
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-metal with metal behavior: metal-free coordination-insertion ring-opening polymerization†
Xin
Wang
 ac,
Jiaxi
Xu
ac,
Zhenjiang
Li
a,
Jingjing
Liu
ac,
Jie
Sun
b,
Nikos
Hadjichristidis
*c and
Kai
Guo
*a
ac,
Jiaxi
Xu
ac,
Zhenjiang
Li
a,
Jingjing
Liu
ac,
Jie
Sun
b,
Nikos
Hadjichristidis
*c and
Kai
Guo
*a
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, China. E-mail: guok@njtech.edu.cn
bCollege of Food Science and Light Industry, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, China
cPhysical Sciences and Engineering Division, KAUST Catalysis Center, Polymer Synthesis Laboratory, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: nikolaos.hadjichristidis@kaust.edu.sa
First published on 15th June 2021
Abstract
The “coordination-insertion” ring-opening polymerization (ROP) mechanism has so far been the monopoly of metal catalysts. In this work, we present a metal-free “coordination-insertion” ROP of trimethylene carbonate (TMC) and ε-caprolactone (ε-CL), as well as their sequential block copolymerization, with N-trimethylsilyl-bis (trifluoromethanesulfonyl)imide (TMSNTf2) as the non-metallic initiator/catalyst. TMSNTf2 was proposed to work through an unprecedented metal-free “coordination-insertion” mechanism, which involves the coordination of monomer to the Si atom of TMSNTf2, the nucleophilic attack of the –NTf2 group on the coordinated monomer, and the cleavage of the acyl–oxygen bond of the monomer. The proposed metal-free “coordination-insertion” ROP was studied by NMR, SEC, and MALDI-TOF analyses. In addition, the TMSNTf2-mediated ROP of TMC and ε-CL led to linear and cyclic polymers following two-stage first-order polymerization processes, as evidenced by structural analyses and kinetics study, which further demonstrated the metal-free “coordination-insertion” mechanism.
Introduction
A primary axiom of the definition of Lewis acid–base is the notion that the combination of a simple Lewis acid and Lewis base results in the neutralization reaction to form “classical” Lewis acid/Lewis base adducts.1 An exception is the transition metal–ligand complex,2 which is a single molecule that contains both Lewis acid and Lewis base activation sites and can simultaneously carry out both electrophilic and nucleophilic activation in chemical reactions (Scheme 1). The most representative example is the transition metal alkoxides,2d,3 which possess metal Lewis acid centers with vacant orbitals and Lewis base ligands with lone pair electrons. The metal alkoxides can initiate/catalyze the ring-opening polymerization (ROP) of cyclic esters through a “coordination-insertion” mechanism, which has been extensively used in the aliphatic polyesters industrial production. However, so far, the metal-free “coordination-insertion” ROP has not been reported.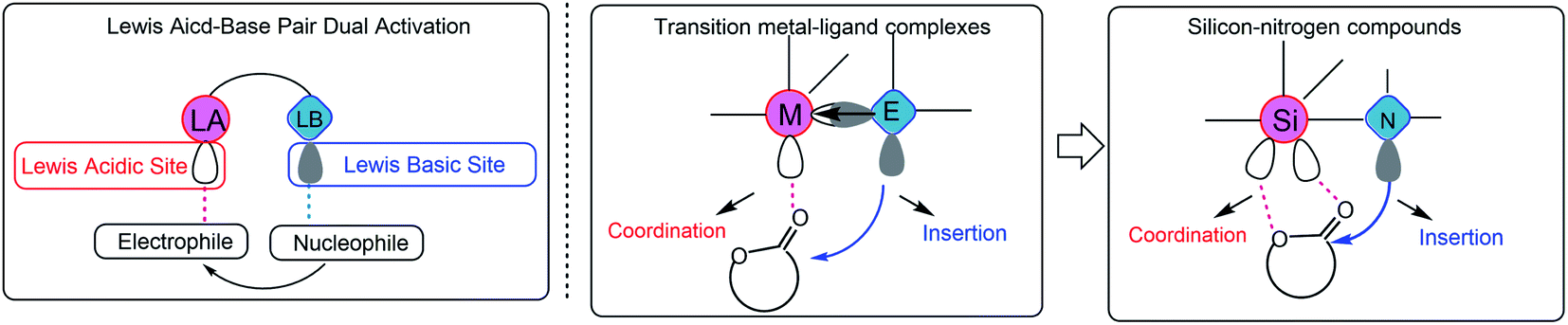 | ||
| Scheme 1 Possible “coordination-insertion” mechanism in ROP of cyclic esters by silicon–nitrogen compound, inspired by the transition metal–ligand complexes. | ||
Numerous recent discoveries have shown that several non-metallic main group elements behave like metals (metalloid).4 The non-metallic elements possess open/stable low-valent derivatives with either open5 or quasi-open coordination sites.6 Among the metalloid elements, silicon (the second richest element on earth, only after oxygen) is a suitable candidate, since it can expand its coordination sphere and reach a “hypervalent” state and thus giving rise to five- and six-coordinate intermediates. The reported various Lewis base donor-stabilized Si compounds demonstrate the unsaturated coordination character of the silicon atom.7 At present, we selected a silicon–nitrogen compound [N-trimethylsilyl-bis(trifluoromethanesulfonyl)imide (TMSNTf2)] to test the possibility of metal-free “coordination-insertion” ROP. The selected silicon–nitrogen compound has a lone pair electron center (tertiary amine) and vacant orbitals (Si atom), similar to metal alkoxide compounds, as shown in Scheme 1. In the case of metal-alkoxides, the “coordination-insertion” mechanism involves the coordination of the monomer with the metal Lewis-acid center, and the insertion of the monomer into the metal-alkoxide bond via the nucleophilic addition of Lewis base ligands (Scheme 1). Therefore, the silicon–nitrogen compound is a promising candidate to promote metal-free “coordination-insertion” in ROP.
In this work, trimethylene carbonate (TMC) was chosen as a model monomer to perform the ROP with TMSNTf2 to verify the metal-free “coordination-insertion” mechanism (Scheme 2). The synthesis of TMSNTf2 was achieved by reaction of bis(trifluoromethanesulfonyl)imide (HNTf2) with allyltrimethylsilane.8 However, if HNTf2 is not consumed completely, the residual HNTf2 interferes, since it also polymerizes TMC by a cationic mechanism leading to polycarbonates with ether units (decarboxylation) as detected by nuclear magnetic resonance (NMR) spectroscopy.9 The absence of the ether units excludes the presence of HNTf2. In contrast, TMSNTf2 initiates/catalyzes the ROP of TMC while preventing undesirable decarboxylation reactions. Besides, the ROP of ε-caprolactone and the block ring-opening sequential copolymerization of ε-CL and TMC were studied with TMSNTf2 as initiator/catalyst. To shed light on the polymerization mechanism, we studied: (a) the kinetics of ROP by NMR and size-exclusion chromatography (SEC), (b) the structure of the products by NMR and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry, (c) the structure of the growing chain, and (d) the monomer activation by TMSNTf2 with NMR titration experiments.
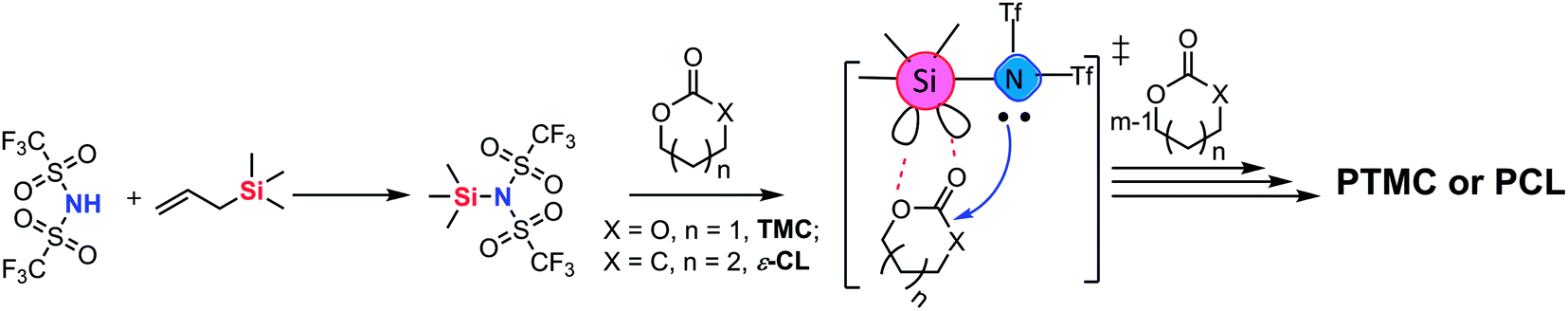 | ||
| Scheme 2 ROP of TMC and ε-CL by the in situ generated TMSNTf2. | ||
Results and discussion
Ring-opening polymerization
Initially, to exclude the possibility that the super-strong Brønsted acid HNTf2 promotes the ROP, we investigated the catalytic performance of HNTf2 in the ROP of TMC (Table 1, entry 1). As anticipated, HNTf2 showed weak control over TMC polymerization with apparent decarboxylation. Due to the extremely strong Brønsted acidity of HNTf2, the undesirable elimination of carbon dioxide during the polymerization seemed to be unavoidable under room temperature. From the 1H NMR spectrum, the ether unit was determined by the triplet signal at 3.47 ppm attributed to methylene protons next to ether oxygen, as shown in Fig. S1.†| Entry | Monomer | Catalyst and initiator | [M]0/[I]0 | Time/h | Conv.c/% | M n (theor)d /kg mol−1 | M n (NMR)e /kg mol−1 | M n (SEC)f /kg mol−1 | Đ (Mw/Mn) |
|---|---|---|---|---|---|---|---|---|---|
a Room temperature, [TMC]0 = 0.7 mol L−1, [ε-CL]0 = 1.0 mol L−1.
b [HNTf2]0/[BnOH]0 = 1/1.
c Determined by 1H NMR in CDCl3 using integrals of the characteristic signals.
d Theoretical molecular weights were calculated based on conversion and the [M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]0 ratio.
e Determined by 1H NMR in CDCl3 by comparing the integration area of the chain end methylene group with that of the corresponding methylene at the repeating units.
f Determined by a tandem SEC-MALS-DRI system using the dn/dc [0.0409 mL g−1] of PTMC and dn/dc [0.070 mL g−1] of PCL in THF at room temperature.
g Determined by SEC in THF using PSt standards. :[I]0 ratio.
e Determined by 1H NMR in CDCl3 by comparing the integration area of the chain end methylene group with that of the corresponding methylene at the repeating units.
f Determined by a tandem SEC-MALS-DRI system using the dn/dc [0.0409 mL g−1] of PTMC and dn/dc [0.070 mL g−1] of PCL in THF at room temperature.
g Determined by SEC in THF using PSt standards.
|
|||||||||
| 1 | TMC | HNTf2 + BnOHb | 20/1 | 3 | 96 | 1.9 | n.d. | 1.0 | 1.29 |
| 2 | TMC | TMSNTf2 | 20/1 | 10 | 95 | 1.9 | 2.1 | 3.6 | 1.22 |
| 3 | TMC | TMSNTf2 | 50/1 | 15 | 97 | 5.0 | 5.4 | 6.1 | 1.17 |
| 4 | TMC | TMSNTf2 | 70/1 | 18 | 95 | 6.8 | 6.6 | 7.2 | 1.10 |
| 5 | TMC | TMSNTf2 | 100/1 | 24 | 94 | 9.6 | 9.7 | 9.0 | 1.12 |
| 6 | ε-CL | TMSNTf2 | 20/1 | 9 | 98 | 2.2 | 2.9 | 4.0 | 1.25 |
| 7 | ε-CL | TMSNTf2 | 30/1 | 14 | 98 | 3.3 | 4.1 | 5.8 | 1.22 |
| 8 | ε-CL | TMSNTf2 | 50/1 | 21 | 95 | 5.4 | 5.8 | 6.5 | 1.18 |
| 9 | ε-CL | TMSNTf2 | 70/1 | 26 | 96 | 7.6 | 7.3 | 7.4 | 1.09 |
| 10 | ε-CL | TMSNTf2 | 100/1 | 32 | 93 | 10.6 | 9.8 | 9.2 | 1.15 |
| 11 | δ-VL | TMSNTf2 | 50/1 | 13 | 91 | 4.8 | 4.7 | 5.4g | 1.21g |
| 12 | L-LA | TMSNTf2 | 50/1 | 48 | 0 | n.d. | n.d. | n.d. | n.d. |
Surprisingly, the ROP of TMC by the in situ generating TMSNTf2 reached 95% in 10 h when the acidity of HNTf2 was regulated by allyltrimethylsilane (ATMS), and TMC was completely converted to poly(trimethylene carbonate) (PTMC) without any decarboxylation (Table 1, entry 2). The chemical structure of PTMC was confirmed by NMR (Fig. 1). There is no triplet signal of polyether at 3.47 ppm (Fig. 1a), which suggests that the TMSNTf2-mediated polymerization proceeded homogeneously without the elimination of CO2. Also, the peaks of the polymer main chain appeared at 2.04 and 4.20–4.24 ppm, the methylene protons adjacent to the ω-chain end of the hydroxyl group appeared at 3.73 ppm. In addition, the 13C NMR spectrum (Fig. 1b) shows all the characteristic signals of the PTMC, thus further confirming that the TMSNTf2-mediated polymerization of TMC proceeds without decarboxylation with complete conversion of HNTf2 to TMSNTf2. TMSNTf2-mediated ROP of ε-CL was also carried out with [ε-CL]0/[TMSNTf2]0 = 20/1 (Table 1, entry 6). The conversion of monomer reached 98.1% in 9 h. From the NMR analysis (Fig. S2†), the chemical structure of PCL was confirmed, revealing that the TMSNTf2 successfully mediated the ROP of ε-CL. Taking into account all the above results, we conclude that only TMSNTf2 and no residual HNTf2 are involved in the metal-free ROP of both cyclic carbonate and lactone monomers.
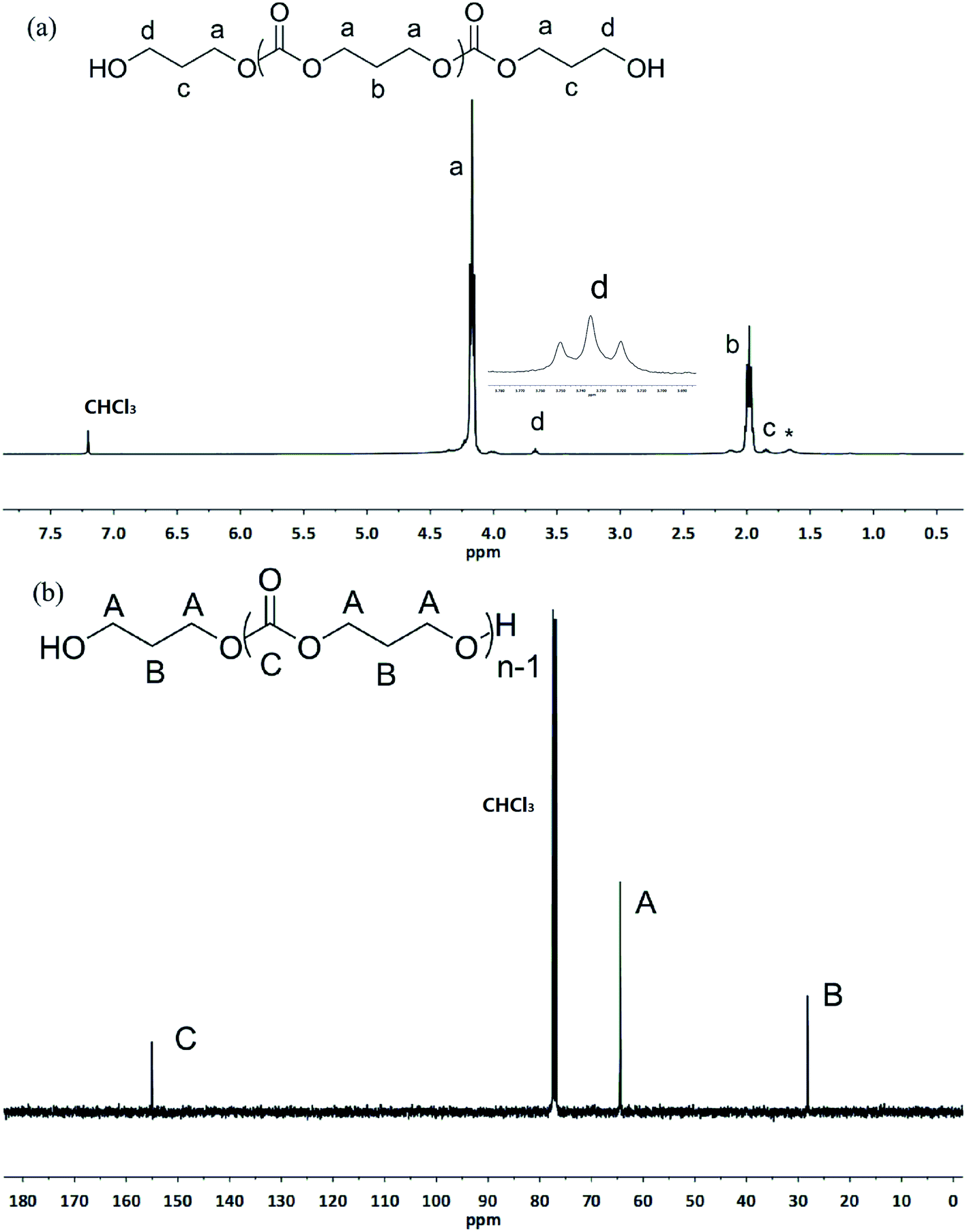 | ||
| Fig. 1 1H NMR (a) and 13C NMR (b) spectra of the obtained PTMC ([M]0/[I]0 = 20, Table 1, entry 2) initiated by TMSNTf2 (asterisk refers to the residual water in polymers). | ||
Mechanism
To obtain an insight into the TMSNTf2-mediated ROP mechanism, we determined the end-chain structure of the synthesized PTMC and PCL by MALDI-TOF MS. In the MALDI-TOF mass spectra of PTMC (Fig. 2), three series of molecular ion peaks are present, a major linear PTMCs with hydroxyl end group (HO-PTMC-H), and two minors, one of which is cyclic (C-PTMC-C) and the other one linear PTMCs with methoxyl end group (MeO-PTMC-H). In the MALDI-TOF spectra of PCL (Fig. S4†), two series of molecular ion peaks appear, a major which is consistent with linear PCLs having hydroxyl end groups (HO-PCL-H) and a minor corresponding cyclic PCLs (C-PCL-C). The mass difference between the neighboring peaks corresponds to the ε-CL monomeric unit of m/z = 114.07. However, the already reported mechanisms for another silicon Lewis acid-initiated ROP of cyclic carbonates and lactones could not explain our results.10 In the case of TMC, the silicon Lewis acid can participate in two pathways (Fig. S3†): in the first one, the monomer carbonyl oxygen attacks its cationic silicon site (“cationic mechanism”), and in the second one, it functions as a ligand coordinating with the carbonyl oxygen via the vacant d orbitals.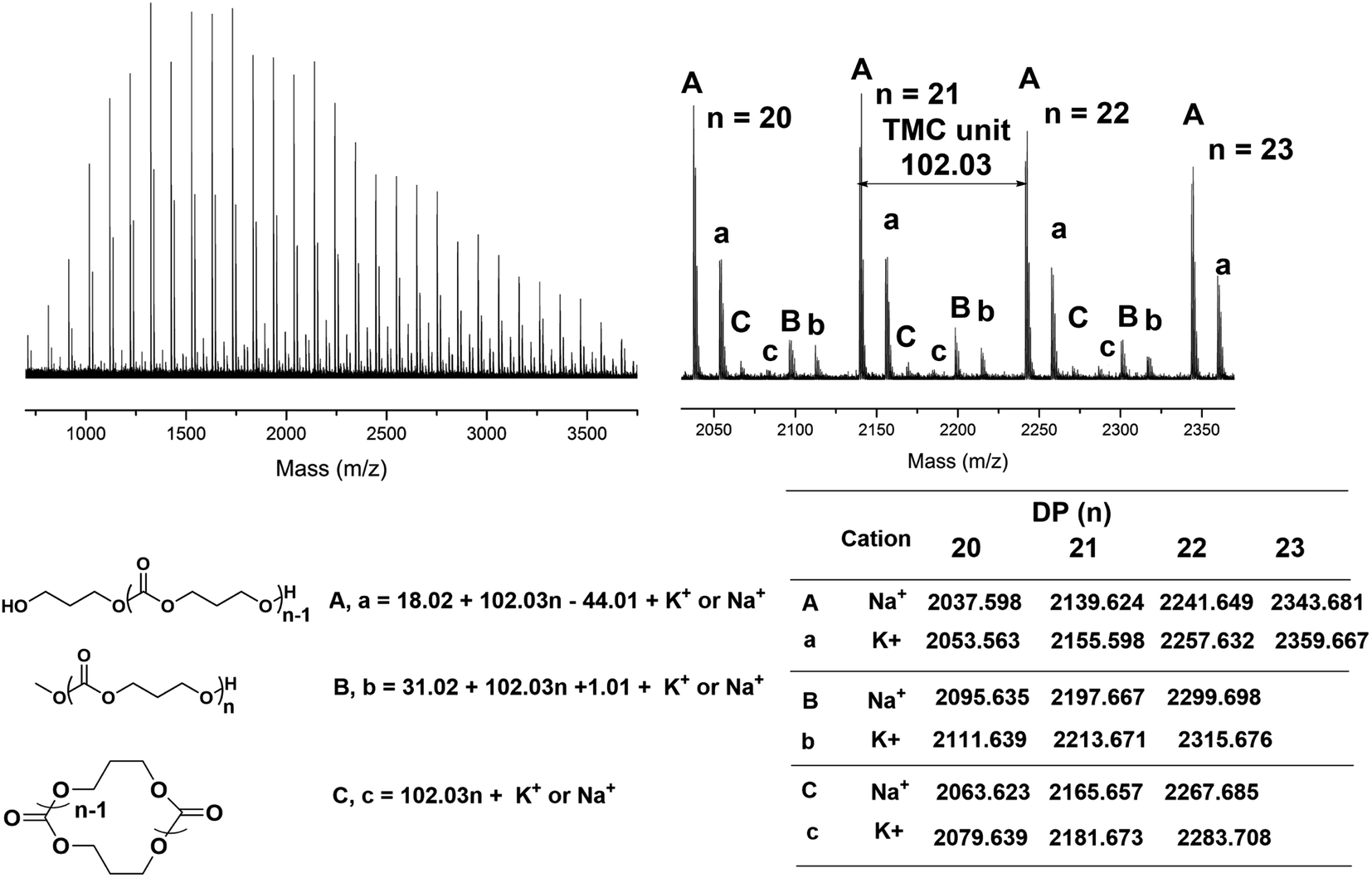 | ||
| Fig. 2 MALDI-TOF MS spectra of the obtained PTMC ([M]0/[I]0 = 20, Table 1, entry 2) initiated by TMSNTf2. | ||
Nevertheless, according to both reported mechanisms, it will be impossible to achieve controlled TMC polymerization without ether units in the backbone, since the formed silicon ester (Fig. S3†) leads to an inevitable decarboxylation and generation of silylether unit (m/z = 58.04).10 In our MALDI-TOF spectra of PTMC, the mass differences between the neighboring peaks of each series of molecular ion peaks were identical to the molecular weight of the TMC monomeric unit (m/z = 102.03). Such results exclude the “cationic mechanism” promoted by HNTf2.
Inspired by the coordination-insertion mechanism of the metallic complexes initiated/catalyzed ROP of cyclic esters,2d,11 we propose a metal-free “coordination-insertion” mechanism of TMSNTf2 for the ROP of cyclic carbonates and lactones. Based on the extension of the donor–acceptor concept proposed by Viktor Gutmann,12 in a molecule with the intramolecular donor–acceptor interaction, the interaction will lead to the enhanced polarity and electrophilicity of the acceptor atom and the enhanced nucleophilicity property of the donor atom. The longer bond between the acceptor atom and the donor atom, the greater the electrophilicity and the nucleophilicity of the corresponding atom will be. In our case, TMSNTf2 has the intramolecular donor–acceptor interaction, N is the electron donor, Si is the electron acceptor, and the N–Si of TMSNTf2 is a relatively long bond.13 Therefore, both the enhanced nucleophilicity of the tertiary amine and the enhanced electrophilicity of the silicon group in TMSNTf2 can take place. When TMSNTf2 is used as initiator/catalyst in ROP, the Si-group may act as a coordination center and the –NTf2 as a unique nucleophile. In a coordination-insertion mechanism, besides the formation of the expected linear polymer, chain termination frequently occurs via intramolecular back-biting reactions to yield a cyclic polymer,14 which explains the existence of cyclic PTMC and PCL in our MALDI-TOF MS spectra. Besides, we even speculate that –SiMe3 is attached to the O-end forming (Me)3Si-polymer-NTf2 with weak interaction. Then upon methanolysis or hydrolysis,15 the Si-alkoxide group is transformed into hydroxyl end-group. Moreover, due to the easy leaving character of –NTf2,16 H2O or alcohols can react (nucleophilic attack) with the carbonyl-NTf2 to generate polymers with alkoxyl end-group. Based on the metal-free “coordination-insertion” mechanism, it is reasonable to explain the existence of several series of molecular ion peaks corresponding to linear polymers and cyclic polymers in the MALDI-TOF spectra of PTMC and PCL.
As suggested by the MALDI-TOF spectra of PTMC (Fig. 2), peaks corresponding to linear PTMCs (HO-PTMC-H) are the most intensive ones, following by the linear PTMCs (MeO-PTMC-H) and the cyclic PTMCs (C-PTMC-C). The formation of MeO-PTMC-H and HO-PTMC-H may be due to the water coming from the air. The water enters the mixture when the reaction tube is opened to the air after the polymerization is completed. Consequently, the water attacks the carbonyl-NTf2 of PTMC before the addition of methanol, leading to hydrolysis/CO2 release and thus producing the major linear HO-PTMC-H component. From the MALDI-TOF spectra of PCL (Fig. S4†), only a major family of linear PCL with hydroxyl end group (HO-PCL-H) and a minor one of cyclic PCL (C-PCL-C) are apparent. The carbonyl-NTf2 connection in (Me)3Si-PCL-NTf2 is more stable than the ester-NTf2 one in (Me)3Si-PTMC-NTf2, and therefore, the methanol cannot replace –NTf2 in (Me)3Si-PCL-NTf2 in the very short precipitation time. But the hydrolysis of the obtained polymer by the inevitable moisture in the air occurs in a relatively long storage time after precipitation. Therefore, only HO-PCL-H appears in the MALDI-TOF spectra of PCL. Besides, the intramolecular back-biting reactions are inevitable in the “coordination-insertion” polymerization, the reason that in the MALDI-TOF spectra of both PTMC and PCL, cyclic structures are present even though in very low content.
Since the water coming from the air enters the polymerization mixture, which leads to linear polymers with hydroxyl end group (major family), we then investigated the polymers produced by TMSNTf2-mediated ROP of TMC and ε-CL, without quenching, and precipitated by super dry methanol in the glovebox. In the MALDI-TOF mass spectra (Fig. 3A) of the PTMC, the peaks of PTMCs shifted from the predominantly linear PTMCs with hydroxyl end group to the mostly linear PTMCs with methoxyl end group instead. As we assumed, the MALDI-TOF spectra consist of two series of molecular ion peaks, with the major series (A, a) assigning to the linear PTMCs (MeO-PTMC-H) and the minor series (B, b) to the cyclic PTMCs. Furthermore, the structure of the linear PTMC (MeO-PTMC-H) was also confirmed by 1H and 13C NMR (Fig. S5†), as evidenced by the resonance at δ 3.77 (CH3OCOO–) in 1H NMR spectrum and the resonance at δ 51.1 (CH3OCOO–) in the 13C NMR spectrum. These results indicate that methanol behaves as a nucleophile to attack the carbonyl-NTf2 of PTMC during the precipitation procedure when H2O was excluded.
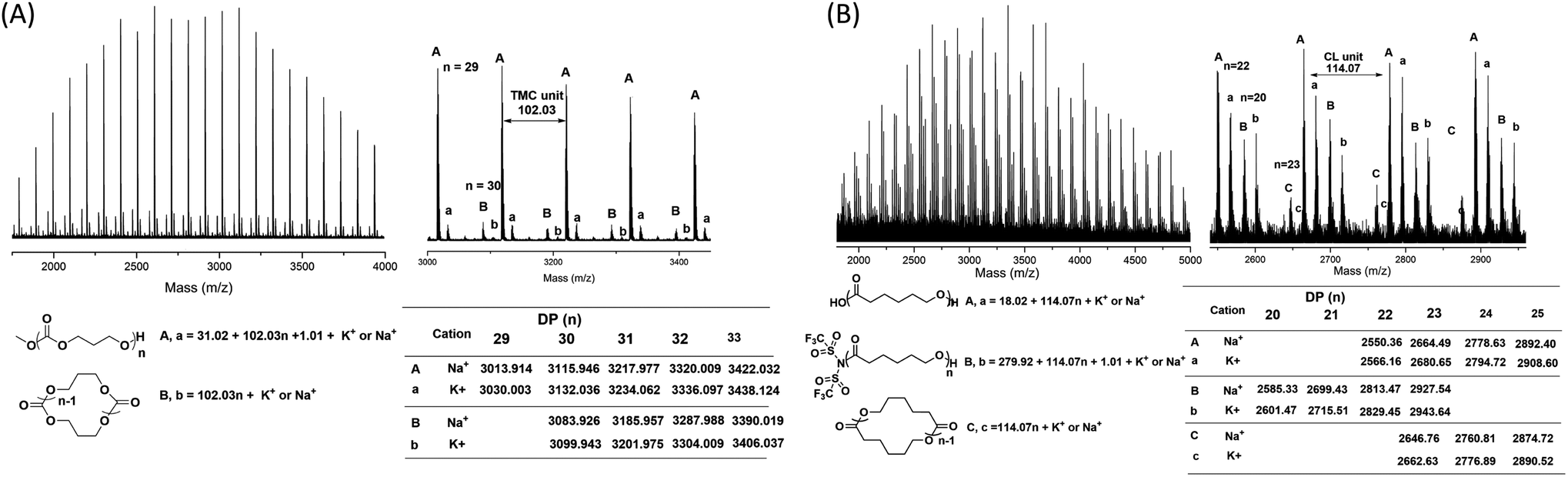 | ||
| Fig. 3 MALDI-TOF MS spectra of (A) PTMC ([M]0/[I]0 = 20) and (B) PCL ([M]0/[I]0 = 20) initiated by TMSNTf2, without quenching, and precipitated by super dry methanol in the glovebox. | ||
As shown in Fig. 3B, the series of molecular ion peaks of Tf2N-PCL-H appear clearly in the MALDI-TOF MS spectra of PCL, which directly evidenced that the –NTf2 nucleophilically attacks the carbonyl carbon of monomer in the initiation step of the ring-opening process. Further confirmation is provided by the 13C NMR and 19F NMR, as evidenced by the resonance at δ 124.8–128.7 ((CF3SO2)2N–) in the 13C NMR spectrum (Fig. S6†) and the resonance at δ −72.02 ((CF3SO2)2N–) in the 19F NMR spectrum (Fig. S7†). In contrast, Tf2N-PTMC-H could never be captured by both MALDI-TOF MS and NMR measurements, which verifies the more stability of the Tf2N-CO– in Tf2N-PCL-H than that of Tf2N-OCO– in Tf2N-PTMC-H. Additionally, major HO-PCL-H still appears, but MeO-PCL-H is not present in the MALDI-TOF MS spectra of PCL (Fig. 3B). These results confirmed that the methanol cannot break the ester-NTf2 connection of (Me)3Si-PTMC-NTf2 in a very short precipitation time. The major HO-PCL-H also comes from the hydrolysis of Tf2N-PCL-H by the inevitable moisture in the air after precipitation. Besides, in the MALDI-TOF MS spectra of PTMC (Fig. 2 and 3) and PCL (Fig. S4† and 3), the presence of small amount of cyclic polymers proves that the intramolecular back-biting reactions17 are not dominant during the TMSNTf2-mediated ROP. Similar phenomena were also observed in other metal-complex initiated ring-opening polymerization.18
Since nucleophiles (methanol and water) can replace the Tf2N– end group of PCL, benzyl alcohol and 2,2-diphenylethanol were also carried out to replace the Tf2N– end group of PCL. Excess benzyl alcohol or 2,2-diphenylethanol was added at the end of the polymerization of ε-CL ([ε-CL]0/[TMSNTf2]0 = 20/1). The reaction was then kept 4 h at room temperature under stirring. Then PCL was obtained from precipitation in methanol three times and drying by vacuum oven for 24 h. The 1H NMR spectra (Fig. S8†) proved that benzyl alcohol or 2,2-diphenylethanol successfully replaced the end group –NTf2 in PCL. Therefore, the easily replaceable –NTf2 provides a method for post-modification of polymer end groups.
In addition, the TMSNTf2 mediated-ROP of δ-VL ([TMSNTf2]0/[δ-VL]0 = 1/50) was carried out (Table 1, entry 11). The conversion of monomer reached 90.6% in 13 h. The polymer was also obtained from the precipitation in super dry methanol. The structure of PCL with the methoxy end group was confirmed by 1H NMR (Fig. S10†). According to the MALDI-TOF mass spectra (Fig. S11†) of the obtained PVL, the series of molecular ion peaks of Tf2N-PVL-H appear clearly. Furthermore, the existence of –NTf2 at the end of the polymer was further evidenced by 19F NMR with the resonance at δ −72.00 ((CF3SO2)2N–) in the 19F NMR spectrum (Fig. S12†). The –NTf2 nucleophilically attacks the carbonyl carbon of monomer in the initiation step of the ring-opening process was again confirmed in the TMSNTf2 mediated-ROP. We also carried out the TMSNTf2 mediated-ROP of L-lactide (Table 1, entry 12). Unfortunately, no polymer was obtained after 48 h, since the first attempt of metal-free “coordination-insertion” catalyst does not have strong enough activation to the carbonyl group of lactide in ROP. Above all, the nucleophilic capacity of the –NTf2 group in TMSNTf2 was proved.
To explain the coordination of Si atom in TMSNTf2 with the monomer, the interaction between TMSNTf2 and monomer was studied by NMR. Unfortunately, when TMSNTf2 was titrated with TMC or ε-CL, the adduct monomer/TMSNTf2 was difficult to observe since propagation occurred immediately. An excellent alternative monomer to study the interaction at initiation is γ-butyrolactone (γ-BL) since it has a positive enthalpy of polymerization and does not polymerize at room temperature.2b When TMSNTf2 (0.25 equiv.) was titrated with γ-BL (1 equiv.), the 13C chemical shift of the carbonyl carbon (γ-BL) moved downfield from 177.83 ppm to 178.62 ppm (Fig. 4), due to its coordination with the Si of TMSNTf2 (deshielding). When TMSNTf2 was increased (0.5–2 equiv.), keeping the same γ-BL (1 equiv.), their chemical shifts moved downfield constantly. Furthermore, methylene carbon adjacent to ester oxygen was moved downfield with the increasing molar ratios of TMSNTf2 to γ-BL (Fig. 4). These results suggest that Si atom coordinates to ester oxygen, and therefore, the electronic density is depressed, rendering the methylene carbon also deshielded. In addition, the mixture of TMSNTf2 (1.0 equiv.) and γ-BL (1.0 equiv.) was studied by 29Si NMR. As shown in Fig. S13,† when TMSNTf2 was titrated with γ-BL, the 29Si chemical shift of TMSNTf2 moved to highfield relative to pure TMSNTf2. This is because the electron density of silicon atom is enhanced (shielding) through the coordination to the oxygen of γ-BL. All these data indicate that TMSNTf2 activates the monomer by coordination with both carbonyl oxygen and ester oxygen.
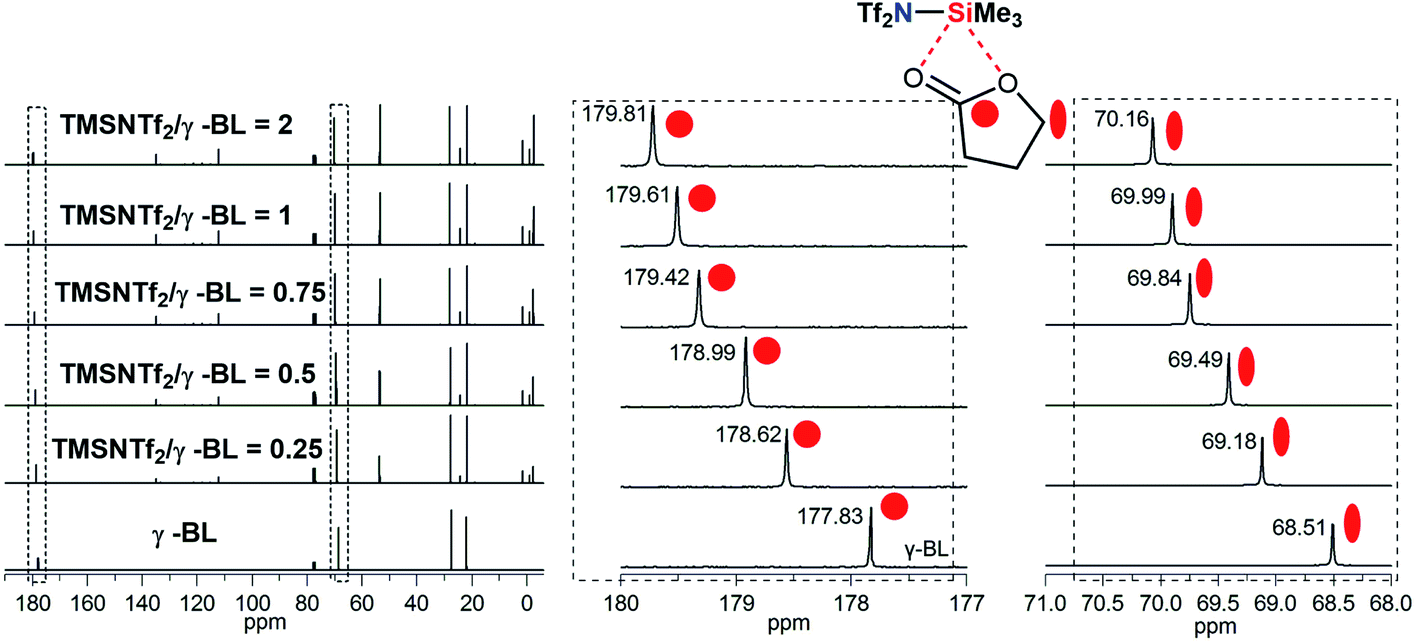 | ||
| Fig. 4 Chemical shifts of cyclic ester methylene and carbonyl carbon in the 13C NMR spectra observed by NMR titration of γ-BL with TMSNTf2 in CDCl3. | ||
Based on the above results, we proposed the first metal-free “coordination-insertion” mechanism of TMSNTf2-mediated ROP of cyclic carbonates and lactones presented in Scheme 3, for TMC as a model monomer. The Si atom carries four sp3 orbitals and five empty electron-accepting d orbitals, similar to Sn atom, used in the metallic coordination-insertion mechanism.4 Two of the vacant orbitals coordinate with the carbonyl and ester oxygen of the TMC, simultaneously, followed by a nucleophilic attack of –NTf2 on the carbonyl carbon and acyl–oxygen cleavage to form a silicon–alkoxo active end group. This initiation step is followed by continuous TMC insertion into the silicon–alkoxo bond. During the polymerization, a small amount of intramolecular transesterification occurs leading to the formation of cyclic structures. When the monomer is totally consumed, the polymer is precipitated in methanol. The nucleophile MeOH or H2O attacks the carbonyl carbon connected to –NTf2 leading to the elimination of –NTf2 end-group. Both initiation and propagation follow the pattern of the “coordination-insertion” mechanism, which in the past was exclusively the monopoly of metal complexes.
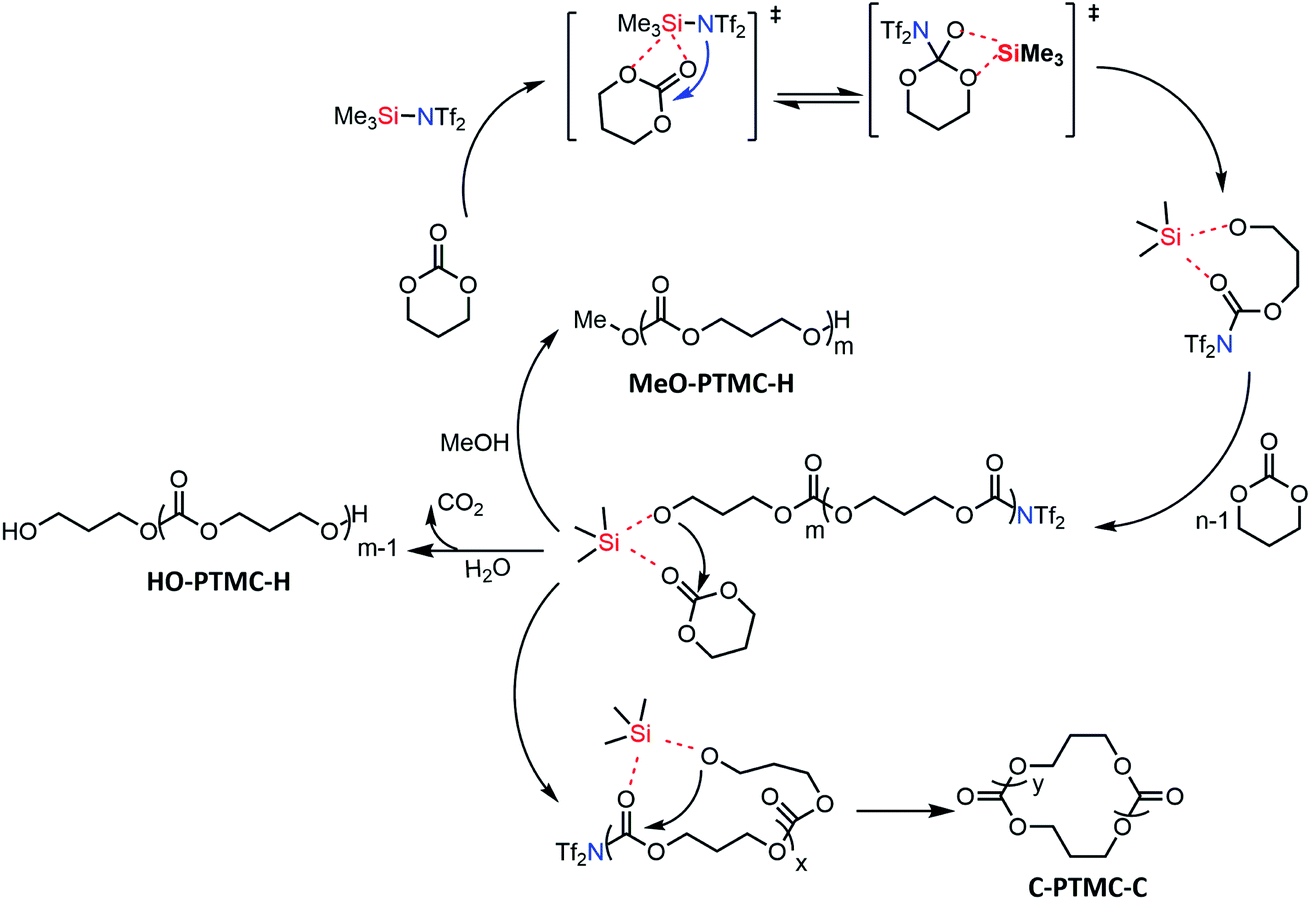 | ||
| Scheme 3 Proposed plausible metal-free “coordination-insertion” mechanism for the ROP mediated by TMSNTf2 (TMC as a model monomer). | ||
Below additional evidence is provided to support our proposed metal-free “coordination-insertion” ROP-mediated by TMSNTf2. The ROP of TMC/TMSNTf2 in a ratio of 10:1 before quenching was studied by 13C NMR conducted in Teflon-valve-sealed J. Young-type NMR tubes, which fully excluded the influence of H2O and other nucleophiles. The presence of the end group corresponding to –CH2CH2CH2O-Si(CH3)3 was detected, as shown in Fig. S14.† Moreover, in Teflon-valve-sealed J. Young-type NMR tubes, ε-CL/TMSNTf2 in a ratio of 1:1 without quenching was also studied by 13C NMR. As shown in Fig. S15b,† the end group corresponding to –CH2CH2CH2CH2CH2O-Si(CH3)3 was observed at 81.2 ppm, in comparison with the characteristic signal of –CH2CH2CH2CH2CH2OH at 63.8 ppm which was produced by ε-CL/TMSNTf2 in a ratio of 1:1 quenched by methanol (Fig. S15a†). Besides, Kricheldorf et al. proposed that the ring-opening of lactones and carbonates proceeds through a transition state, in which the carbonyl oxygen is complexed to silicon via the vacant orbitals.10 The above data further confirmed the coordination activation of Si in the TMSNTf2-mediated ROP.
Kinetics
Polymerization kinetics of TMSNTf2-mediated ROP of TMC were investigated under standard conditions (see Experimental part), where the initial ratios of monomer to initiator were systematically varied ([TMC]0:[TMSNTf2]0 = 20:1–100:1), while the initial monomer concentration was kept constant ([TMC]0 = 0.7 M). Lines consisting of two linear steps for each case were obtained (Fig. 5A). As shown in Fig. 5B, magnification of the first linear steps of Fig. 5A, the polymerizations exhibit a first-order dependence of the monomer concentration with time (i.e., d[M1]/dt = kobs[M1]), which is the fingerprint of living polymerization. As the initial TMSNTf2 concentration increases from 7 to 10, 14 and 32 mM, the observed rate constant (kobs) increases linearly (i.e., kobs = kp[TMSNTf2]0) from 0.16 to 0.23, 0.32 and 0.71 h−1 (Fig. 5C) giving a kp = 22 M−1 h−1. The polymerization kinetics of TMSNTf2-mediated ROP of ε-CL were also investigated ([ε-CL]0:[TMSNTf2]0 = 20:1–100:1, [M]0 = 1.0 M). As shown in Fig. S16A,† all the semilogarithmic plots reveal the same two linear steps with TMC, in which the first step corresponds to a first-order dependence of monomer concentration with time (i.e., d[M1]/dt = kobs[M1]). The plot of the observed rate constant (kobs) versus the initial TMSNTf2 concentration of the first step is shown in Fig. S16B,† in which the kobs increases from 0.12 to 0.7 h−1 when the initial TMSNTf2 concentration increases from 10 to 60 mM. Moreover, the plot of kobsversus [TMSNTf2]0 shows a linear relationship (i.e., kobs = kp[TMSNTf2]0, kp = 11.6 M−1 h−1). All the above data indicate that in the first step of ROP of TMC and ε-CL, TMSNTf2 acts as a single-site initiator.19
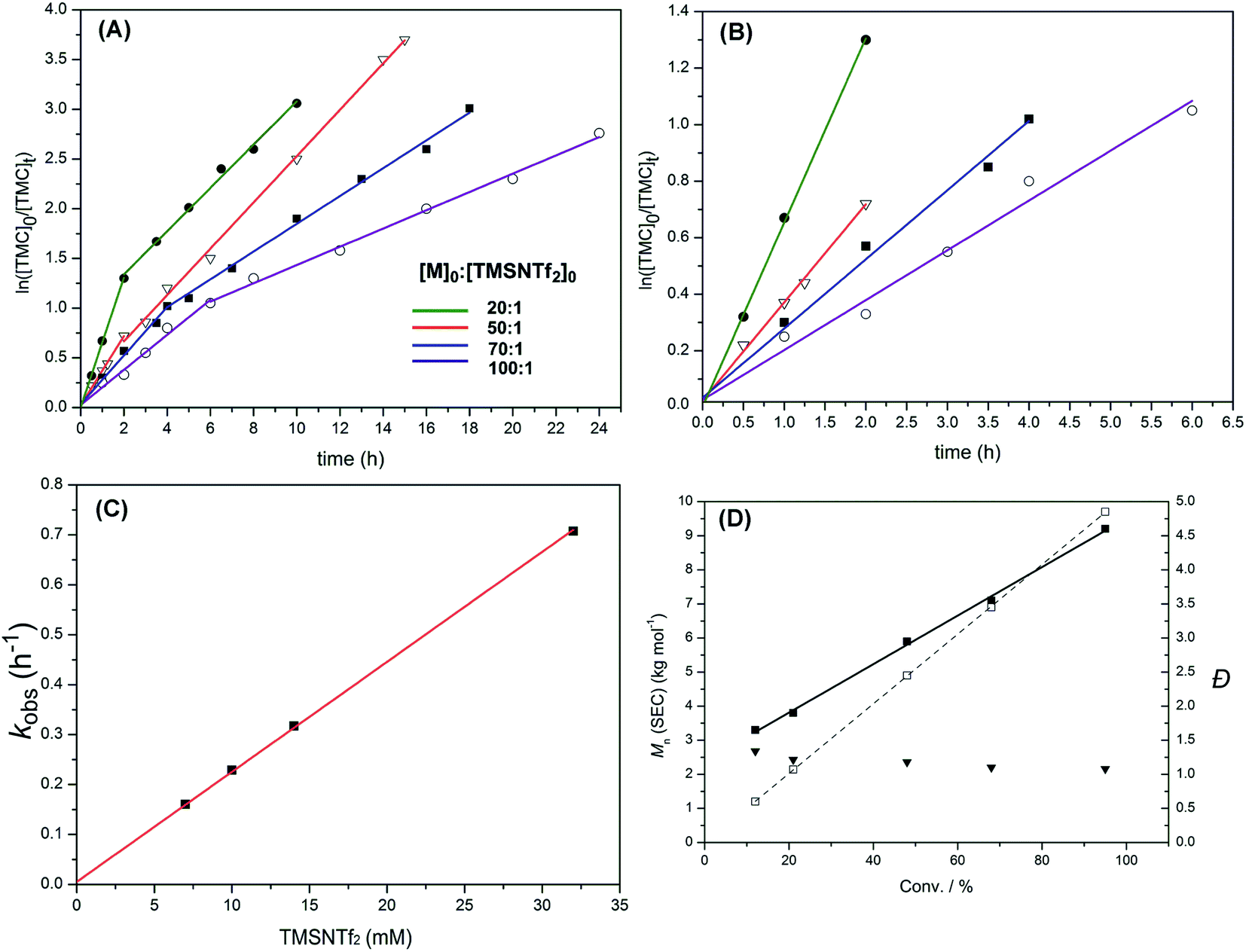 | ||
| Fig. 5 (A) Plots of ln([TMC]0/[TMC]t) versus reaction time for the ROP of TMC initiated by TMSNTf2 in CH2Cl2 at various ratios of TMC to TMSNTf2 ([TMC]0:[TMSNTf2]0 = 20:1–100:1, [M]0 = 0.7 M, 25 °C). (B) Enlargement of the initial linear parts of (A). (C) Plot of the observed rate constant (kobs) versus the initial TMSNTf2 concentration for the ROP of TMC. (D) Plots of Mn,SECversus monomer conversion for the ROP of TMC initiated by TMSNTf2 (Table 1, entry 5): (■) Mn (SEC, using the dn/dc [0.0409 mL g−1]); (□) Mn (theory); (▼) Đ (Mw/Mn, from SEC). | ||
After the first step, the plots of ln([TMC]0/[TMC]t) versus reaction time are linear until monomer conversion up to 90%, but with reduced observed rate constants compared to the first step. The fractional dependence with monomer concentration has also been observed for aluminum, yttrium alkoxides, and other systems.11d,20 According to the previous report, an aggregation process may occur in the second step. The active species of the propagating polymer chain (active silicon group) is stabilized by the excess monomer at the beginning of polymerization, but when the monomer conversion is >50–60%, the concentration of carbonyl group of the monomer decreases a lot accordingly; meanwhile, the carbonyl group of the propagating polymer chain grows a lot. The carbonyl group of polymer will compete with the monomer to occupy the vacant orbitals of the active silicon group at the high monomer conversion, which will reduce the activity of the active silicon group and lead to aggregation of the active silicon group with the polymer chain. The activity loss of the active silicon group results in a slower propagation rate, as shown in Fig. 5A. Similarly, as shown in Fig. S16A,† the aggregation process of TMSNTf2-mediated ROP of ε-CL also leads to a slower propagation rate at the high monomer conversion. Therefore, we speculate that the aggregation process is happening in TMSNTf2-mediated ROP
All polymers produced by various initial feeding ratios of monomer to TMSNTf2 ([M]0:[TMSNTf2]0 = 20:1–100:1, Table 1), show monomodal SEC traces (PTMC: Fig. S17; PCL: Fig. S18†). The plots of polymer Mn(SEC)s as a function of monomer conversion give a linear relationship but somewhat deviate from the theoretical dependencies (TMC, Fig. 5D; ε-CL, Fig. S19†). From the plot of PTMC, the polymer propagating chains formed in the first step continue growing in the second step. However, the molecular weights of PTMC were higher than the theoretical ones at the beginning and then lower than the theoretical ones. This is due to the weak nucleophilic attack of –NTf2 leading to a relatively slower initiation rate as compared to the propagation rate leading to higher molecular weights. Besides, the slower initiation rate than the propagation rate resulted in the broader molecular weight distributions (Đ), as shown in Fig. 5D. When the monomer was consumed, the aggregation of active species to dimeric structure and large clusters occurred, which caused the lower molecular weights. From Fig. S19,† the plot of PCL shows nearly the same phenomenon as PTMC. These results followed the character of a typical metal-complex “coordination-insertion” mechanism in the ROP of cyclic esters for the initiation and propagation steps.
TMSNTf2-mediated diblock copolymerization of ε-CL and TMC
Accordingly, we carried out a sequential block copolymerization of ε-CL and trimethylene carbonate (TMC) with the initial ratio of [ε-CL]0/[TMC]0/[TMSNTf2]0 = 20/30/1 to produce the well-defined diblock polymer (PCL-b-PTMC). The whole process was completed in 22 h, and the chemical structure of the obtained polymer was confirmed by 1H NMR (Fig. S20†). From the 1H NMR spectrum, all the resonance signals could be assigned to the corresponding protons of the main body of PCL-b-PTMC. Fig. S21† shows the SEC traces of the PCL homopolymer macroinitiator and the PCL-b-PTMC diblock copolymer. After the homopolymerization, the molecular weight (Mn,SEC) of polymer increased from 4.0 kg mol−1 (Đ = 1.25) to 5.4 kg mol−1 (Đ = 1.19). When the initial ratio of [ε-CL]0/[TMC]0/[TMSNTf2]0 = 100/100/1 was used, the conversion of TMC in the second polymerization was just 51%, even after 100 h. In this case, the molecular weight of the first block is much higher, resulting in reduced propagating reactivity.Conclusions
In summary, the first metal-free “coordination-insertion” ring-opening polymerization (ROP) of cyclic carbonate and lactone mediated by TMSNTf2 was proposed. The metal-free “coordination-insertion” mechanism, monopoly until now of the metal-complexes, is significantly favorable for TMSNTf2-mediated ROP of TMC and ε-CL, with Si atom acting as a vacant orbitals center (corresponding to the metal) and tertiary amine as an electronegative center (corresponding to the ligand). In the mechanism, the coordination of monomer to Si atom of TMSNTf2 occurs, followed by a nucleophilic attack on the carbonyl carbon of the coordinated monomer by the –NTf2 group, acyl–oxygen cleavage, and insertion of monomer to the Si–N bond of TMSNTf2. ROP of TMC and ε-CL mediated by TMSNTf2 yielded linear and cyclic polymers with two-stage polymerization processes and weak control of molecular weights. In the first stage, the polymerization was initiated by TMSNTf2 with single-site activation. By detailed NMR studies, including NMR titration experiments and NMR analyses of the intermediate reaction mixture, the metal-free “coordination-insertion” mechanism was proved. In conclusion, we proposed a new initiation/catalytic path of TMSNTf2 and the concept of a metal-free “coordination-insertion” mechanism, which is unprecedented in the ROP field. Current efforts focus on polymerizations performed with co-initiators and extending variously metal-free coordination-insertion ROPs.Data availability
The electronic supplementary information include experimental detail, additional NMR data, SEC data, kinetics data and MALDI-ToF data.Author contributions
Zhenjiang Li and Kai Guo conceived the idea and led the project. Xin Wang and Zhenjiang Li designed the experiments. Xin Wang performed the experiments and prepared the ESI.† Xin Wang, Jiaxi Xu, Jingjing Liu, and Zhenjiang Li were involved in the analysis of data. Xin Wang wrote the manuscript. Xin Wang, Zhenjiang Li, Jie Sun, and Nikos Hadjichristidis participated in the discussions and revised the manuscript. All of the authors reviewed, approved, and contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078150), the National Key Research and Development Program of China (2017YFC1104802), the Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), the project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Top-Notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP). N. H. acknowledges the support of King Abdullah University of Science and Technology.Notes and references
- W. B. Jensen, Chem. Rev., 1978, 78, 1–22 Search PubMed.
- (a) R. Ligny, M. Hänninen, S. M. Guillaume and J. F. Carpentier, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 Search PubMed; (b) M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 Search PubMed; (c) E. Martin, P. Dubois and R. Jerome, Macromolecules, 2000, 33, 1530–1535 Search PubMed; (d) H. Y. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 Search PubMed; (e) M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 Search PubMed.
- (a) K. M. Stridsberg, M. Ryner and A. C. Albertsson, in Adv Polym Sci, ed. A. C. Albertsson, Springer-Verlag Berlin, Berlin, 2002, vol. 157, pp. 41–65 Search PubMed; (b) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 Search PubMed; (c) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 Search PubMed.
- (a) P. P. Gaspar and R. West, Silylene chemistry of organic silicon compounds 2, Part 3, ed. Z. Rappoport and Y. Apeloig, Wiley, 1998, vol. 2, pp. 2463–2568 Search PubMed; (b) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 Search PubMed; (c) G. Linti and H. Schnockel, Coord. Chem. Rev., 2000, 206, 285–319 Search PubMed.
- (a) D. W. Stephan, Dalton Trans., 2009, 3129–3136 Search PubMed; (b) H. Lu and J. J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 Search PubMed; (c) H. Y. Zhang, Y. Z. Nie, X. M. Zhi, H. F. Du and J. Yang, Chem. Commun., 2017, 53, 5155–5158 Search PubMed.
- (a) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 Search PubMed; (b) Z. D. Wang, J. Y. Zhang, J. F. Li and C. M. Cui, J. Am. Chem. Soc., 2016, 138, 10421–10424 Search PubMed; (c) F. M. Muck, B. Forster, J. A. Baus, M. Nutz, C. Burschka, R. Bertermann and R. Tacke, Eur. J. Inorg. Chem., 2016, 2016, 3246–3252 Search PubMed; (d) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 Search PubMed; (e) T. W. Bai and J. Ling, J. Phys. Chem. A, 2017, 121, 4588–4593 Search PubMed.
- B. Mathieu and L. Ghosez, Tetrahedron Lett., 1997, 38, 5497–5500 Search PubMed.
- H. R. Kricheldorf and J. Jenssen, J. Macromol. Sci., Part A: Pure Appl.Chem., 1989, 26, 631–644 Search PubMed.
- (a) R. Dunsing and H. R. Kricheldorf, Eur. Polym. J., 1988, 24, 145–150 Search PubMed; (b) H. R. Kricheldorf, Angew. Chem., Int. Ed., 1979, 18, 689–690 Search PubMed.
- (a) H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 Search PubMed; (b) P. Dubois, P. Degee, R. Jerome and P. Teyssie, Macromolecules, 1992, 25, 2614–2618 Search PubMed; (c) H. R. Kricheldorf, M. V. Sumbel and I. Kreisersaunders, Macromolecules, 1991, 24, 1944–1949 Search PubMed; (d) P. Dubois, C. Jacobs, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 2266–2270 Search PubMed.
- (a) V. Gutmann, Electrochim. Acta, 1976, 21, 661–670 Search PubMed; (b) V. Gutmann, The donor-acceptor approach to molecular interactions, Springer, 1978 Search PubMed; (c) V. Gutmann, Pure Appl. Chem., 1979, 51, 2197–2210 Search PubMed.
- W. N. Ottou, E. Conde-Mendizabal, A. Pascual, A. L. Wirotius, D. Bourichon, J. Vignolle, F. Robert, Y. Landais, J. M. Sotiropoulos, K. Miqueu and D. Taton, Macromolecules, 2017, 50, 762–774 Search PubMed.
- G. Montaudo, M. S. Montaudo, C. Puglisi, F. Samperi, N. Spassky, A. LeBorgne and M. Wisniewski, Macromolecules, 1996, 29, 6461–6465 Search PubMed.
- M. H. Chisholm and E. E. Delbridge, New J. Chem., 2003, 27, 1177–1183 Search PubMed.
- S. Antoniotti, V. Dalla and E. Dunach, Angew. Chem., Int. Ed., 2010, 49, 7860–7888 Search PubMed.
- N. Spassky, V. Simic, M. S. Montaudo and L. G. Hubert-Pfalzgraf, Macromol. Chem. Phys., 2000, 201, 2432–2440 Search PubMed.
- (a) A. Kowalski, J. Libiszowski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 1964–1971 Search PubMed; (b) Y. Takashima, Y. Nakayama, K. Watanabe, T. Itono, N. Ueyama, A. Nakamura, H. Yasuda, A. Harada and J. Okuda, Macromolecules, 2002, 35, 7538–7544 Search PubMed; (c) M. Save and A. Soum, Macromol. Chem. Phys., 2002, 203, 2591–2603 Search PubMed.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 Search PubMed.
- (a) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 Search PubMed; (b) C. X. Cai, A. Amgoune, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2004, 330–331 Search PubMed; (c) A. Duda and S. Penczek, Macromol. Rapid Commun., 1994, 15, 559–566 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02551a |
| This journal is © The Royal Society of Chemistry 2021 |