
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving highly efficient aggregation-induced emission, reversible and irreversible photochromism by heavy halogen-regulated photophysics and D–A molecular pattern-controlled photochemistry of through-space conjugated luminogens†
Zuping
Xiong
 ,
Xiaoxiao
Zhang
,
Longxiang
Liu
,
Qiaozhi
Zhu
,
Zhenni
Wang
,
Hui
Feng
and
Zhaosheng
Qian
*
,
Xiaoxiao
Zhang
,
Longxiang
Liu
,
Qiaozhi
Zhu
,
Zhenni
Wang
,
Hui
Feng
and
Zhaosheng
Qian
*
Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Normal University, Yingbin Road 688, Jinhua 321004, People's Republic of China. E-mail: qianzhaosheng@zjnu.cn
First published on 5th July 2021
Abstract
It is extremely challenging but desirable to regulate the photophysical and photochemical processes of aggregation-induced emission luminogens (AIEgens) in distinct states in a controllable manner. Herein, we design two groups of AIEgens based on a triphenylacrylonitrile (TPAN) skeleton with through-space conjugation (TSC) property, demonstrate controlled regulation of photophysical emission efficiency/color and photochemical photochromic and photoactivatable fluorescence behaviours of these compounds, and further validate design principles to achieve highly efficient and emission-tuning AIEgens and to accomplish photo-dependent color switches and fluorescence changes. It is surprisingly found that the introduction of heavy halogens like bromine into a TPAN skeleton dramatically enhances the emission efficiency, and such an abnormal phenomenon against the heavy-atom effect is attributed to the specific through-space conjugation nature of the AIE-active skeleton, effective intermolecular halogen-bond-induced restriction of intramolecular motions, and heavy atom-induced vibration reduction. The incorporation of two electron-donating amino groups into the TPAN skeleton cause the luminogens to undergo a bathochromic shifted emission due to the formation of a D–A pattern. Apart from the regulation of photophysical processes in the solid state, the construction of the D–A pattern in luminogens also results in extremely different photochemical reactions accompanying reversible/irreversible photochromism and photoactivatable fluorescence phenomena in a dispersed state. It is revealed that photo-triggered cyclization and decyclization reactions dominantly contribute to reversible photochromism of the TPAN family, and the photo-induced cyclization–dehydrogenation reaction is responsible for the irreversible color changes and photoactivatable fluorescence behaviours of the NTPAN family. The demonstrations of multiple-mode signaling in photoswitchable patterning and information encryption highlight the importance of controlled regulation of photophysics and photochemistry of fused chromic and AIE-active luminogens in distinct states.
Introduction
Rational regulation of photophysical processes of a luminogen through structural design and functional modification is still challenging but desirable to greatly improve the emission efficiency, alter the emission color and tune the emission lifetime making the luminogen more suited for distinct environments and diverse applications. A great achievement has been made on regulating the photophysics of traditional dye molecules in a controlled way,1,2 and a diversity of dye molecules have been broadly exploited in various applications such as in life science, sensors and optoelectronics.3–6 These dye molecule-based fluorophores with a rigid structure and through-bond conjugation (TBC) property are broadly employed in dilute solutions, but they have to face a great challenge in use in their solid or aggregation state due to their notorious photophysical process of aggregation-caused quenching (ACQ). A new variety of luminogens with aggregation-induced emission (AIEgens) developed recently provide a great opportunity to overcome the ACQ behaviours of traditional TBC dye molecules.7–9 In contrast to through-bond conjugated dyes, these AIEgens intensely fluoresce in the solid or aggregation state but exhibit almost no emission in solutions, which can be attributed to the restriction of intramolecular motions (RIM) in space-restricted environments.10–12 The reverse photophysical behavior of AIEgens to TBC dyes originates from their distinct through-space conjugation (TSC) in the molecular structure and conformation in nature.13–15 Although diverse applications of AIEgen-based materials have been accomplished,16–20 this growing research area is facing several important challenges, one of which is the much lower emission efficiency of AIEgens (frequently <0.5) than the TBC dyes (mostly >0.8).21 Recent advances have revealed that the key factor for AIE is the control over the non-radiative decay (deactivation) pathway to improve the emission efficiency,22 but operable and effective design strategies are still extremely lacking now. Tailoring alkyl linkages has been proved to dramatically enhance the emission efficiency of tetraphenylethylene-based AIEgens without changing their emission color; however, this strategy is only limited to the AIE-active tetraphenylethylene (TPE) skeleton without evidence of expanding its effectiveness to other AIEgens.23 Our recent paper on multi-halogenated TPE derivatives shows that even introducing multiple halogens like bromine into a TPE skeleton can greatly improve their emission efficiencies with the highest value of 0.75, which is largely attributed to the effective restriction of intramolecular motions due to strong intermolecular halogen–halogen interactions.24 However, general design principles with broad applicability are still rare but desperately needed to dramatically increase the emission efficiency of AIEgens equivalent to those of TBC dyes.Photochemical reactions of a luminogen generally compete with its photophysical processes like fluorescence and thus only one aspect of them frequently dominates the photo-triggered behaviours of a luminogen. Photochromism originating from a specific photochemical reaction refers to a reversible transformation of a chemical compound between two forms with different absorption spectra and colors upon photoirradiation.25 Among various organic photochromic materials, the diarylethene family is the most famous and valuable because of their excellent photochromic performance such as the outstanding thermal stability of both isomers, good fatigue resistance and rapid response.26 They can also be lighted up with bright fluorescence in solutions with remote photoirradiation showing great potential in advanced anti-counterfeiting, super-resolution imaging and rewritable photonic devices.27–29 In addition to classic photochromic molecules, recent advances demonstrate that AIEgens are able to undergo photochromic changes upon irradiation after proper modification, and the incorporation of photochromism and aggregation-induced emission can realize photo-controlled dual-channel signaling.30 Direct linkage of traditional photochromic units and classic AIEgens is simple, but the interactions between the two units in such a molecule frequently bring an apparently negative impact on photo-controlled dual absorption/emission changes.31,32 Fusion of a photo-active unit to a tetraarylethene skeleton provides a promising strategy to effectively integrate photochromism and photo-triggered fluorescence switching.33,34 Several recent studies reported that tetraarylethenes after proper modification demonstrate photoinduced absorption and fluorescence changes in the solid state; however, most of these molecules only undergo observable solid-state photochromism with low efficiency accompanying solid-state fluorescence quenching, and almost no photochromic changes can be observed in solution.35–39 Our recent studies reveal that photochromic and solid-state fluorescent molecules can be rationally designed by introducing a traditional photoactive group such as a thienyl or furyl group into a typical AIE molecular skeleton, and the shift of the dominant process between photophysics and photochemistry can be achieved by regulating the position of introducing units.40,41 However, it is still a formidable challenge to achieve simultaneous photophysical fluorescence enhancement in the solid state and photochemistry-controlled chromism and fluorescence switch based on a single AIEgen with relatively bistable isomers in solution or dispersed state upon photoirradiation.
Halogen bonds are broadly used to regulate molecular self-assembly processes in crystal engineering42 and thus are frequently exploited to control the photophysical behaviors of organic optical materials involving halogen bonds.43–45 Inspired by such more ubiquitous halogen-induced interactions and the RIM mechanism of AIEgens, we expect that it could be an effective and general way to tremendously enhance the emission efficiency by introducing halogens to restrain intermolecular motions of AIEgens due to the construction of halogen-induced intermolecular interactions in the aggregation state. The cyano group generally acts as an effective chromophore in luminogens and an electron-acceptor in TICT molecules,46 thus we aim to incorporate a cyano group into the classic AIEgen tetraphenylethene skeleton to activate photochemical activity and reduce intermolecular motions and also to readily construct donor–acceptor patterns by introducing electron-donating amino groups. To this end, we design two groups of AIEgens based on a triphenylacrylonitrile skeleton, explore their photophysical photochemical behaviours, and validate general design principles to achieve high emission efficiency and regulate the photophysical process and photochemical reactions in different states. As shown in Scheme 1a, these designed luminogens consist of the 2,3,3-triphenylacrylonitrile (TPAN) family and the 3,3-bis(4-(dimethylamino)phenyl)-2-phenylacrylonitrile (NTPAN) family, which contain TPAN, NTPAN and their respective halogenated compounds. As shown in Scheme 1b, both TPAN and NTPAN families show a sharply increasing trend in the emission efficiency upon introducing a heavier halogen from fluorine to iodine, and the largest emission efficiency is up to 0.73, validating that it is an effective strategy to regulate the photophysics and enhance the emission efficiency in the solid state by the introduction of simple halogen atoms into a through-space conjugated AIEgen. An apparent red-shift in emission color for the NTPAN family is accomplished as compared to the TPAN family through introducing electron-donating amino groups forming D–A patterned molecules, but highly efficient solid-state fluorescence dominates the photophysical processes of these compounds in the aggregation state for the two groups. In a photochemical aspect, the TPAN family can undergo reversible photochromism upon UV irradiation through photo-triggered cyclization and decyclization reactions in solution. However, the existence of D–A pattern in the NTPAN family greatly changes their photochemical behaviors; NTPAN compounds have UV-dependent photochromic behaviours and therefore undergo irreversible photochromism and exhibit an interesting photoactivatable fluorescence phenomenon upon UV irradiation. The underlying photophysical processes and photochemical reactions are systematically examined by experimental and theoretical analyses to validate the regulation of photophysics and photochemistry by molecular design. These unique dual-mode, reversible and irreversible signaling behaviors controlled by external irradiation make them suitable for photoswitchable patterning of various surfaces and advanced information encryption applications in a dual-channel way.
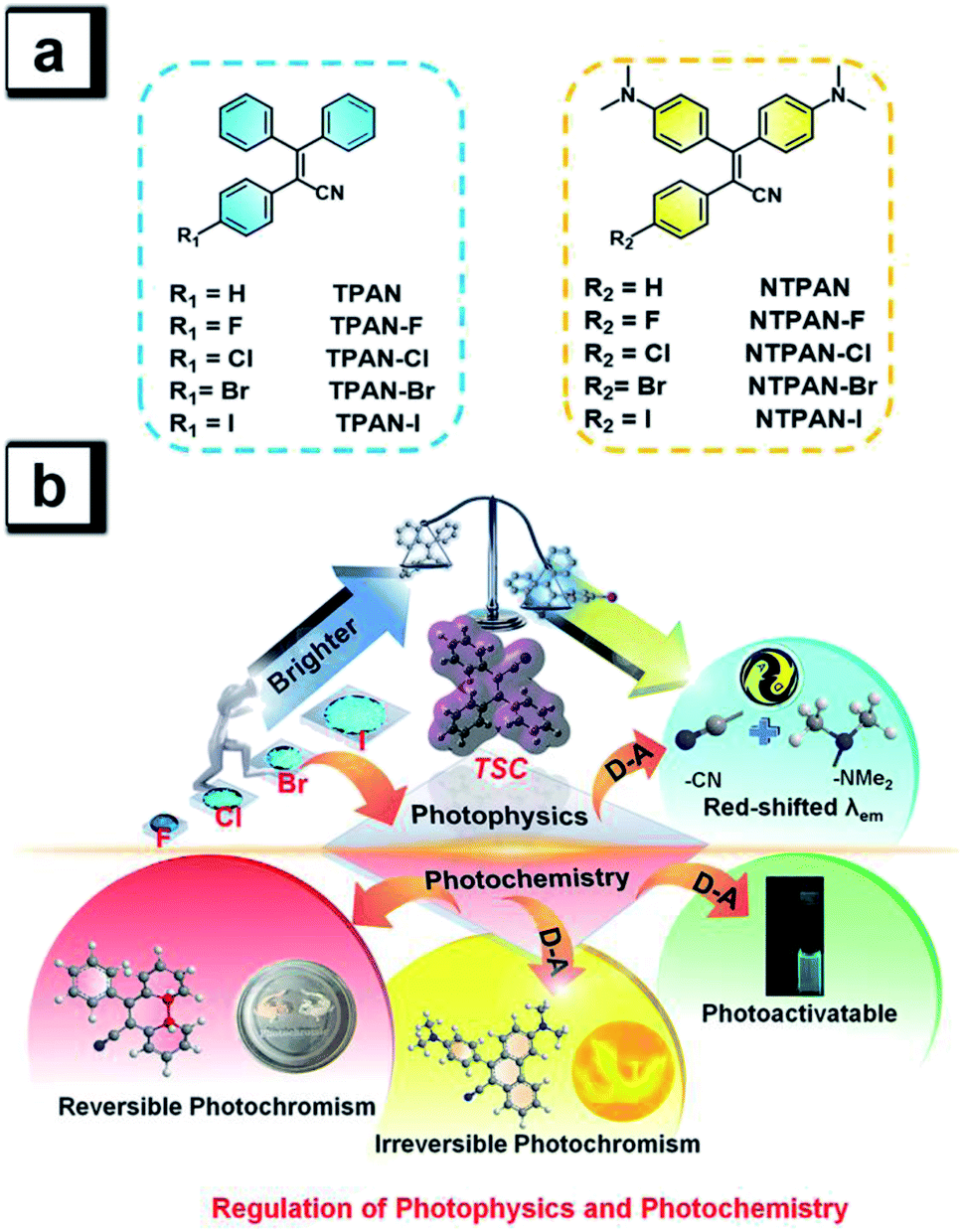 | ||
| Scheme 1 Structures of halogenated TPAN/NTPAN derivatives (a), and schematic illustration of controllable regulation of photophysics and photochemistry of an AIE-active luminogen (TPAN) with through-space conjugation by introducing halogens and constructing donor–acceptor patterns (b). Introducing a heavier halogen gradually increases the emission efficiency; constructing donor–acceptor patterns tremendously alters the emission colors; TPAN without a D–A pattern undergoes reversible photochromism upon irradiation; TPAN with a D–A pattern (NTPAN) undergoes irreversible photochromism and photoactivatable fluorescence upon UV irradiation. | ||
Results and discussion
Synthesis and photophysical properties of triphenylacrylonitrile (TPAN), bis(dimethylamino)triphenylacrylonitrile (NTPAN) and their halogenated derivatives
The classic Knoevenagel condensation reaction47,48 was employed to synthesize TPAN and NTPAN derivatives using a strong base such as sodium hydride, and all the ten compounds were obtained in good yields. They consist of two groups of compounds based on the molecular skeletons TPAN and NTPAN. The TPAN group contains 2,3,3-triphenylacrylonitrile (TPAN), 2-(4-fluorophenyl)-3,3-diphenylacrylonitrile (TPAN-F), 2-(4-chlorophenyl)-3,3-diphenylacrylonitrile (TPAN-Cl), 2-(4-bromophenyl)-3,3-diphenylacrylonitrile (TPAN-Br) and 2-(4-iodophenyl)-3,3-diphenyl-acrylonitrile (TPAN-I), while the NTPAN group is composed of 3,3-bis(4-(dimethylamino)-phenyl)-2-phenylacrylonitrile (NTPAN), 3,3-bis(4-(dimethylamino)phenyl)-2-(4-fluorophenyl)-acrylonitrile (NTPAN-F), 2-(4-chlorophenyl)-3,3-bis(4-(dimethylamino)phenyl)acrylonitrile (NTPAN-Cl), 2-(4-bromophenyl)-3,3-bis(4-(dimethylamino)phenyl)acrylonitrile (NTPAN-Br) and 3,3-bis(4-(dimethylamino)phenyl)-2-(4-iodophenyl)acrylonitrile (NTPAN-I). The detailed synthesis routes are depicted in Scheme S1.† All the compounds were strictly purified using column chromatography and further characterized by 1H NMR and 13C NMR spectroscopy and high-resolution mass spectrometry. To validate their absolute structures and packing patterns in crystals, their absolute structures were determined by the single-crystal X-ray diffraction technique using their respective single crystal. Their determined absolute conformations and crystallographic data are shown in Fig. S1 and Tables S1, S2,† respectively, which absolutely confirm the designed structures in Scheme 1. Through-space conjugation refers to the process of the intramolecular adjacent conjugated subunits significantly interacting with each other through space instead of merely forming covalent bonds in an organic molecule.13–15 From Fig. S1,† it can be readily noted that these molecules show similarly twisted conformations and three phenyl rings in contact with each other or lying in a short distance (around 4.8 Å), which indicates that through-space conjugation occurs for these molecules. Taking TPAN and NTPAN as examples, the independent gradient model (IGM) method is used on the basis that a single crystal structure can be used to visualize such through-space interactions. It is found that the apparent van der Waals interactions shown in the green section occur among three phenyl rings, further validating the occurrence of through-space conjugation in these molecules. Their single-crystal structures and packing patterns are valuable to analyze the relationship between their solid-state fluorescence properties and structural conformations/stacking patterns in addition to verifying their absolute structures and through-space conjugation features.Colorless solid forms and colorless solutions of TPAN groups suggest that these five compounds have selective absorption in UV regions, which is verified by their UV-visible absorption spectra in DCM solvent (Fig. S2†). The major absorption peaks are located around 240 nm and 312 nm for all of them, and almost no absorption above 400 nm can be observed. Their colorless solutions exhibit almost no emission upon UV excitation, but their solids intensively emit blue fluorescence, as shown in Fig. 1a. The major emission peak is located at 462 nm for TPAN, and its halogenated derivatives exhibit a similar blue emission upon UV excitation without obvious change in emission maxima. Their time-resolved decay curves in Fig. S3† indicate that they have short-lived lifetimes in the range of 0.7–1.3 ns, suggesting that these emissions come from singlet states with fluorescent nature. Aggregation-induced emission behaviors of all the five TPAN derivatives were verified by poor solvent-induced aggregation experiments (Fig. S4†), in which the blue emission was found to be progressively enhanced as the water content was increased in the mixed solution for each of them. However, it is surprisingly noted that their fluorescence quantum yields are gradually increased from 10.9% to 49.8% as the atomic number of halogen increases from TPAN-F to TPAN-I. This unusual heavy halogen-caused fluorescence enhancement phenomenon disagrees with the traditional heavy atom effect which describes that the introduction of a heavy atom like bromine generally leads to weakening or quenching of the fluorescence for conventional planar aromatic luminogens due to the heavy atom-induced notable spin–orbit coupling (SOC) effect,49 and this observation also differs slightly from the anti-heavy atom effect of halogenated tetraphenylethene proposed in our previous paper.24 Compared to the TPAN group, all NTPAN derivatives exhibit yellow color in daylight, suggesting that they have apparent absorption in the visible region in addition to UV absorption. Fig. S5† verifies this implication from their major absorption peaks around 270 nm and 400 nm. Resembling TPAN compounds, all NTPAN derivatives have almost negligible emission in solution upon UV excitation but emit intense green/yellow fluorescence in the solid state. Their lifetimes fall in the range of 0.9–3.1 ns (Fig. S6†), and their aggregation experiments also confirmed similar aggregation-induced emission behaviors to TPAN compounds (Fig. S7†). However, the emission maxima of the NTPAN compounds have a huge red-shift relative to the TPAN counterparts after two dimethylamino groups are introduced. All TPAN solids emit a blue emission upon excitation, whereas all NTPAN solids exhibit green-to-yellow fluorescence under UV light. As shown in Fig. 1b, the emission maximum of NTPAN in the solid state is located at 580 nm, which is red-shifted by nearly 100 nm relative to that of TPAN; the smallest difference in the emission peak between NTPAN-I and TPAN-I is also up to 44 nm. These observations suggest that introducing dimethylamino groups into the TPAN skeleton is able to largely tune the emission to long-wavelength regions, and this effect can be attributed to the formation of donor–π–acceptor patterns in NTPAN compounds where two dimethylamino groups act as the donor, while the cyan group functions as the acceptor. Differing from TPAN compounds, NTPAN-Cl has the largest fluorescence quantum yield (73.1%) among the NTPAN derivatives, and the quantum yield gradually decreased from NTPAN-Cl to NTPAN-I, indicating that the heavy atom effect plays an important role in NTPAN compounds. The main photophysical parameters of TPAN and NTPAN compounds in different states are summarized in Table S3,† which visually presents an abnormal photophysical phenomenon for TPAN and NTPAN compounds that the introduction of a heavy halogen greatly enhances the emission efficiencies of these AIEgens in the solid or aggregation state.
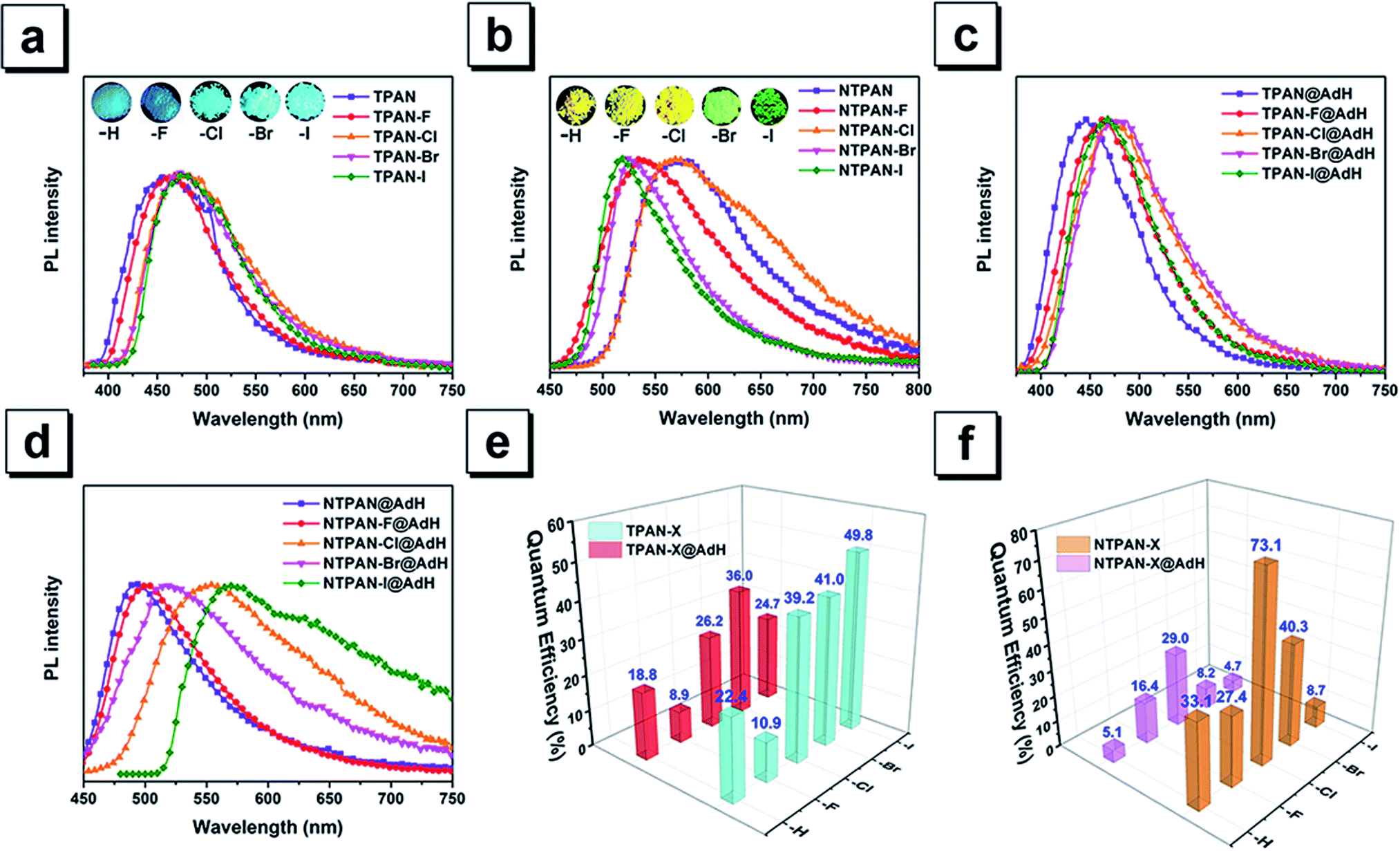 | ||
| Fig. 1 (a) PL spectra and images of TPAN, TPAN-F, TPAN-Cl, TPAN-Br and TPAN-I in the solid state. (b) PL spectra and images of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I in the solid state. (c) PL spectra of TPAN, TPAN-F, TPAN-Cl, TPAN-Br and TPAN-I homogeneously dispersed in adamantane (AdH). (d) PL spectra and images of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I homogeneously dispersed in adamantane (AdH). (e) Quantum efficiencies of TPAN, TPAN-F, TPAN-Cl, TPAN-Br and TPAN-I in the solid state and dispersed in adamantane. (f) Quantum efficiencies of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I in the solid state and dispersed in adamantane. | ||
Mechanistic exploration of halogen-induced solid-state fluorescence enhancement and donor–π–acceptor-caused red-shifted emission
The heavy-atom effect predicts that the substitution of a heavy atom with a large atomic number like bromine generally leads to the fluorescence quenching and simultaneous phosphorescence enhancement in solution for conventional dye molecules because the heavy-atom effect greatly contributes to notable spin–orbit coupling (SOC) and resultantly promotes intersystem crossing processes.50,51 To test the impact of the classic heavy atom effect on halogenated TPAN and NTPAN derivatives, low-temperature experiments were first performed as shown in Fig. 2a and b. For the halogenated TPAN family, each of them in THF at 77 K has only one emission peak at a location close to their respective emission in the solid state without appearance of new peaks; similarly, all halogenated NTPAN compounds in THF at 77 K do not exhibit phosphorescence peaks. Major emissions at 77 K for all halogenated derivatives have short-lived lifetimes in the range of 0.9–6.0 ns (Fig. S8 and S9†). These observations clearly suggest that these TPAN and NTPAN compounds with heavy halogens do not follow the heavy atom effect principle, and the introduction of heavy halogens plays a negligible impact on promoting intersystem crossing and the following phosphorescence, which is similar to the finding in halogenated tetraphenylethene derivatives.24 To understand the heavy halogen-induced solid-state fluorescence enhancement of these luminogens, some important visual information can be obtained from NBO analysis first. It is found that only a small fraction of atomic components from halogens contributes to HOMOs of TPAN and NTPAN derivatives, while almost no contributions from halogens are observed in their LUMOs (Fig. S10†), implying that halogens including heavy bromine and iodine have a minor impact on transitions between HOMOs and LUMOs. This deduction was further verified by quantitative analysis of atomic contributions in Tables S4 and S5.† It is noted that the atomic orbitals of all halogens contribute mainly to the HOMOs of the halogenated TPAN, and their contributions progressively increased in the order of F (2.26%), Cl (5.45%), Br (7.60%) and I (14.73%); with respect to the HOMOs of the halogenated NTPAN, the contributions increased slightly in the order of F (0.67%), Cl (1.24%), Br (1.38%) and I (1.95%). However, the contributions of halogens are still minor relative to other moieties such as phenyl and acrylonitrile groups, whose contributions to HOMOs/LUMOs generally range between 20% and 50%. Thus, a far-reaching enlightenment that can be drawn here is that the introduction of halogens in these AIE molecules would not greatly enhance the spin–orbit coupling effect dominating the promotion of states with different multiplicities. This fact was further explained using energy gaps between S1 and its nearest triplet state with TD-DFT calculations. According to the perturbation theory,52 the intersystem crossing rate (kisc) is proportional to the formula of the spin–orbit coupling constant (ξST) and the energy gap (ΔEST):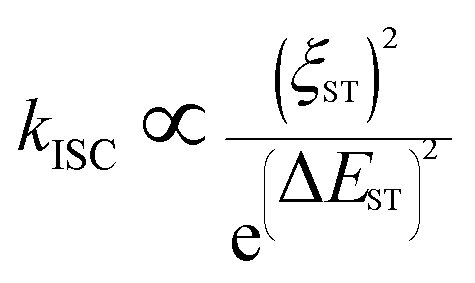
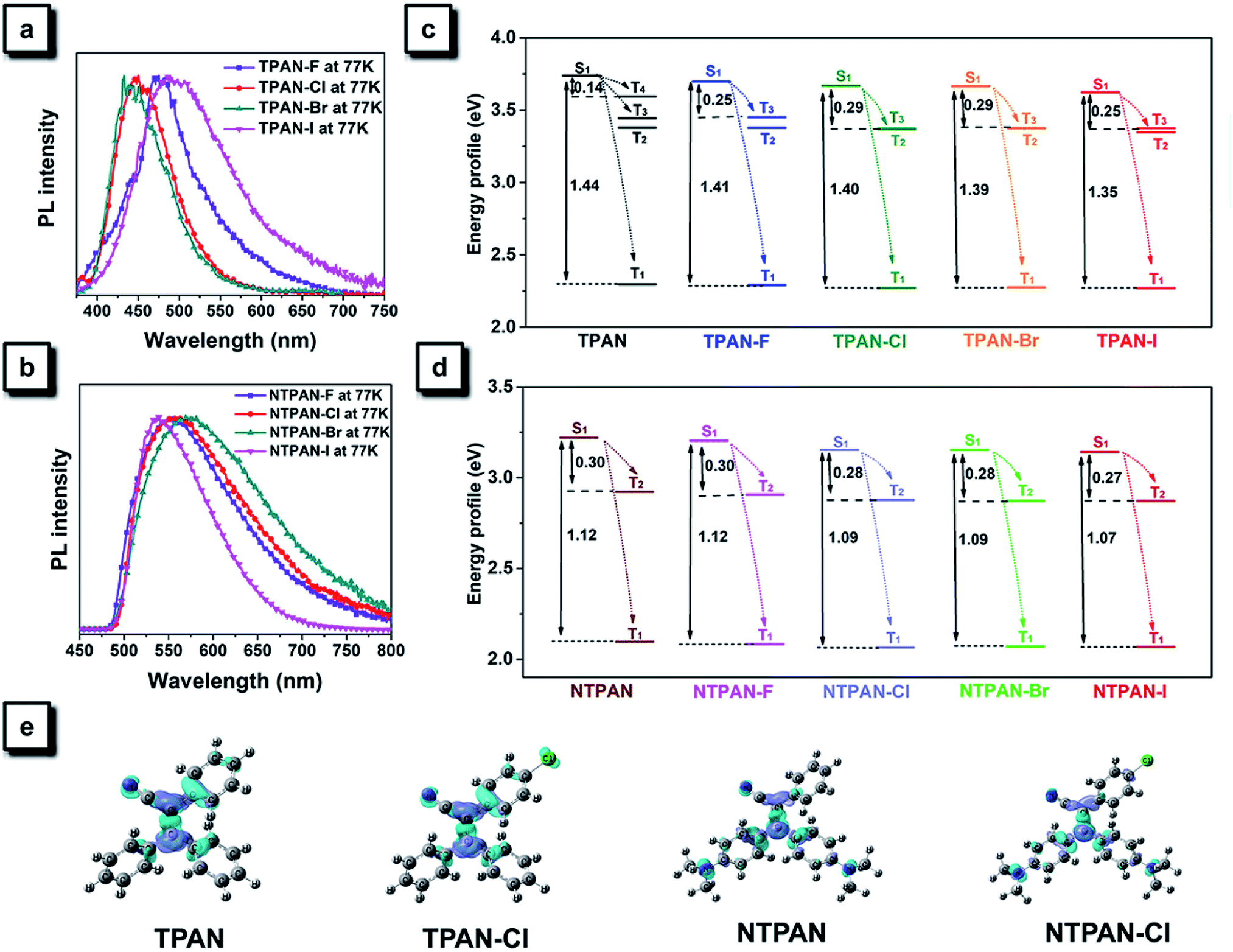 | ||
| Fig. 2 (a) PL spectra of TPAN, TPAN-F, TPAN-Cl, TPAN-Br and TPAN-I in THF at 77 K. (b) PL spectra of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I in THF at 77 K. (c) Energy profiles of excited states of TPAN, TPAN-F, TPAN-Cl, TPAN-Br and TPAN-I calculated with TDDFT. (d) Energy profiles of excited states of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I calculated with TDDFT. (e) Electron density difference maps of TPAN, TPAN-Cl, NTPAN and NTPAN-Cl at the S1 state. | ||
The excitation energies of the first 10 excited states for all TPAN and NTPAN derivatives were examined (Tables S6 and S7†). Fig. 2c shows that the most efficient ISC of TPAN occurs between S1 and T4, but the halogenated TPAN molecules undergo the most efficient transitions from S1 to T3. The energy gaps for halogenated TPAN molecules are slightly higher than those of ΔEST for TPAN, whereas all the values for ΔEST of halogenated TPAN are very close, ranging from 0.25 to 0.29 eV, which clearly suggests that the introduction of heavy halogens does not cause any apparent enhancement of ISC processes for TPAN derivatives. Fig. 2d also shows a slightly declining tendency in ΔEST following the order of NTPAN, NTPAN-F, NTPAN-Cl, NTPAN-Br and NTPAN-I, and their differences in ΔEST between the most efficient ISC process (S1 → T2) are really small, which is similar to that of the TPAN group. More specifically, the largest difference in ΔEST is only 0.03 eV for NTPAN-I as compared to NTPAN (Table S6, ESI†). As for these AIE molecules, the presence of heavier halogens like bromine or iodine has little effect on the spin–orbit coupling constant (ξST) and the involved energy gap (ΔEST), which leads to an insignificant increment of the intersystem crossing rate (kISC). All the theoretical data demonstrate that the introduction of heavy halogens in TPAN and NTPAN molecules does not greatly increase the intersystem crossing process, and no phosphorescence from the triplet state T1 can occur, which is consistent with the experimental observation. Other parameters are also critical to the fluorescence quantum yield of luminogens, such as the radiative constant (kr) and the nonradiative constant (knr), as well as the intersystem crossing rate (kISC). Table S3 (ESI†) reveals that the radiative constant (kr) gradually increased from 1.6 × 108 s−1 for TPAN-F to 3.8 ×108 s−1 for TPAN-I and the nonradiative constant (knr) experienced a sharp decline, whereas it decreased from 12.7 × 108 s−1 for TPAN-F to 3.9 × 108 s−1 for TPAN-I; the radiative constant (kr) gradually increased from 1.1 × 108 s−1 for NTPAN to 2.2 × 108 s−1 for NTPAN-Br and the nonradiative constant (knr) of NTPAN-F is minimum (only 9 × 107 s−1), but the kr decreased unceasingly to 3.9 × 108 s−1 for NTPAN-I. All these parameters indicate that the introduction of heavier halogens can effectively promote the radiative process from S1 to S0 and can mostly prohibit the nonradiative process in TPAN derivatives, which is also applicable to most NTPAN compounds (from NTPAN to NTPAN-Br). One possible reason for such fluorescence enhancement probably comes from intermolecular interactions such as halogen-induced weak interactions in crystals restricting the molecular motion, which can refer to the role of halogen–halogen interaction in multi-halogenated tetraphenylethene molecules.24 For these monohalogenated compounds, the halogen bonds are relatively not obvious, so the contribution of those to the intermolecular interactions cannot be accurately given just according to the distance between atoms. Herein, the intermolecular interactions in the crystal structures of monohaologenated TPAN and NTPAN derivatives are investigated via Hirshfeld surface analysis. Fig. 3a–d first represents the Hirshfeld surfaces of monohalogenated TPAN derivatives mapped over dnorm, where the larger red spots in map indicate the strong intermolecular interactions between the molecules, but the smaller spots are due to weak interactions. Clearly, the Hirshfeld surfaces of these TPAN derivatives are almost blue, and this denotes that most intermolecular contacts are at a distance longer than the van der Waals distance. As a result, weak intermolecular interactions among these monohalogenated molecules may contribute to this anomalous heavy atom-induced fluorescence enhancement rather than the hardly observable strong interactions. Then, the important quantitative information on the hydrogen–halogen weak interactions in these monohalogenated crystals was obtained by plotting two-dimensional (2D) fingerprint plots. It is found that a portion of the Hirshfeld surface area is covered by X (F, Cl, Br and I)⋯H intermolecular contacts. These contacts comprise 10.5%, 12.1%, 12.5% and 13.1% to the total Hirshfeld surfaces for TPAN derivatives, respectively. In order to further investigate the differences in the weak intermolecular interactions caused by the change of halogens, four pie charts are depicted in the bottom of Fig. 3a–d, which represent the percentage contributions of various intermolecular interactions on the basis of Hirshfeld surface analysis for the TPAN group. Noticeably, the contribution of halogen to all other atoms (including H, C and N) increases significantly from 10.5% for TPAN-F to 17.8% for TPAN-I, which coincides with the gradual increase of quantum efficiency from TPAN-F to TPAN-I. It implies that the halogen-induced weak intermolecular forces may promote the restriction of intramolecular motions (RIM), which lead to the solid-state fluorescence enhancement.22 For NTPAN-Cl, the fluorescence enhancement may also be due to the enhancement of the contribution of halogen-induced intermolecular interactions (Fig. S11†). However, the relatively significant decrease in fluorescence for NTPAN-Br and NTPAN-I may be attributed to some degree of heavy-atom effect. On the other hand, the emission efficiencies are apparently decreased when these as-prepared molecules are dispersed in adamantine compared to those in crystals, as shown in Fig. 1. These compounds existing in a pure solid form or homogeneously dispersed in adamantane have similarly short-lived lifetimes in a ns scale (Fig. S12 and S13†); however, comparing their small differences in emission spectra (Fig. 1c and d), the quantum yields of both TPAN and NTPAN compounds dispersed in adamantane become much lower than those in crystalline form, which implies that various intermolecular interactions are important to restrict the molecular motion and enhance the solid-state fluorescence. This observation cannot exclude the possibility of the change in the emission efficiency caused by the subtle variation in molecular conformation. In addition, this specific heavy halogen-induced fluorescence enhancement may be partly attributed to the effective restriction of intramolecular vibrations and rotations due to the large mass effect of heavy halogens.53,54 We compared the vibration intensities of TPAN and TPAN-Br using the same vibration mode, as shown in Fig. 3e. Due to the introduction of heavy atoms, the vibration frequency of TPAN (710.92 cm−1/709.79 cm−1) becomes slightly smaller than that of TPAN-Br (771.83 cm−1/834.69 cm−1) at the same vibration modes involving a vibration mode with bromine and a vibration mode without bromine. The vibration intensities of non-heavy-atom-substituted benzene rings for TPAN (43.19) and TPAN-Br (36.42) are approximate, but there is a significant decrease between the vibration intensity of heavy-atom-substituted benzene rings for TPAN (36.62) and TPAN-Br (8.80). It provides solid evidence that the occurrence of heavy atoms like bromine can effectively lower the molecular vibration strength and thus promote the emission of the luminogen. In brief, three important points including the negligible heavy-atom effect, halogen-induced weak interactions and heavy-atom-induced restriction of intramolecular vibration contribute to the unusual heavy-halogen-induced solid-state fluorescence enhancement and an enormous increase in emission efficiencies.
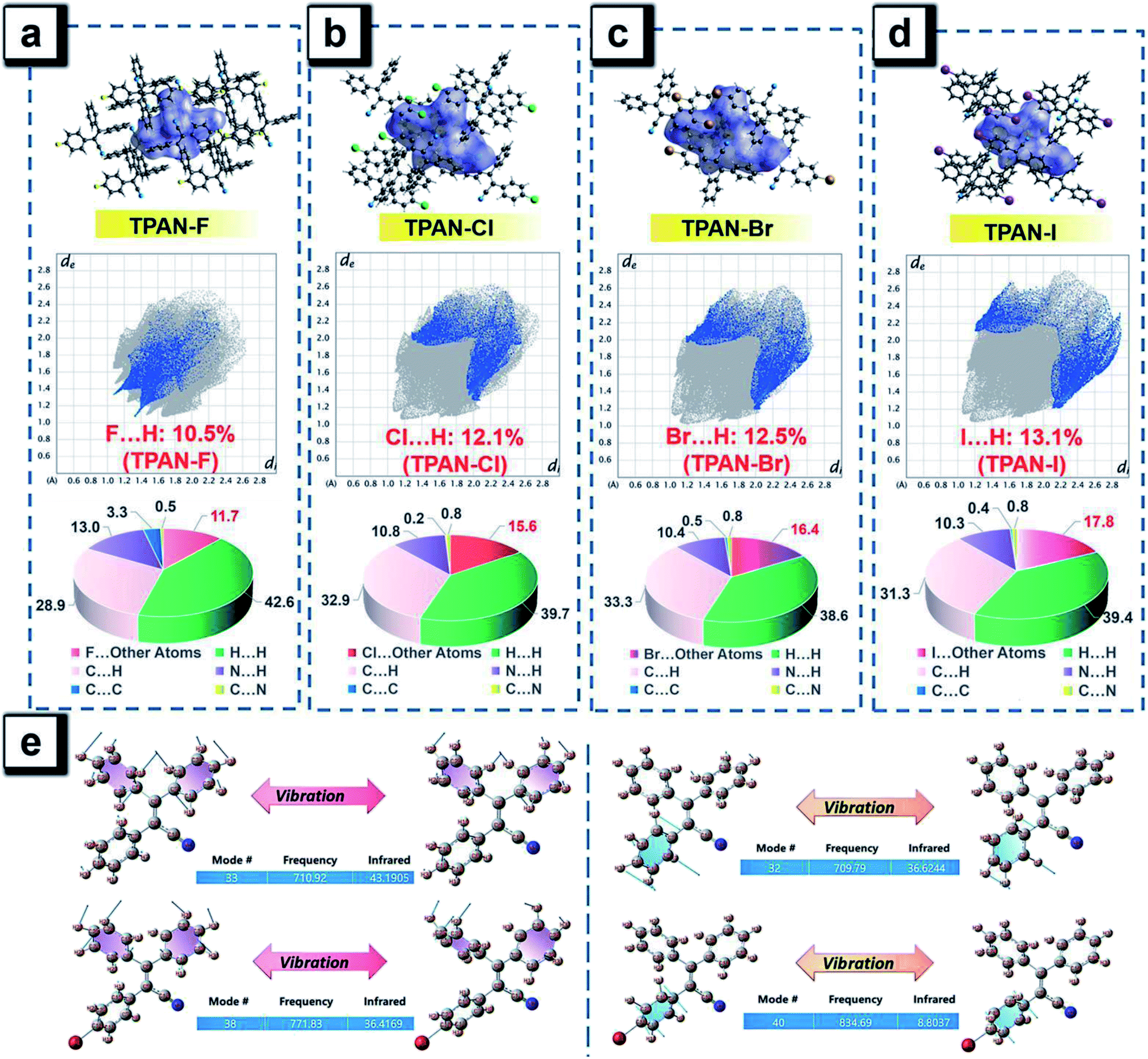 | ||
| Fig. 3 Hirshfeld surfaces of TPAN-F (a), TPAN-Cl (b), TPAN-Br (c) and TPAN-I (d) mapped with the parameter dnorm along with 2-D fingerprint plots with a specific interaction. Pie-charts: relative contributions of various intermolecular interactions to the Hirshfeld surface area. (e) A comparison between TPAN and TPAN-Br at the same vibration mode: vibrational intensity differences of non-heavy-atom-substituted benzene rings (left) and heavy-atom-substituted benzene rings (right). | ||
It is obvious that the emission colors of NTPAN compounds are largely red-shifted compared to those of the TPAN group. This regulation of emission color can be attributed to the construction of a donor–π–acceptor pattern in NTPAN compounds, where the cyano group acts as the donor and the dimethylamino groups function as the acceptor. Such a donor–π–acceptor pattern often leads to a twisted charge-transfer state, which has a longer emission maximum and is usually sensitive to the external environment.46Fig. 2e displays electron density difference maps of TPAN, NTPAN, TPAN-Cl and NTPAN-Cl at the S1 state. It is found that an apparent charge separation between the cyano group and the dimethylamino group takes place for NTPAN and NTPAN-Cl relative to those of TPAN and TPAN-Cl. This difference in electron density at the S1 state between TPAN and NTPAN series indicates that the emission of TPAN stemmed from the local excited state, whereas the emission of NTPAN comes from the charge transfer state, thus the emission of NTPAN compounds should be sensitive to their structural conformations in different packing crystals. Fortunately, we obtained two kinds of crystals for NTPAN-I from two different solvents (Fig. S14†). The NTPAN crystal cultured in DCM was denoted G-type crystal due to its bright green fluorescence, while the crystal grown in acetone was denoted Y-type due to its yellow colored emission. After analyzing their structures, we found that their absolute structures are the same, but their structural conformations are different. In G-type conformation, the angles of three phenyl groups relative to the central ethane plane are 62.7°, 30.2° and 36.8°, whereas those angles changed to 54.0°, 41.8° and 30.5° in Y-type conformation. Such a subtle change in structural conformation probably leads to their distinct emission colors with a large difference of 43 nm in the emission maximum. As a result, the introduction of electron-donating dimethylamino groups into the triphenylacrylonitrile skeleton is capable of tuning the emission colors and is valuable to design mechanochromic materials with color regulation induced by the conformational change.55,56
Reversible photochromism behaviours of triphenylacrylonitrile (TPAN) derivatives and inreversible photochromism behaviours of bis(dimethylamino)triphenylacrylonitrile (NTPAN) compounds
TPAN and its halogenated derivatives exist as white solids, indicating that no significant absorption occurs in the visible range, and this observation is consistent with their major absorption bands (around 240 nm and 310 nm) in DCM (Fig. S2 and S5†). However, their solutions quickly changed from colorless to orange after continuous irradiation with a hand-held UV lamp emitting 365 nm UV light, and such color changes can be easily observed by the naked eye (Fig. 4a–e). Time-dependent UV-visible spectra show that each of them exhibits a new visible absorption peak around 490 nm after continuously irradiating with 365 nm UV light for 60 s. Resembling those furan-/thienyl-containing tetraarylethene compounds in our previous paper,40,41 these TPAN compounds also show a reversible photochromic behavior upon ceasing UV irradiation. After various periods of standing, the orange colors of all solutions can fade out to their original colorless state. Among these compounds, the TPAN-F undergoes significant coloration under the same UV irradiation conditions, and TPAN shows the fast fading time of only 75 s, while the others require 100 s to restore their original colorless state (Fig. 4f–j). The fading of the colored solutions can also be achieved using visible light treatment, and it needs a shorter time of only 5 s for total recovery. All these coloration and fading processes are reversible, and no significant fatigue can be observed after 10 cycles in solution (Fig. 4k). Such a reversible photochromism of TPAN compounds demonstrates that these molecules undergo a UV-triggered cyclization reaction and the following ring–opening reaction.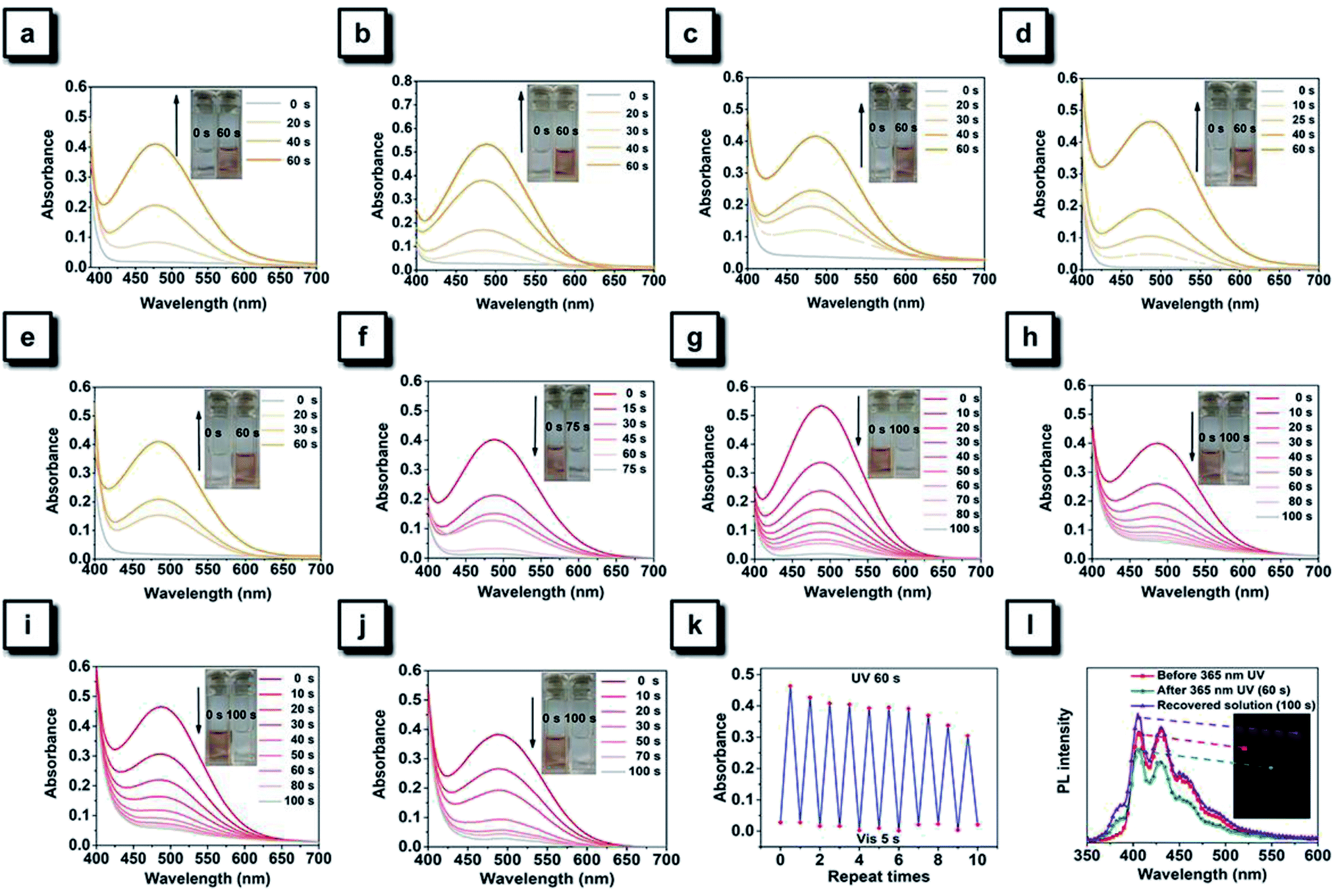 | ||
| Fig. 4 (a–e) Time-dependent UV-vis absorption spectra of TPAN (a), TPAN-F (b), TPAN-Cl (c), TPAN-Br (d), and TPAN-I (e) in DCM (2.0 mM) under continuous UV irradiation at 365 nm. (f–j) Time-dependent UV-vis absorption spectra of colored solutions of TPAN (f), TPAN-F (g), TPAN-Cl (h), TPAN-Br (i), and TPAN-I (j) in DCM (2.0 mM) under natural standing conditions. (k) Recycling times of UV-triggered coloration and white-light-induced decoloration of TPAN-F in DCM (2.0 mM). (l) PL spectra of TPAN-F in DCM (2.0 mM): the original solution (before 365 nm UV), the photochromic solution (after 365 nm UV irradiation) and the complete decoloration solution (placed naturally for 100 s) respectively. Fluorescence photographs of these solutions were taken upon UV excitation. | ||
Among TPAN compounds, TPAN-Cl was used as an example to probe their photochromic mechanisms experimentally and theoretically. To explore the underlying reaction nature, the 1H NMR spectra of TPAN-Cl in CDCl3 before and after continuous irradiation of 365 nm UV light for 10 s and 20 s were compared first, as shown in Fig. 5a. Before UV irradiation, all TPAN-Cl molecules exist in their original structures, and thus all their 1H MNR signals fall in the scope of 6.8–7.8 ppm originating from aromatic hydrogen atoms. However, four additional 1H NMR peaks appear after 20 s of UV irradiation, and they are located at 3.8, 6.1, 6.2 and 6.3 ppm. It is evident that the two peaks at 3.8 ppm do not belong to the aromatic hydrogens but to the alkyl counterparts, suggesting the generation of alkyl carbons and the formation of the cyclized ring under UV irradiation. Due to the cyclization of the central ethane and two neighboring benzene units, three new signals of aromatic hydrogens of the product also emerge around 6.0 ppm. These results provide solid evidence that a portion of molecules of TPAN-Cl are cyclized after UV irradiation. To further demonstrate the underlying nature of cyclization-depending photochromism of these TPAN compounds, the calculated absorption wavelengths for S0 → S1 before and after UV irradiation were compared to those values calculated with DFT calculations. Table S8† depicts the experimental and calculated S0 → S1 absorption wavelengths of TPAN compounds and their cyclized products in DCM. The calculated values of S0 → S1 absorption wavelengths for TPAN compounds in DCM are very close to those experimental data observed in DCM, and the differences are only about 40 nm, indicating that these calculations are accurate to estimate the absorption wavelengths. By comparison, the calculated S0 → S1 absorption wavelengths for cyclized products of TPAN compounds are much longer than those of the respective TPAN compounds and are consistent with those data observed after photochromism. These observations further verify that TPAN compounds can cyclize to their cyclized products under UV irradiation, and these cyclized products can also turn back to their original structures.
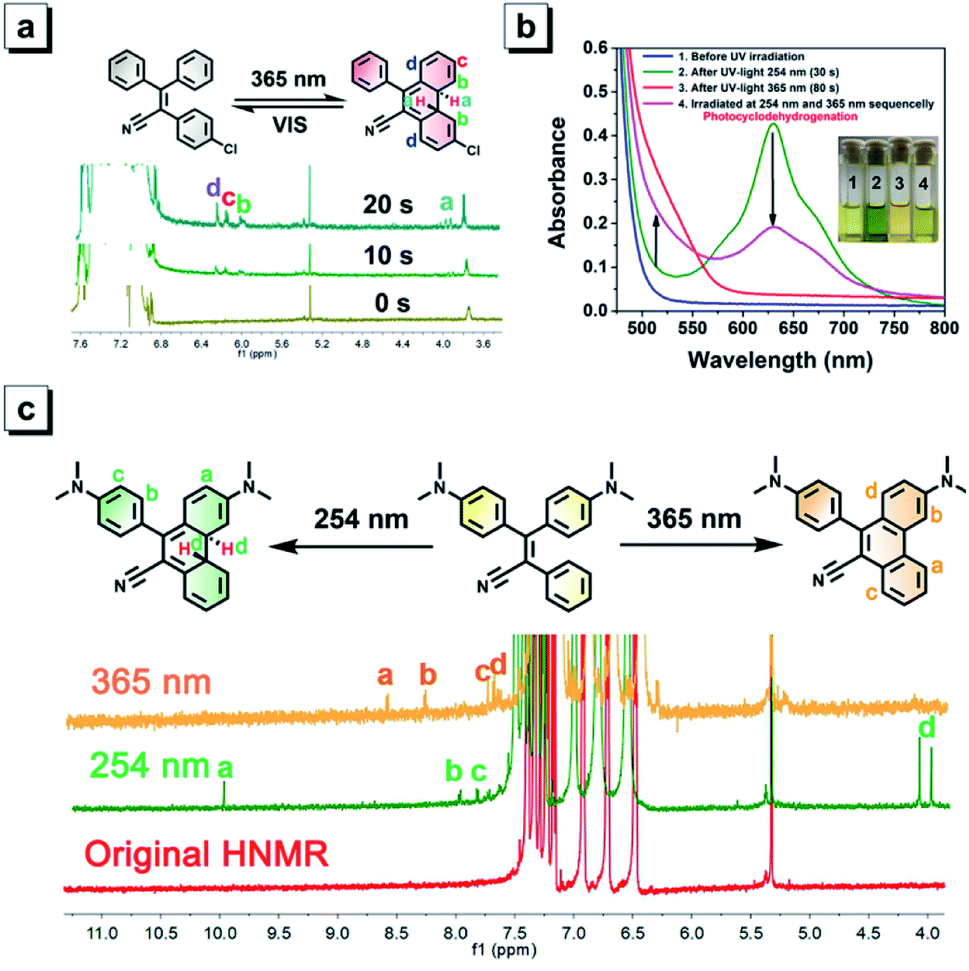 | ||
| Fig. 5 (a) Comparison of 1H NMR spectra of TPAN-Cl in CDCl3 before (0 s) and after UV irradiation (10 s and 20 s). Inset: demonstration of photo-controlled cyclization and ring-opening reactions using TPAN-Cl as an example. (b) Time-dependent UV-vis absorption spectra of NTPAN in dichloromethane with a concentration of 2 mM; (1) before UV irradiation; (2) after irradiation with 254 nm UV light for 30 s; (3) after irradiation with 365 nm UV light for 80 s; and (4) after irradiation with 254 nm and 365 nm UV light successively. Inset: the corresponding pictures of photochromic solutions in daylight. (c) Demonstration of the UV-dependent cyclization reaction and the cyclization–dehydrogenation reaction using NTPAN as an example and their changes in 1H NMR spectra. | ||
For NTPAN group, their photochromic behaviours are apparently different from those of TPAN compounds due to the introduction of the D–A pattern in the NTPAN structure. Under 365 nm UV irradiation, NTPAN shows a slight color change from yellow to orange, which is consistent with the observation that only a slight change in UV-vis absorption can be detected, as shown in Fig. 5b. Such a slight red-shift in absorption peaks implies that the molecular change might occur under 365 nm UV irradiation. However, when the UV light changed to 254 nm from 365 nm, an apparent color change from light yellow to green can be readily observed. Their UV-vis spectra show that a new absorption peak at 625 nm appears after 254 nm UV irradiation, and the large absorbance at the new absorption band is in accord with the deep green color of the irradiated solution. The green colored solution can also fade apparently out when irradiated by white light, which suggests that UV irradiation with 254 nm makes possible the NTPAN cyclization accompanying the color change. After irradiated with 254 nm and 365 nm light, it can be observed that the yellow color of the solution first turns to deep green and then to a green-yellow color. It is predicted that a different photo-induced reaction from the cyclization reaction occurs after 365 nm UV irradiation for NTPAN compounds, and a dehydrogenation reaction followed the cyclization might take place with 365 nm UV irradiation, as depicted in Fig. 5c. Using NTPAN as an example, most proton signals of the original molecule fall in the range of 6–8, and no other peaks can be detected except for the signal from methyl groups. After irradiation with 254 nm UV light, at least four new small signals can be identified, especially the two signals at 4.0 and 4.1 ppm belong to alkyl hydrogens, which provide solid evidence for the occurrence of alkyl hydrogens of the resulting product after UV irradiation. The appearance of two alkyl hydrogen atoms in different chemical environments clearly proves that the cyclization reaction takes place to transform the aromatic hydrogens to alkyl hydrogens under such UV excitation. Compared to the 1H NMR spectrum of NTPAN irradiated with 254 nm UV light, no observable signals at around 4.0 ppm appear in the spectrum of NTPAN solution after 365 nm irradiation, indicating that no alkyl hydrogens are generated during such a process. Accordingly, the new signals at 8.6, 8.3, 7.8 and 7.7 ppm are in accord with the structure of the dehydrogenated product of NTPAN, suggesting that the original NTPAN molecules undergo consecutive cyclization and dehydrogenation reactions to produce a small portion of the dehydrogenated product. To further verify this hypothesis, the absorption maxima for S0 → S1 of NTPAN compounds, their cyclized products and their dehydrogenated products were calculated and compared with their respective experimental data. It is obvious that the new absorption maxima of NTPAN compounds irradiated with 365 nm UV light (at around 520 nm) are much shorter than those after 254 nm UV irradiation (at around 625 nm) from the experimental data. Table S9† displays the comparison between experimental and calculated data for S0 → S1 absorption wavelengths of NTPAN compounds, their cyclized products and their dehydrogenated products. The small differences in the first absorption peaks between the observed and calculated data for NTPAN compounds in DCM solution validate the TDDFT calculations. Although a direct comparison between experimental and calculated data for the cyclized products and dehydrogenated products shows apparent variations varying in tens of nm in wavelength, it is absolutely evident that the calculated S0 → S1 absorption wavelengths of the cyclized products of NTPAN are much longer than those of their respective dehydrogenated products, which is consistent with the experimental observations that the absorption of NTPAN compounds irradiated with 254 nm UV light is remarkably red-shifted relative to those irradiated with 365 nm UV light. This consistence in absorption peaks observed in the experiment and predicted by theory further validates our prediction that UV-dependent photochromic behaviours of NTPAN compounds originate from two distinct reactions, i.e. the cyclization reaction and the cyclization–dehydrogenation reaction.
The 254 nm UV light was first selected to perform photochromic experiments for all NTPAN compounds. As shown in Fig. 6a–e, a deep green color can be observed for all NTPAN compounds after 110 s of UV irradiation, and new absorption peaks at around 625 nm can be detected in their UV-vis spectra. Interestingly, the colored solutions of NTPAN compounds after UV irradiation with 254 nm hardly fade out, keeping the green color even after a long time of standing. Only a very slight decrease at 625 nm can be detected in the UV-vis spectra during the process, providing an implication that the products during such a cyclization reaction are quite stable. It is interesting to note that these green solutions exhibit bright blue fluorescence under UV excitation, as shown in Fig. 6h–l. The generation of bright blue fluorescence of irradiated NTPAN solution can be attributed to the cyclization-induced rigidification of large conjugates containing two phenyl rings and a central ethane. The conjugation of most parts of these molecules enables the transformation of their photophysical behaviours from AIE to ACQ, which indirectly proves that the regulation of intramolecular motions determines the photophysical properties of the luminogens and the RIM mechanism of AIEgens.10–12 The green color of NTPAN and NTPAN-I is much deeper than the others, indicating that the transformation rates for NTPAN and NTPAN-I are greater; however, the fluorescence of irradiated NTPAN solution is much brighter than that of the irradiated NTPAN-I solution. This is due to the fluorescence quenching induced by the heavy atom effect of iodine in the cyclized product of NTPAN-I compound. The photochromic behaviours of NTPAN compounds irradiated with 365 nm light were also examined to demonstrate UV-triggered irreversible coloration and the accompanying fluorescence turn-on phenomenon. Fig. S15† shows photochromic changes of NTPAN compounds under the continuous irradiation of 365 nm light. It is observed that new absorption bands emerge at around 520 nm after 120 s of UV irradiation on the solutions of NTPAN compounds. Although the color of the solution only slightly changed from yellow to orange, such a change can also be identified by the naked eye for most solutions. Among these compounds, NTPAN has the sharpest color change from its large absorption enhancement at the new absorption band, suggesting that NTPAN is favorable to undergo the successive cyclization–dehydrogenation reaction. It is also easily concluded that the introduction of heavy halogens counteracts such chemical changes and progressively decreasing absorbance at the new absorption bands from NTPAN-F to NTPAN-I. Similar to the cyclized products, these dehydrogenated products also exhibit bright blue fluorescence, and as the irradiation increases the fluorescence intensity is gradually enhanced. By comparison, the enhanced fluorescence of the dehydrogenated product progressively declined from NTPAN to NTPAN-I, and this fact resembles the observation of the cyclized products, both of them highlighting the fluorescence quenching effect on planar conjugated luminogens due to the heavy atom effect. After a long time of monitoring of fluorescence brightness, it is found that the fluorescence can always be kept, and no obviously detectable fluorescence decrease can be recorded, indicating that these dehydrogenated products are extremely stable. In comparison with the fast recovery of colored solutions of TPAN compounds after irradiation with UV light, the colored solutions of NTPAN compounds almost barely fade out under various conditions, suggesting that their photochromic behaviours are irreversible relative to those of TPAN compounds. These observations demonstrate that the introduction of dimethylamino groups into the TPAN skeleton not only constructs a unique D–π–A pattern and red-shifted emissions but also greatly turns their photochemical behaviours from unstable and reversible cyclization to UV-dependent stable and irreversible cyclization and dehydrogenation.57,58
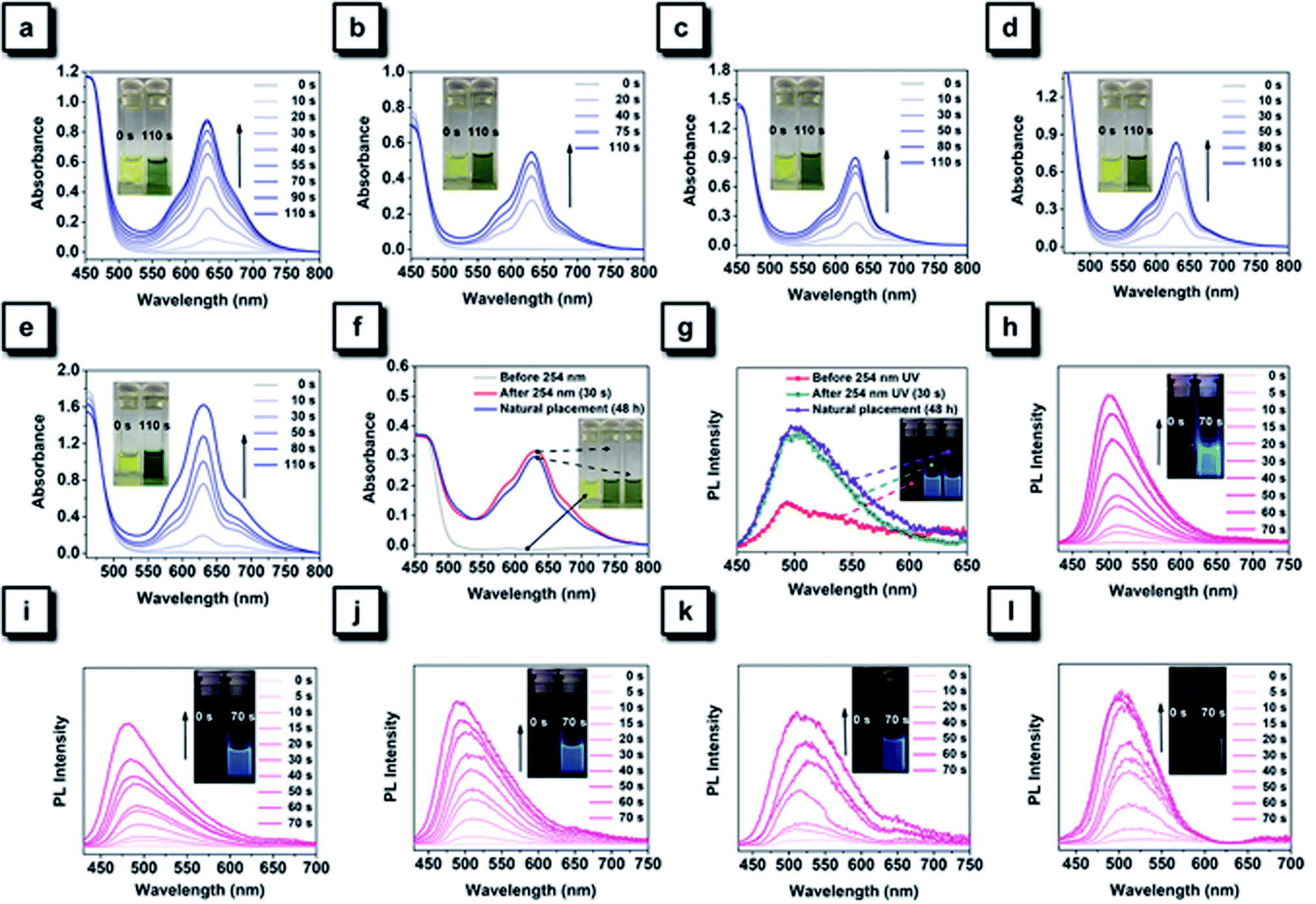 | ||
| Fig. 6 (a-e) Time-dependent UV-vis absorption spectra of NTPAN (a), NTPAN-F (b), NTPAN-Cl (c), NTPAN-Br (d), NTPAN-I (e) in DCM (2.0 mM) under continuous UV irradiation at 254 nm. (f) Change in the UV-vis absorption spectra of NTPAN-F in DCM (2.0 mM) under various conditions: the original solution (before 254 nm UV irradiation), the photochromic solution (after 254 nm UV irradiation) and the photochromic solution (left standing under natural light for 10 min), respectively. (g) Change in the PL spectra and images of NTPAN-F in DCM (2.0 mM) under various conditions. (h–l) Time-dependent PL spectra of NTPAN (h), NTPAN-F (i), NTPAN-Cl (j), NTPAN-Br (k), and NTPAN-I (l) in DCM (2.0 mM) under continuous UV irradiation at 254 nm. | ||
Photoswitchable reversible and irreversible patterning on various surfaces and multiple information encryption
Most diarylethene-based photochromic molecules with fluorescence turn-on behaviors were examined in their solution states, and a few of them exhibited photoswitchable patterning on the solid surface upon irradiation.26–29 Herein, we demonstrate that TPAN compounds with reversible photochromism possess the ability to undergo photo-triggered reactions and the resulting sharp color changes in the diverse matrix and on various surfaces. TPAN was used as an example to show this unique reversible photochromism in various solid environments including in porous cotton, in a cellulose blend and on a polymer surface (Fig. 7a). In such distinct solid environments, the TPAN molecule can also respond to two different irradiation conditions showing two sharply different colors, indicating that irradiation-triggered cyclization and decyclization reactions of these TPAN molecules can efficiently and easily take place in different solid matrixes and surfaces, which will greatly expand their practical applications in various photo-controlled processes. The fast response to external stimuli is also illustrated in Fig. 7b, where TPAN molecules were homogeneously distributed on a paper-based bird model. Upon only 5 s of UV irradiation, the white bird quickly changes to an orange bird; and after 6 min of standing under an ambient environment, the orange bird gradually turns back to its original white. This color shifting can be run many times in a reversible way, which grants these TPAN molecules great potential in photoswitchable patterning and information storage.59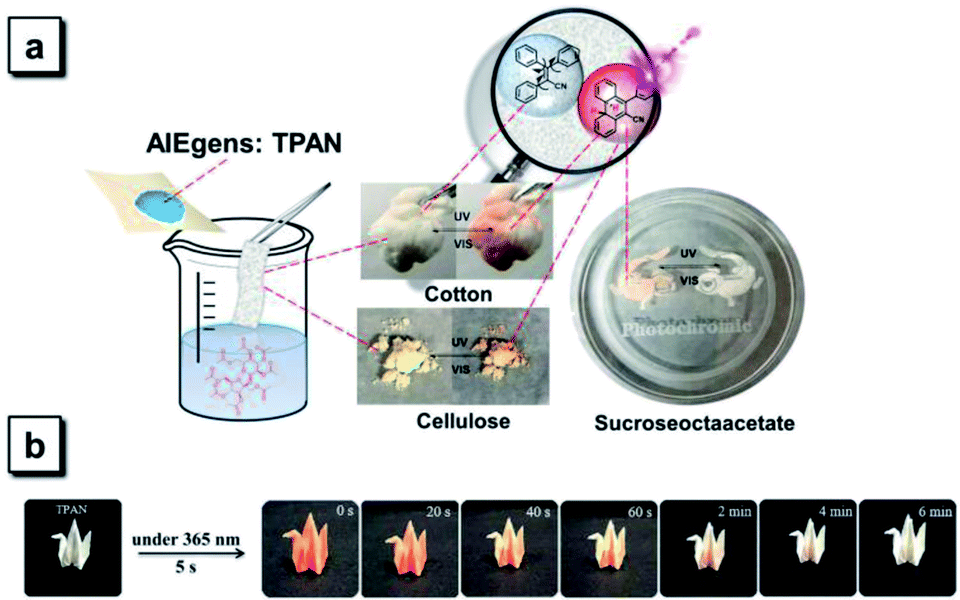 | ||
| Fig. 7 (a) Demonstration of reversible photochromism of TPAN compounds in various environments including in porous cotton, in a solid blend and on a polymer surface using TPAN as an example. (b) Demonstration of the fast response of reversible photochromism of TPAN compounds on a paper-based bird model. | ||
The experimental and theoretical results indicate that the TPAN groups are able to toggle conditionally between two discrete open- and closed-ring-form states upon alternate irradiation with UV and visible light; due to stable cyclized products, the NTPAN compounds can undergo irreversible photochromic transformation under UV light. Thus, these two classes of similar compounds can be used to combine reversible and irreversible photochromic behaviours as key elements of various light-driven information encryption and molecular switches. A combination of TPAN and NTPAN was first used as an example to demonstrate the photo-controlled patterning on a solid matrix. Fig. 8a shows a successive change in a phoenix pattern upon irradiation of different lights from an unobservable state to full emergence and finally to a half pattern. Half of the paper was treated with TPAN and the other was distributed with NTPAN; when a phoenix mask was subject to 30 s of irradiation of 365 nm UV light, a full orange phoenix pattern emerged on the paper. After only 5 s of irradiation of white light, one half of the pattern disappeared and the other half remained orange. Such different responses to distinct irradiation lights using the combined reversible and irreversible photochromism are valuable for multiple information encryption. A demonstration of information encryption on the geographic coordinates of our university is depicted in Fig. 8b, where two papers treated with reversibly photochromic TPAN and irreversibly photochromic NTPAN were used to decode the true geographic coordinates. The misleading geographic coordinates (23.1° N, 13.5° E) on a yellow paper emerges first after 365 nm UV irradiation, the false coordinates directing to some place in Africa. The key information to decode the true coordinates (29.1° N, 119.6° E) can be exhibited upon 365 nm UV irradiation on a white paper. When the two papers are combined, the true geographic coordinates (29.1° N, 119.6° E) for ZJNU can be read. Such a true coordinate pattern can be easily encrypted using white light irradiation and also decoded by UV light, illustrating the multiple information encryption ability by the combination of reversible and irreversible photochromism behaviours of the two groups of molecules. In comparison with the photo-controlled patterning in color, NTPAN compounds have special photo-triggered fluorescence turn-on behaviours and valuable use in photoactivatable fluorescent patterning applications. Fig. 8c shows a demonstration of photoactivatable fluorescent patterning using NTPAN as an example, where a viscous solution of NTPAN containing a great amount of sucrose octaacetate was used to retain the designed patterns. When the masks of designed letters such as ZJNU were used to block part of a beam of UV light, the other parts of the solutions fluoresce in green with gradually increasing intensity. Then, the designed letters ZJNU clearly appear on the bright green fluorescent background under the UV light and becomes clearer as the irradiation time increases. Such a dual-mode signaling of these compounds gives them great advantage to be used in applications of advanced encryption and storage of information due to their specific reversible and irreversible photochromism and photoactivation of fluorescence. These demonstrations illustrate the remarkable potential of these TPAN-based/NTPAN-based AIEgens with photocontrolled photochromism and photoactivatable fluorescence in optical memory media and information encryption.60
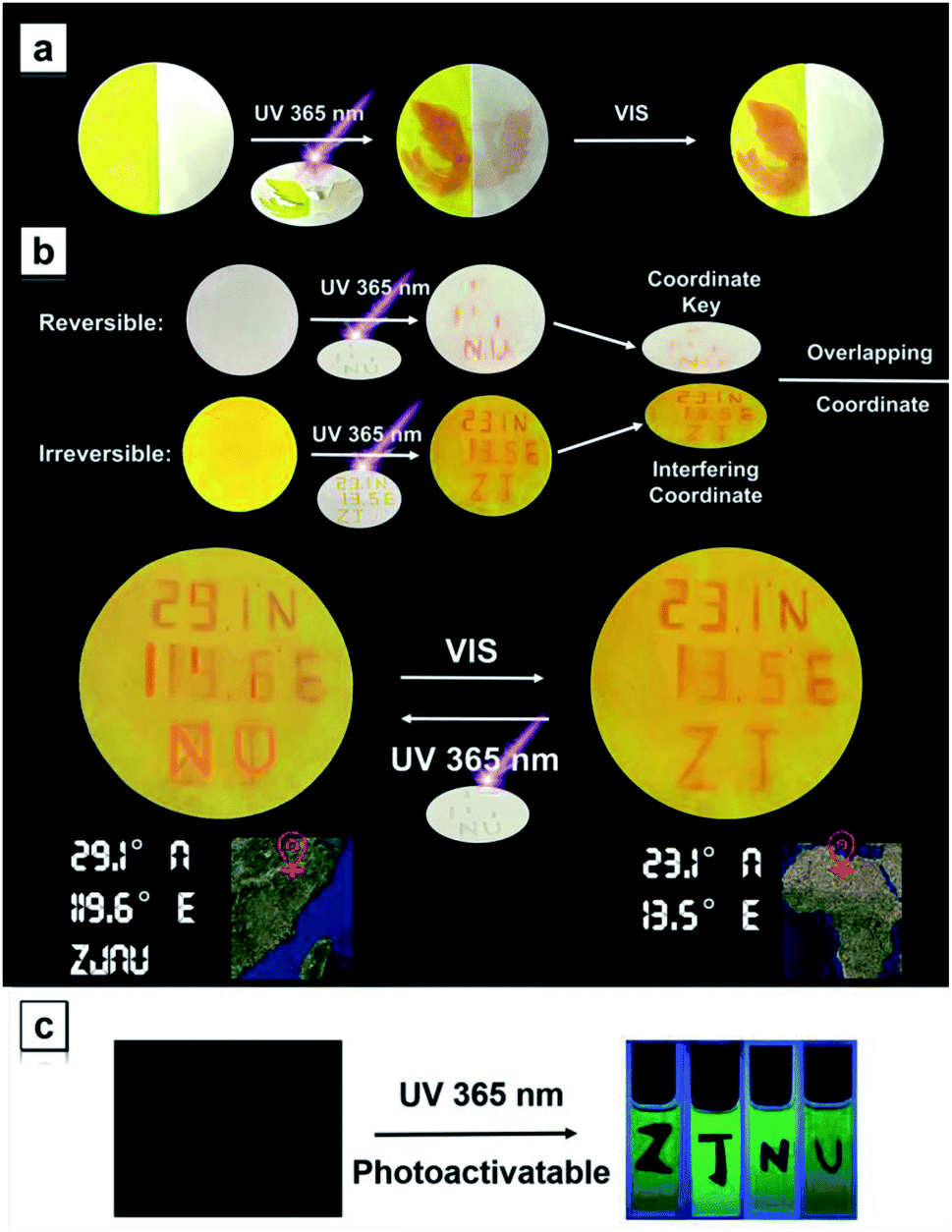 | ||
Fig. 8 (a) A demonstration of photo-controlled patterning application on a filter paper by combining reversible and irreversible photochromism using the blend of TPAN and NTPAN as an example. The right half was treated with TPAN, while the left half was distributed with NTPAN; a phoenix pattern was used to generate controlled patterning, and 365 nm UV light and white light were employed to trigger the color change or erase the generated color. (b) Illustration of the geographic coordinate encryption as a prototype combining reversible (TPAN)/irreversible (NTPAN) photochromism based on a code encryption design. (c) Demonstration of photoactivatable fluorescent patterning using NTPAN as an example. A viscous solution containing NTPAN and sucrose octaacetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) in DCM was used as the background and then the background was irradiated with a beam of UV light at 365 nm through different letter masks (ZJNU). :100) in DCM was used as the background and then the background was irradiated with a beam of UV light at 365 nm through different letter masks (ZJNU). | ||
Conclusions
In summary, two groups of triphenylacrylonitrile derivatives with different functionalities were rationally designed based on an AIE-active skeleton with through-space conjugation property, and controllable regulation of photophysical and photochemical processes in distinct states were achieved by altering the functionals and molecular polarities of the skeleton molecule. Since the discovery of the first AIEgen, AIEgens have been facing a great challenge of much lower emission efficiency than their counterparts through-bond conjugated dyes, but few strategies were proposed to resolve this problem in a general and effective way. In this work, we propose a new strategy of introducing heavy halogens into suitable positions to dramatically improve the emission efficiency in a widely applicable manner, which is expected to be expanded to other AIE-active systems. This anomalous emission enhancement induced by heavy halogens against the traditional heavy-atom effect on through-bond conjugated dyes is attributed to the specific through-space conjugation nature of the AIE-active skeleton, effective intermolecular halogen-bond-induced restriction of intramolecular motions, and heavy atom-induced vibration reduction by evident data and rational analyses. The construction of a donor–acceptor pattern in the AIE skeleton not only leads to photophysical emission alteration to a red-shifted direction but also tremendously changes the dominant photochemical reactions and the accompanying photochromism and photoactivatable fluorescence behaviours. It has been proved that the reversible photochromism of the TPAN family originated from photo-triggered cyclization and decyclization reactions, but the irreversible photochromism and photoactivatable fluorescence of the NTPAN family with a D–A pattern stemmed from the photo-induced cyclization–dehydrogenation reactions upon UV-dependent irradiation in solution. Compared to the previous fused molecules of AIE and photochromism, these molecules show distinct dominant photophysical emission enhancement in the solid state and photochemistry-dominating color and emission changes in the dispersed state to accomplish controllable regulation of photophysics and photochemistry of luminogens. The demonstration of controlled regulation of photophysics and photochemistry of fused chromic and AIE-active luminogens in distinct states provide strong evidence that they have great potential for various applications involved in multiple-mode signaling and diverse surfaces such as photoswitchable patterning and information encryption.Data availability
The datasets supporting this article have been uploaded as part of the ESI.Author contributions
Z. Xiong conceived of the study, collected the experimental data and wrote the initial draft of this article; Z. Qian contributed to the design of central ideas, carried out additional analyses and finalized this article; and H. Feng contributed to the revisions. The remaining authors contributed to the discussion of the results and refining of the ideas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21775139 and 21675143) and Natural Science Foundation of Zhejiang Province (Grant No. LR18B050001).Notes and references
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- F. Moliner, N. Kielland, R. Lavilla and M. Vendrell, Angew. Chem., Int. Ed., 2017, 56, 3758–3769 CrossRef PubMed.
- O. Kocaoglu and E. E. Carlson, Nat. Chem. Biol., 2016, 12, 472–478 CrossRef CAS PubMed.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lan and B. Z. Tang, Chem.–Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- J. Guan, R. Wei, A. Prlj, J. Peng, K. H. Lin, J. Liu, H. Han, C. Corminboeuf, D. Zhao, Z. Yu and J. Zheng, Angew. Chem., Int. Ed., 2020, 59, 14903–14909 CrossRef CAS PubMed.
- Y. Tu, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Natl. Sci. Rev., 2021 DOI:10.1093/nsr/nwaa260.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, CCS Chem., 2019, 1, 181–196 CAS.
- H. Zhang, X. Zheng, N. Xie, Z. He, J. Liu, N. L. C. Leung, Y. Niu, X. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. Qin, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- F. Saal, F. Zhang, M. Holzapfel, M. Stolte, E. Michail, M. Moos, A. Schmiedel, A. Krause, C. Lamber, F. Wurthner and P. Ravat, J. Am. Chem. Soc., 2020, 142, 21298–21303 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 11, 2441–2453 CrossRef PubMed.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, ACS Appl. Mater. Interfaces, 2018, 10, 12189–12216 CrossRef CAS PubMed.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS PubMed.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- B. Z. T. Kenry and B. Liu, Chem, 2020, 6, 1195–1198 Search PubMed.
- S. Suzuki, S. Sasaki, A. S. Sairi, R. Iwai, B. Z. Tang and G. Konishi, Angew. Chem., Int. Ed., 2020, 59, 9856–9867 CrossRef CAS PubMed.
- D. Dang, Z. Qiu, T. Han, Y. Liu, M. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1707210 CrossRef.
- P. Xu, Q. Qiu, X. Ye, M. Wei, W. Xi, H. Feng and Z. Qian, Chem. Commun., 2019, 55, 14938–14941 RSC.
- H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- J. Li, H. K. Bisoyi, S. Lin, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2009, 58, 16052–16056 CrossRef PubMed.
- K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS PubMed.
- Q. Yan and S. Wang, Mater. Chem. Front., 2020, 4, 3153–3175 RSC.
- S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed.
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- Q. Luo, F. Cao, C. Xiong, Q. Dou and D. H. Qu, J. Org. Chem., 2017, 82, 10960–10967 CrossRef CAS PubMed.
- P. Wei, J. X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 1966–1975 CrossRef CAS PubMed.
- Z. He, L. Shan, J. Mei, H. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, X. Gu, Q. Miao and B. Z. Tang, Chem. Sci., 2015, 6, 3538–3543 RSC.
- D. Qu, T. Yu, Z. Yang, T. Luan, Z. Mao, Y. Zhang, S. Liu, J. Xu, Z. Chi and M. R. Bryce, Chem. Sci., 2016, 7, 5302–5306 RSC.
- T. Yu, D. Ou, L. Wang, S. Zheng, Z. Yang, Y. Zhang, Z. Chi, S. Liu, J. Xu and M. P. Aldred, Mater. Chem. Front., 2017, 1, 1900–1904 RSC.
- L. Wang, T. Yu, Z. Xie, X. Chen, Z. Yang, Y. Zhang, M. P. Aldred and Z. G. Chi, J. Mater. Chem. C, 2018, 6, 8832–8838 RSC.
- G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS PubMed.
- S. Zhou, S. Guo, W. Liu, Q. Yang, H. Sun, R. Ding, Z. Qian and H. Feng, J. Mater. Chem. C, 2020, 8, 13197–13204 RSC.
- S. Guo, S. Zhou, J. Chen, P. Guo, R. Ding, H. Sun, H. Feng and Z. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 42410–42419 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 27, 9952–9967 CrossRef PubMed.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. LI, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS.
- Z. Cheng, H. Shi, H. Ma, L. Bian, Q. Wu, L. Gu, S. Cai, X. Wang, W. Xiong, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 678–682 CrossRef CAS PubMed.
- Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- T. C. Owyong, P. Subedi, J. Deng, E. Hinde, J. J. Paxman, J. M. White, W. Chen, B. Heras, W. W. H. Wong and Y. Hong, Angew. Chem., Int. Ed., 2020, 59, 10129–10135 CrossRef CAS PubMed.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- D. S. McClure, J. Chem. Phys., 1952, 20, 682–686 CrossRef CAS.
- M. A. Ei-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef.
- W. Zhao, Z. He, Q. Peng, J. W. Y. Lam, H. Ma, Z. Qiu, Y. Chen, Z. Zhao, Z. Shuai, Y. Dong and B. Z. Tang, Nat. Commun., 2018, 9, 3044 CrossRef PubMed.
- K. H. Park, J. Jeon, Y. Park, S. Lee, H. Kwon, C. Joo, S. Park, H. Han and M. Cho, J. Phys. Chem. Lett., 2013, 4, 2105–2110 CrossRef CAS.
- J. M. Rodgers, W. Zhang, C. G. Bazewicz, J. Chen, S. H. Brewer and F. Gai, J. Phys. Chem. Lett., 2016, 7, 1281–1287 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- M. I. Khazi, W. Jeong and J. M. Kim, Adv. Mater., 2018, 30, 1705310 CrossRef PubMed.
- M. N. Tran and D. M. Chenoweth, Angew. Chem., Int. Ed., 2015, 54, 6442–6446 CrossRef CAS PubMed.
- J. V. Jun, C. M. Haney, R. J. Karpowicz, S. Giannakoulias, V. M. Lee, E. J. Petersson and D. M. Chenoweth, J. Am. Chem. Soc., 2019, 141, 1893–1897 CrossRef CAS PubMed.
- H. Nie, J. L. Self, A. S. Kuenstler, R. C. Hayward and J. R. Alaniz, Adv. Opt. Mater., 2019, 7, 1900224 CrossRef.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, UV-visible and PL spectra, NBOs, crystallographic and photophysical data, calculated data, 1H NMR and 13C NMR spectra and mass spectra. CCDC 2054237, 2054250, 2054251, 2054252, 2054253, 2054256, 2054258, 2054257, 2054254, 2054255 and 2054898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02168k |
| This journal is © The Royal Society of Chemistry 2021 |