
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenetically-targeted photorelease of endocannabinoids enables optical control of GPR55 in pancreatic β-cells†
Janelle M.
Tobias
 abc,
Gabriela
Rajic
a,
Alexander E. G.
Viray
ab,
David
Icka-Araki
abd and
James A.
Frank
*ab
abc,
Gabriela
Rajic
a,
Alexander E. G.
Viray
ab,
David
Icka-Araki
abd and
James A.
Frank
*ab
aVollum Institute, Oregon Health & Science University, Portland, OR, USA. E-mail: frankja@ohsu.edu
bDepartment of Chemical Physiology & Biochemistry, Oregon Health & Science University, Portland, OR, USA
cGraduate Program in Physiology & Pharmacology, Oregon Health & Science University, Portland, OR, USA
dGraduate Program in Biomedical Sciences, Oregon Health & Science University, Portland, OR, USA
First published on 15th September 2021
Abstract
Fatty acid amides (FAAs) are a family of second-messenger lipids that target cannabinoid receptors, and are known mediators of glucose-stimulated insulin secretion from pancreatic β-cells. Due to the diversity observed in FAA structure and pharmacology, coupled with the expression of at least 3 different cannabinoid G protein-coupled receptors in primary and model β-cells, our understanding of their role is limited by our inability to control their actions in time and space. To investigate the mechanisms by which FAAs regulate β-cell excitability, we developed the Optically-Cleavable Targeted (OCT)-ligand approach, which combines the spatial resolution of self-labeling protein (SNAP-) tags with the temporal control of photocaged ligands. By linking a photocaged FAA to an o-benzylguanine (BG) motif, FAA signalling can be directed towards genetically-defined cellular membranes. We designed a probe to release palmitoylethanolamide (PEA), a GPR55 agonist known to stimulate glucose-stimulated insulin secretion (GSIS). When applied to β-cells, OCT-PEA revealed that plasma membrane GPR55 stimulates β-cell Ca2+ activity via phospholipase C. Moving forward, the OCT-ligand approach can be translated to other ligands and receptors, and will open up new experimental possibilities in targeted pharmacology.
Introduction
Endocannabinoid signalling is mediated by cannabinoid receptors, lipid-derived ligands, and the metabolic enzymes that control their synthesis and degradation. The most well-studied component of the endocannabinoid system is cannabinoid receptor 1 (CB1), an inhibitory G protein-coupled receptor (GPCR) that is highly expressed in the central nervous system and responsible for the psychotropic effects of marijuana.1 Besides the canonical receptors CB1 and CB2, recent studies have revealed a diverse collection of proteins that respond to cannabinoid ligands—including other GPCRs, ion channels, and nuclear receptors—birthing the concept of an “extended endocannabinoid system”.2 Similarly, our understanding of cannabinoid ligands has broadened beyond the second-messenger lipids anandamide and 2-arachidonylglycerol. One of the major ligand classes, fatty acid amides (FAAs), are composed of a fatty acyl chain linked by an amide bond to a polar head group. Slight modifications to the acyl chain length, saturation, or head group can profoundly affect their pharmacological profile.3 Coupled with heterogeneous cannabinoid receptor expression at both the tissue and subcellular levels, the mechanisms by which FAAs affect physiology remain poorly understood.Endocannabinoid signalling modulates many metabolic processes in the periphery, including insulin secretion from pancreatic β-cells, which controls blood glucose levels. Glucose-stimulated insulin secretion (GSIS) is initiated by increased glucose uptake and glycolysis, which elevates ATP levels, affecting K+ and Ca2+ channels to drive oscillations in the intracellular Ca2+ concentration ([Ca2+]i).4 The frequency and amplitude of these oscillations correlate tightly to insulin secretion.5 Notably, β-cells express multiple cannabinoid-sensitive receptors and channels that affect β-cell excitability and GSIS,6,7 including CB1, CB2, transient receptor potential cation channel subfamily V member 1 (TRPV1),8 peroxisome proliferator-activated receptors,9 and the atypical cannabinoid receptor GPR55,6,10,11 which is a stimulatory GPCR. In contrast to the inhibitory CB1 and CB2 receptors, GPR55 activation increases [Ca2+]ivia Gαq/11 and phospholipase C (PLC).12 GPR55 is known to enhance GSIS in β-cells,6 making it a potential therapeutic target for diabetes. Yet, our knowledge of GPR55's precise function is hindered by a lack of tools that enable its targeted manipulation using endogenous ligands with spatiotemporal precision.
Advances in chemical biology have provided a collection of tools to interface with endocannabinoid signalling, including ligands that are activated on an optical stimulus.13–15 As light can be applied with unmatched spatiotemporal precision, optical tools like photocaged endocannabinoids have illuminated the function of CB1 and CB2 receptors in excitable cells.16,17 These probes contain a photolabile protecting group (cage) which masks the endocannabinoids' activity until exposed to irradiation, triggering the release of endogenous ligands within milliseconds. However, a limitation of this approach is that the cage's structure impacts the probe's subcellular localization before irradiation, making the signal difficult to control within the cell. Although this spatial restriction has been exploited to release ligands on specific organelles by modifying the cage's charge or aromaticity,18,19 this approach is quite restricted in its application. Adding an element of genetic control, such as a genetically-encoded bioorthogonal protein-tag (SNAP-tag), would allow us to direct caged signalling molecules toward any membrane or protein of interest. While (photo)cleavable substrates for protein tags have been utilized to turn signalling off,20–22 or release reactive moieties for profiling protein–protein interactions,23 applying genetic targeting to activate a GPCR using a photocaged endogenous ligand has yet to be reported.
To this end, this work introduces a novel chemigenetic technique to place cannabinoid signalling under optical control. Coined Optically-Cleavable Targeted (OCT)-ligands, our approach tethers photocaged FAAs to genetically-encoded SNAP-tags, permitting the photo-release of endocannabinoid ligands on genetically-defined subcellular membranes. We introduce an OCT-ligand capable of targeting GPR55 to investigate the downstream mechanisms by which this atypical cannabinoid receptor affects β-cell excitability. More broadly, this work serves as a proof-of-principle for a platform that will advance our understanding of endocannabinoid signalling with subcellular resolution.
Results and discussion
Design and synthesis of an OCT-ligand for GPR55
We designed Optically-Cleavable Targeted (OCT-) ligands to combine a photocaged FAA with a biorthogonal handle, which can be recognized by genetically-encoded protein tags (Fig. 1a). Membrane-anchored SNAP-tags can be expressed on specific subcellular compartments, allowing us to spatially enrich the photocaged (inactive) FAA at a genetically-defined target.24,25 The tethered probe can then be quickly uncaged (activated) by a flash of light, generating bursts of the active molecule at the site of SNAP-tag expression. OCT-ligands are small molecules composed of four units: the FAA ligand (purple), photocage (orange), linker (black), and bioconjugation motif (blue) (Fig. 1b). One of the endogenous ligands for GPR55 is palmitoylethanolamide (PEA), a FAA composed of an ethanolamine head group attached to a palmitate (16:0) lipid chain (Fig. 1b, bottom).26,27 PEA activates GPR55 selectively over CB1 and CB2, and potentiates β-cell excitability and GSIS both in vitro and in vivo.28,29 The activity of OCT-PEA is masked by an ortho-nitrobenzyl photocage,16 and the distal end of the cage is linked to an O6-(4-aminomethyl-benzyl)guanine (BG) motif for covalent tethering to SNAP-tags.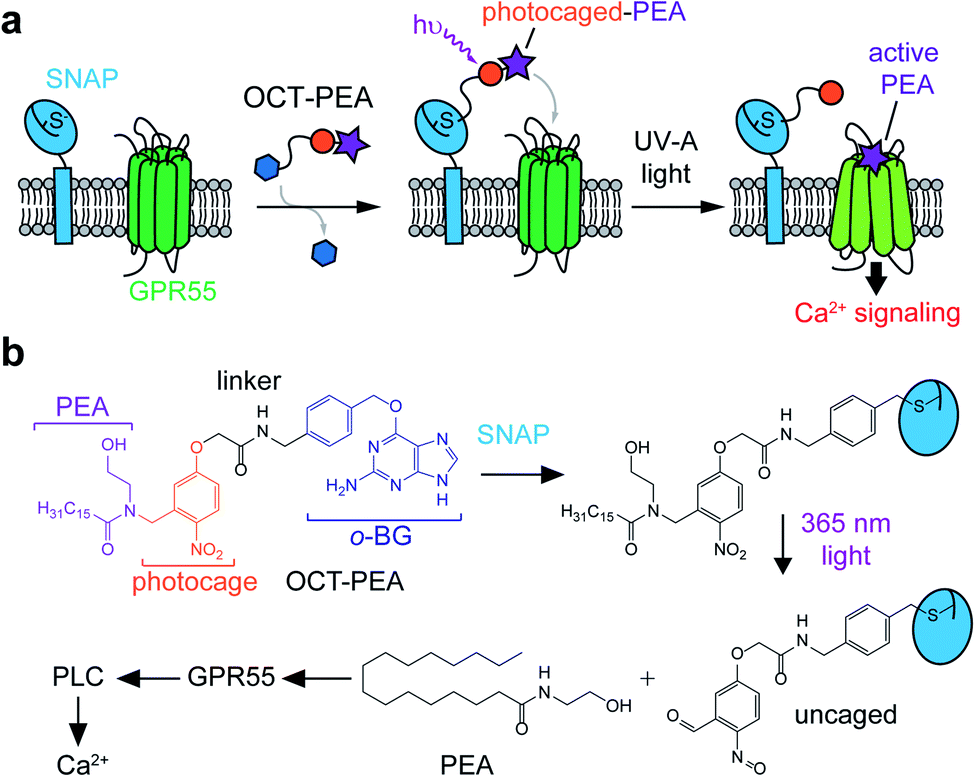 | ||
| Fig. 1 OCT-ligands allow genetic targeting of photocaged FAAs. (a) Schematic depiction of the OCT-ligand approach. Photocaged FAAs can be spatially enriched at the site of SNAP-tag expression and then released on demand to activate nearby GPR55 receptors and their downstream effector pathways. (b) Molecular schematic of OCT-PEA tethering and photolysis. OCT-PEA can be tethered to SNAP-tags and uncaged with UV-A (365 nm) irradiation, releasing PEA to activate GPR55. This stimulation increases intracellular Ca2+ levels via Gαq/11 and PLC. | ||
The synthesis of OCT-PEA (Fig. 2a) commenced with the reaction of 5-hydroxy-2-nitrobenzaldehyde and ethyl bromoacetate to form the phenolic ether 1. A reductive amination with 2-(t-butyloxy)-ethanamine formed the amine 2, and then acylation with palmitoyl chloride led to the tertiary amide 3. Ester hydrolysis under basic conditions afforded carboxylic acid 4, which was then coupled to 6-((4-(aminomethyl)benzyl)oxy)-9H-purin-2-amine (BG-NH2)24 to produce 5. Finally, tBu-ether deprotection using BBr3 at −78 °C afforded OCT-PEA in six steps and 26% overall yield. We confirmed that OCT-PEA could be uncaged with 365 nm LED irradiation (∼35 mW cm−2) using HPLC-MS, to release PEA and the nitroso-benzaldehyde (Fig. S1†). UV-VIS spectroscopy was used to further characterize the photochemical properties of OCT-PEA (in DMSO), which possessed a λmax at 287 nm and an extinction coefficient of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 345 (mol−1 cm−1) (Fig. 2b). 365 nm irradiation (∼35 mW cm−2) resulted in a bathochromic wavelength shift to 354 nm (Fig. 2b), consistent with the expanded electron π-network present in the uncaged product (Fig. 1b, bottom right). Longer wavelengths—such as 415 nm (Fig. 2c, magenta, ∼31 mW cm−2), 470 nm (Fig. 2c, blue, ∼35 mW cm−2) and 565 nm (Fig. 2c, green, ∼31 mW cm−2)—did not uncage OCT-PEA as efficiently (Fig. 2c, S2†). This feature is advantageous for applications in fluorescence microscopy, since the ligand will not be released during imaging of blue and green fluorescent reporters. We also conjugated OCT-PEA to purified SNAP-tags in vitro, which allowed the probe to be uncaged in aqueous buffer. In this case, 365 nm irradiation induced uncaging with a τ ∼ 46 s (Fig. S3†), demonstrating OCT-PEA's ability to be released from SNAP-tags in a physiological environment.
345 (mol−1 cm−1) (Fig. 2b). 365 nm irradiation (∼35 mW cm−2) resulted in a bathochromic wavelength shift to 354 nm (Fig. 2b), consistent with the expanded electron π-network present in the uncaged product (Fig. 1b, bottom right). Longer wavelengths—such as 415 nm (Fig. 2c, magenta, ∼31 mW cm−2), 470 nm (Fig. 2c, blue, ∼35 mW cm−2) and 565 nm (Fig. 2c, green, ∼31 mW cm−2)—did not uncage OCT-PEA as efficiently (Fig. 2c, S2†). This feature is advantageous for applications in fluorescence microscopy, since the ligand will not be released during imaging of blue and green fluorescent reporters. We also conjugated OCT-PEA to purified SNAP-tags in vitro, which allowed the probe to be uncaged in aqueous buffer. In this case, 365 nm irradiation induced uncaging with a τ ∼ 46 s (Fig. S3†), demonstrating OCT-PEA's ability to be released from SNAP-tags in a physiological environment.
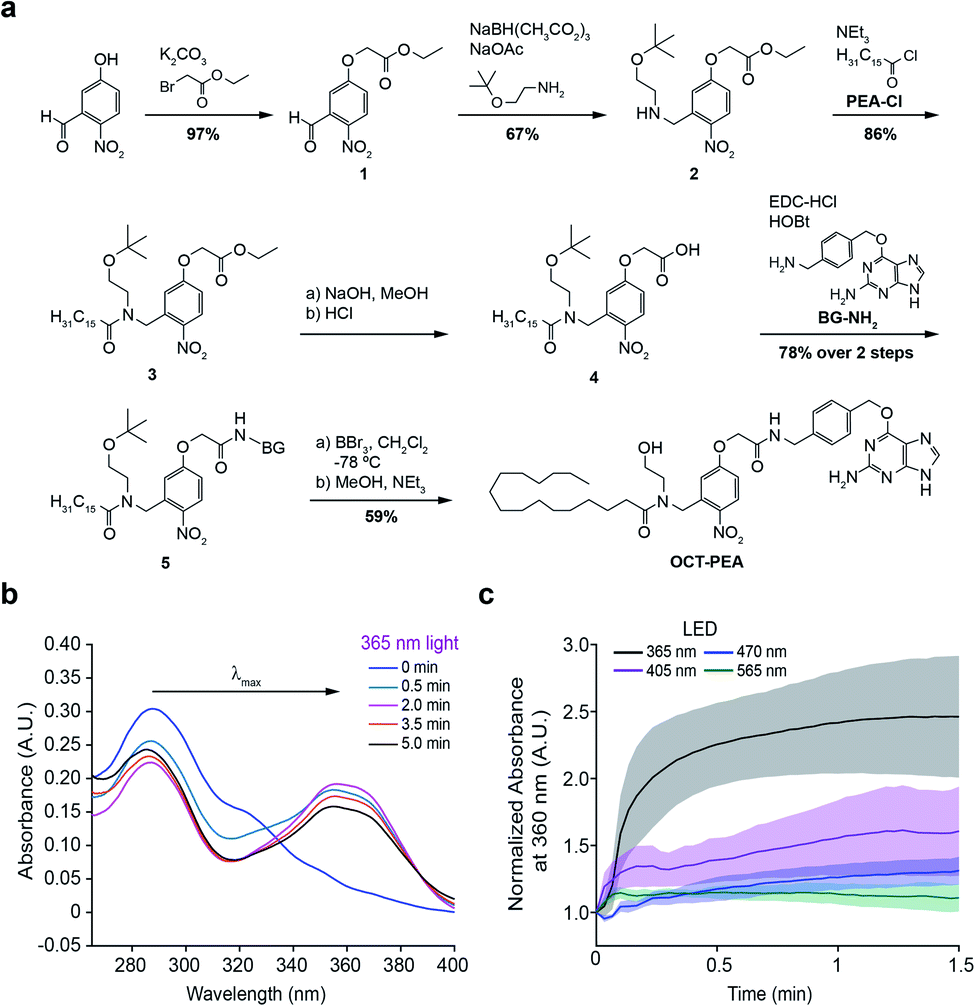 | ||
| Fig. 2 Synthesis and characterization of OCT-PEA. (a) Chemical synthesis of OCT-PEA. (b) UV-VIS absorbance scan showing a time-course of OCT-PEA (20 μM in DMSO) uncaging with 365 nm LED irradiation. A rightward (bathochromic) shift in the main absorbance peak (λmax) was observed as illumination generated the uncaged species. (c) Absorbance at 360 nm over time of OCT-PEA (20 μM in DMSO) uncaging with 365 nm LED (black), 405 nm (magenta), 470 nm (blue), and 565 nm (green) LEDs. N = 3 samples for each. Shaded error bars = mean ± SEM. | ||
GPR55 and PEA mediate Ca2+ signalling in INS-1 β-cells
We used the rat insulinoma INS-1 cell line to evaluate the effect of OCT-PEA on β-cell excitability.30,31 Although other β-cell lines (MIN6, BRIN-D11) and primary islets (human, mouse, rat) are known to express GPR55,6,10,32 its expression and function in INS-1 cells remain uncharacterized.8,33 Therefore, we used immunofluorescence microscopy to confirm the expression and localization of GPR55. INS-1 cells were co-stained with anti-GPR55 and anti-insulin antibodies (Fig. 3a and S4a†). GPR55 immunoreactivity was observed on the INS-1 plasma membrane, while insulin was observed throughout the cell and enriched near the perimeter (Fig. 3b). In control experiments, removal of the primary antibody abolished GPR55 immunofluorescence, ruling out the possibility of nonspecific secondary antibody binding (Fig. S4b†). | ||
| Fig. 3 INS-1 cells express GPR55 and respond to PEA. (a) Immunofluorescence images of fixed INS-1 cells stained for GPR55 (green) and insulin (red). Nuclei were labelled with DAPI (blue). Scale bar = 10 μm. (b) Fluorescence intensity profile plotted across the yellow arrow shown in panel A. (c) Fluorescent Ca2+ imaging using R-GECO showed that PEA addition (5 μM) increased [Ca2+]i. Displayed as Ca2+ traces from five representative cells. (d) Average [Ca2+]i traces for PEA addition under standard conditions (black, 5 μM, N = 612, T = 10), overlaid with average [Ca2+]i traces for PEA addition following pre-incubation with a GPR55 antagonist (CID16020046, 5 μM, blue, N = 449, T = 6) or PLC inhibitor (U73122, 5 μM, red, N = 296, T = 4), which blocked PEA's effect. UV-A irradiation (375 nm) shown as purple bars, and KCl (25 mM) was applied at the end of each experiment. (e) Bar graph displaying the fold change in Ca2+ oscillation frequency induced by compound stimulation. Vehicle addition (DMSO, 0.1% v/v, grey, N = 418, T = 6) did not stimulate [Ca2+]i. The inactive PLC inhibitor analogue (U73343, 5 μM, green, N = 511, T = 7) did not block the effect of PEA. Error bars = mean ± SEM. **P < 0.01, *P < 0.05, ns = P > 0.05, P values reported in Table S2.† | ||
To determine the effect of freely diffusible PEA application on the β-cell [Ca2+]i, INS-1 cells were transfected with the fluorescent biosensor R-GECO,34 which allows for visualization of [Ca2+]i in real-time. Under high glucose conditions (20 mM), we observed characteristic oscillations in [Ca2+]i (Fig. 3c and d). Similar to other β-cell lines,10 PEA increased both the overall [Ca2+]i level (Fig. 3d) and oscillation frequency (Fig. 3e, black) in a dose-dependent manner (Fig. S5†). Pretreatment with either the GPR55 antagonist CID16020046 (Fig. 3d and e, blue and S6a†) or PLC inhibitor U73122 (Fig. 3d and e, red and S6b†) abolished PEA's action.35,36 In control experiments, the effect of PEA remained in the presence of an inactive PLC inhibitor analogue (U73343) (Fig. 3e, green, and S6c and d†).36 Ca2+ levels were not sensitive to 375 nm irradiation alone, nor vehicle addition (Fig. 3e, and S6e and f†). At lower glucose concentrations (11 mM and 3 mM), PEA's effect on [Ca2+]i was reduced (Fig. S7†). Combined with the results above, our experiments confirm that GPR55 is expressed on the surface of INS-1 β-cells and that it responds to PEA to mediate glucose-stimulated Ca2+ oscillations via PLC.
Expression and labelling of cell surface SNAP-tags in β-cells
To localize OCT-PEA nearby GPR55 receptors, we transfected INS-1 cells with a plasmid encoding pDisplay™-SNAP, a plasma membrane-anchored SNAP-tag. We then performed a competition labelling assay between OCT-PEA and a non-permeable SNAP-Surface® Alexa Fluor® 488 (A488) dye to optimize the conditions for OCT-PEA tethering. Transfected cells were labelled with either a vehicle or increasing concentrations and duration of OCT-PEA. They were then thoroughly washed and exposed to A488 to detect any un-reacted SNAP-tags. When preceded with the vehicle, A488 fluorescence was observed on the perimeter of transfected cells (Fig. 4a, left). Pretreatment with OCT-PEA blocked A488 labelling in a dose- and time-dependent manner (Fig. S8†). SNAP-tag labelling was abolished most consistently when cells were labelled with 5 μM OCT-PEA for 2 h (Fig. 4a, right). To assess OCT-PEA's ability to label intracellular SNAP-tags, a similar competition labelling was performed using the above-optimized labelling conditions, except using the cell-permeable dye, SNAP-Cell® Oregon Green®. When pre-treated with vehicle, we observed Oregon Green® fluorescence on intracellular compartments, presumably representing pDisplay™-SNAP in the secretory pathway (Fig. S9a†). When pretreated with OCT-PEA (5 μM, 2 h), dye labelling of intracellular SNAP-tags was still observed (Fig. S9b†). This indicates that OCT-PEA is not cell permeable under these conditions, and primarily labels surface-expressed SNAP-tags. To assess the extent of SNAP-tag internalization during this labelling time, pDisplay™-SNAP-expressing cells were incubated with the non-permeable A488 for 2 h (Fig. S10a†). Although most of the fluorescence remained enriched at the plasma membrane, small puncta appeared inside the cells slowly over time (Fig. S10b†). This finding confirms that the labelled SNAP-tags remain enriched on the plasma membrane, with a fraction of internalized SNAP-tags present. | ||
| Fig. 4 Targeted uncaging of OCT-PEA on the INS-1 cell surface. (a) Pre-incubation with DMSO (0.1% v/v, 2 h) maintains surface SNAP-tag labelling (A488, green, left), whereas pre-incubation with OCT-PEA (5 μM, 2 h) abolished dye labelling (right). Nuclei stained with Hoechst-33342 (blue). Scale bar = 10 μm. (b) Fluorescent Ca2+ imaging using R-GECO showed that OCT-PEA (5 μM, 2 h, black, N = 352, T = 8) increased the average [Ca2+]i in INS-1 cells. Overlaid with averages in the presence of a GPR55 antagonist (CID16020046, 5 μM, blue, N = 212, T = 4) or PLC inhibitor (U73122, 5 μM, red, N = 173, T = 4), which reduced the effect of OCT-PEA. UV-A irradiation (375 nm) shown as purple bars, and KCl (25 mM) was applied at the end of each experiment. (c) Heat map showing individual Ca2+ traces from fifty representative cells which were pre-incubated with OCT-PEA (5 μM, 2 h). Cells normalized to the KCl response. (d) Comparison bar graph of fold change in oscillation frequency in response to OCT-PEA uncaging across different conditions. Uncaging in the presence of GPR55 antagonist (CID16020046, 5 μM, purple, N = 212, T = 4), PLC inhibitor (U73122, 5 μM, blue, N = 173, T = 4), SNAP-Surface® Block (20 μM, yellow, N = 278, T = 5), SNAP-Cell® Block (10 μM, orange, N = 248, T = 4), pDisplay™-SNAPC145A (red, N = 244, T = 5) or pDisplay™-HALO (magenta, N = 330, T = 4) reduced the probe's effect on oscillation frequency. The inactive PLC inhibitor analogue (U73343, 5 μM, green, N = 357, T = 6) did not block the effect of OCT-PEA. Vehicle treatment (DMSO, 0.1% v/v, grey, N = 247, T = 4) did not change the oscillation frequency in response to UV-A irradiation. (e and f) Uncaging OCT-PEA in INS-1 cells transfected with pDisplay™-SNAPC145A reduced the effect of the probe, displayed as (e) average trace of all cells and (f) heat map of representative traces from fifty cells, normalized to the KCl response. (g–i) Targeted uncaging on half of the field of view, displayed as (g) uncaging ROI, (h) bar graph comparing fold change in oscillation frequency between irradiated (N = 145, T = 5) and nonirradiated regions (N = 132, T = 5), and (i) heat map of representative traces from one trial, normalized to KCl response. Error bars = mean ± SEM. *P < 0.05, **P < 0.01, ns = P > 0.05, P values reported in Table S3.† | ||
To evaluate the cytotoxicity of our probe, INS-1 cells were incubated with caged or uncaged OCT-PEA for 24 h and then subjected to a cell viability assay. We did not observe any effect on cell viability induced by the caged or uncaged OCT-PEA up to its solubility limit in the physiological buffer (Fig. S11†). PEA also did not affect cell viability,10,37 confirming that our probes are not cytotoxic to INS-1 cells.
Plasma membrane OCT-PEA uncaging mediates INS-1 Ca2+ levels via GPR55
We evaluated how uncaging OCT-PEA on the cell surface influenced [Ca2+]i dynamics in β-cells under high-glucose conditions (20 mM). INS-1 cells expressing R-GECO and pDisplay™-SNAP were labelled with OCT-PEA, followed by a washing step to remove any untethered probe. In contrast to the large Ca2+ spike observed from bath-applied PEA, OCT-PEA uncaging on the cell surface caused a more subtle increase in [Ca2+]i (Fig. 4b, black). However, we observed that OCT-PEA stimulation strongly increased the Ca2+ oscillation frequency (Fig. 4c and d). Following probe labelling, treatment with the GPR55 antagonist CID16020046 or PLC inhibitor U73122 blocked the effect of OCT-PEA uncaging (Fig. 4b and d and S12a and b†), confirming the involvement of GPR55 and PLC. In control experiments, the inactive PLC inhibitor analogue (U73343) did not significantly block the effect of OCT-PEA uncaging (Fig. 4d and S12c and d†). GPR55 knockdown using two independent siRNAs blocked the effect of OCT-PEA uncaging on oscillation frequency, further supporting GPR55 involvement for mediating the probe's response (Fig. S13†). INS-1 cells that were exposed to a vehicle did not respond to 375 nm irradiation (Fig. 4d and S12e and f†).To confirm that covalent attachment of OCT-PEA to the SNAP-tags on the cell surface is necessary for our probe's mechanism of action, we applied OCT-PEA under conditions in which the tethering reaction could not occur. First, cells were pretreated with SNAP-Cell® Block to prevent subsequent OCT-PEA labelling. This pharmacological approach greatly diminished the impact of OCT-PEA uncaging on [Ca2+]i (Fig. 4d, S14a and b†). We performed similar experiments with SNAP-Surface® Block, which is non-permeable and only blocks SNAP-tags on the cell surface, and not intracellular pools. Again, this blocked the effect of OCT-PEA uncaging, confirming that cell-surface uncaging is the main driver of our observed effect (Fig. 4d and S14c and d†). Alternatively, we transfected INS-1 cells with pDisplay™-HALO, which does not react with the BG moiety on OCT-PEA. This biorthogonal tag exchange also reduced OCT-PEA's effect (Fig. 4d and S13e and f†). Finally, we performed site-directed mutagenesis to our standard pDisplay™-SNAP construct to remove the reactive cysteine. Using immunohistochemistry, we confirmed that this C145A mutation prevents it from reacting with a BG, but did not affect SNAP-tag trafficking or localization (Fig. S15†). Consistent with our other control experiments, this mutation blocked the effect of OCT-PEA on INS-1 [Ca2+]i (Fig. 4d–f). Combined, these results unambiguously confirm that background activity from untethered OCT-PEA was not driving our observed response to uncaging, and that SNAP-tags effectively deliver OCT-PEA to the INS-1 cell surface. Targeted compound uncaging activates GPR55 receptors and PLC near the plasma membrane to stimulate β-cell Ca2+ activity.
Finally, we sought to demonstrate the spatial utility of our OCT-ligand approach. SNAP-tag expressing INS-1 cells were labelled with OCT-PEA, and this time only half the field of view was exposed to 375 nm irradiation. As expected, we observed that only the irradiated cells increased in their Ca2+ oscillation frequency, while those that were not irradiated remained at a constant oscillation frequency (Fig. 4g–i). This provides a mechanism for researchers to directly compare cells in which GPR55 was activated to those which were not, under the exact same experimental conditions.
Conclusion
This study presents a new chemical technology for manipulating cannabinoid signalling on a genetically-defined cellular target. We introduce OCT-PEA, the first photochemical tool that has been shown to target GPR55 in living cells. A photocaged PEA was tethered to the plasma membrane of INS-1 β-cells using membrane-anchored SNAP-tags, and uncaging OCT-PEA on the cell surface stimulated [Ca2+]i oscillations. These results confirm that INS-1 cells express functional GPR55 on their plasma membrane, which is activated by PEA to increase β-cell excitability through PLC activation. Similar to other approaches which utilize membrane-anchored SNAP-tags to localize pharmacologically-active ligands,38,39 this approach does not require the overexpression of signalling proteins and enables the targeting of endogenous receptors. Because SNAP-tags are inactive until conjugated with a pharmacophore, OCT-ligands allow us to acutely manipulate receptor function while avoiding compensatory artefacts. Because OCT-PEA is only uncaged with UV-A irradiation, we can combine OCT-PEA with other blue/green fluorescent biosensors (i.e. Ca2+, cAMP, kinase activity). Reflected by the more subtle effect of OCT-PEA stimulation on the overall Ca2+ level when compared to the application of freely diffusible PEA, we hypothesize that our approach releases PEA similar to how endogenous FAAs are typically generated transiently during periods of cell stimulation.40Because we observed that GPR55 is expressed on the INS-1 plasma membrane, OCT-PEA was targeted to the cell surface. While similar results could also be achieved using other specialized photocages,18,19 future applications of our approach with alternative SNAP-tag targeting sequences will allow us to direct FAA signalling to a variety of subcellular locations without requiring the synthesis of new compounds. Moreover, an increasing body of work indicates that CBRs can be found on internal membranes, even in β-cells.7,41 Thus, applying OCT-ligands to target organelles such as mitochondria or the endoplasmic reticulum will allow us to investigate the function of intracellular receptor pools. Our probes will also be useful to study cannabinoid signalling in intact tissues. Leveraging Cre-recombination technology to facilitate cell-type selective SNAP-tag expression, OCT-ligands will allow us to focus ligand release on β- or α-cells in the intact pancreatic islet.
Our future efforts will aim to develop OCT-ligands with alternate pharmacological or photophysical properties. For example, probes capable of releasing different FAAs will help us dissect the contributions of specific CBR subtypes. Red-shifting the photocage toward longer visible wavelengths will also help translate our strategy in vivo, as lower-energy photons are less phototoxic and penetrate deeper into tissue. Combined with the expansion of our approach to utilize alternative bio-conjugation tags (i.e. HALO-, CLIP-tags),42 OCT-ligands will allow us to generate sophisticated patterns of FAA signalling orthogonally, with unprecedented precision.
Beyond applying OCT-ligands to study CBR signalling in β-cells, this approach will be generalizable to other cell types, ligands, and signalling pathways. These advances will enhance the acuity with which we can manipulate signalling in time and space, setting the stage for the next generation of targeted pharmacology.
Data availability
All MATLAB scrips were written in house and are available upon request. Please see the ESI† for more information.Author contributions
J. A. F. conceived and coordinated the study. J. M. T. synthesized and characterized the compounds. J. M. T., G. R., and A. E. G. V. carried out imaging experiments in cultured cells. A. E. G. V. carried out the cytotoxicity assay. J. T., J. F., and D. I. A. performed UV-VIS and HPLC-MS experiments. J. M. T. and J. A. F. wrote the manuscript with contributions from all other co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Carsten Schultz and Dr Marc Freeman for providing microscopy resources, and Dr Philipp Leippe for helpful discussions. We also thank Dr Show-Ling Shyng for providing the INS-1 cells and input on this study. This work was funded in part by the Medical Research Foundation of Oregon (MRF) New Investigator grant (to J. A. F.), the Vollum Institute Fellowship (to J. A. F.), as well as the NIH Ruth L. Kirschstein T32 Program in Enhanced Research Training (to J. M. T.), and National Science Foundation Graduate Research Fellowship under Grant No. 1937961 (to J. M. T.).Notes and references
- R. G. Pertwee, Pharmacol. Ther., 1997, 74, 129–180 CAS.
- L. Cristino, T. Bisogno and V. Di Marzo, Nat. Rev. Neurol., 2020, 16, 9–29 CrossRef PubMed.
- E. K. Farrell and D. J. Merkler, Drug Discovery Today, 2008, 13, 558–568 CrossRef CAS PubMed.
- M. S. Islam, Adv. Exp. Med. Biol., 2020, 1131, 943–963 CrossRef CAS PubMed.
- J. C. Henquin, Diabetologia, 2009, 52, 739–751 CrossRef CAS PubMed.
- S. Y. Romero-Zerbo, A. Rafacho, A. Díaz-Arteaga, J. Suárez, I. Quesada, M. Imbernon, R. A. Ross, C. Dieguez, F. Rodríguez de Fonseca, R. Nogueiras, Á. Nadal and F. J. Bermúdez-Silva, J. Endocrinol., 2011, 211, 177–185 CAS.
- A. Laguerre, K. Keutler, S. Hauke and C. Schultz, Cell Chem. Biol., 2021, 28, 88–96 CrossRef CAS PubMed.
- K. Malenczyk, M. Jazurek, E. Keimpema, C. Silvestri, J. Janikiewicz, K. Mackie, V. Di Marzo, M. J. Redowicz, T. Harkany and A. Dobrzyn, J. Biol. Chem., 2013, 288, 32685–32699 CrossRef CAS PubMed.
- Y. T. Zhou, M. Shimabukuro, M. Y. Wang, Y. Lee, M. Higa, J. L. Milburn, C. B. Newgard and R. H. Unger, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8898–8903 CrossRef CAS PubMed.
- A. M. McKillop, B. M. Moran, Y. H. A. Abdel-Wahab and P. R. Flatt, Br. J. Pharmacol., 2013, 170, 978–990 CrossRef CAS PubMed.
- A. G. McCloskey, M. G. Miskelly, C. B. T. Moore, M. A. Nesbit, K. A. Christie, A. I. Owolabi, P. R. Flatt and A. M. McKillop, Peptides, 2020, 125, 170251 CrossRef CAS PubMed.
- J. E. Lauckner, J. B. Jensen, H. Y. Chen, H. C. Lu, B. Hille and K. Mackie, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2699–2704 CrossRef CAS PubMed.
- M. V. Westphal, M. A. Schafroth, R. C. Sarott, M. A. Imhof, C. P. Bold, P. Leippe, A. Dhopeshwarkar, J. M. Grandner, V. Katritch, K. Mackie, D. Trauner, E. M. Carreira and J. A. Frank, J. Am. Chem. Soc., 2017, 139, 18206–18212 CrossRef CAS PubMed.
- R. C. Sarott, A. E. G. Viray, P. Pfaff, A. Sadybekov, G. Rajic, V. Katritch, E. M. Carreira and J. A. Frank, J. Am. Chem. Soc., 2021, 143, 736–743 CrossRef CAS PubMed.
- D. Dolles, A. Strasser, H.-J. Wittmann, O. Marinelli, M. Nabissi, R. G. Pertwee and M. Decker, Adv. Ther., 2018, 1, 1700032 CrossRef.
- T. Heinbockel, D. Brager, C. Reich, J. Zhao, S. Muralidharan, B. Alger and J. Kao, J. Neurosci., 2005, 25, 9449–9459 CrossRef CAS PubMed.
- A. Laguerre, S. Hauke, J. Qiu, M. J. Kelly and C. Schultz, J. Am. Chem. Soc., 2019, 141, 16544–16547 CrossRef CAS PubMed.
- A. Nadler, D. A. Yushchenko, R. Muller, F. Stein, S. Feng, C. Mulle, M. Carta and C. Schultz, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS PubMed.
- T. Podewin, J. Ast, J. Broichhagen, N. H. F. Fine, D. Nasteska, P. Leippe, M. Gailer, T. Buenaventura, N. Kanda, B. J. Jones, C. M'Kadmi, J. L. Baneres, J. Marie, A. Tomas, D. Trauner, A. Hoffmann-Röder and D. J. Hodson, ACS Cent. Sci., 2018, 4, 166–179 CrossRef CAS PubMed.
- D. Ollech, T. Pflästerer, A. Shellard, C. Zambarda, J. P. Spatz, P. Marcq, R. Mayor, R. Wombacher and E. A. Cavalcanti-Adam, Nat. Commun., 2020, 11, 1–13 Search PubMed.
- Z. Fang, S. Chen, P. Pickford, J. Broichhagen, D. J. Hodson, I. R. Corrê, S. Kumar, F. Gö Rlitz, C. Dunsby, P. M. W. French, G. A. Rutter, T. Tan, S. R. Bloom, A. Tomas and B. Jones, ACS Pharmacol. Transl. Sci., 2020, 2020, 360 Search PubMed.
- D. C. McCutcheon, G. Lee, A. Carlos, J. E. Montgomery and R. E. Moellering, J. Am. Chem. Soc., 2020, 142, 146–153 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- E. Prifti, L. Reymond, M. Umebayashi, R. Hovius, H. Riezman and K. Johnsson, ACS Chem. Biol., 2014, 9, 606–612 CrossRef CAS PubMed.
- F. A. J. Kuehl, T. A. Jacob, O. H. Ganley, R. E. Ormond and M. A. P. Meisinger, J. Am. Chem. Soc., 1957, 79, 5577–5578 CrossRef CAS.
- L. Rankin and C. J. Fowler, Int. J. Mol. Sci., 2020, 21, 7942 CrossRef CAS PubMed.
- G. Donvito, I. Bettoni, F. Comelli, A. Colombo and B. Costa, CNS Neurol. Disord.–Drug Targets, 2015, 14, 452–462 CrossRef CAS PubMed.
- E. Ryberg, N. Larsson, S. Sjögren, S. Hjorth, N. O. Hermansson, J. Leonova, T. Elebring, K. Nilsson, T. Drmota and P. Greasley, Br. J. Pharmacol., 2007, 152, 1092–1101 CrossRef CAS PubMed.
- M. Asfari, D. Janjic, P. Meda, G. Li, P. A. Halban and C. B. Wollheim, Endocrinology, 1992, 130, 167–178 CrossRef CAS PubMed.
- H. E. Hohmeier, H. Mulder, G. Chen, R. Henkel-Rieger, M. Prentki and C. B. Newgard, Diabetes, 2000, 49, 424–430 CrossRef CAS PubMed.
- B. Liu, S. Song, I. Ruz-Maldonado, A. Pingitore, G. C. Huang, D. Baker, P. M. Jones and S. J. Persaud, Diabetes, Obes. Metab., 2016, 18, 1263–1273 CrossRef CAS PubMed.
- Y. J. Cho, S. H. Choi, R. Lee, H. Hwang, H. Rhim, I. H. Cho, H. C. Kim, J. I. Lee, S. H. Hwang and S. Y. Nah, Molecules, 2020, 25, 1102 CrossRef CAS PubMed.
- Y. Zhao, S. Araki, J. Wu, T. Teramoto, Y. F. Chang, M. Nakano, A. S. Abdelfattah, M. Fujiwara, T. Ishihara, T. Nagai and R. E. Campbell, Science, 2011, 333, 1888–1891 CrossRef CAS PubMed.
- J. Kargl, A. J. Brown, L. Andersen, G. Dorn, R. Schicho, M. Waldhoer and A. Heinemann, J. Pharmacol. Exp. Ther., 2013, 346, 54–66 CrossRef CAS PubMed.
- J. E. Bleasdale, N. R. Thakur, R. S. Gremban, G. L. Bundy, F. A. Fitzpatrick, R. J. Smith and S. Bunting, J. Pharmacol. Exp. Ther., 1990, 255, 756–768 CAS.
- E. R. Nestmann, Food Sci. Nutr., 2017, 5, 292–309 CrossRef CAS PubMed.
- P. C. Donthamsetti, J. Broichhagen, V. Vyklicky, C. Stanley, Z. Fu, M. Visel, J. L. Levitz, J. A. Javitch, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2019, 141, 11522–11530 CrossRef CAS PubMed.
- B. C. Shields, E. Kahuno, C. Kim, P. F. Apostolides, J. Brown, S. Lindo, B. D. Mensh, J. T. Dudman, L. D. Lavis and M. R. Tadross, Science, 2017, 356, 42 CrossRef CAS PubMed.
- V. Di Marzo, Pharmacol. Res., 2009, 60, 77–84 CrossRef CAS PubMed.
- G. Bénard, F. Massa, N. Puente, J. Lourenço, L. Bellocchio, E. Soria-Gómez, I. Matias, A. Delamarre, M. Metna-Laurent, A. Cannich, E. Hebert-Chatelain, C. Mulle, S. Ortega-Gutiérrez, M. Martín-Fontecha, M. Klugmann, S. Guggenhuber, B. Lutz, J. Gertsch, F. Chaouloff, M. Luz López-Rodríguez, P. Grandes, R. Rossignol and G. Marsicano, Nat. Neurosci., 2012, 15, 558–564 CrossRef PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures, tables, methods, and detailed synthetic methods. See DOI: 10.1039/d1sc02527a |
| This journal is © The Royal Society of Chemistry 2021 |