
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom helices to superhelices: hierarchical assembly of homochiral van der Waals 1D coordination polymers†
Guo-Guo
Weng
a,
Ben-Kun
Hong
 b,
Song-Song
Bao
a,
Yujie
Wen
c,
Lan-Qing
Wu
a,
Xin-Da
Huang
a,
Jia-Ge
Jia
a,
Ge-Hua
Wen
a,
Shu-Hua
Li
b,
Luming
Peng
c and
Li-Min
Zheng
*a
b,
Song-Song
Bao
a,
Yujie
Wen
c,
Lan-Qing
Wu
a,
Xin-Da
Huang
a,
Jia-Ge
Jia
a,
Ge-Hua
Wen
a,
Shu-Hua
Li
b,
Luming
Peng
c and
Li-Min
Zheng
*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Centre of Advanced Microstructures, Nanjing University, Nanjing 210023, People's Republic of China. E-mail: lmzheng@nju.edu.cn
bInstitute of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, People's Republic of China
cKey Laboratory of Mesoscopic Chemistry of MOE, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, People's Republic of China
First published on 18th August 2021
Abstract
Chiral transcription from the molecular level to the macroscopic level by self-organization has been a topic of considerable interest for mimicking biological systems. Homochiral coordination polymers (CPs) are intriguing systems that can be applied in the construction of artificial helical architectures, but they have scarcely been explored to date. Herein, we propose a new strategy for the generation of superhelices of 1D CPs by introducing flexible cyclohexyl groups on the side chains to simultaneously induce interchain van der Waals interactions and chain misalignment due to conformer interconversion. Superhelices of S- or R-Tb(cyampH)3·3H2O (S-1H, R-1H) [cyampH2 = S- or R-(1-cyclohexylethyl)aminomethylphosphonic acid] were obtained successfully, the formation of which was found to follow a new type of “chain-twist-growth” mechanism that had not been described previously. The design strategy used in this work may open a new and general route to the hierarchical assembly and synthesis of helical CP materials.
Introduction
The helical architectures of biomolecules play indispensable roles in biological functions, such as molecular recognition, enzyme catalysis and duplication.1 Chiral transcription from the molecular level to the macroscopic level by self-assembly in artificial systems has been a topic of considerable interest not only for increasing the understanding of construction processes in nature but also for developing new functional materials. In recent decades, numerous chiral helical structures based on supramolecular assemblies,2–4 organic polymers,5–7 and inorganic materials8,9 have been generated via chiral recognition and transcription processes.Coordination polymers (CPs), which are composed of metal ion nodes and organic linkers, exhibit many advantages over pure organic systems. Homochiral CPs are of particular interest because of their potential applications in chiral recognition and separation, asymmetric catalysis and multifunctional materials.10–14 Constructing homochiral CPs with helical morphologies is an intriguing route for the development of new materials that possess both chirality and interesting physical and chemical properties. However, although metal ions have been found to trigger the formation of helical structures in supramolecular systems and affect the handedness of these structures,15–18 homochiral CPs with helical morphologies remain elusive.19–22 Only a few examples have been described to date, including helical ribbons based on Ca(II)/Ln(III)–cholate systems,19,20 helical nanobelts based on the Ag(I)–cysteine system,21 and superhelices based on the Tb(III)–pempH2 system, where pempH2 represents R- or S-(1-phenylethylamino)methylphosphonic acid.22 Understanding the formation mechanism of macroscopic helical CPs is, therefore, highly desirable for the development of this field.
The formation mechanisms of the known helical structures of CPs can be classified into two types. First, the “layer-curl-growth” mechanism concerns the formation of coordination or supramolecular layers, followed by distortion of the layer strips to form helical structures. This mechanism has been observed for Ca/Ln–cholate and Ag–cysteine systems.19–21 Second, the “chain-twist-growth” mechanism involves the formation of one-dimensional (1D) coordination chains, followed by twisting of the chain as a consequence of geometric incompatibility. This geometric incompatibility can be caused by the presence of two closely related but different kinds of chains in proper proportion, for example, one positively charged and one neutral, as observed for the Tb/pempH2 system.22 In principle, despite the limited examples available, both mechanisms may be extended to other coordination systems. However, with respect to the chain-twist-growth mechanism, it is extremely challenging to control the proportion of the two closely related but different kinds of chains to achieve twist growth. This raises the question of whether a helical architecture can form when there exists only one kind of chiral chain, either charged or neutral, in the system.
By referring to different levels of chirality in polymers,23 we propose the chiral transcription of homochiral 1D CPs from the molecular to the morphological level to occur in four stages: (1) generation of a primary chiral structure (configuration chirality) from a ligand that contains stereogenic centres; (2) formation of a secondary chiral structure (helix, conformational chirality) from helical chains via metal–ligand coordination; (3) formation of a tertiary chiral structure (superhelix, phase chirality) through packing of the helical chains; and (4) generation of a quaternary chiral structure upon further aggregation of the superhelices. In reality, the helical CP chains are prone to pack in a parallel manner close to each other, leading to highly ordered crystalline materials. In this case, the transcription from conformational chirality (helix) to phase chirality (superhelix) is blocked. To disrupt the parallel packing of the chiral chains and hence encourage twisted chain growth, at least two requirements must be satisfied. First, the interchain interactions must be sufficiently weak to prevent highly ordered and close packing of the helical chains. Second, a slight mismatch between neighbouring chains may provide a driving force to induce twisted packing of the chains.
Previous work has demonstrated that helical assemblies of supramolecules or polymers result from the synergistic effect of various intermolecular non-covalent interactions,24 such as hydrogen bonding, π–π stacking, and electrostatic and van der Waals (vdW) interactions.25–28 While hydrogen bonding, π–π stacking and electrostatic interactions have been well recognized in the formation and stabilization of artificial superhelices, including CPs, vdW forces are scarcely the predominant force controlling helix self-assembly.29 The vdW forces refer to weak but general forces acting between neutral atoms or molecules. They arise from spontaneous charge oscillations that induce fluctuations in the electron distribution of closely spaced molecules or atoms. Recent studies have reported the formation of twisted vdW heterostructures of layered inorganic materials driven by a small misalignment between adjacent layers.30,31 Neutral 1D coordination polymers with dominant vdW interactions can be viewed as van der Waals CP chains. Such chains experience very weak interchain interactions and thus meet the first requirement for helical growth. The next question is how to achieve a slight misalignment between neighbouring neutral chains during the growth process.
In chemistry, conformational isomerism is a form of stereoisomerism in which conformers are interconverted by rotation around single bonds. The different conformers often cannot be isolated because they interconvert too rapidly, but specific conformers may be preferred when the rotational energy barriers are high enough to restrict some rotations.32 For example, cyclohexane has three classical conformations, namely, chair, boat and twist-boat, with the chair as the most stable form. We propose that by appending cyclohexane rings along the sides of chiral CP chains, the two requirements for helical chain assembly, i.e., weak vdW interactions between the chains and instant chain misalignment due to the rapid interconversion of cyclohexane conformers, will be satisfied simultaneously.
Herein, we present an unusual example of superhelix formation via self-assembly of homochiral 1D coordination polymers, S- and R-Tb(cyampH)3·3H2O (S-1H, R-1H) [cyampH2 = S- or R-(1-cyclohexylethyl)aminomethylphosphonic acid,33Scheme 1], driven by a small mismatch between neighbouring van der Waals chains. The mismatch arises from the dynamic cyclohexyl groups appended to the chain sides, which show flexible conformations. Inhibition of the conformational flexibility of the cyclohexyl groups by guest molecules such as acetic acid promotes the formation of crystalline S- or R-Tb(cyampH)3·HOAc·2H2O (S-2C, R-2C). Theoretical calculations confirm the indispensable role of HOAc in stabilizing the crystalline material through hydrogen bonds. The design strategy used in this work may open a new and general route to the hierarchical assembly of helical CP materials.
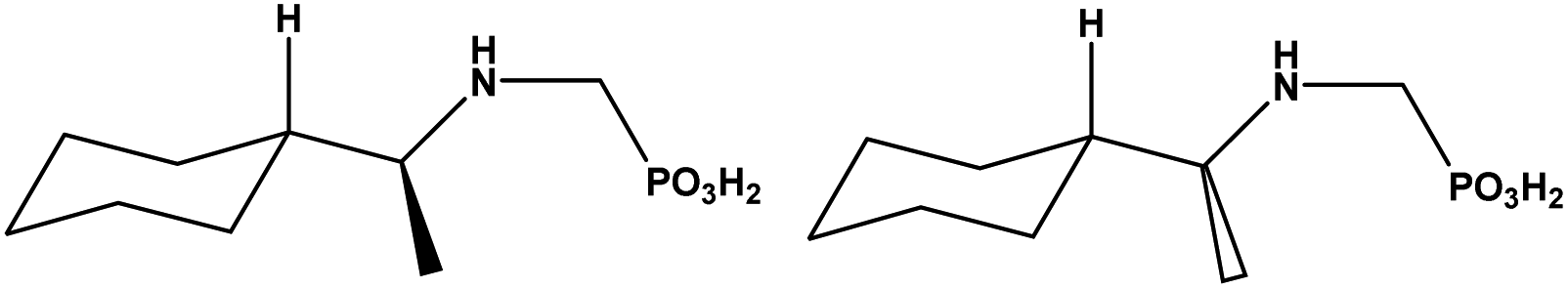 | ||
| Scheme 1 The molecular structures of S-cyampH2 (left) and R-cyampH2 (right). | ||
Results and discussion
Homochiral superhelices: formation, morphology and chirality
Superhelices of S- and R-Tb(cyampH)3·3H2O, named S-1H and R-1H (H refers to helices), were obtained by hydrothermal treatment of a mixture of Tb(OAc)3 and S- or R-cyampH2 (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) in 8.5 mL H2O (initial pH ca. 4.0), adjusted to pH 4.5 using 0.5 M NaOH, at 100 °C for 1 day. After cooling to room temperature, the flocculent precipitates were collected by centrifugation, washed with water and dried in air. The formation of superhelices was found to be highly pH dependent (Fig. S1–S3†). Pure phases of superhelices could be isolated at pH = 4.3–6.5. A lower pH of 3.5–3.7 resulted in rod-like crystals of S- and R-Tb(cyampH)3·HOAc·2H2O, named S-2C and R-2C, where C indicates the crystalline nature. When the pH was in the range of 3.8–4.2, a mixture of rod-like crystals and superhelices was obtained. When the pH was lower than 3.5 or higher than 7, only clear solutions or unidentified powders were observed. The chemical compositions of the superhelices of S-1H and R-1H were characterized by elemental analysis, energy dispersive X-ray (EDX) analyses (Fig. S4†), and TG analyses (Fig. S5†), and the results were in full agreement with the proposed formulae. The formation of helical structures of S-1H and R-1H was anion independent and occurred over the same pH range when other terbium salts, such as Tb(NO3)3 and TbCl3, were used as metal sources (Fig. S6–S9†). The influence of the molar ratio of the starting materials was also studied. A Tb/P molar ratio of 1:5 or 1:6 resulted in superhelices with the same composition and similar morphologies. When the Tb/P molar ratio was 1:1, however, only nanowires were isolated, the composition of which was different from those of S-1H and R-1H (Fig. S10 and S11†).
:3) in 8.5 mL H2O (initial pH ca. 4.0), adjusted to pH 4.5 using 0.5 M NaOH, at 100 °C for 1 day. After cooling to room temperature, the flocculent precipitates were collected by centrifugation, washed with water and dried in air. The formation of superhelices was found to be highly pH dependent (Fig. S1–S3†). Pure phases of superhelices could be isolated at pH = 4.3–6.5. A lower pH of 3.5–3.7 resulted in rod-like crystals of S- and R-Tb(cyampH)3·HOAc·2H2O, named S-2C and R-2C, where C indicates the crystalline nature. When the pH was in the range of 3.8–4.2, a mixture of rod-like crystals and superhelices was obtained. When the pH was lower than 3.5 or higher than 7, only clear solutions or unidentified powders were observed. The chemical compositions of the superhelices of S-1H and R-1H were characterized by elemental analysis, energy dispersive X-ray (EDX) analyses (Fig. S4†), and TG analyses (Fig. S5†), and the results were in full agreement with the proposed formulae. The formation of helical structures of S-1H and R-1H was anion independent and occurred over the same pH range when other terbium salts, such as Tb(NO3)3 and TbCl3, were used as metal sources (Fig. S6–S9†). The influence of the molar ratio of the starting materials was also studied. A Tb/P molar ratio of 1:5 or 1:6 resulted in superhelices with the same composition and similar morphologies. When the Tb/P molar ratio was 1:1, however, only nanowires were isolated, the composition of which was different from those of S-1H and R-1H (Fig. S10 and S11†).
Although S-1H and R-1H gave rise to identical powder X-ray diffraction (PXRD) patterns and infrared (IR) spectra (Fig. S12 and S13†), their mirror image relationship was revealed by their morphologies. The scanning electron microscopy (SEM) images of S-1H showed an exclusively left-handed helical morphology, but superhelices with opposite handedness could not be discerned by SEM (Fig. 1a and S14†). Superhelix aggregates were formed by the intertwining of multiple superhelices. The diameters of the helical aggregates were approximately 2–7 μm, and the lengths ranged from 40 to 100 μm. In contrast, R-1H formed purely right-handed helical structures with similar size parameters (Fig. 1b and S14†). Superhelices of the two materials could also be visualized under an optical microscope, but the handedness could not be clearly identified (Fig. S15†).
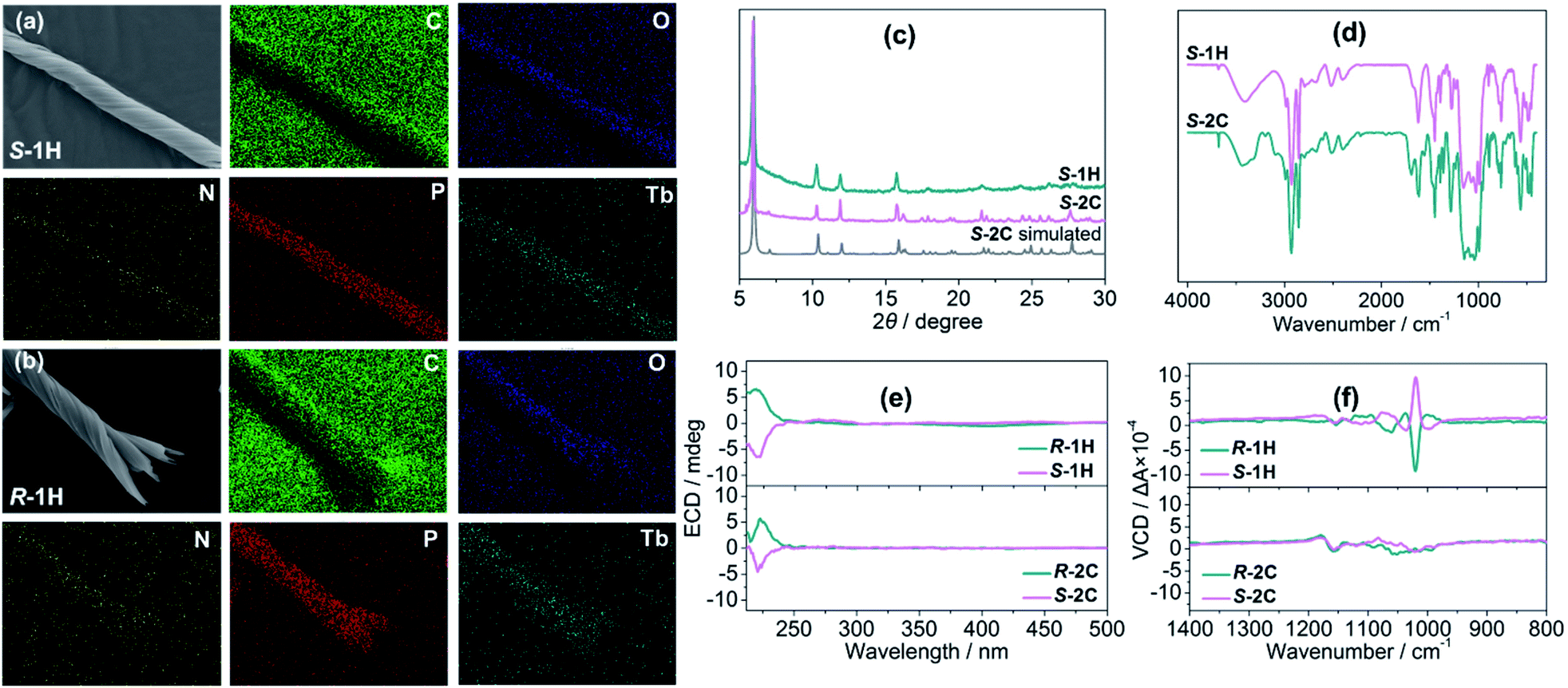 | ||
| Fig. 1 EDX maps of (a) S-1H and (b) R-1H. (c) PXRD patterns of superhelices S-1H and crystals S-2C and S-2C simulated from single crystal data. (d) IR spectra of superhelices S-1H and crystals S-2C, and (e) ECD and (f) VCD spectra of superhelices of R-1H and S-1H and crystals of R-2C and S-2C. | ||
The mirror image relationship of S-1H and R-1H could also be identified from their optical properties. In this context, circular dichroism (CD) is ideal for providing information not only on the molecular scale but also about morphologies.34 Electronic circular dichroism (ECD), which uses electronic transitions as a probe, is often the method of choice. Although the ligands of R- and S-cyampH2 contain no aromatic rings, there appears a weak absorption peak near 220 nm, assigned to the n–π* transitions of the phosphonate oxygen atoms. Both superhelices and crystals of S- and R-isomers show mirror dichroic signals at ca. 220 nm, corroborating the enantiomeric nature of these compounds at the molecular level (Fig. 1e and S16†). Vibrational circular dichroism (VCD), which uses vibrational transitions to probe structures, is the preferred tool to distinguish the chirality of these helical structures. For the S-1H and R-1H helices, the infrared spectra exhibited identical characteristics, with sharp peaks in the 900–1200 cm−1 region, attributed to the stretching vibrations of the –PO3 group (Fig. S13†). The solid-state VCD spectra revealed opposite symmetries at 1190, 1108, 1095, 1074, 1060, 1039, 1020 and 1000 cm−1 (Fig. 1f). In contrast, similar stretching vibrations of opposite symmetries were not observed in the VCD spectra of the rod-like crystals S-2C and R-2C (Fig. 1f); however their IR spectra were similar to those of the helical compounds, except a few additional peaks present at 1690, 1549, and 1357 cm−1, which were attributed to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(C–O), and ν(CH3) vibrations of the lattice acetic acid molecule (Fig. 1d). Thus, the VCD spectra could not identify the opposite symmetries of the R- and S-cyampH2 ligands (Fig. S17†). These results imply that the chirality was transferred and amplified from the optically pure ligand to the macroscopic tertiary helical structure.
O), ν(C–O), and ν(CH3) vibrations of the lattice acetic acid molecule (Fig. 1d). Thus, the VCD spectra could not identify the opposite symmetries of the R- and S-cyampH2 ligands (Fig. S17†). These results imply that the chirality was transferred and amplified from the optically pure ligand to the macroscopic tertiary helical structure.
Crystal structures of S-, R-Tb(cyampH)3·HOAc·2H2O (S-2C, R-2C)
Accurate structural analysis can provide insight into the formation mechanism of superhelices. Note that crystalline compounds S-2C and R-2C were obtained under the same reaction conditions as the superhelices except the pH; thus, their structures were expected to be closely related to each other. Single-crystal structural analyses performed at 193 K revealed that the S- and R-isomers are enantiomeric, crystallizing in a hexagonal crystal system with space groups P61 and P65, respectively. Therefore, only the structure of S-2C is described in detail as a representative. The asymmetric unit of S-2C is composed of one TbIII ion, three S-cyampH− ligands, one acetic acid molecule, and two water molecules (Fig. 2a). Tb1 adopts a distorted dodecahedral geometry, coordinated by eight oxygen atoms (O1, O2, O1A, O4, O5, O5B, O7, and O8B) from six phosphonate groups (Fig. 2a). The Tb–O bond lengths fall in the range of 2.280(4)–2.518(4) Å, and the O–Tb–O bond angles are in the range of 58.5(1)–156.9(1)°. Two of the three crystallographically distinguished S-cyampH− ligands (P1 and P2) are tridentate, chelating and bridging the Tb atoms through two of the three phosphonate oxygen atoms (O1 and O2, and O4 and O5). The third ligand (P3) is bidentate, bridging the Tb atoms via two phosphonate oxygen atoms (O7 and O8). The uncoordinated phosphonate oxygen atoms (O3, O6, and O9) and the protonated amino groups (N1, N2, and N3) are involved in a hydrogen bonding network. Consequently, the equivalent Tb atoms are connected by two μ3-O(P) (O1 and O5) and one O–P–O unit (O7–P3–O8), forming an infinitely extended helical chain running along the c-axis. The chain contains right-handed triple helical strands composed of –Tb1–O1–P1–O2–Tb1– (purple), –Tb1–O4–P2–O5–Tb1– (pink), and –Tb1–O7–P3–O8–Tb1– (blue) (Fig. 2b). The cyclohexyl groups are attached as pendants on the chain sides. The pitch and diameter of the helical chain are 23.53 and 18.56 Å, respectively. The spacing between the chains is 17.031 Å. Neutral acetic acid and lattice water molecules are embedded in the chain between the cyclohexyl rings (Fig. 2c). Extensive intrachain hydrogen bonds are found among amino nitrogen atoms, phosphonate oxygen atoms, acetic acid and lattice water molecules (Table S3†). Notably, the guest acetic acid molecules are anchored to the chain by the formation of moderately strong H-bonds with phosphonate oxygen O9 (O10⋯O9: 2.517) and weak H-bonds with the cyclohexyl group (C9⋯O11: 3.434 Å and C2⋯O11: 3.418 Å). The latter could reduce the conformation flexibility of the cyclohexyl groups. The interchain interactions are dominated by vdW interactions with the shortest C⋯C distances of 3.76–3.80 Å between cyclohexyl moieties and 3.83–3.95 Å between the methyl group of acetic acid and cyclohexyl group (Fig. 2d).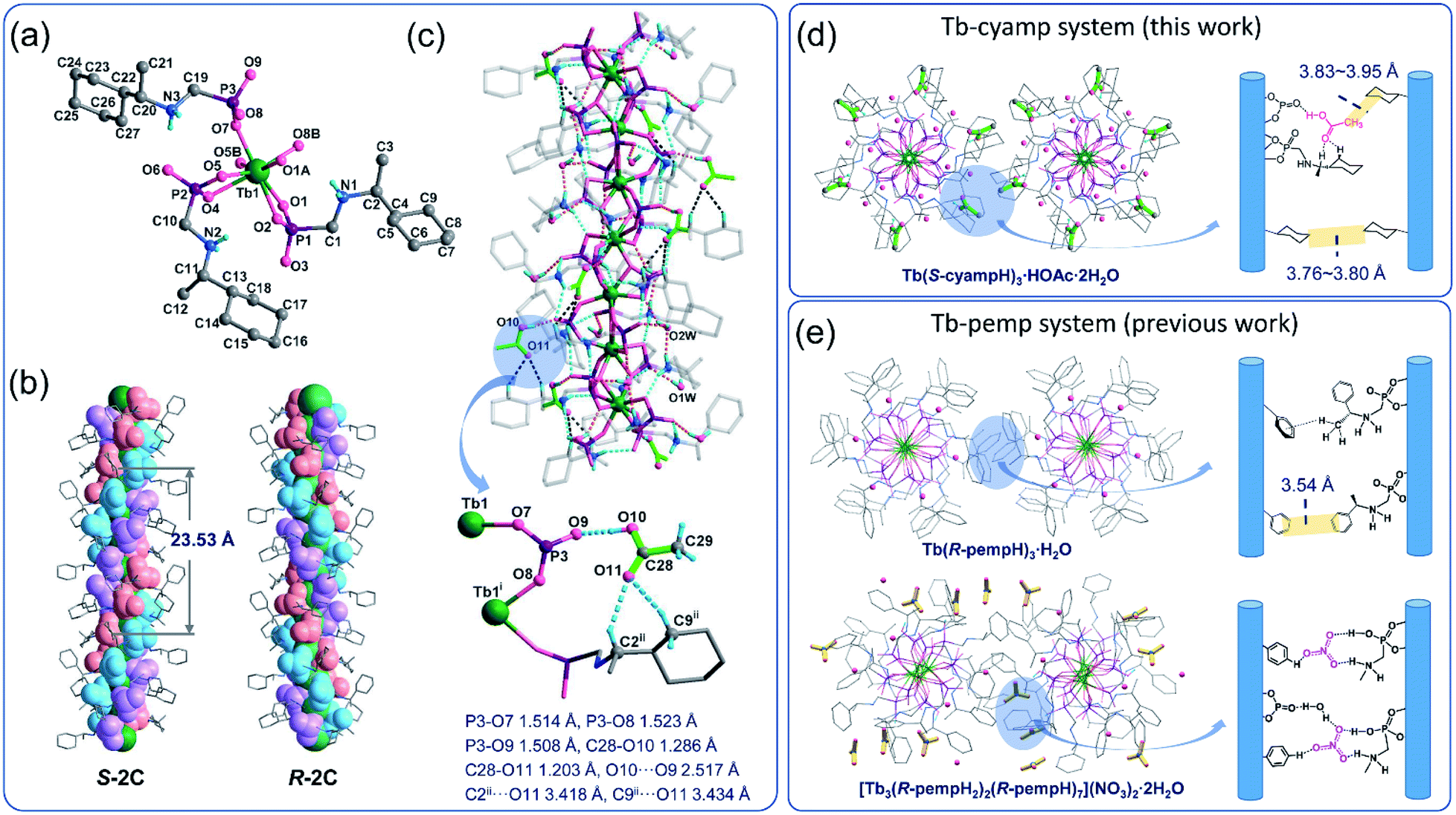 | ||
| Fig. 2 Crystal structure of S-2C from coordinate linkages to weak interactions. (a) The asymmetric unit with atomic labelling. The lattice HOAc and water molecules are omitted for clarity. Symmetry codes: A: x − y, x, z + 1/6; B: y, −x + y, z − 1/6. (b) Side views of the chains in structures S-2C and R-2C which contain right-handed and left-handed triple strands, respectively. (c) One chain of S-2C showing intrachain hydrogen bonds as dotted lines. Symmetry codes: i: x − y, x, z + 1/6; ii: −y, x − y, z + 1/3. (d and e) Comparison of interchain interactions of two adjacent equivalent chains with a list of shortest C⋯C distances between two sides of cyclohexyl or phenyl moieties in Tb–cyamp and Tb–pemp systems, respectively. The HOAc molecules and NO3− anions are highlighted in green and yellow colours, respectively. | ||
To visualize the interactions surrounding the acetic acid molecule in S-2C, the Hirshfeld surface (with respect to the normalized distance) has been generated using Multiwfn,35,36 as well as the corresponding fingerprint plot. As shown in Fig. 3a, the hydrogen bond is the only important interaction between acetic acid and the surrounding molecules; the CO⋯H interactions between carboxyl of acetic acid and the surrounding molecules are weak, and the interactions between the methyl group of acetic acid and the adjacent chains are mainly van der Waals interactions. These conclusions can be confirmed from the fingerprint plot. As shown in Fig. 3b, the fingerprint plot can be roughly decomposed into 3 regions. Combined with Fig. 3c, we can learn how the surface was made. Region 1 corresponds to the strongest hydrogen bond, which makes a total of 4.2% of the surface. Region 2 corresponds to hydrogen bonds generated by the carboxyl and the surrounding molecules, which makes a total of 30.2% of the surface. Region 3 corresponds to the interactions between the H atoms of acetic acid and all the H atoms in the environment, which makes a total of 57.5% of the surface. These conclusions based on Hirshfeld analysis are in good agreement with the quantum chemical calculations (described below) or chemical intuition.
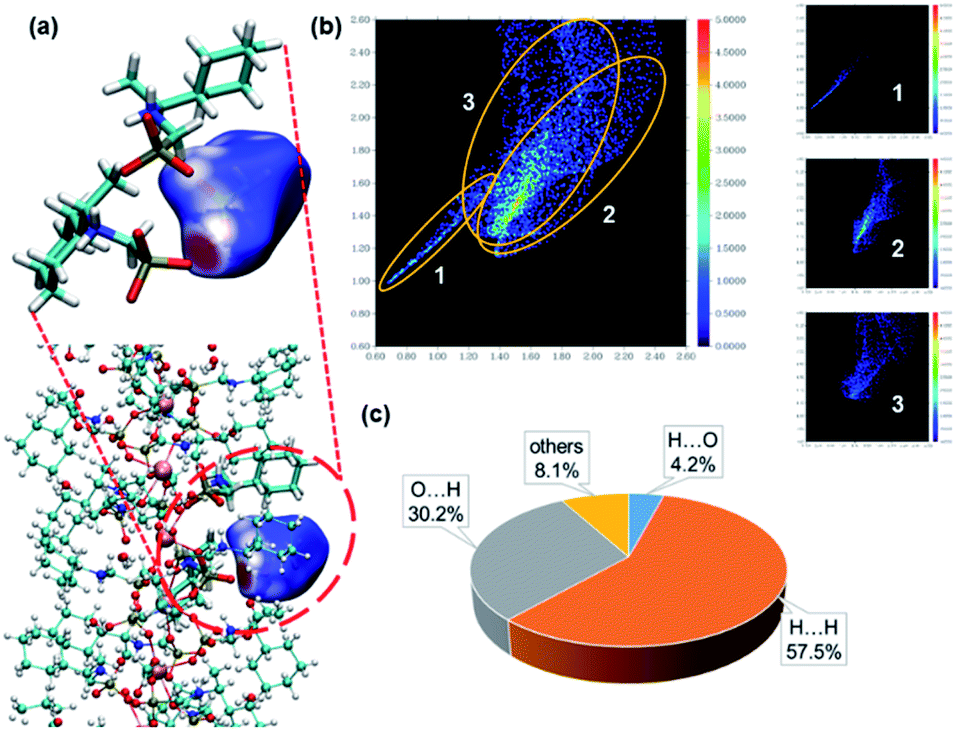 | ||
| Fig. 3 (a) Hirshfeld surface of the acetic acid molecules in S-2C. Colours show the normalized distance (d_norm) to the neighbouring molecules, with red being shorter than the sum of the van der Waals radii, white being approximately the sum and blue being longer than the sum. (b) The two-dimensional Hirshfeld surface fingerprint plot for the acetic acid molecule in S-2C crystal. (c) Individual atomic contact percentage contribution to the Hirshfeld surface of acetic acid. X⋯Y means the interactions, where X atoms are inside the surface (on the acetic acid molecule) and the Y atoms are outside the surface. | ||
The R-isomer R-2C shows a similar chain structure except that each chain contains left-handed triple helical strands (Fig. 2b) in contrast to the right-handed strands in S-2C. The encapsulation of acetic acid molecules induces the weak C–H⋯O hydrogen bonds (C2⋯O11: 3.418 Å, C9⋯O11: 3.441 Å), hence eliminating the conformational flexibility of the cyclohexyl groups and stabilizing the overall crystal lattice. This finding may explain the formation of crystalline R-2C and S-2C.
Comparison of S-2C crystals and S-1H superhelices
The enantiomeric crystalline compounds S-2C and R-2C gave rise to identical PXRD patterns and IR spectra as expected (Fig. S18 and S19†). Interestingly, the PXRD patterns of the crystals were, to a large extent, consistent with those of the helices (Fig. 1c), although their chemical compositions differ in the number of lattice water and acetic acid molecules. A careful comparison between the S-1H superhelices and S-2C crystals revealed that the diffraction peaks of the superhelices at 6.1°, 10.3°, 12.1°, 15.5°, and 15.8° matched well with those of S-2C (6.1°, 10.4°, 12.0°, 15.4°, and 15.9°). However, the width of the diffraction peaks was larger for S-1H (Fig. 1c). In addition, the overall IR spectra of the crystals were also similar to those of the superhelices except for the presence of additional vibrational peaks attributed to the acetic acid guest molecules (Fig. 1d). All these results suggest that the S-1H superhelices are composed of chains similar to those in S-2C, containing right-handed triple helical strands that are entangled to form left-handed superhelices. The broadening of the diffraction peaks was attributed to a decrease in the crystallinity of the superhelices. The diffraction peaks were indexed by using the TOPAS 5.0 program,37 yielding a set of unit cell parameters in space group P61 of a = 17.21 Å, c = 23.77 Å and V = 6094.3 Å3 for S-1H (Fig. S20†). These parameters are similar to those of the crystalline counterpart of S-2C (a = 17.13 Å, c = 23.67 Å and V = 6016.9 Å3), which were indexed using the powder diffraction data at room temperature, but the cell volume of S-1H is much larger than that of S-2C. The increase in the cell volume of S-1H superhelices was related to the larger interchain distance in the superhelices than in the crystalline S-2C, which was possibly caused by the misalignment of the chains during the self-assembly process.A slight difference in the chain packing of the two compounds may be reflected by the motion of hydrogen atoms in their structures. Thus, we performed 1H magic angle spinning (MAS) NMR experiments using the solid samples of S-1H and S-2C. The major resonance in the spectrum of S-2C (Fig. S21†) is a very broad peak (FWHM of ∼50 ppm) centred at approx. 0 ppm, presumably arising from the hydrogen species in the cyclohexyl groups. A large set of spinning sidebands can be observed, indicating the large hyperfine interactions between the 1H species and the unpaired electrons of Tb in S-2C. The centre band of the 1H MAS NMR spectrum of S-1H is broad and similar to that of S-2C; however, the spinning sideband manifolds are much weaker, suggesting that the hyperfine interactions between 1H and the unpaired electrons are significantly reduced. In S-2C, the large hyperfine interactions can explain that acetic acid molecules are anchored to the structure and moderately strong hydrogen bonds are formed. In contrast, the lack of hydrogen bonds in S-1H leads to stronger motion (e.g., rotation of the cyclohexyl groups), decreasing the sizes of hyperfine interactions.381H → 13C cross polarization NMR experiments were also attempted.39 However, no signal was observed for S-2C or S-1H, which can be ascribed to inefficient polarization transfer from 1H to 13C due to the presence of paramagnetic Tb and thus short relaxation times.40
Formation mechanism of superhelices
To study the formation mechanism of these well-defined superhelices, the self-assembly process of the superhelices was explored using both spectroscopy and microscopy by monitoring the products of the hydrothermal reaction of Tb(OAc)3 and S-cyampH2 (pH = 4.5) at 100 °C for different periods of time. Surprisingly, the reaction was very quick. We noticed that a white turbid material formed immediately after mixing solutions of the ligand and metal salt. The amount of white flocculent increased with increasing reaction time. Interestingly, PXRD measurements showed identical patterns for all products (Fig. S22†), although the peaks at 2θ > 10° were very weak for the products at 0 min and 5 min. The IR spectra supported the same conclusion (Fig. S23†). The EDX measurements further confirmed that the molar ratio of Tb:P was 1:3 in all cases (Fig. S24†). All these results suggest the formation of the same material, e.g., superhelices, from the very beginning of the reaction.
SEM images revealed that the turbid material that formed immediately after mixing the two solutions was composed of nanorods with a width and length of approximately 70 nm and 460–470 nm, respectively (Fig. S25†). The nanorods did not have a smooth surface and were determined to have a random-coil conformation. The product at 5 min consisted of similar nanorods without distinct helical morphologies, but the size increased to approximately 100 nm in width and 450–500 nm in length (Fig. 4). Apparently, an increase in the reaction time led to the growth of nanorods both in width and in length.
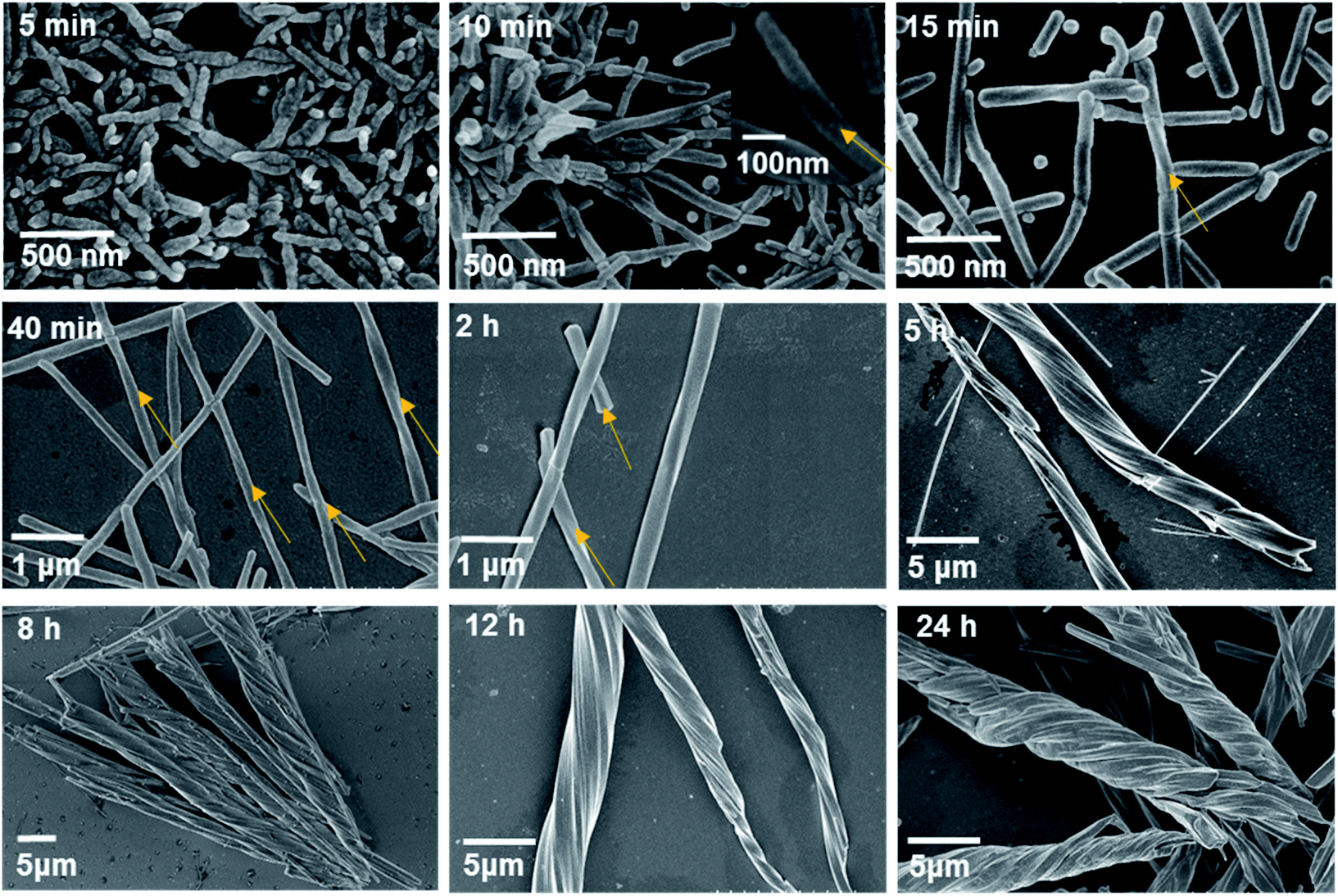 | ||
| Fig. 4 SEM images of the products after hydrothermal reactions of Tb(OAc)3 and S-cyampH2 at pH 4.5 and 100 °C for different periods of time. The yellow arrows represent the direction of chirality. | ||
Longitudinal growth was more favourable than lateral growth, such that the aspect ratio increased with increasing reaction time to approximately 5 at 5 min and 10 at 10 min. This finding is reasonable considering that the interactions within the metal–organic coordination chain are much stronger than those between the chains, which are dominated by vdW interactions.41 Interestingly, a clear twist was observed after reaction for 10 min in the nanorods with diameters of approximately 70–100 nm and lengths of approximately 0.5–1 μm. The helical nanorods grew continuously with the extension of the reaction time. The width and length reached approximately 100 nm and 2 μm for the 15 min product, 140–180 nm and 8 μm for the 40 min product, and approximately 400 nm and 30 μm for the 2 h product. The aspect ratio of the products also increased with increasing reaction time, following the sequence of approximately 20 (at 15 min) <57 (at 40 min) <75 (at 2 h). The final product could be viewed as helical nanofibrils. It was noted that these helical nanorods or nanofibrils have obvious left-handed characteristics, which is contrary to the chiral features at the molecular level of the right-handed three-stranded helix in the crystal structure of S-2C. This phenomenon is similar to the hierarchical structure of collagen formation, the right-handed superhelix of which is composed of three polypeptides with left-handed helical conformations.42 The results show that chirality is transcribed successfully from the secondary chiral structure to the tertiary chiral structure, e.g., from the helix to the superhelix. Moreover, it seems that the superhelices with larger aspect ratios had a strong tendency to cluster together, as evidenced by the formation of branched helical structures with the same handedness after heating for 5 h. Even longer reaction times did not change the helical morphology but resulted in thicker superhelices. After hydrothermal reaction for 8 h and 24 h, the diameters and lengths of the superhelices were approximately 4–6 μm and 46–55 μm, and approximately 4.5–6 μm and 45–60 μm, respectively. The clustering of superhelices may be a result of maximizing the van der Waals contacts, thus minimizing the free energy of the system.43,44
Based on the above experimental results, we propose a possible formation mechanism of the superhelices of S-1H that follows a hierarchical assembly process (Fig. 5). First, a secondary chiral structure containing right-handed three-stranded helices of Tb(S-cyampH)3 is formed. Second, twisted packing of the helices results in the formation of left-handed superhelices. Finally, the superhelices are clustered to form branched helical structures of the same handedness. On a deeper level, the formation of superhelices of S-1H is a natural result of two combined effects: weak vdW interactions between the chiral chains on the one hand and mismatch of chain alignments caused by rapid interconversion of different conformers of nonplanar cyclohexyl groups on the other hand. Therefore, this superhelix formation process follows a new kind of “chain-twist-growth” mechanism concerning only one kind of neutral chain. When guest molecules such as acetic acid were introduced, the conformational flexibility of the cyclohexyl groups was largely restricted due to the presence of hydrogen bonds between the carboxylate oxygen atoms and the cyclohexyl groups. As a consequence, ordered parallel accumulation of the chiral chains was favoured, leading to the formation of rod-like crystals of S-2C.
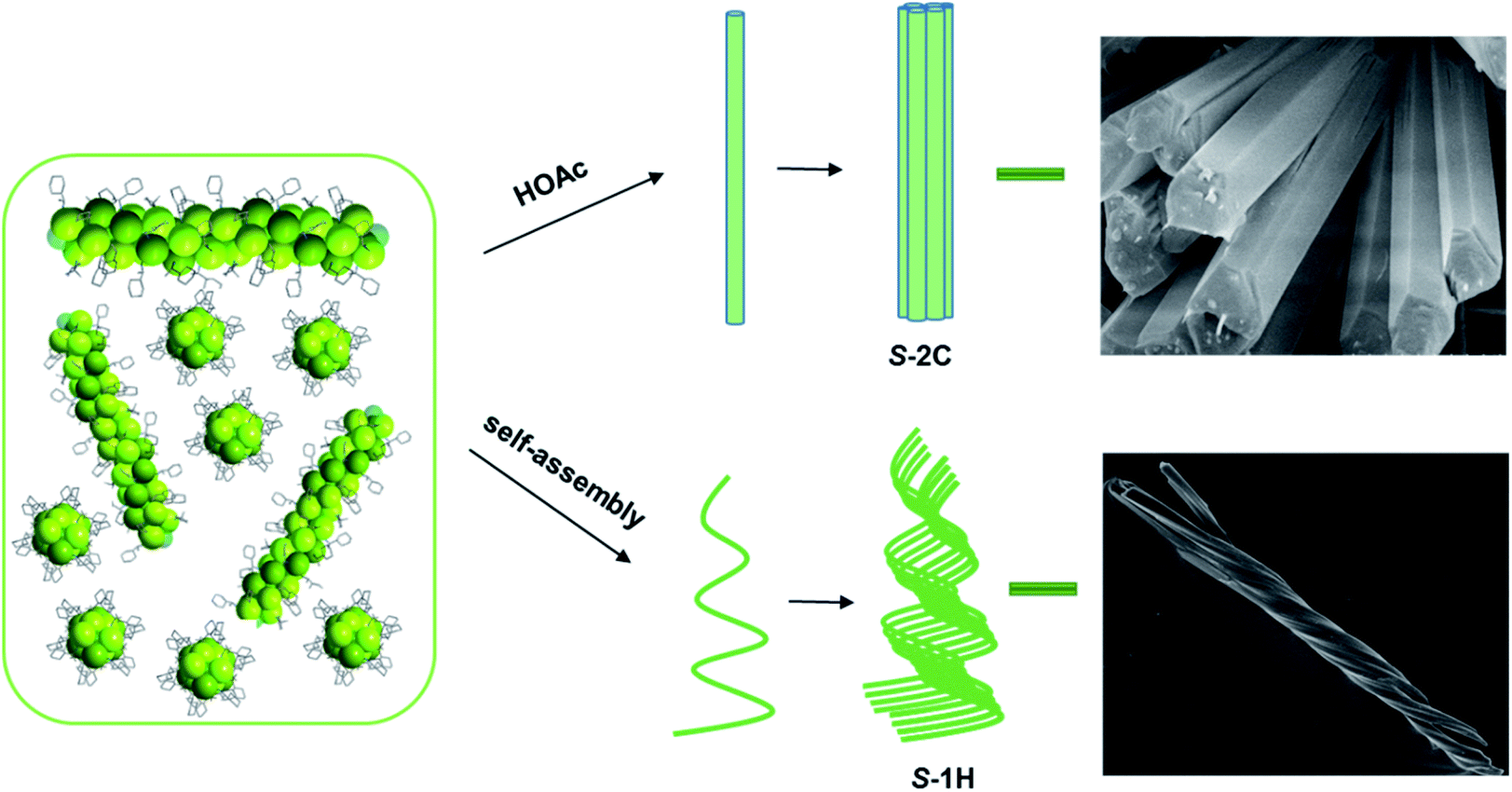 | ||
| Fig. 5 Proposed formation mechanism of the crystalline materials of S-2C and superhelices of S-1H. | ||
The van der Waals interactions are temperature dependent, decreasing with increasing temperature. Temperature can also alter the interconversion rate of conformers and the growth rate of superhelices. Thus, higher temperature would reduce the tendency of parallel packing of the chains but enhance the tendency to form superhelices due to the misalignment of the chains and to facilitate the growth of superhelices.
To examine the temperature influence on the growth of superhelices, we performed hydrothermal reactions of Tb(OAc)3 and S-cyampH2 (molar ratio 1:3, pH = 4.5) for 24 h at different temperatures (40–180 °C). As shown in Fig. 6, the SEM images revealed that only nanorods of approximately 60–70 nm in width and 230–300 nm in length without distinct helical morphology appeared at 40 °C. An increase in temperature accelerated the growth of nanorods, with longitudinal growth increasing much faster than lateral growth. The nanorod sizes were approximately 150 nm in width and 2–3 μm in length at 60 °C and approximately 200 nm in width and 5–6 μm in length at 80 °C. The aspect ratios were approximately 20 at 60 °C and approximately 30 at 80 °C. The helical morphology could be clearly identified above 60 °C. When the temperature reached 100 °C and 120 °C, clustering of the superhelices was evident, resulting in branched helical structures of the same handedness. Interestingly, a further increase in temperature reduced the aggregation tendency of the superhelices, and only left-handed helical fibrils without branching were observed at 140 °C. Reactions at even higher temperatures (160 °C and 180 °C) resulted in unrecognized new phases. Although no significant difference could be identified from the IR spectra (Fig. S26†), the PXRD patterns and SEM and EDX spectra were clearly different from those of S-1H (Fig. S27–S31†). The EDX measurements corroborated the Tb:P molar ratio of 1:3 for the reaction products at 40–140 °C (Fig. S32†). The results showed that superhelices of S-1H could form in the temperature range of 40–140 °C as a result of a compromise between the two tendencies mentioned above, e.g., parallel packing of chains via vdW interactions and mismatched alignment of chains due to conformer interconversion. The most appropriate temperature to obtain superhelices of S-1H with distinct helical morphologies is 80–140 °C. The formation of branched structures at 100 °C and 120 °C suggested that the van der Waals interaction and growth rate of helical chains at these temperatures approached a suitable balance in which clustering of superhelices was favoured. In other words, the branched structure may be suppressed efficiently by increasing the reaction temperature.
 | ||
| Fig. 6 SEM images of products after hydrothermal reactions of Tb(OAc)3 and S-cyampH2 at pH 4.5 for 24 h at different reaction temperatures (40–140 °C). The yellow arrows represent the direction of chirality. | ||
Finally, one may ask about what happens when racemic R/S-cyampH2 ligands are employed to react with Tb(OAc)3, a mixture of left- and right-handed superhelices or others. To answer this question, we carried out similar hydrothermal reactions of Tb(OAc)3 and racemic R/S-cyampH2 at different pH (3.0–7.0) at 100 °C for 24 h. The SEM images revealed the formation of nanospheres at pH 3.0, nanorods without helical morphologies at pH 3.5–5.5, a mixture of nanoparticles and nanorods at pH 6.0–6.5, and nanoparticles at pH 7.0 (Fig. S33†). The PXRD patterns and IR spectra of the products at pH 3.0–6.5 are similar to those of S-1H (Fig. S34†). But the ECD and VCD spectra obtained for the products at pH = 3.5 and 4.5 are silent (Fig. S35 and S36†). All these results indicate that the resulting nanorods were achiral due to the presence of both R- and S-cyampH2 ligands.
Theoretical calculations
Pure phases of superhelices of S-1H and crystals of S-2C were isolated by reacting Tb(OAc)3 and S-cyampH2 (initial pH ca. 4.0) under similar hydrothermal conditions except pH, 4.3–6.5 for the superhelices and 3.5–3.7 for the rod-like crystals. When the pH was in between (3.8–4.2), a mixture of S-1H and S-2C was obtained. The experimental results demonstrated that S-1H and S-2C are very similar in structure but different in chemical composition with an additional guest acetic acid in the latter. Acetic acid plays a critical role in crystal formation. The coordination polymer chains may be supported and fixed by the acetic acid molecules so that they can pile up well into a crystal. To better understand the underlying mechanism of superhelix formation, some quantum chemistry calculations (at the M06-2X45/aug-cc-pVTZ46 level with Grimme's D3 dispersion correction47) were performed using the Gaussian16 program.48 The counterpoise correction49,50 for the basis set superposition error (BSSE) has been applied to obtain more accurate intermolecular interaction energies.To understand the possible role of the acetic acid molecules in the formation of the crystal structure, we focused on the hydrogen bonds between acetic acid and the CP chains. As the crystals were formed at pH 3.5–4.2 and the acetic acid should be deprotonated at pH = 4.3–6.5 because its pKa is 4.76,51 a simplified model including one acetic acid and one S-cyampH− was constructed, as shown in Fig. 7a. The calculated interaction energy is −22.3 kcal mol−1. To understand what will happen at higher pH values, a similar model was constructed without the proton, as shown in Fig. 7b. The interaction energy is calculated to be 89.1 kcal mol−1 in the same position as the first model. It is clear that only at lower pH can the acetic acid molecules be wedged in the CP chains by hydrogen bonds.
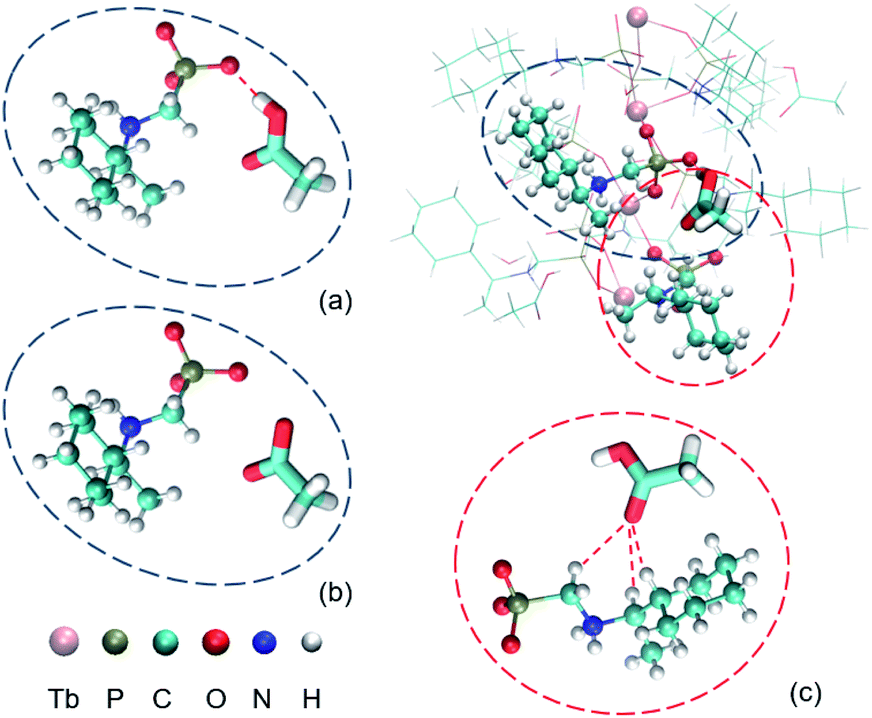 | ||
| Fig. 7 The environment of an acetic acid molecule. (a) The hydrogen bond between acetic acid and one S-cyampH− molecule. (b) The deprotonated structure of (a). (c) The C–H⋯O interactions between acetic acid and another S-cyampH− molecule. | ||
Do the acetic acid molecules obstruct the conformational transitions of the cyclohexyl groups? It is obviously true because the existence of acetic acid does provide the steric hindrance. To further investigate other mechanisms, another model was constructed using one acetic acid and one relevant S-cyampH− as shown in Fig. 7c. The calculated interaction energy is −3.6 kcal mol−1. So, we believe that acetic acid can fix the cyclohexyl group and can reduce its conformational flexibility. The energy barrier of the conformational transitions is increased by the two factors mentioned above, so the cyclohexyl groups tend to stay in the same dominant conformation.
In short, the acetic acid molecules stick to the CP chains of S-Tb(cyampH)3 at relatively lower pH and hinder the conformational transitions of the cyclohexyl groups. This promotes the regular stacking of the chains in the lattice, and hence the formation of ordered crystals of S-2C. Considering that the conformational flexibility of cyclohexyl groups is the key factor in the formation of superhelices in the system, our simulation results here could also clarify the role of cyclohexyl groups in the experiments.
Comparison of superhelix formation mechanisms of Tb/cyampH2 and Tb/pempH2 systems
R-, S-pempH2 differ from R-, S-cyampH2 by phenyl instead of the cyclohexyl group. In previous studies, we found that R-, S-pempH2 can react with terbium nitrate forming superhelices of R-, S-Tb(pempH)3·2H2O. Noting that the superhelices of R-, S-Tb(cyampH)3·3H2O (R-1H, S-1H) in the current study are similar to those of R-, S-Tb(pempH)3·2H2O in chemical composition (except the pending organic groups), chain structures and morphologies, it is naturally expected that they may follow a similar “chain-twist-growth” mechanism. However, careful analyses reveal that the inherent helix formation mechanisms of the two systems are completely different.For the Tb/R-pempH2 system, the twist-growth of chains is concerned with the coexistence of two closely related but different kinds of chains, one positively charged and the other neutral.22 Block-like crystals of the positively charged chain compound R-(H3O)[Tb3(pempH2)2(pempH)7][Tb3(pempH2) (pempH)8](NO3)4·11H2O were isolated as a pure phase at pH 2.5–2.7, while rod-like crystals of the neutral chain compound R-Tb(pempH)3·2H2O were obtained as a pure phase at pH 3.7–4.5. The pure phase of superhelices of R-Tb(pempH)3·2H2O formed in a narrow pH range (ca. 3.0–3.2) has the same structure at the molecular level as that of rod-like crystals. Below or above this pH range, a mixture of superhelices and block- or rod-like crystals was obtained. Theoretical calculations indicated that the curved or twisted chains were found only when the two kinds of chains (positively charged and neutral) reached a suitable ratio (1:4 in this case), which was the prerequisite for helix formation. The ratio of the two kinds of chains can be precisely modulated by pH. This may explain why superhelices of R-Tb(pempH)3·2H2O were formed in such a narrow pH range. The nitrate anions played a key role in promoting the aggregation of the positively charged and neutral chains and hence induced the twisted growth of the chains. When nitrate anions were substituted by chloride or acetate, no superhelices were observed.
Clearly, the requirements for the helix formation in previous studies are quite critical. It needs coexistence of two kinds of different but closely related chains in suitable proportion, as well as nitrate anions to connect the two kinds of chains to induce the twisted growth of the chains. These two requirements are not satisfied for the current Tb/cyampH2 system. First, noting that the phosphonate groups in the Tb/pempH2 system are fully deprotonated at pH ≥ 3.7 with the formation of purely neutral chain compounds, we expect that the phosphonate groups in the Tb/cyampH2 system should be fully deprotonated at pH 4.3–6.5. Thus, the formation of positively charged chains of Tb(cyamHx)3 (x > 1) in this pH range would not be possible. Second, it is difficult for the acetate anions to connect two different kinds of chains like nitrate anions. Structural analyses showed that the polar acetic acid in S-2C inserted into the chain forming moderately strong O–H⋯O hydrogen bonds with phosphonate oxygen atoms and weak C–H⋯O contacts with the cyclohexyl groups from the same chain (Fig. 2c). But the interactions between the methyl groups of acetic acid and the adjacent chains are extremely weak (Fig. 2d). The Hirshfeld surface analysis gives the same conclusion (Fig. 3). In contrast, all three oxygen atoms of the nonpolar NO3- anion in the Tb/pempH2 system can involve in the formation of H-bond networks and thus are able to connect different kinds of chains by forming O–H⋯O, N–H⋯O and C–H⋯O hydrogen bonds with the ligands from adjacent chains (Fig. 2e). More detailed structural differences of the two systems can be found in Table S6.†
To illustrate the specific role of nitrate anions in the formation of superhelices of the Tb/pempH2 system, we re-conducted similar hydrothermal reactions using Tb(OAc)3 instead of Tb(NO3)3 to react with S-pempH2 at different pH. Only crystals were obtained in the pH range of 2.9–6.0. When the pH was lower than 2.9 or higher than 6.0, clear solutions or unidentified powders were observed (Fig. S37–S39†). As a comparison, the helix formation of the Tb/cyampH2 system is anion independent. Superhelices of S-1H and R-1H can be isolated in the same pH range using other terbium salts such as Tb(NO3)3 and TbCl3. These observations indicate that the formation of superhelices of S-1H and R-1H would not follow the similar chain-twist-growth mechanism to the previous study.
Nevertheless, the presence of only one kind of neutral chain with extremely weak interchain interactions cannot give a guarantee of helix formation. In most cases, needle-like crystals with a large aspect ratio would be obtained. The formation of superhelices of S-1H and R-1H is attributed to the conformer interconversion of the cyclohexyl groups in S-, R-cyampH2, which provides the driving force of twist-growth of the chiral neutral chains due to the mismatch in chain stacking. This may explain the fact that superhelices of S-1H and R-1H formed in a wide pH range (4.3–6.5). In contrast, conformer interconversion does not occur for phenyl groups in S-, R-pempH2, and thus rod-like crystals of R-, S-Tb(pempH)3·2H2O dominated above pH 3.3. To this end, this work provides a new type of “chain-twist-growth” mechanism which is very different from the one observed in previous studies.
Conclusions
In this work, we demonstrated that chiral transcription from the molecular level to the morphological level can be achieved in homochiral CPs with neutral chain structures. There are two key factors: (1) weak interchain interactions to avoid tight and parallel packing of the chains and (2) slight misalignment of the chains to drive twist growth of the chains. The two requirements are satisfactorily fulfilled for superhelices of the metal phosphonate compounds S- and R-Tb(cyampH)3·3H2O (S-1H, R-1H), in which the cyclohexyl groups hanging on the chains provide not only a weak vdW interaction between the chains but also misalignment of the chains due to the fast interconversion of the conformers. Such a “chain-twist-growth” mechanism is very different from the one previously proposed by us for superhelices of R-, S-Tb(pempH)3·2H2O, where coexistence of different kinds of chains (positively charged and neutral) and presence of nitrate anions were essential for helix formation.22 Therefore, this work provides a new type of “chain-twist-growth” mechanism.Superhelices S-1H and R-1H are rare examples of neutral CP chains with chirality expressed at both the molecular and macroscopic levels. Superhelices could also be obtained when the TbIII ion was replaced by other lanthanide ions, such as SmIII, EuIII, GdIII, DyIII and HoIII ions (Fig. S40–S43†). The delicate balance of conditions for superhelix formation may be broken by external factors such as guest molecules. Consequently, crystals of S- or R-Tb(cyampH)3·HOAc·2H2O (S-2C, R-2C) were obtained by introducing acetic acid as a guest at lower pH. The solid-state 1H magic angle spinning (MAS) NMR measurements revealed stronger motion of the cyclohexyl groups in S-1H superhelices than in S-2C crystals. Theoretical calculations confirmed that acetic acid plays a critical role in inhibiting the conformer interconversion of the cyclohexyl rings and promoting crystal formation. The design idea of this work is universal in principle and can be extended to other CP systems. This work may also shed light on the development of molecular materials with helical morphologies for applications in asymmetric catalysis, nonlinear optical materials, molecular devices and sensors.
Experimental
Materials and methods
R- and S-1-cyclohexylethylamine were purchased from TCI without further purification, and all the other starting materials were of reagent grade quality. R- and S-(1-cyclohexylethyl) aminomethylphosphonic acid (cyampH2) were prepared according to the literature method.33 The pH value was measured by using a Sartorius PB-10 pH metre. Infrared spectra were measured on a Bruker TENSOR 27 IR spectrometer with pressed KBr pellets in the range of 400–4000 cm−1. Elemental analyses for C, N, and H were performed with a PerkinElmer 240C elemental analyzer. Thermogravimetric analysis (TGA) was performed on a Mettler-Toledo TGA/DSC STARe thermal analyzer in the range of 25–600 °C under a nitrogen flow at a heating rate of 5 °C min−1. Powder X-ray diffraction (PXRD) data were recorded on a Bruker D8 ADVANCE X-ray powder diffractometer (Cu-Kα) at room temperature (λ = 1.5406 Å). Ultraviolet-visible absorption spectra were recorded on a PerkinElmer Lambda 950 spectrometer. Scanning electron microscopy (SEM) measurements were performed on a Hitachi S-4800. ECD spectra were measured on a JASCO J-720W spectrophotometer using KCl pellets at room temperature. Vibrating circular dichroism spectra were recorded on a Bruker PMA50 spectrophotometer using KBr pellets at room temperature. 1H magic angle spinning nuclear magnetic resonance (MAS NMR) experiments were performed on a Bruker Avance III 400 spectrometer equipped with an 89 mm wide-bore 9.4 T superconducting magnet yielding a Larmor frequency of 400 MHz for 1H. The samples were packed into 3.2 mm rotors and spun at 21 kHz. A single pulse NMR sequence was used for data acquisition and the 1H chemical shifts were referenced to adamantane at 1.92 ppm.Synthetic procedures
For R-1H. Yield: 66.0% based on Tb. Elemental analysis (%) calcd for C27H63N3O12P3Tb: C 37.12, H 7.27, and N 4.81; found: C 37.09, H 7.09, and N 4.67. IR (KBr, cm−1): 3416(m), 2983(m), 2926(s), 2855(s), 2792(w), 2673(w), 2519(m), 2403(m), 1669(w), 1616(m), 1448(m), 1390(m), 1349(w), 1310(w), 1275(m), 1232(w), 1155(s), 1069(s), 1025(s), 983(s), 892(m), 762(m), 613(w), 567(m), and 484(m).
For R-2C. Yield: 38.0% based on Tb. Elemental analysis (%) calcd for C29H65N3O13P3Tb: C 38.04, H 7.16, and N 4.59; found: C 38.12, H 6.96, and N 4.59. IR (KBr, cm−1): 3432(m), 2989(m), 2926(s), 2855(s), 2787(w), 2667(w), 2511(m), 2396(m), 1700(w), 1617(m), 1448(m), 1385(m), 1352(w), 1280(m), 1231(w), 1146(s), 1073(s), 1035(s), 986(s), 887(m), 762(m), 617(w), 560(m), and 483(m). The number of lattice acetic acid and water molecules in S-2C and R-2C was also confirmed by thermal analyses (Fig. S44†).
Crystallography
Single crystals of dimensions 0.30 × 0.15 × 0.10 mm3 for S-2C and 0.25 × 0.10 × 0.10 mm3 for R-2C were used for data collection on a Bruker D8 Liquid with Metal-jet diffractometer using graphite-monochromated Ga Kα radiation (λ = 1.34139 Å) (for S-2C) or a Bruker D8 Venture with a TXS diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) (for R-2C). The data were integrated using the Siemens SAINT program.52 Adsorption corrections were applied. The structures were solved by a direct method and refined on F2 by full-matrix least squares using SHELXTL.53 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were either put in calculated positions or found from the difference Fourier maps and refined isotropically. CCDC 2034186 and 2034187 contain the supplementary crystallographic data for this paper.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Guo-Guo Weng: PhD student in charge of the project. Ben-Kun Hong: PhD student in charge of the theoretical calculations and Hirshfeld surface analysis. Song-Song Bao: Associate Prof. in charge of crystal structure analysis. Yujie Wen: PhD student in charge of NMR measurement and analysis. Lan-Qing Wu: M. Sc. student in charge of the VCD test. Xin-Da Huang: post-doc involved in the basic characterization of compounds. Jia-Ge Jia: post-doc participating in tests of chiral properties. Ge-Hua Wen: PhD student in charge of the crystal structure test. Shu-Hua Li: Prof. in charge of the theoretical calculations and Hirshfeld surface analysis. Luming Peng: Prof. in charge of the solid-state NMR data. Li-Min Zheng: Prof. conceptor of the study, corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (91956102 and 21731003), and the National Key R&D Program of China (2017YFA0303203, 2018YFA0306004) . We thank Prof. Yu-qiang Ma of Nanjing University and Prof. Hong-ming Ding of Soochow University for valuable discussions.Notes and references
- J. Adamcik, J.-M. Jung, J. Flakowski, P. De Los Rios, G. Dietler and R. Mezzenga, Nat. Nanotechnol., 2010, 5, 423–428 CrossRef CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242–1271 CrossRef CAS.
- T. Leigh and P. Fernandez-Trillo, Nat. Rev. Chem., 2020, 4, 291–310 CrossRef CAS.
- Y. Wang, J. Xu, Y. Wang and H. Chen, Chem. Soc. Rev., 2013, 42, 2930–2962 RSC.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS.
- Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS.
- J. Rocha, L. D. Carlos, F. A. Almeida Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3297–3312 RSC.
- G.-G. Weng and L.-M. Zheng, Sci. China: Chem., 2020, 63, 619–636 CrossRef CAS.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692–11696 CrossRef CAS.
- R. Sun, L. Wang, J. Tian, X. Zhang and J. Jiang, Nanoscale, 2012, 4, 6990–6996 RSC.
- F. Wang and C.-L. Feng, Angew. Chem., Int. Ed., 2018, 57, 5655–5659 CrossRef CAS PubMed.
- H. Choi, S. Heo, S. Lee, K. Y. Kim, J. H. Lim, S. H. Jung, S. Sung Lee, H. Miyake, J. Y. Lee and J. H. Jung, Chem. Sci., 2020, 11, 721–730 RSC.
- Y. Qiao, Y. Lin, Y. Wang, Z. Yang, J. Liu, J. Zhou, Y. Yan and J. Huang, Nano Lett., 2009, 9, 4500–4504 CrossRef CAS PubMed.
- Y. Qiao, Y. Lin, Z. Yang, H. Chen, S. Zhang, Y. Yan and J. Huang, J. Phys. Chem. B, 2010, 114, 11725–11730 CrossRef CAS.
- C. Li, K. Deng, Z. Tang and L. Jiang, J. Am. Chem. Soc., 2010, 132, 8202–8209 CrossRef CAS.
- J. Huang, H.-m. Ding, Y. Xu, D. Zeng, H. Zhu, D.-M. Zang, S.-S. Bao, Y.-q. Ma and L.-M. Zheng, Nat. Commun., 2017, 8, 2131 CrossRef PubMed.
- C. Y. Li, S. Z. D. Cheng, J. J. Ge, F. Bai, J. Z. Zhang, I. K. Mann, L.-C. Chien, F. W. Harris and B. Lotz, J. Am. Chem. Soc., 2000, 122, 72–79 CrossRef CAS.
- (a) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS; (b) L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. d. Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS; (c) V. Stepanenko, R. Kandanelli, S. Uemura, F. Würthner and G. Fernández, Chem. Sci., 2015, 6, 5853–5858 RSC; (d) J. Matern, K. K. Kartha, L. Sánchez and G. Fernández, Chem. Sci., 2020, 11, 6780–6788 RSC; (e) M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148–2151 CrossRef CAS.
- A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 1998, 37, 63–68 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS.
- X. Dou, N. Mehwish, C. Zhao, J. Liu, C. Xing and C. Feng, Acc. Chem. Res., 2020, 53, 852–862 CrossRef CAS.
- Y. Wang, X. Li, F. Li, W.-Y. Sun, C. Zhu and Y. Cheng, Chem. Commun., 2017, 53, 7505–7508 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- P. Sutter, S. Wimer and E. Sutter, Nature, 2019, 570, 354–357 CrossRef CAS PubMed.
- Y. Liu, J. Wang, S. Kim, H. Sun, F. Yang, Z. Fang, N. Tamura, R. Zhang, X. Song, J. Wen, B. Z. Xu, M. Wang, S. Lin, Q. Yu, K. B. Tom, Y. Deng, J. Turner, E. Chan, D. Jin, R. O. Ritchie, A. M. Minor, D. C. Chrzan, M. C. Scott and J. Yao, Nature, 2019, 570, 358–362 CrossRef CAS.
- R. Chen, Y. Shen, S. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14198–14210 CrossRef CAS.
- Z.-S. Cai, S.-S. Bao, M. Ren and L.-M. Zheng, Chem.–Eur. J., 2014, 20, 17137–17142 CrossRef CAS PubMed.
- M. A. J. Koenis, A. Osypenko, G. Fuks, N. Giuseppone, V. P. Nicu, L. Visscher and W. J. Buma, J. Am. Chem. Soc., 2020, 142, 1020–1028 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS.
- TOPAS, version 5.0, Bruker AXS Inc., Madison, WI, 2014 Search PubMed.
- G. W. Peterson, G. W. Wagner, A. Balboa, J. Mahle, T. Sewell and C. J. Karwacki, J. Phys. Chem. C, 2009, 113, 13906–13917 CrossRef CAS.
- A. Pinest, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1973, 59, 569–590 CrossRef.
- (a) I. Bertini, C. Luchinat and G. Parigi, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 40, 249–273 CrossRef CAS; (b) G. Kervern, G. Pintacuda, Y. Zhang, E. Oldfield, C. Roukoss, E. Kuntz, E. Herdtweck, J.-M. Basset, S. Cadars, A. Lesage, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2006, 128, 13545–13552 CrossRef CAS PubMed.
- Y. Xu, Y.-S. Yu, X.-D. Huang, S.-S. Bao, H.-m. Ding, Y.-q. Ma and L.-M. Zheng, Inorg. Chem., 2018, 57, 12143–12154 CrossRef CAS PubMed.
- J. Engel and H. P. Bächinger, Top. Curr. Chem., 2005, 247, 7–33 CrossRef CAS.
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661–2670 RSC.
- F. Hanke, C. J. Pugh, E. F. Kay, J. B. Taylor, S. M. Todd, C. M. Robertson, B. J. Slater and A. Steiner, Chem. Commun., 2018, 54, 6012–6015 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- R. A. Kendall and T. H. Dunning Jr, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Companies, New York, 15th edn, 1999, Sect. 8.25, Table 8.8 Search PubMed.
- SAINT, Program for Data Extraction and Reduction, Siemens Analytical X-ray Instruments, Madison, WI, 1994–1996 Search PubMed.
- SHELXTL (version 5.0), Reference Manual, Siemens Industrial Automation, Analytical Instruments, Madison, WI, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2034186 and 2034187. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01913a |
| This journal is © The Royal Society of Chemistry 2021 |