
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combination of solid-state NMR and MD simulations reveals the binding mode of a rhomboid protease inhibitor†
Claudia
Bohg‡
a,
Carl
Öster‡
 a,
Tillmann
Utesch‡
b,
Susanne
Bischoff
a,
Sascha
Lange
a,
Chaowei
Shi
ac,
Han
Sun
*b and
Adam
Lange
*ad
a,
Tillmann
Utesch‡
b,
Susanne
Bischoff
a,
Sascha
Lange
a,
Chaowei
Shi
ac,
Han
Sun
*b and
Adam
Lange
*ad
aDepartment of Molecular Biophysics, Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Straße 10, 13125 Berlin, Germany. E-mail: alange@fmp-berlin.de
bStructural Chemistry and Computational Biophysics Group, Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Straße 10, 13125 Berlin, Germany. E-mail: hsun@fmp-berlin.de
cHefei National Laboratory for Physical Sciences at the Microscale, School of Life Sciences, University of Science and Technology of China, Huangshan Road 443, Hefei 230027, People's Republic of China
dInstitut für Biologie, Humboldt-Universität zu Berlin, Invalidenstraße 42, 10115 Berlin, Germany
First published on 1st September 2021
Abstract
Intramembrane proteolysis plays a fundamental role in many biological and pathological processes. Intramembrane proteases thus represent promising pharmacological targets, but few selective inhibitors have been identified. This is in contrast to their soluble counterparts, which are inhibited by many common drugs, and is in part explained by the inherent difficulty to characterize the binding of drug-like molecules to membrane proteins at atomic resolution. Here, we investigated the binding of two different inhibitors to the bacterial rhomboid protease GlpG, an intramembrane protease characterized by a Ser–His catalytic dyad, using solid-state NMR spectroscopy. H/D exchange of deuterated GlpG can reveal the binding position while chemical shift perturbations additionally indicate the allosteric effects of ligand binding. Finally, we determined the exact binding mode of a rhomboid protease-inhibitor using a combination of solid-state NMR and molecular dynamics simulations. We believe this approach can be widely adopted to study the structure and binding of other poorly characterized membrane protein–ligand complexes in a native-like environment and under physiological conditions.
Introduction
Rhomboid proteases are intramembrane serine proteases that cleave transmembrane substrates within the lipid bilayer. Although the structural features of rhomboid proteases have been thoroughly studied,1–7 a 3D structure of an enzyme-substrate complex is still a major goal in the field. A large number of investigations have focused so far on E. coli GlpG, which has become a model system for rhomboid proteases. Its crystal structure reveals a six transmembrane helix (TM) core2 connected by several mechanistically important loops (Fig. 1A). The catalytic dyad composed of serine 201 (TM4) and histidine 254 (TM6) is deeply buried in the enzyme facing the extracellular side. It is still under debate whether TM5 acts as a mobile substrate gate,5 an idea which is supported by several studies revealing L4, TM5 and L5 as dynamic hotspots.8–11 The L1 loop is thought to be involved in the orientation of the enzyme within the lipid bilayer,4,12 whereas the L5 loop acts as a cap above the active site and possibly contributes to substrate binding.13 Models of substrate cleavage have been proposed based on crystal structures of GlpG with small peptide inhibitors11,12,14 and MD simulations10 of substrate binding to the exosite. The exact mechanisms of substrate recognition, binding, and cleavage are, however, still under debate. | ||
| Fig. 1 (A) Crystal structure of GlpG (PDB ID: 2IC8) with highlighted structural features (active site green, loop regions yellow, transmembrane helices 2 and 5 blue). (B) and (C) Chemical structures of 2-styryl-4H-3,1-benzoxazin-4-one (SBO, B) and 7-amino-4-chloro-3-methoxy-1H-2-benzopyran (JLK6, C). SBO and JLK6 inhibit GlpG reversibly and irreversibly, respectively. | ||
Rhomboid proteases fulfill a wide range of biological functions and therefore represent potential new drug targets for the treatment of several life-threatening diseases, including malaria, diabetes, and Parkinson's disease.15 Phosphonofluoridates16 and isocoumarins,3 were discovered as the first inhibitors of GlpG, followed by β-lactams17 and β-lactones,18 however they turned out to be non-selective weak binders and unfavorable for cell biology applications. More recently, saccharins19 and benzoxazinones20 were shown to exhibit stronger binding affinities. Additionally, peptidyl ketoamides14 have been suggested as selective and promising inhibitors for drug development.
Inhibitor research in a structural biology context often relies on crystallization of the protein–ligand complexes, which is highly challenging for membrane proteins and requires detergents to mimic the lipid environment. Quite often crystallization is also hindered by addition of an inhibitor that may disrupt the crystal. Solid-state NMR, however, does not require crystallization. The protein and its interactions with a ligand can be studied under native-like conditions in a lipid bilayer, under physiological temperatures and buffer conditions.21–26 This is of particular importance since rhomboid proteases are highly dependent on their lipid environment, which can alter their cleavage activity and specificity.4,27,28
Previously, we have shown that we can achieve high quality, 1H-detected solid-state NMR spectra of GlpG in liposomes, enabling a residue-specific structural and dynamic investigation.91H-detected solid-state NMR has become a valuable approach to study membrane proteins due to the higher sensitivity compared to 13C detection and the additional information available on H/D exchange and water accessibility.29–32 Here we used it in combination with molecular docking and molecular dynamics (MD) simulations to investigate the effects of inhibitor binding on GlpG.
Results and discussion
To investigate ligand binding in GlpG we chose 7-amino-4-chloro-3-methoxy-1H-2-benzopyran (referred to as JLK6), which is an isocoumarin-derived inhibitor33 (Fig. 1C). It binds irreversibly by forming covalent bonds to the two catalytic dyad residues of GlpG.34 We also investigated a reversibly binding inhibitor, 2-styryl-4H-3,1-benzoxazin-4-one (referred to as SBO),20 which is a structural homologue of JLK6 (Fig. 1B). Previously, a crystallographic study revealed the binding mode of JLK6 (ref. 34) (PDB ID: 2XOW, see Fig. S1†). A docking study suggested the binding pose of SBO,20 but no co-crystal structure has been determined so far.GlpG cleaves rhomboid protease substrates in a highly unspecific manner. No endogenous substrate is known, but GlpG has been shown to cleave several other substrates.35–37 To verify the inhibitory effects of the two substances JLK6 and SBO, we used TatA of P. stuartii as a model substrate. We incubated purified GlpG and TatA in liposomes with inhibitors SBO or JLK6 at different concentrations (5–50× molar excess, see methods). Both inhibitors bind the catalytic dyad (S201 and H254) and prevent substrate interaction (Fig. S2; see also Fig. S3† for a scheme).
We then recorded a series of 1H–15N 2D solid-state NMR spectra of GlpG with and without inhibitors. Notably, the spectral quality of the GlpG–inhibitor complexes is slightly reduced compared to the one of apo-GlpG (Fig. S4†), which may be attributed to conformational heterogeneity of the protein caused by ligand binding. We also observed that some peaks are missing in the spectra of inhibitor-bound GlpG. To identify which residues are affected by inhibitor binding we acquired, in addition to the 1H–15N 2D spectra, three 3D spectra: (H)CαNH, (H)CONH and (H)Cα(CO)NH.38 This was sufficient to transfer our previously published assignments (BMRB ID: 27776)9 of apo-GlpG to GlpG-JLK6 and GlpG-SBO (Fig. S5†). As JLK6 binds irreversibly to GlpG, we can either add the ligand before the dialysis step used for reconstitution or after GlpG has been inserted into a lipid bilayer. By comparing samples prepared in these two different ways we obtained valuable information about residues that were protected from H/D exchange due to inhibitor binding. In the sample where JLK6 is added during the dialysis step, peaks corresponding to the catalytic dyad (S201 and H254) and residues close by (G199, L200, G202, V203, I255 and G257) are not visible. Additionally, peaks are missing for all residues in the gating helix (TM5), the residues at the top of TM3 (K191, F192 and S193), H150 (TM2, part of the oxyanion hole), W196 (L3) and parts of L4 (Q220 and L225). Unexpectedly, the peaks of two residues that are far away from the binding site of JLK6, I113 at the top of TM1 and W136 in loop L1, are also missing. It should be noted that W136 is a highly conserved residue in rhomboid proteases, forming the WR motif together with R137. It was shown that a mutation of W136 led to a significant reduction of the enzyme activity.39
When JLK6 is added after the dialysis step some of these peaks have “re-appeared” because during the time in which H/D exchange takes place no inhibitor was present (indicated on the structure in Fig. 2B). Fig. 2C shows the 2D NCα projections from the 3D (H)CαNH spectra of apo-GlpG (left) and GlpG bound to JLK6 (right) with disappearing and reappearing peaks indicated. All the residues of and around the catalytic dyad, except for H254 and I255 are visible in the sample where JLK6 was added after dialysis. The catalytic residue S201, that is only visible as a weak peak in apo-GlpG, appears stronger in GlpG bound to JLK6 suggesting that the inhibitor stabilizes the backbone of S201 when binding (see also Fig. S6†). F192 at the top of TM3 is visible as a very weak and noisy peak, but K191 and S193 are still not visible. For TM5 only I237 and L229 are visible, suggesting that inhibitor binding affects the dynamics of this region.
 | ||
| Fig. 2 Inhibitor binding observed by solid-state NMR. (A) HNCα chemical shift perturbations (CSPs) between GlpG alone and GlpG bound to JLK6 plotted as a function of residue. Dark blue bars indicate residues not visible in spectra of GlpG bound to JLK6. (B) Residues that are protected from H/D exchange by JLK6 (H150, F192, W196, S201, L229, I237 and G257) are plotted onto the crystal structure of GlpG bound to JLK6 (PDB ID: 2XOW). Sidechains that are close in space to JLK6 are shown as sticks (H150, Q189, F197, S201, W236, F245 and H254). (C) 2D NCα projections from 3D (H)CαNH experiments of GlpG alone (left) and GlpG bound to JLK6 (right), with disappearing and reappearing peaks indicated. (D) Crystal structure of GlpG bound to JLK6 (PDB ID: 2XOW) with HNCα CSPs between GlpG alone and GlpG bound to JLK6 plotted onto the structure. The CSPs are colour coded by increasing value. Residues for which HN assignments are missing are shown in grey and residues for which the peaks have disappeared in the spectra of GlpG bound to JLK6 are shown in dark blue. | ||
In addition to the different H/D exchange patterns, we analysed the chemical shift changes caused by inhibitor binding (see Fig. 2A, D and Table S4† for H, N and Cα chemical shift perturbations (CSPs)). The lower parts of TM2 (G163 to L169), TM3 (G170 to I175) and TM4 (L213 to D218) are unaffected by inhibitor binding. The strongest effect is seen for residue I223 in loop L4, but the rest of L4 is mostly unaffected. Several residues surrounding the binding site of JLK6 show significant chemical shift changes (Q189, F192, G199, L200, S201, N251, G252, A253 and A256). Side chains of A182, S185, Q189, F197 and A253 form the S1 subsite, the water retention site of the enzyme channelling the water and thereby enabling the cleavage mechanism.34 Residues of TM2 and TM5 are part of the exosite of the enzyme that facilitates the first contact with the substrate.40 Other important contact points can be found in L5 (e.g. L244–M247)11 which are unfortunately not visible in our spectra, most likely due to structural dynamics at the μs to ms time scale causing conformational heterogeneity. Additionally, strong effects are seen for several residues in L1. Larger CSPs suggest a change in the local environment caused by inhibitor binding. This can either be through direct interactions with the inhibitor or by allosteric effects. Only a minor structural rearrangement of TM5 was observed in the crystal structure of GlpG bound to JLK6, but according to our data the dynamics of TM5 are affected. Since most residues in TM5 are not visible in our spectra we cannot conclude whether any structural changes occur for TM5, but the ones that are visible (L229 and I237) have rather small CSPs. In contrast to the crystal structure, where no significant structural changes were observed for L1, we observe large chemical shift changes for several residues in the L1 loop. Mutations of the hydrophobic residues (F133, F135, Y138, F139 and L143) in L1 with direct lipid contact were previously shown to decrease the activity of the enzyme, probably through a destabilization of the enzyme within the lipid bilayer.8 It was also observed that many residues of the L1 loop are involved in stabilizing hydrogen bonds e.g. H141 with G199. Furthermore, it was suggested that the L1 loop is involved in substrate interaction, forming the S4 subsite together with residues of L3.12 It is possible that the chemical shift changes we observe for L1 stem from rearrangements of the interactions between L1 and lipids in the bilayer. Such a rearrangement would not be observed in crystal structures of detergent solubilized membrane proteins that lack a lipid bilayer, highlighting the importance of studies under more native-like conditions. However, further investigations are needed to clarify, with certainty, the biological importance of the structural rearrangement observed in L1. Finally, we noticed a few additional peaks in the inhibitor bound samples that were not visible in GlpG alone. It is unfortunately not possible to unambiguously assign these peaks but it seems likely that they originate from flexible regions of GlpG that become more rigid upon inhibitor binding, e.g. loop L5.
Using molecular docking, we next predicted the covalent binding mode of the reversible inhibitor SBO to GlpG using JLK6 in complex with GlpG (PDB ID: 2XOW) as a structural template (Fig. S3†). This approach was reassured by the highly similar chemical shifts observed in GlpG-SBO and GlpG-JLK6 (Tables S2 to S4†). The two best docking poses of SBO (poseA and poseB) (Fig. 3A) were further refined in atomistic MD simulations. Three independent replicas of 500 ns MD simulations for each pose were performed using the Amber force field. Root-mean-square deviation (RMSD) analysis of the protein backbone revealed that GlpG remained stable in both poses (Fig. S7†). The stability of the inhibitor in the binding pocket, however, was strongly dependent on the starting pose. While poseB stayed nearly unchanged (Fig. 3B, yellow), the inhibitor in poseA changed its overall orientation and ended in a binding pose similar to poseB (Fig. 3B, titled poseB-like, red). Therefore, we identify poseB as the preferred binding pose of SBO in GlpG. In this binding mode, the ligand is embedded in a hydrophobic pocket created by F146, H150, Q189, F197, W236, and F245, while a weak hydrogen bond may be formed between the carbonyl oxygen close to the styryl group of SBO and the ε-nitrogen of H254 (Fig. 3C and S8†). Note however, that the exact protonation state of H254 is unclear.
 | ||
| Fig. 3 Computational investigation of the GlpG-SBO complex. (A) Two docking poses, poseA (red) and poseB (yellow), were identified by molecular docking. (B) Selected snapshots after 500 ns MD simulations starting from the poseA and poseB structures and ending with poseB-like (red) and poseB (yellow) binding modes. (C) Key protein–ligand interactions in the poseB binding mode. Amino acids adjacent to the SBO ligand are highlighted as sticks. Other residues of the protein are indicated as grey surface volume. (D) Changes in the water accessibility of the backbone amide protons derived from the MD simulations with and without the binding of SBO. For the evaluation of water accessibility only the last 100 ns of the individual trajectories were included. (E) The difference of root-mean-square deviation per residue, and (F) the difference of root-mean-square fluctuation of the Cα atoms are highlighted on the protein structure. For (D–F), red and blue coloured regions indicate a decrease and increase of inhibitor-bound GlpG (poseB) compared to apo-GlpG (starting structure PDB ID: 2NRF), respectively. All values are averaged over three inhibitor-bound MD simulations. For the apo-GlpG, 10 MD simulations at the same timescale were performed. The maximum and minimum values used for the colour coding were kept at ±0.1 (D) and ±0.03 (E) for better visualization. The L5 cap gave significantly larger ΔRMSD (>1 nm) and ΔRMSF (>0.15 nm) values since it adopts the open cap conformation in the apo state and the closed conformation in the inhibitor bound state. | ||
In order to further validate the predicted binding mode of SBO (poseB), we related the calculated water accessibility of the backbone amide protons (N–H) to the experimental H/D exchange pattern (Fig. 2B). For this, changes in the water accessibility between poseB and apo-GlpG in the closed conformation were determined (Fig. 3D). These differences describe the effect on the water accessibility upon ligand binding and can be thus directly compared to the H/D exchange pattern derived from the solid-state NMR experiments. Nevertheless, it should be noted that the experimental H/D exchange difference could only be determined for the ligand JLK6, which is a structural homologue of SBO but binds irreversibly to the protein. Water adjacent to the N–H of F192, S201, and L229 was only detected in the absence of SBO in the simulations, which agrees well with the protection from H/D exchange observed experimentally (Fig. 2B). Furthermore, the water accessibility of G252 slightly decreased after the inhibitor was added but remained solvent accessible. No contact to water was observed for the N–H of I237 for either of the simulations (with or without SBO). The H/D exchange observed experimentally for I237 and other TM5 residues in apo-GlpG was previously attributed to opening and closing of the TM5 gate,9 which is too slow to be observed in the MD simulations. The only discrepancy between the simulations and the solid-state NMR data was found for H150 and W196. While experimentally both residues were protected from H/D exchange after JLK6 binding, in the simulations the water accessibility was increased when SBO was bound. This disagreement between the simulations and the NMR experiments might be a result of the structural differences between SBO and JLK6 and the minor differences in their binding modes. This includes, among other things, a rotation of H150 and the orientation of W196, which faces SBO but does not point towards JLK6.
Additionally, we calculated the differences in root-mean-square deviations (ΔRMSD, Fig. 3E) and root-mean-square fluctuations (ΔRMSF, Fig. 3F) between simulations of apo-GlpG and SBO-bound GlpG. The ΔRMSD and ΔRMSF can be compared to the CSPs derived from the solid-state NMR experiments (see Fig. S9 and S10† for a direct comparison). In general, the CSP pattern between apo-GlpG and SBO-bound GlpG is remarkably congruent with the residue-wise ΔRMSD and ΔRMSF values. Specifically, TM5, the extracellular parts of TM2 and TM3, and the loops L1 and L5 showed the largest structural mobility changes, which is in accordance with the determined CSPs. More precisely, residues D116–E118 in L1 showed a significantly higher fluctuation (RMSF) in the SBO-bound state compared to the apo state, while the central L1 region including residues F133–F135 underwent structural changes in both states compared to the corresponding initial X-ray structures (ΔRMSD ∼ 0.3 nm). Furthermore, TM3 (Q189–F192) showed decreased RMSD and RMSF values in the SBO bound state compared to the apo state, which could be caused by direct contacts between this region and the inhibitor leading to immobilization. The inhibitor binding pocket (G198–V203) did not exhibit prominent structural and dynamic differences between apo-GlpG and SBO-bound GlpG, suggesting that the CSPs observed in this region are directly caused by interactions with the inhibitor. The extracellular part of TM6 (N251–H254) also showed a drastic decrease in mobility in SBO-bound GlpG, which could be explained by direct interactions between SBO and L5 and TM6, where H254 may form a weak hydrogen bond to SBO (Fig. 3C).
The previous docking model of GlpG bound to a benzoxazinone similar to SBO20 is more in agreement with the second docking pose (poseA, red in Fig. 3A), which covered a different area with its styryl group, than our final models proposed by MD simulations (poseB, yellow in Fig. 3A and B, and poseB-like, red in Fig. 3B). According to our solid-state NMR data SBO interacts with residues in TM3 and L3. In the poseA docking model interactions between SBO and residues of TM3 and L3 (specifically Q189 and F197 in the binding pocket) are missing, indicating that poseA is not consistent with our solid-state NMR data. Therefore, we propose poseB as the most probable binding pose of SBO in complex with GlpG. Furthermore, the observed instability of poseA and the transformation to a poseB-like conformation agrees very well with the binding pose of JLK6 in the crystal structure. Nevertheless, there are some slight differences between the binding modes of JLK6 and SBO in complex with GlpG: the styryl group of SBO is pointing towards Q189 and F197 (see Fig. 3C) but for JLK6 the amino group that is in close proximity with F197 does not directly interact with Q189 (see Fig. S8† for a comparison of the key ligand–protein interactions between JLK6 and SBO). A direct comparison of the chemical shifts between JLK6 bound and SBO bound GlpG reveals that the largest differences are seen for residues around Q189, with a chemical shift difference of 0.84 ppm for the Cα of Q189 being the most pronounced (see Fig. S11, Tables S2 and S3†).
Conclusions
In summary, we have shown that solid-state NMR spectroscopy provides valuable information on inhibitor binding to membrane embedded enzymes. So far, there are few examples where the power of solid-state NMR has been demonstrated in determining the exact binding mode of drug-like molecules with membrane proteins.41,42 Especially, we could show in this study that H/D exchange patterns enable mapping of the interaction site, while CSPs give information on both binding interactions and allosteric effects. Furthermore, it is often difficult to obtain crystal structures of small molecules bound to membrane embedded proteins and we have shown here that solid-state NMR experiments of GlpG–inhibitor complexes are straight-forward for both reversible (SBO) and irreversible (JLK6) inhibitors. The combination of solid-state NMR and MD simulations constitutes a promising approach to study substrate binding to membrane proteins.41,42 We employed this combined strategy here to investigate the structural and dynamic effects caused by inhibitor binding to a membrane-embedded enzyme on the atomic level. There are many examples of membrane proteins with excellent solid-state NMR spectral features and for those systems the applied approach should be similarly useful.Experimental
Expression & purification of isotopically labeled GlpG samples
The GlpG core domain (GlpGΔN, i.e. residues 87–276) with a hexahistidine-tag was expressed and purified as described before.9 Briefly, GlpGΔN in pet15b (Novagen) was transformed into Tuner™ (DE3) pLysS competent cells (Novagen). To produce the [2H, 13C, 15N]-labelled GlpGΔN samples, 15NH4Cl and 13C6–D7-glucose (Cambridge Isotope Laboratories, USA) were used as the sole nitrogen and carbon sources in a perdeuterated M9 medium.Cells were grown in M9 medium at 37 °C until an OD of 0.8 was reached and overexpressed after induction with 500 μM isopropyl-β-D-thiogalactopyranosid (IPTG) for 15 h at 25 °C. Cells were harvested, resuspended in lysis buffer and lysed using an LM10 microfluidizer (Microfluidics, USA) with 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi working pressure. Insoluble parts were removed by centrifugation and the supernatant was incubated with 2% (w/v) n-decyl-β-maltoside (DM, Glycon, Germany) for 2 h at 4 °C. The solubilized protein was purified via cobalt-based affinity chromatography on an ÄKTA™ Pure 25 System (GE Healthcare, Germany). Note that fully protonated buffers were used after the cells were harvested, leading to back-exchange of exchangeable protons in the [2H, 13C, 15N]-labelled GlpGΔN samples.
000 psi working pressure. Insoluble parts were removed by centrifugation and the supernatant was incubated with 2% (w/v) n-decyl-β-maltoside (DM, Glycon, Germany) for 2 h at 4 °C. The solubilized protein was purified via cobalt-based affinity chromatography on an ÄKTA™ Pure 25 System (GE Healthcare, Germany). Note that fully protonated buffers were used after the cells were harvested, leading to back-exchange of exchangeable protons in the [2H, 13C, 15N]-labelled GlpGΔN samples.
Reconstitution
The rhomboid protease was reconstituted into E. coli total lipid extract liposomes. For this, E. coli total lipid extract (Avanti Polar Lipids, USA) was dissolved in dialysis buffer (50 mM Tris, 150 mM NaCl, pH 7.4) containing 3% DM and added to the purified GlpGΔN sample at a lipid/protein ratio of 30:1 (mol/mol). Detergent was removed by dialysis (Spectra/Por® 1 dialysis tubing, MWCO 6000–8000) against 100-fold dialysis buffer over the course of 10 days and with buffer exchanges every 2 days until the sample was completely turbid. To facilitate detergent removal 0.5 g L−1 Bio-Beads SM-2 resin (Biorad) were added to the dialysis buffer.
Treatment with inhibitors
7-Amino-4-chloro-3-methoxy-1H-2-benzopyran (JLK6, Tocris Bioscience, UK) and 2-styryl-4H-3,1-benzoxazin-4-one (SBO, AKos Consulting & Solutions GmbH, Germany) were dissolved in dimethyl sulfoxide (DMSO) to a 100 mM stock solution. JLK6 was added to GlpG after dialysis (when the protein was in a lipid environment) at an inhibitor:protein molar ratio of 10:1. SBO was added to GlpG after dialysis at an inhibitor:protein molar ratio of 5:1. A molar ratio of 50:1 was also tested to check whether the NMR spectra would change but no such change was observed. The final DMSO concentration was less than 0.2% (v/v). To test whether the addition of an inhibitor had any effect on the H/D exchange of GlpG, an additional sample was prepared where JLK6 (in a 10:1 inhibitor:protein molar ratio) was added directly after purification, before the dialysis step, when the protein was in a detergent environment. The GlpG/JLK6 mixture was incubated for 1 h and then dialyzed for 10 days. Samples were centrifuged for 2 h at 300000×g and packed into 1.9 mm rotors (Bruker, USA) with a few crystals of sodium trimethylsilylpropanesulfonate (DSS).
Activity assessment
TatA, the natural substrate of AarA in Providencia stuartii,43 was used as the substrate for the GlpG activity assay as described before.9 A synthesized hexahistidine–SUMO–TatA–FLAG DNA sequence was cloned into a pET15b vector (Novagen). The recombinant substrate protein was overexpressed in a GlpG knock-out E. coli Bl21 (DE3) strain and purified and reconstituted as described for GlpGΔN. 25 μL of substrate-containing proteoliposomes (∼5 mg mL−1 protein concentration, 30:1 (mol/mol) lipid/protein ratio) was mixed with 50 μL of protease-containing proteoliposomes (∼0.5 mg mL−1 protein concentration, 30:1 (mol/mol) lipid/protein ratio) in the presence of detergent DM (0.2% w/v) at 37 °C overnight. This corresponds to a molar ratio of 1:5:180 (GlpG:substrate:lipid). To verify the inhibitory effects, 5–50× molar excess of SBO or JLK6 was added. The reaction was stopped by the addition of SDS-PAGE loading buffer and observed via SDS-PAGE. Due to the highly negative charge of the protein, the hexahistidine–SUMO–TatA–FLAG tandem protein and its cleavage products are less mobile in the SDS-PAGE and run higher than the expected 24 kDa, 11 kDa or 13 kDa.
Solid-state NMR spectroscopy and analysis
Proton-detected solid-state NMR experiments were performed on a Bruker 600 MHz (1H Larmor frequency) wide-bore spectrometer equipped with a 1.9 mm probe, operating at an MAS rate of 40 kHz. The sample temperature was kept around 25 °C, estimated from the chemical shift of water relative to the DSS peak.44 Chemical shift assignments were transferred from the previous assignments (performed at a lower sample temperature) of GlpG9 and only peaks that could be identified in CP based 3D hCαNH and hCONH experiments were used for comparison with the samples containing inhibitors. For the sample where JLK6 was added during dialysis, 2D (H)NH and 3D (H)CαNH, (H)CONH and (H)Cα(CO)NH spectra were recorded in a uniform fashion, following a previously published protocol.38 For the sample with SBO, the same experiments were recorded and for the sample where JLK6 was added after dialysis, only a 2D (H)NH and a 3D (H)CαNH spectrum were recorded. For these samples the 3Ds were recorded using non-uniform sampling with a sampling scheme (generated from http://gwagner.med.harvard.edu/intranet/hmsIST/)45,46 where 35% of the points were recorded. Uniformly acquired spectra were processed using TopSpin 4 (Bruker) and non-uniformly sampled spectra were reconstructed using compressed sensing with the iterative re-weighted least squares algorithm in the qMDD software47–49 with 10 iterations and processed using nmrPipe.50 CcpNmr Analysis51 was used for spectral analysis. 1H – 15N – 13Cα chemical shift perturbations were calculated as Euclidian distances (eqn (1)):52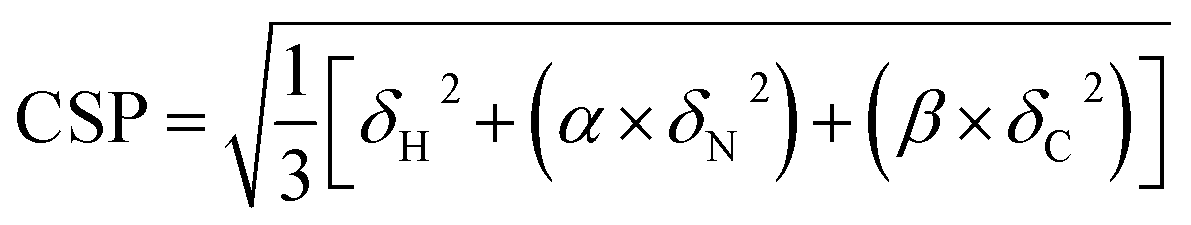 | (1) |
Docking
The crystallographic structure of GlpG in complex with the inhibitor JLK6 (PDB ID: 2XOW)34 served as initial state for the docking studies. All non-protein molecules, including the inhibitor JLK6, were removed. The initial 3D structure of the SBO inhibitor was generated with Gaussview 6.53 Docking with the Schrödinger 2019.3 suite was performed in a two-step procedure.First, the SBO inhibitor was non-covalently docked to the active site pocket of GlpG choosing the side chain oxygen of S201 as centre of the grid (Fig. S3†). The resulting poses were then scored using the Glide routine.54 The ten best poses ranked by the Glide score54 were visually inspected and the pose with the smallest S201(OG) – inhibitor(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) distance (0.283 nm) was chosen for the following step.
O) distance (0.283 nm) was chosen for the following step.
Second, the selected pose was used for covalent docking with CovDock.55 In this step, the ring opening and the bond formation was “alchemically” modelled. The best (called: poseA) and second best poses according to the Prime score56 showed only minor structural differences. Therefore, only poseA (red, Fig. 3A) was selected. The third best pose (called: poseB), however, showed a differently embedded ligand (yellow, Fig. 3A and B) and was also selected. To rule out artificial poses induced by the rigid protein during the docking, the stability of both poses, the best (poseA) and the third best (poseB), was investigated in subsequent all-atom molecular dynamics simulations.
Molecular dynamics simulations
The two covalently linked protein–ligand complexes, poseA and poseB, were inserted in a pre-equilibrated and solvated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane with the g_membed routine.57 The resulting models consisted of 60836 atoms. All-atom molecular dynamics (MD) simulations were carried out with Gromacs 2019.5.58 The Amber99SB*-ILDN force field59 was used for the protein. The POPC lipid membrane and ions were simulated with the parameters derived by Berger et al.60 and Joung et al.,61 respectively. Water was modelled with the SPC/E water potential.62 The parameters of S201 covalently linked to the inhibitor were derived as follows. The geometry of the ligand was optimized and the electrostatic potential was calculated at the Hartree-Fock/6-31G* level using Gaussian09.63 The generalized amber force field (GAFF)64 topology of the inhibitor was generated with the Antechamber software65 using partial charges from the preceding quantum mechanics calculations according to the restrained electrostatic potential (RESP) approach.66 Charges on the backbone were restrained to default values used in the force field to maintain the general scheme.
Preceding the 500 ns long production dynamics, the models were energy minimized and thermally equilibrated at 300 K. A time step of 2 fs was enabled by constraining all bonds to hydrogen atoms with the Lincs algorithms.67 Short-ranged electrostatics and van der Waals interactions were truncated at 1.2 nm. Long-ranged electrostatics were calculated with the Particle-Mesh Ewald summation.68 Temperature and pressure coupling were treated with the V-rescale69 scheme and the Parrinello–Rahman barostate,70 respectively. The temperature was set to 300 K and the pressure to 1 bar. Fluctuations of the periodic cell were only allowed in z-direction, normal to the membrane surface, keeping the density of the membrane unchanged. Both simulations were repeated three times using different starting velocities to get better statistics and to check the reproducibility.
The same procedure was repeated for the closed GlpG structure without ligand (PDB ID: 2NRF, chain B). For the apo-GlpG, however, ten independent productions runs (replicas) of 500 ns were performed.
Data analysis
All MD simulations were analysed by root-mean-square-deviation (RMSD), root-mean-square-fluctuation (RMSF), and RMSD per residue analysis. For this, the protein backbones were aligned to the X-ray structure and all translational and rotational motions were eliminated. For clarity and for catching trends in the two poses, RMSD, RMSF, and RMSD per residue values were averaged over all replicas.The water accessibility of the amide backbone proton was analysed for all amino acids. For the evaluation only the last 100 ns of each individual trajectory were utilized to calculate the average values. The amide backbone proton was defined as accessible if at least one water oxygen was within 3 Å.
Graphical images depicting the structures were generated in ChimeraX.71,72 Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Data availability
Raw data can be obtained from the authors upon reasonable request.Author contributions
C. B., C. Ö. and T. U. contributed equally to this work. C. Ö., C. B. and A. L. performed solid-state NMR experiments and analysed NMR data. T. U. and H. S. performed and analysed docking experiments and MD simulations. C. B., S. B. and S. L. prepared NMR samples. C. B. and C. S. developed and performed the activity assay. C. B., C. Ö., T. U., H.S. and A. L. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 (UniSysCat) – 390540038 (to T. U., H. S. and A. L.). C. Ö. acknowledges funding from the Human Frontier Science Program LT000303/2019-L. C. S. acknowledges funding from the National Natural Science Foundation of China (22077117). Computational resources were granted by the North-German Supercomputing Alliance (HLRN).Notes and references
- Z. Wu, N. Yan, L. Feng, A. Oberstein, H. Yan, R. P. Baker, L. Gu, P. D. Jeffrey, S. Urban and Y. Shi, Nat. Struct. Mol. Biol., 2006, 13, 1084–1091 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang and Y. Ha, Nature, 2006, 444, 179–180 CrossRef CAS PubMed.
- S. Urban, J. R. Lee and M. Freeman, Cell, 2001, 107, 173–182 CrossRef CAS PubMed.
- A.-N. Bondar, C. del Val and S. H. White, Structure, 2009, 17, 395–405 CrossRef CAS PubMed.
- Y. Xue and Y. Ha, J. Biol. Chem., 2013, 288, 16645–16654 CrossRef CAS PubMed.
- S. Urban and S. W. Dickey, Genome Biol., 2011, 12, 231 CrossRef CAS PubMed.
- A. Tichá, B. Collis and K. Strisovsky, Trends Biochem. Sci., 2018, 43, 726–739 CrossRef PubMed.
- R. P. Baker, K. Young, L. Feng, Y. Shi and S. Urban, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8257–8262 CrossRef CAS PubMed.
- C. Shi, C. Öster, C. Bohg, L. Li, S. Lange, V. Chevelkov and A. Lange, J. Am. Chem. Soc., 2019, 141, 17314–17321 CrossRef CAS PubMed.
- M. Shokhen and A. Albeck, Protein Sci., 2017, 26, 2355–2366 CrossRef CAS PubMed.
- S. Cho, R. P. Baker, M. Ji and S. Urban, Nat. Struct. Mol. Biol., 2019, 26, 910–918 CrossRef CAS PubMed.
- S. Zoll, S. Stanchev, J. Began, J. Škerle, M. Lepšík, L. Peclinovská, P. Majer and K. Strisovsky, EMBO J., 2014, 33, 2408–2421 CrossRef CAS PubMed.
- S. Cho, S. W. Dickey and S. Urban, Mol. Cell, 2016, 61, 329–340 CrossRef CAS PubMed.
- A. Tichá, S. Stanchev, K. R. Vinothkumar, D. C. Mikles, P. Pachl, J. Began, J. Škerle, K. Švehlová, M. T. N. Nguyen, S. H. L. Verhelst, D. C. Johnson, D. A. Bachovchin, M. Lepšík, P. Majer and K. Strisovsky, Cell Chem. Biol., 2017, 24, 1523–1536 CrossRef PubMed.
- S. Düsterhöft, U. Künzel and M. Freeman, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 2200–2209 CrossRef PubMed.
- Y. Xue, S. Chowdhury, X. Liu, Y. Akiyama, J. Ellman and Y. Ha, Biochemistry, 2012, 51, 3723–3731 CrossRef CAS PubMed.
- K. R. Vinothkumar, O. A. Pierrat, J. M. Large and M. Freeman, Structure, 2013, 21, 1051–1058 CrossRef CAS PubMed.
- E. V. Wolf, A. Zeißler, O. Vosyka, E. Zeiler, S. Sieber and S. H. L. Verhelst, PLoS One, 2013, 8, e72307 CrossRef CAS PubMed.
- P. Goel, T. Jumpertz, D. C. Mikles, A. Tichá, M. T. N. Nguyen, S. Verhelst, M. Hubalek, D. C. Johnson, D. A. Bachovchin, I. Ogorek, C. U. Pietrzik, K. Strisovsky, B. Schmidt and S. Weggen, Biochemistry, 2017, 56, 6713–6725 CrossRef CAS PubMed.
- P. Goel, T. Jumpertz, A. Tichá, I. Ogorek, D. C. Mikles, M. Hubalek, C. U. Pietrzik, K. Strisovsky, B. Schmidt and S. Weggen, Bioorg. Med. Chem. Lett., 2018, 28, 1417–1422 CrossRef CAS PubMed.
- V. Ladizhansky, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1577–1586 CrossRef CAS PubMed.
- V. S. Mandala, J. K. Williams and M. Hong, Annu. Rev. Biophys., 2018, 47, 201–222 CrossRef CAS PubMed.
- M. R. Elkins and M. Hong, Curr. Opin. Struct. Biol., 2019, 57, 103–109 CrossRef CAS PubMed.
- S. Tapaneeyakorn, A. D. Goddard, J. Oates, C. L. Willis and A. Watts, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1462–1475 CrossRef CAS PubMed.
- S. G. Zech, E. Olejniczak, P. Hajduk, J. Mack and A. E. McDermott, J. Am. Chem. Soc., 2004, 126, 13948–13953 CrossRef CAS PubMed.
- V. S. Mandala, M. J. McKay, A. A. Shcherbakov, A. J. Dregni, A. Kolocouris and M. Hong, Nat. Struct. Mol. Biol., 2020, 27, 1202–1208 CrossRef CAS PubMed.
- S. Urban and S. M. Moin, Cell Rep., 2014, 8, 1241–1247 CrossRef CAS PubMed.
- S. Paschkowsky, F. Oestereich and L. M. Munter, J. Membr. Biol., 2018, 251, 369–378 CrossRef CAS PubMed.
- C. Öster, K. Hendriks, W. Kopec, V. Chevelkov, C. Shi, D. Michl, S. Lange, H. Sun, B. L. de Groot and A. Lange, Sci. Adv., 2019, 5, eaaw6756 CrossRef PubMed.
- S. Jekhmane, J. Medeiros-Silva, J. Li, F. Kümmerer, C. Müller-Hermes, M. Baldus, B. Roux and M. Weingarth, Nat. Commun., 2019, 10, 123 CrossRef PubMed.
- T. Schubeis, T. L. Marchand, C. Daday, W. Kopec, K. T. Movellan, J. Stanek, T. S. Schwarzer, K. Castiglione, B. L. de Groot, G. Pintacuda and L. B. Andreas, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21014–21021 CrossRef CAS PubMed.
- D. Good, C. Pham, J. Jagas, J. R. Lewandowski and V. Ladizhansky, J. Am. Chem. Soc., 2017, 139, 9246–9258 CrossRef CAS PubMed.
- A. Petit, A. Pasini, C. A. da Costa, E. Ayral, J. F. Hernandez, C. Dumanchin-Njock, C. J. Phiel, P. Marambaud, S. Wilk, M. Farzan, P. Fulcrand, J. Martinez, D. Andrau and F. Checler, J. Neurosci. Res., 2003, 74, 370–377 CrossRef CAS PubMed.
- K. R. Vinothkumar, K. Strisovsky, A. Andreeva, Y. Christova, S. Verhelst and M. Freeman, EMBO J., 2010, 29, 3797–3809 CrossRef CAS PubMed.
- M. K. Lemberg, J. Menendez, A. Misik, M. Garcia, C. M. Koth and M. Freeman, EMBO J., 2005, 24, 464–472 CrossRef CAS PubMed.
- S. Urban, J. R. Lee and M. Freeman, EMBO J., 2002, 21, 4277–4286 CrossRef CAS PubMed.
- L. G. Stevenson, K. Strisovsky, K. M. Clemmer, S. Bhatt, M. Freeman and P. N. Rather, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1003–1008 CrossRef CAS PubMed.
- P. Fricke, V. Chevelkov, M. Zinke, K. Giller, S. Becker and A. Lange, Nat. Protoc., 2017, 12, 764–782 CrossRef CAS PubMed.
- Y. Wang, S. Maegawa, Y. Akiyama and Y. Ha, J. Mol. Biol., 2007, 374, 1104–1113 CrossRef CAS PubMed.
- K. Strisovsky, H. J. Sharpe and M. Freeman, Mol. Cell, 2009, 36, 1048–1059 CrossRef CAS PubMed.
- A. A. Shcherbakov, G. Hisao, V. S. Mandala, N. E. Thomas, M. Soltani, E. A. Salter, J. H. Davis, K. A. Henzler-Wildman and M. Hong, Nat. Commun., 2021, 12, 172 CrossRef CAS PubMed.
- M. Yi, T. A. Cross and H.-X. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13311–13316 CrossRef CAS PubMed.
- U. Gohlke, L. Pullan, C. A. McDevitt, I. Porcelli, E. de Leeuw, T. Palmer, H. R. Saibil and B. C. Berks, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10482–10486 CrossRef CAS PubMed.
- A. Böckmann, C. Gardiennet, R. Verel, A. Hunkeler, A. Loquet, G. Pintacuda, L. Emsley, B. H. Meier and A. Lesage, J. Biomol. NMR, 2009, 45, 319–327 CrossRef PubMed.
- S. G. Hyberts, A. G. Milbradt, A. B. Wagner, H. Arthanari and G. Wagner, J. Biomol. NMR, 2012, 52, 315–327 CrossRef CAS PubMed.
- S. G. Hyberts, K. Takeuchi and G. Wagner, J. Am. Chem. Soc., 2010, 132, 2145–2147 CrossRef CAS PubMed.
- V. Y. Orekhov and V. A. Jaravine, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 271–292 CrossRef CAS PubMed.
- X. Qu, M. Mayzel, J. F. Cai, Z. Chen and V. Orekhov, Angew. Chem., Int. Ed., 2015, 54, 852–854 CrossRef CAS PubMed.
- K. Kazimierczuk and V. Y. Orekhov, Angew. Chem., Int. Ed., 2011, 50, 5556–5559 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- S. P. Skinner, R. H. Fogh, W. Boucher, T. J. Ragan, L. G. Mureddu and G. W. Vuister, J. Biomol. NMR, 2016, 66, 111–124 CrossRef CAS PubMed.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, 2019 Search PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- K. Zhu, K. W. Borrelli, J. R. Greenwood, T. Day, R. Abel, R. S. Farid and E. Harder, J. Chem. Inf. Model., 2014, 54, 1932–1940 CrossRef CAS PubMed.
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2004, 55, 351–367 CrossRef CAS PubMed.
- M. G. Wolf, M. Hoefling, C. Aponte-Santamaría, H. Grubmüller and G. Groenhof, J. Comput. Chem., 2010, 31, 2169–2174 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- O. Berger, O. Edholm and F. Jähnig, Biophys. J., 1997, 72, 2002–2013 CrossRef CAS PubMed.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian-09 Revision E.01, 2016 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graph. Model., 2006, 25, 247–260 CrossRef CAS PubMed.
- R. J. Woods and R. Chappelle, J. Mol. Struct.: THEOCHEM, 2000, 527, 149–156 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and tables. See DOI: 10.1039/d1sc02146j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |