
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure, and reactivity of uranium(VI) nitrides†
Luciano
Barluzzi
a,
Fang-Che
Hsueh
a,
Rosario
Scopelliti
 a,
Benjamin E.
Atkinson
b,
Nikolas
Kaltsoyannis
b and
Marinella
Mazzanti
*a
a,
Benjamin E.
Atkinson
b,
Nikolas
Kaltsoyannis
b and
Marinella
Mazzanti
*a
aInsititut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 30th April 2021
Abstract
Uranium nitride compounds are important molecular analogues of uranium nitride materials such as UN and UN2 which are effective catalysts in the Haber–Bosch synthesis of ammonia, but the synthesis of molecular nitrides remains a challenge and studies of the reactivity and of the nature of the bonding are poorly developed. Here we report the synthesis of the first nitride bridged uranium complexes containing U(VI) and provide a unique comparison of reactivity and bonding in U(VI)/U(VI), U(VI)/U(V) and U(V)/U(V) systems. Oxidation of the U(V)/U(V) bis-nitride [K2{U(OSi(OtBu)3)3(μ-N)}2], 1, with mild oxidants yields the U(V)/U(VI) complexes [K{U(OSi(OtBu)3)3(μ-N)}2], 2 and [K2{U(OSi(OtBu)3)3}2(μ-N)2(μ-I)], 3 while oxidation with a stronger oxidant (“magic blue”) yields the U(VI)/U(VI) complex [{U(OSi(OtBu)3)3}2(μ-N)2(μ-thf)], 4. The three complexes show very different stability and reactivity, with N2 release observed for complex 4. Complex 2 undergoes hydrogenolysis to yield imido bridged [K2{U(OSi(OtBu)3)3(μ-NH)}2], 6 and rare amido bridged U(IV)/U(IV) complexes [{U(OSi(OtBu)3)3}2(μ-NH2)2(μ-thf)], 7 while no hydrogenolysis could be observed for 4. Both complexes 2 and 4 react with H+ to yield quantitatively NH4Cl, but only complex 2 reacts with CO and H2. Differences in reactivity can be related to significant differences in the U–N bonding. Computational studies show a delocalised bond across the U–N–U for 1 and 2, but an asymmetric bonding scheme is found for the U(VI)/U(VI) complex 4 which shows a U–N σ orbital well localised to U![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and π orbitals which partially delocalise to form the U–N single bond with the other uranium.
N and π orbitals which partially delocalise to form the U–N single bond with the other uranium.
Introduction
Metal nitride complexes continue to attract significant attention because of their key roles in the catalytic N2 hydrogenation for ammonia production and in N-transfer processes.1 Uranium nitride compounds provide molecular analogues of inorganic uranium nitride materials (UN, UN2, U2N3) which are effective catalysts in the Haber–Bosch synthesis of ammonia2 and are considered as potential fuel for nuclear and space-power reactors.3 Two examples of molecular uranium nitrides, both showing a diamond shaped arrangement of the two nitrides, were obtained from the cleavage of dinitrogen in ambient conditions.4However, the development of rational synthetic routes to molecular uranium nitrides and the study of U–N bonding is still at an early stage.2b,5a–e
In recent years, a few mononuclear5e,6,7a,7b,5c,7c uranium terminal nitrides have been synthesised. A larger number of polynuclear uranium complexes containing bridging nitride has been reported. Most of the nitride bridged uranium complexes isolated so far contain uranium in the +IV oxidation state2b,4a,5b,8a–g with fewer examples containing U(III)9a,1c,9b or U(V).4a,8d,10 Both terminal and bridging uranium nitrides have demonstrated high reactivity towards dinitrogen reduction, CO and CO2 cleavage, and C–H activation.9b,11a–f
The reactivity of high valent uranium nitrides with H2 is also of high current interest for modelling the activity of metal nitride catalysts in N2 hydrogenation. The previous reports of hydrogenolysis by a terminal U(V) nitride,11e U(IV) nitride bridged complexes,9b,12 and a U(V) bis-nitride10a are particularly remarkable considering the paucity of examples of direct hydrogenolysis by metal nitride complexes reported in the literature.1a,13a–f
After the first terminal U(VI) nitride was reported more than ten years ago,7b only two additional examples of U(VI) nitride complexes5e,14 have been isolated very recently. Therefore, the knowledge of U(VI) nitrides lags far behind that of the oxide analogue uranyl(VI) (UO22+) which is the prevalent uranium form in nature. The scarce information available on U(VI) nitrides, mostly limited to gas phase and computational studies,15 is due to the high chemical and photochemical reactivity of the U(VI) fragment.16,7b,11f In particular the diatomic rhombic (UN)2 and (UN2)2 fragments were so far only identified in matrix isolation experiments.15f
Oxidation of isolated U(V) nitrides, when available, allowed the synthesis of the analogous terminal7b or boron-capped U(VI) nitrides,17 but demonstrated significant sensitivity to the reaction conditions7b and failed, so far, to produce U(VI)/U(VI) nitride bridged complexes.
Here we report the synthesis and characterization of the first example of a dinuclear U(VI) nitride which was obtained by oxidation of the previously reported U(V)/U(V) bis-nitride8f,10a [K2{U(OSi(OtBu)3)3(μ-N)}2], 1, with the strong oxidant tris(4-bromophenyl)ammoniumyl hexachloroantimonate (“magic blue”). The use of more common milder oxidants resulted in the isolation of the U(V)/U(VI) analogue.
Studies of the molecular and electronic structure show very different bonding schemes for the two species that result in different reactivity.
Results and discussion
Synthesis of bis-nitride complexes
The synthesis of the targeted bis-nitride complexes of U(VI) was pursued by the oxidation of the previously reported complex 1 (ref. 8f) with different oxidizing agents (Scheme 1).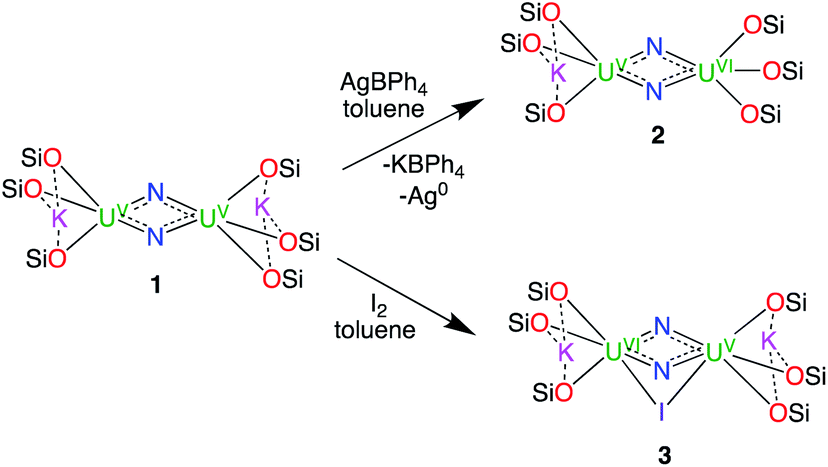 | ||
| Scheme 1 Oxidation of complex 1 affording the complexes [K{U(OSi(OtBu)3)3(μ-N)}2], 2 and [K2{U(OSi(OtBu)3)3}2(μ-N)2(μ-I)], 3. | ||
Reaction of 1 with 1 equiv. of AgBPh4 in d8-toluene led to the full consumption of the starting material over the course of 5 days and the formation of a new species showing a broad resonance between 1.5 and −0.5 ppm. The reaction of complex 1 with 2 equiv. of AgBPh4 resulted in the formation of the same species but with a faster consumption of the starting material (24 h). Cooling down the concentrated reaction mixture at −40 °C resulted in the formation of dark orange crystals of the mixed-valent complex [K{U(OSi(OtBu)3)3(μ-N)}2], 2 in 65% yield. The reaction of complex 1 with 0.5 or 1 equiv. of I2 in toluene led to the formation of the same U(V)/U(VI) complex as observed by 1H NMR spectroscopy. Cooling down the reaction mixture at −30 °C afforded ochre yellow crystals of the complex [K2{U(OSi(OtBu)3)3}2(μ-N)2(μ-I)], 3. The molecular structure of 3 indicates that the KI byproduct remains bound in the crystallised complex. The oxidation of 1 with larger excess of AgBPh4 or I2 failed to produce the desired U(VI)/U(VI) nitride and resulted only in the formation of intractable mixtures of unidentified products.
Thus, in order to obtain a U(VI)/U(VI) bis-μ-nitride complex, we decided to use the stronger oxidant [N(C6H4Br)3][SbCl6] also known as “magic blue”.18 Notably, we recently found that “magic blue” could be used to reach the high oxidation state +IV in lanthanide complexes supported by siloxide ligands.19 Gratifyingly, the reaction of complex 1 with 2 equiv. of [N(C6H4Br)3][SbCl6] in thf resulted in the immediate formation of a single new species resonating at 1.44 ppm with a conversion of 94% measured by 1H NMR spectroscopy. Cooling down the reaction mixture in toluene at −40 °C afforded orange crystals of the U(VI)/U(VI) complex [{U(OSi(OtBu)3)3}2(μ-N)2(μ-thf)], 4 in 35% yield. The low yield of the reaction, despite the almost quantitative conversion, reflects the high solubility of complex 4 in toluene and the separation steps necessary for the removal of the reaction byproducts. These results demonstrate that the use of a strong oxidizing agent such as an ammonium cation is necessary to oxidise the bis-nitride U(V)/U(V) to the U(VI)/U(VI) analogue. It should be noted that complex 4 undergoes decomposition if left in the reaction mixture for a long time before isolation, as indicated by 1H NMR spectroscopy. The formation of new species is observed after 6 h with full consumption of complex 4 after 5 days. Cooling down the resulting decomposed reaction mixture in toluene at −40 °C overnight afforded light green crystals of the complex [U(OSi(OtBu)3)3Cl(thf)2], 5. The molecular structure of 5 and its description are reported in the ESI.† The presence of the chloride ligand suggests that the oxidised nitride reacts with the SbCl6− anion. The non-innocence of the SbCl6− anion in the oxidation of organometallic substrates was previously demonstrated.20 Similar U(IV) halide species were also isolated upon reaction of the U(V) terminal nitride [U(TrenTIPS)N]7b with halide-containing oxidizing agents.7b
The molecular structure of complex 2 (Fig. 1) was determined by XRD studies and shows a dimeric U(V)/U(VI) complex bridged by two nitride ligands. Each U centre is pentacoordinated in a slightly distorted square pyramidal geometry and is bound to the two bridging nitrides and to three siloxide ligands. One set of siloxide ligands provides a O6 coordination pocket for the K+ counterion. The average U1–Osil distance of 2.17(1) Å is slightly longer than the average U2–Osil of 2.11(1) probably due to the presence of the K+ bound to the siloxide ligands. The U–N bond distances are all very similar (U1–N1 = 2.020(4) Å, U1–N2 = 2.067(4) Å, U2–N1 = 2.091(4) Å, and U2–N2 = 2.046(4) Å) and the average U–N bond distance of 2.06(3) Å is statistically equivalent to the one previously reported for complex 1 (2.06(6) Å).8f These values are consistent with the presence of U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond character similar to that found in mono-nitride bridged uranium compounds (2.032(2)–2.080(4) Å).8d,8f,12b Overall, the metrical parameters indicate the presence of a charge delocalised species.
N double bond character similar to that found in mono-nitride bridged uranium compounds (2.032(2)–2.080(4) Å).8d,8f,12b Overall, the metrical parameters indicate the presence of a charge delocalised species.
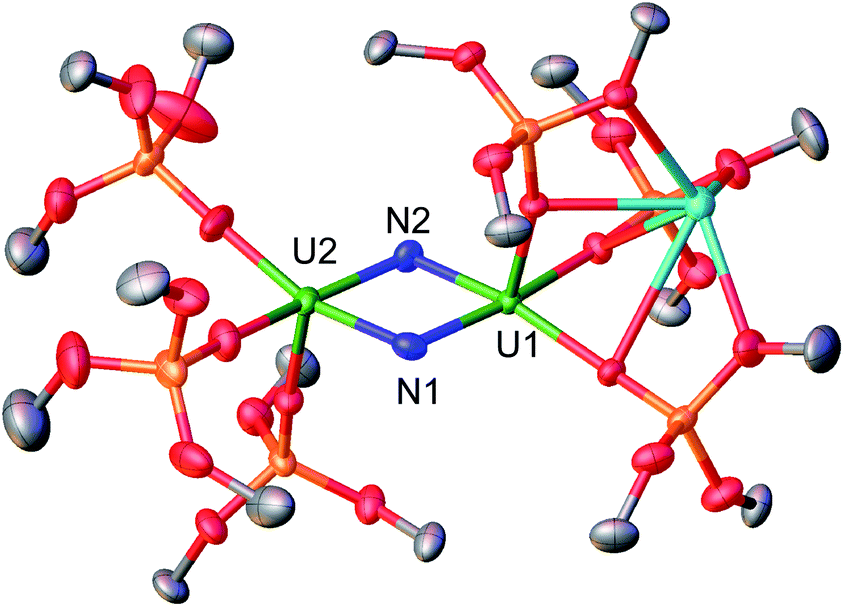 | ||
| Fig. 1 Molecular structure of the complex [K{U(OSi(OtBu)3)3(μ-N)}2], 2 with thermal ellipsoids drawn at the 50% probability level. The methyl groups of the tert-butyl moieties and the co-crystallized solvent molecules have been omitted for clarity. | ||
The molecular structure of 3 (Fig. S40†), as determined by XRD studies, shows the presence of a bis-nitride bridged dimeric U(V)/U(VI) complex which differs from 2 only by the presence of an additional bridging iodide ligand and an additional bound potassium cation. In 3, two potassium cations are bound in each O6 coordination pocket formed by three siloxide ligands. The U–Osil distances (mean U–O = 2.190(5) Å) and the U–N bond distances (mean U–N = 2.13(5) Å) are nearly equivalent for the two U centres, suggesting no localization of the charge. The values of the U–N bond distances in 2 and 3 are only slightly longer than those found in 1 (2.022(5)–2.101(6) Å) and are comparable to the longer values reported for UVI = NR groups (2.034–2.068 Å).21 In both complexes the two nitrides bind the uranium centres in a quite symmetric fashion with similar values of U–N distances.
The molecular structure of complex 4, as determined by XRD studies (Fig. 2), shows a dimeric U(VI) complex bridged by two nitride and one thf ligand. The bridging mode adopted by the thf ligand is not common, but was previously reported in complexes of metal ions of the d, s and f block.22 Each U atom is hexacoordinated in a distorted octahedral geometry bound by the three bridging ligands and three siloxide ligands. Asymmetric U–N bond distances are observed in complex 4.
 | ||
| Fig. 2 Molecular structure of the complex [{U(OSi(OtBu)3)3}2(μ-N)2(μ-thf)], 4 with thermal ellipsoids drawn at the 50% probability level. The methyl groups of the tert-butyl moieties have been omitted for clarity. | ||
In particular, short U–N distances (U1–N1 = 1.966(16) Å and U2–N2 = 1.850 (12) Å) alternate with much longer distances (U1–N2 = 2.292(13) Å and U2–N1 = 2.252(16) Å). The longer distances are comparable to those found in uranium nitride clusters containing U–N single bonds (2.154(7)–to 2.399(5) Å).23,8b,4b The values of the shorter U–N distances in 4 are longer than those found in the only two previously reported terminal U(VI) nitrides (1.769(2) Å and 1.799(7) Å)7b,5e but similar to those found in borane capped U(VI) nitrides (1.880(4)–1.916(4) Å)17,6b and in tris- and tetrakis(imido) imido complexes (2.034–2.068 Å),21b consistent with the presence of UN multiple bond. The observed bonding scheme suggests that each nitride ligand binds one uranium cation with a triple bond and the other one with a single dative bond (Fig. 3). A similar alternate asymmetric bonding scheme was calculated for the matrix isolated U(VI) bis-μ-nitride [UN(μ-N)]2 (U1–N1 = 1.859 Å, U1–N2 = 2.281 Å).15f An asymmetric binding mode is also found in the only two reported polynuclear U(VI) nitrides.14,8c In the trinuclear [(NH3)8UIV(μ-N)(NH3)5UVI(μ-N)UIV(NH3)8]Br8·26NH3 a UVIN2 fragment binds two [(NH3)8UIV]4+ moieties through the bridging nitrides with UVIN bond distances of 1.853(3) and 1.854(3) Å and UIV–N distances of 2.304(3) and 2.300(3) Å.14 In the dinuclear [Na(DME)2][(NR2)2(O)U(μ-N)(CH2SiMe2NR)U(NR2)2], the NUVIO fragment binds the [UIV(CH2SiMe2NR)U(NR2)2] moiety trough a bridging nitride with a UVIN bond distance of 1.818(9) Å and a UIV–N distance of 2.284(8) Å.8c
 | ||
| Fig. 3 U2N2 cores and structural parameters of the complexes 1, 2, and 4. | ||
For U2 all the U–Osil bond distances are similar (2.09(1), 2.10(1) and 2.11(1) Å). In contrast, for U1 we observe a small shortening of the U–Osil bond trans to the nitride (2.068(9) Å; N–U–O = 161.8(6)°) compared the U–Osil bonds cis to the nitride (2.15(1) and 2.12(1) Å). Such shortening could be interpreted in terms of the inverse trans influence (ITI), a phenomenon previously observed for terminal U(VI) oxo and nitride complexes supported by strong σ and π donor ligands.24
Thus, the asymmetric binding mode found in 4 is very different from the more symmetric binding found both in the mixed valent U(VI)/U(V) bis nitride complexes 2 and 3 and in the previously reported U(V)/U(V) analogue 1.
Stability and reactivity of the bis-nitride complexes
With these bis-nitride complexes in hand, their stability and reactivity were investigated and compared to that of recently isolated terminal U(V) and U(VI) nitrides.The U(V)/U(V) bis-nitride 1 was reported to be stable both in solid state and in solution at room temperature for at least 5 days.
Similarly, once isolated the terminal U(VI) nitride [NBu4][U(OSi(OtBu)3)4(N)]5e was reported to be stable in solution for at least three weeks.5e
Complex 2 was also found to be stable in solid and in solution for at least 5 days.
Complexes 1, 2, and the terminal nitride [NBu4][U(OSi(OtBu)3)4(N)] showed no sign of decomposition when put under dynamic vacuum suggesting that nitride coupling and release of N2 do not occur for these systems.
A significantly lower solution stability was found for complex 4 which slowly decomposes in a d8-thf solution under an N2 or an Ar atmosphere to afford a complex mixture of products (Scheme 2).
 | ||
| Scheme 2 Synthesis of complex [{U(OSi(OtBu)3)3}2(μ-N)2(μ-thf)], 4 and its decomposition. | ||
The formation of new species was detected after 6 h and full decomposition was observed after 5 days giving rise to an intractable mixture of species. The decomposition of complex 4 was greatly affected by its exposure to dynamic vacuum. Full consumption of complex 4 was, indeed, observed after exposure to dynamic vacuum for 15 minutes. The reaction mixture obtained upon decomposition of complex 4 under Ar or under vacuum presents the same unidentified products as indicated by 1H NMR spectroscopy. The observed low stability under dynamic vacuum of 4 is suggestive of dinitrogen release through nitride coupling. To test this possibility, the headspace of the reaction mixture after full decomposition of complex 4 under an Ar atmosphere was analyzed by GC-MS. The presence of N2 was detected. Moreover, we found that addition of excess HCl to the reaction mixture obtained after exposure of 4 to vacuum showed only traces of NH4Cl, suggesting that the decomposition products do not contain nitride ligands (see below for observed production of NH4Cl from addition of acid to 4). This suggests that the decomposition of complex 4 occurs via oxidative coupling of the nitride ligand.
Nitride coupling has not been reported in f-element chemistry, but slow N2 release was previously observed for a tetrameric Fe bis-μ-nitride complex supported by a β-diketiminate ligand in a benzene solution.25 Electron rich transition metal terminal nitride species have also been reported to undergo spontaneous nitride coupling reactivity because of the presence of d electrons that populate the M–N π* orbitals.1b,26
Density functional theory calculations (vide infra) identified two isomers of 4, a U(V)/U(V) isomer bridged by N24− and a U(IV)/U(IV) N22− isomer, which are possible intermediates in a dinitrogen-evolution mechanism.
The reactivity of the U(V)/U(VI) and U(VI)/U(VI) bis-nitride towards CO, H2, and H+ was investigated and compared with previous studies performed on the U(V)/U(V) bis-nitride10a and terminal U(VI) nitrides.11a,5e,11e The addition of 1 atm of CO to a degassed d8-toluene solution of complex [K{U(OSi(OtBu)3)3(μ-N)}2], 2 led to a colour change from brown-orange to light green over the course of 30 minutes and to the formation of an intractable mixture as observed by 1H NMR spectroscopy. When the reaction was repeated with 13CO, the 13C NMR spectrum of the reaction mixture showed the presence of two paramagnetically shifted peaks at 215.71 and −162.60 ppm attributed to two U-bound 13C-containing species. The observed resonances fall in the broad range of chemical shifts previously reported for U(IV) and U(III) bound isocyanate ligands (492.4 to −164 ppm).1c,27 When the reaction residue was hydrolysed with pD = 12 D2O, N13CO− was observed as the only 13C-containing species by 13C NMR spectroscopy. Quantitative 13C NMR spectroscopy measured with respect to an added standard of CH313COONa revealed the formation of 2 equiv. of N13CO− per complex, thus proving that both nitride ligands are converted to isocyanate. The two-electron oxidation of two molecules of CO requires the two-electron reduction of both uranium centres in the U(V)/U(VI) complex 2, involving the formation of U(III)–U(IV) species. Ligand redistribution is probably at the origin of the presence of multiple species. Similar reductive carbonylation reactivity was observed in U(V)11a and U(VI)11a,5e terminal nitride complexes. Such reactivity differs from that reported for the U(V)/U(V) complex 1. Upon reaction with 13CO, the two nitride ligands of complex 1 are instead converted to equimolar amounts of 13CN− and N13CO− to yield the U(IV)–U(IV) complex [K2{U(OSi(OtBu)3)3}2(μ-O)(μ-CN)(μ-NCO)].10a Therefore in complex 1 one nitride undergoes reductive carbonylation but the second nitride acts as a strong nucleophile and effects the cleavage of the CO molecule. The different reactivity observed for the mixed valent complex 2 can be assigned to its higher oxidation state that facilitates the reductive carbonylation process.
However, surprisingly exposure of d8-thf or d8-toluene solutions of the complex 4 to 1 atm of CO did not result in any reaction even after 6 days. Monitoring the evolution of the reactions by 1H NMR spectroscopy revealed only the presence of decomposition products and no 13C-containing species could be detected by 13C NMR spectroscopy after hydrolysis of the reaction mixture with pD = 12 D2O.
The lack of reactivity of the U(VI)/U(VI) bis-nitride also contrasts with the high reactivity towards CO of the analogous terminal U(VI) nitride complex supported by four siloxide ligands [NBu4][U(OSi(OtBu)3)4(N)], bearing a U–N triple bond.5e
The only other reported terminal U(VI) nitride, the [U(VI)(TrenTIPS)N] complex,7b was found to react slowly with CO over the course of 16 h while the analogous [U(V)(TrenTIPS)N]− reacts immediately with the substrate. Such slower reaction observed for the U(VI) complex was interpreted in terms of sterical congestion at the U(VI) centre preventing binding of CO to the metal in the proposed reaction intermediate.11a A similar reasoning could be used to explain, at least in part, the lack of reactivity of the U(VI) bis-nitride complex 4 compared to the fast reaction of the U(V) analogue, complex 1, with CO.
Complex 4 reacts rapidly with CO2 in ambient conditions but the resulting products could not be identified.
Since we recently reported the rare ability of complex 1 to promote the oxidative cleavage of H2 in ambient conditions, affording the reduced imido-bridged U(IV)/U(IV) complex [K2{U(OSi(OtBu)3)3(μ-NH)}2], 6, we set out to investigate the reactivity of complexes 2 and 4 with H2 (Scheme 3). Addition of 1 atm of H2 to a d8-toluene solution of complex 2 led to its slow conversion over the course of 2 days to an equimolar mixture of two species resonating at 0.27 and −0.87 ppm. Monitoring the evolution of the reaction mixture via1H NMR spectroscopy did not reveal the presence of intermediate species. The resonance at −0.87 ppm was assigned to the previously reported complex [K2{U(OSi(OtBu)3)3(μ-NH)}2], 6. The presence of 6 was also confirmed by single crystal X-ray diffractometry. Cooling down a concentrated toluene/thf reaction mixture at −40 °C led to the formation of light yellow crystals of complex 6 and light green crystals of the complex [{U(OSi(OtBu)3)3}2(μ-NH2)2(μ-thf)], 7. The similar solubility of the complexes 6 and 7 prevented the isolation of analytically pure amounts of the latter from the reaction of complex 2 with H2.
 | ||
| Scheme 3 Reactivity of complex 2 with H2 yielding an equimolar mixture of the complexes [K2{U(OSi(OtBu)3)3(μ-NH)}2], 6 and [U(OSi(OtBu)3)3(μ-NH2)]2, 7. | ||
Nevertheless, complex 7 could be independently synthesised upon protonation of complex [K2{U(OSi(OtBu)3)3(μ-NH)}2], 6 with 2 equiv. of the weak acid NEt3HBPh4, confirming the presence of amido bridging ligands in complex 7.
The molecular structure of complex 7, as determined by XRD studies (Fig. 4), shows a dimeric U(IV) complex bridged by two amido and one thf ligand. Each U centre is hexacoordinated in a distorted octahedral geometry. The bridging NH2 linkage is unprecedented in uranium complexes. The U–N bond distances (U1–N1 = 2.441(7), U1–N2 = 2.443(7), U2–N1 = 2.436(10), U2–N2 = 2.455(8) Å) are elongated compared to the U–Nimido distances in complex 6 (2.192(3)–2.273(3) Å) and are longer than the distances found in the rare examples of terminal U–NH2 complexes (2.183(6)–2.335(3) Å).7b,11e,28 The reaction of complex 2 with H2 is likely to involve first the oxidative cleavage of one molecule of H2 to afford a transient U(IV)–U(V) bis-μ-NH followed by ligand scrambling affording complex 6 and a transient U(V)–U(V) bis-μ-NH complex. The latter immediately reacts with a second molecule of H2 to afford complex 7 (Scheme S2†).
 | ||
| Fig. 4 Molecular structure of the complex [{U(OSi(OtBu)3)3}2(μ-NH2)2(μ-thf)], 7 with thermal ellipsoids drawn at the 50% probability level. The methyl groups of the tert-butyl moieties have been omitted for clarity. | ||
It should be noted that bis-imido U(IV)/U(IV) complex 6 does not react further with H2 suggesting that a high oxidation state of the metal centre is required for an imido complex to effect oxidative cleavage of H2. Similar oxidative cleavage of H2 was reported very recently for a terminal U(V)11e but terminal U(VI) nitrides did not react. The analogous nitride bridged complex U(VI)/U(VI) 4 also did not react with 1 atm of H2. Monitoring the evolution of the reaction of 4 with H2 by 1H NMR spectroscopy revealed only the presence of decomposition products.
These results seem to indicate that the presence of U(V) in the charge delocalised complex 2, best described as a U 5.5+/U 5.5+ complex, results in a higher reactivity towards H2 compared to the U(V)/U(V) and U(VI)/U(VI) analogues.
The computed mechanism of hydrogenolysis by the terminal U(V) complex [UV(TrenTIPS)(N)][K(B15C5)2] (TrenTIPS = N(CH2CH2NSiiPr3)33−; B15C5 = benzo-15-crown-5-ether) involves an heterolytic cleavage of H2 and formation of a hydride imide U(V) intermediate followed by a 1,2 addition to yield the final U(III)–NH2 complex. The observed reactivity of the terminal U(V) and lack of reactivity of the analogous U(VI) was explained in terms of a combination of the 5f-electron not being fully nonbonding, and the uranium(V)-nitride bond being highly polar.
A mechanism involving an heterolytic cleavage of H2 to yield an imide hydride followed by hydride migration to the second nitride is likely to occur in both hydrogenation steps of the nitrides in complex 2. A similar mechanism has also been computed for the hydrogenolysis effected by an iron bis-nitride complex very recently reported by Walter.13e
Finally, despite being unreactive towards CO and H2, the nitride ligands in complex 4 react readily with H+ to afford ammonia. A 100% conversion to NH4Cl (2 equiv. of NH4Cl per complex) was observed by 1H NMR analysis of the products in d6-dmso (dimethylsulfone was added as an internal standard for quantification) upon reaction of complex 4 with excess of a 2 M solution of HCl in diethylether.
Similarly, a 93% conversion to NH4Cl was observed upon reaction of 2 with a 2 M solution of HCl in diethylether. A lower yield to NH4Cl (78%) was observed10a upon the addition of HCl in the same conditions to complex 1.
Computational analysis
We performed DFT calculations on several of the diuranium species identified here to compare the uranium–nitrogen bonding across this set of similar complexes, i.e. the bis-nitrides 1, 2 and 4, and the terminal uranium nitride we recently reported, [NBu4][U(OSi(OtBu)3)4(N)],5e8. PBE0 calculations were performed at geometries in which the heavy atom positions were taken from experiment, with hydrogen positions optimised. Full computational methodology is given in the ESI.† For 1 (a U(V)/U(V) triplet) and 2 (a U(VI)/U(V) doublet, with the U(V) centre proximal to K+), singly occupied molecular orbitals are predominantly 5fU in character.Natural Bond Orbital (NBO) and Quantum Theory of Atoms in Molecules (QTAIM) analyses were performed to obtain bond orders (the Wiberg bond index, BOW(U,N), in NBO and the QTAIM delocalisation index δ(U|N)) and atomic charges, shown in Table 1. For 8, BOW(U,N) = 2.76 and δ(U|N) = 2.60, indicating a strong triple bond. For 4, the shorter pair of bonds have BOW(U,N) of 2.16 and 2.10 for the 1.85 and 1.97 Å bonds respectively, and the longer pair of bonds have BOW(U,N) = 0.85 and 0.78, i.e. the shorter pair of bonds have orders c. 2.6 times larger than the longer pair. A similar picture emerges from the QTAIM data, with δ(U|N) for the shorter pair of bonds c. 2.5 times greater than for the longer bonds, 2.01 and 1.95 vs. 0.79 and 0.73, respectively. By contrast, the U–N BOW(U,N) in 1 vary from 1.17 to 1.47 and in 2 from 1.16 to 1.65, and the QTAIM varies from 1.20 to 1.45 in 1 and from 1.14 to 1.55 in 2, i.e. the variation in bond orders in 1 and 2 is appreciably smaller than the substantial alternation observed in 4.
| QTAIM | NBO | |||||
|---|---|---|---|---|---|---|
| q(U) | q(N) | δ(U|N) | q(U) | q(N) | BOW(U,N) | |
| 1 | 2.64, 2.64 | −1.33, −1.33 | 1.45, 1.45, 1.20, 1.20 | 1.87, 1.87 | −1.34, −1.34 | 1.47, 1.47, 1.19, 1.19 |
| 2 | 2.81, 2.66 | −1.24, −1.21 | 1.14, 1.28, 1.39, 1.55, | 1.88, 1.76 | −0.91, −0.88 | 1.66, 1.48, 1.31, 1.16 |
| 4 | 2.85, 2.84 | −1.11, −1.07 | 2.01, 1.95, 0.79, 0.73 | 1.88, 1.80 | −0.72, −0.70 | 2.16, 2.10, 0.85, 0.78 |
| 8 | 2.78 | −0.95 | 2.60 | 1.93 | −0.69 | 2.76 |
The QTAIM charges for the U(V) centres in 1 and 2 are between 2.64–2.66, and the U(VI) centres in 2, 4 and 8 between 2.78–2.85. Calculated charges are rarely as large as formal oxidation states, particularly for high oxidation state systems, but the QTAIM charges for uranium atoms in the same formal oxidation state are in quite a small range, and distinguish U(V) from U(VI). By contrast, NBO charges are smaller than the analogous QTAIM values, and span a larger, overlapping range, with U(V) centres lying between 1.76 and 1.87, and U(VI) centres between 1.80 and 1.88.
We have also performed Intrinsic Bonding Orbital (IBO) analyses.29 This technique localises orbitals via a projection onto a minimal quality basis set (Intrinsic Atomic Orbitals, IAOs), minimising the number of atoms the orbital is distributed over. Sets of the IBOs in the U2N2 ring for the studied complexes are shown in Fig. 5. Orbitals are well localised onto one half of the U2N2 ring (at least 99% in the IAO basis), with a similar set of orbitals being localised onto the opposite half (shown in Fig. S45 of the ESI†). The interpretation of the IBOs of 8 is unambiguous, with three bonding orbitals localised to UN, and a full σ2π4 triple bond, consistent with the high values of BOW(U,N) and δ(U|N). The much smaller values of the bond order metrics for 1 and 2 are supported by the bonding IBOs, which are well delocalised across U–N–U. The significantly different bond orders for the shorter and longer bonds in 4 are also consistent with its IBOs, which show a U–N σ orbital well localised to UN but with π orbitals which partially delocalise to form the U–N single bond with the other uranium. The σ and two π orbitals have 3.8, 16.0 and 9.7% contribution respectively from the singly bound uranium (average 9.8%), and 39.2, 22.8 and 27.7% character from the triply bound uranium. By contrast, across the three bonding IBOs the delocalised 2 has an average character of 25.6% on one uranium (U–N = 2.02 Å) and 16.3% on the other (U–N = 2.09 Å). This change in bonding from 4 to 1 and 2 is similar to that calculated for the matrix isolated U(VI) bis-μ-nitride [UN(μ-N)]2 described as a dimer of two UN2 units, while in the same study the U(III) species [U(μ-N)]2 was found to be highly delocalised.15f
 | ||
| Fig. 5 α spin IBOs of the U2N2 ring in 1, 2 and 4, and for the UN bond in 8. The isosurfaces enclose 90% of the orbital. | ||
Calculations using the PBE0 functional (full methodology is given in the ESI†) identified a U(V)/U(V) isomer bridged by N24− with an energy of +0.5 kJ mol−1, and a U(IV)/U(IV) N22− isomer with an energy of −19.1 kJ mol−1, relative to the U(VI)/U(VI) bis-nitride isomer. The N–N bond lengths are 1.44 and 1.25 Å respectively, i.e. single and double bonds as expected for the N24− and N22− bridging ligands, and distinct from the observed N–N distance in 4, 2.47(2) Å where the N–N bond is completely broken. Due to the challenges presented by the size of the molecule and multiple spin-crossovers we have not identified transition states linking these isomers, and our calculations were performed in the gas phase, but do suggest an energetically plausible route to oxidation of the nitrides, leading to N2 evolution.
Conclusions
In conclusion we have been able to synthesise and characterise the first example of a molecular compound containing the cyclic (U(VI)N)2 core previously only identified in gas phase experiments. We showed that the U(VI)/U(VI) bis-nitride (4) can be prepared from the oxidation of the U(V)/U(V) analogue (1) using a strong oxidizing agent, while weaker oxidizing agents only afford the U(V)/U(VI) analogues (2 and 3). The three compounds show different stability and reactivity. Notably, both 1 and 2 are stable in solution while 4 rapidly releases dinitrogen probably through U(V)/U(V)N24− and U(IV)/U(IV)N22− intermediates. The reactivity of 2 and 4 with CO, H2, and H+ was explored and compared. Both complexes react with strong acids to yield NH4+ quantitatively. Complex 4 does not react with CO or H2 while both nitrides in 2 undergo reductive carbonylation to yield 2 NCO−. The lack of reactivity of 4 with CO contrasts with the high reactivity with CO reported for the terminal U(VI) nitride (8).5e Complex 2 also effects the cleavage of H2 to yield the bis-imido, 6 and bis-amido 7 providing a rare example of direct hydrogenolysis by a metal nitride. Computational studies show a very different bonding scheme for complexes 1 and 2 compared to the U(VI) bis-nitride 4; the former shows a delocalised π bonding system while the latter has a U–N σ orbital well localised to UN but with π orbitals which partially delocalise to form the U–N single bond with the other uranium.
These results indicate that the delocalised π bonding system found in the complexes 1 and 2 stabilise the cyclic (UN)2 structures defavouring N2 release compared to 4 and results in an higher nucleophilic reactivity of the nitrides towards CO and H2.
Authors contributions
L. B. carried out the synthetic experiments and analysed the experimental data; R. S. carried out the X-ray single crystal structure analyses; H.S synthesised and characterised complexes 6 and 7; B. E. A. and N. K. carried out and analysed the computational data. M. M. originated the central idea, coordinated the work, and analysed the experimental data. M. M., L. B., and B. E. A. wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200021_178793 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank R. Moinat and F. Sepulveda for carrying out the elemental and mass spectrometry analyses, F. Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses. BEA and NK acknowledge the assistance given by Research IT, and the use of the Computational Shared Facility and the HPC Pool funded by the Research Lifecycle Programme at The University of Manchester.Notes and references
- (a) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed; (b) A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed. Engl., 2013, 52, 12510–12529 CrossRef CAS PubMed; (c) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed; (d) N. B. Thompson, M. T. Green and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 15312–15315 CrossRef CAS PubMed; (e) L. R. Doyle, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2019, 58, 6674–6677 CrossRef CAS PubMed; (f) M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed; (g) S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed; (h) J. Sun, J. Abbenseth, H. Verplancke, M. Diefenbach, B. de Bruin, D. Hunger, C. Wurtele, J. van Slageren, M. C. Holthausen and S. Schneider, Nat. Chem., 2020, 12, 1054–1059 CrossRef CAS PubMed.
- (a) F. Haber, Angew. Chem., 1914, 27, 473–477 CrossRef CAS; (b) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266, 2–15 CrossRef.
- (a) R. B. Matthews, K. M. Chidester, C. W. Hoth, R. E. Mason and R. L. Petty, J. Nucl. Mater., 1988, 151, 334–344 CrossRef CAS; (b) G. W. C. Silva, C. B. Yeamans, A. P. Sattelberger, T. Hartmann, G. S. Cerefice and K. R. Czerwinski, Inorg. Chem., 2009, 48, 10635–10642 CrossRef CAS PubMed.
- (a) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed. Engl., 2002, 41, 3433–3436 CrossRef CAS; (b) X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- (a) L. Andrews, X. Wang, R. Lindh, B. O. Roos and C. J. Marsden, Angew. Chem., Int. Ed. Engl., 2008, 47, 5366–5370 CrossRef CAS PubMed; (b) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (c) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7 Search PubMed; (d) B. E. Atkinson, H. S. Hu and N. Kaltsoyannis, Chem. Commun., 2018, 54, 11100–11103 RSC; (e) L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed.
- (a) A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed; (b) M. A. Boreen, G. D. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed; (c) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed; (b) G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2008, 47, 3040–3042 CrossRef CAS PubMed; (c) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed; (d) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed; (e) T. K. Todorova, L. Gagliardi, J. R. Walensky, K. A. Miller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 12397–12403 CrossRef CAS PubMed; (f) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (g) L. Maria, I. C. Santos, V. R. Sousa and J. Marcalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed.
- (a) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed; (b) C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed.
- (a) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (b) J. Du, D. M. King, L. Chatelain, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- (a) P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2014, 53, 10412–10415 CrossRef CAS PubMed; (b) M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2016, 55, 4074–4078 CrossRef CAS PubMed; (c) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2016, 55, 12290–12294 CrossRef CAS PubMed; (d) K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M. H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed; (e) L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11 Search PubMed; (f) M. Yadav, A. Metta-Magana and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC.
- (a) M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2018, 57, 3697–3700 CrossRef CAS PubMed; (b) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- (a) S. D. Brown, M. P. Mehn and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 13146–13147 CrossRef CAS; (b) J. Schoffel, A. Y. Rogachev, S. D. George and P. Burger, Angew. Chem., Int. Ed. Engl., 2009, 48, 4734–4738 CrossRef; (c) F. S. Schendzielorz, M. Finger, C. Volkmann, C. Wurtele and S. Schneider, Angew. Chem., Int. Ed. Engl., 2016, 55, 11417–11420 CrossRef CAS PubMed; (d) S. Kim, H. Y. Zhong, Y. Park, F. Loose and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 9518–9524 CrossRef CAS PubMed; (e) M. Reiners, D. Baabe, K. Munster, M. K. Zaretzke, M. Freytag, P. G. Jones, Y. Coppel, S. Bontemps, I. del Rosal, L. Maron and M. D. Walter, Nat. Chem., 2020, 12, 740–746 CrossRef PubMed; (f) D. Sengupta, C. Sandoval-Pauker, E. Schueller, A. M. Encerrado-Manriquez, A. Metta-Magana, W. Y. Lee, R. Seshadri, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2020, 142, 8233–8242 CrossRef CAS PubMed.
- S. S. Rudel, H. L. Deubner, M. Muller, A. J. Karttunen and F. Kraus, Nat. Chem., 2020, 12, 962–967 CrossRef CAS PubMed.
- (a) D. W. Green and G. T. Reedy, J. Chem. Phys., 1976, 65, 2921–2922 CrossRef CAS; (b) R. D. Hunt, J. T. Yustein and L. Andrews, J. Chem. Phys., 1993, 98, 6070–6074 CrossRef CAS; (c) P. Pyykko, J. Li and N. Runeberg, J. Phys. Chem., 1994, 98, 4809–4813 CrossRef; (d) X. F. Wang, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Inorg. Chem., 2011, 50, 3826–3831 CrossRef CAS PubMed; (e) L. Andrews, X. F. Wang, Y. Gong and G. P. Kushto, J. Phys. Chem. A, 2014, 118, 5289–5303 CrossRef CAS PubMed; (f) B. Vlaisavljeyich, L. Andrews, X. F. Wang, Y. Gong, G. P. Kushto and B. E. Bursten, J. Am. Chem. Soc., 2016, 138, 893–905 CrossRef PubMed.
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- (a) C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS PubMed; (b) A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS PubMed.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 11732–11743 CrossRef CAS PubMed.
- (a) N. H. Anderson, S. O. Odoh, Y. Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (b) N. H. Anderson, J. Xie, D. Ray, M. Zeller, L. Gagliardi and S. C. Bart, Nat. Chem., 2017, 9, 850–855 CrossRef CAS PubMed.
- (a) T. Chlupaty, M. Bilek, J. Merna, J. Brus, Z. Ruzickova, T. Strassner and A. Ruzicka, J. Chem. Soc., Dalton Trans., 2019, 48, 5335–5342 RSC; (b) J. X. Liu, W. F. Chen, J. F. Li and C. M. Cui, ACS Catal., 2018, 8, 2230–2235 CrossRef CAS; (c) S. Komorski, M. K. Leszczynski, I. Justyniak and J. Lewinski, Inorg. Chem., 2016, 55, 5104–5106 CrossRef CAS PubMed.
- W. J. Evans, K. A. Miller, J. W. Ziller and J. Greaves, Inorg. Chem., 2007, 46, 8008–8018 CrossRef CAS PubMed.
- H. S. La Pierre and K. Meyer, Inorg. Chem., 2013, 52, 529–539 CrossRef CAS PubMed.
- (a) K. C. MacLeod, D. J. Vinyard and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 10226–10229 CrossRef CAS PubMed; (b) E. O'Grady and N. Kaltsoyannis, J. Chem. Soc., Dalton Trans., 2002, 1233–1239 RSC.
- (a) M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed; (b) M. G. Scheibel, Y. l. Wu, A. C. Stueckl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135, 17719–17722 CrossRef CAS PubMed; (c) J. Abbenseth, M. Finger, C. Wurtele, M. Kasanmascheff and S. Schneider, Inorg. Chem. Front., 2016, 3, 469–477 RSC.
- C. Camp, L. Chatelain, C. E. Kefalidis, J. Pecaut, L. Maron and M. Mazzanti, Chem. Commun., 2015, 51, 15454–15457 RSC.
- (a) G. F. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS; (b) J. A. H. Frey, F. G. N. Cloke and S. M. Roe, Organometallics, 2015, 34, 2102–2105 CrossRef.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, NMR spectra, computational details, crystallographic data. CCDC numbers: 2072156–2072160; 2080129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01796a |
| This journal is © The Royal Society of Chemistry 2021 |