
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective rhodium-catalyzed 2-C–H 1,3-dienylation of indoles: dual functions of the directing group†
Yizhan
Zhai
ab,
Xue
Zhang
 *a and
Shengming
Ma
*ac
*a and
Shengming
Ma
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai, 200032, P. R. China. E-mail: masm@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
cResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China
First published on 21st July 2021
Abstract
A rhodium-catalyzed intermolecular highly stereoselective 1,3-dienylation at the 2-position of indoles with non-terminal allenyl carbonates has been developed by using 2-pyrimidinyl or pyridinyl as the directing group. The reaction tolerates many functional groups affording the products in decent yields under mild conditions. In addition to C–H bond activation, the directing group also played a vital role in the determination of Z-stereoselectivity for the C–H functionalization reaction with 4-aryl-2,3-allenyl carbonates, which is confirmed by the E-selectivity observed with 4-alkyl-2,3-allenyl carbonates. DFT calculations have been conducted to reveal that π–π stacking involving the directing 2-pyrimidinyl or pyridinyl group is the origin of the observed stereoselectivity. Various synthetic transformations have also been demonstrated.
Introduction
Installing directing groups (DGs) is a versatile and reliable strategy for selective C–H functionalization.1 Generally, directing groups are installed onto aromatic rings or other systems to ensure reactivity and regioselectivity by their coordination with metal and they are structurally left unchanged after the corresponding reactions (eqn (a-1) in Scheme 1).1 In some cases, directing groups could also serve as internal oxidants or participate in the reaction directly to generate cyclic2,3 and acyclic4 compounds (eqn (a-2) and (a-3) in Scheme 1). With continuous interest on C–H functionalization with allenes,5,6 we wish to report here the first example of dual functions of the directing group in the rhodium(III)-catalyzed C–H functionalization of indoles with 4-aryl-2,3-allenyl carbonates generating unexpected thermodynamically unstable 2-(1(Z)-aryl-1,3-dienyl) indoles dictated by the aryl group and the directing group via π–π stacking (Scheme 1b).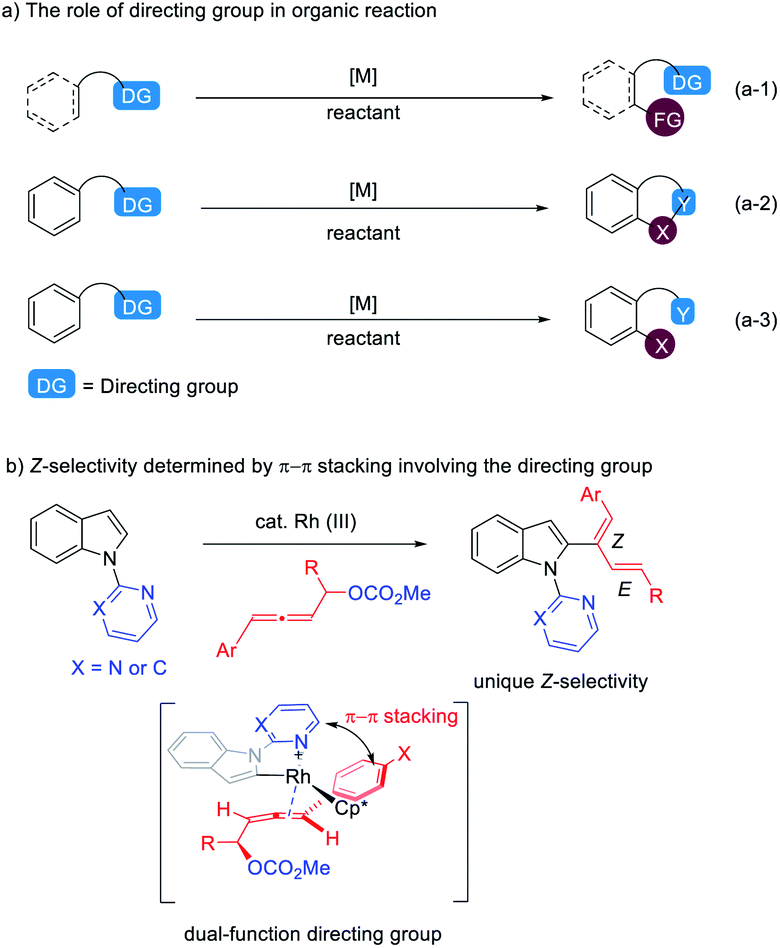 | ||
| Scheme 1 Novel function of directing groups in C–H functionalization reactions. | ||
Results and discussion
After some trial and error, it was observed that the [Cp*RhCl2]2-catalyzed reaction of indole 1a and 4-phenyl-2,3-allenyl carbonate 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 in the presence of AgSbF6 and PivOH produced 2-(1(Z),3-dienyl)indole 3aa in toluene at room temperature in 80% yield with an unexpected Z-selectivity of 96:4 (Table 1, entry 1), which is very different from what was observed by Shi and his coworkers with alkylidenecyclopropanes affording a 1:1 E/Z mixture of 1,3-dienes.8a The reaction in dioxane afforded the product in a much lower yield (Table 1, entry 2) while no expected product was formed in CH3CN with 75% recovery of 1a (Table 1, entry 3). When AgBF4 was used instead of AgSbF6, the reaction was much slower (Table 1, entry 4). The control experiment showed that no product was generated in the absence of AgSbF6 (Table 1, entry 5).
7 in the presence of AgSbF6 and PivOH produced 2-(1(Z),3-dienyl)indole 3aa in toluene at room temperature in 80% yield with an unexpected Z-selectivity of 96:4 (Table 1, entry 1), which is very different from what was observed by Shi and his coworkers with alkylidenecyclopropanes affording a 1:1 E/Z mixture of 1,3-dienes.8a The reaction in dioxane afforded the product in a much lower yield (Table 1, entry 2) while no expected product was formed in CH3CN with 75% recovery of 1a (Table 1, entry 3). When AgBF4 was used instead of AgSbF6, the reaction was much slower (Table 1, entry 4). The control experiment showed that no product was generated in the absence of AgSbF6 (Table 1, entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Variation from the standard conditions | NMR recovery of 1ab (%) | NMR yield of 3aab(%) | Z/E of 3aac |
| a The reactions were carried out using 0.2 mmol of 1a, 1.1 equiv. of 2a, 2.5 mol% of [Cp*RhCl2]2, 30 mol% of [Ag], and 1 equiv. of PivOH in 1 mL of solvent at room temperature. b Determined by 1H NMR analysis using CH3NO2 as an internal standard. c The ratio of Z/E was determined by 1H NMR analysis of the crude reaction mixture before chromatographic separation. | ||||
| 1 | None | — | 80 | 96/4 |
| 2 | Dioxane as solvent | — | 62 | 96/4 |
| 3 | CH3CN as solvent | 75 | — | — |
| 4 | AgBF4 replacing AgSbF6 | 60 | 20 | 96/4 |
| 5 | No [Ag] | 78 | — | — |
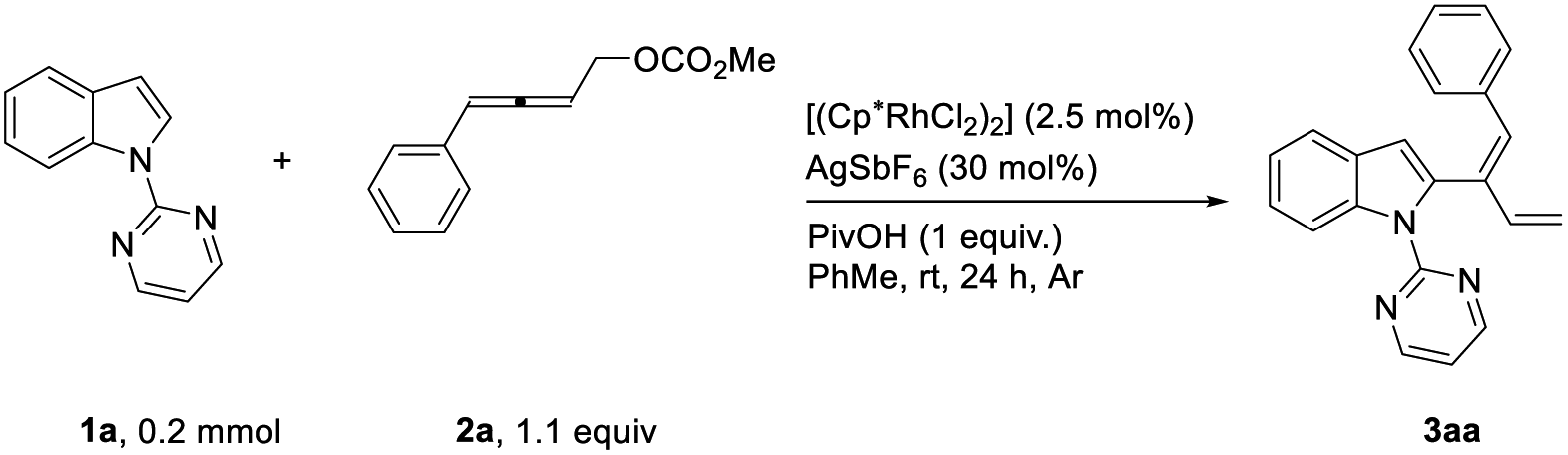
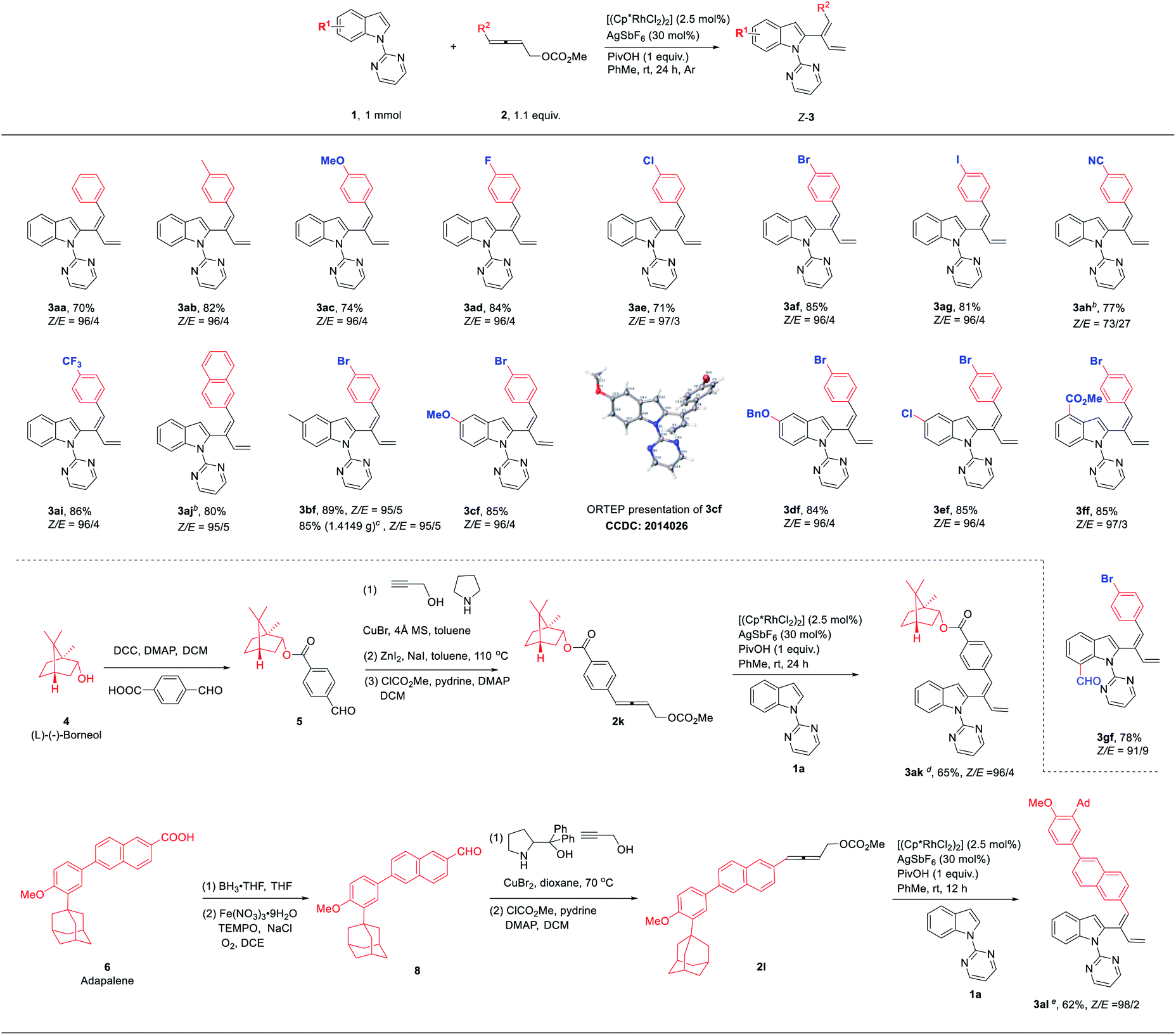
With the optimized reaction conditions in hand, we set out to examine the scope of the reaction (Table 2). Firstly, differently substituted 4-aryl-2,3-allenyl carbonates were applied with 1-(2-pyrimidinyl)indole 1a. Allenes with the 4-phenyl groups substituted with electron-donating (4-Me (3ab) and 4-MeO (3ac)) and electron-withdrawing synthetically versatile groups (4 F-, 4-Cl-, 4-Br-, 4-I-, 4-CN- and 4-CF3-) (3ad–3ai) could react smoothly. 4-Naphthyl-2,3-butadienyl carbonate also underwent the reaction although it should be conducted at 70 °C (3aj). The scope of indoles was demonstrated using 4-(p-bromophenyl)-2,3-butadienyl carbonate 2f: 5-Me, 5-MeO, 5-BnO, 5-Cl, 4-CO2Me and 7-CHO could be tolerated, affording the corresponding 1,3-dienylation products 3bf–3gf in decent yields and an excellent stereoselectivity. The stereoselectivity of this reaction was further established by the X-ray diffraction study of compound 3cf. The functional groups highlighted in blue proved that this methodology tolerates many reactive functionalities and, thus, is very powerful for the syntheses of other indole derivatives. To further demonstrate the synthetic and medicinal potential of the reaction, allenyl carbonates derived from natural products or drug molecules were prepared via the ATA reaction of the corresponding aldehydes:7 Under standard conditions, a range of such allenyl carbonates derivatized from (L)-(−)-borneol (4) and adapalene (6) could be effectively applied affording the related products with an excellent stereoselectivity (3ak–3al).
| a The reactions were carried out using 1 mmol of 1, 1.1 equiv. of 2, 2.5 mol% of [Cp*RhCl2]2, 30 mol% of AgSbF6, and 1 equiv. of PivOH in 5 mL of toluene at room temperature. Yields of isolated products were given. The ratio of Z/E was determined by 1H NMR analysis. b The reaction was carried out at 70 °C. c The reaction was carried out on a 4 mmol scale. d The reaction was carried out on a 0.2 mmol scale. e The reaction was carried out on a 0.4 mmol scale. |
|---|
|
In addition to primary 4-aryl-2,3-butadienyl carbonates, secondary 4-aryl-2,3-butadienyl carbonate 2m was also tested, affording 2-(1(Z),3(E)-dienyl)indole 3am with a Z-selectivity of 99:1 for the remaining double bond and complete E-selectivity for the newly formed double bond, respectively (eqn (1)).

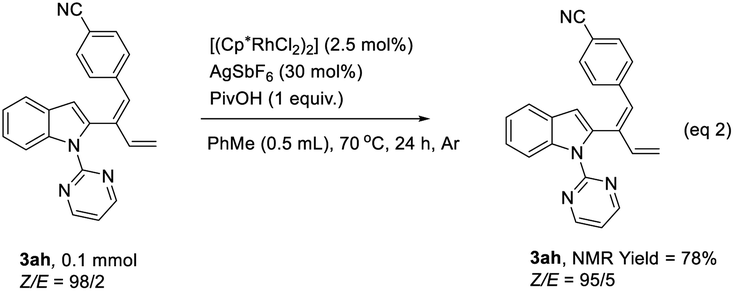
In order to clarify the role of directing groups, some control experiments were carried out (Table 3). When the 2-pyrimidinyl group was replaced with Me (1h), no product was generated under standard conditions (Table 3, entry 1). Furthermore, directing groups such as Piv (1i)9a and t-Bu2P (1j)9b failed to afford the corresponding products (Table 3, entries 2 and 3). When this directing group was replaced with the pyridinyl group (1k), the reaction still worked, indicating the importance of the nitrogen-containing directing group (Table 3, entry 4). On the other hand, in order to figure out the reason for the observed poor stereoselectivity of compound 3ah, some control experiments were also conducted. The Z/E-selectivity of 3ah remained almost unchanged regardless of lowering the reaction temperature, shortening the reaction time, or running the reaction in the absence of light (Table 3, entries 5–7). In addition, when compound 3ah with a Z/E ratio of 98/2 obtained by preparative HPLC was treated under the standard condition at 70 °C, the ratio of Z/E slightly decreased (eqn (2)). These experimental results could rule out the possibility of isomerization from Z-3ah to E-3ah. The reason for the relatively lower stereoselectivity of 3ah remained unclear.
|
|
|||||
|---|---|---|---|---|---|
| Entry | DG | R | NMR recovery of 1b (%) | NMR yield of 3b (%) | Z/E of 3 |
| a The reactions were carried out using 0.2 mmol of 1, 1.1 equiv. of 2, 2.5 mol% of [Cp*RhCl2]2, 30 mol% of AgSbF6, and 1 equiv. of PivOH in 1 mL of toluene at room temperature. b Determined by 1H NMR analysis using CH3NO2 as an internal standard. c 0.5 mmol scale. d Isolated yield. e The ratio of Z/E was determined by 1H NMR analysis. f At 50 °C. g 1 mmol scale for 7 h. h At 70 °C. i The reaction was run in the absence of light. | |||||
| 1 | Me (1h) | H (2a) | 83 | — | — |
| 2 | Piv (1i) | (2a) | 76 | — | — |
| 3 | t-Bu2P (1j) | (2a) | 85 | — | — |
| 4c | 2-pyridinyl (1k) | (2a) | — | 3ka 60(52d) | 90/10e |
| 5f | 2-pyrimidinyl (1a) | CN (2h) | 25 | 3ah 60 | 75/25 |
| 6g,h | (1a) | (2h) | 28 | 3ah 57 | 74/26 |
| 7h,i | (1a) | (2h) | — | 3ah 66 | 73/27 |
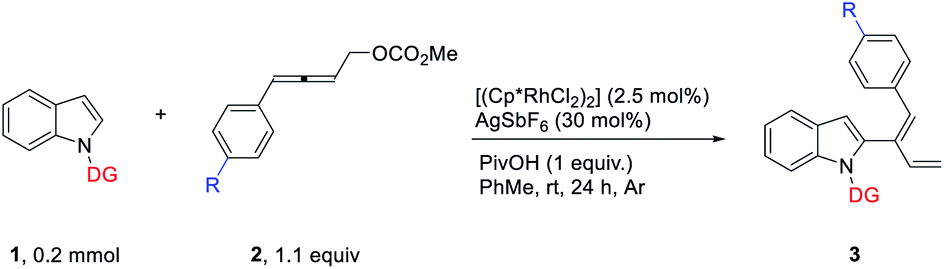
DFT calculations have been performed to investigate the mechanism of this rhodium-catalyzed 1,3-dienylation of indoles. Fig. 1 presents the energetic profile for the most favorable pathway of the reaction of indole 1a and 4-phenyl-2,3-allenyl carbonate 2a. The pivalate-ligated species Cp*Rh(OPiv)2 is generally regarded as the active catalyst, which may be generated via Rh dimer dissociation and ligand exchange starting from [Cp*RhCl2]2 and PivOH.2k Cp*Rh(OPiv)2 firstly dissociates one pivalate ligand and coordinates with the N atom of the pyrimidine moiety in 1a, resulting in the formation of the complex INT1, which is selected as the free energy reference. Subsequent indole 2-position C–H activation is realized via a six-membered cyclic transition state TS1, in which the resting pivalate ligand acts as the base to deprotonate the ortho aromatic proton to form the Rh–C bond simultaneously. This concerted metalation-deprotonation (CMD)10,11 process requires an energy barrier of 10.2 kcal mol−1 to afford the five-membered rhodacycle INT2. Subsequently, the dissociation of the PivOH molecule from the Rh(III) center and the coordination of the allene moiety of 2a provides INT3. Then the coordinated allenic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond connected with the aryl group in 2a undergoes an insertion into the Rh–C bond viaTS2_a to provide the π-allyl Rh(III) complex INT4, which may easily isomerize to the η1-allyl Rh(III) complex INT5 with the help of the coordination of the carbonate carbonyl group. This cyclic carborhodation step is computed to be exergonic (ΔGsol = −10.5 kcal mol−1) requiring an activation barrier of 14.6 kcal mol−1 (TS2, Fig. 1). Subsequent β-oxygen elimination of INT5 needs to overcome an energy barrier of 12.8 kcal mol−1 (TS3), providing the final product and regenerating the Rh(III) catalyst. Overall, the current reaction is found to occur irreversibly, as the energy of the final product INT6 is about 20 kcal mol−1 below the energy of the starting materials, and the carborhodation step is not only the rate-limiting but also the selectivity-determining step.
C double bond connected with the aryl group in 2a undergoes an insertion into the Rh–C bond viaTS2_a to provide the π-allyl Rh(III) complex INT4, which may easily isomerize to the η1-allyl Rh(III) complex INT5 with the help of the coordination of the carbonate carbonyl group. This cyclic carborhodation step is computed to be exergonic (ΔGsol = −10.5 kcal mol−1) requiring an activation barrier of 14.6 kcal mol−1 (TS2, Fig. 1). Subsequent β-oxygen elimination of INT5 needs to overcome an energy barrier of 12.8 kcal mol−1 (TS3), providing the final product and regenerating the Rh(III) catalyst. Overall, the current reaction is found to occur irreversibly, as the energy of the final product INT6 is about 20 kcal mol−1 below the energy of the starting materials, and the carborhodation step is not only the rate-limiting but also the selectivity-determining step.
 | ||
| Fig. 1 Free energy profile (kcal mol−1) for the most favorable pathway of the reaction of indole 1a with 4-phenyl-2,3-allenyl carbonate 2a. Bond lengths are given in angstroms. | ||
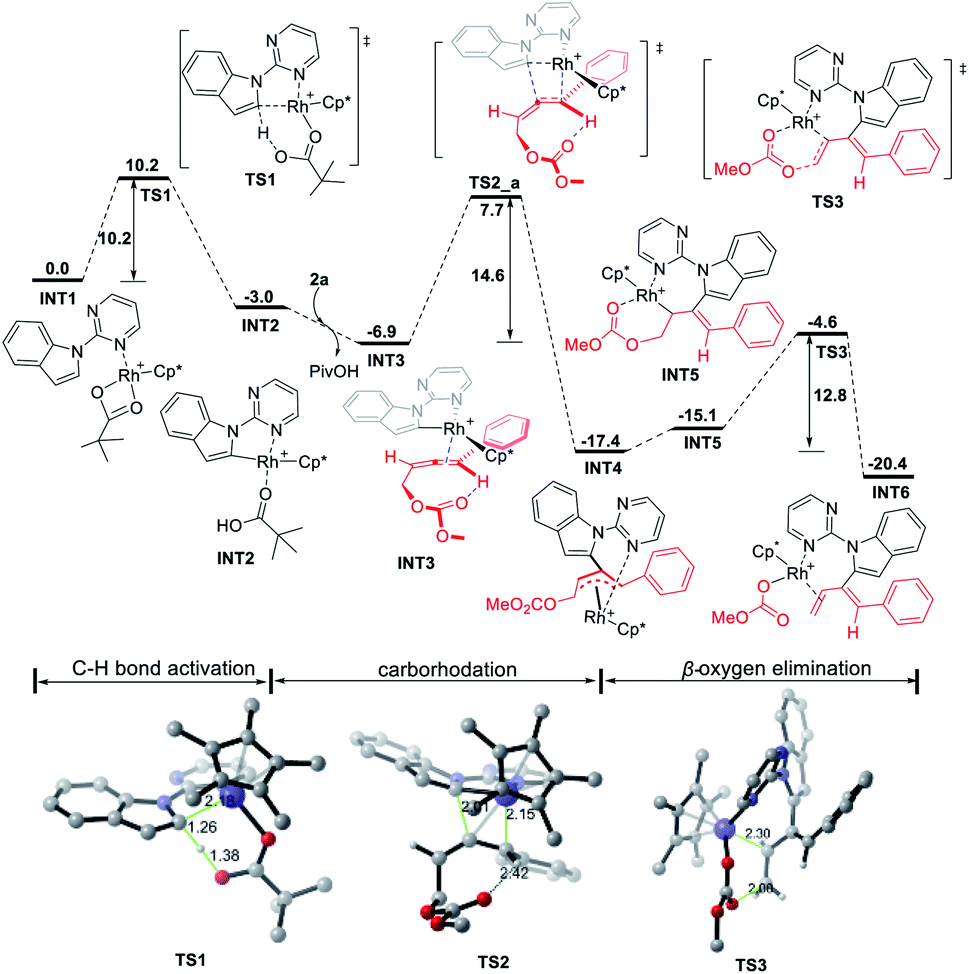
The stereoselectivity of the reaction of indole 1a with 4-phenyl-2,3-allenyl carbonate 2a was further explored by DFT calculations. Due to the possibility of the participation of both CC bonds of allenyl carbonates 2a, there are totally eight competing transition states referring to the stereoselectivity of the carborhodation step (TS2_a–d and TS2_a′–d′Fig. 2). Nevertheless, in order to avoid the severe steric interactions, the Rh–C bond would prefer to approach from the opposite side of the larger substituents (the methyl carbonate or the phenyl group) and, thus, TS2_a/TS2_b referring to the upper-face insertion of the C2C3 bond and TS2_c/TS2_d referring to the front-face insertion of the C1C2 bond are relatively more favorable. Due to the steric hinderace, the other four transition structures (TS2_a′–d') are all disfavorable. TS2_a, which leads to the formation of the Z-isomer, was found to be the most stable, thus, accounting for the domination of the Z products in the reaction with 4-aryl-2,3-allenyl carbonates. TS2_a and TS2_b associated with the upper-face insertion of the Rh–C bond into the phenyl substituted C2C3 bond are more favorable than the other two (TS2_c/TS2_d). Meanwhile, the intramolecular hydrogen bond, formed between the carbonyl oxygen with the sp2 hydrogen stabilized both structures of TS2_a and TS2_b. The π–π stacking interaction between the phenyl substituent of 2a with the pyrimidine group further increases the stability of TS2_a, which is also proven by the non-covalent interaction (NCI) analysis (performed by Multiwfn12 and VMD13 software). However, the structure of TS2_b suffers from unfavorable steric repulsions between the phenyl group and Cp* ligand and also between the methyl carbonate and the indole group. Thus, both factors account for the preference of TS2_a over TS2_b by 4.1 kcal mol−1. Furthermore, no hydrogen bonds exist in TS2_c and TS2_d associated with the front-face insertion of the Rh–C bond into the C1C2 bond. Without the hydrogen bond and the obvious hindrance, TS2_c presents similar stability to TS2_b (18.7 and 18.9 kcal mol−1). However, severe steric interactions between the methyl carbonate and the Cp* ligand destabilize TS2_d, making it even more unfavorable (6.5 kcal mol−1 higher than that of TS2_a). Therefore, not only the steric effect but also the π–π stacking interaction are operative in the control of the Z-stereoselectivity.
 | ||
| Fig. 2 Origin of the Z-stereoselectivity for the reaction of 1a with 4-phenyl-2,3-allenyl carbonate 2a. Free energies are given in kcal mol−1 with respect of INT3. | ||
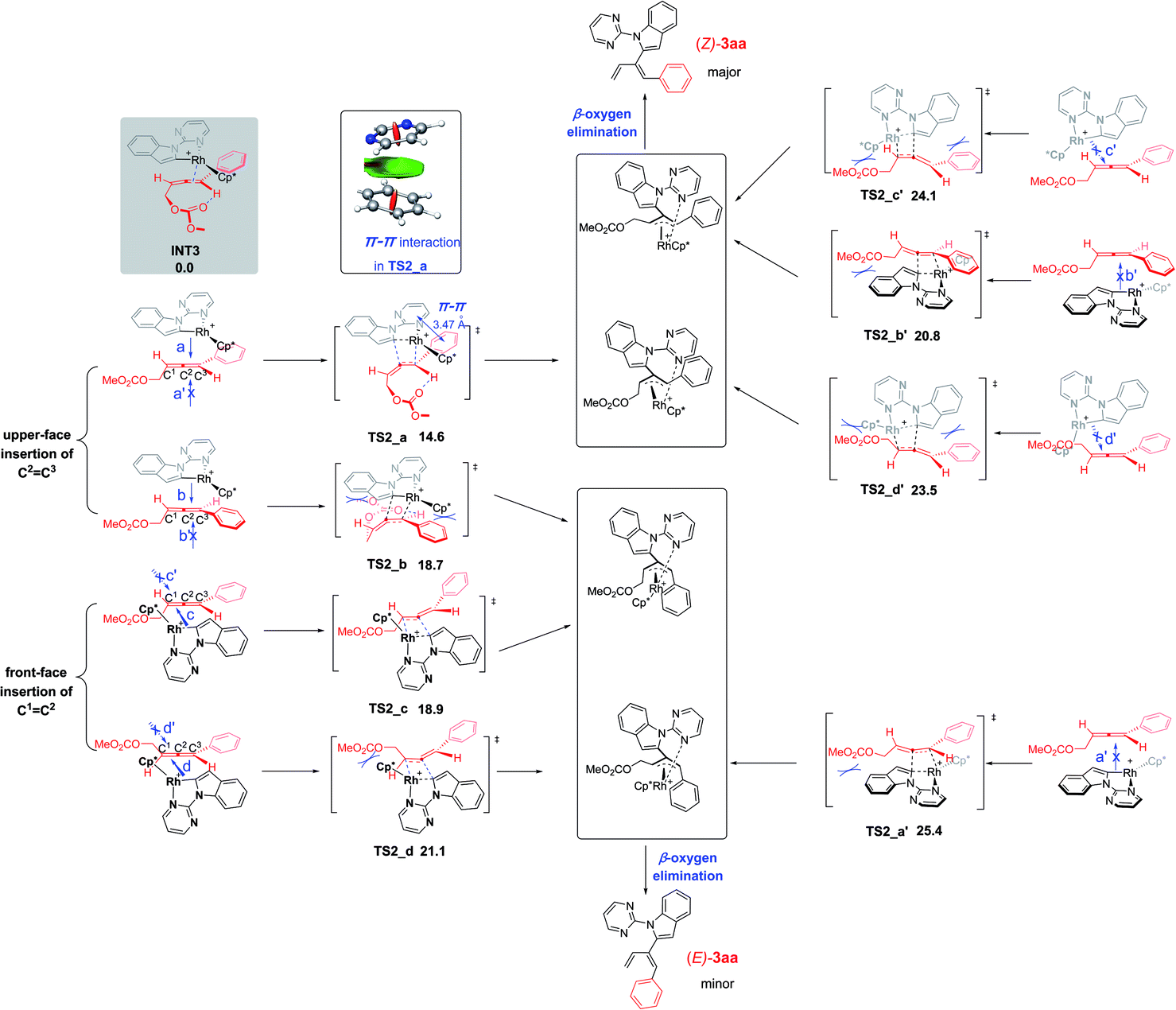
A further question is how about the selectivity of the 4-alkyl-2,3-allenyl carbonates without the π–π stacking interaction. Similar calculations were then conducted on the reaction of 4-cyclohexyl-2,3-allenyl carbonates 2n. The four competing transition structures TS2_a1–d1, associated with 2n participating in the carborhodation step, are listed in Fig. 3 (below) together with the corresponding transition structures of the 2a system (above) for comparison. Due to the lack of the π–π stacking interaction, TS2_a1 suffers from the steric interactions between the cyclohexyl substituent of 2n with the pyrimidine group, leading to the decrease of the stability of TS2_a1. Moreover, unfavorable steric repulsions in TS2_b1 and TS2_d1 destabilize both structures. With no obvious hindrance, TS2_c1 becomes the most favorable one among the four transition structures. Thus, the reactions of alkyl-substituted allenyl carbonates should present different stereoselectivity as compared to the aryl-substituted allenyl carbonates to yield the major E products.
 | ||
| Fig. 3 Origin of the different stereoselectivity for the reaction of 4-phenyl- and 4-cyclohexyl-2,3-allenyl carbonates (2a and 2n). Free energies are given in kcal mol−1 with respect to INT3 or INT3_1. | ||
In order to further investigate the factors controlling the stereoselectivity, we also analyzed the frontier molecular orbitals for the carborhodation step, which mainly involve a nucleophilic attack of the C–Rh σ bond on the coordinated CC bond. The C–Rh σ bond orbital in INT3 corresponds to the HOMO, while the π* orbital of the coordinated CC double bond of allenyl carbonate 2 corresponds to the LUMO. Fig. 4 shows the spatial plots and the orbital energies for the LUMOs of 4-phenyl-2,3-allenyl carbonate 2a and 4-cyclohexyl-2,3-allenyl carbonate 2n. The LUMO of 2a is mainly the π* orbital of the phenyl substituted C2C3 bond, suggesting the preference of the C2C3 bond over the other C2C1 bond in the carborhodation step. Nevertheless, in the LUMO of 2n, the π* orbital of the C2C1 bond contributes significantly, indicating that 2n and 2a prefer to participate in the carborhodation with different CC bonds, which has already been proven by the results shown in Fig. 2 and 3. Further examination reveals that a conjugative effect between the phenyl substituent and the C2C3 bond stabilizes the LUMO of 2a, making the orbital energy of the LUMO in 2a lower than that in 2n, and thus 2a would be more favourable to participate in the carborhodation reaction.
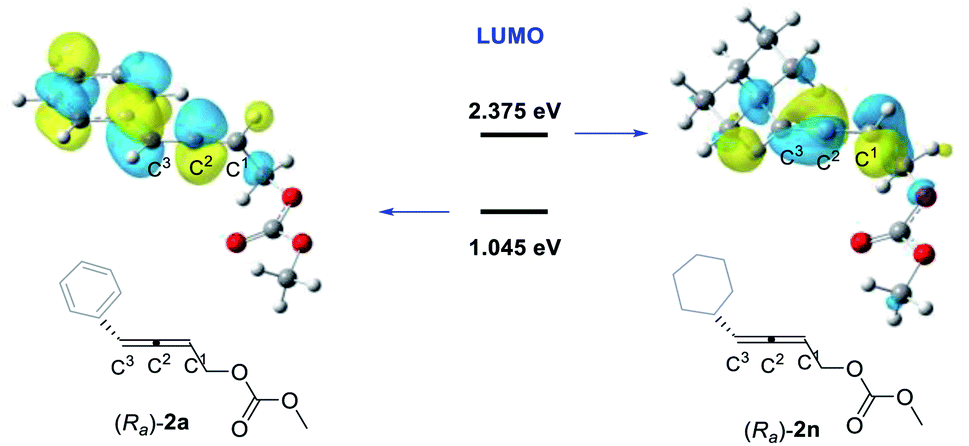 | ||
| Fig. 4 LUMOs calculated for (Ra)-2a and (Ra)-2n (R enantiomer is selected as the example). The orbital energies are given in eV. | ||
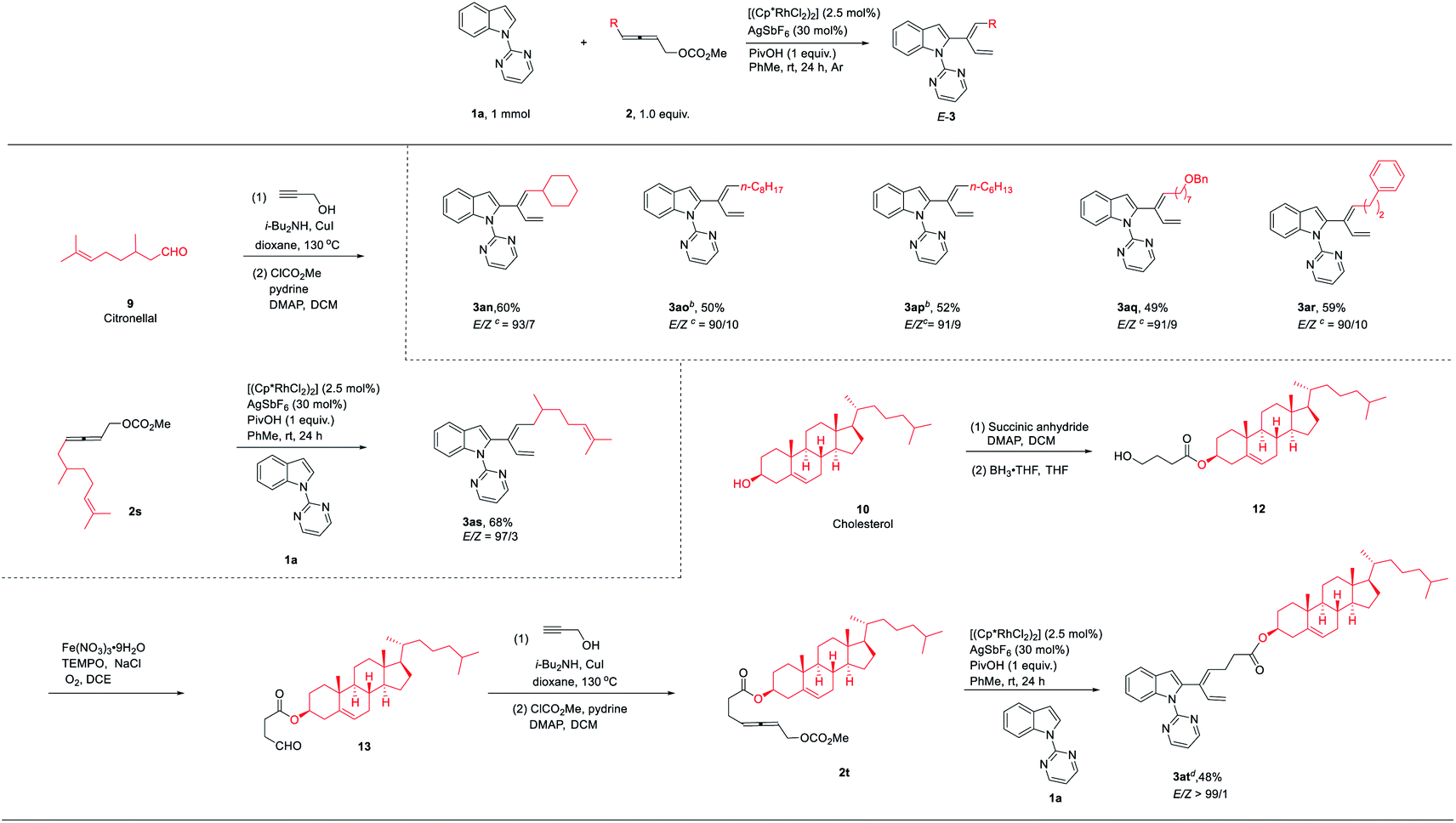
Encouraged by the above DFT calculation results, 4-alkyl-2,3- allenyl carbonates were tested under standard conditions (Table 4). As expected, the stereoselectivity was reversed, affording 2-(1E,3-dienyl)indoles 3an–3ar smoothly in good yields and an E-stereoselectivity of 90:10–93:7. The CC configuration of compound 3ao was established by NOESY studies (see the ESI for details). Therefore, the experimental results were in line with DFT calculations. To illustrate the utility of this catalytic system, the scope could be extended to allenyl carbonates derived from natural products or drug molecules; 2s and 2t derived from citronellal (9) and cholesterol (10) could provide the corresponding products with an excellent stereoselectivity (3as–3at) under standard conditions. It is interesting to observe that the E-stereoselectivity for 3as and 3at has been greatly improved obviously due to the increased bulkiness of the R group.
| a The reactions were carried out using 1 mmol of 1a, 1.0 equiv. of 2, 2.5 mol% of [Cp*RhCl2]2, 30 mol% of AgSbF6, and 1 equiv. of PivOH in 5 mL of toluene at room temperature. Yields of isolated products were given. The ratio of Z/E was determined by 1H NMR analysis. An unidentified unknown product was formed. b 1.1 equiv. of 2 was added. c The yield and ratio of Z/E were calculated based on E-3 and the Z/E mixture of 3 after column chromatography according to 1H NMR analysis. d 0.5 mmol scale. |
|---|
|
The synthetic potential of the diene products has also been demonstrated (Scheme 2); compound 3bf could undergo the Diels–Alder reaction with N-methylmaleimide and diethyl acetylenedicarboxylate to yield corresponding compounds 14 and 15. The Suzuki coupling reaction with PhB(OH)2 generated compound 16.14a The directing group 2-pyrimidinyl was further applied to execute subsequent C–H functionalization at C-7 of indole with methyl acrylate to afford compound 17.14b The directing group could be removed to generate corresponding indole 18 in the presence of NaOEt and DMSO at room temperature. Furthermore, the Diels–Alder reaction of 3am with diethyl acetylenedicarboxylate under the catalysis of Sc(OTf)3 at 100 °C afforded cis-19 in 55% yield. Similarly, when cyclohexyl-substituted 2-(1E,3-dienyl)indole 3an was treated with NaOEt and DMSO at 40 °C, corresponding indole E-20 could be produced without isomerization.
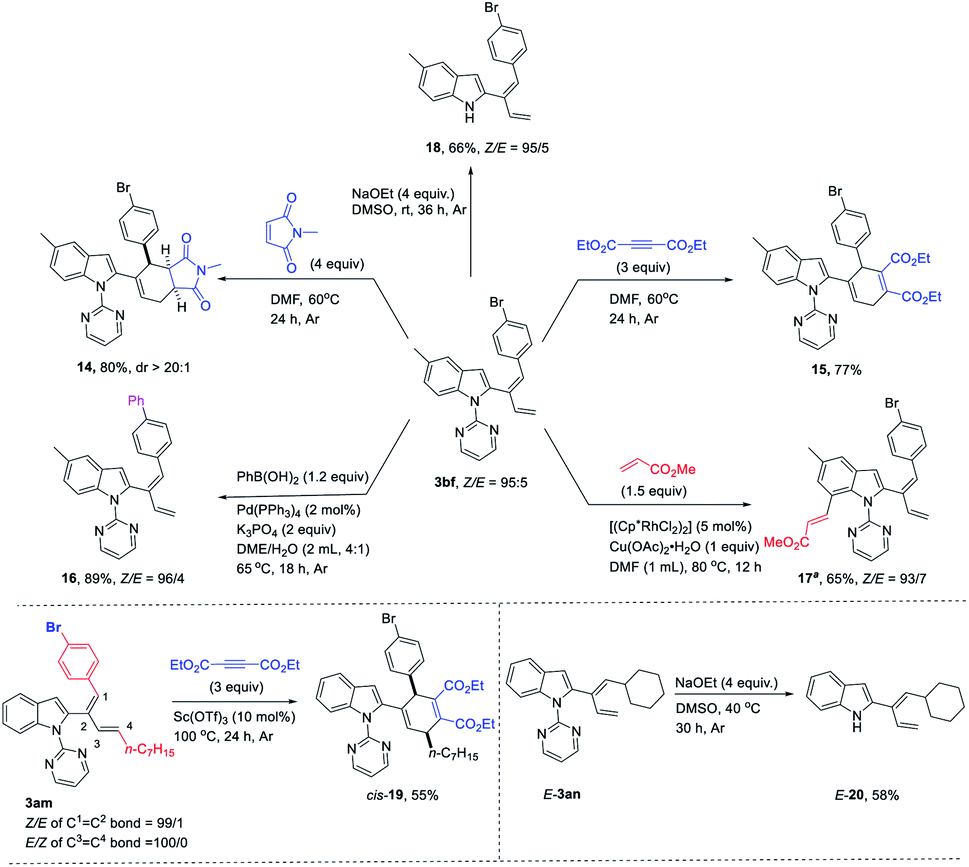 | ||
| Scheme 2 Synthetic applications of 3bf, 3am, and E-3an. | ||
Conclusions
In summary, we have developed a rhodium-catalyzed C–H functionalization of indoles with 4-aryl-2,3-allenol carbonates to access 2-(1,3-diene)-substituted indoles with the assistance of directing groups leading to unique thermodynamically unfavored Z-stereoselectivity. In the reaction, the directing group not only plays a vital role in showing basic directing effects, but also has distinctive π–π stacking interactions with the aromatic ring of allene substrates. DFT calculations rationalized the Z selectivity of the reaction for the aryl substrate and forecasted an E selectivity for the alkyl-substituted allene substrate, which was confirmed by the experimental results. The synthetic potential of the products has been demonstrated. Due to the importance of the indole derivatives, this method will be of high interest for organic and medicinal chemists. Further studies are ongoing in our laboratory.Data availability
Crystallographic data for 3cf has been deposited at the CCDC with the number of 2014026 and can be obtained from http://www.ccdc.cam.ac.uk. The ESI include experimental detail, analytical data, computational details, and all the spectra.Author contributions
Y. Z. performed the experiments. X. Z. performed DFT calculations. S. M. directed the project. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (grant no. 21690063 and 21988101 to S. M.) and Natural Science Foundation of Shanghai (grant no. 19ZR1468500 to X. Z.) is greatly appreciated. We also thank Mr Jie Wang in this group for reproducing the results for 3ap and 3as presented in Table 3 and 3am in eqn (1), and we thank Mr Yifan Cui and Mr Weiyi Wang in this group for providing 2s and 2l′ (see the ESI) respectively.Notes and references
- For selected reviews on C-H activation, see: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (e) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC; (f) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (g) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (h) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (i) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS PubMed; (j) L. Ping, D. S. Chung, J. Bouffard and S. Lee, Chem. Soc. Rev., 2017, 46, 4299 RSC; (k) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (l) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (m) Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. Madron du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516 CrossRef.
- For selected examples on the use of directing groups as an internal oxidant to construct cyclic compounds, see (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed; (b) Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed; (c) B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS; (d) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS; (e) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS; (f) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed; (g) R. B. Dateer and S. Chang, J. Am. Chem. Soc., 2015, 137, 4908 CrossRef CAS PubMed; (h) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS; (i) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (j) W. Zhen, F. Wang, M. Zhao, Z. Du and X. Li, Angew. Chem., Int. Ed., 2012, 51, 11819 CrossRef CAS PubMed; (k) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed; (l) S. Wu, R. Zeng, C. Fu, Y. Yu, X. Zhang and S. Ma, Chem. Sci., 2015, 6, 2275 RSC; (m) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed.
- For selected examples on directing groups participating in the reaction directly to construct cyclic compounds, see: (a) M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211 CrossRef CAS; (b) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS PubMed; (c) W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560 CrossRef CAS PubMed; (d) N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636 CrossRef CAS PubMed; (e) X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (f) Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed; (g) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407 CrossRef CAS; (h) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS; (i) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS PubMed; (j) X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163 CrossRef CAS PubMed; (k) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (l) S. C. Reddy, I. Khan and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 12115 CrossRef PubMed; (m) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS; (n) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS PubMed; (o) H. Chen, Y.-X. Wang, Y.-X. Luan and M. Ye, Angew. Chem., Int. Ed., 2020, 59, 9428 CrossRef CAS; (p) C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 1762 CrossRef CAS; (q) Z. Zhou, G. Liu and X. Lu, Org. Lett., 2016, 18, 5668 CrossRef CAS PubMed; (r) S. Wu, X. Wu, C. Fu and S. Ma, Org. Lett., 2018, 20, 2831 CrossRef CAS; (s) J. Karthikeyan, R. Haridharan and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 12343 CrossRef CAS PubMed; (t) Y. Lian, R. G. Bergman, L. D. Lavis and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 7122 CrossRef CAS PubMed; (u) Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520 CrossRef CAS PubMed; (v) B. Su and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 12137 CrossRef CAS PubMed; (w) W. Du, Q. Gu, Z. Li and D. Yang, J. Am. Chem. Soc., 2015, 137, 1130 CrossRef CAS PubMed.
- For the use of directing group as an internal oxidant to construct acyclic compounds, see (a) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed; (b) R. Zeng, S. Wu, C. Fu and S. Ma, J. Am. Chem. Soc., 2013, 135, 18284 CrossRef CAS PubMed; (c) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed; (d) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970 CrossRef CAS PubMed; (e) X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794 CrossRef CAS PubMed; (f) J. Zhou, J. Shi, Z. Qi, X. Li, H. E. Xu and W. Yi, ACS Catal., 2015, 5, 6999 CrossRef CAS; (g) B. Li, J. Lan, D. Wu and J. You, Angew. Chem., Int. Ed., 2015, 54, 14008 CrossRef CAS; (h) X. Wang, T. Gensch, A. Lerchen, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 6506 CrossRef CAS PubMed; (i) Y. Wu, Z. Chen, Y. Yang, W. Zhu and B. Zhou, J. Am. Chem. Soc., 2018, 140, 42 CrossRef CAS; (j) W. Yi, W. Chen, F.-X. Liu, Y. Zhong, D. Wu, Z. Zhou and H. Gao, ACS Catal., 2018, 8, 9508 CrossRef CAS; (k) G. Zheng, Z. Zhou, G. Zhu, S. Zhai, H. Xu, X. Duan, W. Yi and X. Li, Angew. Chem., Int. Ed., 2020, 59, 2890 CrossRef CAS; (l) K. Ozols, S. Onodera, Ł. Wožniak and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 655 CrossRef CAS.
- For as seminal report, see: Y. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248 CrossRef CAS PubMed.
- For selected reports with Rh-catalysis, see: (a) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181 CrossRef CAS PubMed; (b) R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597 CrossRef CAS PubMed; (c) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed; (d) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636 CrossRef CAS PubMed; (e) H. Wang, B. Beiring, D.-G. Yu, K. D. Collins and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12430 CrossRef CAS PubMed; (f) T.-J. Gong, W. Su, Z.-J. Liu, W.-M. Cheng, B. Xiao and Y. Fu, Org. Lett., 2014, 16, 330 CrossRef CAS PubMed; (g) C. Ghosh, P. J. Nagtilak and M. Kapur, Org. Lett., 2019, 21, 3237 CrossRef CAS.
- X. Huang and S. Ma, Acc. Chem. Res., 2019, 52, 1301 CAS.
- (a) R. Liu, Y. Wei and M. Shi, Chem. Commun., 2019, 55, 7558 RSC; (b) Z. Shen, I. Maksso, R. Kuniyil, T. Rogge and L. Ackermann, Chem. Commun., 2021, 57, 3668 RSC.
- (a) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072 CrossRef CAS; (b) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062 CrossRef CAS.
- For selected recent reviews concerning CMD processes, see: (a) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114 CrossRef CAS PubMed; (b) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799 CrossRef CAS PubMed; (c) Y.-F. Yang, X. Hong, J.-Q. Yu and K. N. Houk, Acc. Chem. Res., 2017, 50, 2853 CrossRef CAS PubMed; (d) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed; (e) W. Zhu and T. B. Gunnoe, Acc. Chem. Res., 2020, 53, 920 CrossRef CAS PubMed.
- For selected recent examples concerning CMD processes, see: (a) P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231 CrossRef CAS PubMed; (b) S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623 CrossRef CAS PubMed; (c) M. Simonetti, G. J. P. Perry, X. C. Cambeiro, F. Juliá-Hernández, J. N. Arokianathar and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 3596 CrossRef CAS PubMed; (d) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278 CrossRef CAS PubMed; (e) H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909 CrossRef CAS; (f) W. Liu, J. Zheng, Z. Liu, W. Hu, X. Wang and Y. Dang, ACS Catal., 2018, 8, 7698 CrossRef CAS; (g) D. D. Beattie, A. C. Grunwald, T. Perse, L. L. Schafer and J. A. Love, J. Am. Chem. Soc., 2018, 140, 12602 CrossRef CAS PubMed; (h) W. Li, D. Yuan, G. Wang, Y. Zhao, J. Xie, S. Li and C. Zhu, J. Am. Chem. Soc., 2019, 141, 3187 CrossRef CAS PubMed; (i) L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821 CrossRef CAS PubMed; (j) Y. Luo, C. Shan, S. Liu, T. Zhang, L. Zhu, K. Zhong, R. Bai and Y. Lan, ACS Catal., 2019, 9, 10876 CrossRef CAS; (k) Z. Fan, K. L. Bay, X. Chen, Z. Zhuang, K.-S. Park, H. S. Yeung, K. N. Houk and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 4770 CrossRef CAS; (l) B. A. Suslick and T. D. Tilley, J. Am. Chem. Soc., 2020, 142, 11203 CrossRef CAS.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed; (b) T. Lu, Multiwfn, version 3.5, 2018 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- (a) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS; (b) J. Shi, Y. Yan, Q. Li, H. E. Xu and W. Yi, Chem. Commun., 2014, 50, 6483 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds, DFT calculations, NMR spectra of substrates and products. CCDC [2014026]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02167b |
| This journal is © The Royal Society of Chemistry 2021 |