
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinol-containing ligands enable high superoxide dismutase activity by modulating coordination number, charge, oxidation states and stability of manganese complexes throughout redox cycling†
Laura
Senft
 a,
Jamonica L.
Moore
c,
Alicja
Franke
a,
Katherine R.
Fisher
a,
Andreas
Scheitler
b,
Achim
Zahl
b,
Ralph
Puchta
b,
Dominik
Fehn
b,
Sidney
Ison
c,
Safaa
Sader
c,
Ivana
Ivanović-Burmazović
*a and
Christian R.
Goldsmith
*c
a,
Jamonica L.
Moore
c,
Alicja
Franke
a,
Katherine R.
Fisher
a,
Andreas
Scheitler
b,
Achim
Zahl
b,
Ralph
Puchta
b,
Dominik
Fehn
b,
Sidney
Ison
c,
Safaa
Sader
c,
Ivana
Ivanović-Burmazović
*a and
Christian R.
Goldsmith
*c
aDepartment of Chemistry, Ludwig-Maximilian-University, Butenandtstr. 5-13, D 81377 Munich, Germany. E-mail: ivana.ivanovic-burmazovic@cup.uni-muenchen.de
bDepartment of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen-Nuremberg, 91058 Erlangen, Germany
cDepartment of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA. E-mail: crgoldsmith@auburn.edu
First published on 13th July 2021
Abstract
Reactivity assays previously suggested that two quinol-containing MRI contrast agent sensors for H2O2, [Mn(H2qp1)(MeCN)]2+ and [Mn(H4qp2)Br2], could also catalytically degrade superoxide. Subsequently, [Zn(H2qp1)(OTf)]+ was found to use the redox activity of the H2qp1 ligand to catalyze the conversion of O2˙− to O2 and H2O2, raising the possibility that the organic ligand, rather than the metal, could serve as the redox partner for O2˙− in the manganese chemistry. Here, we use stopped-flow kinetics and cryospray-ionization mass spectrometry (CSI-MS) analysis of the direct reactions between the manganese-containing contrast agents and O2˙− to confirm the activity and elucidate the catalytic mechanism. The obtained data are consistent with the operation of multiple parallel catalytic cycles, with both the quinol groups and manganese cycling through different oxidation states during the reactions with superoxide. The choice of ligand impacts the overall charges of the intermediates and allows us to visualize complementary sets of intermediates within the catalytic cycles using CSI-MS. With the diquinolic H4qp2, we detect Mn(III)-superoxo intermediates with both reduced and oxidized forms of the ligand, a Mn(III)-hydroperoxo compound, and what is formally a Mn(IV)-oxo species with the monoquinolate/mono-para-quinone form of H4qp2. With the monoquinolic H2qp1, we observe a Mn(II)-superoxo ↔ Mn(III)-peroxo intermediate with the oxidized para-quinone form of the ligand. The observation of these species suggests inner-sphere mechanisms for O2˙− oxidation and reduction that include both the ligand and manganese as redox partners. The higher positive charges of the complexes with the reduced and oxidized forms of H2qp1 compared to those with related forms of H4qp2 result in higher catalytic activity (kcat ∼ 108 M−1 s−1 at pH 7.4) that rivals those of the most active superoxide dismutase (SOD) mimics. The manganese complex with H2qp1 is markedly more stable in water than other highly active non-porphyrin-based and even some Mn(II) porphyrin-based SOD mimics.
Introduction
High concentrations of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and superoxide (O2˙−), are known to oxidatively degrade biomolecules, and their over-accumulation is believed to be involved in the pathology of several severe health conditions, including COVID-19.1–9 The potential negative impacts of ROS overproduction on human health have motivated extensive efforts to develop methods to detect and detoxify ROS in biological environments.10–13 One attractive antioxidant design strategy is to synthesize small molecules that resemble the enzymes that the body itself uses to regulate ROS concentrations. A small dose of such an antioxidant would alleviate oxidative stress by catalytically degrading ROS. Investigated antioxidants include functional mimics of superoxide dismutases (SODs), which are manganese-, iron-, nickel-, or copper-zinc-containing enzymes that catalyze the degradation of O2˙− to O2 and H2O2 (eqn (1)).14–21 The enzymatic catalysis relies upon the metal center cycling between two oxidation states; there are no known SODs that instead use an organic redox partner for the disproportionation of O2˙−.| 2O2˙− + 2H+ → O2 + H2O2 | (1) |
Previously, our labs developed and characterized three water- and air-stable Mn(II) complexes with redox-active organic ligands (Scheme 1) that respond to H2O2 with changes in their T1-weighted relaxivity (r1).22–24 Consequently, the Mn(II) complexes can serve as redox-responsive contrast agents for magnetic resonance imaging (MRI). Notably, the MRI responses result from the oxidation of the organic component, rather than the metal, distinguishing these from redox-responsive probes developed by the Caravan group.25–29 The first sensor, [Mn(Hptp1)(MeCN)]2+ (1), reacts with H2O2 to form a binuclear Mn(II) species; the reaction with H2O2 oxidatively couples the para-methyl phenol groups from two equiv. of the pre-activated contrast agent.22 The other two compounds, [Mn(H2qp1)(MeCN)]2+ (2, MeCN = acetonitrile) and [Mn(H4qp2)Br2] (3), feature quinol subunits which are oxidized to para-quinone groups upon their reactions with H2O2 (Scheme 2).23,24 In the reduced form of the ligands, the quinols can deprotonate and bind tightly to Mn(II) ions as quinolates; in the oxidized form, conversely, the neutral para-quinones have much weaker metal binding affinities and appear to be displaced by water molecules.24 The better aquation of the Mn(II) ion is proposed to increase r1 and thereby enhance MRI contrast.
 | ||
| Scheme 1 Redox-active ligands discussed in this study. | ||
 | ||
| Scheme 2 Redox reactions involving metal-bound quinols. | ||
Curiously, prolonged exposure of 2 and 3 to a large excess of H2O2 converts only 70% of the quinols to para-quinones.23,24 The existence of a possible equilibrium mixture of the reduced and oxidized forms of the ligands in the presence of excess ROS led us to speculate that the ligands were cycling between these different oxidation states; this could potentially allow 2 and 3 to catalyze ROS degradation with or without a classical metal-centered redox pathway. We initially screened the antioxidant activity of the three manganese complexes using the xanthine oxidase/hypoxanthine/lucigenin assay.30,31 Our results suggested that 1, 2, and 3 all behave as SOD mimics.23,24 The SOD activity of 1 was then confirmed using more quantitatively accurate stopped-flow kinetics measurements.32 Comprehensive study of 1 revealed that in contrast to the majority of non-porphyrin-based manganese-containing SOD mimics, its SOD activity mainly proceeds through an outer-sphere pathway, with additional superoxide and oxygen reductase activity operating through an inner-sphere mechanism involving a Mn(IV)-oxo intermediate and the generation of OH˙ radicals.32 As was observed with H2O2, these reactive species oxidatively couple the phenol groups of separate molecules to dimerize 1. The dimerization reaction has little impact on the SOD activity and essentially protects the catalyst from inadvertently generated ROS. Much like most other non-porphyrin-based Mn SOD mimics,14–16,33–371 is more active under more acidic and phosphate-free conditions.
More recently, we found that a Zn(II) complex with the H2qp1 ligand, [Zn(H2qp1)(OTf)](OTf) (4), efficiently catalyzes the degradation of O2˙−.38 These results demonstrate that the quinol in the H2qp1 ligand is, by itself, a viable redox partner for superoxide dismutation. The free H2qp1 ligand, however, cannot act as a SOD mimic, demonstrating that a metal ion is essential for the observed catalysis.
Complexes 2 and 3 contain manganese centers in addition to quinol moieties, and catalytic superoxide degradation could involve either one or both of these as redox partners. It is also possible that the two redox-active components may act synergistically to improve either the rate of the reactions with O2˙− or the stability of the catalyst; the latter effect was observed with 1, which channels other ROS towards the functionally innocuous dimerization reaction.32 In order to determine the roles that the metal and ligand have in catalysis, we have investigated the (de)protonation equilibria, water exchange processes, and solution stability of 2 and have used stopped-flow kinetics techniques to confirm and quantitatively assess the abilities of 2 and 3 to act as SOD mimics. We have also identified several catalytically relevant intermediates by cryo-ionization mass spectrometry (CSI-MS). We observe that the ligand influences the catalysis through (A) its ability to access multiple oxidation states, (B) its charge, and (C) its ability to control the accessibility and rate of ligand exchange at the metal center through steric and conformational effects. Despite the high structural similarity of 1 to 2, we find that 2 and 3 both react with O2˙− through more efficient inner-sphere mechanisms, rather than the predominantly outer-sphere pathway observed for 1.32 Our results have allowed us to identify some of the key factors that modulate SOD catalysis by manganese complexes with this class of redox-active ligand.
Experimental section
Materials
All chemicals and solvents were purchased from Sigma-Aldrich and used as received unless otherwise noted. All deuterated solvents were bought from Cambridge Isotopes. Diethyl ether (ether) and methanol (MeOH) were bought from Fisher. Methylene chloride (CH2Cl2) was purchased from Mallinckrodt Baker. N-(2,5-Dihydroxybenzyl)-N,N′,N′-tris(2-pyridinylmethyl)-1,2-ethanediamine (H2qp1), N,N′-bis(2,5-dihydroxybenzyl)-N,N′-bis(2-pyridinylmethyl)-1,2-ethanediamine (H4qp2), [Mn(H2qp1)(MeCN)](OTf)2 (2), and [Mn(H4qp2)Br2] (3) were prepared through previously described procedures.23,24Instrumentation
All 1H and 13C NMR spectra were recorded on a 400 MHz or 600 MHz AV Bruker NMR spectrometer at 293 K. In each spectrum, the reported NMR resonance peak frequencies were referenced to internal standards. 17O NMR data were collected on a Bruker AVANCE DRX 400WB spectrometer with a superconducting wide-bore magnet operating at a 54.24 MHz resonance frequency and a 9.4 T magnetic induction. A Varian Cary 50 spectrophotometer was used to collect optical data, which were then processed using software from the WinUV Analysis Suite.Potentiometric titrations
The speciation chemistry of 2 in water was assessed using a METROHM 765 Dosimat with a jacketed, airtight glass titration vessel. A Fisher Scientific Accumet Research AR15 pH meter was used to determine the pH of the sample solutions during the titrations. The electrode was calibrated before each titration using commercially available standard solutions buffered to pH 4.0, 7.0, and 10.0. All samples were purged with argon prior to analysis and subsequently analyzed under an argon atmosphere at 25 °C. All solution samples were prepared in solutions of 100 mM KCl in deionized Millipore water. The titrations investigating metal–ligand speciation were run with solutions that contained a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of the ligand and MnCl2·(4H2O). Carbonate-free solutions of 0.10 M KOH and 0.10 M HCl were prepared using argon-saturated deionized Millipore water. These data were analyzed and fit to speciation models using the Hyperquad2006 program.39
:1 molar mixture of the ligand and MnCl2·(4H2O). Carbonate-free solutions of 0.10 M KOH and 0.10 M HCl were prepared using argon-saturated deionized Millipore water. These data were analyzed and fit to speciation models using the Hyperquad2006 program.39
17O NMR measurements of aquation numbers (q) and water exchange processes
17O NMR spectra were recorded on a Bruker AVANCE DRX 400WB spectrometer equipped with a spectrospin superconducting widebore magnet operating at a resonance frequency of 54.24 MHz at a magnetic induction of 9.4 T. The measurements at atmospheric pressure were performed with a commercial 5 mm Bruker broad band probe thermostated with a Bruker B-VT 3000 variable temperature unit. Relaxation rates were measured for both the paramagnetic solutions and metal-free aqueous buffer solutions. The line widths at half the maximum height of the signal were determined by a deconvolution procedure on the real part of the Fourier transformed spectra with a Lorentzian shape function in the data analysis module of Bruker Topspin 1.3 software. Pressure-dependent measurements were done with a custom-made thermostated high-pressure probe. The sample was measured in a standard 5 mm NMR tube cut to a length of 50 mm. To enable pressure transmittance to the solution, the NMR tube was closed with a moveable Macor piston. The advantage of this method is that oxygen-sensitive samples can be easily placed in the NMR tube and sealed with the Macor piston under an argon atmosphere. The pressure was applied to the high-pressure probe via a perfluorinated hydrocarbon (hexafluoropropylene oxide, Hostinert 175, Hoechst) and measured by a VDO gauge with an accuracy of ±1%. The temperature was adjusted with circulating, thermostated water (Colora thermostat WK 16) to ±0.1 K of the desired value and monitored before each measurement with an internal Pt-resistance thermometer with an accuracy of ±0.2 K. Enriched (10%) 17O-labeled water (D-Chem Ltd Tel Aviv, Israel) was used for the 17O NMR water exchange experiments. Samples were prepared by dissolving solid Mn(II) complexes in MeCN and refilling the sample with degassed buffer solution (60 mM acetate buffered to pH 4.0 or 60 mM HEPES buffered to pH 7.4 or 7.0) to get a 4–5 mM solution of the complex containing 10% MeCN. 10%-enriched 17O-labeled water was added to the solution (1:10 v/v) which was then transferred to a NMR tube. The temperature-dependence of the 17O-line broadening was studied from 274.2 to either 343.2 or 348.2 K. A detailed description of the experiments and general data treatment can be found in the ESI.†
Aquation numbers (q) were calculated from the maximum 17O transverse relaxivity,  , and the equation:
, and the equation:  ; Gale, Zhu, and Caravan previously used this relationship to estimate the inner-sphere hydration state of Mn(II) in coordination complexes.40 Relaxation rates were measured both for aqueous solutions containing 2 and for metal-free solutions buffered to pH 4.0 and 7.4.
; Gale, Zhu, and Caravan previously used this relationship to estimate the inner-sphere hydration state of Mn(II) in coordination complexes.40 Relaxation rates were measured both for aqueous solutions containing 2 and for metal-free solutions buffered to pH 4.0 and 7.4.
Cryospray-ionization mass spectrometry
Cryospray-ionization mass spectrometry (CSI-MS) measurements were performed on a UHR-TOF Bruker Daltonik maXis plus, an ESI-quadrupole time-of-flight (qToF) mass spectrometer capable of a resolution of at least 60.000 (FWHM), which was coupled to a Bruker Daltonik Cryospray unit. Detection was in positive ion mode with a source voltage of 3.5 kV and a flow rate of 240 μL per hour. The temperatures of the spray gas (N2) and the dry gas used for solvent removal were maintained at −40 °C and −35 °C, respectively. The mass spectrometer was calibrated prior to every experiment via direct infusion of an Agilent ESI-TOF low concentration tuning mixture, which provided a m/z range of singly charged peaks up to 2700 Da in both ion modes. For the reactions with O2˙−, 1 mM solutions of either 2 or 3 in MeCN (1% DMF) were cooled to −40 °C and mixed with excess solid KO2. The mixture was diluted to roughly 1 × 10−5 M and quickly injected into the mass spectrometer. Applied solvents were not extra dry in order to provide a source of protons. The measured data were processed and analyzed with Bruker Data Analysis. The complete CSI-MS spectra recorded for the reactions of 2 and 3 with excess superoxide are shown in Fig. S1 and S2, respectively, in ESI.†Determination of the SOD activity
We previously assessed the abilities of 2 and 3 to catalytically degrade superoxide using the xanthine oxidase/hypoxanthine/lucigenin assay.23,24,31 Each assay was carried out in a total volume of 1 mL containing 50 mM Tris (pH 8.0), 50 μM hypoxanthine, 0.005 U mL−1 xanthine oxidase (Calbiochem), and 5 μM dark adapted lucigenin in the presence of either 0.1 nM or 10 μM of the Mn(II) complex or its vehicle.The ability of the Mn(II) complexes to catalytically degrade superoxide was tested by a direct method using stopped-flow techniques as described elsewhere.34 Experiments were carried out using syringes 1, 2, and 3 on a Biologic SFM-400 instrument that was equipped with an Energetiq LDLS ENQ EQ-99-FC laser driven light source and a J&M TIDAS diode array detector (integration time = 0.5 ms, λ = 180–724 nm). The source of superoxide was commercially available KO2 dissolved in dry and non-buffered DMSO ([O2˙−] ≈ 1–2 mM). Complexes 2 and 3 were each tested at four different concentrations between 0.9 and 9 μM in aqueous solutions buffered with HEPES or sodium phosphate to either pH 7.4 or pH 8.1. The ionic strength of all solutions was 111 mM. The aqueous solution containing the studied Mn(II) complex was mixed in a 9:1 ratio with the superoxide solution in DMSO using a high-density mixer. In each experiment, the concentration of superoxide exceeded that of the metal-containing catalyst by at least ten-fold to ensure catalytic conditions. Millipore water was used for the preparation of the buffer solutions. All of the prepared buffers were treated with Chelex 100 sodium exchange resin for at least 12 h before use in order to remove adventitious metal ions. The data analysis was performed using the BioKine V4.66 software. Each reported kobs value is the average of at least 10 measurements. The reported kcat values were determined from the slope of the kobsvs. [SODm] plot. The presence of H2O2 was qualitatively confirmed using Baker Testrips for peroxides.
Cryo UV/vis spectroscopy measurements
Cryo UV/vis experiments were performed with the use of low temperature stopped-flow techniques involving a biologic cryo SFM-4000 four-syringe stopped-flow combined with a 150 W xenon lamp and a J&M TIDAS diode array detector (200–724 nm) with an integration time of 0.5 ms. A Huber C-905 cryostat filled with silicon oil was used to cool the samples. Specfit, Global Analysis System software (version 3.0.38 for 32 bit Windows system) was used to analyze the spectral changes and perform a global analysis of kinetic traces at various wavelengths.EPR spectroscopy measurements
EPR spectra were recorded on a JEOL continuous wave spectrometer JES-FA200, equipped with an X-band Gunn diode oscillator bridge, a cylindric mode cavity, and a helium cryostat. The samples were measured in the solid state in quartz glass EPR tubes at 95 K and 7 K. The spectra shown were measured using the following parameters: microwave frequency = 8.959 GHz, modulation width = 1.0, and 0.5 mT, microwave power = 1.0 mW, modulation frequency = 100 kHz, time constant = 0.1 s.DFT calculations
All calculations were done by applying the B3LYP functional41–43 in combination with the def2svp basis set.44–46 The characterization as minima or transition states was done by computation of vibrational frequencies at the same level. All wavefunctions were tested successfully for stability.47–49 Relative energies were corrected for zero-point vibrational energies (ZPE). All calculations were performed using the Gaussian 16 program package.50Results and discussion
Before one can identify the factors that govern the ability of manganese complexes with polydentate ligands (Scheme 1) to catalytically decompose superoxide, one needs to understand how they behave in water. We have therefore characterized the pH-dependent speciation, stability, and water exchange kinetics of [Mn(H2qp1)(MeCN)](OTf)2 (2) in aqueous solutions using potentiometric and spectrophotometric pH titrations and variable temperature and pressure 17O NMR spectroscopy. We previously reported analogous studies for [Mn(Hptp1)(MeCN)]2+ (1) and [Mn(H4qp2)Br2] (3).24,32 The new data enable us to fully compare the aqueous chemistries of 1, 2, and 3. Further, we determined catalytic rate constants for superoxide dismutation by 2 and 3 using the same techniques that were used for 1, again enabling the three to be compared.32 Mechanistic insights were gained by characterizing reactive intermediates using low-temperature UV/vis and EPR spectroscopies and CSI-MS.Protonation equilibria and solution stability
Potentiometric and spectrophotometric pH titration data were collected to assess the stability and ligand protonation states of 2 in water. Previously, we found that the H2qp1 ligand itself undergoes two ionization events consistent with pKL values of 7.24 and 4.72 which were assigned to the (de)protonation of one of the tertiary amines (H3qp1+) and one of the pyridine rings (H4qp12+), respectively.38 We observed precipitation upon raising the pH above 10; consequently, we were unable to measure reliable pKa values for the quinolic O–H groups of H2qp1, which likely fully deprotonate at pH values higher than 10. Similar ligand instability above pH 9 was observed for H4qp2.24By comparison, complex 2 displays ionization events with pKa values of 6.3 and 3.1 (Fig. S3, S4,† and Table 1). The pKa value of 6.3 for the Mn(II) complex is consistent with the (de)protonation of a M(II)-bound phenolate.26,51 The assignment of the 6.3 pKa value to the (de)protonation of a metal-bound quinol is supported by spectrophotometric pH titration data. At pH 6.0 and above, there is a strong peak at 305 nm; the energy of this band is consistent with an intraligand electronic transition for a phenolate, rather than a phenol (Fig. S5†).24,26 As the pH drops below 6, this band disappears and is replaced by a prominent shoulder at 295 nm, an energy which is more consistent with the conjugate acid phenol. At physiologically relevant pH 7.4, 93% of the complex has a [Mn(Hqp1)]+ core, with the other 7% existing as the dicationic [Mn(H2qp1)]2+ (Fig. 1). Additional aquation number (q) measurements were performed to fully describe the water speciation (vide infra).
| a Ligand pKa values correspond to the following equilibrium constants: Hptp1 KL1 = [Hptp1][H+]/[H2ptp1+], from ref. 32, KL2 = [ptp1−][H+]/[Hptp1], from ref. 32, H2qp1 KL1 = [H3qp1+][H+]/[H4qp12+], from ref. 38, KL2 = [H2qp1][H+]/[H3qp1+], from ref. 38, H4qp2 KL1 = [(H5qp2)+][H+]/[(H6qp2)2+], from ref. 24, KL2 = [(H4qp2)][H+]/[(H5qp2)+], from ref. 24. b Metal complex pKa values correspond to the following equilibrium constants: 1Ka(Mn(H2ptp1)3+) = [Mn(Hptp1)2+][H+]/[Mn(H2ptp1)3+], from ref. 32, Ka(Mn(Hptp1)2+) = [Mn(ptp1)+][H+]/[Mn(Hptp1)2+], from ref. 32, 2Ka(Mn(H3qp1)3+) = [Mn(H2qp1)2+][H+]/[Mn(H3qp1)3+], Ka(Mn(H2qp1)2+) = [Mn(Hqp1)+][H+]/[Mn(H2qp1)2+], 3Ka(Mn(H5qp2)3+) = [Mn(H4qp2)2+][H+]/[Mn(H5qp2)3+], from ref. 24, Ka(Mn(H4qp2)2+) = [Mn(H3qp2)+][H+]/[Mn(H4qp2)2+], from ref. 24, Ka(Mn(H3qp2)+) = [Mn(H2qp2)][H+]/[Mn(H3qp2)+], from ref. 24. c pMn = −log[Mn(II)]free calculated for [Mn(II)] = 1.0 mM and [L] = 1.0 mM at 25 °C and pH 7.4. | |||
|---|---|---|---|
| Hptp1 | 1 | ||
| pKL1a | 4.11 | pKa(Mn(H2ptp1)3+)b | 3.93 |
| pKL2a | 10.01 | pKa(Mn(Hptp1)2+)b | 6.16 |
| pMn (pH 7.4)c | 5.40 | ||
|
|||
| H2qp1 | 2 | ||
| pKL1a | 4.72 (±0.08) | pKa(Mn(H3qp1)3+)b | 3.1 |
| pKL2a | 7.24 (±0.03) | pKa(Mn(H2qp1)2+)b | 6.3 |
| pMn (pH 7.4)c | 7.25 | ||
|
|||
| H4qp2 | 3 | ||
| pKL1a | 4.47 (±0.08) | pKa(Mn(H5qp2)3+)b | 5.53 |
| pKL2a | 7.18 (±0.03) | pKa(Mn(H4qp1)2+)b | 5.82 |
| pKa(Mn(H3qp1)+)b | 7.14 | ||
| pMn (pH 7.4)c | 5.36 | ||
 | ||
| Fig. 1 Species distribution for solutions of 1:1 H2qp1/MnCl2·(4H2O) in water containing 100 mM KCl as a function of pH at 25 °C and [H2qp1] and [MnCl2·(4H2O)] both equal to 1.0 mM. | ||
Analogous studies for 1, which features a para-methyl phenol group in place of the quinol, revealed that 1 is slightly more acidic, and the metal-bound phenol has a pKa value of 6.16.32 Consequently, the conjugate base phenolate should be a slightly weaker base and electron pair donor than the quinolate in 2. To our initial surprise, 2 is much more stable in water than 1 as evidenced by their respective pMn52 values of 7.25 and 5.40 (Table 1).32A priori, the hydroxyl group para to the coordinating O atom in H2qp1 would be anticipated to be a better electron donor than the methyl group in Hptp1,53 possibly explaining why H2qp1 coordinates more tightly to Mn(II) than Hptp1. We recently found that N,N′-bis(2,5-dihydroxybenzyl)ethanediamine-N,N′-diacetic acid formed a more stable Mn(II) complex than N,N′-bis(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid, providing another example of a quinol-for-phenol substitution improving complex stability.54 That quinols/quinolates are indeed better ligands than phenols/phenolates is supported by previously obtained crystallographic data. In the structures of 1 and 2 obtained from MeCN, Hptp1 and H2qp1 remain protonated and coordinate to the Mn(II) centers in a hexadentate fashion.22,23 The Mn–OH(quinol) bond (2.319 Å) in [Mn(H2qp1)(MeCN)]2+ is markedly shorter than the Mn–OH(phenol) bond (2.366 Å) in [Mn(Hptp1)(MeCN)]2+,22,23 demonstrating that quinol binds to Mn(II) more tightly than para-methyl phenol.
At pH 7.4, the H2qp1 ligand is also found to coordinate Mn(II) more tightly than H4qp2. The pMn value of 3 was determined to be 5.36.24 Although over 99% of both the H2qp1 and H4qp2 ligands are bound to the Mn(II) above pH 7.0 (Fig. 1),24H4qp2 protonates and dissociates from the metal ion to a greater extent than H2qp1 under acidic conditions. Less than 10% of the Mn(II) is ligand-free at pH 3 with 1.0 mM amounts of metal and H2qp1; conversely, over 80% of the Mn(II) dissociates from H4qp2 under the same conditions.24 The pKa values of the first and the second Mn(II)-quinol groups in 3 were found to be 5.82 and 7.14, respectively, resulting in 66% of species with a doubly deprotonated [Mn(H2qp2)] core and 33% of species with a singly deprotonated [Mn(H3qp2)]+ core at pH 7.4.24 The crystallographic data that we have obtained thus far with the H2qp1 and H4qp2 ligands suggest that protonated quinols bind less avidly to metal ions than pyridine rings.23,24,38 Whenever these ligands have not been fully coordinated to the metal centers in the solid-state structures, it is always the quinol groups that are not ligated. The stronger metal-binding affinity of H2qp1 relative to H4qp2 is therefore likely a consequence of replacing one of the H4qp2 quinols with a pyridine. The singly deprotonated H3qp2− thereby acts more like a pentadentate ligand at physiological pH; Hqp1−, conversely, behaves more fully like a hexadentate ligand.
Aquation and water exchange processes
The aquation number (q) and rate of water exchange at 298 K (k298ex) for 2 were determined from variable temperature 17O NMR data at pH 4.0 and 7.4 using a protocol developed by Gale et al. (Fig. 2).40 The aquation of 2 is not as extensive as it is for either 1 or 3 (Table 2). The simplest interpretation of the 0.7 value of q at pH 7.4 for 2 suggests that approximately 70% of the Mn(II) binds a single water molecule, with the other 30% without coordinated water molecules. The overall composition of the major component is therefore proposed to be [Mn(Hqp1)(OH2)]+. The pH 7.4 solutions structures of 1, 2, and 3 are depicted on Scheme 3. The metal center in this species could be heptacoordinate; this is plausible since the previously obtained crystal structure of [Mn(H2qp1)(MeCN)]2+ contains a seven-coordinate metal center.23 The minor component, [Mn(Hqp1)]+, most likely features a hexacoordinate metal center. Complexes 1 and 3, which respectively exist as [Mn(ptp1)]+ or a mixture of [Mn(H2qp2)] and [Mn(H3qp2)]+ at pH 7.0–7.4, have higher aquation numbers (Table 2).24,32 That the aquation number for 2 is lower than it is for 1 and 3 under similar conditions may suggest that the monoaqua species for 2 is thermodynamically favored to a lesser degree than the analogous species for the Hptp1 and H4qp2 ligands. Between pH 7.0 and 7.4, H2qp1 appears to be more strongly electron-donating than the other two ligands, as evident from its stronger chelating ability, vide supra. The more electron-rich metal center in 2 may thereby have a weaker affinity for the additional aqua ligand. | ||
Fig. 2 Plots of  as a function of temperature for 2 at pH = 4 (black squares) and pH = 7.4 (red squares). Experimental conditions: [2] = 4.0 mM in 60 mM acetate buffer (pH = 4.0) or in 60 mM HEPES buffer (pH = 7.4) with 10% (v/v) of 10% 17OH2, B = 9.4 T. as a function of temperature for 2 at pH = 4 (black squares) and pH = 7.4 (red squares). Experimental conditions: [2] = 4.0 mM in 60 mM acetate buffer (pH = 4.0) or in 60 mM HEPES buffer (pH = 7.4) with 10% (v/v) of 10% 17OH2, B = 9.4 T. | ||
| Parameter | 1 (pH 4.8)a | 1 (pH 7.4)a | 2 (pH 4.0) | 2 (pH 7.4) | 3 (pH 7.0)b |
|---|---|---|---|---|---|
| a From ref. 32. b From ref. 24. | |||||
| q | 1 | 1 | 1.3 (±0.3) | 0.7 (±0.2) | 0.9 (±0.2) |
| k 298ex (s−1) | 1.9 × 107 | 1.3 × 108 | 2.9 (±0.2) × 107 | 1.1 (±0.1) × 108 | 4.9 (±1.4) × 106 |
| ΔH‡ (kJ mol−1) | 26.4 (±0.2) | 26.6 (±0.5) | 13.6 (±0.9) | 28.7 (±0.7) | 22.0 (±1.7) |
| ΔS‡ (J K−1 mol−1) | −17.0 (±0.7) | −0.65 (±1.68) | −56.3 (±3.3) | +5.4 (±2.2) | −41.0 (±2.0) |
| ΔV‡ (cm3 mol−1) | −10.9 (±0.4) | +8.1 ± (0.4) | −15.2 (±0.9) | +10.9 (±0.6) | nd |
 | ||
| Scheme 3 Proposed solution state structures of 1, 2, and 3 at pH 7.4 based on titration and 17O NMR data. Note that 3 exists as a mixture of [Mn(H2qp2)(OH2)] and [Mn(H3qp2)(OH2)]+. | ||
That less of the Mn(II) in 2 is heptacoordinate suggests that water exchange proceeds through a dissociative mechanism (D), which is supported by the highly positive activation volume of +10.9 (±0.6) cm3 mol−1 as well as a positive activation entropy (Fig. S6, S7,† and Table 2). These temperature- and pressure-dependent 17O NMR data confirm that Hqp1− labilizes the coordinated water molecule. They also explain the co-existence of the seven-coordinate [Mn(Hqp1)(OH2)]+ and the six-coordinate [Mn(Hqp1)]+ species at pH 7.4, with the latter being the intermediate for the dissociative water exchange mechanism. DFT calculations (B3LYP/def2svp; Fig. 3) support the operation of a D mechanism and predict that the energy difference between [Mn(Hqp1)(OH2)]+ and [Mn(Hqp1)]+ is only 2.2 kJ mol−1, with the seven-coordinate structure being slightly more stable. These calculations also predict a low activation barrier for ligand exchange (8.6 kJ mol−1, Fig. 3), explaining the high experimentally observed lability of [Mn(Hqp1)(OH2)]+.
 | ||
| Fig. 3 Energy profile for water exchange on seven-coordinate [Mn(Hqp1)(OH2)]+ with six-coordinate [Mn(Hqp1)]+ as an intermediate species (relative energies: B3LYP/def2svp). | ||
At pH 4.0, the number of coordinated water molecules is higher (q = 1.3), suggesting an equilibrium between two Mn(II) species: [Mn(H3qp1)(H2O)2]3+ and [Mn(H2qp1)(H2O)]2+. The corresponding values of ΔS‡ and ΔV‡ become substantially more negative with a concomitant decrease in k298ex (Table 2, Fig. S8 and S9†). The same trends were seen for complex 1 at pH = 4.8.32 The substantially more negative values for the activation entropy and activation volume at pH 4 than at pH 7.4 indicate an associative mechanism of water exchange, consistent with both [Mn(H3qp1)(H2O)2]3+ and [Mn(H2qp1)(H2O)]2+ being hexacoordinate; the lower coordination numbers would be more amenable to associative pathways for water exchange. Water exchange for 2 is almost four-fold faster at pH 7.4 than at pH 4. The more positive charges of [Mn(H3qp1)(H2O)2]3+ and [Mn(H2qp1)(H2O)]2+ relative to [Mn(Hqp1)(H2O)]+ also contribute to the slower rate of water exchange at the lower pH. The faster rate and a dissociative character of the exchange mechanism under more basic conditions suggest that the coordination of quinolate groups increases the kinetic lability of the Mn(II) center. The higher ligand substitution reactivity of 2 at pH 7.4, may be beneficial for catalytic activity involving an inner-sphere mechanism.
SOD activity of complexes 2 and 3
The SOD activities of complexes 2 and 3 were initially assessed using an established procedure that uses xanthine oxidase and hypoxanthine to produce O2˙− and a subsequent reaction with lucigenin to detect it.23,24,30,31 The concentrations at which half of the sensing reaction was blocked (IC50) were found to be 11.3 nM (2) and 18.2 nM (3). Based on the IC50 values and the 7.7 × 105 M−1 s−1 rate constant reported for the reaction between O2˙− and lucigenin,55 one would estimate kcat values of 3.4 × 108 M−1 s−1 (2) and 2.1 × 108 M−1 s−1 (3) from these data.Side reactions between the various components of the assays have been hypothesized to influence the measured IC50, and these assays frequently provide inaccurate accounts of antioxidant activity.17,34,56–59 In order to obtain the best possible assessment of the SOD mimicry of 2 and 3, we performed an extensive kinetic analysis of the reactions between O2˙− and the complexes in aqueous solutions buffered to pH 7.4 and 8.1 (Table 3). We recently reported similar studies for complexes 1 and 4, the latter being the Zn(II) complex [Zn(H2qp1)(OTf)]+.32,38 The H2qp1 ligand by itself was previously tested and found to be inactive as a catalyst for the decomposition of O2˙− at either pH 7.4 or 8.1.38 The degradation of superoxide is second-order in O2˙− in the absence of added catalyst, confirming that treatment of the solutions through a Chelex exchange matrix successfully removed all potentially catalytic metal impurities. The decomposition of O2˙− in solutions containing either 2 or 3, conversely, were found to be first-order with respect to both O2˙− and the manganese catalyst. The kcat values for 2 and 3 are both within the ranges seen for manganese-containing SOD mimics, with the most efficient of the complexes having kcat values ranging from 107 to 108 M−1 s−1.15,18,19,30,32,33,35,60–70
Complex 2 is especially effective in HEPES buffer, with a catalytic activity (logkcat = 7.99) that rivals those of cationic porphyrin complexes.18,19 One particularly effective porphyrin-based SOD mimic is MnIIBr8TM-4-PyP4+, which has both a highly positive charge and a large catalytic rate constant (logkcat = 8.34, determined by the cytochrome c assay) that approaches the 8.84–9.30 values measured for SODs.71–73 However, MnIIBr8TM-4-PyP4+ is unstable at neutral pH and releases free manganese; approximately 30% of the Mn is estimated to be lost.74 The decomposition of the complex has a substantial impact on the reactivity; if the MnIIBr8TM-4-PyP4+ catalyst were fully intact, its estimated logkcat value would exceed 8.67.74 The release of neurotoxic Mn(II) ions would severely limit the ability of this and similarly unstable complexes to serve as pharmaceuticals.71 Complex 2 also compares well to manganese complexes with pentaazamacrocyclic ligands, which comprise the other major class of highly active SOD mimetics. Arguably the best of these mimics is M40401, which has a catalytic rate of approximately 1.5 × 109 M−1 s−1.68,69 As with MnIIBr8TM-4-PyP4+, M40401 is markedly less stable in water than 2, with a pMn = 4.7 versus the 7.25 value measured for 2.75 Although complex 2 is about an order of magnitude less active than M40401, its much greater aqueous stability may allow it to engage in longer-lived catalysis.
Much like most other manganese-containing SOD mimics, complex 2 displays lesser activity at pH 8.1 and in phosphate buffer.14–16,32–37 The inhibition of the SOD activity by phosphate through its competitive binding to the metal center34 is usually indicative of an inner-sphere pathway for superoxide decomposition.14 The activity of complex 3, conversely, is less sensitive to the composition of the solution; despite this, the data suggest that 3 likewise reacts with superoxide through inner-sphere pathways. The second quinol in 3 does not enhance the catalysis. To the contrary, 2 is a superior catalyst to 3 in two of the investigated aqueous solutions and is approximately equivalent to 3 in phosphate buffer. The second quinol in 3 partly deprotonates to a quinolate, which reduces the overall charge of the complex in water. Complex 2 largely exists as [Mn(Hqp1)(H2O)]+ with some [Mn(H2qp1)]2+ at pH 7.4, whereas complex 3 is a mixture of 32% [Mn(H3qp2)(H2O)]+ and 66% [Mn(H2qp2)(H2O)].24 The lesser average positive charge on the H4qp2 complexes is anticipated to weaken their abilities to attract and bind anions, both slowing the rate of superoxide complexation and decomposition and attenuating the inhibitory effect of phosphate. The lessened positive charge for 3 also accounts for its slower rate of water exchange which unlike 2, appears to proceed through an associative mechanism on the basis of its highly negative entropy of activation.24 These results and the intermediate species characterized within the catalytic cycles (vide infra) are consistent with both 2 and 3 degrading O2˙− through inner-sphere pathways.
Despite its strong structural similarity to 2, complex 1 exhibits much lower SOD activity than either 2 or 3 and catalytically degrades O2˙− through mostly outer-sphere pathways.32 These differences can be rationalized by considering the Mn(III/II) reduction potentials. Engagement of the coordinated water molecule in proton coupled electron transfer (PCET) lowers the Mn(III/II) reduction potential (466 mV vs. Ag/AgCl)32 and allows an outer-sphere reduction of O2˙−, yielding a Mn(III)–OH species that can then oxidize superoxide to O2 and regenerate the Mn(II)–OH2 complex. The calculated structure of the Mn(III) form of 1, [MnIII(ptp1)(OH)]+, features a strong bond between the OH− and the metal center. Although the Mn(III) center in [MnIII(ptp1)(OH)]+ is predicted to be pentacoordinate with three neutral N-donors and an anionic O-donor from the ptp1− ligand and a second anionic O-donor from the hydroxide, its distorted square pyramidal structure, the diamine backbone of the ligand, and the negatively charged phenolate and hydroxide groups block access to the only accessible coordination site for potential sixth ligands.32 Therefore, our calculations suggest that the Mn(III) form of 1 is inaccessible to O2˙− and cannot oxidize it through an inner-sphere mechanism. Conversely, the Mn(III/II) reduction potentials of 2 (645 mV vs. Ag/AgCl)23 and 3 (725 mV vs. Ag/AgCl)24 are both too high to support efficient reduction of O2˙− through an outer-sphere catalytic pathway; 2 and 3 are instead proposed to catalyze superoxide degradation through inner-sphere pathways that involve the oxidation and reduction of the quinol/para-quinone groups (vide infra). Complexes 2 and 3 differ from 1 in that the stabilizations of their Mn(III) forms do not require additional anionic ligands. The Mn(III) state of 3 is stabilized by the deprotonation of the second quinol group, which yields species b in Scheme 4. The Mn(III) form of 2 can be stabilized via inner-sphere electron transfer from the quinolate to the Mn(III) center, yielding the Mn(II)-quinoxyl radical valence tautomer which can then undergo deprotonation of the unbound OH group to produce species d in Scheme 4. The additional acid/base and redox transformations available to quinols are important factors that allow 2 and 3 to perform efficient catalytic inner-sphere decomposition of superoxide.
 | ||
Scheme 4 Proposed mechanism of catalytic superoxide removal by 2 and 3. Depicted are two quinol moieties of H4qp2 within the structure of 3 (the rest of the pyridylamine ligand is omitted for clarity). In the case of 2, the leftmost quinol moiety is replaced by a pyridine ring from the ligand. Within the cycle of 3, the intermediates marked with  could not be observed by CSI-MS due to their neutral charge. Note that species presented as Mn–O–O− refer to formally coordinated superoxide and those presented as Mn–O−–O− to formally coordinated peroxide. could not be observed by CSI-MS due to their neutral charge. Note that species presented as Mn–O–O− refer to formally coordinated superoxide and those presented as Mn–O−–O− to formally coordinated peroxide. | ||
Observation of reaction intermediates and mechanistic analysis of SOD mimicry based on the CSI-MS results
The reactions between KO2 and the manganese complexes were probed by cryo-MS, rapid-scan stopped-flow and cryo UV/vis techniques, and EPR spectroscopy. Based on our results, we propose a detailed common mechanism for complexes 2 and 3 (Scheme 4).We were able to identify most of the reactive and volatile intermediates participating in redox catalysis during the reactions between 2 and 3 with excess superoxide (species a–k in Scheme 4) by analyzing these reactions in MeCN at −40 °C with ultra-high resolution cryo mass spectrometry. The initial reaction between the starting Mn(II) species with the ligand in a fully reduced state (a) and O2˙− oxidizes the metal center to Mn(III) (b, Fig. 4), with concomitant reduction of O2˙− to H2O2. For compound 3, the presence of two negative charges from the two quinolates in the coordination sphere stabilizes the +3 oxidation state of the metal. Since an external anion, such as OH−, is not needed to stabilize the complex, the metal center remains accessible to additional equivalents of O2˙−, unlike the Mn(III) form of 1.32 Species b can therefore coordinate O2˙− (c, Fig. S10 in ESI†) and continue to participate in inner-sphere catalytic cycles (red cycle in Scheme 4). This type of reactivity is well-established for many Mn-containing SOD mimics with redox-innocent polydentate ligands.15–17,30,61 However, the introduction of redox-active, non-innocent ligands enables more versatile reactivity, introducing pathways involving ligand oxidation and reduction (d and e) in addition to traditional metal-based redox catalysis.
 | ||
| Fig. 4 CSI-MS spectrometry of 3 upon reaction with KO2. The graphics depict possible structures; we cannot preclude other modes of ligand coordination. The m/z = 538.1404 feature is assigned to the Mn(II)-quinol-quinone intermediate (e in Scheme 4). The other m/z peaks for e, overlap with those of the Mn(III)-diquinolate species b, such as the one at 539.1482. The m/z = 542.1584 feature is unique for species b. Experimental conditions: 1 mM solutions of 3 in MeCN (1% DMF) were cooled to −40 °C and then mixed with an excess of solid KO2. Subsequently, the mixture was diluted in a pre-cooled syringe with pre-cooled MeCN to approximately 1 × 10−5 M and quickly injected into the mass spectrometer. | ||
The inclusion of redox-active ligands also appears to allow the generation of a higher-valent Mn(IV)-oxo intermediate (i). The observation of this species would imply that the Mn(III)-superoxo species formed with 3 (c) can itself undergo heterolytic O–O bond cleavage, with a two-electron oxidation of the hydroquinone ligand to a para-quinone and loss of water. Similar reactivity was recently reported for an iron porphyrin complex with a pendant quinol group; in this work, an Fe(III)-superoxo reacts to form a ferryl species (Fe(IV)-oxo) with an oxidized para-quinone moiety.76 The reaction goes through an Fe(III)-semiquinone (quinoxyl radical) species as an intermediate.76 This path (upper orange arrow in Scheme 4) might contribute to our detection of a quinoxyl-radical by the UV/vis spectroscopy (vide infra) during the catalytic degradation of O2˙− by 3.
In the case of 2, the initial oxidation by O2˙− appears to generate a Mn(II)-quinoxyl radical species (d) through tautomerization of Mn(III)-quinolate since one quinolate anion is not sufficient to stabilize the formally +3 oxidation state of the metal center. The quinoxyl radical is a stronger acid than quinolate, which facilitates its deprotonation (d, Fig. 5). Thus, 2 probably does not enter the metal-only cycle observed for 3 (red cycle), and its SOD activity relies more on the ligand's ability to undergo redox transformation. Although intermediate d was not directly observed for 3, it too may be capable of forming a Mn(II) intermediate with a quinoxyl radical through either a valence tautomerism or direct oxidation of the quinols. We find this likely since Mn(II)-para-quinone species (e in Fig. 4 and S11†) are observed with both 2 and 3; the para-quinone results from further oxidation of the quinoxyl radical in species d.38
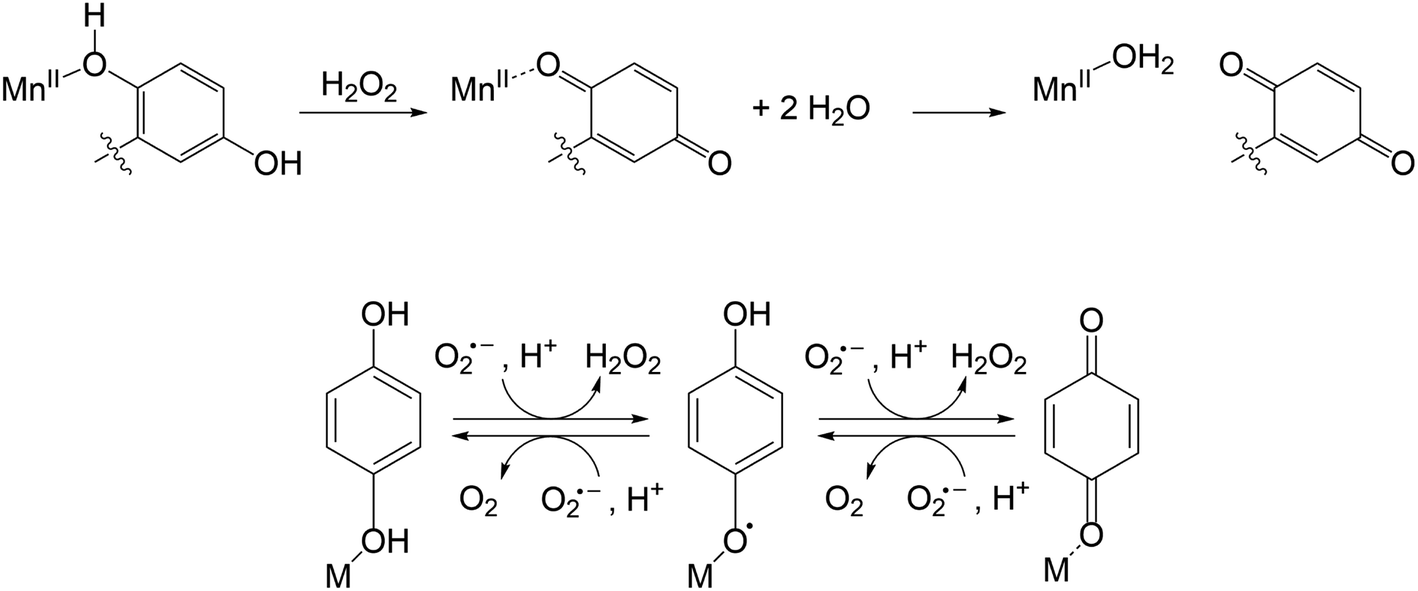
 | ||
| Fig. 5 CSI-MS of 2 upon reaction with KO2. The graphics depict possible structures; we cannot preclude other modes of ligand coordination. The m/z = 508.1535 peak is assigned to the Mn(II)-quinoxyl radical species (d). The other m/z peaks for d overlap with those of the initial Mn(II)-quinolate complex (a), such as that at 509.1620. The m/z = 512.1699 feature is unique to a. Experimental conditions: 1 mM solutions of 2 in MeCN (1% DMF) were cooled to −40 °C and then mixed with an excess of solid KO2. Subsequently, the mixture was diluted in a pre-cooled syringe with pre-cooled MeCN to approximately 1 × 10−5 M and quickly injected into the mass spectrometer. | ||
Cycling between a metal-coordinated quinolate, quinoxyl radical, and para-quinone (violet cycle) was the sole possible mechanism for the SOD activity of the Zn(II) complex with H2qp1 (4). With 2 and 3, however, the ligand-only redox cycle is not likely to be a major contributor to the overall superoxide degradation since the catalysis exhibited by 4 is much slower (kcat = 3.4 × 106 M−1 s−1 for 4 at pH 7.4 in HEPES)38 than what was detected for 2 and 3. The Mn(II)-quinone species of 2 and 3 (e) can enter alternative metal-based catalytic cycles for superoxide degradation (blue cycle; for CSI-MS spectra of f/g see Fig. S11†). We speculate that these pathways are more efficient than those with the reduced ligands for two reasons. First, the oxidized para-quinone forms of the ligands, qp1 and H2qp2, are effectively pentadentate ligands, with either N5 or N4O donor sets respectively. The removal of a chelating arm improves the accessibility of the metal center to O2˙− in both the Mn(II) and Mn(III) states. The N5 coordination provided by qp1 is stronger than that of the N4O coordination by H2qp2, maintaining the higher stability of 2 under catalytic conditions (vide infra) and bolstering its SOD activity. Second, the transformation of the quinolate to a neutral para-quinone increases the overall positive charge of the catalyst, which would more readily attract and bind O2˙−.
An intermediate that is noticeably unobservable during the catalysis is the Mn(III)-para-quinone species j, which is either tricationic (for 2) or dicationic (for 3). We believe that j rapidly reacts with O2˙− to yield species k (Fig. S12†), which then oxidizes O2˙− to O2 and regenerates the Mn(II) species e. The heightened positive charges of j species would attract O2˙− more efficiently and enable its stronger coordination to the metal center due to the stronger coulombic interactions. The inability to detect intermediates j with cryo-MS would be consistent with the heightened positive charge increasing the rate of reactivity to the extent that these species are too reactive to observe by CSI-MS. The ability of 2 to attain a tricationic form would partially explain its high activity, particularly relative to that of 3. Notably, the most active reported manganese-containing SOD mimic M40401 (based on the catalytic rate constants determined by direct stopped-flow measurements)68,69 features the neutral N5 pentaazamacrocyclic ligand. M40401 likewise has a +2 or +3 overall charge depending on the manganese redox state during the catalytic cycling and predominantly exists as a pentacoordinate species in solution, supporting our observed correlations between catalyst charge, the accessibility of the metal center for superoxide binding, and the rate of superoxide decomposition.
The hydroperoxo species h from the blue cycle in Scheme 4 can either get protonated to release H2O2 or undergo homolytic cleavage to generate a high-valent Mn(IV)-oxo species i (Fig. 6) and a hydroxyl radical. The ability of the H4qp2 to provide a dianionic coordination sphere in its reduced form (H2qp22−) and a monoanionic one in its two-electron oxidized form (Hqp2−) allows this ligand system to better stabilize the higher-valent intermediate than H2qp1. We can therefore observe i for 3, but not 2, using CSI-MS. We note that Mn(IV)-oxo intermediates could also form directly from the Mn(III)-superoxo species c (vide supra; orange paths). Although i could not be detected under CSI-MS experimental conditions in the reaction with 2, perpendicular EPR measurements suggest that this species is formed transiently with the H2qp1 system (vide infra). The end-products of the reactions between 2 and O2˙− provide further, but indirect, evidence for the generation of the OH˙ by-products anticipated from the formation of Mn(IV)-oxo species. Upon prolonged reaction times, we observe Mn(II) complexes with oxygenated ligand backbones and missing quinol/para-quinone groups by CSI-MS (Fig. 7). We believe that O2˙− will normally intercept and react with Mn(IV)-oxo and OH˙ oxidants quickly enough to prevent them from oxidizing the organic ligands. When the catalysis has depleted the O2˙−, however, this protective benefit is lost, resulting in ligand oxidation. The benzylic carbons that connect the quinols/para-quinones to the rest of the ligand are particularly susceptible to oxidation due to their weak C–H bonds.38,77 With 3, the long-term decomposition renders the ligand tetradentate. In water, neutral and acyclic N4 coordination environments cannot out-compete water molecules as ligands; consequently, the end-products of the reactions between 3 and O2˙− consist of a mixture of organic ligand fragments and no distinct Mn(II) or Mn(III) species. With 2, the loss of a quinol group from H2qp1 yields a pentadentate ligand with a N5 coordination environment. This decomposition product can still bind Mn(II) tightly enough to allow us to observe discrete Mn(II) complexes with the degraded ligand by CSI-MS (Fig. 7). The m/z features of the decomposition products are less prominent for 2 than those for 3; this is consistent with the H2qp1 system generating fewer Mn(IV)-oxo and OH˙ species during catalysis. Importantly, the species with the oxygenated H2qp1 ligand could still be catalytically active even without its quinol/para-quinone group (vide supra). Lastly, we also detect a Mn(II) species with an oxygenated benzylic carbon of H2qp1 where the quinol is still attached to the framework (Fig. 7B). The quinol is in its reduced form, which suggests that the ligand indeed cycles between its quinol and para-quinone oxidation states during catalysis (purple pathway) in parallel with the metal redox cycling (red and blue cycles).
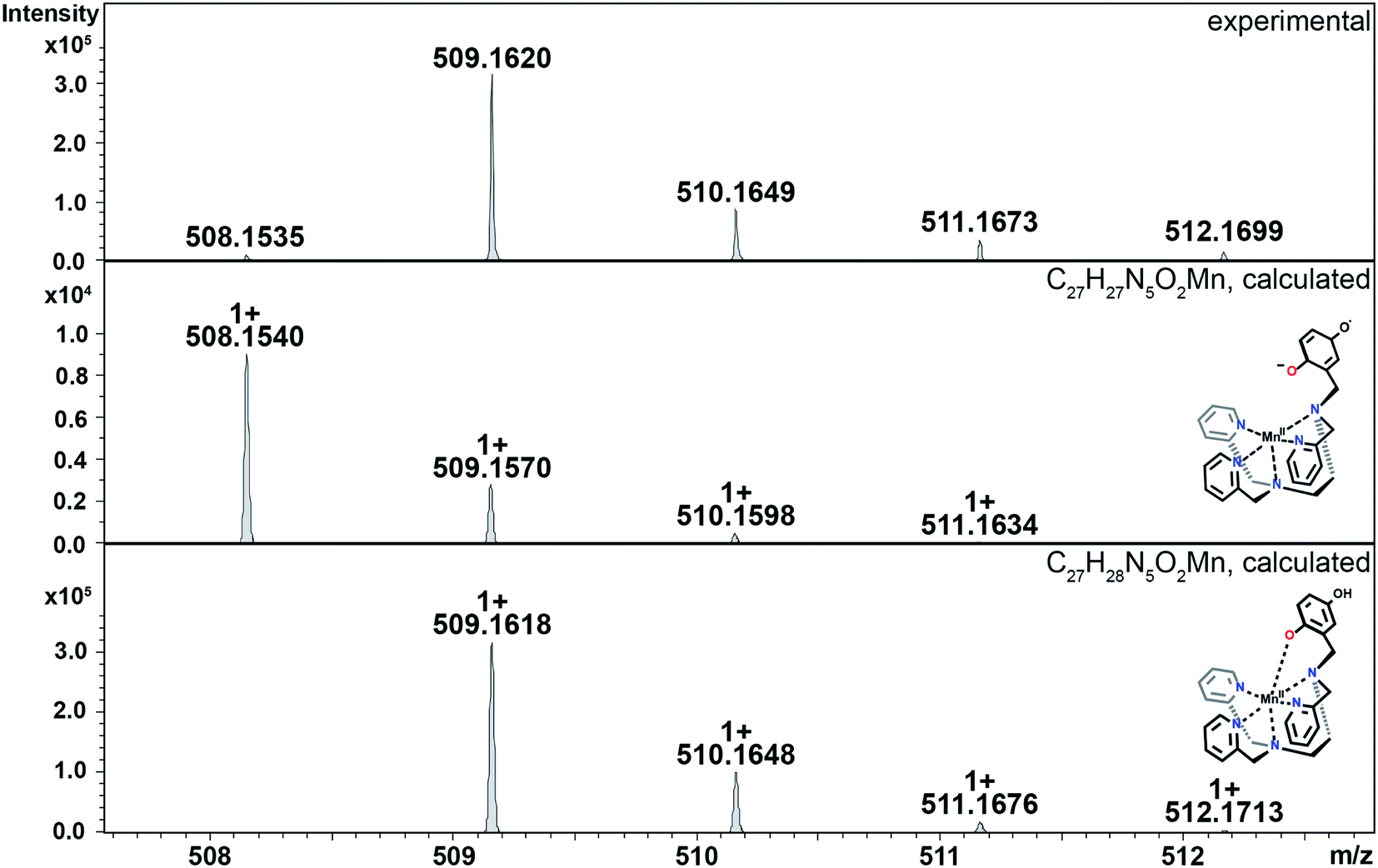
 | ||
| Fig. 6 CSI-MS of 3 upon reaction with KO2. Observed products include: (A) the Mn(III)-hydroperoxo-para-quinone complex h for 3, which has a m/z = 571.1378, and (B) the Mn(IV)-oxo species i for 3, which has a m/z = 554.1353. Experimental conditions: 1 mM solutions of 3 in MeCN (1% DMF) were cooled to −40 °C and then mixed with an excess of solid KO2. Subsequently, the mixture was diluted in a pre-cooled syringe with pre-cooled MeCN to approximately 1 × 10−5 M and quickly injected into the mass spectrometer. | ||
 | ||
| Fig. 7 CSI-MS spectra of end-products of 2 after reaction with O2˙−. Species A (m/z = 387.1248), C (m/z = 433.1305) and D (m/z = 445.135) show a complete loss of the quinol/para-quinone moiety. Species B (m/z = 523.1411) shows an oxygenated ligand backbone. | ||
Although parallel MS experiments using a 18O-labeled source of superoxide would be invaluable for corroborating the identities of many of the detected intermediates, such studies were impractical due to the nature of the available resources. We were unable to find a reliable and economic source of 18O-labeled KO2 but were previously able to synthesize this reagent.78 Regrettably, the synthetic procedure contaminates the product with substantial amounts of basic impurities that have proven to be difficult to remove. These basic impurities are not present in commercially available K16O2. In prior studies, we found that the buffer solutions used under the required CSI-MS experimental conditions cannot sufficiently mitigate the added basicity. As a consequence of the more basic conditions that arise from the use of the 18O-labeled oxidant, the data are dominated by metal-OH species which yield m/z features that overlap with and obscure the regions corresponding to the target intermediates.38
Observation of reaction intermediates by low-temperature UV/vis and EPR
When 0.1 mM 2 is rapidly mixed with an excess of superoxide (0.5–1 mM) in MeCN at −40 °C, three bands at 422, 448, and 595 nm appear within 3 s (Fig. 8, green line). Over the next 30 s, the features at 422 and 448 nm begin to diminish while the broad band at 595 nm continues to intensify (Fig. 8, blue line). The appearance of an isosbestic point at 474 nm indicates that the observed spectral changes can be ascribed to two distinct species that are formed as intermediates in the reaction between 2 and O2˙− at low temperature. When the reaction is monitored at −40 °C for 1200 s, the 595 nm band slowly disappears as the absorbance below 400 nm increases without the formation of distinct peaks (Fig. S13 in ESI†). The long-term spectra likely correspond to a complex mixture of products (vide supra). | ||
| Fig. 8 (A) Time-resolved UV/vis spectra measured after stopped-flow mixing 0.1 mM 2 with 0.5–1 mM KO2 in MeCN at −40 °C. 1 equiv. of dibenzo-18-crown-6 was added to solubilize the KO2. Red line: the first spectrum observed after mixing 2 and O2˙−. Green and blue lines: spectra recorded 3 s and 30 s after mixing, respectively. (B) Kinetic traces recorded for this reaction within the first 60 s depicting the formation of two reaction intermediates. | ||
Although absorption bands above 400 nm may be consistent with higher-valent manganese intermediates formed from the oxidation of the metal center in 2 by O2˙−, the energies and characteristic shapes of the features at 422 and 448 nm resemble those found in the spectrum of the semiquinone radical anion.79,80 When the H2qp1 ligand by itself is allowed to react with an excess of O2˙− in MeCN, we observe similar bands by UV/vis (Fig. S14†), confirming that the species observed as the first intermediate in the low-temperature reaction between 2 and O2˙− is best interpreted as a Mn(II) complex with a semiquinone radical anion ligand. The UV/vis changes observed at low temperature therefore result from ligand-centered, rather than metal-centered, oxidation. The UV/vis data corroborate the identity of species d (Scheme 4 and Fig. 5).
The formation of the semiquinone radical anion is accompanied by an additional process that produces the species associated with the broad band centered at 595 nm. This secondary reaction is also observed in the reaction between O2˙− and the free H2qp1 ligand; the peak at 595 nm therefore cannot be ascribed to a low-energy charge transfer band between the metal and the ligand. The most probable explanations for the 595 nm absorbance band involve the occurrence of either intermolecular self-association between semiquinone radicals or an electron transfer equilibrium between semiquinone radical electron donors and para-quinone electron acceptors to form corresponding π-dimers. Such π-association charge transfer equilibria of organic radical ions have been the subject of many investigations.81,82 They have been shown to be thermochromic systems which display a strong absorption band at ∼600 nm at lower temperatures. Unfortunately, these intermolecular self-association processes could not be observed with the extremely dilute solutions and low-pressure conditions associated with the CSI-MS experiments.
Mn(III)-peroxo complexes with amino-pyridine ligands have been documented to have UV/vis bands between 550 and 600 nm.83–85 We cannot preclude the possibility that these are partly contributing to the 595 nm band observed for 2. To further investigate the potential contribution of multiple species to the 595 nm band, we compared the kinetic traces for the reaction between KO2 and the H2qp1 ligand and that between KO2 and complex 2 (Fig. S15†). Although the kinetic trace obtained for the ligand reaction can be satisfactorily fit to a one-exponential function, the kinetic trace for the reaction with complex 2 cannot, suggesting that more than one process is contributing to the increased absorbance at ∼600 nm.
The subsequent slow disappearance of the 595 nm band over 10 min at −40 °C indicates that further oxidation processes occur. Unfortunately, these processes cannot be well resolved from the UV/vis data since (A) the more intense absorption bands of the oxidized ligands dominate the spectra and (B) the high reactivities of higher-valent manganese intermediates preclude their accumulation to concentrations high enough to be clearly observed by UV/vis spectroscopy under the experimental conditions. In the reactions between 2 and O2˙−, we observe a small but significant increase in the absorbance at 700 nm that is not duplicated in the reactions between free H2qp1 and O2˙− (Fig. S16†). This may be consistent with the presence of a small amount of a higher-valent manganese species, but the data do not unambiguously support this.
Additional evidence of high-valent manganese species in the reaction cycles of 2 was provided by EPR analysis of freeze-quenched samples. To prepare the samples, an MeCN solution of 2 was rapidly mixed with an excess of O2˙− at 233 K and then rapidly cooled to 9 K after 30 s. The X-band EPR spectrum recorded in frozen MeCN at 9 K in the perpendicular mode displays the characteristic features of the high-spin d3 electronic configuration anticipated for a monomeric Mn(IV) complex, with a strong signal at g ∼ 2 and weak broad signals at g ∼ 4 and g ∼ 1.23 (Fig. S17†). The EPR spectra of most Mn(IV) complexes tend to follow one of two patterns, depending on the magnitude of the zero-field splitting. When the axial parameter D is much larger than the energy provided by the spectrometer (0.31 cm−1 with X-band), a weak signal is found at g ∼ 2 and a strong signal is observed at g ∼ 4. When D is much smaller than the excitation energy, the predominant peak instead occurs at g ∼ 2 with weaker features at g ∼ 4; this tends to occur in high symmetry environments.86–92 With complex 2, the more intense signal at g ∼ 2 indicates that the Mn(IV) species has a small zero-field splitting, with 2|D| ≪ hν.
The spectrum shown in Fig. S17† was analyzed and fitted using EasySpin in MATLAB®,93 and the values of the obtained parameters are given in Table S3.† The linewidth (fitted using a Gaussian distribution) is relatively large; most probably due to unresolvable hyperfine coupling between the manganese nucleus and nitrogen atoms in the surrounding ligand. The magnitude of the D value was determined to be 0.048 cm−1 (note: the sign of D is unknown), hence confirming that 2|D| ≪ hν. The E/D is approximately equal to zero (0.05), showing that there is negligible rhombicity in the system. These values suggest that the effective electronic environment of the Mn(IV) closely approximates an ideal octahedral configuration. The N5 core of the two electron-oxidized para-quinone form of the ligand (qp1) and the oxo group would provide such a coordination environment.
It should be noted that it is theoretically possible to model the spectrum for 2 as a Mn(II) species. However, although residual Mn(II) species, superoxide, and/or ligand radicals could contribute to the strong signal at g ∼ 2, our control experiments analyzing the EPR spectra of MeCN solutions of 2, the semiquinone radical ligand, and O2˙− (Fig. S18†) demonstrate that none of these three species can account for the feature at g ∼ 2 shown in Fig. S17.† The vast majority of the superoxide has been consumed by the 30 s timepoint. In principle, the broad feature at g ∼ 2 can also be ascribed to another Mn(II) species generated in the catalytic cycle (Scheme 4). This species should display high symmetry, regardless of whether the spectrum is simulated as a Mn(II) or as a Mn(IV) species, since the magnitude of the E/D ratio is small. The only plausible candidate with these characteristics would be a Mn(II) intermediate bearing a quinone ligand (species e shown in Scheme 4). Although we cannot preclude this possibility, we believe that the UV/vis data taken during the catalysis performed by 2 is indicative of a transient high-valent manganese intermediate, and consequently, we assign the feature at g ∼ 2 to a Mn(IV) species. Although the zero-field splitting parameters obtained in the present study are in line with those observed for other Mn(IV) centers,92 the D parameter is significantly smaller than those reported for other Mn(IV)-oxo adducts. The D values for other reported Mn(IV)-oxo complexes are usually much larger than the microwave frequency, resulting in a more intense EPR signal situated near g ∼ 4. A rare counterexample of a Mn(IV)-oxo species with a more prominent signal at g ∼ 2 was described by van Eldik et al. for the oxidation of [MnII(bpy)2Cl2] by H2O2.94 Although the authors did not perform explicit calculations to reveal the value of D for this system, limiting direct comparison, the formation of a Mn(IV)-oxo complex was supported by UV/vis spectroscopy. Consequently, the features at g ∼ 2 observed in the present study could plausibly be assigned to high-valent manganese-oxo intermediates. Very recently, an unprecedented signal at g ∼ 1.8 was observed during water oxidation reactions involving manganese-containing heterogeneous catalysts; the feature was assigned to a low-spin Mn(IV)-oxo species with S = 1/2.95 Although the signal at ∼2 described in the present work could be also generally ascribed to a low-spin Mn(IV)-oxo species, we could not adequately fit the data using a S = 1/2 model; a S = 3/2 model provides a superior fit.
When UV/vis is used to analyze a rapidly mixed MeCN solution containing 0.1 mM 3 and 0.5–1.0 mM O2˙− at −40 °C, we observe bands similar to those found for analogous reactions with 2: sharp features at 423 and 448 nm and a much broader band centered at 595 nm (Fig. 9, green and blue lines). Over longer periods of time, however, a new band appears at 520 nm as the 595 nm feature vanishes, resulting in the solution changing color from blue to purple (Fig. 9, magenta line). Since a 520 nm band was not observed for 2, this suggests that 3 proceeds through an additional intermediate. The 520 nm peak is also observed when the free H4qp2 ligand is oxidized by O2˙− under similar reaction conditions, suggesting that this new feature corresponds to a purely ligand-derived electronic transition (Fig. S19†).
 | ||
| Fig. 9 Time-resolved UV/vis spectra acquired during the reaction between 0.1 mM 3 and excess (0.5–1 mM) O2˙− in MeCN at −40 °C. Red line: the spectrum taken immediately after mixing the solutions of 3 and O2˙−. Green, blue and magenta lines: spectra recorded at 30, 60, and 375 s after mixing, respectively. | ||
As with the other bands, the 520 nm peak observed during oxidation of both 3 and H4qp2 cannot correlate to a charge transfer process between the ligand and the metal center but must instead be associated with a redox process on the ligand itself. The purple color of the solution is reminiscent of the charge-transfer complex quinhydrone, which consists of a reduced quinone interacting with an oxidized para-quinone through ring stacking.96–98 In the presence of light, an electron from the reduced hydroquinone donor transfers to the oxidized benzoquinone acceptor, accounting for the characteristic purple hue of quinhydrone.99,100 This charge-transfer complex can only form when both redox forms of the quinol group – the two-electron oxidized para-quinone and fully reduced quinol – exist in the solution at the same time. That a similar complex is not observed during the oxidation of either H2qp1 or 2 suggests that this is an intramolecular, rather than an intermolecular, process that requires the molecule to have two redox-active quinol/para-quinone subunits. Although the existence of this species cannot be clearly resolved from the UV/vis data due to interference from the strong absorbance bands of the semiquinone radicals, the cryo-MS data demonstrate that a Mn(II)-quinol-para-quinone intermediate e is produced during the oxidation of 3 by O2˙− (Scheme 4, the m/z = 538.1404 species in Fig. 4). In intermediate e, the quinhydrone would form from intramolecular ring stacking interactions between the quinol and para-quinone groups in the coordination sphere.
Since the UV/vis data recorded during the oxidation of 3 by O2˙− are dominated by the strong absorption bands assigned to semiquinone radical anions (422 and 448 nm),79,80 quinhydrone (520 nm),96–98 and also partially to π-association charge transfer equilibria (the red shifted band at 595 nm),81,82 it is difficult to identify additional intermediates using these data by themselves (Fig. S20†). With this system, the CSI-MS results are quite helpful to understanding the overall mechanism (Scheme 4) and, concerning their complexity, are rare for SOD catalytic cycles.38 Lastly, we note that the UV/vis data suggest that the H4qp2 ligand in 3 undergoes secondary oxidation reactions, specifically oxygenation of the methylene positions and cleavage of the quinol groups from the 1,2-ethanediamine backbone, more slowly than the H2qp1 ligand in 2. This can be rationalized by the much higher SOD activity of 2, which more quickly decomposes the O2˙− that could sufficiently scavenge reactive Mn(IV)-oxo and hydroxyl radicals (vide supra).
The reaction between 3 and O2˙− was also characterized by EPR. In these experiments, MeCN solutions of 3 were rapidly mixed with an excess of O2˙− at 233 K, and the resulting mixtures were frozen at 9 K after 30 s and analyzed by X-band EPR (Fig. S21†). Similar to the reactivity of complex 2, the vast majority of the superoxide has been consumed by the 30 s time point. When the reaction is quenched at 10 s, however, we observe an additional sharp signal that can be attributed to residual superoxide (Fig. S22†). The 9 K spectrum obtained at 30 s (Fig. S21†) is complicated and suggests the presence of multiple species. The intense signal centered at g ∼ 2.0 with the partially resolved hyperfine splitting from the manganese nucleus (I = 5/2) is most likely a Mn(II) species. In addition, the spectrum exhibits multiple low-field components below 2000 G and high-field components above 4000 G. These features probably result from the superposition of signals from one or more distinct S = 3/2 species, with the S value being characteristic of a mononuclear Mn(IV) species with D > hν.86–92 Using EasySpin,93 it was possible to simulate the spectrum with two components: a Mn(IV) species (S = 3/2) and a Mn(II) species (S = 5/2) (Fig. S21†). Fitting this spectrum was not trivial due the multiple components and many parameters needed for optimization. As a result, the obtained fit, although reasonable, is not as accurate as that for complex 2. Nevertheless, the model still accounts for the main features and reproduces most of the features observed for a highly complicated spectrum. According to our model, the Mn(II) and Mn(IV) species are present in an approximately 1:1 ratio (Table S3†). The Mn(IV) complex has a D value of magnitude 0.323 cm−1, which is slightly greater than hv. The E/D ratio for the Mn(IV) species generated from 3 is small suggesting that this species, like the one generated from 2, is slightly axially distorted with negligible rhombicity.
An alternative EasySpin model that also provided a reasonable fit to the data included two Mn(IV) species: one with 2|D| ≪ hν and another with a D value greater than hν (Fig. S23† and Table S3†). Although there is little difference between the two models used to fit 3, we believe that, though possible, it is unlikely for two different reactive Mn(IV) species to coexist together under these conditions. As previously mentioned, the hyperfine splitting observed around the central g ∼ 2 feature suggests that a Mn(II) species is more likely, although we were unable to simulate these features using either model. Irrespective of which model is correct, both give evidence for the existence of a Mn(IV) species. It should be noted that other models were also tested that consisted of one or two Mn(IV) species with 2|D| ≪ hν, but these did not yield accurate simulations representative of our spectrum for 3. Models involving Mn(II) alone were also tested, but they likewise did not replicate as many of the spectral features.
Conclusions
Although complexes 1 and 2 are structurally similar, with the only difference being the identity of the para substituent in the phenolic portion of the ligand, the two compounds display markedly different activities with O2˙−. The para-methyl group in 1 provides a Mn(III/II) reduction potential that is low enough to enable outer-sphere electron transfer; whereas, the para-hydroxyl group in 2 increases this reduction potential enough to make this pathway energetically non-competitive with inner-sphere oxidation. The hydroxyl-for-methyl substitution on the phenol also increases the catalytic rate, with 2 displaying activity comparable to those of cationic porphyrin manganese complexes62,71 and potent pentaazamacrocyclic Mn SOD mimetics that have entered clinical trials, but with superior aqueous stability to the latter.101–103 The inclusion of a second quinol in 3 was found to destabilize the Mn(II) complex in water and reduce both the catalytic activity and its substitution reactivity. Both effects are proposed to result from a lesser overall charge on the manganese complex, which should weaken the ability of the complex to attract both O2˙− and competitive inhibitors, such as phosphate. Although a typical counter-ion inhibition is not observed, 3 catalytically degrades O2˙− through an inner-sphere mechanism. The CSI-MS and time resolved UV/vis data provide evidence for a host of reaction intermediates that demonstrate the inner-sphere nature of the catalysis and reveal that both the manganese centers and the ligands are oxidatively active during catalysis. Though involvement of high-valent manganese species is well documented in the catalase-like activity of porphyrin- and non-porphyrin-based enzyme mimetics,67,104 generation of Mn(IV)-oxo in a direct reaction between a monomeric Mn(II) complex and superoxide within a SOD catalytic cycle had not been documented prior to this report. In the case of a polymeric mixed valence Mn(III)/Mn(II) complex, Mn(III)–O–Mn(IV) was identified as an intermediate in a reaction with superoxide by EPR spectroscopy.105 Our explicit observation of Mn(IV)-oxo intermediates generated from mononuclear Mn(II) SOD mimetics by CSI-MS, UV/vis and EPR spectra is unique. Using the SOD activity of the Zn(II) analogue of 2 (4) as an estimate for the ligand-only contribution to the reactivity, we conclude that the metal-based inner-sphere SOD mechanism provides the more efficient pathway for superoxide degradation in Mn-quinol/phenol-based systems. The steric/conformational features enabling stable pentacoordination upon ligand oxidation and higher overall positive charges of 2 allow it to efficiently bind O2˙−. The oxidation of the Hqp1− ligand to neutral qp1 also limits the accumulation of a potentially deleterious Mn(IV)-oxo species, relative to the H4qp2 system. These aforementioned factors combine to make 2 arguably the most effective reported Mn SOD mimic when one accounts for its higher stability. The results here demonstrate that the concerted/complementing redox-activity of manganese and organic ligands can be a highly effective strategy for developing small molecule antioxidants.Data availability
Most of the data that support the findings of this study are present in the article and its ESI. The corresponding authors will provide additional data not available in these documents upon request.Author contributions
L. S., J. L. M., A. F., K. R. F., A. S., A. Z., R. P., D. F., S. I., and S. S. designed and conducted the experiments and collected and analyzed the data. L. S., J. L. M., A. F., K. R. F., A. S., A. Z., and R. P. also wrote/edited portions of the manuscript. L. S., I. I.-B., and C. R. G. conceived the idea. I. I.-B. and C. R. G. also supervised the project, provided the majority of the resources, and wrote/edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Auburn University, the National Science Foundation (NSF-CHE-1662875 and NSF-CHE-1954336), and the German Research Foundation (DFG Research Grant IV 80/13-1) for financial support. The authors acknowledge Prof. Karsten Meyer for enabling EPR measurements in his laboratory and Prof. Lena J. Daumann for the support in the analysis and interpretation of EPR data.References
- R. L. Mosley, E. J. Benner, I. Kadiu, M. Thomas, M. D. Boska, K. Hasan, C. Laurie and H. E. Gendelman, Clin. Neurosci. Res., 2006, 6, 261–281 CrossRef CAS PubMed.
- I. M. Fearon and S. P. Faux, J. Mol. Cell. Cardiol., 2009, 47, 372–381 CrossRef CAS PubMed.
- C. K. Roberts and K. K. Sindhu, Life Sci., 2009, 84, 705–712 CrossRef CAS PubMed.
- G. Eskici and P. H. Axelsen, Biochemistry, 2012, 51, 6289–6311 CrossRef CAS PubMed.
- S. F. AbdulSalam, F. S. Thowfeik and E. J. Merino, Biochemistry, 2016, 55, 5341–5352 CrossRef CAS PubMed.
- M. I. Ahmed, J. D. Gladden, S. H. Litovsky, S. G. Lloyd, H. Gupta, S. Inusah, T. Denney Jr, P. Powell, D. C. McGiffin and L. J. Dell'Italia, J. Am. Coll. Cardiol., 2010, 55, 671–679 CrossRef CAS PubMed.
- V. L. Kinnula, Curr. Drug Targets–Inflamm. Allergy, 2005, 4, 465–470 CrossRef CAS PubMed.
- A. S. Abouhashem, K. Singh, H. M. E. Azzazy and C. K. Sen, Antioxid. Redox Signaling, 2020, 33, 59–65 CrossRef CAS PubMed.
- M. Laforge, C. Elbim, C. Frère, M. Hémadi, C. Massaad, P. Nuss, J.-J. Benoliel and C. Becker, Nat. Rev. Immunol., 2020, 20, 515–516 CrossRef CAS PubMed.
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS PubMed.
- E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652–16659 CrossRef CAS PubMed.
- Y. Koide, Y. Urano, S. Kenmoku, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 10324–10325 CrossRef CAS PubMed.
- H. Maeda, K. Yamamoto, Y. Nomura, I. Kohno, L. Hafsi, N. Ueda, S. Yoshida, M. Fukuda, Y. Fukuyasu, Y. Yamauchi and N. Itoh, J. Am. Chem. Soc., 2005, 127, 68–69 CrossRef CAS PubMed.
- D. P. Riley and R. H. Weiss, J. Am. Chem. Soc., 1994, 116, 387–388 CrossRef CAS.
- D. P. Riley and O. F. Schall, Adv. Inorg. Chem., 2006, 59, 233–263 CrossRef.
- D. P. Riley, P. J. Lennon, W. L. Neumann and R. H. Weiss, J. Am. Chem. Soc., 1997, 119, 6522–6528 CrossRef CAS.
- D. P. Riley, Chem. Rev., 1999, 99, 2573–2588 CrossRef CAS PubMed.
- A. Tovmasyan, S. Carballal, R. Ghazaryan, L. Melikyan, T. Weitner, C. G. C. Maia, J. S. Reboucas, R. Radi, I. Spasojevic, L. Benov and I. Batinic-Haberle, Inorg. Chem., 2014, 53, 11467–11483 CrossRef CAS PubMed.
- I. Batinic-Haberle, J. S. Reboucas and I. Spasojevic, Antioxid. Redox Signaling, 2010, 13, 877–918 CrossRef CAS PubMed.
- I. Batinic-Haberle, Z. Rajic, A. Tovmasyan, J. S. Reboucas, X. Ye, K. W. Leong, M. W. Dewhirst, Z. Vujaskovic, L. Benov and I. Spasojevic, Free Radical Biol. Med., 2011, 51, 1035–1053 CrossRef CAS PubMed.
- I. A. Abreu and D. E. Cabelli, Biochim. Biophys. Acta, 2010, 1804, 263–274 CrossRef CAS PubMed.
- M. Yu, R. J. Beyers, J. D. Gorden, J. N. Cross and C. R. Goldsmith, Inorg. Chem., 2012, 51, 9153–9155 CrossRef CAS PubMed.
- M. Yu, S. L. Ambrose, Z. L. Whaley, S. Fan, J. D. Gorden, R. J. Beyers, D. D. Schwartz and C. R. Goldsmith, J. Am. Chem. Soc., 2014, 136, 12836–12839 CrossRef CAS PubMed.
- M. Yu, M. B. Ward, A. Franke, S. L. Ambrose, Z. L. Whaley, T. M. Bradford, J. D. Gorden, R. J. Beyers, R. C. Cattley, I. Ivanović-Burmazović, D. D. Schwartz and C. R. Goldsmith, Inorg. Chem., 2017, 56, 2812–2826 CrossRef CAS PubMed.
- G. S. Loving, S. Mukherjee and P. Caravan, J. Am. Chem. Soc., 2013, 135, 4620–4623 CrossRef CAS PubMed.
- E. M. Gale, S. Mukherjee, C. Liu, G. S. Loving and P. Caravan, Inorg. Chem., 2014, 53, 10748–10761 CrossRef CAS PubMed.
- E. M. Gale, C. M. Jones, I. Ramsay, C. T. Farrar and P. Caravan, J. Am. Chem. Soc., 2016, 138, 15861–15864 CrossRef CAS PubMed.
- H. Wang, V. C. Jordan, I. A. Ramsay, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, J. Am. Chem. Soc., 2019, 141, 5916–5925 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- O. Iranzo, Bioorg. Chem., 2011, 39, 73–87 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS.
- I. Kenkel, A. Franke, M. Dürr, A. Zahl, C. Dücker-Benfer, J. Langer, M. R. Filipović, M. Yu, R. Puchta, S. R. Fiedler, M. P. Shores, C. R. Goldsmith and I. Ivanović-Burmazović, J. Am. Chem. Soc., 2017, 139, 1472–1484 CrossRef CAS PubMed.
- D. Lieb, F. C. Friedel, M. Yawer, A. Zahl, M. M. Khusniyarov, F. W. Heinemann and I. Ivanović-Burmazović, Inorg. Chem., 2013, 52, 222–236 CrossRef CAS PubMed.
- F. C. Friedel, D. Lieb and I. Ivanović-Burmazović, J. Inorg. Biochem., 2012, 109, 26–32 CrossRef CAS PubMed.
- I. Ivanović-Burmazović, Adv. Inorg. Chem., 2008, 60, 59–100 CrossRef.
- C. M. Weekley, I. Kenkel, R. Lippert, S. Wei, D. Lieb, T. Cranwell, J. L. Wedding, A. S. Zillmann, R. Rohr, M. R. Filipovic, I. Ivanović-Burmazović and H. H. Harris, Inorg. Chem., 2017, 56, 6076–6093 CrossRef CAS PubMed.
- D. P. Riley, S. L. Henke, P. J. Lennon, R. H. Weiss, W. L. Neumann, W. J. Rivers, K. W. Aston, K. R. Sample, H. Rahman, C.-S. Ling, J.-J. Shieh and D. H. Busch, Inorg. Chem., 1996, 35, 5213–5231 CrossRef CAS.
- M. B. Ward, A. Scheitler, M. Yu, L. Senft, A. S. Zillmann, J. D. Gorden, D. D. Schwartz, I. Ivanović-Burmazović and C. R. Goldsmith, Nat. Chem., 2018, 10, 1207–1212 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- E. M. Gale, J. Zhu and P. Caravan, J. Am. Chem. Soc., 2013, 135, 18600–18608 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- R. Seeger and J. A. Pople, J. Chem. Phys., 1977, 66, 3045–3050 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047–9052 CrossRef CAS.
- H. B. Schlegel and J. J. McDouall, in Computational Advances in Organic Chemistry, ed. C. Ögretir and I. G. Csizmadia, Kluwer Academic, The Netherlands, 1991, pp. 167–185 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- S. C. Sahoo, M. Dubey, M. A. Alam and M. Ray, Inorg. Chim. Acta, 2010, 363, 3055–3060 CrossRef CAS.
- B. Drahoš, J. Kotek, P. Hermann, I. Lukeš and É. Tóth, Inorg. Chem., 2010, 49, 3224–3238 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- T. E. Hutchinson, A. Bashir, M. Yu, R. J. Beyers and C. R. Goldsmith, Inorg. Chim. Acta, 2019, 496, 119045 CrossRef CAS.
- D. Taubert, T. Breitenbach, A. Lazar, P. Censarek, S. Harlfinger, R. Berkels, W. Klaus and R. Roesen, Free Radical Biol. Med., 2003, 35, 1599–1607 CrossRef CAS PubMed.
- I. Fridovich, J. Biol. Chem., 1997, 272, 18515–18517 CrossRef CAS PubMed.
- S. I. Liochev, Chem. Res. Toxicol., 2013, 26, 1312–1319 Search PubMed.
- D. P. Riley, W. J. Rivers and R. H. Weiss, Anal. Biochem., 1991, 196, 344–349 CrossRef CAS PubMed.
- S. I. Liochev and I. Fridovich, Arch. Biochem. Biophys., 1997, 337, 115–120 CrossRef CAS PubMed.
- A. Dees, A. Zahl, R. Puchta, N. J. R. van Eikema Hommes, F. W. Heinemann and I. Ivanović-Burmazović, Inorg. Chem., 2007, 46, 2459–2470 CrossRef CAS PubMed.
- I. Ivanović-Burmazović and M. R. Filipović, Adv. Inorg. Chem., 2012, 64, 53–95 CrossRef.
- I. Batinic-Haberle and A. Tovmasyan, in Oxidative Stress and Antioxidant Protection: The Science of Free Radical Biology and Disease, ed. D. Armstrong and R. D. Stratton, John Wiley & Sons, Hoboken, NJ, 2016, ch. 27, pp. 415–470 Search PubMed.
- C. Palopoli, J. Ferreyra, A. Conte-Daban, M. Richezzi, A. Foi, F. Doctorovich, E. Anxolabéhère-Mallart, C. Hureau and S. R. Signorella, ACS Omega, 2019, 4, 48–57 CrossRef CAS PubMed.
- A. Vincent, M. Thauvin, E. Quévrain, E. Mathieu, S. Layani, P. Seksik, I. Batinic-Haberle, S. Vriz, C. Policar and N. Delsuc, J. Inorg. Biochem., 2021, 219, 111431 CrossRef CAS PubMed.
- E. Mathieu, A.-S. Bernard, E. Quévrain, M. Zoumpoulaki, S. Iriart, C. Lung-Soong, B. Lai, K. Medjoubi, L. Henry, S. Nagarajan, F. Poyer, A. Scheitler, I. Ivanović-Burmazović, S. Marco, A. Somogyi, P. Seksik, N. Delsuc and C. Policar, Chem. Commun., 2020, 56, 7885–7888 RSC.
- H. Y. V. Ching, I. Kenkel, N. Delsuc, E. Mathieu, I. Ivanović-Burmazović and C. Policar, J. Inorg. Biochem., 2016, 160, 172–179 CrossRef CAS PubMed.
- G. N. Ledesma, H. Eury, E. Anxolabéhère-Mallart, C. Hureau and S. R. Signorella, J. Inorg. Biochem., 2015, 146, 69–76 CrossRef CAS PubMed.
- D. Salvemini, Z.-Q. Wang, J. L. Zweier, A. Samouilov, H. Macarthur, T. P. Misko, M. G. Currie, S. Cuzzocrea, J. A. Sikorski and D. P. Riley, Science, 1999, 286, 304–306 CrossRef CAS PubMed.
- K. Aston, N. Rath, A. Naik, U. Slomczynska, O. F. Schall and D. P. Riley, Inorg. Chem., 2001, 40, 1779–1789 CrossRef CAS PubMed.
- C. Policar, in Redox-Active Therapeutics. Oxidative Stress in Applied Basic Research and Clinical Practice, ed. I. Batinić-Haberle, J. Rebouças and I. Spasojević, Springer, Cham, 2016 Search PubMed.
- I. Batinic-Haberle and I. Spasojevic, J. Porphyrins Phthalocyanines, 2019, 23, 1326–1335 CrossRef CAS.
- I. Batinic-Haberle, A. Tovmasyan and I. Spasojevic, Antioxid. Redox Signaling, 2018, 29, 1691–1724 CrossRef CAS PubMed.
- I. Batinić-Haberle, S. I. Liochev, I. Spasojević and I. Fridovich, Arch. Biochem. Biophys., 1997, 343, 225–233 CrossRef PubMed.
- G. DeFreitas-Silva, J. S. Rebouças, I. Spasojević, L. Benov, Y. M. Idemori and I. Batinić-Haberle, Arch. Biochem. Biophys., 2008, 477, 105–112 CrossRef CAS PubMed.
- D. Lieb, Ph.D. thesis, University of Erlangen-Nuremberg, 2013.
- A. Singha and A. Dey, Chem. Commun., 2019, 55, 5591–5594 RSC.
- Q. Zhang, A. Bell-Taylor, F. M. Bronston, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2017, 56, 773–782 CrossRef CAS PubMed.
- K. Duerr, J. Olah, R. Davydov, M. Kleimann, J. Li, N. Lang, R. Puchta, E. Hübner, T. Drewello, J. N. Harvey, N. Jux and I. Ivanović-Burmazović, Dalton Trans., 2010, 39, 2049–2056 RSC.
- A. J. Swallow, in Functions of Quinones in Energy Converting Systems, ed. B. L. Trumpower, Elsevier, Inc., New York, NY, 1982, pp. 59–72 Search PubMed.
- X. Zhao, H. Imahori, C.-G. Zhan, Y. Sakata, S. Iwata and T. Kitagawa, J. Phys. Chem. A, 1997, 101, 622–631 CrossRef CAS.
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850–13858 CrossRef CAS PubMed.
- J.-M. Lü, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 12161–12171 CrossRef PubMed.
- R. A. Geiger, D. F. Leto, S. Chattopadhyay, P. Dorlet, E. Anxolabéhère-Mallart and T. A. Jackson, Inorg. Chem., 2011, 50, 10190–10203 CrossRef CAS PubMed.
- S. Groni, G. Blain, R. Guillot, C. Policar and E. Anxolabéhère-Mallart, Inorg. Chem., 2007, 46, 1951–1953 CrossRef CAS PubMed.
- H. Y. V. Ching, E. Anxolabéhère-Mallart, H. E. Colmer, C. Costentin, P. Dorlet, T. A. Jackson, C. Policar and M. Robert, Chem. Sci., 2014, 5, 2304–2310 RSC.
- D. F. Leto, A. A. Massie, H. E. Colmer and T. A. Jackson, Inorg. Chem., 2016, 55, 3272–3282 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, A. S. Borovik and M. P. Hendrich, Inorg. Chem., 2013, 52, 12568–12575 CrossRef CAS PubMed.
- C. Duboc, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- C. Duboc and M.-N. Collomb, Chem. Commun., 2009, 2715–2717 RSC.
- M. Zlatar, M. Gruden, O. Y. Vassilyeva, E. A. Buvaylo, A. N. Ponomarev, S. A. Zvyagin, J. Wosnitza, J. Krzystek, P. Garcia-Fernandez and C. Duboc, Inorg. Chem., 2016, 55, 1192–1201 CrossRef CAS PubMed.
- T. Weyhermüller, T. K. Paine, E. Bothe, E. Bill and P. Chaudhuri, Inorg. Chim. Acta, 2002, 337, 344–356 CrossRef.
- B. C. Dave and R. S. Czernuszewicz, J. Coord. Chem., 1994, 33, 257–269 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. Rothbart, E. Ember and R. van Eldik, Dalton Trans., 2010, 39, 3264–3272 RSC.
- S. Park, K. Jin, H. K. Lim, J. Kim, K. H. Cho, S. Choi, H. Seo, M. Y. Lee, Y. H. Lee, S. Yoon, M. Kim, H. Kim, S. H. Kim and K. T. Nam, Nat. Commun., 2020, 11, 5230–5239 CrossRef CAS PubMed.
- M. A. Slifkin, Charge Transfer Interactions of Biomolecules, Academic Press, London, 1971 Search PubMed.
- M. J. González Moa, M. Mandado and R. A. Mosquera, J. Phys. Chem. A, 2007, 111, 1998–2001 CrossRef PubMed.
- J. Regeimbal, S. Gleiter, B. L. Trumpower, C.-A. Yu, M. Diwakar, D. P. Ballou and J. C. A. Bardwell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13779–13784 CrossRef CAS PubMed.
- N. Li and J. Wang, J. Phys. Chem. A, 2008, 112, 6281–6284 CrossRef CAS PubMed.
- L. Tong, Q. Chen, A. A. Wong, R. Gómez-Bombarelli, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Phys. Chem. Chem. Phys., 2017, 19, 31684–31691 RSC.
- C. M. Anderson, B. G. Allen, W. Sun, C. M. Lee, S. Agarwala, M. Venigalla, L. Greenberg, D. Adkins, Y. Chen, W. Zhen, D. Mould, J. Holmlund, J. Brill, S. Sonis and J. Buatti, Int. J. Radiat. Oncol., Biol., Phys., 2016, 94, 869–870 CrossRef.
- R. Bonetta, Chem.–Eur. J., 2018, 24, 5032–5041 CrossRef CAS PubMed.
- The two clinical trials involving M40403 have been suspended (NCT00033956) or terminated (NCT00101621). The studies on the safety, tolerability, and pharmacokinetics of the derivatives GC00034702 and GC00034711 are being performed (NCT03096756 and NCT03099824).
- M. Chino, L. Leone, G. Zambrano, F. Pirro, D. D. Alonzo, V. Firpo, D. Aref, L. Lista, O. Maglio, F. Nastri and A. Lombardi, Biopolymers, 2018, 109, e23107 CrossRef PubMed , 23101–23119..
- R. O. Costa, S. S. Ferreira, C. A. Pereira, J. R. Harmer, C. J. Noble, G. Schenk, R. W. A. Franco, J. A. L. C. Resende, P. Comba, A. E. Roberts, C. Fernandes and A. Horn Jr, Front. Chem., 2018, 6, 491 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full CSI-MS spectra for the reactions between KO2 and 2 and 3 (Fig. S1 and S2); pH titration data for 2 (Fig. S3–S5); experimental details of water exchange experiments and general treatment of the 17O NMR data (Fig. S6–S9); expanded CSI-MS spectra for the reactions between KO2 and 2 and 3 (Fig. S10–S12); UV/vis low-temperature stopped-flow spectra of the intermediates of the reactions between 2 or 3 and superoxide (Fig. S13–S16, S19, and S20); EPR spectra of the reaction intermediates (Fig. S17, S18, and S21–S23); Tables S1–S3. See DOI: 10.1039/d1sc02465e |
| This journal is © The Royal Society of Chemistry 2021 |