more ordered film packing state and higher film crystallinity with multiple Bragg peaks of the P172 film (Fig. 16b). These results were opposite to the general cognition of the relationship between the polymer backbone planarity and the FET performance. Considering their different film morphologies and electron mobilities, the most key factor in achieving high electron mobilities could be the good film morphology formed during the process of fabricating the device rather than the planar backbone.
The imide groups of the PHI and TPD units were both constructed in a five-membered ring, which had limited π-extension and modifiable position. Therefore, the N-alkyl-2,2′-bithiophene3,3′-dicarboximide (BTI) unit built up its imide structure in a seven-membered ring with two fused thiophene units. Since the first report in 2008,157 the BTI unit has been increasingly investigated and had derived many types of monomers, among which dithienylthienothiophenebisimide (TBI) was the most common one to see. With two fused BTI units, TBI was equipped with two imide groups and the more extended π-conjugation. A series of polymers P173-P176 were prepared based on TBI by Saito and co-workers.158 All the polymers exhibited good FET performances but different charge polarities. P173 and P174 showed p-channel transport behaviors while ambipolar and n-type transport performances were achieved by P175 and P176, respectively. Moreover, the face-on orientation in the P174/PC71BM blend film was beneficial for its hole transport and charge collection, thus facilitating the highest PCE of 8.0% among those polymers. More recently, Shi and co-workers developed two all-acceptor containing polymers P177 and P178 with considerable performances in both OFETs and OSCs.159 The homo-polymer P177 and polymer P178 showed apparently lowered LUMO energy levels relative to P173-P176, which provided them with unipolar n-type transport performances with the respective max electron mobilities of 3.10 and 1.23 cm2 V−1 s−1, respectively (Fig. 16c). Moreover, compared with their neat films, the apparent face-on orientations with stronger (010) scattering peaks were observed in both blend films of P177 and P178. As the electron-accepting materials in the OSC devices, P177 and P178 exhibited a PCE of 6.67% and 8.61%, respectively (Fig. 16d). Compared with the polymer derived from the condensation of BTI and hexamethylditin units, P177 obtained from BTI and distannylated BTI units had a higher Mn of about 35.5 kDa and showed a smoother film morphology with small RMS roughness, thereby reducing the carrier traps in the polymer film.
Besides the fusion strategy in the TBI monomer, two directly connected BTI units were also used in P180.160 When compared with the more rigid TBI-based P179, P180 had more blue-shifted absorption spectra, despite their comparable HOMO and LUMO energy levels. Besides, the fused BTI units enabled the P179 film to strongly aggregate, thereby facilitating continuous domains in its blend film. Such preferable properties led to more efficient exciton dissociation and charge transport. Therefore, P179-based FET devices showed a μe of 1.13 cm2 V−1 s−1 and afforded a PCE up to 6.85% in its OSC devices. Both performances were much better than those of polymer P180. It was not strange for the unideal properties of P180 when considering its single bond connected BTI units, which led to a sine-wave shaped backbone rather than the linear shape of P179 (Fig. 16e). Thereafter, the further fluorine-modified P181 displayed much improved performances in both OFET and OSC devices compared to those of P180.161 The incorporation of fluorine atoms formed multiple S⋯F intramolecular interactions thus promising a more planar backbone like the TBI unit in P179 to afford effective π-conjugation and strong intermolecular packing. Eventually, P181 displayed considerably enhanced OFET performance with a μe of 2.73 cm2 V−1 s−1 and a high PCE of 6.50% in its OSC device, both of which were greatly enhanced in comparison with P180 and comparable to those of P179. Because of the electron-donating nature of the thiophene rings, the TBI unit actually was not electron-deficient as other imide-containing acceptors, which could also be concluded from the high LUMO energy levels of corresponding polymers. Therefore, the polymer P182 was prepared based on the modified building block f-FBTI2-Br, which has a fluorine atom substituted at the β-positions of thiophene in the TBI unit.162 The LUMO energy level was slightly lowered to −3.46 eV and the fluorinated-TBI unit showed improved backbone planarity compared to the unfluorinated one, with a smaller dihedral angle of 0° between two BTI units (Fig. 16f). In comparison with the preferential edge-on orientation of the P179 film, the fluorination provided the P182 film with a face-on dominating bimodal orientation with a distance of 3.6–3.7 Å, thus leading to its further enhanced PCE of 8.1%. As the other most used electron-withdrawing atom, nitrogen was also embedded in the TBI unit of polymer P183.163 The S⋯N intramolecular interaction greatly optimized the planar conformation of the thiazole-based TBI unit thereby facilitating a dense molecular packing state with a π-stacking distance of 3.35 Å (Fig. 16g). Different from the fluorinated TBI unit in polymer P182, the thiazole-based imide in P183 could not only lower the FMO energy levels but also adjust the dihedral angles between two adjacent BTI units to almost 0° without sacrificing the planarity of the BTI core, thus forming a compact π-stacking distance of 3.65 Å. The off-center spin-coated P183 film showed the highest electron mobility of 1.61 cm2 V−1 s−1. The high reactivity of the distannylated BTI unit enabled novel acceptor–acceptor polymers, which were favorable for perovskite solar cells (PVSCs) due to the potentially improved electron extraction and simultaneously optimized hole-blocking property compared with the D–A polymers. Therefore, the NDI and PDI units were chosen to replace one of the BTI units in P177 and finally afforded the polymers P184 and P185, respectively.164 Both polymers were applied in perovskite solar cells (PVSCs) as electron transport materials to replace PC61BM and exhibited remarkable PCE values of 19.5% and 20.8% for the P184- and P185-based devices, respectively. Besides, the high hydrophobicity of two polymers protected the perovskite active layer from the penetration of moisture, thereby promising outstanding device stability, especially for P185, whose PCE maintained 90% of the initial value after 70 hours.
The BTI unit was mostly developed based on the seven-membered imide ring, as shown in Fig. 15, the polymers P186-P188 were built up with tricyclic aromatic six-membered lactam monomers.165 All their acceptors contained six-membered lactam rings with fused benzene or thiophene moieties. Three polymers showed similar FMO energy levels and band gaps but different performances in OSC devices under the same conditions. Attributed to the less bimolecular recombination and more balanced hole/electron mobilities, P187 afforded a higher PCE of 10.16% than 8.61% and 8.47% of P186 and P188, respectively.
 | ||
| Fig. 15 The molecular structures of polymers P159–P191. | ||
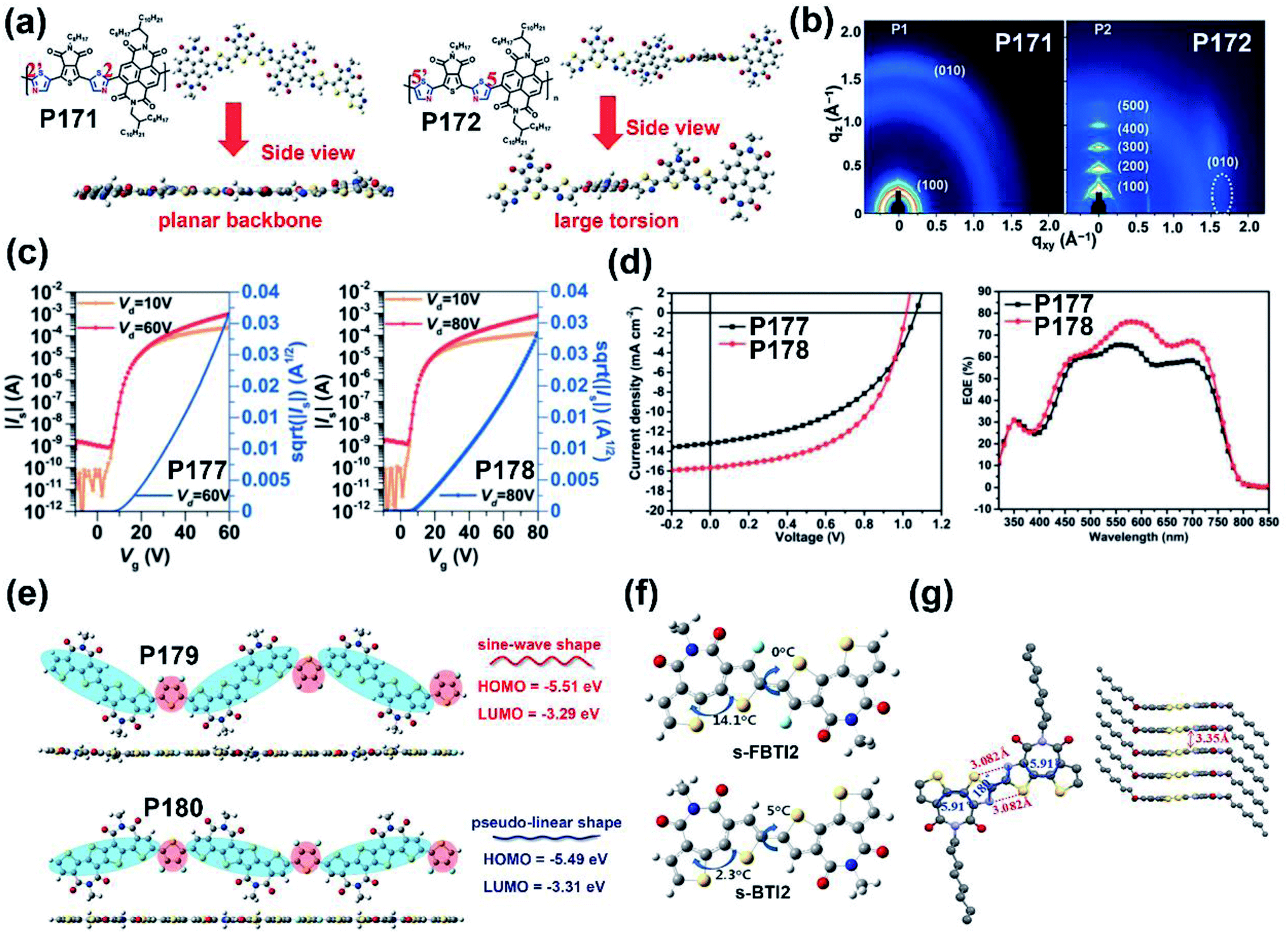 | ||
| Fig. 16 (a) The molecular structures of P171-P172 and their side views of the optimized backbone geometries. Reproduced from ref. 156 with permission from Wiley, copyright 2019. (b) 2D-GIXRD patterns of P171-P172. Reproduced from ref. 156 with permission from Wiley, copyright 2019. (c) The transfer characteristics of P177- and P178-based OFETs. Reproduced from ref. 159 with permission from Wiley, copyright 2020. (d) J–V characteristics and EQE spectra of the PTB7-Th: P177(P178)-based all-PSCs. Reproduced from ref. 159 with permission from Wiley, copyright 2020. (e) The simulated backbone conformation with the minimum energy and the backbone curvatures of the P179 and P180 trimers. Reproduced from ref. 160 with permission from Wiley, copyright 2017. (f) The DFT-calculated structures of the dual BTI monomers before (bottom) and after fluorination (up). Reproduced from ref. 161 with permission from Wiley, copyright 2018. (g) Single crystal structure (left) and the π-stacking distance and packing state (right) of the repeating unit of P183. Reproduced from ref. 163 with permission from Wiley, copyright 2018. | ||
Compared with the seven-membered imide, the above three six-membered building blocks had one less carbonyl group, which led to the much higher FMO energy levels of their corresponding polymers in comparison with most BTI-based polymers. Therefore, Huang and co-workers developed a new building block, EBIQ, based on the acceptor unit of P188.166 The replacement of one fused phenyl ring with the carbonyl group provided the EBIQ with an additional O⋯H hydrogen bond and preferable planarity, promising small π-spacing distances of around 3.50 Å for P189-P191 (Table 1). With the bimodal orientation for their films, all three polymers showed ambipolar transport behaviors, and the highest hole/electron mobilities of 0.138/0.074 cm2 V−1 s−1 were achieved by P190 due to its smallest π–π distance of 3.47 Å provided by enhanced dispersion interactions.
4. Summary and outlook
Recent two and three decades have witnessed the rapid development of semiconductor polymers for applications in OEFTs and OSCs. Among the widely investigated polymers, a large number of D–A alternating polymers have been designed and synthesized, which could be attributed to the prosperous development of donor and acceptor units, especially easily adjustable electron-deficient building blocks.In the first part, we discuss lactam-containing polymers based on electron-withdrawing building blocks, containing DPP, IID, and their derivatives. Because of the variety of adjustable flanking units, DPP-based polymers are equipped with numerous optoelectronic properties, such as good solubility offered by furan, deep-lying FMO energy levels enabled by pyridine, thienothiophene-orientated extended backbone conjugation, individually adjustable LUMO energy levels offered by thiazole, selenophene-induced compact polymer chain packing state, etc. All the above bridges can be incorporated asymmetrically into the DPP core because of their potentially combined favorable properties. Direct modifications of the DPP core, including the exchange of lactam nitrogen and carbonyl groups, insertion of the benzene/naphthalene ring, the introduction of half-fused lactam, and the conversion from the fusion strategy to the double-bond connection, can still exert their potential in optimizing the specific properties of polymers. Different from DPP-based building blocks, most derivatives and analogs of the IID core are developed with relatively better planarities due to many intramolecular interactions (e.g., S⋯O) and O⋯H hydrogen bonds. By structurally adjusting the indolinone units with substituted electron-withdrawing heteroatoms, the obtained polymers have lowered energy levels and achieve high balanced ambipolar transport performances. The extension of the indolinone unit can efficaciously facilitate effective π-orbital overlap and preferable face-on orientation in the polymer films for exhibiting high PCE of OSC devices. Further modification with a variety of inserting units destroys the former planar backbone in IID and constructs new monomers also featuring planar backbones and additionally more delocalized molecular orbitals, more alkylation sites, and decreased sidechain density for an ordered sidechain self-assembly process.
In the second section, we discuss the recent structural evolution of NDI, PDI, BTI, and other imide-functionalized building blocks, as well as the corresponding polymers. In comparison, NDI and PDI units have more rigid backbones than those of DPP and IID cores. Precisely because of their special structures, the laterally extended conjugation can promise good planarity and relieve the steric effect between NDI/PDI and the adjacent units at the same time. This provides those polymers with improved mainchain stacking and more percentage of face-on orientation in the films, let alone their enhanced OSC performances. For the backbone extension in the vertical direction, it shows effective adjustment in achieving a narrow band gap with the relatively low-lying LUMO energy level, which favors ambipolar charge carrier performances. Moreover, PHI, BTI, and other imide-containing units are also introduced. Whether the bonding or the fusion strategy, e.g., BiTPD and TBI, polymers can be easily constructed to have more imide moieties in their backbones, due to the structural simplicity of those single imide-functionalized units. This provides the polymer with a staggered sidechain arrangement, which is different from the symmetrical alkylating position in NDI/PDI units. Such a combination contributes to ordered layer-to-layer lamellar stacking and interchain packing simultaneously.
To sum up, several methods have been developed to promote the evolution of those polymer backbones: (i) incorporating electron-withdrawing atoms or groups, (ii) extending the polymeric π-conjugation length, (iii) side chain engineering and (iv) choosing the appropriate donor–acceptor pair. All these structural modifications are mainly focused on two aspects, one of which is decreasing the charge carrier injection barrier and the other one is promoting the charge carrier transport in the polymer film.
Despite the numerous kinds of polymers and their enhancing device performances in OFETs and OSCs, there are still several things that need to be settled down in the next few years. First, the dominant packing state of the polymer chain is uncontrollable. The available sidechain and mainchain orientations, which are in line with the expectations, help investigate the relationship between the chemical structure and the intra- or interchain charge transport. Second, the flexible high-performance semiconductor polymers are still lower in number. The flexibility and conductivity of semiconducting polymers get all the attention. Though there are several pieces of research on flexible polymers and corresponding flexible devices that have been reported, OFET or OSC devices based on intrinsically flexible semiconductor polymers can hardly afford comparable performances with their unstretched or unbending counterparts. So far, long side chain substitution has been proved to be able to improve polymer flexibility due to the ability to counteract the strain energy during the deformation process. Such modification neither affects intrachain carrier transport properties nor prevents interchain aggregation and it deserves further study. Finally, the shape of the polymer backbone, which is easily ignored, should also be considered as another key factor affecting the device performance. Many aspects are associated with the shape formation of polymer backbones, such as the shape of the building block, the bonding method and relative position, and the conformational stability. All those adjustable aspects can be fine-tuned for numerous shape-changeable polymers.
In addition to those mentioned above, the mobility hype is another key point worthy of being paid attention to. As is known, an ideal OFET device should contain multiple features, like negligible metal/semiconductor contact resistance, no diffusion, and biasing-independent mobility. Nevertheless, it is hard to fabricate such a perfect FET device due to the existence of Schottky barriers at the metal/semiconductor contact and other commonly existing device disorders. It is widely accepted that most OFET charge carrier mobilities (μ) are mainly extracted by the conventional method in linear and saturation regions. Owing to the respective underestimated and overestimated mobilities obtained from linear and saturation regions, such swinging characteristics of the extracted mobility make the analysis of intrinsic charge carrier mobilities difficult.167 Therefore, neither of the two mobilities reflects the intrinsic μ of semiconductors, especially when a non-ideal electrical behavior existed. It is noted that we should make a reasonable assessment of those seemed “ultra-high” mobilities probably extracted from the “kink” or “double-sloped” transfer curves, whose drain-source current is not changing linearly as the gate voltage (VG) increasing. Such non-ideal transfer curves are usually caused by the aforementioned contact resistance and always lead to a substantially inaccurate μ. According to the relative values of contact resistance (Rc) and channel resistance (Rch), the extracted μ can be different. When Rc is larger than Rch, the rapidly decreased Rc in the low VG region leads to a steep increase in the drain-source current, which means that μ obtained from the low VG with the steeper slope can hardly reflect the intrinsic transfer behavior and is overestimated. In this situation, the more valid μ should be extracted from the high VG region. On the other hand, when Rc is smaller than Rch, the obtained μ is always underestimated whether in the low or high VG region.168 Despite their referential functions, such pitfall of mobility should be avoided for the improved reliability of the mobility. To reduce the impact of Rc on the valid μ, we should pay more attention to the electrode materials and the FMO energy levels of polymer semiconductors. Apart from these two key points, the gate dielectric is another noteworthy factor in the futural optimization of more reliable mobilities. It can affect the polymer film morphology to achieve different charge trapping therefore shielding the gate field, which leads to greatly varied Rc.169 In addition to making changes at the experimental level, another extracted μ called the low-field mobility extracted by the Y-function method appeared to achieve less mobility misinterpretation and higher reliability. It has been pointed out in recent years that the mobilities obtained from the Y-function method are closer to the effective mobilities in comparison with those extracted by the conventional method whether in linear or saturation regions.170 It will be more delightful to build up strong reliability in the field than chasing the so-called “ultra-high performance” by incorrect methods.
Author contributions
G. Y. worked out the overall plan of this review article. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Key Research and Development Program of China (2017YFA0204703 and 2016YFB0401102), the National Natural Science Foundation of China (22021002, 21673258, 21774134, and 22075294), and the Beijing National Laboratory for Molecular Sciences (BNLMS – CXXM-202101).Notes and references
- H. Akamatu, H. Inokuchi and Y. Matsunaga, Nature, 1954, 173, 168–169 CrossRef.
- H. Akamatu, H. Inokuchi and Y. Matsunaga, Bull. Chem. Soc. Jpn., 1956, 29, 213–218 CrossRef.
- A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210–1212 CrossRef CAS.
- N. M. Randell, P. C. Boutin and T. L. Kelly, J. Mater. Chem. A, 2016, 4, 6940–6945 RSC.
- B. Nketia-Yawson, H.-S. Lee, D. Seo, Y. Yoon, W.-T. Park, K. Kwak, H. J. Son, B. Kim and Y.-Y. Noh, Adv. Mater., 2015, 27, 3045–3052 CrossRef CAS PubMed.
- H. Chen, A. Wadsworth, C. Ma, A. Nanni, W. Zhang, M. Nikolka, A. M. T. Luci, L. M. A. Perdigão, K. J. Thorley, C. Cendra, B. Larson, G. Rumbles, T. D. Anthopoulos, A. Salleo, G. Costantini, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2019, 141, 18806–18813 CrossRef CAS PubMed.
- Z. Chen, M. Li, M. Hu, S. Wang, Z. Miao, S. Xu, C. Chen, H. Dong, W. Huang and R. Chen, J. Mater. Chem. C, 2020, 8, 2094–2101 RSC.
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 CrossRef CAS.
- B. Fan, K. Zhang, X. F. Jiang, L. Ying, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1606396 CrossRef PubMed.
- F. P. Wu, H. I. Un, Y. Li, H. Hu, Y. Yuan, B. Yang, K. Xiao, W. Chen, J. Y. Wang, Z. Q. Jiang, J. Pei and L. S. Liao, Chemistry, 2017, 23, 14723–14727 CrossRef CAS PubMed.
- H. C. Wu, C. C. Hung, C. W. Hong, H. S. Sun, J. T. Wang, G. Yamashita, T. Higashihara and W. C. Chen, Macromolecules, 2016, 49, 8540–8548 CrossRef CAS.
- K. Q. Huang, X. Zhao, Y. C. Du, S. Kim, X. H. Wang, H. B. Lu, K. Cho, G. B. Zhang and L. Z. Qiu, J. Mater. Chem. C, 2019, 7, 7618–7626 RSC.
- X. Chen, Z. Zhang, Z. Ding, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 10376–10380 CrossRef CAS PubMed.
- B. Kang, R. Kim, S. B. Lee, S.-K. Kwon, Y.-H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- F. Wang, K. Nakano, H. Segawa and K. Tajima, Chem. Mater., 2019, 31, 2097–2105 CrossRef CAS.
- Y. Yang, Z. Liu, L. Chen, J. Yao, G. Lin, X. Zhang, G. Zhang and D. Zhang, Chem. Mater., 2019, 31, 1800–1807 CrossRef CAS.
- J. Tian, L. Fu, Z. Liu, H. Geng, Y. Sun, G. Lin, X. Zhang, G. Zhang and D. Zhang, Adv. Funct. Mater., 2019, 29, 1807176 CrossRef.
- C. Grand, S. Baek, T.-H. Lai, N. Deb, W. Zajaczkowski, R. Stalder, K. Müllen, W. Pisula, D. G. Bucknall, F. So and J. R. Reynolds, Macromolecules, 2016, 49, 4008–4022 CrossRef CAS.
- S. Liu, Y. Firdaus, S. Thomas, Z. Kan, F. Cruciani, S. Lopatin, J.-L. Bredas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2018, 57, 531–535 CrossRef CAS PubMed.
- G. G. Jeon, M. Lee, J. Nam, W. Park, M. Yang, J. H. Choi, D. K. Yoon, E. Lee, B. Kim and J. H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 29824–29830 CrossRef CAS PubMed.
- Q. Liu, Y. Wang, A. Kohara, H. Matsumoto, S. Manzhos, K. Feron, S. E. Bottle, J. Bell, T. Michinobu and P. Sonar, Adv. Funct. Mater., 2020, 30, 1907452 CrossRef CAS.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, M. J. Dyson, N. Stingelin, W. Chen, H.-J. Ju, G.-X. Zhang and D.-Q. Zhang, Adv. Sci., 2017, 4, 1700048 CrossRef PubMed.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
- A. C. Rochat, L. Cassar and A. Iqbal, US Pat., 4579949, 1986 Search PubMed.
- W. K. Chan, Y. Chen, Z. Peng and L. Yu, J. Am. Chem. Soc., 1993, 115, 11735–11743 CrossRef CAS.
- J. C. Bijleveld, A. P. Zoombelt, S. G. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- Z. Fei, L. Chen, Y. Han, E. Gann, A. S. R. Chesman, C. R. McNeill, T. D. Anthopoulos, M. Heeney and A. Pietrangelo, J. Am. Chem. Soc., 2017, 139, 8094–8097 CrossRef CAS PubMed.
- H. Luo, C. Yu, Z. Liu, G. Zhang, H. Geng, Y. Yi, K. Broch, Y. Hu, A. Sadhanala, L. Jiang, P. Qi, Z. Cai, H. Sirringhaus and D. Zhang, Sci. Adv., 2016, 2, 1600076 CrossRef PubMed.
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. Frechet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS PubMed.
- J. C. Bijleveld, B. P. Karsten, S. G. J. Mathijssen, M. M. Wienk, D. M. de Leeuw and R. A. J. Janssen, J. Mater. Chem., 2011, 21, 1600–1606 RSC.
- P. Sonar, J. Chang, Z. Shi, E. Gann, J. Li, J. Wu and C. R. McNeill, J. Mater. Chem. C, 2015, 3, 9299–9305 RSC.
- W. Zhou, C. Yu, H. Chen, T. Jia, W. Zhang, G. Yu and F. Li, J. Phys. Chem. B, 2016, 120, 4824–4832 CrossRef CAS PubMed.
- M. Shahid, T. McCarthy-Ward, J. Labram, S. Rossbauer, E. B. Domingo, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2012, 3, 181–185 RSC.
- Q. Liu, Y. Wang, A. Kohara, H. Matsumoto, S. Manzhos, K. Feron, S. E. Bottle, J. Bell, T. Michinobu and P. Sonar, Adv. Funct. Mater., 2019, 30, 1907452 CrossRef.
- C. Kanimozhi, P. Balraju, G. D. Sharma and S. Patil, J. Phys. Chem. B, 2010, 114, 3095–3103 CrossRef CAS PubMed.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Chem. Commun., 2013, 49, 8495–8497 RSC.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- Q. Liu, S. Kumagai, S. Manzhos, Y. Q. Chen, I. Angunawela, M. M. Nahid, K. Feron, S. E. Bottle, J. Bell, H. Ade, J. Takeya and P. Sonar, Adv. Funct. Mater., 2020, 30, 2000489 CrossRef CAS.
- X. Yan, M. Xiong, J. T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z. Y. Wang, Z. F. Yao, J. Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed.
- W. Li, W. S. Roelofs, M. Turbiez, M. M. Wienk and R. A. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- Z. Chen, D. Gao, J. Huang, Z. Mao, W. Zhang and G. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 34725–34734 CrossRef CAS PubMed.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- H.-Y. Chen, M. Nikolka, A. Wadsworth, W. Yue, A. Onwubiko, M. Xiao, A. J. P. White, D. Baran, H. Sirringhaus and I. McCulloch, Macromolecules, 2017, 51, 71–79 CrossRef.
- Z. Ni, H. Dong, H. Wang, S. Ding, Y. Zou, Q. Zhao, Y. Zhen, F. Liu, L. Jiang and W. Hu, Adv. Mater., 2018, 30, 1704843 CrossRef PubMed.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- Y. Yu, Y. Wu, A. Zhang, C. Li, Z. Tang, W. Ma, Y. Wu and W. Li, ACS Appl. Mater. Interfaces, 2016, 8, 30328–30335 CrossRef CAS PubMed.
- G. G. Qiu, Z. Y. Jiang, Z. J. Ni, H. L. Wang, H. L. Dong, J. Q. Zhang, X. T. Zhang, Z. B. Shu, K. Lu, Y. G. Zhen, Z. X. Wei and W. P. Hu, J. Mater. Chem. C, 2017, 5, 566–572 RSC.
- Z. Y. Jiang, Z. J. Ni, H. L. Wang, Z. Wang, J. Q. Zhang, G. G. Qiu, J. Fang, Y. J. Zhang, H. L. Dong, K. Lu, W. P. Hu and Z. X. Wei, Polym. Chem., 2017, 8, 5603–5610 RSC.
- H. Song, Y. F. Deng, Y. Gao, Y. Jiang, H. K. Tian, D. H. Yan, Y. H. Geng and F. S. Wang, Macromolecules, 2017, 50, 2344–2353 CrossRef CAS.
- S. Ding, Z. Ni, M. Hu, G. Qiu, J. Li, J. Ye, X. Zhang, F. Liu, H. Dong and W. Hu, Macromol. Rapid Commun., 2018, 39, 1800225 CrossRef PubMed.
- H. Tanaka, S. Kawamura, P. Sonar, Y. Shimoi, T. T. Do and T. Takenobu, Adv. Funct. Mater., 2020, 30, 2000389 CrossRef CAS.
- W. Li, M. Kelchtermans, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2013, 1, 15150–15157 RSC.
- J. S. Lee, S. K. Son, S. Song, H. Kim, D. R. Lee, K. Kim, M. J. Ko, D. H. Choi, B. Kim and J. H. Cho, Chem. Mater., 2012, 24, 1316–1323 CrossRef CAS.
- B. C. Schroeder, T. Kurosawa, T. Fu, Y.-C. Chiu, J. Mun, G.-J. N. Wang, X. Gu, L. Shaw, J. W. E. Kneller, T. Kreouzis, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2017, 27, 1701973 CrossRef.
- H. Song, Y. Gao, W. Li, H. Tian, D. Yan, Y. Geng and F. Wang, J. Mater. Chem. C, 2015, 3, 11135–11143 RSC.
- P. J. Leenaers, M. M. Wienk and R. A. J. Janssen, Org. Electron., 2020, 86, 105914 CrossRef CAS.
- K. Aoshima, M. Nomura and A. Saeki, ACS Omega, 2019, 4, 15645–15652 CrossRef CAS PubMed.
- P. J. Leenaers, H. van Eersel, J. Li, M. M. Wienk and R. A. J. Janssen, Macromolecules, 2020, 53, 7749–7758 CrossRef CAS PubMed.
- P. Sonar, J. Chang, Z. Shi, J. Wu and J. Li, J. Mater. Chem. C, 2015, 3, 2080–2085 RSC.
- S. Song, S.-J. Ko, H. Shin, Y. Jin, I. Kim, J. Y. Kim and H. Suh, Synth. Met., 2012, 162, 2288–2293 CrossRef CAS.
- S. F. Lu, M. Drees, Y. Yao, D. Boudinet, H. Yan, H. L. Pan, J. Q. Wang, Y. N. Li, H. Usta and A. Facchetti, Macromolecules, 2013, 46, 3895–3906 CrossRef CAS.
- K. Iijima and T. Mori, Chem. Lett., 2017, 46, 357–359 CrossRef CAS.
- X. Guo, S. R. Puniredd, B. He, T. Marszalek, M. Baumgarten, W. Pisula and K. Mullen, Chem. Mater., 2014, 26, 3595–3598 CrossRef CAS PubMed.
- W. Cui, J. Yuen and F. Wudl, Macromolecules, 2011, 44, 7869–7873 CrossRef CAS.
- G. W. P. van Pruissen, E. A. Pidko, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. C, 2014, 2, 731–735 RSC.
- W. Hong, B. Sun, C. Guo, J. Yuen, Y. Li, S. Lu, C. Huang and A. Facchetti, Chem. Commun., 2013, 49, 484–486 RSC.
- H. C. Zhang, J. M. Neudorfl and B. Tieke, Polym. Chem., 2014, 5, 3754–3757 RSC.
- H. C. Zhang, S. Zhang, Y. F. Mao, K. W. Liu, Y. M. Chen, Z. Jiang, J. Strzalka, W. J. Yang, C. L. Wang and Y. Zhu, Polym. Chem., 2017, 8, 3255–3260 RSC.
- H. C. Zhang, K. Yang, Y. M. Chen, R. Bhatta, M. Tsige, S. Z. D. Cheng and Y. Zhu, Macromol. Chem. Phys., 2017, 218, 1600617 CrossRef.
- Z. X. Cai, Y. L. Guo, S. F. Yang, Q. Peng, H. W. Luo, Z. T. Liu, G. X. Zhang, Y. Q. Liu and D. Q. Zhang, Chem. Mater., 2013, 25, 471–478 CrossRef CAS.
- Z. Cai, H. Luo, P. Qi, J. Wang, G. Zhang, Z. Liu and D. Zhang, Macromolecules, 2014, 47, 2899–2906 CrossRef CAS.
- S. F. Yang, Z. T. Liu, Z. X. Cai, H. W. Luo, P. L. Qi, G. X. Zhang and D. Q. Zhang, Macromolecules, 2016, 49, 5857–5865 CrossRef CAS.
- K. C. Lee, H. R. Lee, S. H. Kang, J. Lee, Y. I. Park, S. M. Noh, J. H. Oh and C. Yang, Polym. Chem., 2018, 9, 5234–5241 RSC.
- J. G. Wang, X. Chen, Z. X. Cai, H. W. Luo, Y. H. Li, Z. T. Liu, G. X. Zhang and D. Q. Zhang, Polym. Chem., 2013, 4, 5283–5290 RSC.
- D. D. Shi, Z. T. Liu, J. Ma, Z. Y. Zhao, L. X. Tan, G. B. Lin, J. W. Tian, X. S. Zhang, G. X. Zhang and D. Q. Zhang, Adv. Funct. Mater., 2020, 30, 1910235 CrossRef CAS.
- F. Trilling, O. Sachnik and U. Scherf, Polym. Chem., 2019, 10, 627–632 RSC.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- S. Liu, Y. Firdaus, S. Thomas, Z. Kan, F. Cruciani, S. Lopatin, J. L. Bredas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2018, 57, 531–535 CrossRef CAS PubMed.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- S. F. Liao, C. F. Lu, A. D. Fenta, C. T. Chen, C. Y. Chao and W. F. Su, J. Mater. Chem. A, 2019, 7, 21309–21320 RSC.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 CrossRef PubMed.
- J. Yang, Z. Zhao, H. Geng, C. Cheng, J. Chen, Y. Sun, L. Shi, Y. Yi, Z. Shuai, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1702115 CrossRef PubMed.
- J. Y. Huang, Z. P. Mao, Z. H. Chen, D. Gao, C. Y. Wei, W. F. Zhang and G. Yu, Chem. Mater., 2016, 28, 2209–2218 CrossRef CAS.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- G. Kim, H. Kim, M. Jang, Y. K. Jung, J. H. Oh and C. Yang, J. Mater. Chem. C, 2016, 4, 9554–9560 RSC.
- G. Kim, S. J. Kang, G. K. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- C. Li, H. I. Un, J. Peng, M. Cai, X. Wang, J. Wang, Z. Lan, J. Pei and X. Wan, Chemistry, 2018, 24, 9807–9811 CrossRef CAS PubMed.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J. C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 CAS.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H. Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702–4707 CrossRef CAS PubMed.
- M. Neophytou, D. Bryant, S. Lopatin, H. Chen, R. K. Hallani, L. Cater, I. McCulloch and W. Yue, Macromol. Rapid Commun., 2018, 39, 1700820 CrossRef PubMed.
- W. S. Yoon, D. W. Kim, M. W. Choi, J. M. Park and S. Y. Park, Adv. Energy Mater., 2018, 8, 1701467 CrossRef.
- M. A. Cai, X. C. Bao, X. Wang, H. R. Zhang, M. Qiu, R. Q. Yang, C. M. Yang and X. B. Wan, Chem. Mater., 2016, 28, 6196–6206 CrossRef CAS.
- M. Cai, Z. Y. Zhao, Y. F. Liu, X. Wang, Y. Q. Liu, Z. G. Lan and X. B. Wan, Macromolecules, 2017, 50, 8497–8504 CrossRef CAS.
- S. Chen, H. J. Cho, J. Lee, Y. K. Yang, Z. G. Zhang, Y. F. Li and C. Yang, Adv. Energy Mater., 2017, 7, 1701125 CrossRef.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Frechet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS.
- B. Shahid, D. Q. Zhu, Q. Wang, X. Y. Yuan, I. Ismail, Y. Wu, Z. R. Du and R. Q. Yang, Polym. Int., 2020, 69, 564–570 CrossRef CAS.
- M. Tomassetti, F. Ouhib, A. Wislez, A. S. Duwez, H. Penxten, W. Dierckx, I. Cardinaletti, R. A. A. Bovee, G. W. P. van Pruissen, C. Jerome, J. Manca, W. Maes and C. Detrembleur, Polym. Chem., 2015, 6, 6040–6049 RSC.
- J. L. Li, Y. F. Chai, W. V. Wang, Z. F. Shi, Z. G. Xu and H. L. Zhang, Chem. Commun., 2017, 53, 5882–5885 RSC.
- Y. Kim, H. Hwang, N. K. Kim, K. Hwang, J. J. Park, G. I. Shin and D. Y. Kim, Adv. Mater., 2018, 30, 1706557 CrossRef PubMed.
- S. G. Li, Z. C. Yuan, J. Y. Yuan, P. Deng, Q. Zhang and B. Q. Sun, J. Mater. Chem. A, 2014, 2, 5427–5433 RSC.
- W. F. Zhang, N. H. Zheng, C. Y. Wei, J. Y. Huang, D. Gao, K. L. Shi, J. Xu, D. H. Yan, Y. C. Han and G. Yu, Polym. Chem., 2016, 7, 1413–1421 RSC.
- Y. K. Zhou, S. Y. Zhang, W. F. Zhang, J. Huang, C. Y. Wei, H. Li, L. P. Wang and G. Yu, Macromolecules, 2018, 51, 7093–7103 CrossRef CAS.
- Y. H. He, J. T. E. Quinn, D. L. Hou, J. H. L. Ngai and Y. N. Li, J. Mater. Chem. C, 2017, 5, 12163–12171 RSC.
- X. Zhou, N. Ai, Z. H. Guo, F. D. Zhuang, Y. S. Jiang, J. Y. Wang and J. Pei, Chem. Mater., 2015, 27, 1815–1820 CrossRef CAS.
- G. B. Zhang, Z. W. Ye, P. Li, J. H. Guo, Q. H. Wang, L. X. Tang, H. B. Lu and L. Z. Qiu, Polym. Chem., 2015, 6, 3970–3978 RSC.
- Y. Z. Dai, N. Ai, Y. Lu, Y. Q. Zheng, J. H. Dou, K. Shi, T. Lei, J. Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- K. Shi, W. Zhang, D. Gao, S. Zhang, Z. Lin, Y. Zou, L. Wang and G. Yu, Adv. Mater., 2018, 30, 1705286 CrossRef PubMed.
- Y. Q. Zheng, T. Lei, J. H. Dou, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, Adv. Mater., 2016, 28, 7213–7219 CrossRef CAS PubMed.
- Y. H. He, J. Quinn, Y. F. Deng and Y. N. Li, Org. Electron., 2016, 35, 41–46 CrossRef CAS.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Chem. Commun., 2015, 51, 13515–13518 RSC.
- J. T. E. Quinn, C. Guo, F. Haider, H. Patel, D. A. Khan and Y. N. Li, J. Mater. Chem. C, 2017, 5, 5902–5909 RSC.
- Y. He, C. Guo, B. Sun, J. Quinn and Y. Li, Chem. Commun., 2015, 51, 8093–8096 RSC.
- Y. Cao, J. S. Yuan, X. Zhou, X. Y. Wang, F. D. Zhuang, J. Y. Wang and J. Pei, Chem. Commun., 2015, 51, 10514–10516 RSC.
- Y. Jiang, Y. Gao, H. K. Tian, J. Q. Ding, D. H. Yan, Y. H. Geng and F. S. Wang, Macromolecules, 2016, 49, 2135–2144 CrossRef CAS.
- H. Song, Y. Deng, Y. Jiang, H. Tian and Y. Geng, Chem. Commun., 2018, 54, 782–785 RSC.
- A. Karki, G. J. A. H. Wetzelaer, G. N. M. Reddy, V. Nádaždy, M. Seifrid, F. Schauer, G. C. Bazan, B. F. Chmelka, P. W. M. Blom and T. Q. Nguyen, Adv. Funct. Mater., 2019, 29, 1901109 CrossRef.
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H. Y. Chen, M. K. Ravva, D. A. Hanifi, A. C. Knall, B. Purushothaman, M. Nikolka, J. C. Flores, A. Salleo, J. L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 416 CrossRef PubMed.
- Y. Lu, Z. D. Yu, R. Z. Zhang, Z. F. Yao, H. Y. You, L. Jiang, H. I. Un, B. W. Dong, M. Xiong, J. Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2019, 58, 11390–11394 CrossRef CAS PubMed.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- B. Fan, W. Zhong, L. Ying, D. Zhang, M. Li, Y. Lin, R. Xia, F. Liu, H. L. Yip, N. Li, Y. Ma, C. J. Brabec, F. Huang and Y. Cao, Nat. Commun., 2019, 10, 4100 CrossRef PubMed.
- Y. Han, Y. Liu, J. Yuan, H. Dong, Y. Li, W. Ma, S. T. Lee and B. Sun, ACS Nano, 2017, 11, 7215–7222 CrossRef CAS PubMed.
- Z. Wu, C. Sun, S. Dong, X. F. Jiang, S. Wu, H. Wu, H. L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS PubMed.
- D. Shukla, S. F. Nelson, D. C. Freeman, M. Rajeswaran, W. G. Ahearn, D. M. Meyer and J. T. Carey, Chem. Mater., 2008, 20, 7486–7491 CrossRef CAS.
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 CrossRef PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS PubMed.
- Y. Fukutomi, M. Nakano, J. Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 CrossRef CAS PubMed.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. L. Tang and E. J. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- Y. Wang and K. Takimiya, Adv. Mater., 2020, 32, 2002060 CrossRef.
- Z. J. Li, K. Feng, J. Liu, J. Mei, Y. Li and Q. Peng, J. Mater. Chem. A, 2016, 4, 7372–7381 RSC.
- X. Zhao, C. Ge, X. Xu, J. Huang, X. Yang, Q. Peng, W. S. Li and X. Gao, Chem. Commun., 2019, 55, 10234–10237 RSC.
- X. Li, J. Guo, L. Yang, M. Chao, L. Zheng, Z. Ma, Y. Hu, Y. Zhao, H. Chen and Y. Liu, Front. Chem., 2019, 7, 362 CrossRef CAS PubMed.
- J. Chen, X. Zhuang, W. Huang, M. Su, L.-w. Feng, S. M. Swick, G. Wang, Y. Chen, J. Yu, X. Guo, T. J. Marks and A. Facchetti, Chem. Mater., 2020, 32, 5317–5326 CrossRef CAS.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- S. S. Li, H. Zhang, W. C. Zhao, L. Ye, H. F. Yao, B. Yang, S. Q. Zhang and J. H. Hou, Adv. Energy Mater., 2016, 6, 1501991 CrossRef.
- I. H. Jung, D. L. Zhao, J. Jang, W. Chen, E. S. Landry, L. Y. Lu, D. V. Talapin and L. P. Yu, Chem. Mater., 2015, 27, 5941–5948 CrossRef CAS.
- H. Usta, C. Newman, Z. Chen and A. Facchetti, Adv. Mater., 2012, 24, 3678–3684 CrossRef CAS PubMed.
- J. Yang, F. Chen, H. Ran, J. Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, Macromol. Rapid Commun., 2018, 39, 1700715 CrossRef PubMed.
- M. Liu, J. Yang, C. L. Lang, Y. Zhang, E. J. Zhou, Z. T. Liu, F. Y. Guo and L. C. Zhao, Macromolecules, 2017, 50, 7559–7566 CrossRef CAS.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef PubMed.
- Y. P. Yu, N. Xue, C. Y. Xiao, M. K. Ravva, Y. J. Gu, L. Y. Wu, L. Zhang, Z. K. Li, W. Yue and Z. H. Wang, J. Mater. Chem. C, 2019, 7, 12263–12269 RSC.
- H. Li, F. S. Kim, G. Ren and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14920–14923 CrossRef CAS.
- A. Drewniak, M. D. Tomczyk, L. Hanusek, A. Mielanczyk, K. Walczak, P. Nitschke, B. Hajduk and P. Ledwon, Polymers, 2018, 10, 487 CrossRef.
- X. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206–7207 CrossRef CAS PubMed.
- J. W. Yu, J. Yang, X. Zhou, S. M. Yu, Y. M. Tang, H. Wang, J. H. Chen, S. M. Zhang and X. G. Guo, Macromolecules, 2017, 50, 8928–8937 CrossRef CAS.
- J. Yu, P. Chen, C. W. Koh, H. Wang, K. Yang, X. Zhou, B. Liu, Q. Liao, J. Chen, H. Sun, H. Y. Woo, S. Zhang and X. Guo, Adv. Sci., 2019, 6, 1801743 CrossRef.
- L. Lan, Z. Chen, Q. Hu, L. Ying, R. Zhu, F. Liu, T. P. Russell, F. Huang and Y. Cao, Adv. Sci., 2016, 3, 1600032 CrossRef PubMed.
- B. A. Abdulahi, X. M. Li, M. Mone, B. Kiros, Z. Genene, S. L. Qiao, R. Q. Yang, E. G. Wang and W. Mammo, J. Mater. Chem. A, 2019, 7, 19522–19530 RSC.
- Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394–5404 CrossRef CAS PubMed.
- W. F. Shen, W. C. Chen, D. Q. Zhu, J. D. Zhang, X. F. Xu, H. X. Jiang, T. Wang, E. G. Wang and R. Q. Yang, J. Mater. Chem. A, 2017, 5, 12400–12406 RSC.
- J. H. Kim, J. B. Park, I. H. Jung, A. C. Grimsdale, S. C. Yoon, H. Yang and D. H. Hwang, Energy Environ. Sci., 2015, 8, 2352–2356 RSC.
- S. Q. Zhang, X. L. Qiao, Y. Xiong, Q. H. Wu, Z. Q. Liu, H. X. Li and Q. Fang, Org. Electron., 2018, 56, 146–151 CrossRef CAS.
- J. Zhao, Q. Li, S. Liu, Z. Cao, X. Jiao, Y.-P. Cai and F. Huang, ACS Energy Lett., 2020, 5, 367–375 CrossRef CAS.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, Angew. Chem., Int. Ed., 2019, 58, 11893–11902 CrossRef CAS PubMed.
- J. A. Letizia, M. R. Salata, C. M. Tribout, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 9679–9694 CrossRef CAS PubMed.
- M. Saito, I. Osaka, Y. Suda, H. Yoshida and K. Takimiya, Adv. Mater., 2016, 28, 6921–6925 CrossRef CAS.
- Y. Shi, H. Guo, J. Huang, X. Zhang, Z. Wu, K. Yang, Y. Zhang, K. Feng, H. Y. Woo, R. P. Ortiz, M. Zhou and X. Guo, Angew. Chem., Int. Ed., 2020, 59, 14449–14457 CrossRef CAS PubMed.
- Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS PubMed.
- H. L. Sun, Y. M. Tang, H. Guo, M. A. Uddin, S. H. Ling, R. Z. Wang, Y. F. Wang, X. Zhou, H. Y. Woo and X. G. Guo, Sol. RRL., 2019, 3, 1800265 CrossRef.
- H. Sun, Y. Tang, C. W. Koh, S. Ling, R. Wang, K. Yang, J. Yu, Y. Shi, Y. Wang, H. Y. Woo and X. Guo, Adv. Mater., 2019, 31, 1807220 CrossRef.
- Y. Shi, H. Guo, M. Qin, J. Zhao, Y. Wang, H. Wang, Y. Wang, A. Facchetti, X. Lu and X. Guo, Adv. Mater., 2018, 30, 1705745 CrossRef PubMed.
- Y. Q. Shi, W. Chen, Z. A. Wu, Y. Wang, W. P. Sun, K. Yang, Y. M. Tang, H. Y. Woo, M. Zhou, A. B. Djurisic, Z. B. He and X. G. Guo, J. Mater. Chem. A, 2020, 8, 13754–13762 RSC.
- D. Li, Z. Xiao, S. Z. Wang, X. J. Geng, S. F. Yang, J. F. Fang, H. Yang and L. M. Ding, Adv. Energy Mater., 2018, 8, 1800397 CrossRef.
- J. Y. Huang, Y. C. Pan, Z. H. Chen, W. F. Zhang and G. Yu, Macromolecules, 2019, 52, 8238–8247 CrossRef CAS.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2017, 17, 2–7 CrossRef PubMed.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- C. Liu, Y. Xu, Y. Li, W. Scheideler and T. Minari, J. Phys. Chem. B, 2013, 117, 12337–12345 CAS.
- Y. Xu, H. Sun, A. Liu, H. Zhu, B. Li, T. Minari, F. Balestra, G. Ghibaudo and Y.-Y. Noh, Adv. Funct. Mater., 2018, 28, 1803907 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |