DOI:
10.1039/D1SC01906F
(Edge Article)
Chem. Sci., 2021,
12, 8501-8511
Electrochemical studies of tris(cyclopentadienyl)thorium and uranium complexes in the +2, +3, and +4 oxidation states†
Received
3rd April 2021
, Accepted 6th May 2021
First published on 7th May 2021
Introduction
The redox chemistry of the actinide elements has recently undergone a significant change: the range of oxidation states available in crystallographically-characterizable molecular complexes has been extended to +2. The discovery of the first molecular example of U(II) involved potassium graphite reduction of the tris(cyclopentadienyl) complex 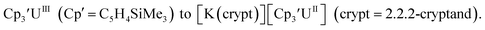 1 Subsequently, the tris(cyclopentadienyl) complexes
1 Subsequently, the tris(cyclopentadienyl) complexes 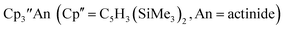 proved to be good precursors for the first examples of crystallographically-characterizable molecular compounds containing Th(II),2 Np(II),3–5 and Pu(II),6eqn (1). Examples of U(II) are now known in different coordination environments beyond the tris(cyclopentadienyl) ligand sets of eqn (1).7–9
proved to be good precursors for the first examples of crystallographically-characterizable molecular compounds containing Th(II),2 Np(II),3–5 and Pu(II),6eqn (1). Examples of U(II) are now known in different coordination environments beyond the tris(cyclopentadienyl) ligand sets of eqn (1).7–9| | 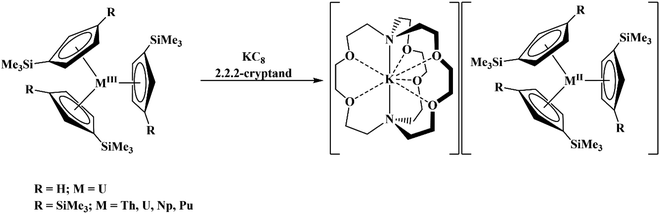 | (1) |
Despite the rapid development of synthetic An(II) chemistry, there have been few electrochemical studies of these low valent systems, although extensive electrochemistry has been reported for the higher oxidation states of the actinides.10–13 This is due in part to the high reactivity of the divalent and trivalent complexes. In addition, actinide electrochemical studies have been challenging because the +3 and +4 metal precursor complexes can react with supporting electrolytes. For example, Inman and Cloke found problems studying (C5Me5)ThIV[C8H6(SiMe2tBu)2]Cl using [nBu4N][PF6] as supporting electrolyte14 as well as with 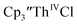 using [nBu4N][B(C6F5)4] as supporting electrolyte.15
using [nBu4N][B(C6F5)4] as supporting electrolyte.15
Although electrochemical data have been reported on two U(II) systems,9,16 analogous studies on Th(II) complexes and on the tris(cyclopentadienyl) systems that led to the first molecular examples of U(II) have been absent. Meyer and coworkers identified the U(III)/U(II) couple in [(Ad,MeArO)3mes]UIII at −2.495 V vs. Fc+/0,16 that guided synthetic efforts and allowed isolation of [K(crypt)]{[(Ad,MeArO)3mes]UII}.7 More recently, Layfield and coworkers reported the U(III)/U(II) couple of (C5iPr5)2UII to be −2.33 V vs. Fc+/0.9 Inman and Cloke studied Th(IV)/Th(III) redox couples and found that [nBu4N][BPh4] was a good supporting electrolyte for their complexes.15,17 Encouraged by their results, we utilized this supporting electrolyte to obtain electrochemical data in this study and on 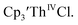 18
18
Due to the importance of the tris(cyclopentadienyl) ligand set in the development of low oxidation state actinide chemistry,19,20 the electrochemistry of a variety of tris(cyclopentadienyl)uranium and thorium complexes using Cp′′, Cp′, and Cptet ligands (Cptet = C5Me4H), Scheme 1, is reported here as well as the first reported electrochemical measurements on isolated Th(II) complexes.2 Also reported are spectroelectrochemical studies on the Th(II) compounds that led to the discovery of new synthetic routes to Th(II) compounds. The results are compared with cyclopentadienyl ligand effects previously examined electrochemically with titanium and zirconium complexes21 and with rare-earth metal reaction chemistry.22–24
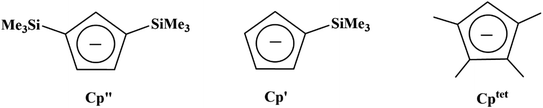 |
| | Scheme 1 Chemical structures of Cp′′, Cp′, and Cptet ligands used in this study. | |
Results
Electrochemical protocol
All data were collected in THF using 100 mM [nBu4N][BPh4] or 200 mM [nBu4N][PF6] supporting electrolyte concentrations. Both [nBu4N][BPh4] and [nBu4N][PF6] were recrystallized three times prior to use. The low polarity of THF leads to large internal resistance in the electrochemical cell with peak separations over 200 mV often observed.15,16 Unless specifically stated, all potentials are referenced to the ferrocenium/ferrocene couple with (C5Me5)2Fe as an internal standard, Fig. S12 and S13.† All electrochemical data were collected with a glassy carbon disc working electrode, platinum wire counter electrode, and silver wire pseudo-reference electrode. All scans were recorded in the cathodic direction except for the isolated U(II) and Th(II) compounds which were recorded in the anodic direction. Representative cyclic voltammograms are shown in Fig. 1–6 and complete details are in the ESI.†
Uranium complexes
Initially, U(III) complexes known to undergo chemical reduction and oxidation were examined to determine if both the U(IV)/U(III) and U(III)/U(II) redox events could be observed electrochemically. Indeed, both redox couples were observed in the voltammograms for the U(III) complexes 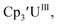 25
25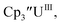 26 and Cptet3UIII,26 and for the isolated U(II) complexes
26 and Cptet3UIII,26 and for the isolated U(II) complexes 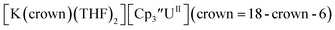 27 and
27 and 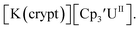 1 These values are summarized in Tables 1 and 2 and highlights are described in the following paragraphs.
1 These values are summarized in Tables 1 and 2 and highlights are described in the following paragraphs.
Table 1 Reduction potentials assigned to U(IV)/U(III) couples in this study and the literature
|
|
E
PA (V) |
E
PC (V) |
U(IV)/U(III) E1/2 (V) |
ΔEpp (C5Me5)2Fe (V) |
|
100 mM [nBu4N][BPh4]/THF.
50 mM [nBu4N][BPh4]/THF.
130 mM [nBu4N][PF6]/THF.
|
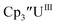
|
−1.04 |
−0.83 |
−0.94a |
0.20 |
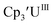
|
−1.33 |
−1.20 |
−1.26b |
0.36 |
| Cptet3UIII |
−1.54 |
−1.39 |
−1.46a |
0.12 |
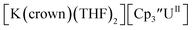
|
−1.09 |
−0.37 |
−0.73a |
0.15 |
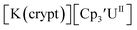
|
−1.45 |
−1.12 |
−1.28a |
0.57 |
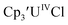
|
|
|
−1.83 (ref. 28)c |
|
| (C5H5)3UIVCl |
|
|
−1.87 (ref. 28 and 29)c |
|
| (C5MeH4)3UIVCl |
|
|
−1.88(ref. 28)c |
|
| (C5tBuH4)3UIVCl |
|
|
−1.93(ref. 28)c |
|
Table 2 Reduction potentials assigned to U(III)/U(II) couples in this study and the literature
|
|
E
PA (V) |
E
PC (V) |
U(III)/U(II) E1/2 (V) |
ΔEpp (C5Me5)2Fe (V) |
|
100 mM [nBu4N][BPh4]/THF.
50 mM [nBu4N][BPh4]THF.
60 mM [nBu4N][BPh4]/THF.
100 mM [nBu4N][PF6]/THF.
|
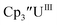
|
−2.79 |
−2.67 |
−2.73a |
0.20 |
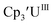
|
−2.43 |
−2.08 |
−2.26b |
0.36 |
| Cptet3UIII |
−3.18 |
−3.04 |
−3.11a |
0.12 |
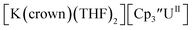
|
−2.77 |
−2.65 |
−2.71a |
0.15 |
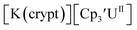
|
−2.50 |
−2.03 |
−2.27b |
0.57 |
| [(Ad,MeArO)3mes]UIII |
|
|
−2.495 (ref. 16)d |
|
| (C5iPr5)2UII |
|
|
−2.33 (ref. 9)c |
|
Cp′′.
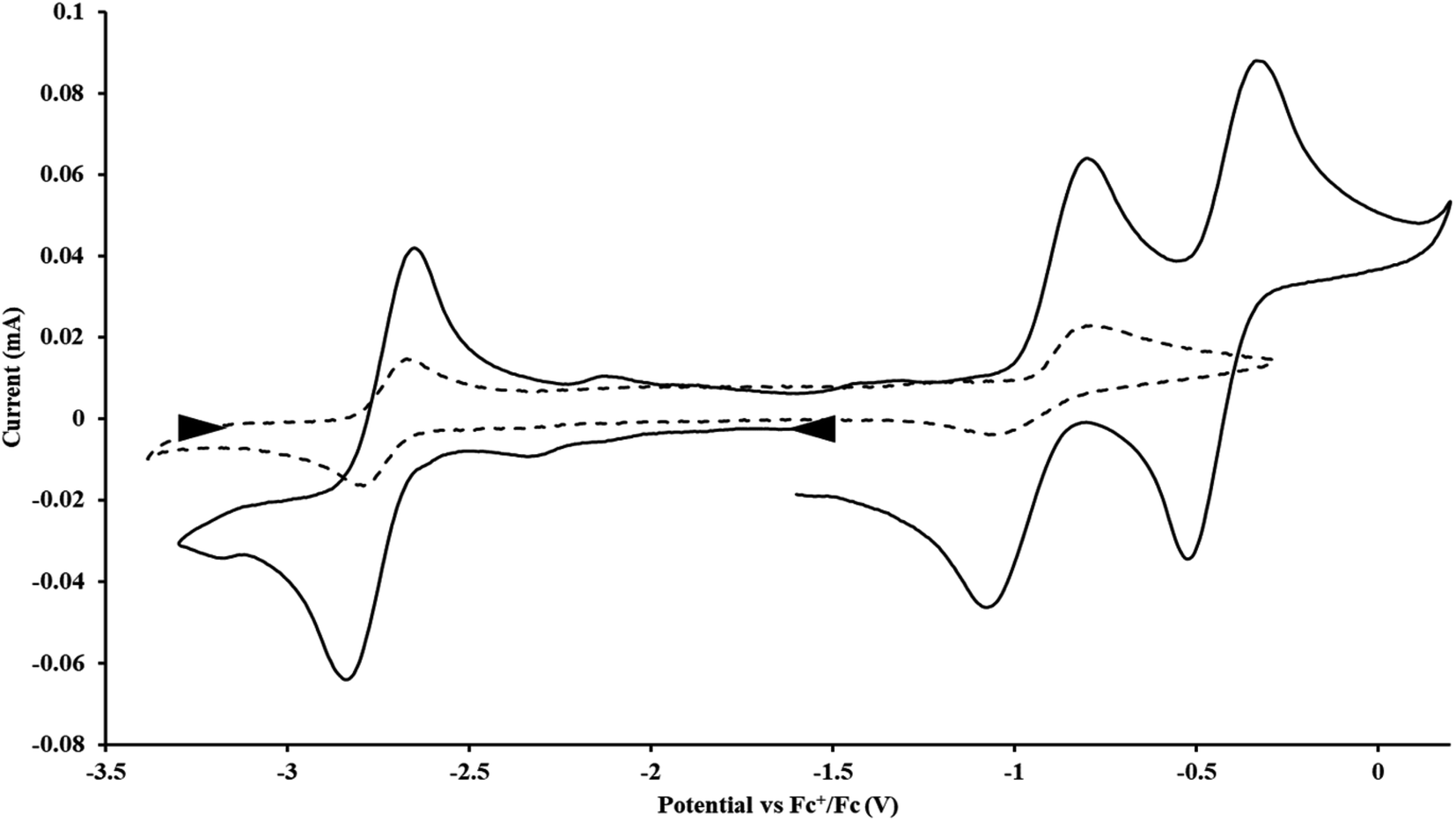
With the bis(trimethylsilyl)cyclopentadienyl ligand, redox couples assigned to U(IV)/U(III) and U(III)/U(II) are observed at −0.94 V and −2.73 V, respectively, for 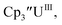 Fig. 1 and S14.† In comparison, the isolated U(II) complex
Fig. 1 and S14.† In comparison, the isolated U(II) complex 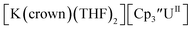 27 displays two redox events at −0.73 V and −2.71 V, Fig. 1 and S25.† The E1/2 values for the U(III)/U(II) couple are nearly identical in both systems and the event centered at −2.71 V only appears when scanning anodically for
27 displays two redox events at −0.73 V and −2.71 V, Fig. 1 and S25.† The E1/2 values for the U(III)/U(II) couple are nearly identical in both systems and the event centered at −2.71 V only appears when scanning anodically for 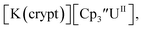 which supports the assignment as the U(III)/U(II) couple.
which supports the assignment as the U(III)/U(II) couple.
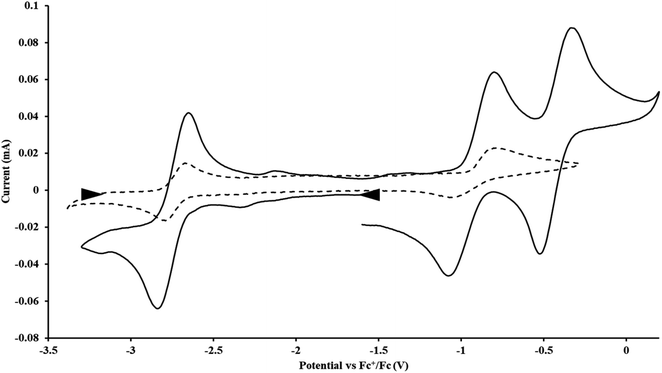 |
| | Fig. 1 Voltammogram of 4.6 mM 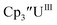 (solid) and 3.0 mM (solid) and 3.0 mM 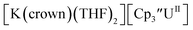 (dashed) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe. (dashed) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe. | |
Cp′.
Similar reproducible data were obtained with the mono(trimethylsilyl)cyclopentadienyl ligand with U(IV)/U(III) and U(III)/U(II) couples at −1.26 V and −2.26 V, respectively, for 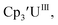 Fig. 2 and S17.† Likewise, the U(IV)/U(III) and U(III)/U(II) couples were observed at −1.28 V and −2.27 V for the U(II) complex
Fig. 2 and S17.† Likewise, the U(IV)/U(III) and U(III)/U(II) couples were observed at −1.28 V and −2.27 V for the U(II) complex 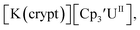 Fig. 2 and S24.† These data were obtained with 50 mM [nBu4N][BPh4] because decomposition occurred at higher electrolyte concentrations. The event at −2.27 V for
Fig. 2 and S24.† These data were obtained with 50 mM [nBu4N][BPh4] because decomposition occurred at higher electrolyte concentrations. The event at −2.27 V for 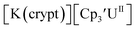 only appears when scanning anodically. The −2.27 V E1/2 value for
only appears when scanning anodically. The −2.27 V E1/2 value for 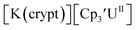 was less negative than the −2.71 V value for
was less negative than the −2.71 V value for 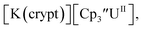 but it is similar to the two previously reported U(III)/U(II) couples for [(Ad,MeArO)3mes]UIII and (C5iPr5)2UII.9,16 The minor unassigned events at about −1.9 V in Fig. 2 and S24† attest to the complexity of the system. They were observed across multiple runs and do not disappear after repeated recrystallization of substrate and electrolyte.
but it is similar to the two previously reported U(III)/U(II) couples for [(Ad,MeArO)3mes]UIII and (C5iPr5)2UII.9,16 The minor unassigned events at about −1.9 V in Fig. 2 and S24† attest to the complexity of the system. They were observed across multiple runs and do not disappear after repeated recrystallization of substrate and electrolyte.
 |
| | Fig. 2 Voltammogram of 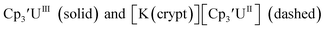 at ν = 200 mV s−1, in 50 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe. at ν = 200 mV s−1, in 50 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe. | |
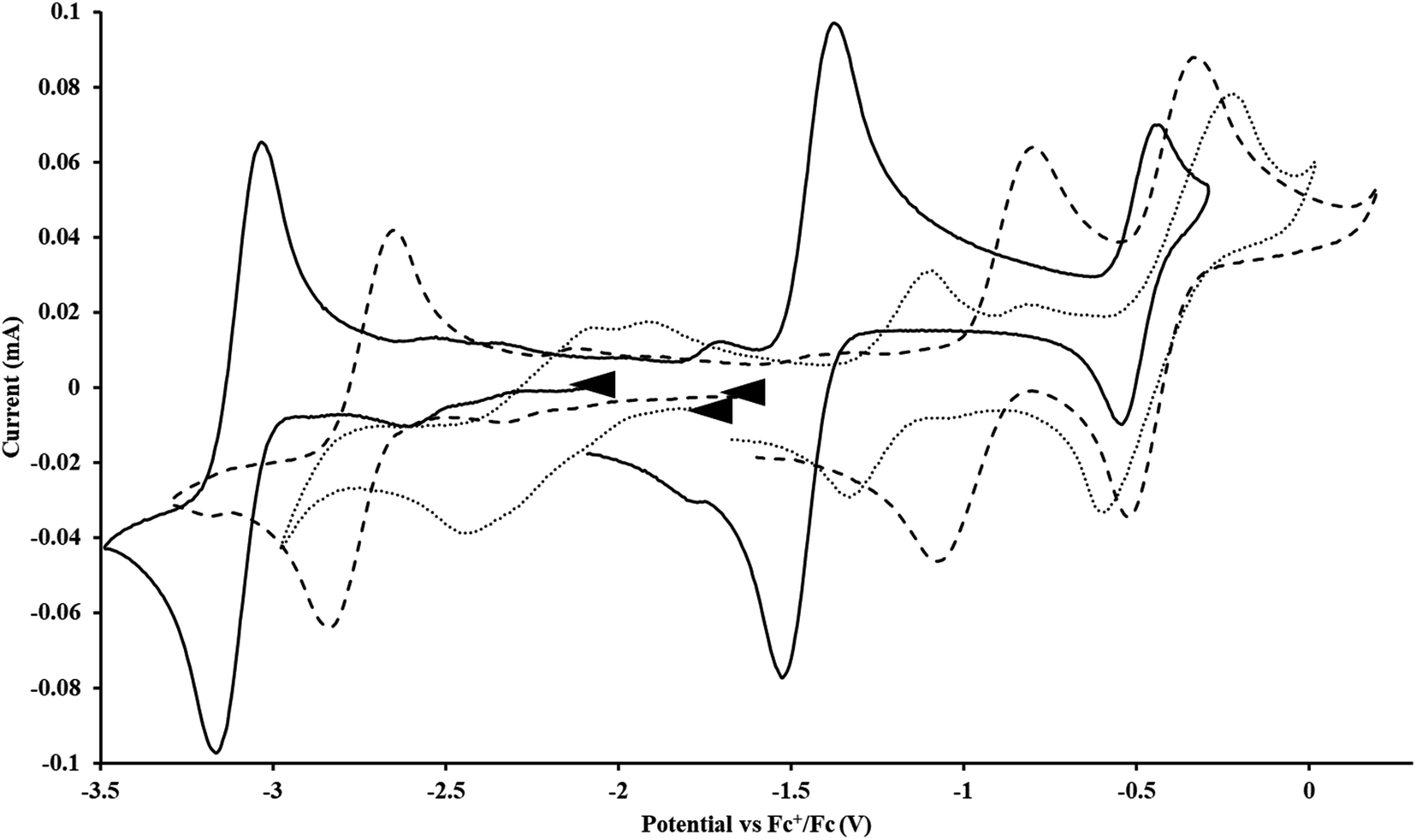
Cptet.
With the tetramethylcyclopentadienyl ligand, the U(IV)/U(III) and U(III)/U(II) couples in Cptet3UIII were more negative than in 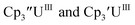 : −1.46 V and −3.11 V, Fig. 3 and S20.† However, data could not be obtained from the isolated U(II) compound [K(crypt)][Cptet3UII] because contact with the supporting electrolyte led to immediate decomposition. The voltammogram obtained from the resulting solution displayed at least five redox events, Fig. S29.† This reactivity is consistent with the more strongly reducing nature of the Cptet complexes as shown by the data in Tables 1 and 2. A third, minor event at −1.7 V was present and cannot be assigned with confidence.
: −1.46 V and −3.11 V, Fig. 3 and S20.† However, data could not be obtained from the isolated U(II) compound [K(crypt)][Cptet3UII] because contact with the supporting electrolyte led to immediate decomposition. The voltammogram obtained from the resulting solution displayed at least five redox events, Fig. S29.† This reactivity is consistent with the more strongly reducing nature of the Cptet complexes as shown by the data in Tables 1 and 2. A third, minor event at −1.7 V was present and cannot be assigned with confidence.
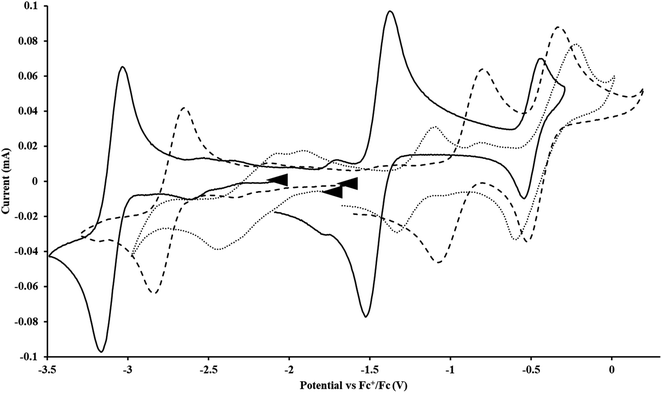 |
| | Fig. 3 Voltammogram of 7.2 mM Cptet3UIII (solid, 100 mM [nBu4N][BPh4]/THF) compared to voltammograms of 4.6 mM 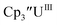 (dashed, 100 mM [nBu4N][BPh4]/THF) and 11 mM (dashed, 100 mM [nBu4N][BPh4]/THF) and 11 mM 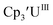 (dotted, 50 mM [nBu4N][BPh4]/THF) at ν = 200 mV s−1. The events centered at −0.495 V are due to internal standard (C5Me5)2FeII. (dotted, 50 mM [nBu4N][BPh4]/THF) at ν = 200 mV s−1. The events centered at −0.495 V are due to internal standard (C5Me5)2FeII. | |
Thorium complexes
Electrochemical data were collected on all the thorium compounds in this study using both [nBu4N][PF6] and [nBu4N][BPh4] despite multiple reports that electrochemical data on organothorium complexes are difficult to obtain using [nBu4N][PF6].11,14,15,30–32 Since the voltammograms do not differ drastically between electrolytes, only the data using [nBu4N][BPh4], Table 3, are discussed below (data with [nBu4N][PF6] are in Table S1†).
Table 3 Reduction potentials of tris(cyclopentadienyl)thorium complexes with 100 mM [nBu4N][BPh4] supporting electrolyte
|
|
Th(IV)/Th(III) |
Th(III)/Th(II) |
|
|
E
PC (V) |
E
PA (V) |
E
1/2 (V) |
E
PC (V) |
E
PA (V) |
E
1/2 (V) |
ΔEpp Fc (V) |
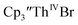
|
−3.00 |
−2.77 |
−2.89 |
|
|
|
0.14 |
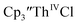
|
−3.04 |
−2.81 |
−2.93 |
|
|
|
0.22 |
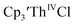
|
−3.38 |
−2.90 |
−3.14 |
|
|
|
0.16 |
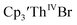
|
−3.17 |
|
|
|
|
|
|
| Cptet3ThIVBr |
−3.48 |
−3.19 |
−3.34 |
|
|
|
0.18 |
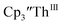
|
|
|
|
−2.92 |
−2.78 |
−2.85 |
0.19 |
| Cptet3ThIII |
|
|
|
−3.33 |
−3.23 |
−3.28 |
0.16 |
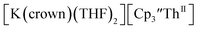
|
|
|
|
−2.89 |
−2.79 |
−2.84 |
0.09 |
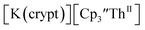
|
|
|
|
−2.90 |
−2.81 |
−2.85 |
0.09 |
Thorium(IV) complexes
Cp′′.
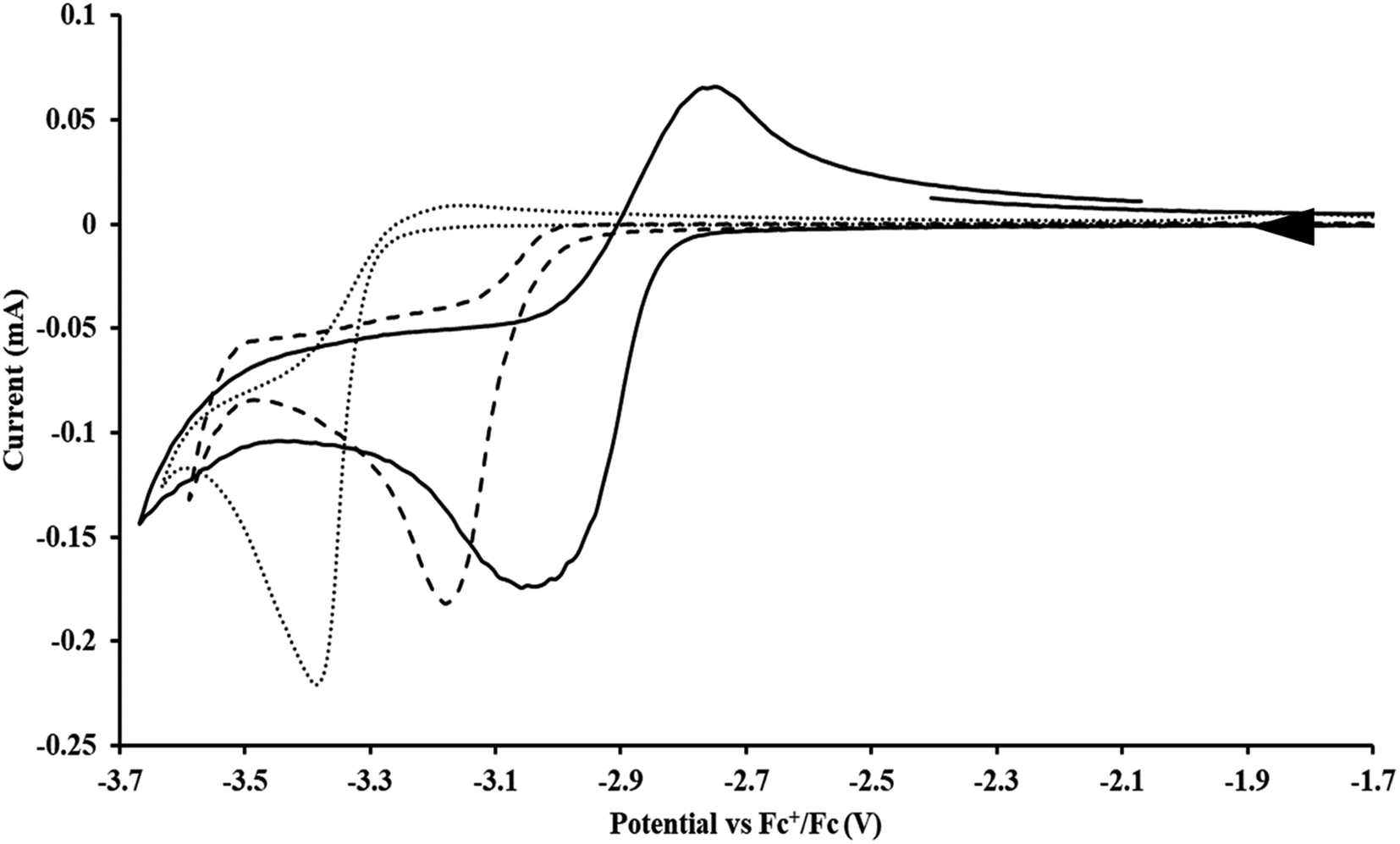
Initially, 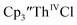 was examined to compare with the values previously reported by Cloke et al.15 The cyclic voltammogram of
was examined to compare with the values previously reported by Cloke et al.15 The cyclic voltammogram of 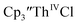 under our conditions shows the Th(IV)/Th(III) couple at −2.93 V, Fig. S34,† which is close to the value of −2.96 V reported for
under our conditions shows the Th(IV)/Th(III) couple at −2.93 V, Fig. S34,† which is close to the value of −2.96 V reported for 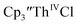 and
and 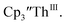 15 Similarly, the cyclic voltammogram of
15 Similarly, the cyclic voltammogram of 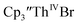 (ref. 2) shows a Th(IV)/Th(III) redox couple at −2.89 V, Fig. 4 and S30.† This suggests that the identity of halide does not significantly affect the reduction potential in this system. This is also consistent with bulk synthetic studies that show that
(ref. 2) shows a Th(IV)/Th(III) redox couple at −2.89 V, Fig. 4 and S30.† This suggests that the identity of halide does not significantly affect the reduction potential in this system. This is also consistent with bulk synthetic studies that show that 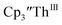 can be synthesized from both
can be synthesized from both 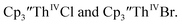 2,33,34
2,33,34
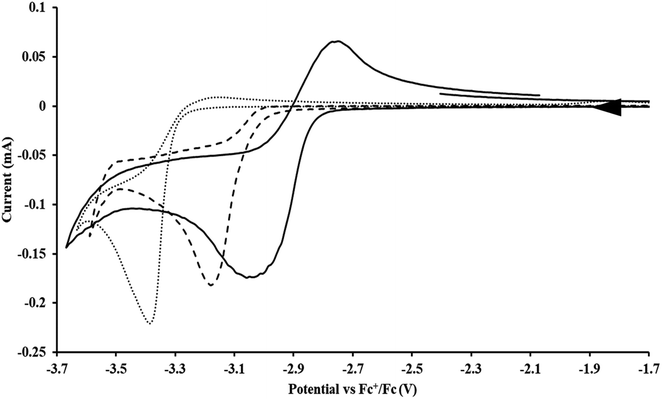 |
| | Fig. 4 Voltammogram of 7.4 mM 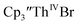 (solid), 15 mM (solid), 15 mM 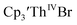 (dashed), and 12 mM Cptet3ThIVBr (dotted) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. (dashed), and 12 mM Cptet3ThIVBr (dotted) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. | |
Cp′ and Cptet.
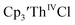 35 and Cptet3ThIVBr (ref. 36) were also examined as each these complexes can be chemically reduced to form tris(cyclopentadienyl)Th(III) species.18,36 The cyclic voltammogram of
35 and Cptet3ThIVBr (ref. 36) were also examined as each these complexes can be chemically reduced to form tris(cyclopentadienyl)Th(III) species.18,36 The cyclic voltammogram of 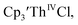 35 Fig. S38,† exhibited a cathodic event at −3.14 V that is 0.21 V more negative than that of
35 Fig. S38,† exhibited a cathodic event at −3.14 V that is 0.21 V more negative than that of 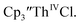 Similarly, the voltammogram of
Similarly, the voltammogram of 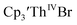 had a cathodic event at −3.17 V, Fig. 4 and S63.† This event was determined to be a one electron process by comparing the current passed to that of the internal standard, Fig. S65.† The voltammogram of Cptet3ThIVBr had a cathodic event at −3.48 V, Fig. 4 and S44.† The events in the voltammograms of
had a cathodic event at −3.17 V, Fig. 4 and S63.† This event was determined to be a one electron process by comparing the current passed to that of the internal standard, Fig. S65.† The voltammogram of Cptet3ThIVBr had a cathodic event at −3.48 V, Fig. 4 and S44.† The events in the voltammograms of 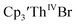 and Cptet3ThIVBr are practically irreversible even at scan rates up to 2000 mV s−1. These results, along with the uranium studies above in Table 1, clearly show that the reduction potential of the actinide complex trends with the electron donation strength of the ligand in the order of Cptet > Cp′ > Cp′′.
and Cptet3ThIVBr are practically irreversible even at scan rates up to 2000 mV s−1. These results, along with the uranium studies above in Table 1, clearly show that the reduction potential of the actinide complex trends with the electron donation strength of the ligand in the order of Cptet > Cp′ > Cp′′.
In addition to the Th(IV)/Th(III) couple, the voltammograms of the Th(IV) compounds showed an irreversible anodic process that could be a cyclopentadienide oxidation, based on the electrochemical data collected on the cyclopentadienyl salts, KCp′, KCp′′, and KCptet, Fig. S66.† These irreversible anodic events were not found in the uranium systems. This difference in Th and U electrochemistry has been previously observed.11,15,37,38 Clearly, the Lewis acidity of the metal influences the potential for these cyclopentadienide oxidations. Cyclopentadienyl rings bound to K+, [K(chelate)]+, or Ann+ could have different oxidation potentials as evidenced by the differing voltammograms of KCp′′, [K(crown)][Cp′′], and [K(crypt)][Cp′′], Fig. S67.†
Th(III) complexes
Cp′′.
There are fewer Th(III) options to study since there are only five crystallographically-characterized tris(cyclopentadienyl) Th(III) complexes, 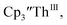 33,34 [C5H3(SiMe2tBu)2]3ThIII,34 Cptet3ThIII,36 (C5tBu2H3)3ThIII,39 and (C5Me5)3ThIII.40 Other Th(III) compounds have been isolated with different ligand environments,41–45 but our initial attempts to collect electrochemical data on (C5Me5)2ThIII[iPrNC(Me)NiPr]44 led to immediate decomposition. Inman and Cloke found that scanning anodically on
33,34 [C5H3(SiMe2tBu)2]3ThIII,34 Cptet3ThIII,36 (C5tBu2H3)3ThIII,39 and (C5Me5)3ThIII.40 Other Th(III) compounds have been isolated with different ligand environments,41–45 but our initial attempts to collect electrochemical data on (C5Me5)2ThIII[iPrNC(Me)NiPr]44 led to immediate decomposition. Inman and Cloke found that scanning anodically on 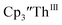 gave a process at −2.96 V that matched the reduction of
gave a process at −2.96 V that matched the reduction of 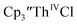 described above and established the Th(IV)/Th(III) couple.15 In our hands, scanning cathodically on
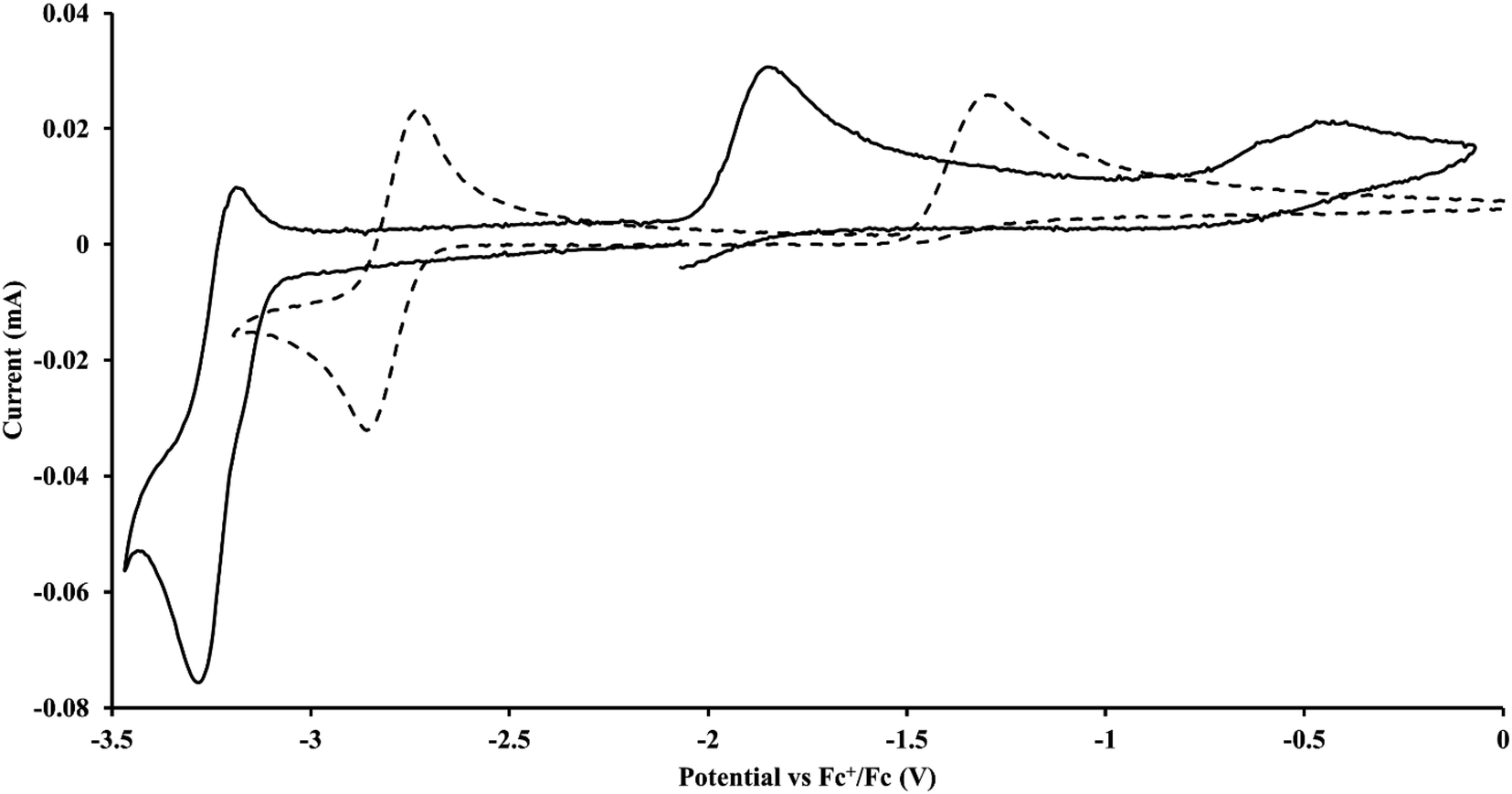
described above and established the Th(IV)/Th(III) couple.15 In our hands, scanning cathodically on 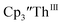 showed a voltammogram with a redox process centered at −2.85 V, Fig. 5 and S40.† A second cathodic event appears after the first cycle at −2.29 V, or when scanning anodically from the open circuit potential, Fig. S40.† The event at −2.29 V was also observed by Cloke and was attributed to a ligand-based event.
showed a voltammogram with a redox process centered at −2.85 V, Fig. 5 and S40.† A second cathodic event appears after the first cycle at −2.29 V, or when scanning anodically from the open circuit potential, Fig. S40.† The event at −2.29 V was also observed by Cloke and was attributed to a ligand-based event.
 |
| | Fig. 5 Voltammogram of 4.9 mM 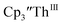 (solid) at ν = 200 mV s−1 and 6.7 mM Cptet3ThIII (dashed) at ν = 400 mV s−1 in 100 mM [nBu4N][BPh4]/THF. (solid) at ν = 200 mV s−1 and 6.7 mM Cptet3ThIII (dashed) at ν = 400 mV s−1 in 100 mM [nBu4N][BPh4]/THF. | |
Cp′ and Cptet.
Since 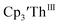 has only been generated in situ,18 it was not studied under the present conditions. The voltammogram of Cptet3ThIII at ν = 200 mV s−1 displays only a cathodic event, but at ν ≥ 400 mV s−1, a return oxidation appears and the Th(III)/Th(II) redox couple is centered at −3.28 V, Fig. 5 and S48.† This value matches the trend observed for the uranium systems in that Cptet complexes of thorium are more difficult to reduce than the silyl-cyclopentadienyl analogs. An anodic event at −1.87 V is present and is attributed to a Cptet-based process.
has only been generated in situ,18 it was not studied under the present conditions. The voltammogram of Cptet3ThIII at ν = 200 mV s−1 displays only a cathodic event, but at ν ≥ 400 mV s−1, a return oxidation appears and the Th(III)/Th(II) redox couple is centered at −3.28 V, Fig. 5 and S48.† This value matches the trend observed for the uranium systems in that Cptet complexes of thorium are more difficult to reduce than the silyl-cyclopentadienyl analogs. An anodic event at −1.87 V is present and is attributed to a Cptet-based process.
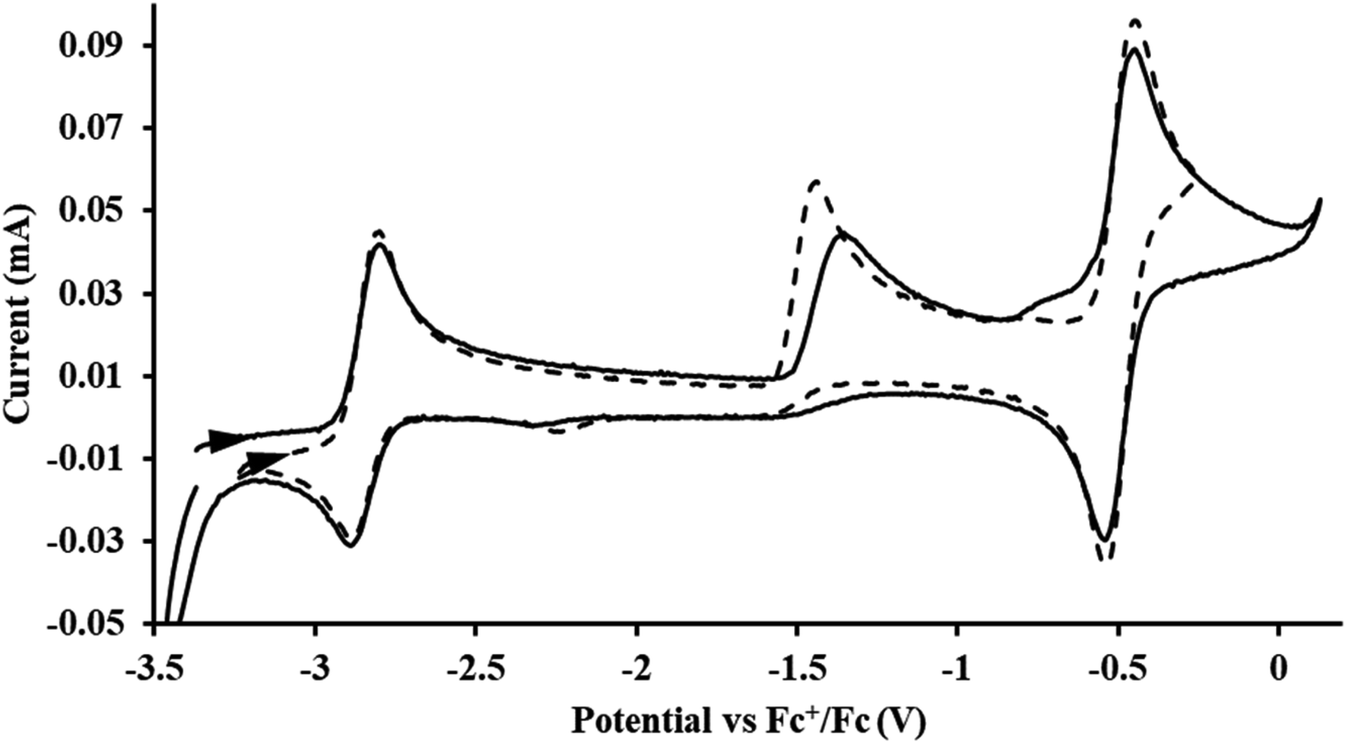
Th(II) complexes
The only isolated Th(II) compounds 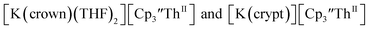 exhibited nearly identical voltammograms. Scanning anodically,
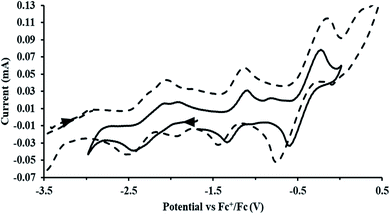
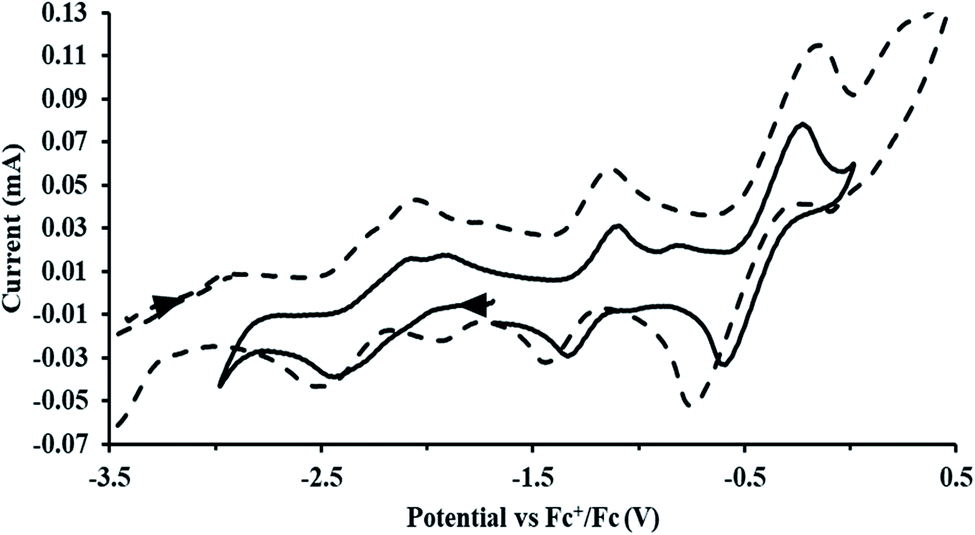
exhibited nearly identical voltammograms. Scanning anodically, 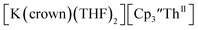 showed a redox process centered at −2.84 V, which is assigned as the Th(III)/Th(II) redox couple, and a second irreversible anodic event at −1.38 V, attributed to ligand-based oxidation, Fig. 6 and S52.† The voltammogram of this Th(II) compound was practically identical over 5 cycles, Fig. S54.†
showed a redox process centered at −2.84 V, which is assigned as the Th(III)/Th(II) redox couple, and a second irreversible anodic event at −1.38 V, attributed to ligand-based oxidation, Fig. 6 and S52.† The voltammogram of this Th(II) compound was practically identical over 5 cycles, Fig. S54.†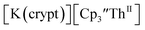 similarly showed a reversible event centered at −2.85 V and a second anodic event at −1.43 V, Fig. 6, S57 and S61.†
similarly showed a reversible event centered at −2.85 V and a second anodic event at −1.43 V, Fig. 6, S57 and S61.†
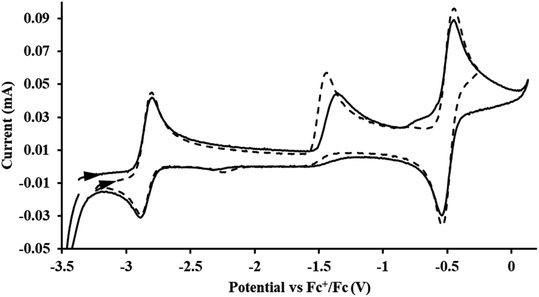 |
| | Fig. 6 Voltammogram of 4.6 mM 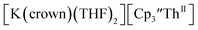 (solid) and 3.1 mM (solid) and 3.1 mM 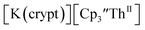 (dashed) with internal standard (C5Me5)2Fe at ν = 200 mV s−1 in 100 mM [nBu4N][BPh4]/THF. (dashed) with internal standard (C5Me5)2Fe at ν = 200 mV s−1 in 100 mM [nBu4N][BPh4]/THF. | |
Thorium spectroelectrochemistry
The data on isolated 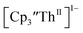 complexes suggested that the Th(III)/Th(II) redox process occurs at about the same potential as the Th(IV)/Th(III) potential of
complexes suggested that the Th(III)/Th(II) redox process occurs at about the same potential as the Th(IV)/Th(III) potential of 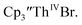 To investigate this further, spectroelectrochemical UV-visible measurements were obtained. A potential of −2.90 V was applied to a solution of
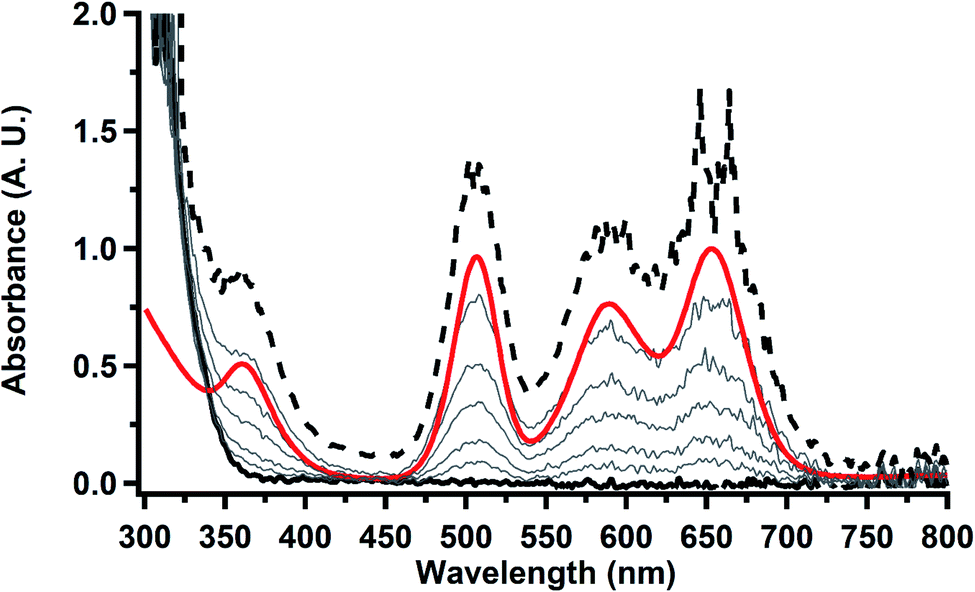
To investigate this further, spectroelectrochemical UV-visible measurements were obtained. A potential of −2.90 V was applied to a solution of 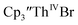 in 200 mM [nBu4N][PF6]/THF and the UV-visible spectrum was recorded approximately every 5 seconds during electrolysis. The formation of
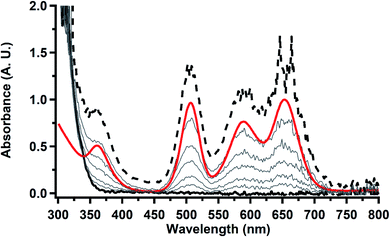
in 200 mM [nBu4N][PF6]/THF and the UV-visible spectrum was recorded approximately every 5 seconds during electrolysis. The formation of 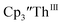 is clearly shown by the growth of four bands at roughly 360, 500, 580, and 680 nm, Fig. 7, which correspond to the absorption spectrum of
is clearly shown by the growth of four bands at roughly 360, 500, 580, and 680 nm, Fig. 7, which correspond to the absorption spectrum of 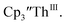 33,34 No further reduction to the
33,34 No further reduction to the 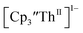 was observed,2 although it cannot be ruled out as the absorbance spectrum reached the maximum of the detector.
was observed,2 although it cannot be ruled out as the absorbance spectrum reached the maximum of the detector.
 |
| | Fig. 7 UV-visible spectrum of 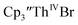 (black, solid) converting to (black, solid) converting to 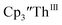 (black, dashed) during electrolysis at −2.90 V with a starting concentration of 7.0 mM in 200 mM [nBu4N][PF6]/THF. The growth of four bands at 365, 510, 590, and 655 nm is indicative of (black, dashed) during electrolysis at −2.90 V with a starting concentration of 7.0 mM in 200 mM [nBu4N][PF6]/THF. The growth of four bands at 365, 510, 590, and 655 nm is indicative of 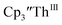 (red).34 (red).34 | |
Electrolysis of a solution of 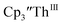 in 200 mM [nBu4N][PF6]/THF at −2.90 V shows clean conversion to the Th(II) species
in 200 mM [nBu4N][PF6]/THF at −2.90 V shows clean conversion to the Th(II) species 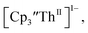 2 as indicated by the growth of the large absorption at 650 nm and the concomitant decrease in absorptions at 360, 500, 580, and 680 nm, Fig. 8. Although the absorption spectrum of
2 as indicated by the growth of the large absorption at 650 nm and the concomitant decrease in absorptions at 360, 500, 580, and 680 nm, Fig. 8. Although the absorption spectrum of 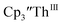 had disappeared, the absorption at 650 nm, indicative of Th(II),2 decreased in intensity as the electrolysis continued. The Th(II) species appears to be unstable under the electrolysis conditions.
had disappeared, the absorption at 650 nm, indicative of Th(II),2 decreased in intensity as the electrolysis continued. The Th(II) species appears to be unstable under the electrolysis conditions.
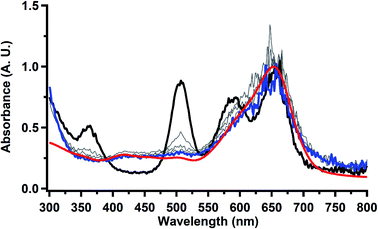 |
| | Fig. 8 UV-visible spectrum of 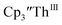 (black) converting to (black) converting to 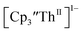 (blue) during electrolysis at −2.90 V with a starting concentration of 1.1 mM in 200 mM [nBu4N][PF6]/THF. The growth of the band at 650 nm is indicative of (blue) during electrolysis at −2.90 V with a starting concentration of 1.1 mM in 200 mM [nBu4N][PF6]/THF. The growth of the band at 650 nm is indicative of 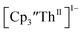 (red).2 (red).2 | |
Chemical synthesis of Th(II) complexes from Th(IV) precursors
The similarity of the Th(IV)/Th(III) couple in 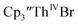 and Th(III)/Th(II) couple in
and Th(III)/Th(II) couple in 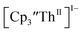 suggested that Th(IV) compounds could be used as the precursors to Th(II) compounds as well as the known Th(III) precursor,
suggested that Th(IV) compounds could be used as the precursors to Th(II) compounds as well as the known Th(III) precursor, 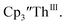 Indeed, reaction of 2.2 equivalents of KC8 to a THF solution of
Indeed, reaction of 2.2 equivalents of KC8 to a THF solution of 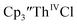 and crown afforded
and crown afforded 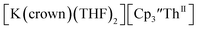 in 50% crystalline yield, with a significant amount of
in 50% crystalline yield, with a significant amount of 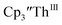 as a byproduct. Previously, Lappert reported that prolonged stirring of a solution of
as a byproduct. Previously, Lappert reported that prolonged stirring of a solution of 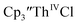 over excess NaK alloy developed a green color,34 which was later confirmed to be the color of Th(II).2
over excess NaK alloy developed a green color,34 which was later confirmed to be the color of Th(II).2
Conversion of Th(IV) to Th(II) was also studied with 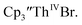 Reaction of
Reaction of 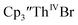 with 2 equivalents of KC8 in THF generated a dark green solution characteristic of Th(II) within 5 minutes, as did reaction of
with 2 equivalents of KC8 in THF generated a dark green solution characteristic of Th(II) within 5 minutes, as did reaction of 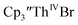 with excess Na and with excess Li. The UV-visible spectra of these solutions have a strong absorption at 650 nm, identical to the previously reported spectra of
with excess Na and with excess Li. The UV-visible spectra of these solutions have a strong absorption at 650 nm, identical to the previously reported spectra of 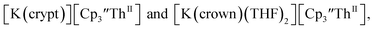 2 but the spectra also show a non-negligible amount of
2 but the spectra also show a non-negligible amount of 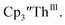 34 Formation of the Th(III) complex is reasonable based on the fact that
34 Formation of the Th(III) complex is reasonable based on the fact that 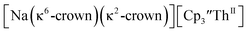 (see below) reacts with
(see below) reacts with 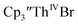 in THF to immediately form
in THF to immediately form 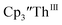 in near quantitative yield.
in near quantitative yield.
These results show that a chelating agent is not necessary for the chemical synthesis of Th(II) species in solution. However, the chelating agent appears necessary for efficient separation of the Th(II) product from the Th(III) starting material, as pure samples of 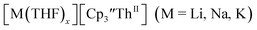 were not isolated even though it is possible to isolate chelate-free examples of
were not isolated even though it is possible to isolate chelate-free examples of 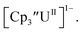 46 Further support for the importance of alkali metal chelates is that addition of 18-crown-6 to the reaction of
46 Further support for the importance of alkali metal chelates is that addition of 18-crown-6 to the reaction of 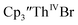 and excess Na provided X-ray quality crystals that were identified as
and excess Na provided X-ray quality crystals that were identified as 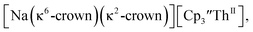 only the third reported crystal structure of a Th(II) complex, Scheme 2, Fig. 9.
only the third reported crystal structure of a Th(II) complex, Scheme 2, Fig. 9.
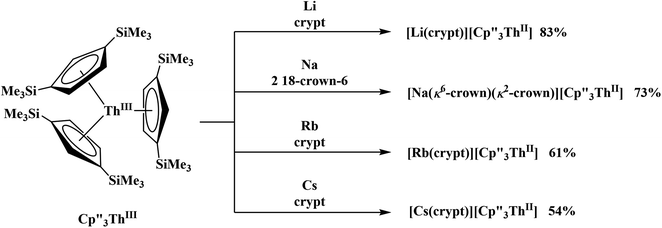 |
| | Scheme 2 Synthesis of new Th(II) compounds. | |
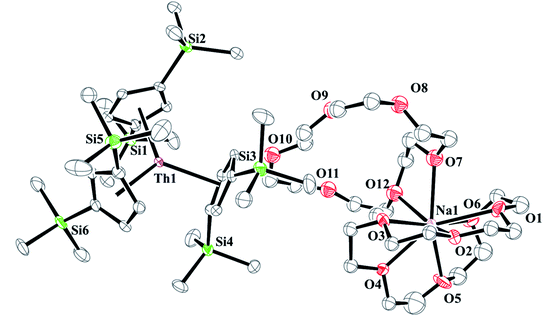 |
| | Fig. 9 Thermal ellipsoid plot of 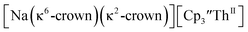 plotted at the 35% probability level. Hydrogen atoms and disorder in the κ2-crown unit have been removed for clarity. plotted at the 35% probability level. Hydrogen atoms and disorder in the κ2-crown unit have been removed for clarity. | |
Similarly, the reaction of  Rb, and crypt in THF afforded dichroic blue/red crystals of
Rb, and crypt in THF afforded dichroic blue/red crystals of  isolated in 61% crystalline yield and identified by X-ray crystallography, Scheme 2, Fig. S69.† In addition, the reaction of
isolated in 61% crystalline yield and identified by X-ray crystallography, Scheme 2, Fig. S69.† In addition, the reaction of 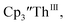 Cs, and crypt afforded dark blue/red crystals of
Cs, and crypt afforded dark blue/red crystals of 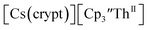 in 54% crystalline yield, Scheme 2, Fig. S70.† The [Rb(crypt)]1+ and [Cs(crypt)]1+ compounds are isomorphous with the [K(crypt)]1+ analog2 and can be easily separated from the
in 54% crystalline yield, Scheme 2, Fig. S70.† The [Rb(crypt)]1+ and [Cs(crypt)]1+ compounds are isomorphous with the [K(crypt)]1+ analog2 and can be easily separated from the 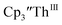 starting material, which was difficult without the use of a chelate. The reaction of
starting material, which was difficult without the use of a chelate. The reaction of 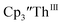 , Li, and crypt formed dark blue-green needles of
, Li, and crypt formed dark blue-green needles of 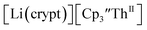 in 83% yield, but the crystals were not suitable for X-ray diffraction, Scheme 2.
in 83% yield, but the crystals were not suitable for X-ray diffraction, Scheme 2.
Since the reaction chemistry and the spectroelectrochemistry suggested that the Th(II) complexes were generated from a Th(IV) precursor through a Th(III) intermediate, reactions with the two-electron reductant Ba were studied. The Ba(II)/Ba(0) reduction potential is nearly identical to that of K(I)/K(0).47 Surprisingly, prolonged stirring of a THF solution of 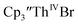 and excess Ba afforded only
and excess Ba afforded only 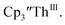 When chelates were added, the reaction of
When chelates were added, the reaction of 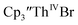 and crown or
and crown or 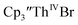 and crypt over excess Ba formed
and crypt over excess Ba formed 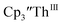 and then the dark green color of Th(II) with UV-visible spectra consistent with
and then the dark green color of Th(II) with UV-visible spectra consistent with 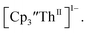 Addition of elemental Hg did not appear to affect the rate of formation of the Th(II) species. These results, coupled with the spectroelectrochemical measurements, strongly suggest that the Th(IV)/Th(II) redox couple is not observed experimentally in these systems and that instead two one-electron processes occur.
Addition of elemental Hg did not appear to affect the rate of formation of the Th(II) species. These results, coupled with the spectroelectrochemical measurements, strongly suggest that the Th(IV)/Th(II) redox couple is not observed experimentally in these systems and that instead two one-electron processes occur.
Discussion
An(IV)/An(III) processes
The trends observed in the U(IV)/U(III) and Th(IV)/Th(III) redox couples in Tables 1–3 indicate that Cptet is more electron donating than Cp′, which is more electron donating than Cp′′. This follows the electron-donating ability of the ligands previously found in studies of (C5R5)2Zr(CO)2 complexes21 and yttrium compounds.22,24 For the zirconium complexes, the CO stretching frequency and the reduction potentials were analyzed to determine electron-donation strength of the cyclopentadienyl ligand. Generally in these An(IV)/An(III) studies, the thorium complexes showed less reversible processes than the uranium compounds. In the 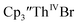 case, UV-visible spectroelectrochemistry measurements show that this compound is reduced under electrochemical conditions to
case, UV-visible spectroelectrochemistry measurements show that this compound is reduced under electrochemical conditions to 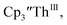 which requires loss of Br1− and geometric reorganization. In the
which requires loss of Br1− and geometric reorganization. In the 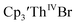 case, density functional theory calculations have shown that the putative initial reduction product,
case, density functional theory calculations have shown that the putative initial reduction product, 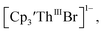 would be unstable with respect to
would be unstable with respect to 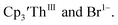 18 These results are consistent with the electrochemical irreversibility of the system.
18 These results are consistent with the electrochemical irreversibility of the system.
An(III)/An(II) processes
To our knowledge, only two other U(III)/U(II) couples have been assigned via electrochemistry: [(Ad,MeArO)3mes]UIII at −2.495 V using [nBu4N][PF6]16 and (C5iPr5)2UII at −2.33 V using [nBu4N][BPh4].9 The −2.26 V value for 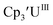 matches well with these two data points, even though
matches well with these two data points, even though 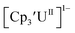 and (C5iPr5)2UII have been assigned 5f36d1 electron configurations,1,9 while {[(Ad,MeArO)3mes]UII}1− is best described as 5f4.7 The −2.73 V reduction potential for
and (C5iPr5)2UII have been assigned 5f36d1 electron configurations,1,9 while {[(Ad,MeArO)3mes]UII}1− is best described as 5f4.7 The −2.73 V reduction potential for 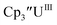 is unexpectedly more reducing than those of these other three complexes. This is also unusual in that solutions of
is unexpectedly more reducing than those of these other three complexes. This is also unusual in that solutions of 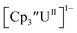 have longer lifetimes than solutions of
have longer lifetimes than solutions of 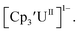 27 The U(III)/U(II) reduction potential for Cptet3UIII was determined to be −3.11 V, which is the most negative reduction potential for these compounds and matches the trend observed for the An(IV)/An(III) couples.
27 The U(III)/U(II) reduction potential for Cptet3UIII was determined to be −3.11 V, which is the most negative reduction potential for these compounds and matches the trend observed for the An(IV)/An(III) couples.
Th(II) complexes were investigated for the first time via electrochemistry and the E1/2 values for the Th(III)/Th(II) couple observed in the isolated Th(II) compounds matched the value observed in 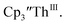 Surprisingly, the Th(IV)/Th(III) couple of
Surprisingly, the Th(IV)/Th(III) couple of 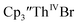 appears to be about the same as the value for the Th(III)/Th(II) couple of
appears to be about the same as the value for the Th(III)/Th(II) couple of 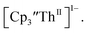 This result was tested chemically and it was found that reduction of Th(IV) with excess reducing agent would form Th(II) compounds directly with KC8, Na, Li, and Ba both with and without the use of a chelating agent. Blue
This result was tested chemically and it was found that reduction of Th(IV) with excess reducing agent would form Th(II) compounds directly with KC8, Na, Li, and Ba both with and without the use of a chelating agent. Blue 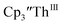 is observed as an intermediate in these reactions which indicates formation of the Th(II) products arises from two one-electron reductions. Furthermore, the E1/2 values for Th(III)/Th(II) match the expected trend compared to uranium based on previously calculated An(III)/An(II) reduction potentials.48–50
is observed as an intermediate in these reactions which indicates formation of the Th(II) products arises from two one-electron reductions. Furthermore, the E1/2 values for Th(III)/Th(II) match the expected trend compared to uranium based on previously calculated An(III)/An(II) reduction potentials.48–50
The thorium electrochemistry was also unusual in that electrochemical data were obtained using [nBu4N][PF6] as supporting electrolyte on isolated Th(IV), Th(III), and Th(II) compounds. This electrolyte has proven to be more reactive than [nBu4N][BPh4] with some complexes11,15 and it may have been expected that Th(II) would react with it. The fact that the Th(III)/Th(II) reduction potentials vary slightly depending on the specific electrolyte highlights the fact the reduction potentials of these systems are very sensitive to experimental conditions.
Conclusion
Electrochemical data on three series of tris(cyclopentadienyl) An(IV), An(III), and An(II) (An = Th, U) complexes, including the first data on Th(II) complexes, complimented by UV-visible spectroelectrochemical measurements, show a direct correlation between reduction potential and the electron-donating ability of the cyclopentadienyl ring. The studies indicate that Th(III) is a stronger reductant than U(III), but the reduction potential of U(II) is similar to that of Th(II). Two unexpected results should stimulate further studies. The U(III)/U(II) reduction potential of 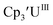 is similar to the two previously reported U(III)/U(II) values, but it is significantly less negative than the Cp′′ analog. The reduction potentials of Th(IV)/Th(III) and Th(III)/Th(II) couples are sufficiently similar that Th(II) complexes can be made directly from Th(IV) precursors without the need to isolate the Th(III) intermediate.
is similar to the two previously reported U(III)/U(II) values, but it is significantly less negative than the Cp′′ analog. The reduction potentials of Th(IV)/Th(III) and Th(III)/Th(II) couples are sufficiently similar that Th(II) complexes can be made directly from Th(IV) precursors without the need to isolate the Th(III) intermediate.
Author contributions
J. C. W. synthesized all compounds and performed the cyclic voltammetry experiments. J. C. W. and J. M. B. performed the spectroelectrochemistry experiments. J. W. Z. analyzed the X-ray diffraction data. All authors analyzed the electrochemistry data. J. C. W. and W. J. E. wrote the manuscript with input from all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences of the Department of Energy (DE-SC0004739 to W. J. E.) and the U.S. National Science Foundation (CHE-1554744 and CHE-2102589 to J. Y. Y.) for support. J. Y. Y. is grateful for support as a Sloan Foundation Fellow, Camille Dreyfus Teacher-Scholar, and CIFAR Azrieli Global Scholar in the Bio-inspired Energy Program. We thank Xinran S. Wang for assistance with the spectroelectrochemistry measurements, Ryan R. Langeslay and Nicholas R. Rightmire for preliminary experimental results, and Kirsten A. Hewitt for assistance with the TOC graphic.
References
- M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Identification of the +2 Oxidation State for Uranium in a Crystalline Molecular Complex, [K(2.2.2-Cryptand)][(C5H4SiMe3)3U], J. Am. Chem. Soc., 2013, 135, 13310–13313, DOI:10.1021/ja406791t.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Synthesis, Structure, and Reactivity of Crystalline Molecular Complexes of the {[C5H3(SiMe3)2]3Th}1− Anion Containing Thorium in the Formal +2 Oxidation State, Chem. Sci., 2015, 6, 517–521, 10.1039/C4SC03033H.
- J. Su, C. J. Windorff, E. R. Batista, W. J. Evans, A. J. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, D. H. Woen and P. Yang, Identification of the Formal +2 Oxidation State of Neptunium: Synthesis and Structural Characterization of {NpII[C5H3(SiMe3)2]3}1–, J. Am. Chem. Soc., 2018, 140, 7425–7428, DOI:10.1021/jacs.8b03907.
- M. S. Dutkiewicz, J. H. Farnaby, C. Apostolidis, E. Colineau, O. Walter, N. Magnani, M. G. Gardiner, J. B. Love, N. Kaltsoyannis, R. Caciuffo and P. L. Arnold, Organometallic Neptunium(III) Complexes, Nat. Chem., 2016, 8, 797–802, DOI:10.1038/nchem.2520.
- M. S. Dutkiewicz, C. Apostolidis, O. Walter and P. L. Arnold, Reduction Chemistry of Neptunium Cyclpentadienide Complexes: From Structure to Understanding, Chem. Sci., 2017, 8, 2553–2561, 10.1039/c7sc00034k.
- C. J. Windorff, G. P. Chen, J. N. Cross, W. J. Evans, F. Furche, A. J. Gaunt, M. T. Janicke, S. A. Kozimor and B. L. Scott, Identification of the Formal +2 Oxidation State of Plutonium: Synthesis and Characterization of {PuII[C5H3(SiMe3)2]3}−, J. Am. Chem. Soc., 2017, 139, 3970–3973, DOI:10.1021/jacs.7b00706.
- H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Synthesis and Characterization of a Uranium(II) Monoarene Complex Supported by δ Backbonding, Angew. Chem., Int. Ed., 2014, 53, 7158–7162, DOI:10.1002/anie.201402050.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. Mccracken, M. P. Shores, J. M. Boncella and A. L. Odom, Synthesis and Characterization of a Neutral U(II) Arene Sandwich Complex, J. Am. Chem. Soc., 2018, 140, 17369–17373, DOI:10.1021/jacs.8b10888.
- F. S. Guo, N. Tsoureas, G. Z. Huang, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Isolation of a Perfectly Linear Uranium(II) Metallocene, Angew. Chem., Int. Ed., 2020, 59, 2299–2303, DOI:10.1002/anie.201912663.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Thorium(iv) and Uranium(IV) Ketimide Complexes Prepared by Nitrile Insertion into Actinide-Alkyl and -Aryl Bonds, Organometallics, 2004, 23, 4682–4692, DOI:10.1021/om0343824.
- D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Trends in Electronic Structure and Redox Energetics for Early-Actinide Pentamethylcyclopentadienyl Complexes, Organometallics, 2004, 23, 5142–5153, DOI:10.1021/om049634v.
- E. J. Schelter, J. M. Veauthier, J. D. Thompson, B. L. Scott, K. D. John, D. E. Morris and J. L. Kiplinger, 4f-5f Heterotrimetallic Complexes Exhibiting Electrochemical and Magnetic Communication, J. Am. Chem. Soc., 2006, 128, 2198–2199, DOI:10.1021/ja057808+.
- K. A. Erickson, B. D. Kagan, B. L. Scott, D. E. Morris and J. L. Kiplinger, Revisiting the Bis(Dimethylamido) Metallocene Complexes of Thorium and Uranium: Improved Syntheses, Structure, Spectroscopy, and Redox Energetics of (C5Me5)2An(NMe2)2 (An = Th, U), Dalton Trans., 2017, 46, 11208–11213, 10.1039/c7dt02373a.
- Z. E. Button, J. A. Higgins, M. Suvova, F. G. N. Cloke and S. M. Roe, Mixed Sandwich Thorium Complexes Incorporating Bis(Tri-Isopropylsilyl)Cyclooctatetraenyl and Pentamethylcyclopentadienyl Ligands: Synthesis, Structure and Reactivity, Dalton Trans., 2015, 44, 2588–2596, 10.1039/c4dt02362e.
- C. J. Inman and F. G. N. Cloke, The Experimental Determination of Th(IV)/Th(III) Redox Potentials in Organometallic Thorium Complexes, Dalton Trans., 2019, 48, 10782–10784, 10.1039/c9dt01553a.
- H. S. La Pierre, H. Kameo, D. P. Halter, F. W. Heinemann and K. Meyer, Coordination and Redox Isomerization in the Reduction of a Uranium(III) Monoarene Complex, Angew. Chem., Int. Ed., 2014, 53, 7154–7157, DOI:10.1002/anie.201402048.
- J. A. Hlina, J. R. Pankhurst, N. Kaltsoyannis and P. L. Arnold, Metal-Metal Bonding in Uranium-Group 10 Complexes, J. Am. Chem. Soc., 2016, 138, 3333–3345, DOI:10.1021/jacs.5b10698.
- J. C. Wedal, S. Bekoe, J. W. Ziller, F. Furche and W. J. Evans, In Search of Tris(Trimethylsilylcyclopentadienyl) Thorium, Dalton Trans., 2019, 48, 16633–16640, 10.1039/C9DT03674A.
- W. J. Evans, Tutorial on the Role of Cyclopentadienyl Ligands in the Discovery of Molecular Complexes of the Rare-Earth and Actinide Metals in New Oxidation States, Organometallics, 2016, 35, 3088–3100, DOI:10.1021/acs.organomet.6b00466.
-
D. H. Woen and W. J. Evans, Expanding the +2 Oxidation State of the Rare-Earth Metals, Uranium, and Thorium in Molecular Complexes, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier B.V., 2016, pp. 1–57, DOI:10.1016/bs.hpcre.2016.08.002.
- C. E. Zachmanoglou, A. Docrat, B. M. Bridgewater, G. Parkin, C. G. Brandow, J. E. Bercaw, C. N. Jardine, M. Lyall, J. C. Green and J. B. Keister, The Electronic Influence of Ring Substituents and Ansa Bridges in Zirconocene Complexes as Probed by Infrared Spectroscopic, Electrochemical, and Computational Studies, J. Am. Chem. Soc., 2002, 124, 9525–9546, DOI:10.1021/ja020236y.
- J. F. Corbey, D. H. Woen, C. T. Palumbo, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Ligand Effects in the Synthesis of Ln2+ Complexes by Reduction of Tris(Cyclopentadienyl) Precursors Including C–H Bond Activation of an Indenyl Anion, Organometallics, 2015, 34, 3909–3921, DOI:10.1021/acs.organomet.5b00500.
- S. A. Moehring, M. Beltran-Leiva, D. Paez-Hernandez, R. Arratia-Perez, J. W. Ziller and W. J. Evans, Rare-Earth Metal(II) Aryloxides: Structure, Synthesis, and EPR Spectroscopy of [K(2.2.2-Cryptand)][Sc(OC6H2tBu2-2,6-Me-4)3], Chem.–Eur. J., 2018, 24, 18059–18067, DOI:10.1002/chem.201803807.
- S. A. Moehring and W. J. Evans, Evaluating Electron Transfer Reactivity of Rare-Earth Metal(II) Complexes Using EPR Spectroscopy, Organometallics, 2020, 39, 1187–1194, DOI:10.1021/acs.organomet.9b00837.
- J. G. Brennan, R. A. Andersen and A. Zalkin, Chemistry of Trivalent Uranium Metallocenes: Electron-Transfer Reactions with Carbon Disulfide. Formation of [(RC5H4)3U]2[μ–η1,η1-CS2], Inorg. Chem., 1986, 25, 1756–1760, DOI:10.1021/ic00231a007.
- M. del Mar Conejo, J. S. Parry, E. Carmona, M. Schultz, J. G. Brennann, S. M. Beshouri, R. A. Andersen, R. D. Rogers, S. Coles and M. Hursthouse, Carbon Monoxide and Isocyanide Complexes of Trivalent Uranium Metallocenes, Chem.–Eur. J., 1999, 5, 3000–3009 CrossRef CAS.
- C. J. Windorff, M. R. MacDonald, K. R. Meihaus, J. W. Ziller, J. R. Long and W. J. Evans, Expanding the Chemistry of Molecular U2+ Complexes: Synthesis, Characterization, and Reactivity of the {[C5H3(SiMe3)2]3U}− Anion, Chem.–Eur. J., 2016, 22, 772–782, DOI:10.1002/chem.201503583.
- C. Clappe, D. Leveugle, D. Hauchard and G. Durand, Electrochemical Studies of Tricyclopentadienyl Uranium IV Chloride Complexes: (RCp)3UCl (RCp = RC5H4 with R = H; Me: CH3; tBu: (CH3)3C; TMS: (CH3)3Si) Evidence of a Disproportionation Mechanism in Oxidation, J. Electroanal. Chem., 1998, 448, 95–103, DOI:10.1016/S0022-0728(98)00029-1.
- D. Hauchard, M. Cassir, J. Chivot and M. Ephritikhine, Electrochemical Study of Uranium(IV) and Uranium(IV) Organometallic Compounds in Tetrahydrofuran by Means of Conventional Microelectrodes and Ultramicroelectrodes. Part I. Application to the Na(Hg) Reduction of Cp3UCl (Cp = η–C5H5), J. Electroanal. Chem., 1991, 313, 227–241, DOI:10.1016/0022-0728(91)85182-O.
- R. G. Finke, G. Gaughan and R. Voegeli, Organoactinide Electrochemistry. A Cyclic Voltammetric and Coulometric Study of (C5Me5)2UCl2, [(C5Me5)2UCl2·THF]−Na+, (C5Me5)2UCl·THF and (C5Me5)2ThCl2, J. Organomet. Chem., 1982, 229, 179–184, DOI:10.1016/S0022-328X(00)90280-8.
- P. L. Watson, T. H. Tulip and I. Williams, Defluorination of Perfluoroolefins by Divalent Lanthanoid Reagents: Activating C–F Bonds, Organometallics, 1990, 9, 1999–2009, DOI:10.1021/om00157a006.
- O. Maury, M. Ephritikhine, M. Nierlich, M. Lance and E. Samuel, Chloride Ion Transfer during the U(IV)/U(III) Reduction of UCl4 in Tetrahydrofuran (THF), Studied by Cyclic Voltammetry; Synthesis and Molecular Structure of [nBu4][UCl5(THF)], Inorg. Chim. Acta, 1998, 279, 210–216, DOI:10.1016/s0020-1693(98)00126-1.
- P. C. Blake, M. F. Lappert, J. L. Atwood and H. Zhang, The Synthesis and Characterisation, Including X-Ray Diffraction Study, of [Th{η–C5H3(SiMe3)2}3]; The First Thorium(III) Crystal Structure, J. Chem. Soc., Chem. Commun., 1986, 453, 1148–1149, 10.1039/C39860001148.
- P. C. Blake, N. M. Edelstein, P. B. Hitchcock, W. K. Kot, M. F. Lappert, G. V. Shalimoff and S. Tian, Synthesis, Properties and Structures of the Tris(Cyclopentadienyl)Thorium(III) Complexes [Th{η5–C5H3(SiMe2R)2-1,3}3] (R = Me or tBu), J. Organomet. Chem., 2001, 636, 124–129, DOI:10.1016/S0022-328X(01)00860-9.
- C. J. Windorff, M. R. MacDonald, J. W. Ziller and W. J. Evans, Trimethylsilylcyclopentadienyl (Cp′) Uranium Chemistry: Synthetic and Structural Studies of Cp4′U and Cp3′UX (X = Cl, I, Me), Z. Anorg. Allg. Chem., 2017, 643, 2011–2018, DOI:10.1002/zaac.201700323.
- N. A. Siladke, C. L. Webster, J. R. Walensky, M. K. Takase, J. W. Ziller, D. J. Grant, L. Gagliardi and W. J. Evans, Actinide Metallocene Hydride Chemistry: C–H Activation in Tetramethylcyclopentadienyl Ligands to Form [μ–η5–C5Me3H(CH2)–κC]2– Tuck-over Ligands in a Tetrathorium Octahydride Complex, Organometallics, 2013, 32, 6522–6531, DOI:10.1021/om4008482.
- C. J. Kuehl, R. E. Da Re, B. L. Scott, D. E. Morris and K. D. John, Toward New Paradigms in Mixed-Valency: Ytterbocene-Terpyridine Charge-Transfer Complexes, Chem. Commun., 2003, 3, 2336–2337, 10.1039/b306484k.
- R. E. Da Re, C. J. Kuehl, M. G. Brown, R. C. Rocha, E. D. Bauer, K. D. John, D. E. Morris, A. P. Shreve and J. L. Sarrao, Electrochemical and Spectroscopic Characterization of the Novel Charge-Transfer Ground State in Diimine Complexes of Ytterbocene, Inorg. Chem., 2003, 42, 5551–5559, DOI:10.1021/ic030069i.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. Mcinnes and D. P. Mills, Actinide Covalency Measured by Pulsed Electron Paramagnetic Resonance Spectroscopy, Nat. Chem., 2016, 9, 578–583, DOI:10.1038/nchem.2692.
- R. R. Langeslay, G. P. Chen, C. J. Windorff, A. K. Chan, J. W. Ziller, F. Furche and W. J. Evans, Synthesis, Structure, and Reactivity of the Sterically Crowded Th3+ Complex (C5Me5)3Th Including Formation of the Thorium Carbonyl, [(C5Me5)3Th(CO)][BPh4], J. Am. Chem. Soc., 2017, 139, 3387–3398, DOI:10.1021/jacs.6b10826.
- A. B. Altman, A. C. Brown, G. Rao, T. D. Lohrey, R. D. Britt, L. Maron, S. G. Minasian, D. K. Shuh and J. Arnold, Chemical Structure and Bonding in a Thorium(III)-Aluminum Heterobimetallic Complex, Chem. Sci., 2018, 9, 4317–4324, 10.1039/c8sc01260a.
- D. N. Huh, S. Roy, J. W. Ziller, F. Furche and W. J. Evans, Isolation of a Square-Planar Th(III) Complex: Synthesis and Structure of [Th(OC6H2tBu2-2,6-Me-4)4]1−, J. Am. Chem. Soc., 2019, 141, 12458–12463, DOI:10.1021/jacs.9b04399.
- J. S. Parry, F. G. N. Cloke, S. J. Coles and M. B. Hursthouse, Synthesis and Characterization of the First Sandwich Complex of Trivalent Thorium: A Structural Comparison with the Uranium Analogue, J. Am. Chem. Soc., 1999, 121, 6867–6871, DOI:10.1021/ja9903633.
- J. R. Walensky, R. L. Martin, J. W. Ziller and W. J. Evans, Importance of Energy Level Matching for Bonding in Th3+–Am3+ Actinide Metallocene Amidinates, (C5Me5)2[iPrNC(Me)NiPr]An, Inorg. Chem., 2010, 49, 10007–10012, DOI:10.1021/ic1013285.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Expanding Thorium Hydride Chemistry Through Th2+, Including the Synthesis of a Mixed-Valent Th4+/Th3+ Hydride Complex, J. Am. Chem. Soc., 2016, 138, 4036–4045, DOI:10.1021/jacs.5b11508.
- D. N. Huh, J. W. Ziller and W. J. Evans, Chelate-Free Synthesis of the U(II) Complex, [(C5H3(SiMe3)2)3U]1−, Using Li and Cs Reductants and Comparative Studies of La(II) and Ce(II) Analogs, Inorg. Chem., 2018, 57, 11809–11814, DOI:10.1021/acs.inorgchem.8b01966.
-
CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Press, 97th edn, 2016 Search PubMed.
-
L. R. Morss, Comparative Thermochemical and Oxidation-Reduction Properties of Lanthanides and Actinides, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneider Jr, L. Eyring, G. R. Choppin and G. H. Lander, Elsevier Science, Amsterdam, 1994 Search PubMed.
- S. G. Bratsch and J. J. Lagowski, Actinide Thermodynamic Predictions. 3. Thermodynamics of Compounds and Aquo Ions of the 2+, 3+, and 4+ Oxidation States and Standard Electrode Potentials at 298.15 K, J. Phys. Chem., 1986, 90, 307–312, DOI:10.1021/j100274a021.
- Q.-Y. Wu, J.-H. Lan, C.-Z. Wang, Z.-P. Cheng, Z.-F. Chai, J. K. Gibson and W.-Q. Shi, Paving the Way for the Synthesis of a Series of Divalent Actinide Complexes: A Theoretical Perspective, Dalton Trans., 2016, 45, 3102–3110, 10.1039/C5DT04540A.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2068541–2068544. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01906f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Jeffrey M.
Barlow
,
Jeffrey M.
Barlow
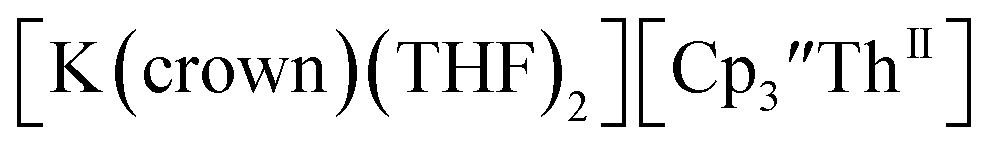 directly from
directly from 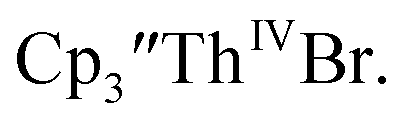 UV-visible spectroelectrochemical measurements and reactions of
UV-visible spectroelectrochemical measurements and reactions of 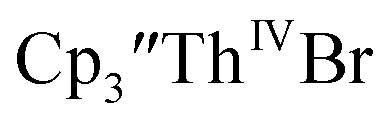 with elemental barium indicated that the thorium system undergoes sequential one electron transformations.
with elemental barium indicated that the thorium system undergoes sequential one electron transformations.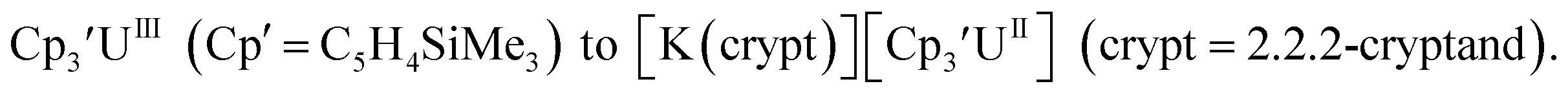 1 Subsequently, the tris(cyclopentadienyl) complexes
1 Subsequently, the tris(cyclopentadienyl) complexes 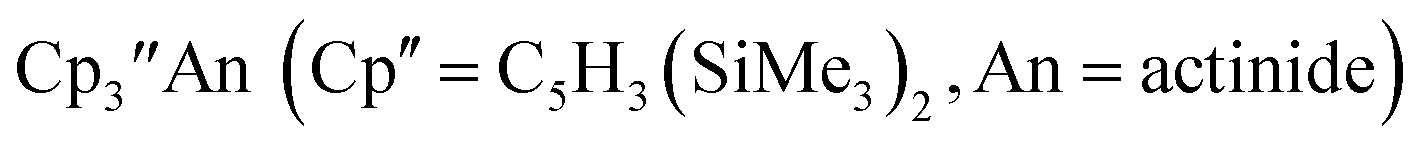 proved to be good precursors for the first examples of crystallographically-characterizable molecular compounds containing Th(II),2 Np(II),3–5 and Pu(II),6eqn (1). Examples of U(II) are now known in different coordination environments beyond the tris(cyclopentadienyl) ligand sets of eqn (1).7–9
proved to be good precursors for the first examples of crystallographically-characterizable molecular compounds containing Th(II),2 Np(II),3–5 and Pu(II),6eqn (1). Examples of U(II) are now known in different coordination environments beyond the tris(cyclopentadienyl) ligand sets of eqn (1).7–9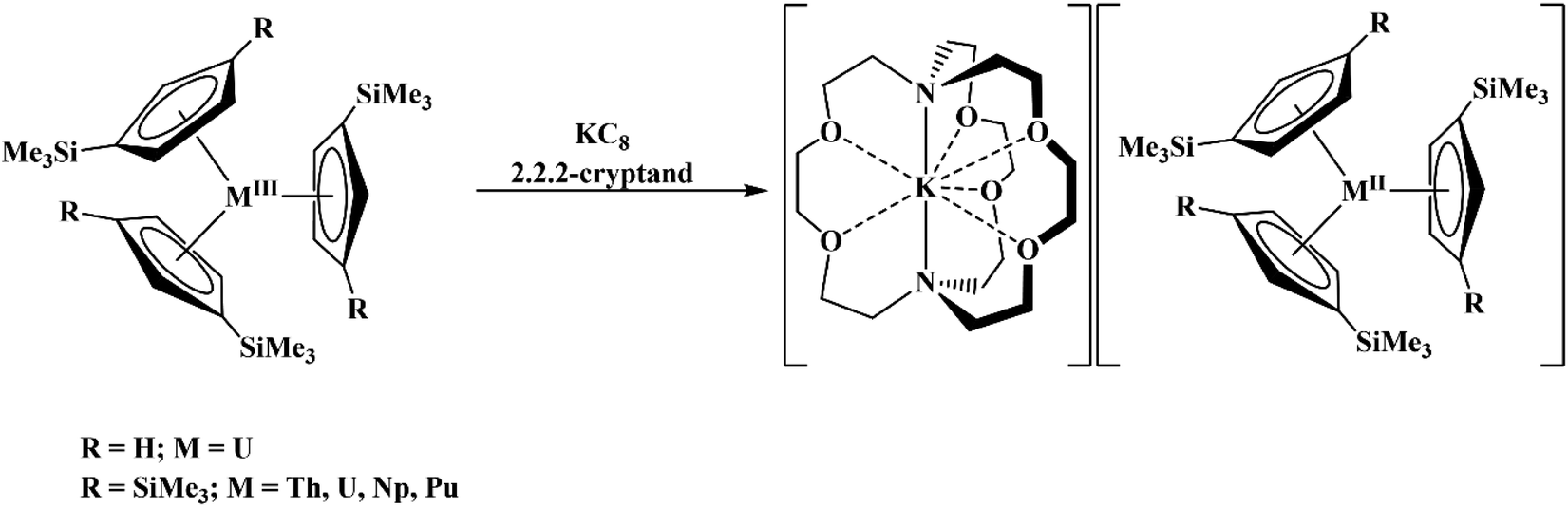
 using [nBu4N][B(C6F5)4] as supporting electrolyte.15
using [nBu4N][B(C6F5)4] as supporting electrolyte.15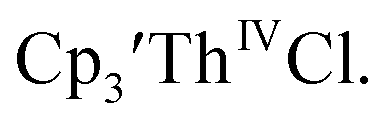 18
18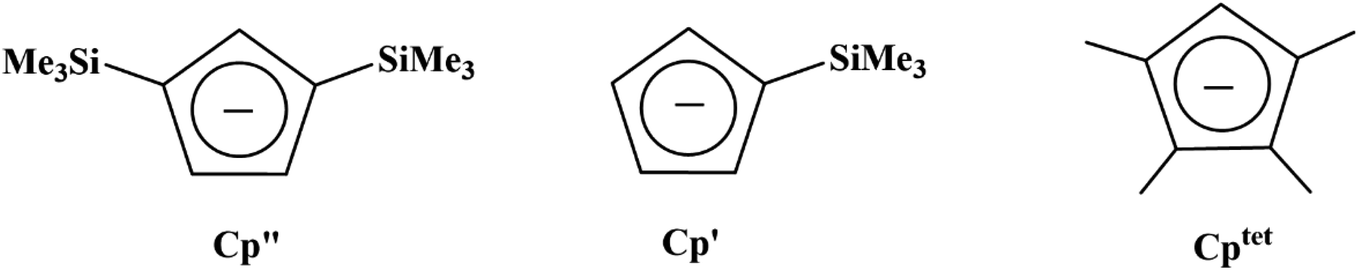
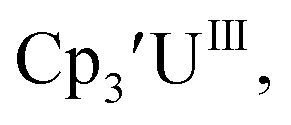 25
25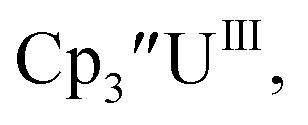 26 and Cptet3UIII,26 and for the isolated U(II) complexes
26 and Cptet3UIII,26 and for the isolated U(II) complexes 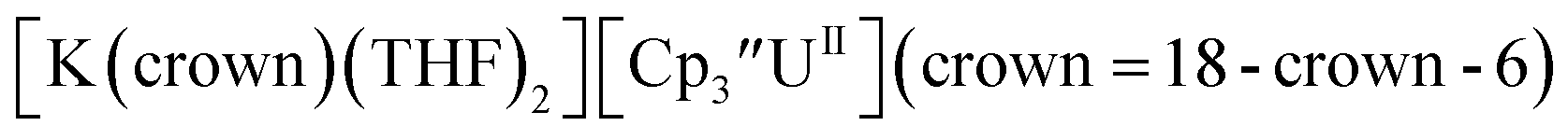 27 and
27 and 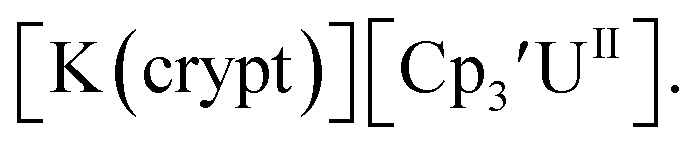 1 These values are summarized in Tables 1 and 2 and highlights are described in the following paragraphs.
1 These values are summarized in Tables 1 and 2 and highlights are described in the following paragraphs.
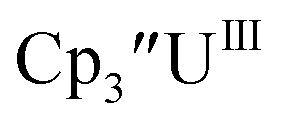
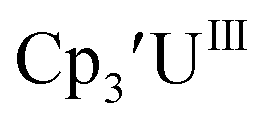

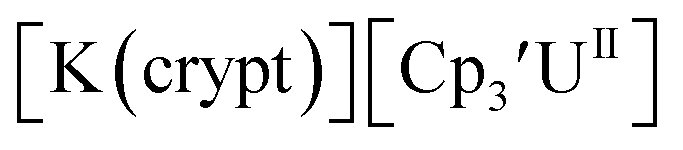
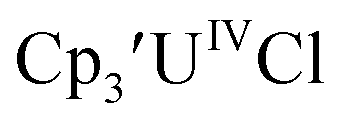
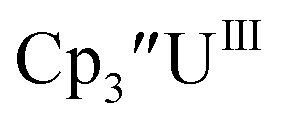
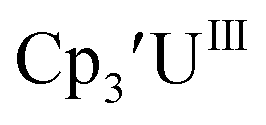

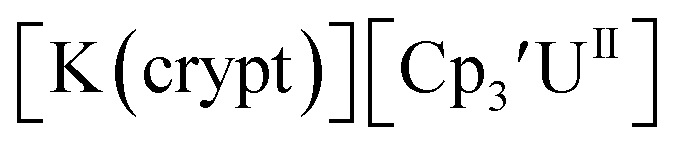
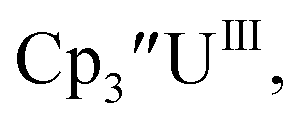 Fig. 1 and S14.† In comparison, the isolated U(II) complex
Fig. 1 and S14.† In comparison, the isolated U(II) complex  27 displays two redox events at −0.73 V and −2.71 V, Fig. 1 and S25.† The E1/2 values for the U(III)/U(II) couple are nearly identical in both systems and the event centered at −2.71 V only appears when scanning anodically for
27 displays two redox events at −0.73 V and −2.71 V, Fig. 1 and S25.† The E1/2 values for the U(III)/U(II) couple are nearly identical in both systems and the event centered at −2.71 V only appears when scanning anodically for 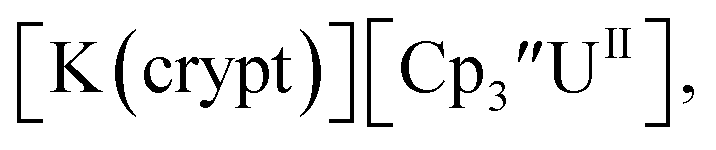 which supports the assignment as the U(III)/U(II) couple.
which supports the assignment as the U(III)/U(II) couple.
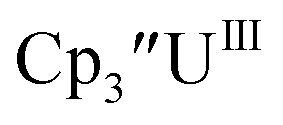 (solid) and 3.0 mM
(solid) and 3.0 mM  (dashed) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe.
(dashed) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe.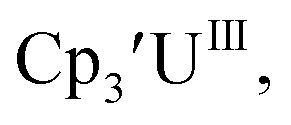 Fig. 2 and S17.† Likewise, the U(IV)/U(III) and U(III)/U(II) couples were observed at −1.28 V and −2.27 V for the U(II) complex
Fig. 2 and S17.† Likewise, the U(IV)/U(III) and U(III)/U(II) couples were observed at −1.28 V and −2.27 V for the U(II) complex 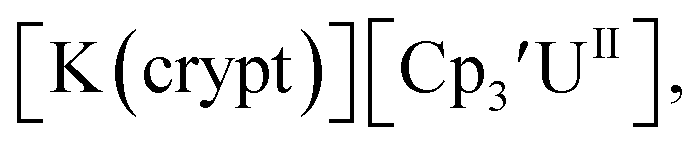 Fig. 2 and S24.† These data were obtained with 50 mM [nBu4N][BPh4] because decomposition occurred at higher electrolyte concentrations. The event at −2.27 V for
Fig. 2 and S24.† These data were obtained with 50 mM [nBu4N][BPh4] because decomposition occurred at higher electrolyte concentrations. The event at −2.27 V for 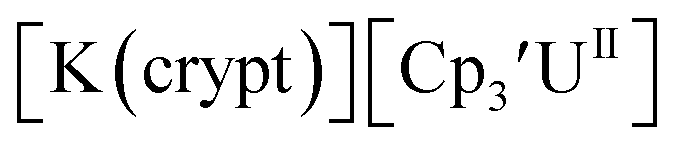 only appears when scanning anodically. The −2.27 V E1/2 value for
only appears when scanning anodically. The −2.27 V E1/2 value for 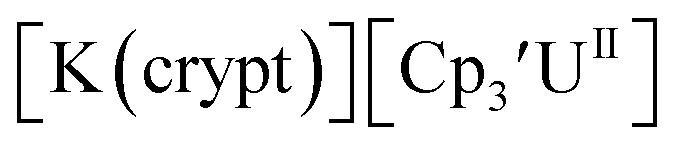 was less negative than the −2.71 V value for
was less negative than the −2.71 V value for 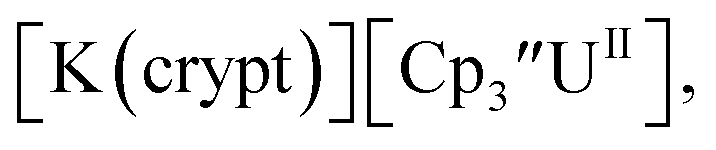 but it is similar to the two previously reported U(III)/U(II) couples for [(Ad,MeArO)3mes]UIII and (C5iPr5)2UII.9,16 The minor unassigned events at about −1.9 V in Fig. 2 and S24† attest to the complexity of the system. They were observed across multiple runs and do not disappear after repeated recrystallization of substrate and electrolyte.
but it is similar to the two previously reported U(III)/U(II) couples for [(Ad,MeArO)3mes]UIII and (C5iPr5)2UII.9,16 The minor unassigned events at about −1.9 V in Fig. 2 and S24† attest to the complexity of the system. They were observed across multiple runs and do not disappear after repeated recrystallization of substrate and electrolyte.
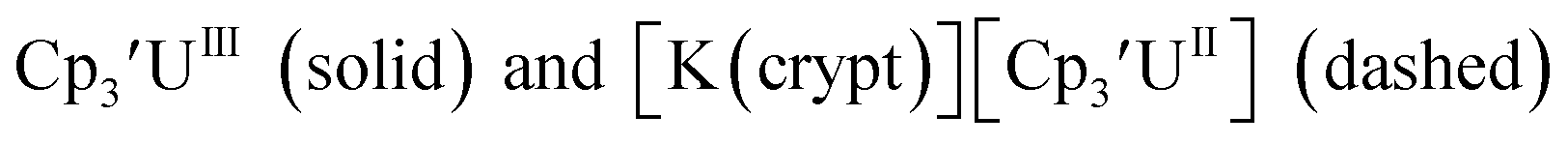 at ν = 200 mV s−1, in 50 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe.
at ν = 200 mV s−1, in 50 mM [nBu4N][BPh4]/THF. The event centered at −0.495 V is due to internal standard (C5Me5)2Fe.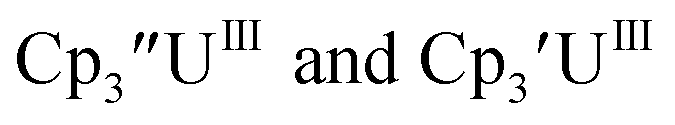 : −1.46 V and −3.11 V, Fig. 3 and S20.† However, data could not be obtained from the isolated U(II) compound [K(crypt)][Cptet3UII] because contact with the supporting electrolyte led to immediate decomposition. The voltammogram obtained from the resulting solution displayed at least five redox events, Fig. S29.† This reactivity is consistent with the more strongly reducing nature of the Cptet complexes as shown by the data in Tables 1 and 2. A third, minor event at −1.7 V was present and cannot be assigned with confidence.
: −1.46 V and −3.11 V, Fig. 3 and S20.† However, data could not be obtained from the isolated U(II) compound [K(crypt)][Cptet3UII] because contact with the supporting electrolyte led to immediate decomposition. The voltammogram obtained from the resulting solution displayed at least five redox events, Fig. S29.† This reactivity is consistent with the more strongly reducing nature of the Cptet complexes as shown by the data in Tables 1 and 2. A third, minor event at −1.7 V was present and cannot be assigned with confidence.
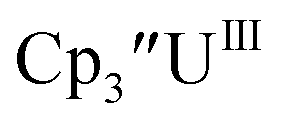 (dashed, 100 mM [nBu4N][BPh4]/THF) and 11 mM
(dashed, 100 mM [nBu4N][BPh4]/THF) and 11 mM 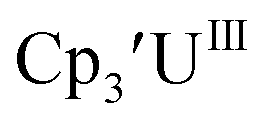 (dotted, 50 mM [nBu4N][BPh4]/THF) at ν = 200 mV s−1. The events centered at −0.495 V are due to internal standard (C5Me5)2FeII.
(dotted, 50 mM [nBu4N][BPh4]/THF) at ν = 200 mV s−1. The events centered at −0.495 V are due to internal standard (C5Me5)2FeII.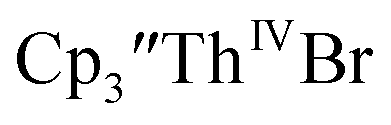
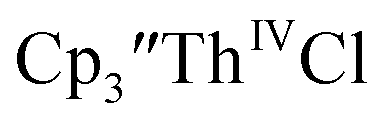
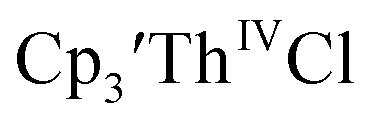
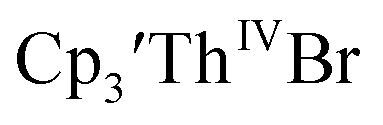
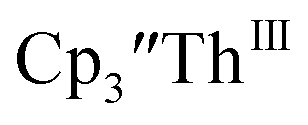
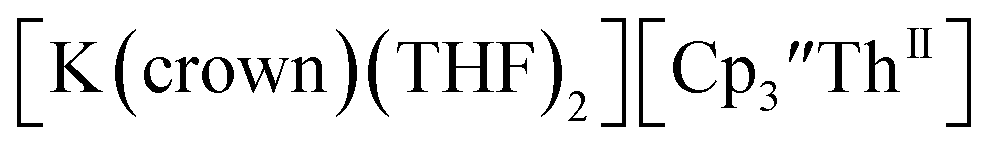
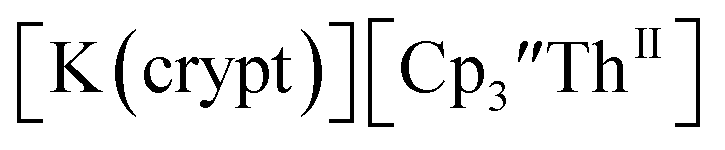
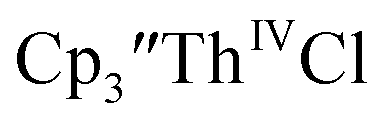 was examined to compare with the values previously reported by Cloke et al.15 The cyclic voltammogram of
was examined to compare with the values previously reported by Cloke et al.15 The cyclic voltammogram of 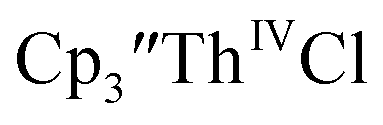 under our conditions shows the Th(IV)/Th(III) couple at −2.93 V, Fig. S34,† which is close to the value of −2.96 V reported for
under our conditions shows the Th(IV)/Th(III) couple at −2.93 V, Fig. S34,† which is close to the value of −2.96 V reported for 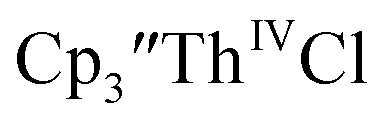 and
and 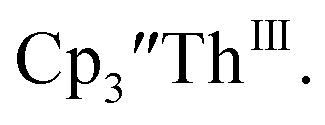 15 Similarly, the cyclic voltammogram of
15 Similarly, the cyclic voltammogram of 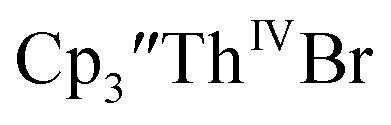 (ref. 2) shows a Th(IV)/Th(III) redox couple at −2.89 V, Fig. 4 and S30.† This suggests that the identity of halide does not significantly affect the reduction potential in this system. This is also consistent with bulk synthetic studies that show that
(ref. 2) shows a Th(IV)/Th(III) redox couple at −2.89 V, Fig. 4 and S30.† This suggests that the identity of halide does not significantly affect the reduction potential in this system. This is also consistent with bulk synthetic studies that show that 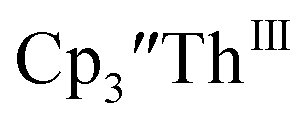 can be synthesized from both
can be synthesized from both 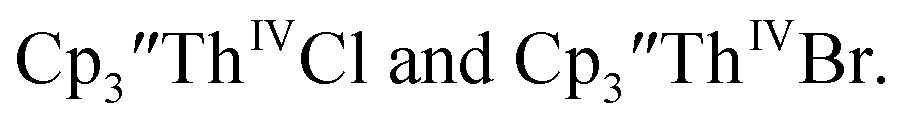 2,33,34
2,33,34
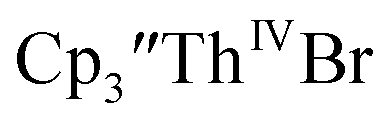 (solid), 15 mM
(solid), 15 mM 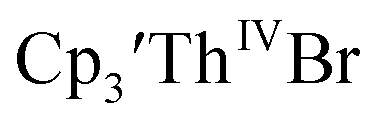 (dashed), and 12 mM Cptet3ThIVBr (dotted) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF.
(dashed), and 12 mM Cptet3ThIVBr (dotted) at ν = 200 mV s−1, in 100 mM [nBu4N][BPh4]/THF.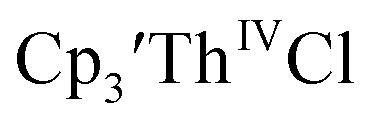 35 and Cptet3ThIVBr (ref. 36) were also examined as each these complexes can be chemically reduced to form tris(cyclopentadienyl)Th(III) species.18,36 The cyclic voltammogram of
35 and Cptet3ThIVBr (ref. 36) were also examined as each these complexes can be chemically reduced to form tris(cyclopentadienyl)Th(III) species.18,36 The cyclic voltammogram of 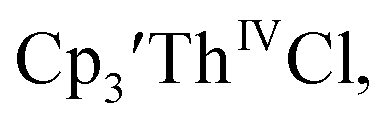 35 Fig. S38,† exhibited a cathodic event at −3.14 V that is 0.21 V more negative than that of
35 Fig. S38,† exhibited a cathodic event at −3.14 V that is 0.21 V more negative than that of 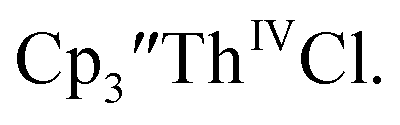 Similarly, the voltammogram of
Similarly, the voltammogram of 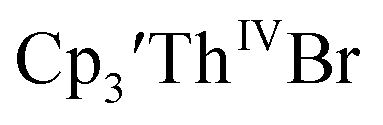 had a cathodic event at −3.17 V, Fig. 4 and S63.† This event was determined to be a one electron process by comparing the current passed to that of the internal standard, Fig. S65.† The voltammogram of Cptet3ThIVBr had a cathodic event at −3.48 V, Fig. 4 and S44.† The events in the voltammograms of
had a cathodic event at −3.17 V, Fig. 4 and S63.† This event was determined to be a one electron process by comparing the current passed to that of the internal standard, Fig. S65.† The voltammogram of Cptet3ThIVBr had a cathodic event at −3.48 V, Fig. 4 and S44.† The events in the voltammograms of 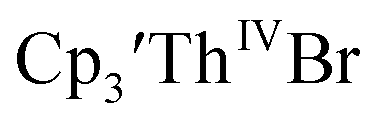 and Cptet3ThIVBr are practically irreversible even at scan rates up to 2000 mV s−1. These results, along with the uranium studies above in Table 1, clearly show that the reduction potential of the actinide complex trends with the electron donation strength of the ligand in the order of Cptet > Cp′ > Cp′′.
and Cptet3ThIVBr are practically irreversible even at scan rates up to 2000 mV s−1. These results, along with the uranium studies above in Table 1, clearly show that the reduction potential of the actinide complex trends with the electron donation strength of the ligand in the order of Cptet > Cp′ > Cp′′.
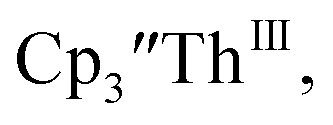 33,34 [C5H3(SiMe2tBu)2]3ThIII,34 Cptet3ThIII,36 (C5tBu2H3)3ThIII,39 and (C5Me5)3ThIII.40 Other Th(III) compounds have been isolated with different ligand environments,41–45 but our initial attempts to collect electrochemical data on (C5Me5)2ThIII[iPrNC(Me)NiPr]44 led to immediate decomposition. Inman and Cloke found that scanning anodically on
33,34 [C5H3(SiMe2tBu)2]3ThIII,34 Cptet3ThIII,36 (C5tBu2H3)3ThIII,39 and (C5Me5)3ThIII.40 Other Th(III) compounds have been isolated with different ligand environments,41–45 but our initial attempts to collect electrochemical data on (C5Me5)2ThIII[iPrNC(Me)NiPr]44 led to immediate decomposition. Inman and Cloke found that scanning anodically on 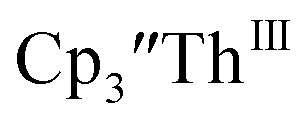 gave a process at −2.96 V that matched the reduction of
gave a process at −2.96 V that matched the reduction of 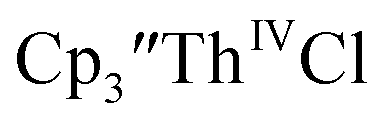 described above and established the Th(IV)/Th(III) couple.15 In our hands, scanning cathodically on
described above and established the Th(IV)/Th(III) couple.15 In our hands, scanning cathodically on 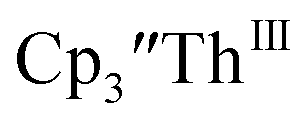 showed a voltammogram with a redox process centered at −2.85 V, Fig. 5 and S40.† A second cathodic event appears after the first cycle at −2.29 V, or when scanning anodically from the open circuit potential, Fig. S40.† The event at −2.29 V was also observed by Cloke and was attributed to a ligand-based event.
showed a voltammogram with a redox process centered at −2.85 V, Fig. 5 and S40.† A second cathodic event appears after the first cycle at −2.29 V, or when scanning anodically from the open circuit potential, Fig. S40.† The event at −2.29 V was also observed by Cloke and was attributed to a ligand-based event.
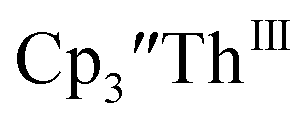 (solid) at ν = 200 mV s−1 and 6.7 mM Cptet3ThIII (dashed) at ν = 400 mV s−1 in 100 mM [nBu4N][BPh4]/THF.
(solid) at ν = 200 mV s−1 and 6.7 mM Cptet3ThIII (dashed) at ν = 400 mV s−1 in 100 mM [nBu4N][BPh4]/THF.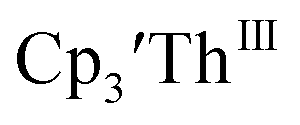 has only been generated in situ,18 it was not studied under the present conditions. The voltammogram of Cptet3ThIII at ν = 200 mV s−1 displays only a cathodic event, but at ν ≥ 400 mV s−1, a return oxidation appears and the Th(III)/Th(II) redox couple is centered at −3.28 V, Fig. 5 and S48.† This value matches the trend observed for the uranium systems in that Cptet complexes of thorium are more difficult to reduce than the silyl-cyclopentadienyl analogs. An anodic event at −1.87 V is present and is attributed to a Cptet-based process.
has only been generated in situ,18 it was not studied under the present conditions. The voltammogram of Cptet3ThIII at ν = 200 mV s−1 displays only a cathodic event, but at ν ≥ 400 mV s−1, a return oxidation appears and the Th(III)/Th(II) redox couple is centered at −3.28 V, Fig. 5 and S48.† This value matches the trend observed for the uranium systems in that Cptet complexes of thorium are more difficult to reduce than the silyl-cyclopentadienyl analogs. An anodic event at −1.87 V is present and is attributed to a Cptet-based process.
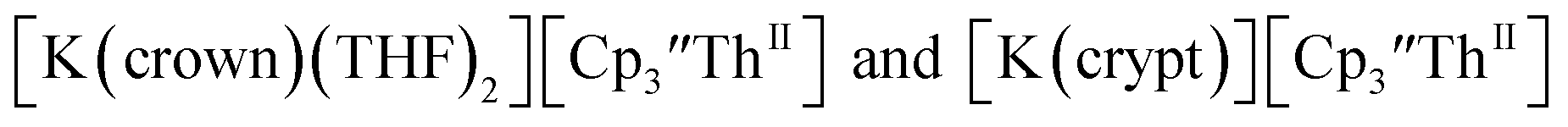 exhibited nearly identical voltammograms. Scanning anodically,
exhibited nearly identical voltammograms. Scanning anodically, 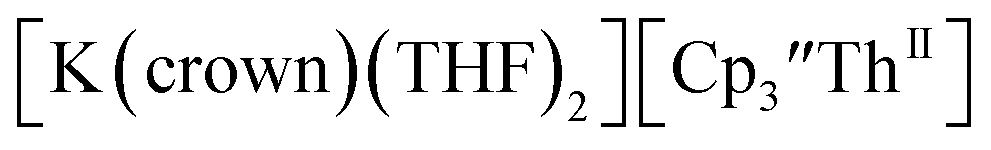 showed a redox process centered at −2.84 V, which is assigned as the Th(III)/Th(II) redox couple, and a second irreversible anodic event at −1.38 V, attributed to ligand-based oxidation, Fig. 6 and S52.† The voltammogram of this Th(II) compound was practically identical over 5 cycles, Fig. S54.†
showed a redox process centered at −2.84 V, which is assigned as the Th(III)/Th(II) redox couple, and a second irreversible anodic event at −1.38 V, attributed to ligand-based oxidation, Fig. 6 and S52.† The voltammogram of this Th(II) compound was practically identical over 5 cycles, Fig. S54.† similarly showed a reversible event centered at −2.85 V and a second anodic event at −1.43 V, Fig. 6, S57 and S61.†
similarly showed a reversible event centered at −2.85 V and a second anodic event at −1.43 V, Fig. 6, S57 and S61.†
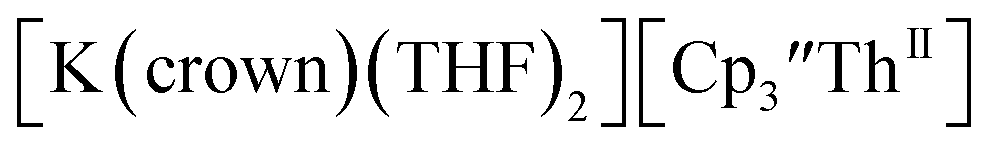 (solid) and 3.1 mM
(solid) and 3.1 mM 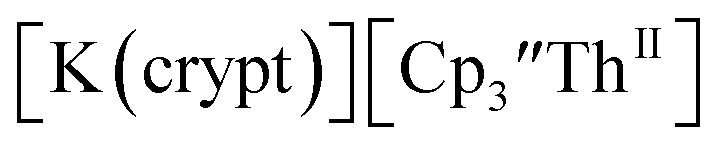 (dashed) with internal standard (C5Me5)2Fe at ν = 200 mV s−1 in 100 mM [nBu4N][BPh4]/THF.
(dashed) with internal standard (C5Me5)2Fe at ν = 200 mV s−1 in 100 mM [nBu4N][BPh4]/THF.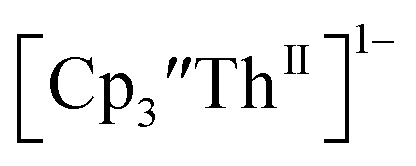 complexes suggested that the Th(III)/Th(II) redox process occurs at about the same potential as the Th(IV)/Th(III) potential of
complexes suggested that the Th(III)/Th(II) redox process occurs at about the same potential as the Th(IV)/Th(III) potential of 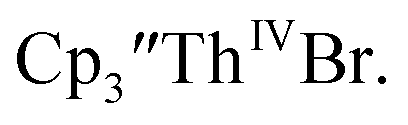 To investigate this further, spectroelectrochemical UV-visible measurements were obtained. A potential of −2.90 V was applied to a solution of
To investigate this further, spectroelectrochemical UV-visible measurements were obtained. A potential of −2.90 V was applied to a solution of 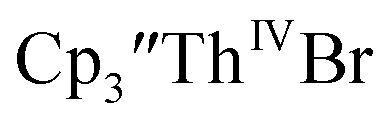 in 200 mM [nBu4N][PF6]/THF and the UV-visible spectrum was recorded approximately every 5 seconds during electrolysis. The formation of
in 200 mM [nBu4N][PF6]/THF and the UV-visible spectrum was recorded approximately every 5 seconds during electrolysis. The formation of 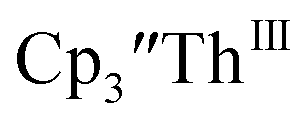 is clearly shown by the growth of four bands at roughly 360, 500, 580, and 680 nm, Fig. 7, which correspond to the absorption spectrum of
is clearly shown by the growth of four bands at roughly 360, 500, 580, and 680 nm, Fig. 7, which correspond to the absorption spectrum of 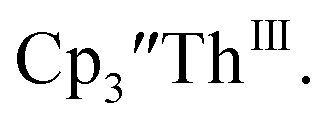 33,34 No further reduction to the
33,34 No further reduction to the 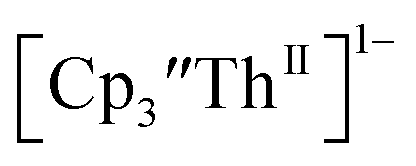 was observed,2 although it cannot be ruled out as the absorbance spectrum reached the maximum of the detector.
was observed,2 although it cannot be ruled out as the absorbance spectrum reached the maximum of the detector.
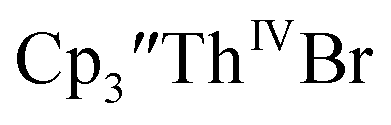 (black, solid) converting to
(black, solid) converting to 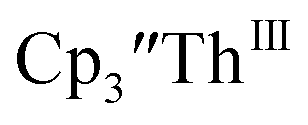 (black, dashed) during electrolysis at −2.90 V with a starting concentration of 7.0 mM in 200 mM [nBu4N][PF6]/THF. The growth of four bands at 365, 510, 590, and 655 nm is indicative of
(black, dashed) during electrolysis at −2.90 V with a starting concentration of 7.0 mM in 200 mM [nBu4N][PF6]/THF. The growth of four bands at 365, 510, 590, and 655 nm is indicative of 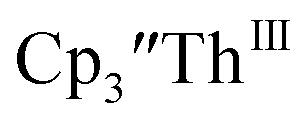 (red).34
(red).34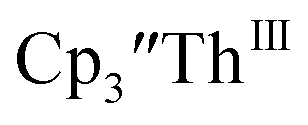 in 200 mM [nBu4N][PF6]/THF at −2.90 V shows clean conversion to the Th(II) species
in 200 mM [nBu4N][PF6]/THF at −2.90 V shows clean conversion to the Th(II) species 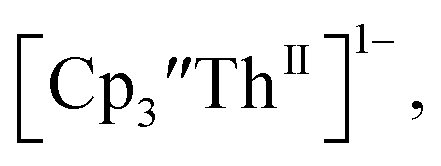 2 as indicated by the growth of the large absorption at 650 nm and the concomitant decrease in absorptions at 360, 500, 580, and 680 nm, Fig. 8. Although the absorption spectrum of
2 as indicated by the growth of the large absorption at 650 nm and the concomitant decrease in absorptions at 360, 500, 580, and 680 nm, Fig. 8. Although the absorption spectrum of 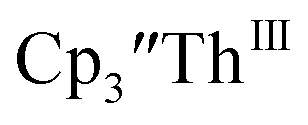 had disappeared, the absorption at 650 nm, indicative of Th(II),2 decreased in intensity as the electrolysis continued. The Th(II) species appears to be unstable under the electrolysis conditions.
had disappeared, the absorption at 650 nm, indicative of Th(II),2 decreased in intensity as the electrolysis continued. The Th(II) species appears to be unstable under the electrolysis conditions.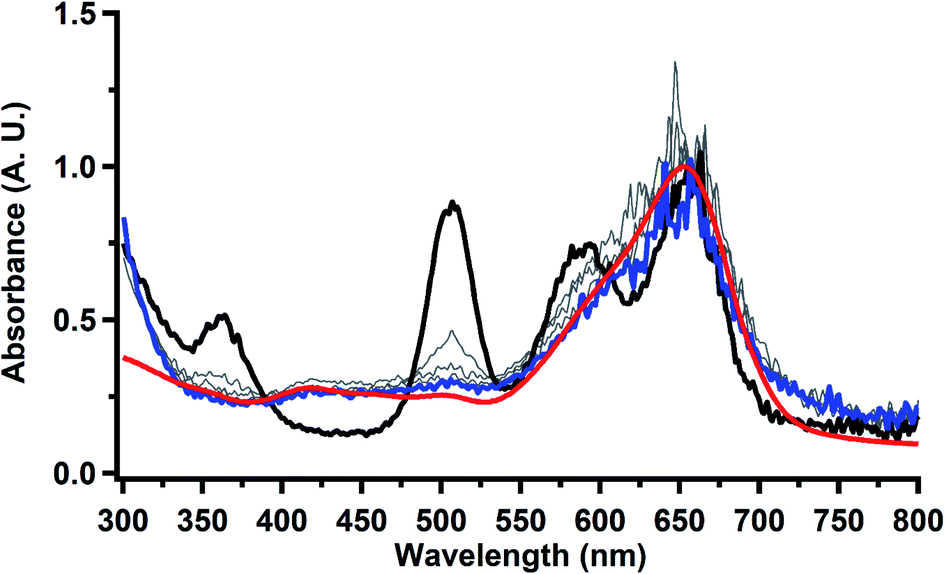
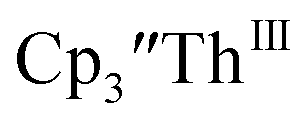 (black) converting to
(black) converting to 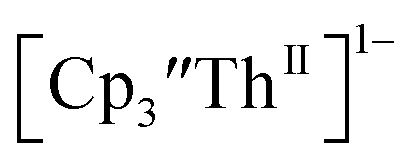 (blue) during electrolysis at −2.90 V with a starting concentration of 1.1 mM in 200 mM [nBu4N][PF6]/THF. The growth of the band at 650 nm is indicative of
(blue) during electrolysis at −2.90 V with a starting concentration of 1.1 mM in 200 mM [nBu4N][PF6]/THF. The growth of the band at 650 nm is indicative of 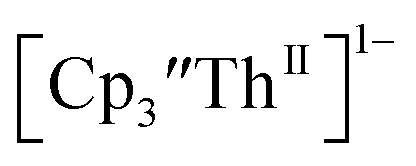 (red).2
(red).2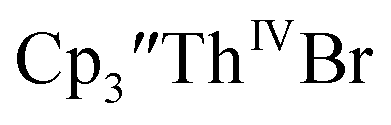 and Th(III)/Th(II) couple in
and Th(III)/Th(II) couple in 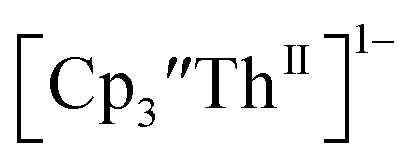 suggested that Th(IV) compounds could be used as the precursors to Th(II) compounds as well as the known Th(III) precursor,
suggested that Th(IV) compounds could be used as the precursors to Th(II) compounds as well as the known Th(III) precursor, 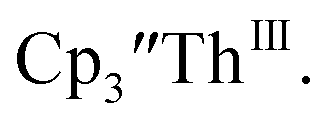 Indeed, reaction of 2.2 equivalents of KC8 to a THF solution of
Indeed, reaction of 2.2 equivalents of KC8 to a THF solution of 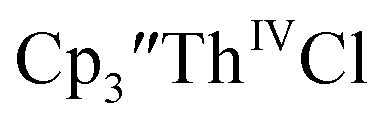 and crown afforded
and crown afforded 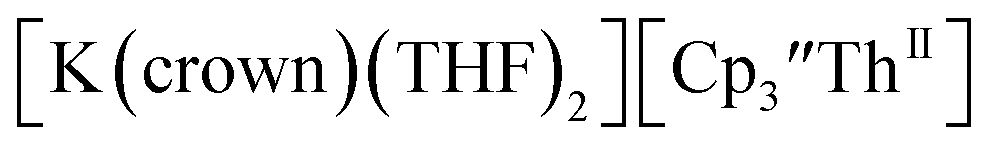 in 50% crystalline yield, with a significant amount of
in 50% crystalline yield, with a significant amount of 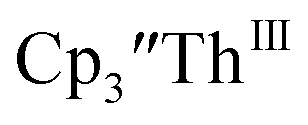 as a byproduct. Previously, Lappert reported that prolonged stirring of a solution of
as a byproduct. Previously, Lappert reported that prolonged stirring of a solution of 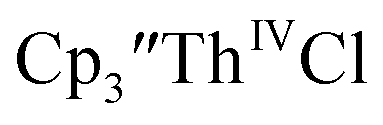 over excess NaK alloy developed a green color,34 which was later confirmed to be the color of Th(II).2
over excess NaK alloy developed a green color,34 which was later confirmed to be the color of Th(II).2
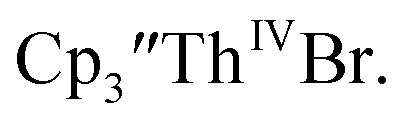 Reaction of
Reaction of 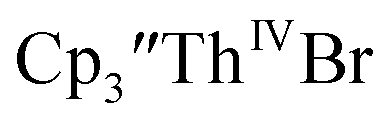 with 2 equivalents of KC8 in THF generated a dark green solution characteristic of Th(II) within 5 minutes, as did reaction of
with 2 equivalents of KC8 in THF generated a dark green solution characteristic of Th(II) within 5 minutes, as did reaction of 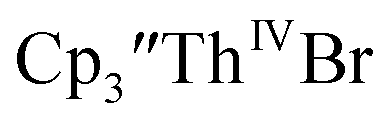 with excess Na and with excess Li. The UV-visible spectra of these solutions have a strong absorption at 650 nm, identical to the previously reported spectra of
with excess Na and with excess Li. The UV-visible spectra of these solutions have a strong absorption at 650 nm, identical to the previously reported spectra of 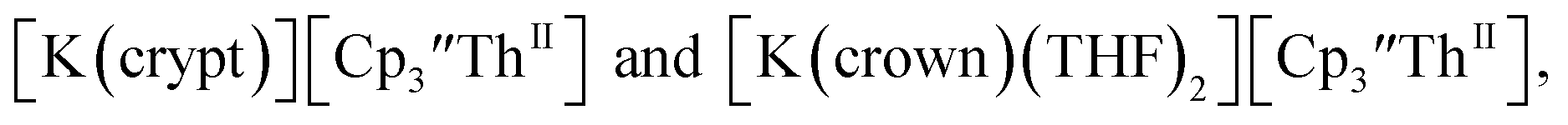 2 but the spectra also show a non-negligible amount of
2 but the spectra also show a non-negligible amount of 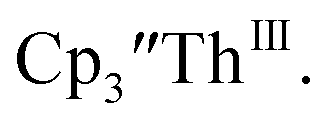 34 Formation of the Th(III) complex is reasonable based on the fact that
34 Formation of the Th(III) complex is reasonable based on the fact that 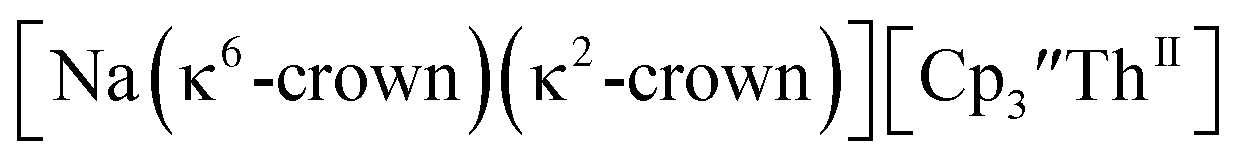 (see below) reacts with
(see below) reacts with 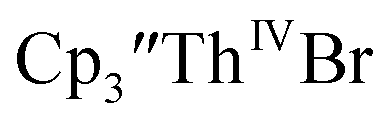 in THF to immediately form
in THF to immediately form 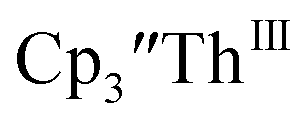 in near quantitative yield.
in near quantitative yield.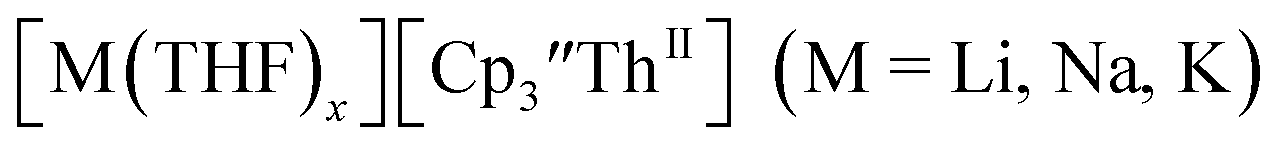 were not isolated even though it is possible to isolate chelate-free examples of
were not isolated even though it is possible to isolate chelate-free examples of 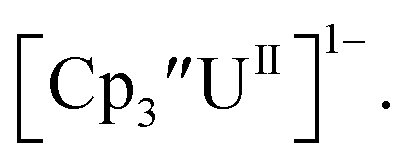 46 Further support for the importance of alkali metal chelates is that addition of 18-crown-6 to the reaction of
46 Further support for the importance of alkali metal chelates is that addition of 18-crown-6 to the reaction of 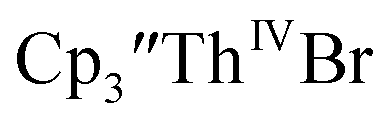 and excess Na provided X-ray quality crystals that were identified as
and excess Na provided X-ray quality crystals that were identified as 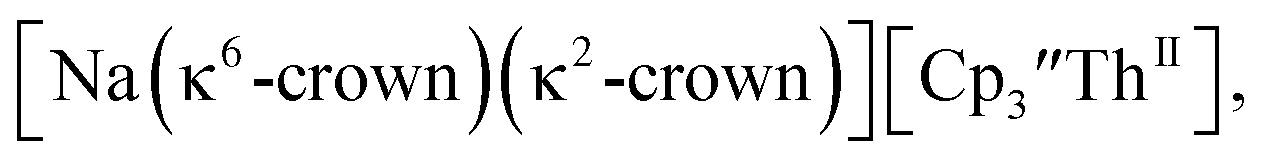 only the third reported crystal structure of a Th(II) complex, Scheme 2, Fig. 9.
only the third reported crystal structure of a Th(II) complex, Scheme 2, Fig. 9.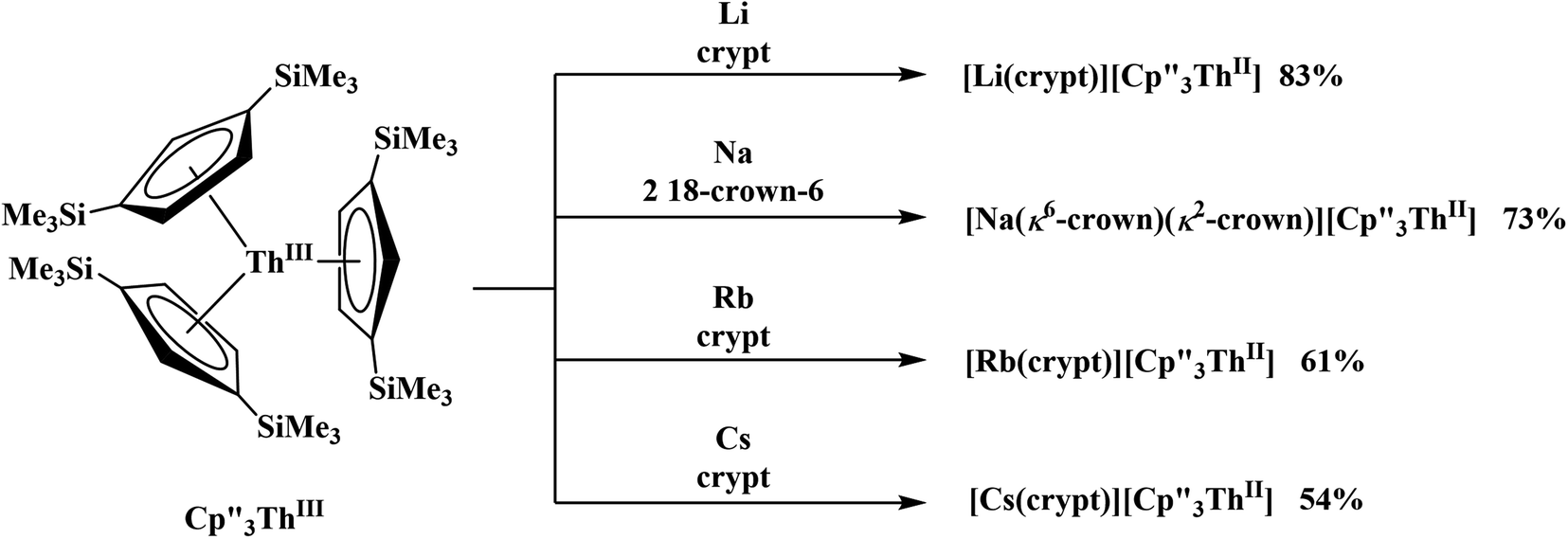
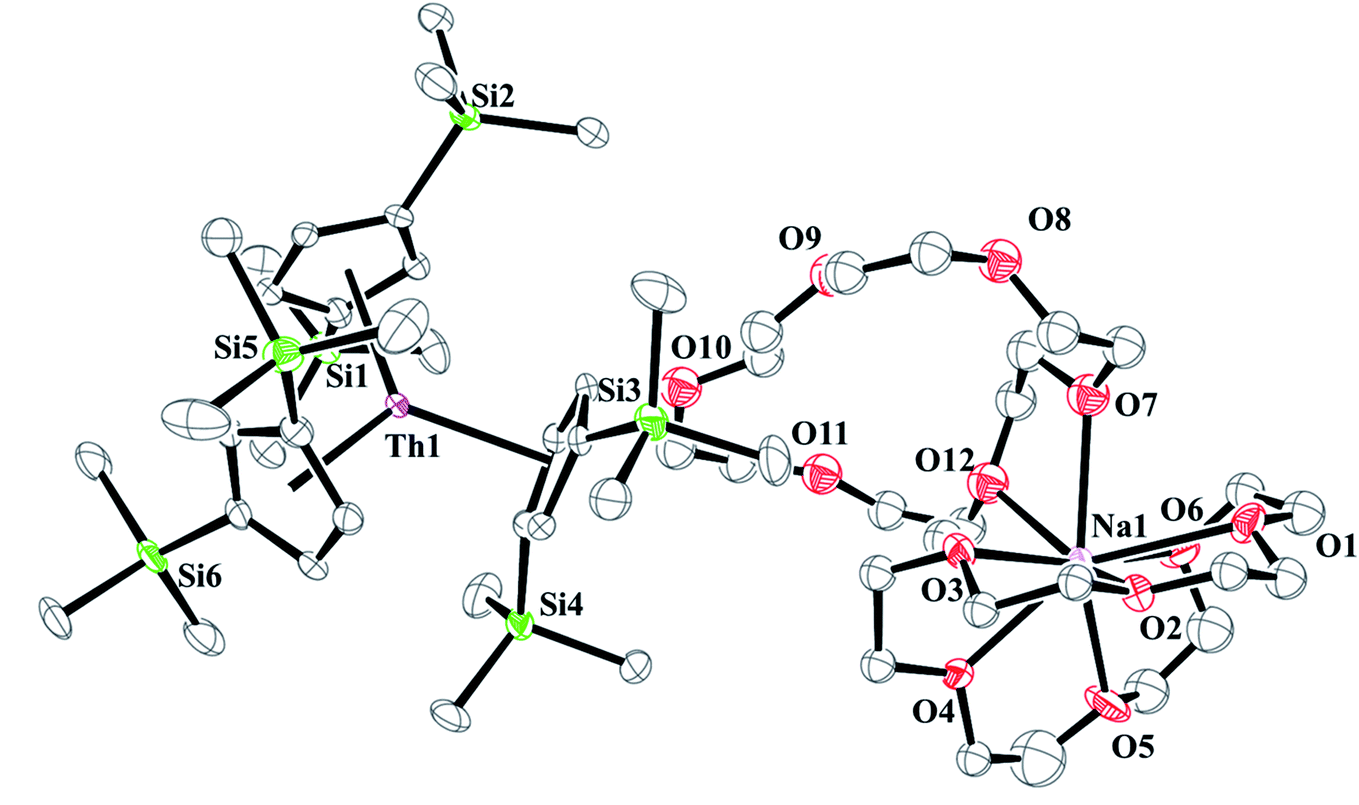
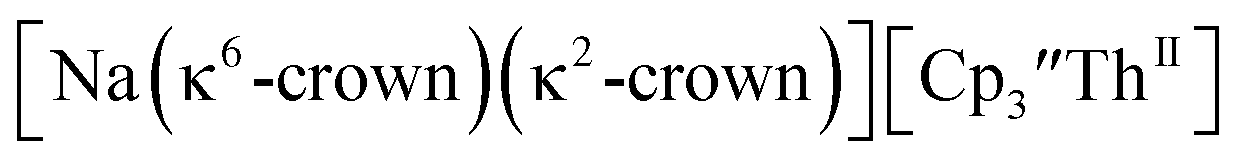 plotted at the 35% probability level. Hydrogen atoms and disorder in the κ2-crown unit have been removed for clarity.
plotted at the 35% probability level. Hydrogen atoms and disorder in the κ2-crown unit have been removed for clarity.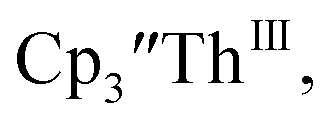 Rb, and crypt in THF afforded dichroic blue/red crystals of
Rb, and crypt in THF afforded dichroic blue/red crystals of 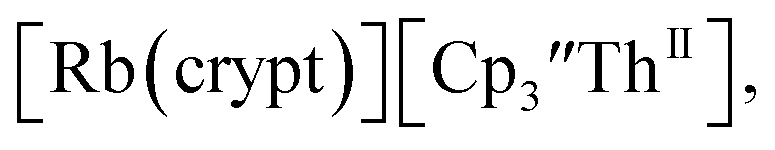 isolated in 61% crystalline yield and identified by X-ray crystallography, Scheme 2, Fig. S69.† In addition, the reaction of
isolated in 61% crystalline yield and identified by X-ray crystallography, Scheme 2, Fig. S69.† In addition, the reaction of 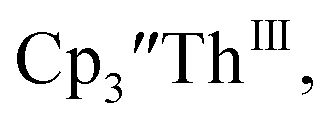 Cs, and crypt afforded dark blue/red crystals of
Cs, and crypt afforded dark blue/red crystals of 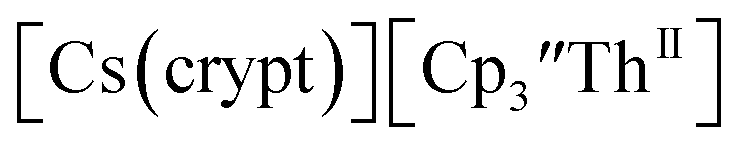 in 54% crystalline yield, Scheme 2, Fig. S70.† The [Rb(crypt)]1+ and [Cs(crypt)]1+ compounds are isomorphous with the [K(crypt)]1+ analog2 and can be easily separated from the
in 54% crystalline yield, Scheme 2, Fig. S70.† The [Rb(crypt)]1+ and [Cs(crypt)]1+ compounds are isomorphous with the [K(crypt)]1+ analog2 and can be easily separated from the 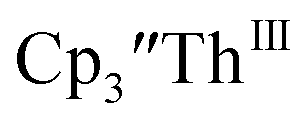 starting material, which was difficult without the use of a chelate. The reaction of
starting material, which was difficult without the use of a chelate. The reaction of 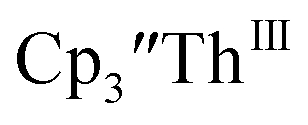 , Li, and crypt formed dark blue-green needles of
, Li, and crypt formed dark blue-green needles of 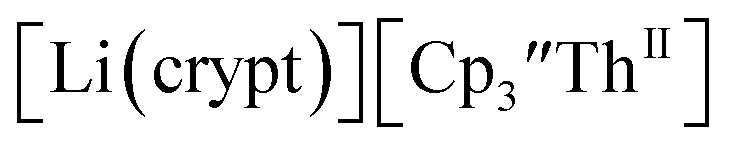 in 83% yield, but the crystals were not suitable for X-ray diffraction, Scheme 2.
in 83% yield, but the crystals were not suitable for X-ray diffraction, Scheme 2.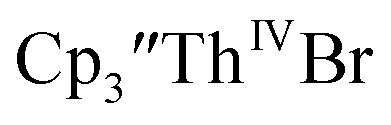 and excess Ba afforded only
and excess Ba afforded only 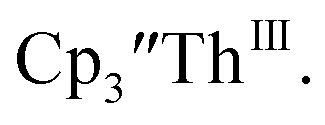 When chelates were added, the reaction of
When chelates were added, the reaction of 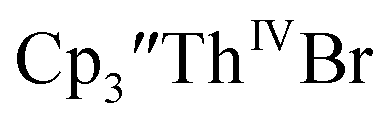 and crown or
and crown or 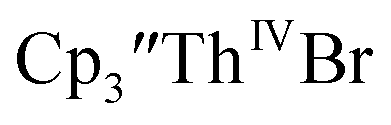 and crypt over excess Ba formed
and crypt over excess Ba formed 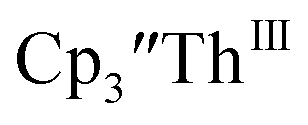 and then the dark green color of Th(II) with UV-visible spectra consistent with
and then the dark green color of Th(II) with UV-visible spectra consistent with 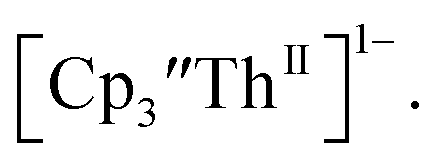 Addition of elemental Hg did not appear to affect the rate of formation of the Th(II) species. These results, coupled with the spectroelectrochemical measurements, strongly suggest that the Th(IV)/Th(II) redox couple is not observed experimentally in these systems and that instead two one-electron processes occur.
Addition of elemental Hg did not appear to affect the rate of formation of the Th(II) species. These results, coupled with the spectroelectrochemical measurements, strongly suggest that the Th(IV)/Th(II) redox couple is not observed experimentally in these systems and that instead two one-electron processes occur.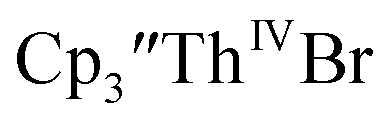 case, UV-visible spectroelectrochemistry measurements show that this compound is reduced under electrochemical conditions to
case, UV-visible spectroelectrochemistry measurements show that this compound is reduced under electrochemical conditions to 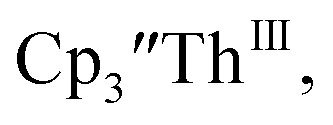 which requires loss of Br1− and geometric reorganization. In the
which requires loss of Br1− and geometric reorganization. In the  case, density functional theory calculations have shown that the putative initial reduction product,
case, density functional theory calculations have shown that the putative initial reduction product, 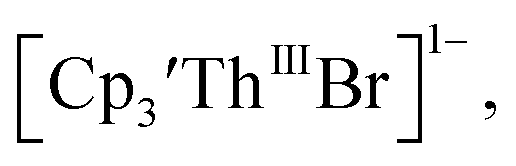 would be unstable with respect to
would be unstable with respect to 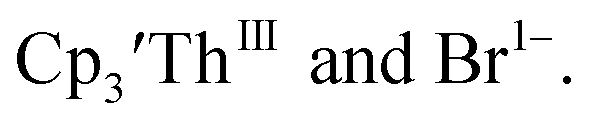 18 These results are consistent with the electrochemical irreversibility of the system.
18 These results are consistent with the electrochemical irreversibility of the system.
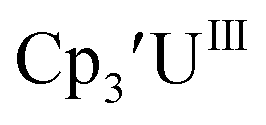 matches well with these two data points, even though
matches well with these two data points, even though 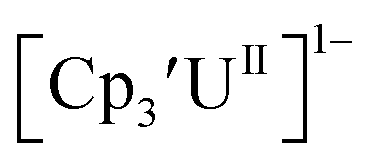 and (C5iPr5)2UII have been assigned 5f36d1 electron configurations,1,9 while {[(Ad,MeArO)3mes]UII}1− is best described as 5f4.7 The −2.73 V reduction potential for
and (C5iPr5)2UII have been assigned 5f36d1 electron configurations,1,9 while {[(Ad,MeArO)3mes]UII}1− is best described as 5f4.7 The −2.73 V reduction potential for 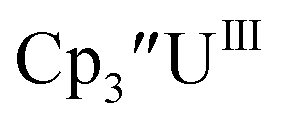 is unexpectedly more reducing than those of these other three complexes. This is also unusual in that solutions of
is unexpectedly more reducing than those of these other three complexes. This is also unusual in that solutions of 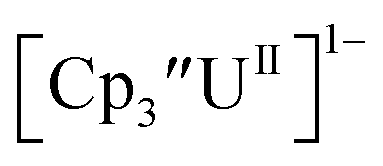 have longer lifetimes than solutions of
have longer lifetimes than solutions of 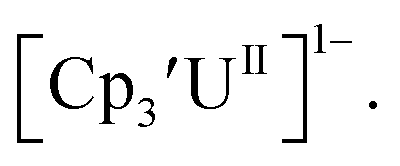 27 The U(III)/U(II) reduction potential for Cptet3UIII was determined to be −3.11 V, which is the most negative reduction potential for these compounds and matches the trend observed for the An(IV)/An(III) couples.
27 The U(III)/U(II) reduction potential for Cptet3UIII was determined to be −3.11 V, which is the most negative reduction potential for these compounds and matches the trend observed for the An(IV)/An(III) couples.
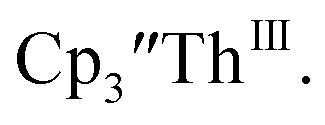 Surprisingly, the Th(IV)/Th(III) couple of
Surprisingly, the Th(IV)/Th(III) couple of  appears to be about the same as the value for the Th(III)/Th(II) couple of
appears to be about the same as the value for the Th(III)/Th(II) couple of 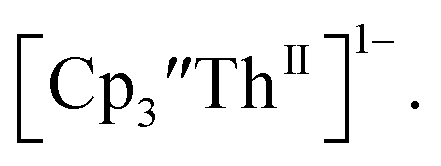 This result was tested chemically and it was found that reduction of Th(IV) with excess reducing agent would form Th(II) compounds directly with KC8, Na, Li, and Ba both with and without the use of a chelating agent. Blue
This result was tested chemically and it was found that reduction of Th(IV) with excess reducing agent would form Th(II) compounds directly with KC8, Na, Li, and Ba both with and without the use of a chelating agent. Blue 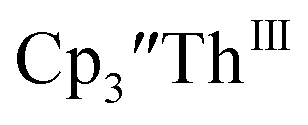 is observed as an intermediate in these reactions which indicates formation of the Th(II) products arises from two one-electron reductions. Furthermore, the E1/2 values for Th(III)/Th(II) match the expected trend compared to uranium based on previously calculated An(III)/An(II) reduction potentials.48–50
is observed as an intermediate in these reactions which indicates formation of the Th(II) products arises from two one-electron reductions. Furthermore, the E1/2 values for Th(III)/Th(II) match the expected trend compared to uranium based on previously calculated An(III)/An(II) reduction potentials.48–50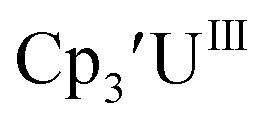 is similar to the two previously reported U(III)/U(II) values, but it is significantly less negative than the Cp′′ analog. The reduction potentials of Th(IV)/Th(III) and Th(III)/Th(II) couples are sufficiently similar that Th(II) complexes can be made directly from Th(IV) precursors without the need to isolate the Th(III) intermediate.
is similar to the two previously reported U(III)/U(II) values, but it is significantly less negative than the Cp′′ analog. The reduction potentials of Th(IV)/Th(III) and Th(III)/Th(II) couples are sufficiently similar that Th(II) complexes can be made directly from Th(IV) precursors without the need to isolate the Th(III) intermediate.