
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule imaging and kinetic analysis of intermolecular polyoxometalate reactions†
Jack W.
Jordan
 a,
Kayleigh L. Y.
Fung
a,
Stephen T.
Skowron
a,
Christopher S.
Allen
be,
Johannes
Biskupek
c,
Graham N.
Newton
d,
Ute
Kaiser
c and
Andrei N.
Khlobystov
*a
a,
Kayleigh L. Y.
Fung
a,
Stephen T.
Skowron
a,
Christopher S.
Allen
be,
Johannes
Biskupek
c,
Graham N.
Newton
d,
Ute
Kaiser
c and
Andrei N.
Khlobystov
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: Andrei.Khlobystov@nottingham.ac.uk
bElectron Physical Science Imaging Center, Diamond Light Source Ltd., Didcot OX11 0DE, UK
cElectron Microscopy Group of Materials Science, Ulm University, 89081 Ulm, Germany
dGSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Nottingham NG7 2TU, UK
eDepartment of Materials, University of Oxford, Oxford OX1 3HP, UK
First published on 26th April 2021
Abstract
We induce and study reactions of polyoxometalate (POM) molecules, [PW12O40]3− (Keggin) and [P2W18O62]6− (Wells–Dawson), at the single-molecule level. Several identical carbon nanotubes aligned side by side within a bundle provided a platform for spatiotemporally resolved imaging of ca. 100 molecules encapsulated within the nanotubes by transmission electron microscopy (TEM). Due to the entrapment of POM molecules their proximity to one another is effectively controlled, limiting molecular motion in two dimensions but leaving the third dimension available for intermolecular reactions between pairs of neighbouring molecules. By coupling the information gained from high resolution structural and kinetics experiments via the variation of key imaging parameters in the TEM, we shed light on the reaction mechanism. The dissociation of W–O bonds, a key initial step of POM reactions, is revealed to be reversible by the kinetic analysis, followed by an irreversible bonding of POM molecules to their nearest neighbours, leading to a continuous tungsten oxide nanowire, which subsequently transforms into amorphous tungsten-rich clusters due to progressive loss of oxygen atoms. The overall intermolecular reaction can therefore be described as a step-wise reductive polycondensation of POM molecules, via an intermediate state of an oxide nanowire. Kinetic analysis enabled by controlled variation of the electron flux in TEM revealed the reaction to be highly flux-dependent, which leads to reaction rates too fast to follow under the standard TEM imaging conditions. Although this presents a challenge for traditional structural characterisation of POM molecules, we harness this effect by controlling the conditions around the molecules and tuning the imaging parameters in TEM, which combined with theoretical modelling and image simulation, can shed light on the atomistic mechanisms of the reactions of POMs. This approach, based on the direct space and real time chemical reaction analysis by TEM, adds a new method to the arsenal of single-molecule kinetics techniques.
Introduction
One of the biggest challenges of chemistry is to reconcile reaction kinetics on the macroscale with molecular dynamics at the atomic scale. Although it is the latter that provides the mechanistic understanding required for a rational approach to synthesis, traditional ensemble kinetics analysis methods operate at the bulk regime and give averaged information that cannot be fundamentally disentangled to the discrete single-molecule level events.1,2 Ensemble kinetics is nonetheless a highly successful field and one of the most powerful tools available to chemists for revealing reaction mechanisms; however, the ensemble-averaged measurements can only confirm rather than prove reaction mechanisms, at worst limiting descriptions to the phenomenological rather than mechanistic.3One aspect of this limitation can be seen with the theoretical divide between classical (deterministic) and stochastic (probabilistic) chemical kinetics, the distinction between the two becoming important for reactions involving small populations.4 Experimentally, if ensemble kinetics methods can be considered ‘top-down’, then over the last four decades a range of techniques have developed which operate in a ‘bottom-up’ approach, in which information is measured from individual molecules and built up statistically to provide equivalent information to that which is normally accessible to ensemble kinetics. The key advantage of such single-molecule kinetics approaches lies with their ability to distinguish information that is in principle unobtainable by ensemble-averaging techniques,5 whether by revealing rare or transient species6,7 or by providing insight into otherwise unobserved population heterogeneities.8 The ability to disentangle the constructed statistical distributions by subpopulations in order to probe subsets of the reaction can reveal large fluctuations of various underlying properties on an individual molecular basis that collapse to a single averaged ensemble measurement.
The field of optical microscopy provides the most mature example of single-molecule kinetics, having been extensively developed since the 1990's following early pioneering studies in the 1960's.9 Primarily in the context of large biomolecules, the use of super resolution and fluorescence based microscopy techniques to track individual molecular positions and intermolecular distances has enabled single-molecule investigations of diffusion and interaction kinetics, and even revealed information about intramolecular processes such as conformational changes, especially important for such large molecules that exhibit both static and dynamic disorder.5,10 More recently, developments following the first electrochemical detection of single molecules in 1995 (ref. 11) have promised to provide a platform for single-molecule measurements for entities ranging from cells to much smaller molecules.8,12,13 The movement of this area of research towards new single-molecule kinetics methods has sparked intense discussion in the electrochemical literature over the last decade.12,14,15 As neither electrochemical nor fluorescence microscopy methods can deliver atomic-level information, further advancement in the chemistry of single molecules relies on scanning probe microscopy (SPM) methods that can potentially provide atomically resolved structural information about the reacting species and their local environment that can be coupled to the statistical bottom-up kinetics measurements for additional insight that would be completely inaccessible to ensemble-averaged methods. For example, force microscopies based on mechanical interaction – in particular atomic force microscopy (AFM) – have been successfully utilised to probe large biologically relevant molecules,5,6 and different SPM methods can now routinely achieve atomic spatial resolution and have recently shed light on the atomic structures of reaction products16–18 and reaction intermediates.19 However, compared to other methods SPM lacks the temporal resolution needed to capture reaction dynamics with spatiotemporal continuity, and conditions required for atomic resolution (e.g. cryogenic temperatures) are often incompatible with the activation of reactions in situ. In the context of single-molecule biology, although high-speed AFM has been developed and can reach approximately 30 fps,5 this comes at the expense of spatial resolution and field of view, and with current technologies appears limited to a maximum frame-rate of approximately 100 fps.20
Transmission electron microscopy (TEM) on the other hand can provide both high spatial and temporal resolution. For example, a 4D TEM methodology was applied to the direct imaging of physicochemical transformations in materials on the nanosecond timescale,21 (but with spatial resolution being significantly degraded to ∼20–30 nm in this specific case). An alternative method is to utilise the TEM electron beam (e-beam) as both a fast imaging tool and a source of energy for stimulating chemical reactions, an approach termed ChemTEM.22,23 Such a dual-use of the e-beam enables the triggering and recording of molecular transformations as they happen, without the need to introduce any additional source of energy (such as heat in SPM or laser pulses in 4D-TEM). Moreover, at different energies available to the e-beam, it can trigger different chemical processes in the same material24 and, in addition, the tuning of this e-beam flux may control the rates of the reactions.25 The ChemTEM methodology therefore provides both of the requirements common to all fields exploring single-molecule kinetics: control of molecular positions (confinement within the SWNT) and control of energy supplied to the molecules (with the e-beam).7,22 However with ChemTEM, as with any local-probe method, the key challenge in studying reactions at the single-molecule level is caused by the primary advantage: each measurement provides observations that are specific to a particular molecule in a specific local environment during that measurement. Therefore, ideally, a reaction should be studied at the single-molecule level but for as many examples of single-molecule events as possible at the same time, thus delivering both (i) detailed knowledge of the reaction mechanism and (ii) confidence in its generality for that class of molecules. Unlike the optical microscopy methods discussed above TEM is still in its infancy in this regard, with very recent progress in the development of fast direct electron detectors26 and machine learning techniques27 only just starting to enable the kinds of high throughput experiments and large datasets required to build up meaningful statistics at the single-molecule level (as distinct from class-averaging techniques used in cryo-EM28 and elsewhere29). Hence, there have been relatively few quantitative studies of single-molecule (or entity) kinetics using TEM;30–33 a notable exception is the work of Isaacson et al. in the Crewe lab in the 1970's shortly following the development of the scanning TEM,34,35 which – akin to Rotman's 1961 study for optical single-molecule kinetics36 – may come to be seen as being decades ahead of its time.
Here, we make progress towards single-molecule kinetics in the TEM by combining single-molecule structural and kinetic information from imaging the transient reactants, intermediates and products of reactions of the Keggin, [PW12O40]3− (Fig. 1A) and Wells–Dawson, [P2W18O62]6− (Fig. 1B) polyoxometalates (POM). Under the electron beam neighbouring POMs quickly react with one another, revealed to be a polycondensation mechanism forming intermediate metal oxide nanowires and resulting in metallic tungsten nanoclusters. By imaging ca. 100 molecules reacting simultaneously using a bundle of parallel carbon nanotubes as reaction vessels and harnessing a key controllable aspect of the e-beam stimulus, we demonstrate that the rate of the reaction is highly dependent upon the e-beam flux. Combining atomic-level structural and kinetic information to determine the reaction mechanism, our measurements indicate that the initial metal–oxygen bond dissociation in POM molecules at the onset of the polycondensation reaction is reversible, and we use this to provide practical guidance to enable the imaging of molecular metal oxides in their native states with TEM.
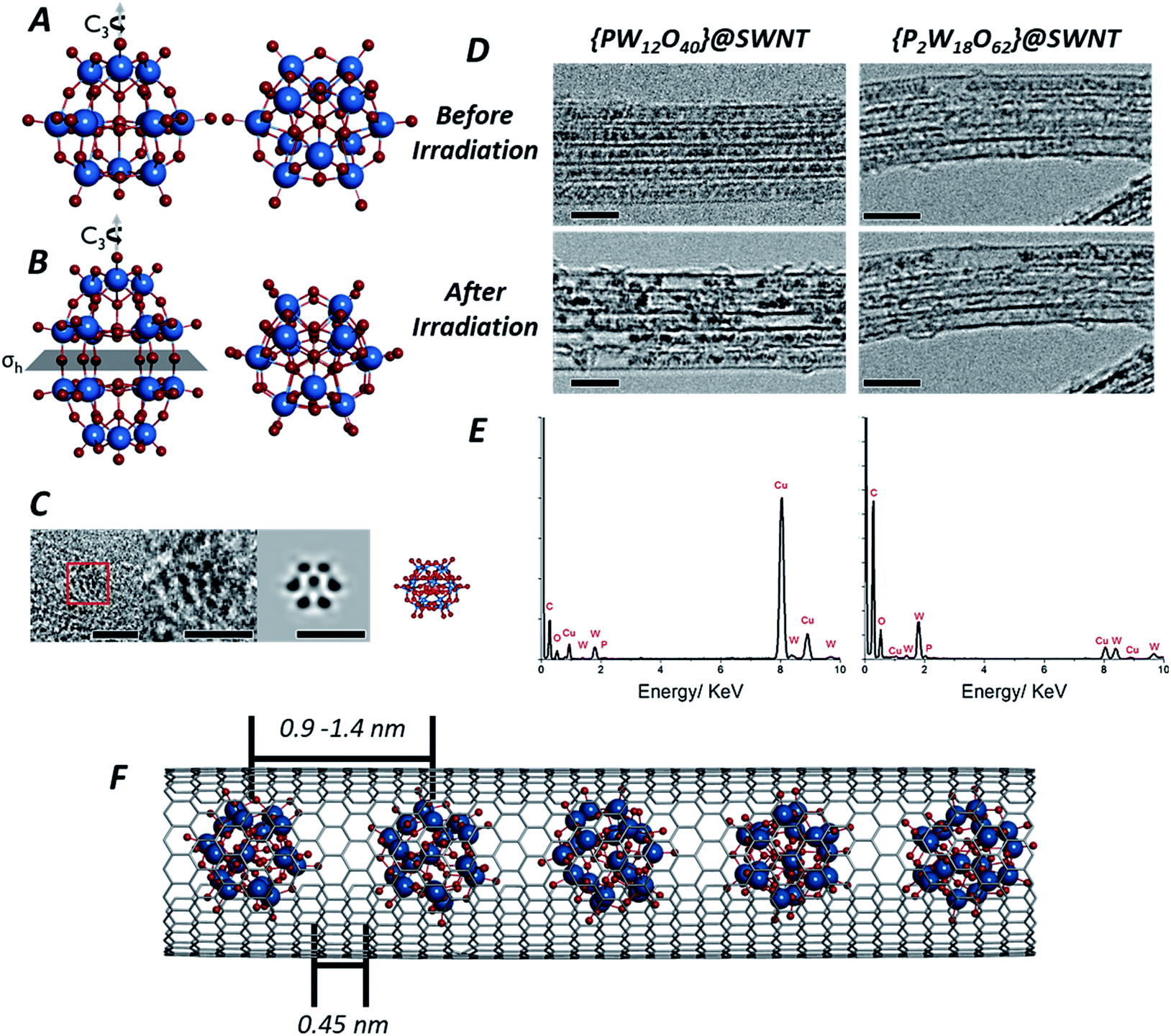 | ||
| Fig. 1 Structures of both the Keggin [PW12O40]3− (A) and Wells–Dawson [P2W18O62]6− (B) anions: the Keggin molecule possesses a Td symmetry, with a set of C3 rotation axes, and the Wells–Dawson molecules possesses D3h symmetry, with a C3 rotation axis and a horizontal mirror plane σh (tungsten atoms are shown in blue, oxygen in red and phosphorous in orange). AC-TEM image of the Wells–Dawson molecule encapsulated within a SWNT and projected along C3 axis (left) with corresponding structural model and simulated TEM image (right). Scale bars are 1 nm (C). TEM images of [PW12O40]@SWNT (left) and [P2W18O62]@SWNT (right) arranged in bundles of ∼5 nanotubes (D): at the beginning of imaging all molecules are close-packed and periodically spaced within the nanotube cavity (top images, D), followed by condensation into larger, aperiodic structures after an exposure of ∼10 min to the electron beam (bottom images, D). Scale bars are 5 nm. Images acquired at 80 keV. EDX spectra of [PW12O40]@SWNT (left) and [P2W18O62]@SWNT (right) (E) confirming the presence of the POM elements, in good agreement with their atomic ratios (copper peaks are due to the TEM grid). Model of [P2W18O62]@SWNT (F). ESI Video 1† demonstrates transformations of [PW12O40]@SWNT under high dose of e-beam. | ||
Experimental and computational methods
Synthesis of POM@SWNT materials
The synthesis of both H3[PW12O40] and K6[P2W18O62] were carried out using synthetic methodologies reported previously.37 Synthesis of the POM@SWNT materials involved heating SWNTs (200 mg) at 600 °C for 45 minutes. The resulting black solid (100 mg) was then added to a rapidly stirred solution of POM (2 g) in water (30 mL). The suspensions were sonicated for 5 minutes and subsequently stirred at room temperature for 2 days. The suspensions were then filtered through PTFE membranes to give black solids (120 mg).TEM imaging
Low magnification TEM images for kinetic analysis were acquired on a JEOL 2100F TEM field emission gun microscope at 80 kV. Atomically resolved AC-TEM at varying accelerating voltages was carried out on a Cs corrected FEI Titan 80-300 TEM operated at 80 kV (images were acquired using a Gatan Ultrascan 1000XP and exposure times of 0.25 to 1.0 s) and a Cs/Cc corrected SALVE microscope at 30 kV and 60 kV (images were acquired using a Gatan Ultrascan 1000XP and exposure times of 0.25 to 1.0 s). High frame rate AC-TEM imaging was carried out on a Cs corrected JEOL ARM300F microscope at 80 kV (fast image acquisition with exposure times of 0.1 s and frame rates of 10 fps were acquired using a Gatan OneView Camera). Samples were prepared by dispersion in isopropyl alcohol and were drop-cast onto a copper grid mounted with lacey carbon film.TEM image simulation
TEM image simulations were produced using the QSTEM multislice code,38 using 20 slices and a simulation pixel size of 0.0052 nm (re-binned 5× to the experimental pixel size of 0.026 nm). The following imaging parameters were used, in order to match the AC-TEM experiments acquired at a low doses per image (JEOL ARM300F): 80 kV accelerating voltage; −0.107 nm defocus (Scherzer); 5 nm defocal spread; 0.517 μm Cs; 1.7 mm Cc; 41.65 mrad α; 3.371 nm, 82.75° C12; 0.0982 μm, 33.55° C21; 0.0595 μm, −16.59° C23. Poisson noise due to a finite electron dose was applied with the intensity of each pixel calculated as I(x,y) = Poisson random[Isim(x,y)DΔxΔy], where Isim(x,y) is the image intensity resulting from the multislice simulation, D is the electron dose per image (4.6 × 104 e− nm−2) and ΔxΔy is the pixel size.Density functional theory
Partial atomic charges of the [P2W18O62]6− anion were calculated using a Mulliken population analysis of density functional theory (DFT) calculations. The Q-Chem 5.0 quantum chemistry software package39 was used to optimise the geometry using the BP86VWN exchange–correlation functional,40,41 the def2-svpd basis set with the associated def2-ecp for tungsten,42 and empirical Grimme dispersion, resulting in a structure with an RMSD of 0.0114 nm compared to the crystal structure.43 These partial atomic charges were used in the multislice image simulations as described in the ESI.† Geometry optimisations of SWNT and oxygen species were performed at an initial B3LYP/6-31G* level of theory,44,45 followed by ωB97X-D/6-31G*.46 A 1.1 nm diameter (8,8) SWNT with dangling bonds terminated by hydrogen atoms (C160H32) was the model SWNT, used to determine the stability of epoxides on the interior surface of the tube and the energetic landscape of a variety of 2O@SWNT species (see ESI for further details†).Results
Owing to their rigid, symmetrical structure and the presence of high atomic number elements, such as W, POM molecules have previously been successfully imaged by TEM. Examples include imaging of the Lindquist [W6O19]2− anion confined within double-walled carbon nanotubes47 (DWNT) or the Keggin48 and its analogues49,50 imaged on graphene supports or encapsulated within SWNTs.51 Recently52 we showed that POMs can be successfully encapsulated within SWNTs, a process driven by coulombic forces and resulting from the simple immersion of carbon nanotubes with an average diameter of 1.4 nm into an aqueous solution containing the Keggin [PW12O40]3− or Wells–Dawson [P2W18O62]6− POM anions. Under these conditions, POM molecules enter the nanotube as naked anions, without cations, with the host-nanotubes effectively balancing their charges and drawing the POMs from solution into the SWNT cavity. The presence of the molecules inside the nanotubes can be directly observed in our lower magnification TEM images (see Experimental methods) as dark circles (Fig. 1D) with diameters of ∼0.7 nm for [PW12O40]3− and ∼0.8 nm for [P2W18O62]6− due to a much higher contrast of the tungsten-containing POM molecules (atomic number Z = 74) in comparison to surrounding carbon (Z = 6). Energy dispersive X-ray (EDX) analysis (Fig. 1E) performed on bundles of {PW12O40}@SWNT and {P2W18O62}@SWNT confirmed the presence of P, W and O in the ratio 0.02![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.18:0.79 and 0.02:0.19:0.80 respectively, in good agreement with calculated 0.02:0.23:0.75 and 0.02:0.22:0.76 for [PW12O40]3− and [P2W18O62]6− respectively (Fig. 1E). This is consistent with the previous works on POMs in nanotubes, all indicating that the structure, composition and chemical state of POMs inside SWNT remains the same as in free molecules.47,52
:0.18:0.79 and 0.02:0.19:0.80 respectively, in good agreement with calculated 0.02:0.23:0.75 and 0.02:0.22:0.76 for [PW12O40]3− and [P2W18O62]6− respectively (Fig. 1E). This is consistent with the previous works on POMs in nanotubes, all indicating that the structure, composition and chemical state of POMs inside SWNT remains the same as in free molecules.47,52
Isolated POM molecules have previously been successfully imaged by TEM both inside nanotubes47,51,52 and on graphene.48,49 However, in our TEM imaging of {PW12O40}@SWNT and {P2W18O62}@SWNT, the molecules are close to one another throughout the sample (Fig. 1D, top), with each molecule surrounded by two nearest neighbours thus allowing intermolecular reactions between the neighbouring molecules. Thermal gravimetric analysis (TGA) of the POM@SWNT materials (Fig. S1, ESI†) indicates a high loading of POM, with a POM molecule for approximately every 100 carbons atoms, consistent with our TEM imaging. Imaging at a low electron flux (∼8 × 104 e− nm−2 s−1) reveals relatively uniform intermolecular distances (centre-to-centre) of 0.9–1.2 nm and 0.9–1.4 nm and an average gap between neighbouring molecules of 0.45 and 0.44 nm for {PW12O40}@SWNT and {P2W18O62}@SWNT respectively. Time series imaging of the same area indicated that the molecules perform translational and rotational motion within the nanotubes53 on the timescale of seconds, such that the intermolecular distances fluctuate and in some cases the shapes of POM molecules appear to blur due to the motion during a single frame exposure (0.5 s). This effect was more pronounced in {P2W18O62}@SWNT due to the cylindrical shape of the [P2W18O62]6− molecule, leading to a greater number of non-equivalent orientations over the course of a single exposure (Fig. S2 and S3, ESI†). Most of these changes appeared to be reversible over the period of several minutes, but irreversible changes in POM@SWNT gradually accumulate after ∼10 min of e-beam irradiation.
Time series TEM imaging at electron fluxes of 105 e− nm−2 s−1 and higher reveal a much faster accumulation of irreversible changes in the POM structures (Fig. 1D, bottom). At this flux, the POM molecules appear to be significantly more reactive under the e-beam than fullerenes54,55 or perchlorocoronene23 reported previously. The accumulation of changes appears to be dependent on the flux rather than the total e-beam dose received by the molecules, such that a doubling of the flux more than doubles the reaction rate. The volume of original POM material appears to contract over the course of seconds, freeing space in the nanotube, and forms structures with lower volume but higher contrast than the original POMs (Fig. 1D, bottom). While still accompanied with a degree of translational movement (reversible dynamics) the overall progression of the coalescence of POM molecules into a polymeric structure is irreversible, and steadily continues throughout the imaging series. The transformation of POM molecules takes place equally in isolated nanotubes and in bundles (which are commonly present in SWNT samples due to strong dispersion interactions between the nanotubes). Thin bundles of SWNTs consisting of 5–10 parallel nanotubes of the same diameter allow the simultaneous observation of the dynamics of ca. 100 POM molecules in each of our time-series imaging experiments, enabling a quantitative comparison of POM behaviour under different conditions as discussed below. Interestingly, variation of the e-beam energy from 80 to 30 keV did not appear to have significant effects on the reaction rate of the POM molecules (Fig. S5, ESI†).
Our lower-magnification ChemTEM imaging provides spatially and temporally continuous data at the few-molecule level. However, aberration corrected TEM (AC-TEM) imaging, performed using the same energy of the e-beam (80 keV), requires a higher flux and was therefore carried out using a more sensitive electron detector camera that enabled the imaging of {P2W18O62}@SWNT at 80 keV using a flux of 2.3 × 105 e− nm−2 s−1 and exposure time of 0.1 s per frame. Although the absolute electron flux remained similar to the lower magnification ‘high flux’ image series, the high frame rate ensured a total electron dose of only 2.3 × 104 e− nm−2 per image. Under these conditions isolated single molecules of [P2W18O62]6− could be clearly observed (Fig. 1C), showing positions of individual W-atoms and W–W distances between 0.31 nm and 0.62 nm close to those observed in [P2W18O62]6− in a crystal form.56 Comparison of the experimental images with the structural model and simulated TEM images suggests that while individual atoms of oxygen (Z = 8) and phosphorus (Z = 15) cannot be observed, they can contribute to the contrast of the tungsten atoms (Z = 74) if they overlap in the projection, elongating the projection of the latter (Fig. S2 and S3, ESI†). Fast image capture rate and low flux AC-TEM imaging of the condensation of POM molecules allows us to observe an extremely transient, early stage of the process (Fig. 2): POM molecules appear to join one another through bridging oxygen atoms on the sides of the molecules, such that the molecular C3-axis is perpendicular to the axis of the nanotube (Fig. 2B). Time-resolved imaging showed that the interlinking of neighbouring molecules is accompanied by a reduction of the intermolecular gap and precedes the collapse of the POM structure into a much denser, higher contrast material, as shown later in the reaction kinetics measurements.
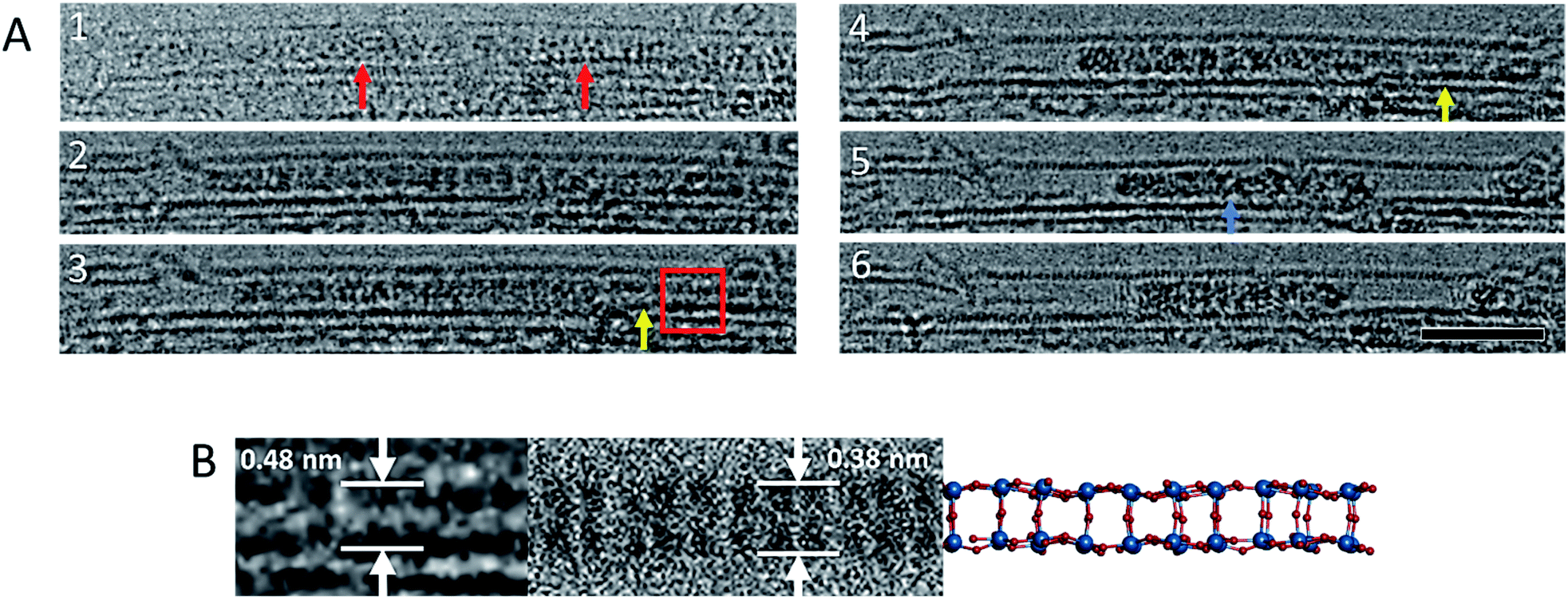 | ||
| Fig. 2 Image sequence showing a reaction between [P2W18O62]6− molecules (red arrows) leading to an ordered WOx nanowire (yellow arrows). This ordered nanowire eventually leads to an amorphous structure of higher contrast (blue arrow) (A). Images acquired at 80 keV. Scale bar is 5 nm. An enlarged image of the oxide nanowire that forms as an intermediate during the polycondensation reaction, and a corresponding simulated TEM image and model of the nanowire (B) derived from bonded [P2W18O62]6− molecules arranged with their C3 axis parallel to each other, and their top and bottom hemispheres removed, with no geometry optimisation or bond relaxation applied (Fig. S4 in the ESI) ESI Video 2.† | ||
To study the atomic structures of the products of the polycondensation reactions, AC-TEM analysis was carried out at high electron flux. AC-TEM of both {PW12O40}@SWNT and {P2W18O62}@SWNT at a flux of approximately 1–2 × 106 e− nm−2 s−1 at 80 keV showed the complete loss of POM molecular structure and formation of nanoclusters (Fig. 3). The metal–metal distances in these nanoclusters ranged between 0.2–0.3 nm, significantly shorter than expected for pristine POMs, and closer to the value of 0.31 nm for bulk tungsten metal. These nanoclusters appear to be much less reactive than the initial POMs, but also showed continued condensation, with neighbouring nanoclusters occasionally merging into one (Fig. 3B). Unlike previous reports of metallic tungsten in nanotubes under the same conditions,57,58 the metal-rich nanoclusters derived from POMs are reactive with the host nanotube, promoting the formation of vacancy defects (Fig. 3C), and appear to generate species containing single W-atoms that also actively bind to the carbon lattice of the SWNT and shuttle between neighbouring nanoclusters. Over time, the vacancy defects grew bigger in the SWNT sidewalls until the nanotube is severed (Fig. 3D). It is important to emphasise that under our imaging conditions carbon atoms of the SWNT exposed to the 80 keV e-beam receive a maximum of ETmax 15.8 eV per atom (eqn(1)), below the atom displacement threshold from the nanotube lattice59 (∼17 eV corresponding to the 86 keV e-beam). This means that the SWNT containers should not be significantly affected by the e-beam while the molecules react, as shown in previous work.60,61 However, in the POM@SWNT materials, particularly towards the end of the time series at high electron flux, the formation of defects in nanotube sidewalls was clearly noticeable (Fig. 3), indicating generation of highly reactive species during the process of POM polycondensation as discussed below.
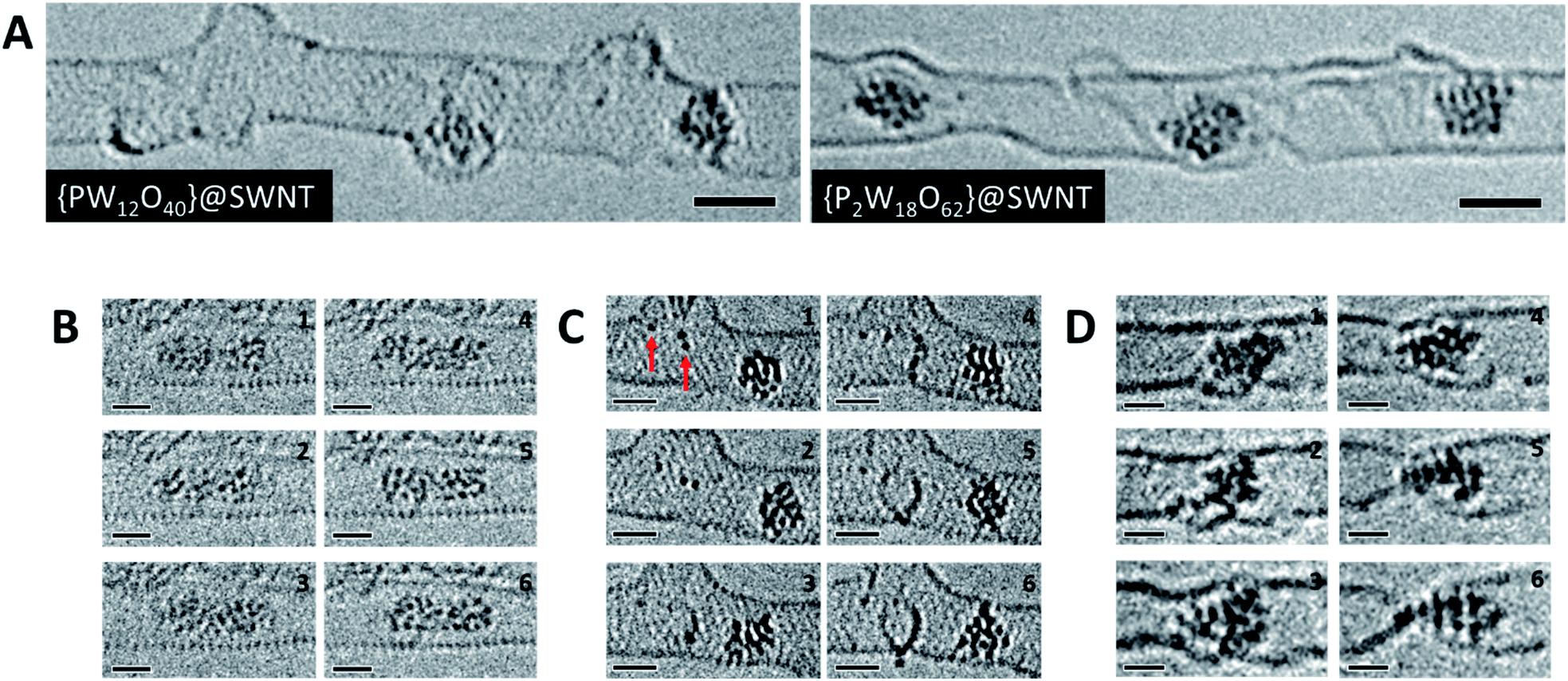 | ||
| Fig. 3 High resolution AC-TEM images of [PW12O40]@SWNT and [P2W18O62]@SWNT recorded at high flux (A). Condensation reaction between two nanoclusters derived from [PW12O40]@SWNT over the course of 10 seconds (B). Observation of species with one or two W atoms (red arrows) within the SWNT interacting with the sidewall and forming vacancy defects over the course of 200 seconds in [PW12O40]@SWNT (C). SWNT cutting observed by a nanocluster derived from [P2W18O62]@SWNT over the course of 150 seconds (D). All image series were captured at 80 keV and at electron fluxes of 1–2 × 106 e− nm−2 s−1. Scale bars are 1 nm. ESI Videos 3 and 4† illustrate motion of an individual POM and a process of defect formation and propagation in a SWNT promoted by the POM, respectively. | ||
Discussion
In order to rationalise the observed molecular dynamics and reactivity, it is important to establish the chemical nature of the POM transformation induced by the e-beam. Although in general the e-beam can induce reactions via many possible mechanisms, confinement of molecules in an atomically thin carbon nanotube mitigates secondary electron processes.62 As evidenced by previous electrochemical studies of POM@SWNT,52 the POMs are also in excellent electronic contact with the SWNT, mitigating any effects of radiolysis by ionisation; being highly conducting the host-nanotube acts as an effective charge and heat sink. POM molecules have a high degree of structural stability across a very wide range of oxidation states,63,64 making it unlikely that structural degradation under the beam will occur via this route. The low e-beam flux used in our experiments limits electronic excitation processes to single excitations, which are also unlikely to trigger the reactions observed, especially considering the use of POMs as photocatalysts.65Under our experimental conditions, the most significant mechanism of energy transfer from the e-beam is the direct knock-on (DKO) effect which causes shifts of atoms from their equilibrium positions within the chemical bond, causing bond dissociation if the transferred energy is greater than the threshold energy, Ed, for the atom displacement from the molecule.22,23,58 The amount of energy that can be transferred to a stationary atom from an incident electron is described by:
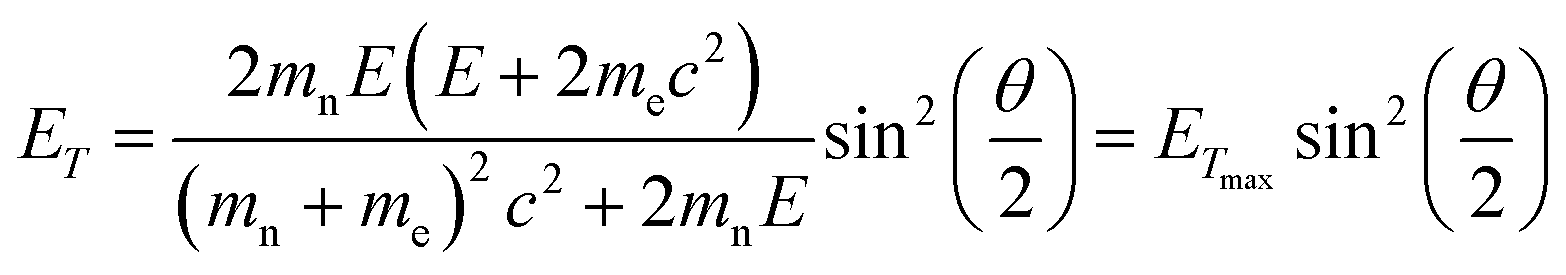 | (1) |
Once a W–O bond is broken, it creates a coordinatively unsaturated, reactive metal centre in the POM molecule, opening a pathway for polycondensation with neighbouring molecules:
| [P2W18O62]6− ⇌ [P2W18O61](6−n)− + On− | (2) |
The two neighbouring POM molecules can react through either a bridging metal-oxo-metal bond or a metal–metal bond, in both cases leading to condensation of discrete molecules into one larger metal oxide cluster, but with a lower content of oxygen (hence higher contrast in TEM images) and shorter W–W distances. Fast AC-TEM imaging captured the initial stages of the condensation, showing that the POM molecules are aligned with their long axis parallel to each other, and therefore indicating that W–O bonds above and beneath the σh mirror plane (Fig. 1B) take part in the first step. This leads to a transiently observed continuous metal oxide wire (Fig. 2), which still retains some molecular features of the POM, shortly before further elimination of oxygen atoms leads to amorphisation of the resultant material in the nanotube.
In a pristine POM molecule, the tungsten atoms are separated by oxygen atoms thus effectively ‘diluting’ TEM contrast of the high-Z metal. The elimination of oxygen will therefore progressively increase the atomic percent of tungsten, hence increasing the TEM contrast of the material in the nanotube as observed in our time series images (Fig. 4A). Another consequence of oxygen elimination is the increase of density and decrease of the molar volume of the material in nanotubes: considering that the approximate length of a [P2W18O62]6− within a SWNT is 0.8 nm and that each POM molecule yields 18 atoms of W in the process, based on the density of tungsten metal (assuming all oxygen is eliminated) there will be a substantial reduction of the volume of material in the nanotube, up to 77% if all oxygen atoms are removed (details in the ESI†). Therefore, as the reaction proceeds, the POM initially fully occupying the nanotube cavity will shrink, producing voids between the molecules (Fig. 4A and B).
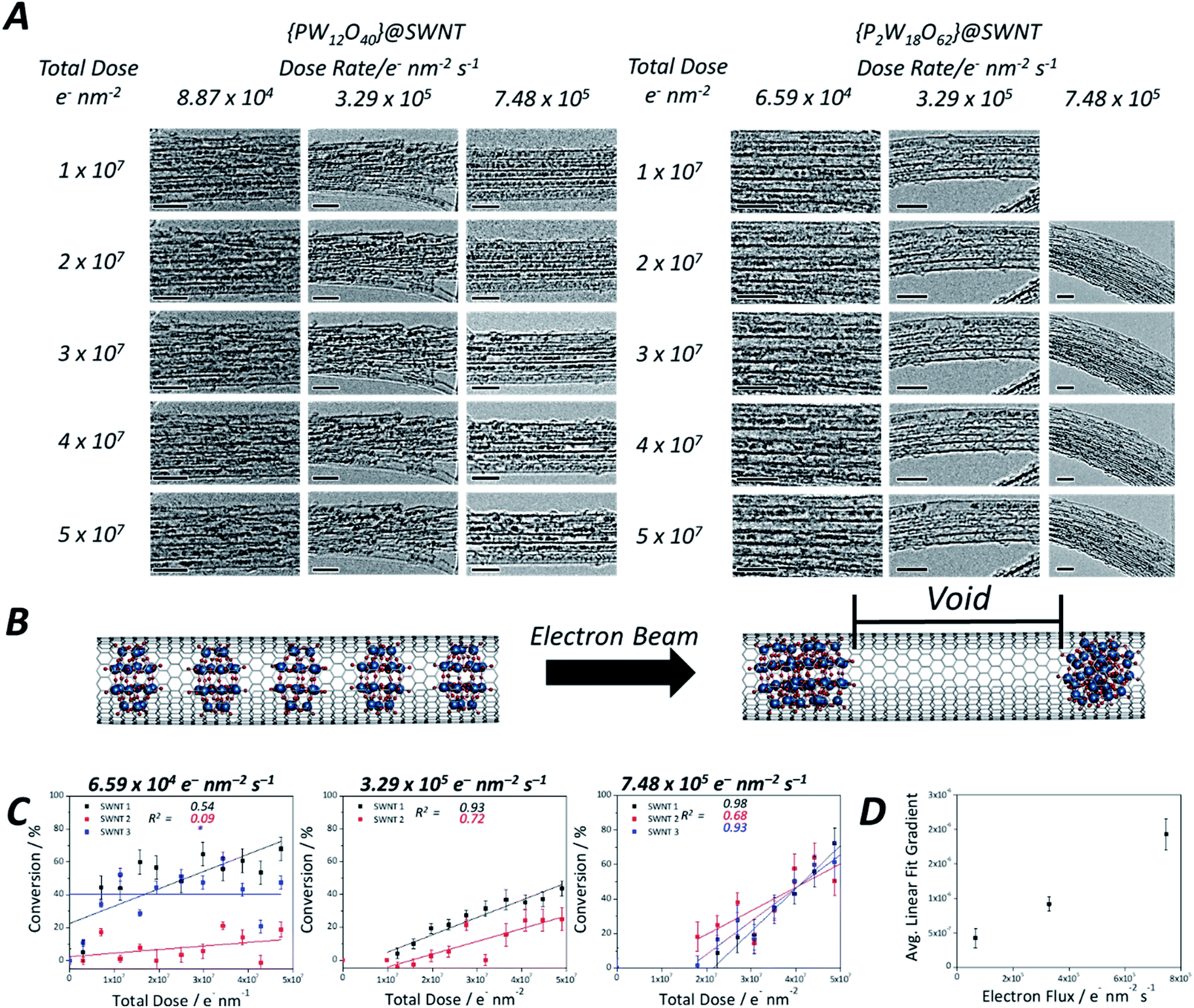 | ||
| Fig. 4 Time series TEM images of {PW12O40}@SWNT and {P2W18O62}@SWNT recorded at increasing flux of the 80 keV e-beam, scale bars 5 nm (A). Structural model demonstrating discrete POM molecules reacting to form larger metal oxide clusters, creating voids in nanotubes observed with TEM (B). Plots showing conversion of POM to tungsten metal against total dose of the 80 keV e-beam for [P2W18O62]@SWNT (R2 shown inset) at 6.59 × 104 e− nm−2 s−1, 3.29 × 105 e− nm−2 s−1 and 7.48 × 105 e− nm−2 s−1 (C). The linear fit for the series indicated by * resulted in a small but negative R2 value (−0.03), indicating no gradient and therefore no overall conversion during the image series. Average linear fit gradients against electron flux demonstrate the rate dependence on electron flux (D). | ||
As the reaction of the POM is accompanied by a significant reduction in volume, creating cylindrically symmetrical space in the host nanotube, a simple measurement of the length of the voids between the molecules can quantify the reaction progress. Such an opportunity to study reaction kinetics quantitatively at the sub-100 molecule level with spatiotemporal continuity has yet to be fully explored with other such methods, as most analytical methods are severely limited either by time resolution or by space resolution, making ChemTEM unique. Moreover, areas of the sample where several nanotubes are positioned parallel to each other (so-called SWNT bundles) were chosen for imaging, allowing quantification of our observations necessary for reaction kinetics analysis, but without losing information about positions of individual molecules. It must be noted, however, that this approach to follow the kinetics of the reaction works best if the molecules are packed densely along the nanotube, with few gaps at the start of the reaction. Any significant gaps due to loose molecular packing would allow translational motion of the molecules masking the progress of the reaction. In this study, [PW12O40]3− molecules translate back and forth more readily due their smaller size; as a result, molecules of this POM exhibit less accurate data (Fig. S6, ESI†), particularly at a low electron flux, and the signal-to-noise ratio in each frame is lower in the early stages of the reaction when individual POMs are still present and can easily translate. In contrast, [P2W18O62]6− molecules fill the host-nanotube more efficiently, resulting in less translation, although similarly displaying high levels of translation at lower electron flux, as evidenced by the low R2 values at lower flux (Fig. 4C). At higher flux, the intermolecular reaction of these molecules is much faster leading to less interference of the molecular motion in the observed reaction kinetics.
Using this approach, we have measured the polycondensation of {P2W18O62}@SWNT in terms of the degree of conversion to tungsten metal and plotted it as a function of the total electron dose on the molecule (Fig. 4C) (details are in the ESI†). The reaction rate for the initial step of pristine POM molecules is breaking the first W–O bond to generate a reactive species:
| v1 = k1[POM] = j × σd × [POM] | (3) |
As previously reported,70 a consideration of the characteristic timescales of electronic excitations and atomic displacements is key to understanding reactions induced by the electron beam. Considering the low electron fluxes and nature of the POM@SWNT system, as discussed above, the most plausible explanation is that the reversible step corresponds to emission and recombination of oxygen atoms with the parent POM molecule rather than electronic excitations and relaxations. The rate of the recombination step can therefore be given as:
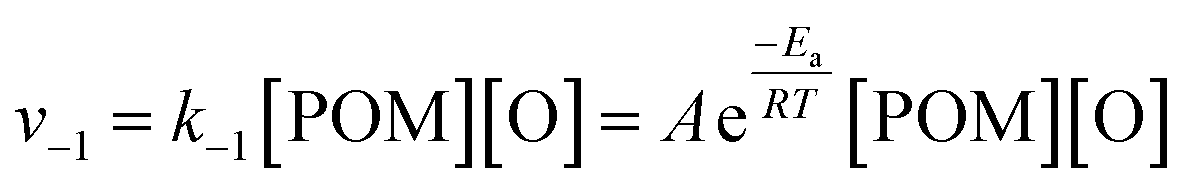 | (4) |
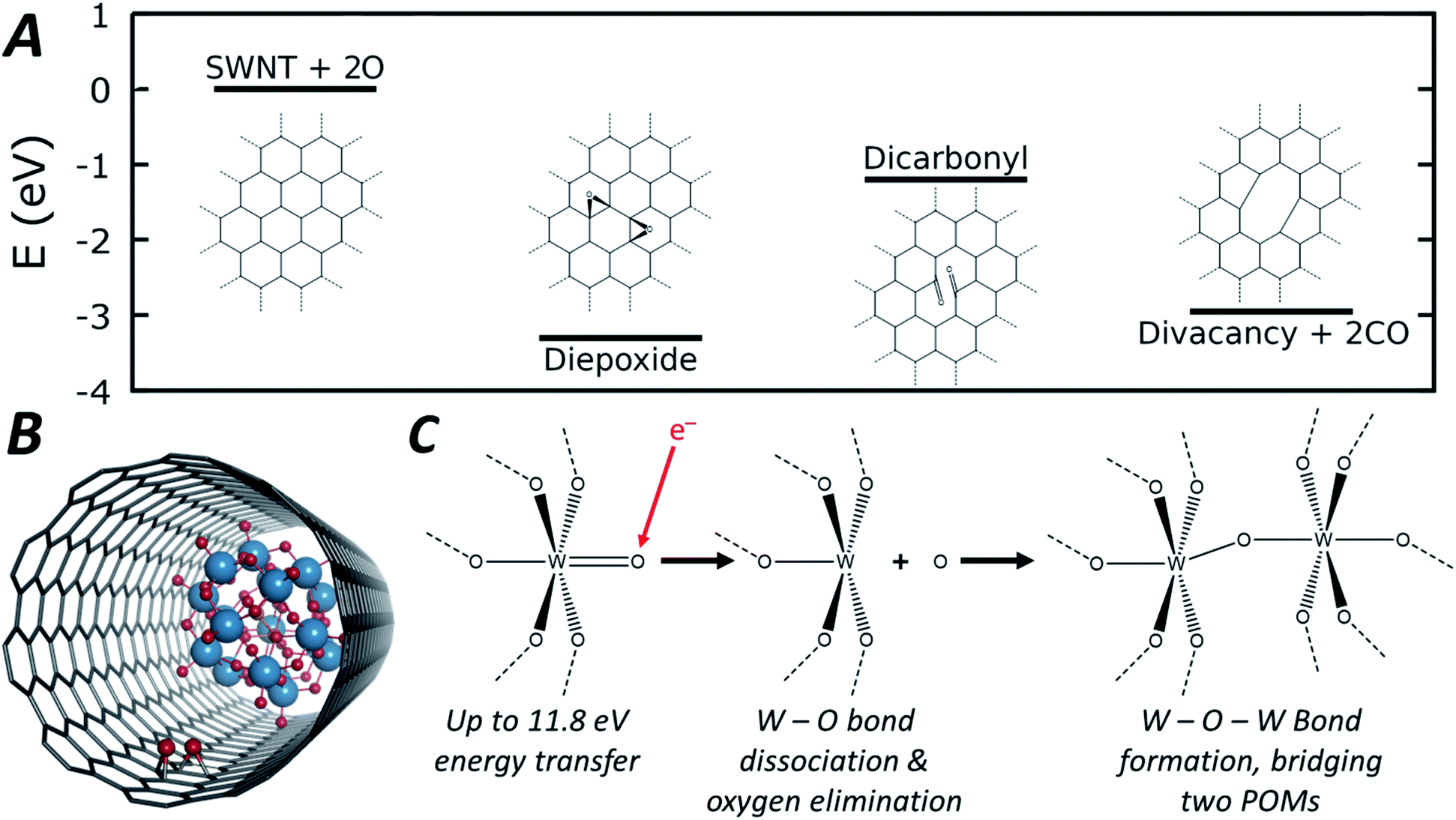 | ||
| Fig. 5 Energetic pathway of di-epoxide formation from atomic oxygen emitted from POM molecules with schematic structures shown inset (A, full structures shown in Fig. S10, ESI†). Diagram depicting the di-epoxide groups formed within the SWNT interior of the POM@SWNT material (B). General reaction scheme depicting oxygen elimination and subsequent W–O–W bond formation that leads to the observed condensation reaction (C). | ||
As the POM molecules undergo reductive condensation with loss of oxygen (Fig. 4), it is important to consider the fate of oxygen atoms generated in the nanotube. Empty nanotubes remain unchanged under the beam at 80 keV, but in POM@SWNT materials vacancy defect formation in the carbon lattice of the nanotube is apparent, particularly at high electron flux (Fig. 3) which is likely to be related to the atomic oxygen produced in the vicinity of POM molecules. The concave side of a SWNT is much less reactive than the convex wall, with only rare examples of internal cavity modifications reported,72 but our DFT calculations show that oxygen is able to bond to the carbon atoms of the SWNT from the inside to form epoxides (schematic shown in Fig. 5B). From oxygen atoms generated by e-beam irradiation of the POM, the formation of two epoxides in close proximity to form a di-epoxide is thermodynamically favourable by 2.4 to 3.3 eV, depending on their exact relative configuration (see ESI for details†), and once two or more epoxide groups are formed on the graphitic lattice, they are known to undergo re-arrangement to carbonyl groups.73 Under our imaging conditions – in which the electron beam can transfer up to 11.8 eV of kinetic energy to a stationary oxygen atom – the formation of carbonyl groups on the SWNT sidewall can be followed by emission of CO to form vacancy defects. Fig. 5A shows the energetic pathway of this process for the lowest energy configuration of di-epoxide found in our calculations. Overall, while the POM is being reduced to metal by losing oxygen, the nanotube sidewall is oxidised by the oxygen emitted from POM molecules, and in turn loses carbon as CO, consistent with our AC-TEM imaging with a higher electron flux. Furthermore, pure tungsten metal is known to be relatively inert towards the nanotube under the 80 keV e-beam,57,58 but our observations show that the intermediate amorphous clusters formed during the POM polycondensation are reactive towards the carbon lattice, therefore indicating that they are not pure tungsten and retain some oxygen (Fig. 3). The final products of the POM condensation appear to be inert to further reactions and SWNT defect formation, and thus closer to the metallic state of tungsten, which is also consistent with the observed interatomic W–W spacing of 0.2 nm indicating metallic bonds (Fig. 3).
Conclusions
Among the methods for single-molecule kinetics analysis, TEM offers both high spatial and temporal resolution. This study utilises the e-beam of TEM as both a fast imaging tool and a source of energy for stimulating the chemical reactions of polyoxometalates directly during the imaging. Imaging the reactions of POM molecules, the ChemTEM approach has progressed from spatiotemporally continuous observations of singular reaction events towards measuring chemical reactions of ca. 100 molecules simultaneously. This advance has allowed statistically meaningful reaction kinetics analysis whilst maintaining spatial resolution at the single-molecule level. Reactions of polyoxometalate molecules confined within SWNTs have been directly observed by TEM operated in a plethora of conditions (energy, total dose and fluxes of the electron beam), revealing structural and dynamic information holding the key to understanding the reaction mechanism. Specifically, this experimental approach, combined with theoretical modelling and image analysis, has revealed that the reactions of POM molecules proceed via a polycondensation pathway, beginning from a reversible W–O bond dissociation step, followed by an intermolecular W–O–W bond formation, bridging the neighbouring molecules and leading to transient tungsten oxide nanowires intermediates, which subsequently transform to amorphous tungsten-rich clusters – the final product of the reductive polycondensation of POM molecules.In addition to the atomistic understanding of POM reactions, our approach has provided important insights into general aspects of imaging of metal oxides at the nanoscale. As metal oxide hybrid nanomaterials, especially metal oxide–carbon heterostructures, are now playing an increasingly important role in catalysis, electrocatalysis and energy storage technologies, accurate TEM characterisation of these materials is becoming critical. By elucidating the pathways and rates of POM transformations under the e-beam, our ChemTEM investigation gives a roadmap towards artefact-free TEM characterisation of these delicate nanoscale metal oxides, by controlling the e-beam flux which has been shown to significantly influence the metal–oxygen bond dissociation step.
Author contributions
J. W. J. prepared and analysed materials, interpreted TEM data, and proposed mechanisms of POM polycondensation; K. L. Y. F. designed and performed TEM imaging for kinetics measurements (JEOL 2100F); S. T. S. carried out DFT and TEM image simulations; J. W. J. and S. T. S. developed chemical kinetics description of reactions of POM in e-beam; C. S. A. carried out high frame rate AC-TEM imaging (JEOL ARM300F); J. B. performed atomically resolved AC-TEM at varying accelerating voltages (FEI Titan 80-300, SALVE microscope); G. N. N. contributed to material design; U. K. contributed to TEM experiment design and method development; A. N. K., S. T. S. and K. L. Y. F. generated the concept and developed methodology for inorganic molecule reaction kinetics measurements using e-beam as a stimulus and a probe at the same time; all authors have contributed to writing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
J. W. J., G. N. N. and A. N. K. thank the EPSRC for funding through the Centre for Doctoral Training in Sustainable Chemistry (EP/L015633/1). U. K. and J. B. thank the Deutsche Forschungsgemeinschaft (DFG) and the State Baden Württemberg for financial support in the frame of the SALVE and project No. 424798828. A. N. K. thanks the EPSRC (Established Career Fellowship). K. L. Y. F. is grateful to the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for access to their facilities. S. T. S. is grateful to the High Performance Computing (HPC) Facility at the University of Nottingham for providing computational time. We thank Diamond Light Source for access and support in use of the electron Physical Science Imaging Centre (Instrument E02, proposal number MG23260) that contributed to the results presented here.Notes and references
- S. R. Logan, Fundamentals of Chemical Kinetics, Longman Group Ltd., England, 1996 Search PubMed.
- L. J. P. Atkins, Chemical Principles: The Quest for Insight, W. H. Freeman & Co, N.Y., 2nd edn, 2002 Search PubMed.
- B. Temel, H. Meskine, K. Reuter, M. Scheffler and H. Metiu, J. Chem. Phys., 2007, 126, 204711 CrossRef PubMed.
- P. Lecca, Biophys. Rev., 2013, 5, 323–345 CrossRef CAS PubMed.
- Handbook of Single-Molecule Biophysics, Springer-Verlag, New York, 1 edn, 2009 Search PubMed.
- N. G. Walter, C.-Y. Huang, A. J. Manzo and M. A. Sobhy, Nat. Methods, 2008, 5, 475–489 CrossRef CAS PubMed.
- K. Qiu, T. P. Fato, B. Yuan and Y.-T. Long, Small, 2019, 15, 1805426 CrossRef PubMed.
- S. G. Lemay, S. Kang, K. Mathwig and P. S. Singh, Acc. Chem. Res., 2013, 46, 369–377 CrossRef CAS PubMed.
- V. I. Claessen, H. Engelkamp, P. C. M. Christianen, J. C. Maan, R. J. M. Nolte, K. Blank and A. E. Rowan, Annu. Rev. Anal. Chem., 2010, 3, 319–340 CrossRef CAS PubMed.
- J. Elf and I. Barkefors, Annu. Rev. Biochem., 2019, 88, 635–659 CrossRef CAS PubMed.
- F.-R. F. Fan and A. J. Bard, Science, 1995, 267, 871–874 CrossRef CAS PubMed.
- L. A. Baker, J. Am. Chem. Soc., 2018, 140, 15549–15559 CrossRef CAS PubMed.
- S. Kang, A. F. Nieuwenhuis, K. Mathwig, D. Mampallil, Z. A. Kostiuchenko and S. G. Lemay, Faraday Discuss., 2016, 193, 41–50 RSC.
- A. J. Bard, ACS Nano, 2008, 2, 2437–2440 CrossRef CAS PubMed.
- A. N. Sekretaryova, M. Y. Vagin, A. P. F. Turner and M. Eriksson, ACS Catal., 2017, 7, 3591–3593 CrossRef CAS.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan and R. J. M. Nolte, et al. , Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- N. Pavliček, P. Gawel, D. R. Kohn, Z. Majzik, Y. Xiong, G. Meyer, H. L. Anderson and L. Gross, Nat. Chem., 2018, 10, 853–858 CrossRef PubMed.
- K. Kaiser, L. Gross and F. Schulz, ACS Nano, 2019, 13, 6947–6954 CrossRef CAS PubMed.
- A. Riss, A. P. Paz, S. Wickenburg, H.-Z. Tsai, D. G. De Oteyza, A. J. Bradley, M. M. Ugeda and P. Gorman, et al. , Nat. Chem., 2016, 8, 678–683 CrossRef CAS PubMed.
- T. Ando, Biophys. Rev., 2018, 10, 285–292 CrossRef CAS PubMed.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS PubMed.
- S. T. Skowron, T. W. Chamberlain, J. Biskupek, U. Kaiser, E. Besley and A. N. Khlobystov, Acc. Chem. Res., 2017, 50, 1797–1807 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, A. V. Markevich, S. Kurasch, O. Reimer, K. E. Walker and G. A. Rance, et al. , ACS Nano, 2017, 11, 2509–2520 CrossRef CAS PubMed.
- T. Zoberbier, T. W. Chamberlain, J. Biskupek, M. Suyetin, A. G. Majouga, E. Besley, U. Kaiser and A. N. Khlobystov, Small, 2016, 12, 1649–1657 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622–629 CrossRef CAS PubMed.
- J. A. Mir, R. Clough, R. MacInnes, C. Gough, R. Plackett, I. Shipsey, H. Sawada and I. MacLaren, et al. , Ultramicroscopy, 2017, 182, 44–53 CrossRef CAS PubMed.
- S. V. Kalinin, O. Dyck, A. Ghosh, Y. Liu, R. Proksch, B. G. Sumpter and M. Ziatdinov, 2020, arXiv:2010.09196.
- K. R. Vinothkumar and R. Henderson, Q. Rev. Biophys., 2016, 49, e13 CrossRef PubMed.
- C. Kramberger and J. C. Meyer, Ultramicroscopy, 2016, 170, 60–68 CrossRef CAS PubMed.
- J. C. Meyer, F. Eder, S. Kurasch, V. Skakalova, J. Kotakoski, H. J. Park, S. Roth and A. Chuvilin, et al. , Phys. Rev. Lett., 2012, 108, 196102 CrossRef PubMed.
- J. Kotakoski, C. Mangler and J. C. Meyer, Nat. Commun., 2014, 5, 3991 CrossRef CAS PubMed.
- S. Skowron, V. Koroteev, M. Baldoni, S. Lopatin, A. Zurutuza, A. Chuvilin and E. Besley, Carbon, 2016, 105, 168–182 CrossRef.
- S. Okada, S. Kowashi, L. Schweighauser, K. Yamanouchi, K. Harano and E. Nakamura, J. Am. Chem. Soc., 2017, 139, 18281–18287 CrossRef CAS PubMed.
- M. S. Isaacson, J. Langmore, N. W. Parker, D. Kopf and M. Utlaut, Ultramicroscopy, 1976, 1, 359–376 CrossRef CAS PubMed.
- M. Isaacson, D. Kopf, M. Utlaut, N. W. Parker and A. V. Crewe, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1802–1806 CrossRef CAS PubMed.
- B. Rotman, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS PubMed.
- C. R. Graham and R. G. Finke, Inorg. Chem., 2008, 47, 3679–3686 CrossRef CAS PubMed.
- C. T. Koch, PhD thesis, Arizona State University, 2002.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange and A. Behn, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- B. Dawson, Acta Crystallogr., 1953, 6, 113–126 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. Sloan, G. Matthewman, C. Dyer-Smith, A. Y. Sung, Z. Liu, K. Suenaga, A. I. Kirkland and E. Flahaut, ACS Nano, 2008, 2, 966–976 CrossRef CAS PubMed.
- N. Vats, S. Rauschenbach, W. Sigle, S. Sen, S. Abb, A. Portz, M. Durr and M. Burghard, et al. , Nanoscale, 2018, 10, 4952–4961 RSC.
- J. Sloan, Z. Liu, K. Suenaga, N. R. Wilson, P. A. Pandey, L. M. Perkins, J. P. Rourke and I. J. Shannon, Nano Lett., 2010, 10, 4600–4606 CrossRef CAS PubMed.
- C. Bosch-Navarro, L. M. Perkins, R. J. Kashtiban, J. P. Rourke, I. J. Shannon and J. Sloan, ACS Nano, 2016, 10, 796–802 CrossRef CAS PubMed.
- B. Fei, H. Lu, W. Chen and J. H. Xin, Carbon, 2006, 44, 2261–2264 CrossRef CAS.
- J. W. Jordan, G. A. Lowe, R. L. McSweeney, C. T. Stoppiello, R. W. Lodge, S. T. Skowron, J. Biskupek and G. A. Rance, et al. , Adv. Mater., 2019, 31, 1904182 CrossRef CAS PubMed.
- A. N. Khlobystov, K. Porfyrakis, M. Kanai, D. A. Britz, A. Ardavan, H. Shinohara, T. J. S. Dennis and G. A. D. Briggs, Angew. Chem., Int. Ed., 2004, 43, 1386–1389 CrossRef CAS PubMed.
- J. H. Warner, Y. Ito, M. Zaka, L. Ge, T. Akachi, H. Okimoto, K. Porfyrakis and A. A. R. Watt, et al. , Nano Lett., 2008, 8, 2328–2335 CrossRef CAS PubMed.
- M. Koshino, Y. Niimi, E. Nakamura, H. Kataura, T. Okazaki, K. Suenaga and S. Iijima, Nat. Chem., 2010, 2, 117–124 CrossRef CAS PubMed.
- C. T. Stoppiello, J. Biskupek, Z. Y. Li, G. A. Rance, A. Botos, R. M. Fogarty, R. A. Bourne and J. Yuan, et al. , Nanoscale, 2017, 9, 14385–14394 RSC.
- T. Zoberbier, T. W. Chamberlain, J. Biskupek, N. Kuganathan, S. Eyhusen, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, J. Am. Chem. Soc., 2012, 134, 3073–3079 CrossRef CAS PubMed.
- K. Cao, T. Zoberbier, J. Biskupek, A. Botos, R. L. McSweeney, A. Kurtoglu, C. T. Stoppiello and A. V. Markevich, et al. , Nat. Commun., 2018, 9, 3382 CrossRef PubMed.
- B. W. Smith and D. E. Luzzi, J. Appl. Phys., 2001, 90, 3509–3515 CrossRef CAS.
- S. T. Skowron, I. V. Lebedeva, A. M. Popov and E. Bichoutskaia, Nanoscale, 2013, 5, 6677–6692 RSC.
- J. H. Warner, M. H. Rümmeli, L. Ge, T. Gemming, B. Montanari, N. M. Harrison, B. Büchner and G. A. D. Briggs, Nat. Nanotechnol., 2009, 4, 500 CrossRef CAS PubMed.
- S. T. Skowron, S. L. Roberts, A. N. Khlobystov and E. Besley, Micron, 2019, 120, 96–103 CrossRef CAS PubMed.
- H. Wang, S. Hamanaka, Y. Nishimoto, S. Irle, T. Yokoyama, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS PubMed.
- Y. Nishimoto, D. Yokogawa, H. Yoshikawa, K. Awaga and S. Irle, J. Am. Chem. Soc., 2014, 136, 9042–9052 CrossRef CAS PubMed.
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47(15), 5120–5136 RSC.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622–629 CrossRef CAS PubMed.
- P. M. F. J. Costa, J. Sloan, T. Rutherford and M. L. H. Green, Chem. Mater., 2005, 17, 6579–6582 CrossRef CAS.
- G. Brown, S. R. Bailey, J. Sloan, C. Xu, S. Friedrichs, E. Flahaut, K. S. Coleman and J. L. Hutchison, et al. , Chem. Commun., 2001, 845–846, 10.1039/B101261O.
- A. W. Robertson, C. S. Allen, Y. A. Wu, K. He, J. Olivier, J. Neethling, A. I. Kirkland and J. H. Warner, Nat. Commun., 2012, 3, 1144 CrossRef PubMed.
- N. Jiang and J. C. H. Spence, Ultramicroscopy, 2012, 113, 77–82 CrossRef CAS.
- N. Jiang, Rep. Prog. Phys., 2015, 79, 016501 CrossRef PubMed.
- T. W. Chamberlain, J. C. Meyer, J. Biskupek, J. Leschner, A. Santana, N. A. Besley, E. Bichoutskaia and U. Kaiser, et al. , Nat. Chem., 2011, 3, 732–737 CrossRef CAS PubMed.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01874d |
| This journal is © The Royal Society of Chemistry 2021 |