
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge breathing effect induced by water sorption in a remarkably stable nonporous cyanide-bridged coordination polymer†
Michał
Magott
 *a,
Bartłomiej
Gaweł
b,
Marcin
Sarewicz
c,
Mateusz
Reczyński
a,
Karolina
Ogorzały
a,
Wacław
Makowski
a and
Dawid
Pinkowicz
*a
*a,
Bartłomiej
Gaweł
b,
Marcin
Sarewicz
c,
Mateusz
Reczyński
a,
Karolina
Ogorzały
a,
Wacław
Makowski
a and
Dawid
Pinkowicz
*a
aFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland. E-mail: michal.magott@uj.edu.pl; dawid.pinkowicz@uj.edu.pl
bDepartment of Materials Science and Engineering, Norwegian University of Science and Technology (NTNU), 7491 Trondheim, Norway
cDepartment of Molecular Biophysics, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Kraków, Poland
First published on 1st June 2021
Abstract
While metal–organic frameworks (MOFs) are at the forefront of cutting-edge porous materials, extraordinary sorption properties can also be observed in Prussian Blue Analogs (PBAs) and related materials comprising extremely short bridging ligands. Herein, we present a bimetallic nonporous cyanide-bridged coordination polymer (CP) {[Mn(imH)]2[Mo(CN)8]}n (1Mn; imH = imidazole) that can efficiently and reversibly capture and release water molecules over tens of cycles without any fatigue despite being based on one of the shortest bridging ligands known – the cyanide. The sorption performance of {[Mn(imH)]2[Mo(CN)8]}n matches or even outperforms MOFs that are typically selected for water harvesting applications with perfect sorption reversibility and very low desorption temperatures. Water sorption in 1Mn is possible due to the breathing effect (accompanied by a dramatic cyanide-framework transformation) occurring in three well-defined steps between four different crystal phases studied structurally by X-ray diffraction structural analysis. Moreover, the capture of H2O by 1Mn switches the EPR signal intensity of the MnII centres, which has been demonstrated by in situ EPR measurements and enables monitoring of the hydration level of 1Mn by EPR. The sorption of water in 1Mn controls also its photomagnetic behavior at the cryogenic regime, thanks to the presence of the [MoIV(CN)8]4− photomagnetic chromophore in the structure. These observations demonstrate the extraordinary sorption potential of cyanide-bridged CPs and the possibility to merge it with the unique physical properties of this class of compounds arising from their bimetallic character (e.g. photomagnetism and long-range magnetic ordering).
Introduction
Metal–organic frameworks (MOFs) dominated the field of porous molecule-based materials due to their outstanding sorption characteristics1,2 including the capture of small molecules (e.g. H2, CO2, CH4),3–7 water,8,9 alcohols10–13 or compounds as large as aromatics14,15 or proteins.16,17 Some of the recent advancements in this field cover exceptional water harvesting applications18–22 and the construction of adsorption-driven heat pumps.23–25 Therefore, it is widely believed that MOFs outperform other water sorption materials, as they can indeed absorb moisture at much lower vapor pressures than active carbons and may be regenerated at significantly lower temperatures than silica gels or zeolites. Nonetheless, MOFs are currently being challenged by covalent organic frameworks (COFs),26–29 which in principle are more stable against H2O, as opposed to the hydrolysis susceptible metal-carboxylate moieties present in MOFs.30In this whirlwind of MOF/COF possibilities, CN-bridged coordination polymers (CN-CPs) – Prussian Blue Analogues (PBAs) and related cyanide-bridged frameworks – remain underrated or even omitted as potential sorbents despite some promising reports of H2 (ref. 31–34) and ammonia35–37 sorption. This is caused by another common belief that these materials show low fatigue resistance limited to several sorption cycles and can release toxic components in the process – like the first examples of MOFs before the seminal work by Yaghi et al.38 and before the demonstration of reversible bond breaking in the water-stable DMOF-TM showing state-of-the-art water sorption capabilities.39 Overcoming this stereotype is crucial for further progress in the field of multifunctional molecular materials as CN-CPs offer the possibility of fine tuning of many different functionalities including magnetism or magnetic and photomagnetic switching upon solvent removal/exchange.40–47 This in turn will potentially lead to ground-breaking magnetic sponge systems48,49 operational at room temperature.
In this work, we address this issue by presenting state-of-the-art water sorption properties and exceptional cyclability of a completely nonporous cyanide-bridged framework {[MnII(imH)]2[MoIV(CN)8]}n (1Mn) (the anhydrous form of {[MnII(imH)(H2O)2]2[MoIV(CN)8]·4H2O}n (1Mn·8H2O), reported by Shen et al. in 2006)50 and the analogue of the low-temperature magnetic sponge-like system {[MnII(imH)]2[NbIV(CN)8]}n (ref. 45) as well as the photomagnetic sponge {[MnII(imH)]2[WIV(CN)8]}n.47 Despite the obvious lack of porosity demonstrated in the nitrogen sorption experiment, 1Mn shows three water-induced quasi-phase transitions, accompanied by a very large breathing effect51–53 and a substantial change in the cyanide bridging pattern fully understood based on the powder X-ray diffraction structural analysis. The three-step breathing process is fully reversible and exceptionally fatigue resistant over tens of adsorption/desorption cycles, with the complete preservation of crystallinity and very high water uptake exceeding 25% w/w.
Apart from the exceptional water sorption and cyclability, 1Mn exhibits marked magnetic and photomagnetic changes upon water capture. Similar effects were previously reported in paramagnetic MOFs where strong interdependence between sorption and other physical properties occurs.54–60 In some cases, the profound structural and electronic changes induced by guest molecules enable the characterization/monitoring of the sorption process using EPR spectroscopy. This can be done directly in the case of paramagnetic guests61–65 or indirectly by analysing the g-factor shift of paramagnetic metal centers as in some CuII-based porous materials.54,66–68 Noteworthy, EPR spectroscopy is commonly applied to study the state of the paramagnetic active sites doped into the mesoporous hosts such as zeolites in various catalytic processes.69–72 In the case of 1Mn, presented herein, the state of the MnII sites was successfully monitored in the real time by employing in situ EPR spectroscopy during the adsorption/desorption of water molecules. Structural and electronic changes within the coordination sphere of MnII affect also the low-temperature photo-induced magnetization of the compound which is evidenced by detailed photomagnetic studies.
Results and discussion
Structure, breathing behaviour and sorption properties
{[MnII(imH)(H2O)2]2[MoIV(CN)8]·4H2O}n (1Mn·8H2O) was prepared according to the modified literature procedure50 by combining water solutions of manganese(II) chloride and imidazole with potassium octacyanomolybdate(IV), affording yellow crystals suitable for single crystal X-ray diffraction (SCXRD; see Experimental section for details). 1Mn·8H2O forms a three-dimensional coordination framework in C2/c space group, with two octahedral manganese(II) cations coordinated by one imidazole ligand (disordered between two positions), three nitrogen atoms of the [MoIV(CN)8]4− in a mer configuration and two aqua ligand in cis disposition (Table 1 and Fig. S1†). Each octacyanidomolybdate(IV) connects six manganese(II) centres and the Mn2Mo unit is accompanied by four crystallization water molecules located in the channels along the crystallographic c direction (Fig. 1c). The 3-D framework of 1Mn·8H2O can be described as a “crossed-ladder” cyanide bridging pattern, similar to that depicted in Fig. 2a and S2 in the ESI,† with two sets of coordination ladders cross-linking at the MoIV centres.| Abbreviation | Chemical formula | Structure determination method | Space group | Coordination geometry of the MnII ions | Coordination numbers of the MnII ions | Number of CN-bridges per MoIV |
|---|---|---|---|---|---|---|
| 1Mn·8H2O | {[MnII(imH)(H2O)2]2[MoIV(CN)8]·4H2O}n | SCXRD | C2/c | Octahedron/octahedron | 6/6 | 6 |
| 1Mn·3H2O | {[MnII(imH)(H2O)2][MnII(imH)(H2O)][MoIV(CN)8]}n | PXRD |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Distorted octahedron/trigonal bipyramid | 6/5 | 6 |
| 1Mn·2H2O | {[MnII(imH)(H2O)]2[MoIV(CN)8]}n | PXRD | C2/c | Trigonal bipyramid/trigonal bipyramid | 5/5 | 6 |
| 1Mn | {[MnII(imH)]2[MoIV(CN)8]}n | PXRD |
P |
Distorted tetrahedron/trigonal bipyramid | 4/5 | 7 |
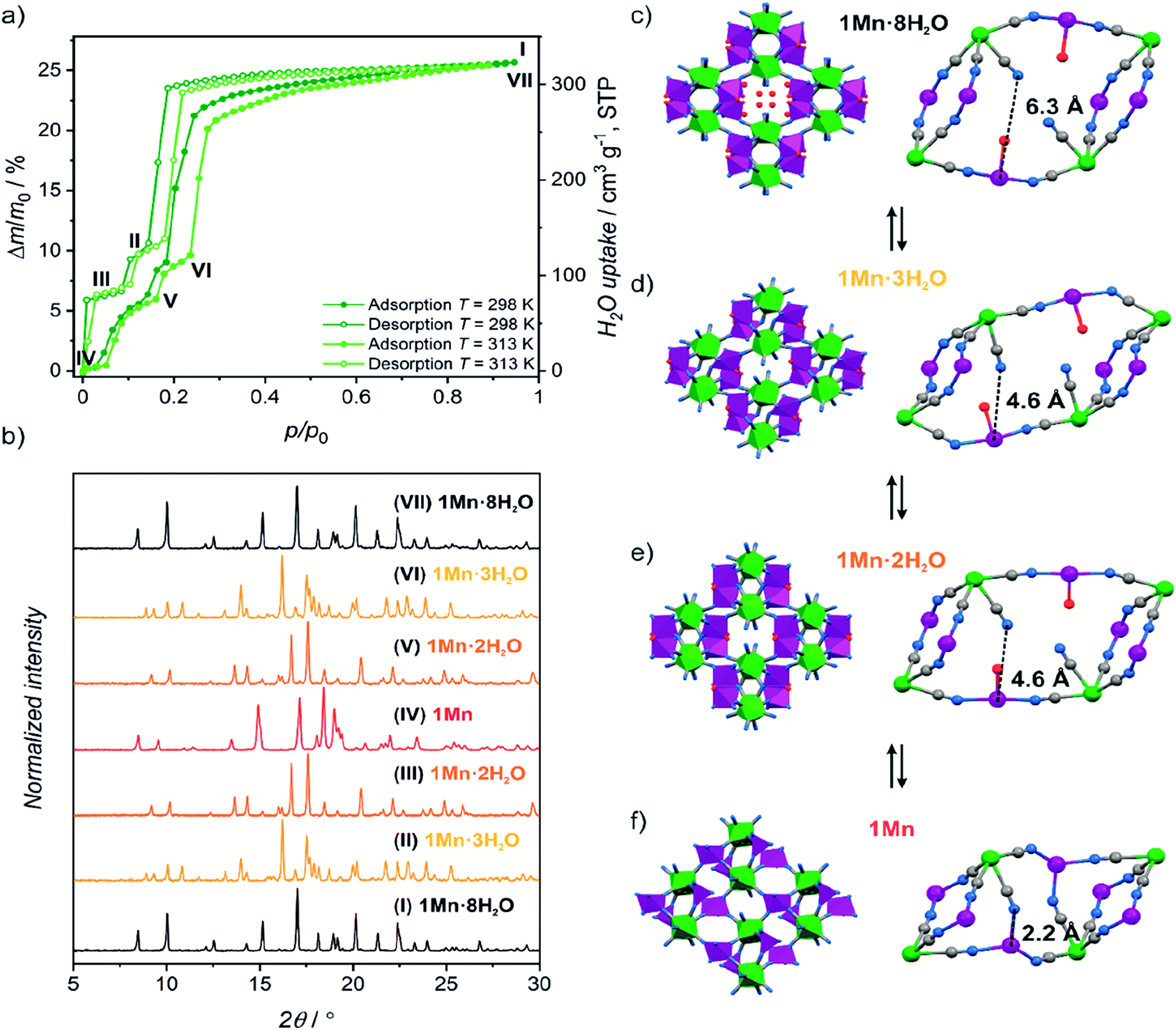 | ||
| Fig. 1 (a) Water sorption/desorption isotherms for 1Mn·8H2O at 298 K and 313 K. Roman numbers enumerate different dehydration/hydration stages. (b) PXRD patterns observed for phases denoted in Fig. 1a. (c–f) Schematic representation of structural changes occurring during 1Mn·8H2O transformation between different crystalline phases. Imidazole molecules and hydrogen atoms were omitted for clarity. | ||
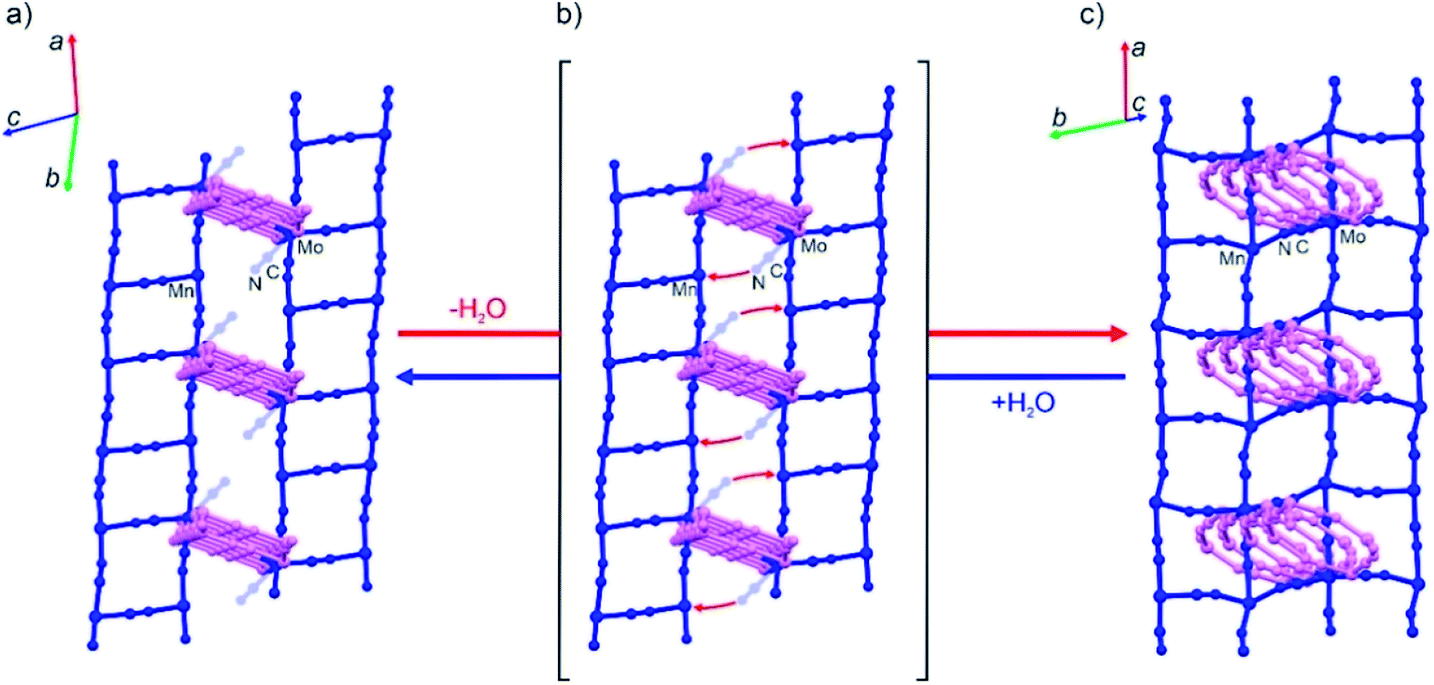 | ||
| Fig. 2 Simplified structural diagrams showing the change of the cyanide bridging pattern in the last step of the dehydration of 1Mn·2H2O (a) accompanied by the additional CN-bridge formation (b) which finally yields anhydrous 1Mn (c). The initial CN-bridged skeleton of 1Mn·2H2O comprises two sets of color-coded (pink and blue) “ladder” motifs intersecting at the MoIV nodes with six bridging CN− ligands (three per each “ladder”) (a). Upon desolvation only the blue “ladders” transform into “square grids” due to the additional CN-bridge formation (b) while the pink “ladders” adapt their geometry in the final CN-bridged scaffold of 1Mn (c). The MoIV nodes in the anhydrous 1Mn utilize seven CN− ligands for bridging towards MnII ions (three per pink “ladder” and four per “blue square grid”). Please see Fig. S2 in the ESI† for the same figure with color-coded atoms. | ||
The coordination and crystallization water molecules can be removed completely by heating 1Mn·8H2O above 60 °C in a dry nitrogen atmosphere, and the coordination skeleton of the resulting {[MnII(imH)]2[MoIV(CN)8]}n (1Mn) (Fig. 1f and 2c) remains stable up to 250 °C (TGA, Fig. S3†). Interestingly, water adsorption/desorption isotherm recorded at 298 K shows that the solvent loss is in fact a three-step process (Fig. 1a), proceeding through two intermediate phases. In the desorption branch, the 1Mn·8H2O phase is stable down to ca. 20% of the relative humidity (RH). The first intermediate mass plateau is observed in the 10–14% RH range corresponding to Δm/m0 of 9.3–10.6% (Fig. 1a, II; Δm/m0 denotes mass change relative to the anhydrous state, m0). This step is in good agreement with {[MnII(imH)(H2O)2][MnII(imH)(H2O)][MoIV(CN)8]}n composition (calculated Δm/m0 = 9.8% corresponds to 3 water molecules), which would account for simultaneous removal of four crystallization water molecules and one coordinated water molecule. Indeed, powder X-ray diffraction (PXRD) confirms the appearance of a new crystalline phase 1Mn·3H2O obtained under conditions corresponding to point II in Fig. 1a (Fig. 1b, (II)).
Upon further decrease in relative humidity down to 1–8% RH range, another mass plateau emerges for Δm/m0 = 6.1–6.6% (Fig. 1a, (III)), which is close to 6.5% predicted for {[MnII(imH)(H2O)]2[MoIV(CN)8]}n (1Mn·2H2O). The PXRD experiment shows complete disappearance of 1Mn·3H2O reflections at this step and the emergence of a new powder pattern for 1Mn·2H2O is depicted in Fig. 1b, (III). Passing dry nitrogen over the sample produces anhydrous 1Mn, accompanied by the final changes in the PXRD pattern (Fig. 1b, (IV)). The rehydration process shows distinct hysteresis in water sorption isotherm, yet powder X-ray diffraction confirms that it proceeds through the same crystalline phases as during dehydration (Fig. 1a and b: (V, VI and VII)). Overall, the stepwise water adsorption isotherm for 1Mn at 298 K shows the water uptake of 0.26 gwater g−1 at p/p0 = 0.9, but already reaches 0.225 g g−1 at p/p0 = 0.3.
Upon solvent removal crystals of 1Mn·8H2O crack, precluding the determination of the crystal structures of the dehydrated phases by means of SCXRD. Therefore structures of compounds 1Mn·3H2O, 1Mn·2H2O and 1Mn were modelled by Rietveld refinement of the respective PXRD patterns (Fig. S4–S6†). The most significant structural features of the three dehydrated phases are summarized in Table 1.
In the first step of dehydration, the water-filled channels of {[MnII(imH)(H2O)2]2[MoIV(CN)8]·4H2O}n1Mn·8H2O are emptied and one coordinated water molecule per formula unit is removed (Fig. 1c and d), which leads to a 14.0% shrinkage of the unit cell. Thus, the initially symmetry equivalent manganese(II) centres are diversified into the distorted octahedral [MnII(imH)(H2O)2(μ-NC)3] and trigonal bipyramidal [MnII(imH)(H2O)(μ-NC)3] moieties in the resulting {[MnII(imH)(H2O)2][MnII(imH)(H2O)][MoIV(CN)8]}n1Mn·3H2O framework (see Table 1) with a similar “crossed-ladder” bridging pattern as that of the parent 1Mn·8H2O. This decreases the lattice symmetry down to P space group. The removal of an additional coordinated water molecule restores the C2/c symmetry, by converting the remaining six-coordinated manganese(II) into a trigonal bipyramidal motif in the {[MnII(imH)(H2O)]2[MoIV(CN)8]}n1Mn·2H2O framework (Fig. 1e and 2a). In this step, only a minimal change of the unit cell volume is observed (−1.6%).
The most pronounced transformation is observed in the last dehydration step. Removal of the remaining two aqua ligands enables the formation of an additional cyanide bridge in {[MnII(imH)]2[MoIV(CN)8]}n1Mn, enforcing further 11.7% decrease in the unit cell volume as well as the dramatic cyanide bridging pattern change depicted in Fig. 2. The “crossed-ladder” pattern changes into a “ladders-crossing-square-grids” coordination scaffold. In the final structure (triclinic, P, Fig. 1f) the tetracoordinate [MnII(imH)(μ-NC)3] adopts distorted vacant trigonal bipyramidal geometry and the pentacoordinate [MnII(imH)(μ-NC)4] centre becomes a distorted trigonal bipyramid. Each octacyanidomolybdate(IV) unit forms seven cyanide bridges to the neighbouring manganese(II) centres in the anhydrous form.
IR spectra recorded during the dehydration experiment reveal changes consistent with those described above – the bands associated with H2O molecules disappear and the bands in the cyanide stretching vibration region change significantly for each phase (please see Fig. S7–S9 as well as the detailed description of the IR spectra in the ESI†).
The aforementioned dehydration-driven stepwise transformations are mirrored in the rehydration experiments, as depicted in Fig. 1a and b. This includes also the bridging pattern changes from “ladders-crossing-square-grids” to “crossed ladders” (Fig. 2). The unit cell volume per formula unit changes from 996 Å3 in 1Mn to 1332 Å3 in 1Mn·8H2O, which accounts for the 34% total change between the anhydrous and the fully hydrated phases, respectively. Such tremendous unit cell expansion is unprecedented among other porous cyanide-bridged polymers46,84–92 and falls within the range observed for flexible MOFs (Table 2). Interestingly, the structure of the anhydrous 1Mn does not show any solvent accessible voids and hence – no permanent porosity. This is evidenced by type II nitrogen adsorption isotherm recorded at 77 K (Fig. 3). The observed N2 uptake of 16 cm3 g−1 at p/p0 = 1.0 is an order of magnitude smaller than observed for MOFs showing similar total water uptake (such as DMOF-TM39,93 or MOF-801 (ref. 9 and 74)) and even smaller than reported for the typically nonporous materials such as Zr6O4(OH)4(SQU)5.25(CH3COO)1.5 MOF based on a squaric acid.94 Therefore combined structural and sorption studies of 1Mn show that initial chemisorption of four water molecules opens the channels in its structure, which only then enables physisorption of the remaining water. Although similar water-induced gate-opening behaviour was previously reported for JUK-8,80 SIFSIX-23-Cu83 and the MIL-53 family,51,95,96 to the best of our knowledge 1Mn constitutes the first case among non-reticular materials.
| Compound | Water uptake (p/p0 = 0.3)/ cm3 g−1, STP | Total water uptake /cm3 g−1, STP | Water-induced breathing behaviour (total volume change: ΔV/Vanhydrous) | Ref. |
|---|---|---|---|---|
| a Values in italic are lower than those reported for 1Mn. b Total water uptake determined only from the amount of water in the crystal structure. | ||||
| CAU-10 | 275 | 365 | No | 9 and 73 |
| MOF-801 | 380 | 450 | No | 9 and 74 |
| UiO-66 | 125 | 535 | No | 9 and 75 |
| MOF-841 | 550 | 640 | No | 9 |
| Mg-MOF-74 | 605 | 750 | No | 9 and 76 |
| Co2Cl2BTDD | 1100 | 1200 | No | 77 |
| Ni2Cl2BTDD | 240 | 1350 | No | 20 |
| Cr-MIL-101 | 150 | 1700 | No | 78 and 79 |
| JUK-8 | 90 | 315 | Yes (24.5%) | 80 |
| 1Mn | 280 | 320 | Yes (34%) | This work |
| Cr-MIL-53 | n/a | 110 | Yes (−47%) | 51 |
| Al-MIL-53 | 90 | 120 | Yes (−49%) | 81 and 82 |
| SIFSYX-23-Cu | 160 | 325 | Yes (53%) | 83 |
| Fe-MIL-88 | n/a | 850b | Yes (85%) | 52 |
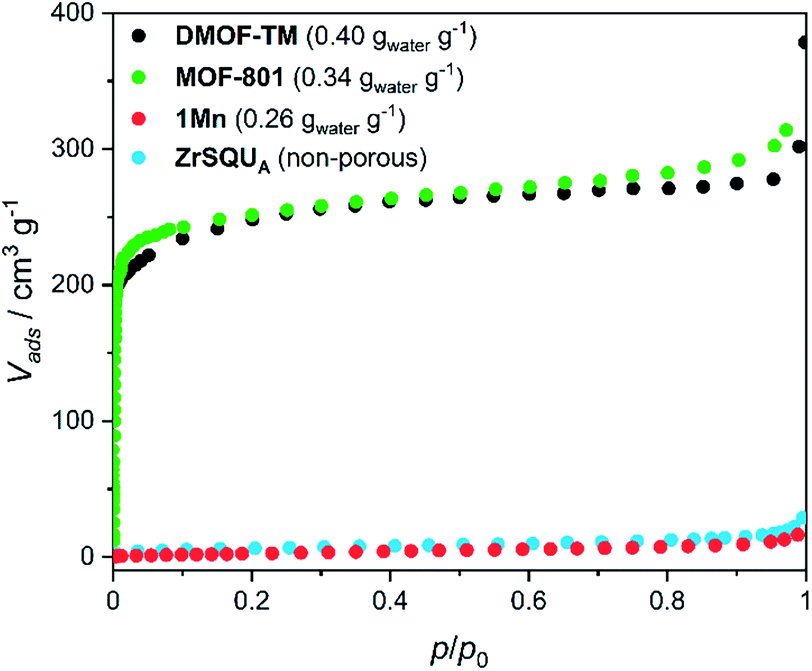 | ||
| Fig. 3 Nitrogen adsorption isotherms at 77 K for 1Mn and selected MOFs. Water loading values at p/p0 = 0.9 and T = 298 K presented in the parentheses were reported in ref. 39 for DMOF-TM and ref. 9 for MOF-801. BET isotherms for MOFs were adapted from ref. 39, 74 and 93. | ||
1Mn·8H2O shows marvellous stability during dehydration–rehydration cycling taking into account the accompanying huge unit cell volume variations. This was confirmed by dynamic vapor sorption (DVS) experiments. The sample mass was monitored as the relative humidity was switched between 0 and 95% at 298 K in 57 cycles (Fig. 4a; see Fig. S10† for full representation of 57 adsorption–desorption cycles). The observed mass change of 26.7% between 0 and 95% RH agrees perfectly with the value of 26.2% expected for transition between 1Mn and 1Mn·8H2O. No change in the curve profile is observed in 57 cycles performed over the course of 430 hours. Powder X-ray diffraction pattern collected for the sample rehydrated after the end of the cycling experiment shows no difference from the one recorded for the pristine sample at the beginning of the experiment (Fig. 4b). Apart from the apparent broadening of the diffraction peaks (see Fig. S11†), the PXRD patterns of the fully hydrated samples before and after the cycling experiment do not show any significant differences. The peak broadening can be explained by the decrease in the grain size resulting from the repetitive breaking of the crystallites in the consecutive dehydration/rehydration cycles, as evidenced by SEM pictures collected for the sample before and after cycling experiments (Fig. S12†). Nonetheless, both DVS and PXRD experiments confirm the perfect stability of the material in water sorption experiments, with full retainment of its water uptake and crystallinity up to 58 cycles.
 | ||
| Fig. 4 (a) Water cycling stability test (0% RH dehydration – 95% RH rehydration) of 1Mn·8H2O at 298 K (numbers above the curve enumerate the consecutive dehydration–rehydration cycles; Fig. S10† shows all 57 cycles). (b) Powder X-ray diffraction patterns for 1Mn·8H2O: calculated from the single-crystal structure (grey), experimental for pristine sample (black) and experimental after 58 dehydration–rehydration cycles at 25 °C (green). | ||
High water-stability and water uptake at low relative pressures are required for application in atmospheric water harvesting.9,97 For this purpose materials with low desorption temperatures are preferable, in order to easily retrieve a liquid condensate from the hydrated sorbent. By comparing the adsorption isotherms at 25 °C and 40 °C (Fig. S13†), we conclude that the material should produce around 0.172 gwater g−1 under 0.95 kPa water pressure (30% RH at 25 °C) upon cycling between these two temperatures. Such a process should switch the material between the 1Mn·8H2O and 1Mn·2H2O hydration states with a simultaneous release of all crystallization water molecules and a half of the coordinated ones at only 40 °C. To confirm that 1Mn can be used for moisture harvesting, we performed a cycling experiment which emulates desert conditions in the daytime (40 °C, 10% RH) and at night (25 °C, 30% RH; Fig. S14†).9 The cycling process revealed a real working capacity of 0.164 g g−1 which approaches the theoretical value deduced from the adsorption isotherms.
The observed release of the chemisorbed water at low temperatures is surprising, as chemisorption is usually associated with high adsorption enthalpy. Therefore we decided to estimate the average adsorption enthalpy of water in 1Mn by employing the van't Hoff equation to the water adsorption isotherms, as well as integrating differential scanning calorimetry (DSC) curve obtained during the sample dehydration (Fig. S15 and 16,† see the Experimental section for details). Both methods give similar results, with effective adsorption enthalpy of 66 kJ mol−1 per water molecule deduced from the van't Hoff equation and 64 kJ mol−1 obtained from the DSC experiment (it is important to note that effective adsorption enthalpy includes energetic effects of water adsorption and cyanide bridge breaking that proceed simultaneously). The small discrepancy results from the difference in desorption temperatures (25–40 °C for the isotherm method, and >70 °C for the DSC experiment). The obtained adsorption enthalpy (64–66 kJ mol−1) is much higher than that reported for hydrophilic MOFs23 and in line with the chemisorption of half of the water molecules. Nonetheless, the strong binding of water does not prevent its easy removal at low temperatures, as demonstrated in the previous parts of the manuscript. This must be associated with fast desorption kinetics, which can be quantified assuming first order kinetics of dehydration and applying the Kissinger equation98,99 to the TGA results at different heating rates (Fig. S17–19†):
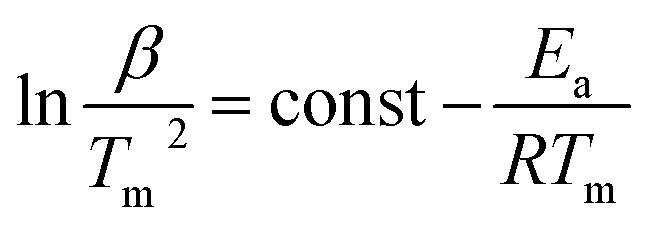
Water sorption in 1Mn was also tested by quasi-equilibrated temperature programmed desorption and adsorption (QE-TPDA).100–102 In this method, the sorption of volatile compounds is studied by cyclic heating and cooling of a quartz tube containing the sample, under the flow of adsorbate (in this case – water vapor) dispersed in a stream of helium gas. The QE-TPDA profiles present positive signals when excess water is desorbed from the sample or negative signals when carrier gas is being depleted of the adsorbate. Recently, the method has been proven useful in studies of water sorption in MOFs103,104 and cyanide-bridged assemblies.105 The QE-TPDA profiles of 1Mn·8H2O demonstrate a three-step desorption process (Fig. 5), which is in line with the aforementioned water desorption isotherm. While the first two steps remain almost unchanged in the 30 consecutive sorption/desorption cycles, the high-temperature one, corresponding to the 1Mn·2H2O → 1Mn transformation, drifts to higher temperatures from 86 °C in the first cycle to 98 °C in cycles 20th–30th. It is worth noting that these temperatures are higher than in the TGA experiments because in QE-TPDA the water vapor pressure at 20 °C is 100% RH (TGA analysis is performed using dry nitrogen).
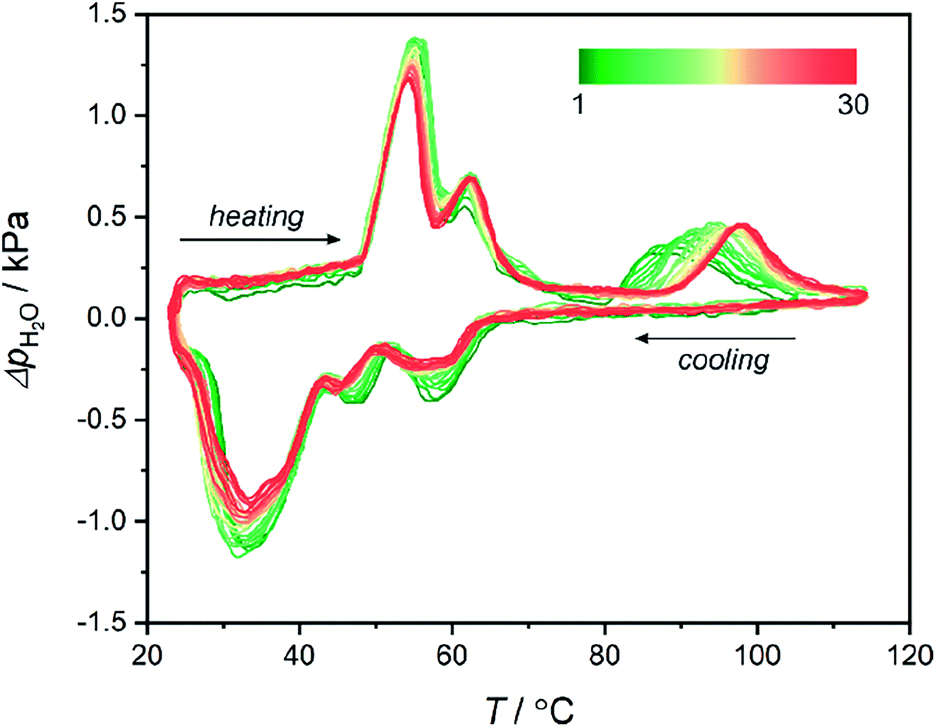 | ||
| Fig. 5 30 heating/cooling QE-TPDA profiles obtained for 1Mn·8H2O at a temperature sweep rate of 0.75 °C min−1 using H2O (2.4 kPa)/He mixture as a carrier gas. | ||
The shift in the final desorption temperature is hard to explain, as no visible decrease in water loading can be deduced from the QE-TPDA signal after 30 cycles of temperature driven desorption. Powder X-ray diffractogram obtained after the experiment (30 adsorption/desorption cycles in the 23–115 °C range) shows that the compound preserves crystallinity, but shows small unidentified reflections indicating some thermal fatigue/damage (Fig. S20†). Possibly, it may indicate minor structural changes, such as the appearance of defects in the structure affecting the last desorption step. Still, the QE-TPDA analysis confirms the exceptionally good stability of 1Mn during 23–115 °C thermal cycling in a humid environment.
EPR spectroscopy
Paramagnetic manganese(II) nodes in the structure of 1Mn enable water sorption studies using electron paramagnetic resonance (EPR) spectroscopy. Manganese(II) cations usually show a negligible contribution from the orbital momentum, sustaining sufficiently long spin relaxation times to observe the EPR spectrum at room temperature. This is accompanied by relatively small zero field splitting (ZFS) effect (in the range of the X-band microwave frequency), which is influenced by the local coordination environment of the metal ions. Therefore EPR may be utilized to track changes in manganese(II) geometry upon solvation/desolvation at room temperature and in real time. In our experiment, powdered 1Mn·8H2O was subjected to a continuous flow of nitrogen (either dry or saturated with water vapours) inside the EPR resonator (for details please refer to Experimental section). 1Mn·8H2O shows a broad signal at g = 2.00 demonstrated in Fig. 6a (scans 0–10) and Fig. S21,† which originates from the overlap of transitions between different mS states for S = 5/2.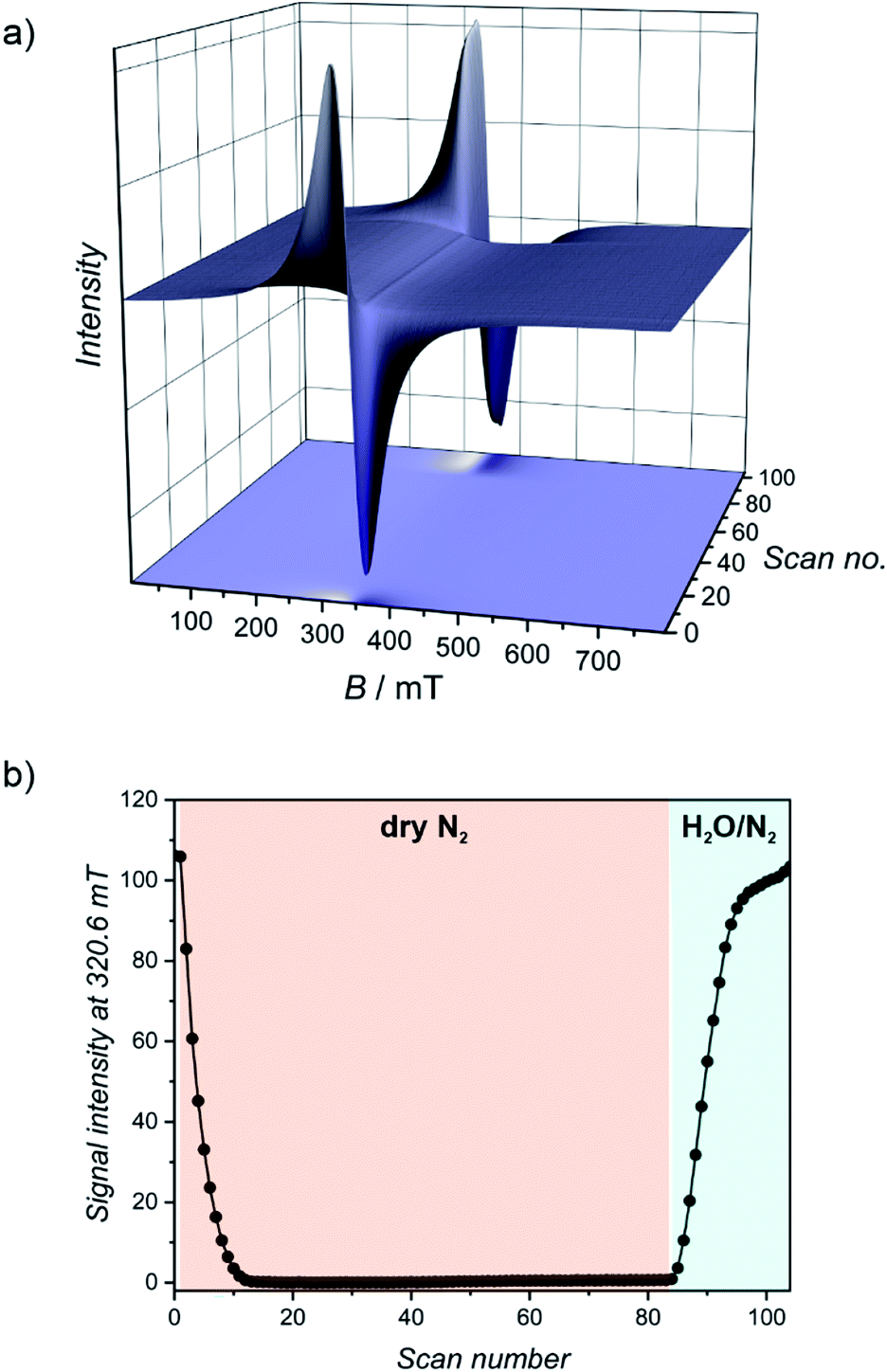 | ||
| Fig. 6 (a) Continuous wave X-band (9.7 GHz) EPR spectra obtained for 1Mn·8H2O during dehydration (scans 0–84) and rehydration (scans 85–105) at room temperature. (b) EPR signal intensity for 1Mn·8H2O upon dry N2 purging (scan numbers 0–84) and rehydration in N2 saturated with water vapor (scan numbers 85–105). | ||
Drying the sample leads to the rapid drop in the signal intensity and its broadening (Fig. 6a). The lowest EPR signal intensity at 320.6 mT is reached around scan no. 25 (Fig. 6b and S22†). The hydration stage at this step can be identified by comparison with the EPR spectra recorded for the isolated phases (Fig. S21 and S23†). It was assigned as a mixture of 1Mn·2H2O and 1Mn·3H2O, in line with the DVS measurement, in which the observed mass change in the intermediate step of dry gas dehydration can be attributed to 1Mn·2H2O (Fig. 4a). Upon further drying, the intensity of the transition increases slightly for over 50 scans (Fig. 6b), which corresponds to a slow formation of the anhydrous 1Mn. Saturating purge gas with water vapours leads to a very fast signal recovery with no observation of the intermediate steps depicted in Fig. 6, exactly as in the DVS experiment.
In order to study phase-dependent variation of ZFS in 1Mn·8H2O, we prepared its isomorphous cadmium analogue {[Cd(imH)(H2O)2]2[MoIV(CN)8]·4H2O}n (2Cd·8H2O) (Fig. S24†). Resolution of six transitions resulting from hyperfine splitting caused by manganese I = 5/2 nucleus was achieved for the diamagnetic matrix of 2Cd·8H2O doped with manganese(II) (2Cd·8H2O:Mn), but left ZFS unresolved (Fig. S25†). This suggests only minimal zero-field splitting, which was estimated using EasySpin software106 to be no larger than |D| = 0.04 cm−1. Unlike 1Mn·8H2O, dehydration of 2Cd·8H2O leads to its amorphization and decomposition, which is observed in the TGA measurement at the temperature ca. 80 °C lower than for the Mn-analogue (Fig. S3†). Nevertheless, the crystallinity of the dry amorphous solid dried under vacuum at room temperature can be restored by rehydration at 100% RH (Fig. S26†). Despite the restoration of the crystallinity, the water sorption capacity of 2Cd decreases significantly in the subsequent QE-TPDA sweeps (Fig. S27†), indicating irreversible damage of the coordination skeleton.
Amorphization is even more pronounced for 2Cd·8H2O:Mn, which after dehydration shows no diffraction peaks in the PXRD experiment (Fig. S28†). Thus, we were unable to determine the exact parameters of ZFS for 1Mn, 1Mn·2H2O and 1Mn·3H2O by studying the Mn-doped Cd-analogues as the corresponding crystalline phases could not be identified. This highlights the key role of manganese(II) for the stabilization and “breathing” performance of the CN-bridged framework upon water adsorption/desorption. Apparently, manganese(II) not only provides paramagnetic properties of the material but is responsible for the structural integrity of the framework.
Magnetic and photomagnetic properties
Results of the EPR spectroscopy are fully supported by magnetometry of 1Mn and its hydrated states, as depicted in Fig. S29 and 30.† Room-temperature χT products for all four compounds are within 8.8–9.0 cm3 K mol−1 range, as expected for two independent manganese(II) cations characterized by g = 2.0. The χT(T) curves show a distinct decrease at the lowest temperatures, which is more pronounced for more dehydrated phases, as quantified by fitting the experimental points to the Curie–Weiss law (Fig. S29,† inset) that yields the following Weiss constants: −0.7 K for 1Mn·8H2O, −1.2 K for 1Mn·3H2O, −1.5 K for 1Mn·2H2O and −3.6 K for 1Mn. It can be ascribed to increasing ZFS and/or stronger antiferromagnetic interactions, due to the decreasing distances between Mn(II) centres associated with dehydration and unit cell volume contraction. The influence of stronger antiferromagnetic interactions explains also the shape of the M(H) dependencies, showing a deviation from the Brillouin function expected for two non-interacting MnII centers which becomes more distinct with the dehydration level in the 1Mn·8H2O → 1Mn·3H2O → 1Mn·2H2O → 1Mn series (Fig. S30†). Nonetheless, all phases behave as paramagnets in the 2–300 K range, as expected for manganese(II) cations separated by the diamagnetic octacyanomolybdate(IV) groups.The octacyanomolybdate(IV) anion was previously demonstrated to behave as an intrinsic photomagnetic chromophore in several bimetallic CN-bridged systems.107–109 Therefore we decided to study the photomagnetic properties of 1Mn and 1Mn·8H2O at low temperatures. Both compounds strongly respond to 450 nm light (irradiation wavelength selected based on the UV-vis spectra; Fig. S31†) with a 13-fold increase of the magnetization in the case of 1Mn and a 16-fold increase for 1Mn·8H2O at 10 K (Fig. S32†). After irradiation, 1Mn shows clear bifurcation of ZFC-FC curves at Tc = 72 K (Fig. 7), which is slightly lower than for its octacyanotungstate(IV) analogue with Tc = 93 K.47 This behaviour is assumed to originate from the photo-induced formation of S = 1 MoIV (ref. 109) which couples magnetically with S = 5/2 MnII centres and enables ferrimagnetic ordering of the network.108 More importantly, photo-induced magnetic ordering at Tc = 40 K is also demonstrated by the 1Mn·8H2O in contrast to the lack of the photomagnetic response in the case of its [WIV(CN)8]-based analogue {[MnII(imH)(H2O)2]2[WIV(CN)8]·4H2O}n.47 However, the photomagnetic behaviour of both compounds 1Mn and 1Mn·8H2O shows signs of irradiation damage. The latter preserves the characteristics of long-range magnetic ordering even after 2 hours of thermal relaxation at 240 K (Fig. S33–36†). As far as we know, 1Mn and {[MnII(imH)]2[WIV(CN)8]}n constitute the first examples of isostructural octacyanometalate(IV)-based photomagnets in which both MoIV and WIV congeners demonstrate photo-induced magnetic ordering.41 This enables comparison of their photomagnetic functionalities, with the former responding to light even in the fully hydrated state and the latter demonstrating significantly higher magnetic ordering temperature and better reversibility of the photomagnetic effect.
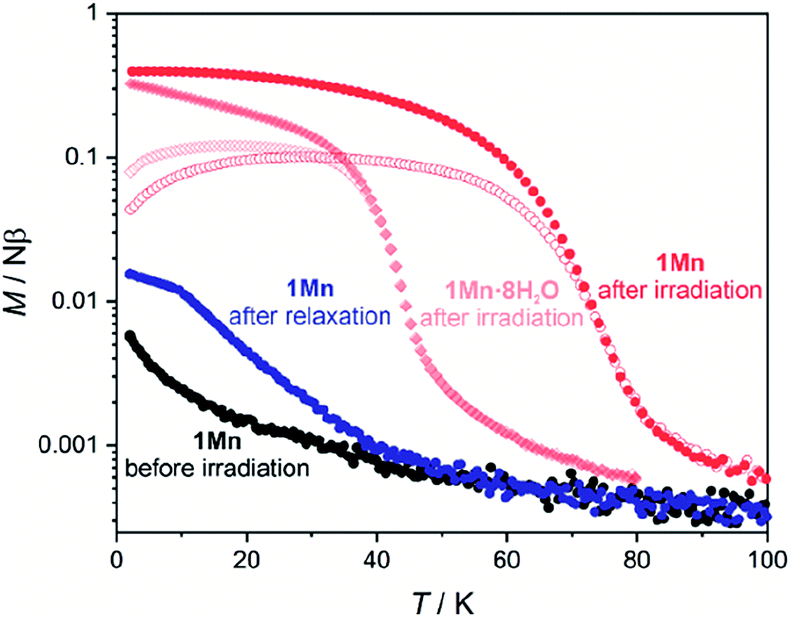 | ||
| Fig. 7 Field-cooled (closed symbols) and zero field-cooled (open symbols) curves for 1Mn (circles) and 1Mn·8H2O (rhombi) under HDC = 0.02 T. Both samples were irradiated with λ = 450 nm and P = 3–5 mW light, thermal relaxation for 1Mn was performed by heating to 315 K and stabilizing at this temperature for 1 hour. Complete magnetic/photomagnetic data for both 1Mn·8H2O and 1Mn are presented in Fig. S33 and S34 in the ESI.† | ||
Conclusions
A completely nonporous cyanide-bridged coordination polymer {[Mn(imH)]2[Mo(CN)8]}n with exceptional water sorption, stability, and cyclability is demonstrated. {[Mn(imH)]2[Mo(CN)8]}n shows outstanding breathing effect with a 34% volume increase as a result of >25% w/w water uptake. The breathing of the framework is accompanied/caused by a reversible breaking/formation of an additional Mn–N coordination bond between MnII centres and CN− ligands. This behaviour occurs in three well-defined steps and is completely reversible over tens of water vapor pressure- or temperature-swing cycles with full retention of the water sorption capacity and the crystallinity of the compound. It clearly shows that cyanide-bridged coordination polymers demonstrate competitive sorption properties, and the alleged instability of their CN-bridged coordination skeleton is merely a common belief. Simultaneous changes in the cyanide bridging pattern as well as the metal centre geometry in the presented systems significantly affect both their magnetic and photomagnetic properties. They can be also tracked in real time using EPR spectroscopy.The presented results demonstrate that cyanide-bridged coordination polymers and PBA analogues can combine exceptional sorption properties with various magnetic functionalities like photo-induced magnetization changes or long-range magnetic ordering affected by guest molecules. They constitute, therefore, a new competitive class of multifunctional sorption materials. This study re-establishes CN-CPs and PBAs as high-performance sorption materials with the potential to challenge current state-of-the-art MOFs.
Experimental section
Synthesis details
All reagents were used as supplied from commercial sources (Alfa Aesar). Potassium octacyanomolybdate(IV) was obtained according to the previously reported procedure.1102Cd and 2Cd:Mn were obtained from 2Cd·8H2O and 2Cd·8H2O:Mn respectively, which were vacuum-dried (p ≈ 10−2 mbar) over P4O10 for 12 hours at room temperature.
Single crystal X-ray diffraction
SCXRD experiments were performed for 1Mn·8H2O and 2Cd·8H2O using Bruker D8 Quest Eco Photon50 CMOS diffractometer (Mo Kα radiation, Triumph® monochromator). Single crystals were moved directly from mother liquor into cryo-oil to avoid solvent loss on air. Absorption corrections, data reduction and unit cell refinements were performed using SADABS and SAINT programs included in the Apex3 suite. The structures were solved using direct methods and refined anisotropically using weighted full-matrix least-squares on F2.112–114 Hydrogen atoms of the ligands were placed in calculated positions and refined as riding on the parent atoms. Structural diagrams were prepared using Mercury CSD 4.0.115Powder X-ray diffraction
PXRD data were obtained at room temperature for ground crystalline samples loaded into glass capillaries (0.5 mm in diameter for structural measurements, 0.7 mm for phase purity measurements). Different hydration states presented in the Fig. 1b were stabilized in the following conditions: I and VII – saturated water vapor at 298 K (RH ≈ 100%, sample in the capillary was always covered with a drop of distilled water to maintain high humidity conditions during the course of the measurement), II – saturated LiCl solution at 298 K (RH = 11.3 ± 0.3%), III – saturated KOH solution at 313 K (RH = 6.3 ± 0.4%), IV – dry argon atmosphere at 298 K, V – saturated LiCl solution at 298 K (RH = 11.3 ± 0.3%) and VI – saturated CH3COOK solution at 313 K (RH = 18.7 ± 0.5%).111 Phase purity measurements were subjected to background correction using the DIFFRAC algorithm implemented in the DIFFRAC.EVA V5 software. The measurements were carried out using Bruker D8 Advance diffractometer (Cu Kα radiation, graphite monochromator). The unit cell parameters of 1Mn, 1Mn·2H2O, 1Mn·3H2O were determined using the Winplotr and DICVOL06 indexing software.116,117 The obtained parameters were refined by fitting the experimental pattern according to the LeBail method in JANA2006.118 The structure determination was performed using a direct-space method FOX software.119 The starting models consisted of three molecular fragments: one [MoIV(CN)8]4− and two [MnII(imH)]2+ (or [MnII(imH)(H2O)]2+/[MnII(imH)(H2O)2]2+ in the case of 1Mn·2H2O and 1Mn·3H2O) with bond distances and angles based on the single crystal model of 1Mn·8H2O. The obtained preliminary models were, subsequently, refined using the JANA2006 Rietveld software keeping the fragments rigid. The final agreement factors are presented in ESI (Tables S1 and S2†).Physical characterization
The dynamic vapor sorption measurements were performed using an SMS DVS Resolution apparatus for an initial sample mass of 5–10 mg. Sample mass at each step of water sorption isotherm was assumed stable after reaching the dm/dt < 0.002 %m0 min−1 limit. The isosteric enthalpy of water adsorption for each point of adsorption isotherm at 298 K was calculated by interpolating data obtained at 313 K to the corresponding data points at 298 K (Table S3†) and then applying the van't Hoff equation: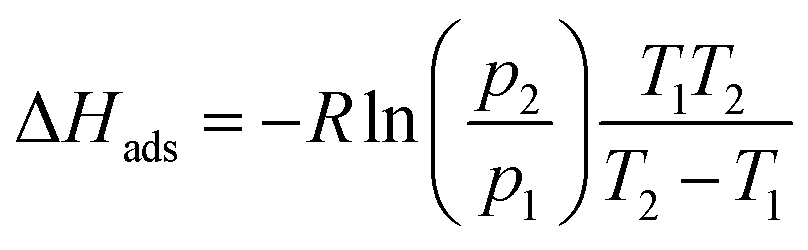
Average adsorption enthalpy was determined by integrating the obtained curve (Fig. S16†) within Δm/m0 = 1.5–25.4% and then dividing by the range. The QE-TPDA measurements were performed using a homemade thermodesorption apparatus (equipped with a VICI Microvolume TCD-2 thermal conductivity detector, electronic mass flow controllers of the carrier gas Brooks 5850, and a passive room temperature saturator) described in details elsewhere.100–104 TGA was performed using a NETZSCH TG 209 F1 Libra under a flow of nitrogen (20 mL min−1). The DSC measurement was performed with the use of a Mettler Toledo DSC 822e. After reaching 200 °C the measurement of the heating curve was repeated and the second measurement for the anhydrous sample was used as background. Average water adsorption enthalpy from calorimetric measurement was calculated by integrating the DSC curve and dividing the obtained value by the total number of water molecules. Elemental analyses were performed using an ELEMENTAR Vario Micro Cube CHNS analyzer. SEM measurements were performed using Hitachi S-4700 FE-SEM scanning electron microscope.
EPR spectroscopy
Continuous-wave EPR spectra in X-band were conducted on a Bruker Elexsys E500 spectrometer (Faculty of Chemistry, Jagiellonian University, Kraków, Poland). In situ dehydration–rehydration experiments in X-band were conducted on a Bruker Elexsys E580 spectrometer (Department of Molecular Biophysics, Jagiellonian University, Kraków, Poland). These measurements were performed in a quartz tube shaped like a Pasteur pipette. The narrow side of the tube was closed with a piece of a KIMTECH SCIENCE* Precision Wipe, on which a small amount (ca. 3 mg) of the sample was placed. Tygon® tubing was used to connect the broad side of the quartz tube with an adapter equipped with a glass stopcock, that was used to deliver purge gas to the sample. In the dehydration experiment, it was connected directly to the source of dry nitrogen gas, while in the rehydration experiment – the nitrogen gas was passed through a water bubbler.Other spectroscopic measurements
Infrared spectra were recorded using a Nicolet iN10 MX FT-IR microscope in the transmission mode (a small amount of powdered sample was spread on BaF2 pellet). Dehydration-rehydration experiments were performed with the use of a Linkam THMS350V stage. UV-vis spectra were measured in transmission mode for samples mixed with paraffin oil between two quartz slides using a PerkinElmer Lambda 35 UV/VIS spectrophotometer equipped with an integrating sphere.Magnetic and photomagnetic measurements
Magnetic susceptibility measurements were performed using a Quantum Design MPMS-3 Evercool magnetometer in magnetic fields up to 7 T for samples packed into Delrin sample holders.120 The experimental data were corrected for the diamagnetism of the sample and the sample holder. Photomagnetic measurements were performed for samples placed between two layers of scotch tape and inserted into the plastic straw. 1Mn was prepared for photomagnetic measurements in the oxygen- and water-free glovebox to prevent its rehydration and 1Mn·8H2O was inserted into the magnetometer and vacuum pumped below 240 K to avoid its dehydration. Irradiation was performed using 450 nm light produced by a laser diode (L450P1600MM; power at the sample position 6–10 mW cm−2).Data availability
CCDC 2046137 (1Mn·8H2O), 2046138 (2Cd·8H2O), 2048781 (1Mn·3H2O), 2048780 (1Mn·8H2O) and 2048779 (1Mn) contain the supplementary crystallographic data for this paper.Author contributions
M. Magott: conceptualization, funding acquisition, investigation (synthesis, X-ray diffraction, TGA, magnetic and photomagnetic measurements, part of EPR and DVS experiments), writing – original draft, writing – review & editing, B. Gaweł: investigation and formal analysis (determination of PXRD structures), M. Sarewicz: investigation and supervision (EPR), M. Reczyński: investigation and supervision (water vapour sorption measurements using DVS), writing – review & editing, K. Ogorzały: investigation (QE-TPDA), W. Makowski: funding acquisition, investigation and supervision (QE-TPDA), D. Pinkowicz: conceptualization, funding acquisition, project administration, supervision (magnetic and photomagnetic measurements), writing – review & editing. All authors reviewed and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Polish Ministry of Science and Higher Education within the Diamond Grant (0192/DIA/2017/46) and the Polish National Science Centre within the Sonata Bis 6 (2016/22/E/ST5/00055). WM gratefully acknowledges Polish National Science Centre for the financial support of the QE-TPDA characterization within the Opus 15 (2018/29/B/ST4/00328) project. The authors gratefully acknowledge Prof. Artur Osyczka for providing access to the EPR facility of the Faculty of Biochemistry, Biophysics and Biotechnology, Department of Molecular Biophysics.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal–Organic Frameworks and Covalent Organic Frameworks, Wiley-VCH Verlag GmbH & Co. KGaA, 2019 Search PubMed.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 Search PubMed.
- D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 Search PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 Search PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 Search PubMed.
- J. Zhang, W. Kosaka, Y. Kitagawa and H. Miyasaka, Nat. Chem., 2021, 13, 191–199 Search PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 Search PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 Search PubMed.
- S. Bourrelly, B. Moulin, A. Rivera, G. Maurin, S. Devautour-Vinot, C. Serre, T. Devic, P. Horcajada, A. Vimont, G. Clet, M. Daturi, J.-C. Lavalley, S. Loera-Serna, R. Denoyel, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2010, 132, 9488–9498 Search PubMed.
- Z. Lin, R. Zou, J. Liang, W. Xia, D. Xia, Y. Wang, J. Lin, T. Hu, Q. Chen, X. Wang, Y. Zhao and A. K. Burrell, J. Mater. Chem., 2012, 22, 7813–7818 Search PubMed.
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 13145–13147 Search PubMed.
- Y. Tang, D. Dubbeldam, X. Guo, G. Rothenberg and S. Tanase, ACS Appl. Mater. Interfaces, 2019, 11, 21126–21136 Search PubMed.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeulen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 2292–2301 Search PubMed.
- J. Navarro-Sánchez, A. I. Argente-García, Y. Moliner-Martínez, D. Roca-Sanjuán, D. Antypov, P. Campíns-Falcó, M. J. Rosseinsky and C. Martí-Gastaldo, J. Am. Chem. Soc., 2017, 139, 4294–4297 Search PubMed.
- V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 Search PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 Search PubMed.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430 Search PubMed.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 Search PubMed.
- A. J. Rieth, A. M. Wright, G. Skorupskii, J. L. Mancuso, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2019, 141, 13858–13866 Search PubMed.
- N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 Search PubMed.
- M. W. Logan, S. Langevin and Z. Xia, Sci. Rep., 2020, 10, 1492 Search PubMed.
- A. Khutia, H. U. Rammelberg, T. Schmidt, S. Henninger and C. Janiak, Chem. Mater., 2013, 25, 790–798 Search PubMed.
- M. F. de Lange, K. J. F. M. Verouden, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Chem. Rev., 2015, 115, 12205–12250 Search PubMed.
- A. J. Rieth, A. M. Wright, S. Rao, H. Kim, A. D. LaPotin, E. N. Wang and M. Dincă, J. Am. Chem. Soc., 2018, 140, 17591–17596 Search PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 Search PubMed.
- B. P. Biswal, S. Kandambeth, S. Chandra, D. B. Shinde, S. Bera, S. Karak, B. Garai, U. K. Kharul and R. Banerjee, J. Mater. Chem. A, 2015, 3, 23664–23669 Search PubMed.
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J.-L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 Search PubMed.
- H. L. Nguyen, N. Hanikel, S. J. Lyle, C. Zhu, D. M. Proserpio and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 2218–2221 Search PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 Search PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 Search PubMed.
- K. W. Chapman, P. D. Southon, C. L. Weeks and C. J. Kepert, Chem. Commun., 2005, 3322–3324 Search PubMed.
- C. P. Krap, J. Balmaseda, L. F. del Castillo, B. Zamora and E. Reguera, Energy Fuels, 2010, 24, 581–589 Search PubMed.
- C. P. Krap, J. Balmaseda, B. Zamora and E. Reguera, Int. J. Hydrogen Energy, 2010, 35, 10381–10386 Search PubMed.
- A. Takahashi, H. Tanaka, D. Parajuli, T. Nakamura, K. Minami, Y. Sugiyama, Y. Hakuta, S.-i. Ohkoshi and T. Kawamoto, J. Am. Chem. Soc., 2016, 138, 6376–6379 Search PubMed.
- Y. Jiang, A. Takahashi, T. Kawamoto, M. Asai, N. Zhang, Z. Lei, Z. Zhang, K. Kojima, K. Imoto, K. Nakagawa, S.-i. Ohkoshi and T. Nakamura, Chem. Commun., 2018, 54, 11961–11964 Search PubMed.
- Y. Jiang, A. Takahashi, T. Kawamoto, M. Asai, N. Zhang, Z. Lei, Z. Zhang, K. Kojima and T. Nakamura, Inorg. Chim. Acta, 2020, 501, 119273 Search PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 Search PubMed.
- N. C. Burtch, I. M. Walton, J. T. Hungerford, C. R. Morelock, Y. Jiao, J. Heinen, Y.-S. Chen, A. A. Yakovenko, W. Xu, D. Dubbeldam and K. S. Walton, Nat. Chem., 2020, 12, 186–192 Search PubMed.
- O. Stefańczyk and S.-i. Ohkoshi, Chem.–Eur. J., 2019, 25, 15963–15977 Search PubMed.
- S. Chorazy, J. J. Zakrzewski, M. Magott, T. Korzeniak, B. Nowicka, D. Pinkowicz, R. Podgajny and B. Sieklucka, Chem. Soc. Rev., 2020, 49, 5945–6001 Search PubMed.
- G. Agustí, R. Ohtani, K. Yoneda, A. B. Gaspar, M. Ohba, J. F. Sánchez-Royo, M. C. Muñoz, S. Kitagawa and J. A. Real, Angew. Chem., Int. Ed., 2009, 48, 8944–8947 Search PubMed.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 Search PubMed.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 Search PubMed.
- D. Pinkowicz, R. Podgajny, M. Bałanda, M. Makarewicz, B. Gaweł, W. Łasocha and B. Sieklucka, Inorg. Chem., 2008, 47, 9745–9747 Search PubMed.
- D. Pinkowicz, R. Podgajny, B. Gaweł, W. Nitek, W. Łasocha, M. Oszajca, M. Czapla, M. Makarewicz, M. Bałanda and B. Sieklucka, Angew. Chem., Int. Ed., 2011, 50, 3973–3977 Search PubMed.
- M. Magott, M. Reczyński, B. Gaweł, B. Sieklucka and D. Pinkowicz, J. Am. Chem. Soc., 2018, 140, 15876–15882 Search PubMed.
- J. Larionova, S. A. Chavan, J. V. Yakhmi, A. G. Frøystein, J. Sletten, C. Sourisseau and O. Kahn, Inorg. Chem., 1997, 36, 6374–6381 Search PubMed.
- O. Kahn, J. Larionova and J. V. Yakhmi, Chem.–Eur. J., 1999, 5, 3443–3449 Search PubMed.
- L. Shen, Y. Zhang and J. Uiu, J. Coord. Chem., 2006, 59, 629–635 Search PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 Search PubMed.
- C. Mellot-Draznieks, C. Serre, S. Surblé, N. Audebrand and G. Férey, J. Am. Chem. Soc., 2005, 127, 16273–16278 Search PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 Search PubMed.
- E. Coronado, M. Giménez-Marqués, G. M. Espallargas and L. Brammer, Nat. Commun., 2012, 3, 828 Search PubMed.
- I.-R. Jeon, B. Negru, R. P. Van Duyne and T. D. Harris, J. Am. Chem. Soc., 2015, 137, 15699–15702 Search PubMed.
- W. Kosaka, Z. Liu, J. Zhang, Y. Sato, A. Hori, R. Matsuda, S. Kitagawa and H. Miyasaka, Nat. Commun., 2018, 9, 5420 Search PubMed.
- L. Liu, J. A. DeGayner, L. Sun, D. Z. Zee and T. D. Harris, Chem. Sci., 2019, 10, 4652–4661 Search PubMed.
- L. Qu, H. Iguchi, S. Takaishi, F. Habib, C. F. Leong, D. M. D'Alessandro, T. Yoshida, H. Abe, E. Nishibori and M. Yamashita, J. Am. Chem. Soc., 2019, 141, 6802–6806 Search PubMed.
- J. Chen, Y. Sekine, A. Okazawa, H. Sato, W. Kosaka and H. Miyasaka, Chem. Sci., 2020, 11, 3610–3618 Search PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 Search PubMed.
- A. M. Sheveleva, D. I. Kolokolov, A. A. Gabrienko, A. G. Stepanov, S. A. Gromilov, I. K. Shundrina, R. Z. Sagdeev, M. V. Fedin and E. G. Bagryanskaya, J. Phys. Chem. Lett., 2014, 5, 20–24 Search PubMed.
- M. Mendt, F. Gutt, N. Kavoosi, V. Bon, I. Senkovska, S. Kaskel and A. Pöppl, J. Phys. Chem. C, 2016, 120, 14246–14259 Search PubMed.
- A. T. Gallagher, C. D. Malliakas and T. D. Harris, Inorg. Chem., 2017, 56, 4654–4661 Search PubMed.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 Search PubMed.
- J. Li, X. Han, X. Zhang, A. M. Sheveleva, Y. Cheng, F. Tuna, E. J. L. McInnes, L. J. McCormick McPherson, S. J. Teat, L. L. Daemen, A. J. Ramirez-Cuesta, M. Schröder and S. Yang, Nat. Chem., 2019, 11, 1085–1090 Search PubMed.
- Y. Jiang, J. Huang, B. Kasumaj, G. Jeschke, M. Hunger, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 2058–2059 Search PubMed.
- A. Kultaeva, V. Bon, M. S. Weiss, A. Pöppl and S. Kaskel, Inorg. Chem., 2018, 57, 11920–11929 Search PubMed.
- A. S. Poryvaev, A. M. Sheveleva, P. A. Demakov, S. S. Arzumanov, A. G. Stepanov, D. N. Dybtsev and M. V. Fedin, Appl. Magn. Reson., 2018, 49, 255–264 Search PubMed.
- M. Hunger and J. Weitkamp, Angew. Chem., Int. Ed., 2001, 40, 2954–2971 Search PubMed.
- A. Brückner, Chem. Soc. Rev., 2010, 39, 4673–4684 Search PubMed.
- K. Sobańska, P. Pietrzyk and Z. Sojka, ACS Catal., 2017, 7, 2935–2947 Search PubMed.
- P. Pietrzyk, K. Góra-Marek, T. Mazur, B. Mozgawa, M. Radoń, M. Chiesa, Z. Zhao and Z. Sojka, J. Catal., 2021, 394, 206–219 Search PubMed.
- H. Reinsch, M. A. van der Veen, B. Gil, B. Marszalek, T. Verbiest, D. de Vos and N. Stock, Chem. Mater., 2013, 25, 17–26 Search PubMed.
- G. Wißmann, A. Schaate, S. Lilienthal, I. Bremer, A. M. Schneider and P. Behrens, Microporous Mesoporous Mater., 2012, 152, 64–70 Search PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 Search PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. L. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 Search PubMed.
- A. J. Rieth, S. Yang, E. N. Wang and M. Dincă, ACS Cent. Sci., 2017, 3, 668–672 Search PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 Search PubMed.
- P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325–330 Search PubMed.
- K. Roztocki, F. Formalik, A. Krawczuk, I. Senkovska, B. Kuchta, S. Kaskel and D. Matoga, Angew. Chem., Int. Ed., 2020, 59, 4491–4497 Search PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 Search PubMed.
- J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102–3111 Search PubMed.
- B.-Q. Song, Q.-Y. Yang, S.-Q. Wang, M. Vandichel, A. Kumar, C. Crowley, N. Kumar, C.-H. Deng, V. GasconPerez, M. Lusi, H. Wu, W. Zhou and M. J. Zaworotko, J. Am. Chem. Soc., 2020, 142, 6896–6901 Search PubMed.
- Q.-L. Wang, H. Southerland, J.-R. Li, A. V. Prosvirin, H. Zhao and K. R. Dunbar, Angew. Chem., Int. Ed., 2012, 51, 9321–9324 Search PubMed.
- S. Tanase, F. Tuna, P. Guionneau, T. Maris, G. Rombaut, C. Mathonière, M. Andruh, O. Kahn and J.-P. Sutter, Inorg. Chem., 2003, 42, 1625–1631 Search PubMed.
- S.-i. Ohkoshi, Y. Tsunobuchi, H. Takahashi, T. Hozumi, M. Shiro and K. Hashimoto, J. Am. Chem. Soc., 2007, 129, 3084–3085 Search PubMed.
- T. Yoshihide, H. Kazuhito, S. Motoo, H. Toshiya and O. Shin-ichi, Chem. Lett., 2007, 36, 1464–1465 Search PubMed.
- M. Reczyński, S. Chorazy, B. Nowicka, B. Sieklucka and S.-i. Ohkoshi, Inorg. Chem., 2017, 56, 179–185 Search PubMed.
- K. Imoto, D. Takahashi, Y. Tsunobuchi, W. Kosaka, M. Arai, H. Tokoro and S.-i. Ohkoshi, Eur. J. Inorg. Chem., 2010, 2010, 4079–4082 Search PubMed.
- K. Komori-Orisaku, O. Stefańczyk, S. Ohishi, N. Ozaki, Y. Miyamoto, K. Imoto and S.-i. Ohkoshi, Chem.–Eur. J., 2019, 25, 11066–11073 Search PubMed.
- B. Nowicka, M. Rams, K. Stadnicka and B. Sieklucka, Inorg. Chem., 2007, 46, 8123–8125 Search PubMed.
- B. Nowicka, M. Bałanda, B. Gaweł, G. Ćwiak, A. Budziak, W. Łasocha and B. Sieklucka, Dalton Trans., 2011, 40, 3067–3073 Search PubMed.
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521–3529 Search PubMed.
- B. Bueken, H. Reinsch, N. Reimer, I. Stassen, F. Vermoortele, R. Ameloot, N. Stock, C. E. A. Kirschhock and D. De Vos, Chem. Commun., 2014, 50, 10055–10058 Search PubMed.
- F.-X. Coudert, A. U. Ortiz, V. Haigis, D. Bousquet, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, N. Geoffroy, J.-P. Bellat, G. Ortiz, G. Chaplais, J. Patarin and A. Boutin, J. Phys. Chem. C, 2014, 118, 5397–5405 Search PubMed.
- S. Devautour-Vinot, G. Maurin, F. Henn, C. Serre and G. Férey, Phys. Chem. Chem. Phys., 2010, 12, 12478–12485 Search PubMed.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 Search PubMed.
- S. Vyazovkin, A. K. Burnham, J. M. Criado, L. A. Pérez-Maqueda, C. Popescu and N. Sbirrazzuoli, Thermochim. Acta, 2011, 520, 1–19 Search PubMed.
- R. L. Blaine and H. E. Kissinger, Thermochim. Acta, 2012, 540, 1–6 Search PubMed.
- W. Makowski, Thermochim. Acta, 2007, 454, 26–32 Search PubMed.
- W. Makowski, B. Gil and D. Majda, Catal. Lett., 2008, 120, 154–160 Search PubMed.
- M. Mańko, B. Gil, R. Janus, P. Kuśtrowski and W. Makowski, Thermochim. Acta, 2010, 511, 82–88 Search PubMed.
- K. Roztocki, M. Lupa, A. Sławek, W. Makowski, I. Senkovska, S. Kaskel and D. Matoga, Inorg. Chem., 2018, 57, 3287–3296 Search PubMed.
- A. Sławek, J. M. Vicent-Luna, B. Marszałek, B. Gil, R. E. Morris, W. Makowski and S. Calero, Chem. Mater., 2018, 30, 5116–5127 Search PubMed.
- R. Jankowski, M. Reczyński, S. Chorazy, M. Zychowicz, M. Arczyński, M. Kozieł, K. Ogorzały, W. Makowski, D. Pinkowicz and B. Sieklucka, Chem.–Eur. J., 2020, 26, 11187–11198 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 Search PubMed.
- N. Bridonneau, J. Long, J. L. Cantin, J. von Bardeleben, S. Pillet, E. E. Bendeif, D. Aravena, E. Ruiz and V. Marvaud, Chem. Commun., 2015, 51, 8229–8232 Search PubMed.
- M. Magott, O. Stefańczyk, B. Sieklucka and D. Pinkowicz, Angew. Chem., Int. Ed., 2017, 56, 13283–13287 Search PubMed.
- X. Qi, S. Pillet, C. de Graaf, M. Magott, E.-E. Bendeif, P. Guionneau, M. Rouzières, V. Marvaud, O. Stefańczyk, D. Pinkowicz and C. Mathonière, Angew. Chem., Int. Ed., 2020, 59, 3117–3121 Search PubMed.
- G. Handzlik, M. Magott, B. Sieklucka and D. Pinkowicz, Eur. J. Inorg. Chem., 2016, 2016, 4872–4877 Search PubMed.
- L. Greenspan, J. Res. Natl. Bur. Stand., Sect. A, 1977, 81, 89–96 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 Search PubMed.
- T. Roisnel and J. Rodríquez-Carvajal, Mater. Sci. Forum, 2001, 378–381, 118–123 Search PubMed.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724–731 Search PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 Search PubMed.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 Search PubMed.
- M. Arczyński, J. Stanek, B. Sieklucka, K. R. Dunbar and D. Pinkowicz, J. Am. Chem. Soc., 2019, 141, 19067–19077 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural diagrams, TGA profiles, PXRD patterns, water adsorption isotherms, DSC curves, EPR, IR and UV-vis spectra, magnetic and photomagnetic data and CIF files. CCDC [2046137, 2046138, 2048781, 2048780 and 2048779]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02060a |
| This journal is © The Royal Society of Chemistry 2021 |