DOI:
10.1039/D1SC02614C
(Perspective)
Chem. Sci., 2021,
12, 10972-10984
Sulfur stereogenic centers in transition-metal-catalyzed asymmetric C–H functionalization: generation and utilization
Received
12th May 2021
, Accepted 7th July 2021
First published on 7th July 2021
Abstract
Transition-metal-catalyzed enantioselective C–H functionalization has emerged as a powerful tool for the synthesis of enantioenriched compounds in chemical and pharmaceutical industries. Sulfur-based functionalities are ubiquitous in many of the biologically active compounds, medicinal agents, functional materials, chiral auxiliaries and ligands. This perspective highlights recent advances in sulfur functional group enabled transition-metal-catalyzed enantioselective C–H functionalization for the construction of sulfur stereogenic centers, as well as the utilization of chiral sulfoxides to realize stereoselective C–H functionalization.
1. Introduction
Transition-metal-catalyzed C–H functionalization has emerged as a powerful tool for the construction of C–C and C–heteroatom bonds owing to its high step and atom economy.1–7 By viewing the C–H bond as “ubiquitous functionality”, synthetic chemists open a new chapter in organic synthesis, which has revolutionised the rules for assembling molecules. In particular, new catalytic transformations based on the enantioselective functionalization of C–H bonds have attracted considerable attention,8–12 given the paramount importance of chirality in organic molecules and growing demand for enantiopure compounds in chemical and pharmaceutical industries. Although asymmetric C–H functionalization is still in its infancy, the recently developed chiral directing groups (cDGs) and chiral ligands have armed this promising methodology to streamline the synthesis of various chiral molecules in an atom- and step-economical manner.
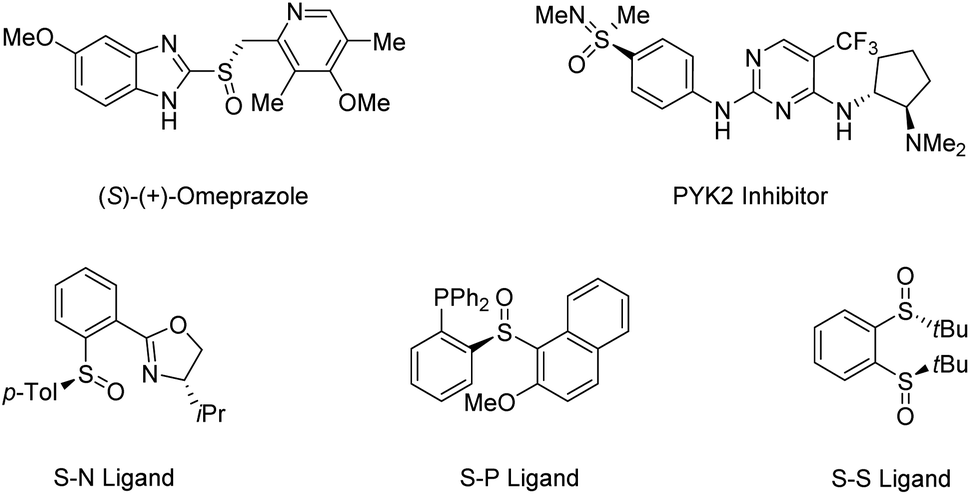
Sulfur-based functionalities are ubiquitous in many of the biologically active compounds, medicinal agents, and functional materials. Biomolecules containing sulfur stereocenters play an important role in the chemical reactions of general metabolism.13 Owing to their significant bioactivity, an increasing number of marketed drugs and bioactive compounds contain sulfoxides and sulfoximines in enantiopure form.14–18 Moreover, organosulfur compounds are commonly utilized as starting materials, reagents, or intermediates in synthetic organic chemistry,19–22 in which chiral sulfoxides are particularly useful in asymmetric catalysis as versatile chiral auxiliaries or ligands due to their high optical stability (Scheme 1).23–27 Traditional strategies toward enantioenriched sulfoxides mainly rely on resolution techniques and diastereoselective transformations, in which the need for stoichiometric amounts of chiral-pool reagents has made their synthetic application cumbersome.28 So far, the most popular routes for the access of enantioenriched sulfoxides have been metal-catalyzed and biocatalytic asymmetric sulfide oxidation processes. However, the requirement for discrimination between the substituents of sulfides usually limits their utility.29
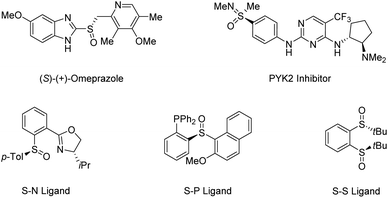 |
| | Scheme 1 Representative bioactive molecules and ligands containing sulfur stereogenic centers. | |
Given the importance of asymmetric C–H functionalization technology in modern synthetic chemistry, as well as the abundance of chiral organosulfur compounds in biologically active compounds, pharmaceuticals and materials, the generation and utilization of sulfur stereogenic centers in transition-metal-catalyzed asymmetric C–H functionalization is undoubtably highly attractive. This Perspective is aimed to comprehensively highlight recent advances in sulfur functional group directed transition-metal-catalyzed enantioselective C–H functionalization for the construction of sulfur stereogenic centers, as well as the utilization of chiral sulfoxides to steer stereoselective C–H functionalization. We hope to shed light on new perspectives, and inspire the use, the completion, and the improvement of this emerging methodology, which could be attractive to practitioners of medicinal chemistry and materials science.
2. Generation of sulfur stereocenters through enantioselective C–H functionalization
In recent years, sulfoxides and sulfoximines have found a unique place in C–H functionalization owing to their various modes of reactivity. A number of sulfoxide and sulfoximine directed C–H functionalization reactions have successfully been demonstrated.30–32 Importantly, an additional advantage of these scaffolds is their potentially stereogenic character, and thus enantioselective C–H functionalization reactions could be explored for the facile generation of sulfur stereogenic centers.
2.1 Sulfoxide-directed enantioselective C–H functionalization
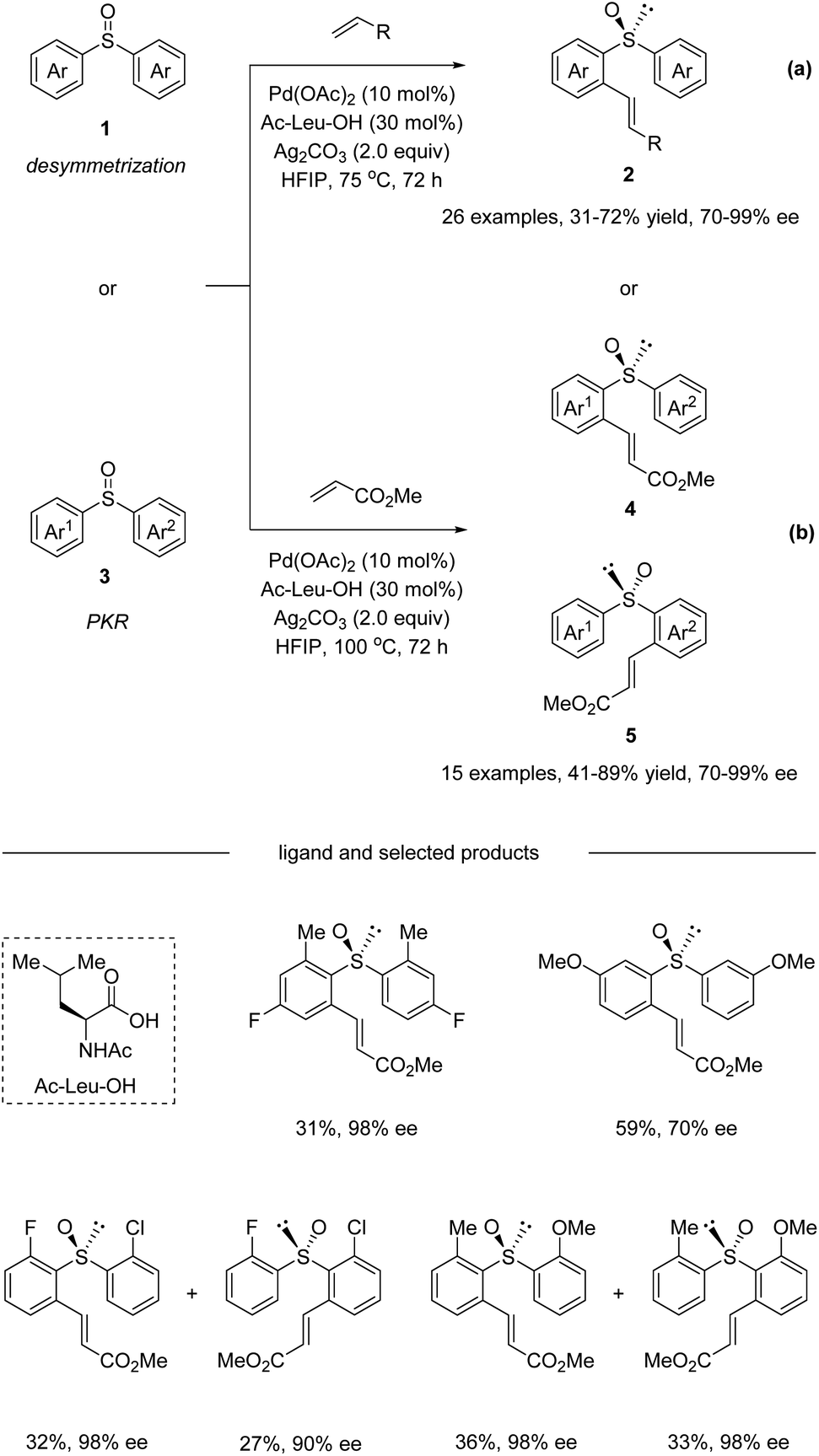
In 2018, Wang and co-workers reported a Pd(II)-catalyzed enantioselective C–H olefination with non-chiral and racemic diaryl sulfoxides via desymmetrization and parallel kinetic resolution (PKR) (Scheme 2).33 The optimization of a range of different monoprotected amino acids revealed that Ac-Leu-OH was the best ligand for this transformation. It is worth mentioning that pre-stirring of the chiral amino acid ligand with Pd(OAc)2 is essential, because the coordination ability of sulfoxide could influence their complexation. Under the optimized conditions, the desymmetrization of symmetric diaryl sulfoxides 1 afforded ortho substituted olefination products 2 in moderate yields with good enantioselectivities (Scheme 2a). Substrates bearing electron-neutral, electron-donating, and weakly electron-withdrawing substituents or para, meta, and ortho substituents were all tolerated. Aside from methyl acrylate, diethyl vinyl phosphonate and pentafluoro-styrene were also competent reaction partners delivering the corresponding products with good ee. Additionally, non-symmetric diaryl sulfoxides 3 were converted in a parallel kinetic resolution pathway (Scheme 2b) to give two products from the racemic starting materials. Diaryl sulfoxides reacted to give products 4 and 5 in excellent enantioselectivity via stereo-divergent olefination.
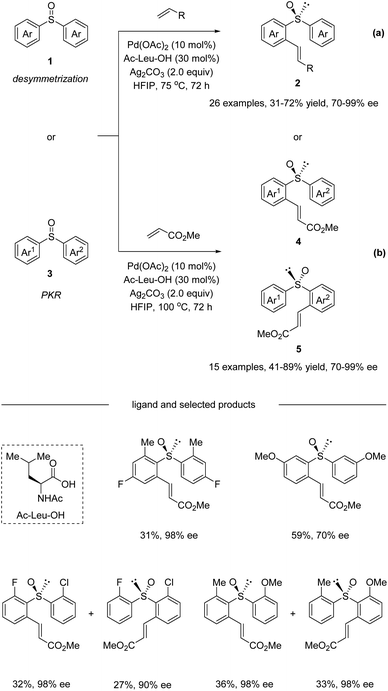 |
| | Scheme 2 Pd(II)-catalyzed enantioselective C–H olefination of diaryl sulfoxides. (a) Desymmetrization pathway. (b) Parallel kinetic resolution pathway. | |
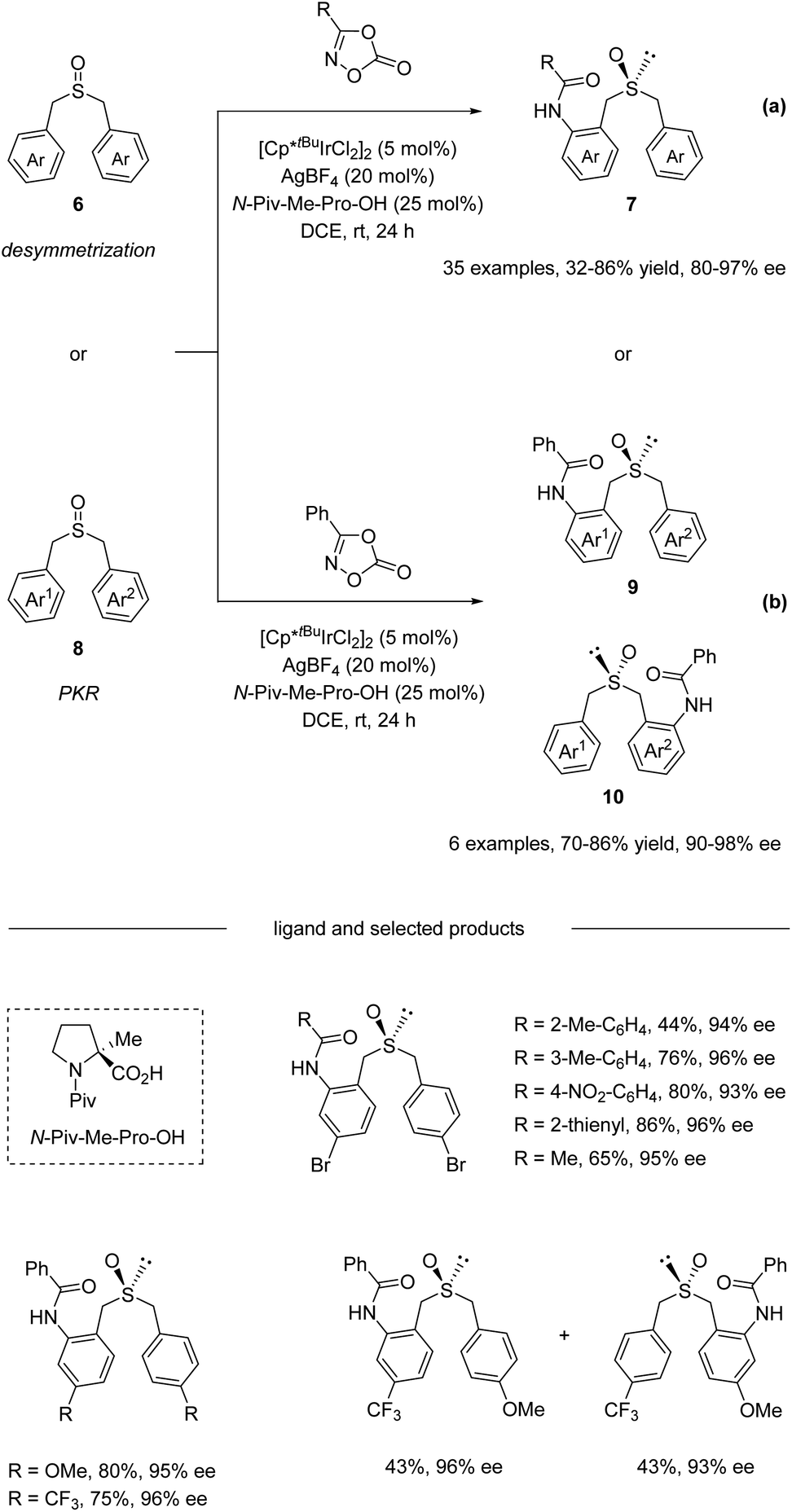
In 2020, He and co-workers reported an Ir(III)-catalyzed asymmetric C–H amidation of pro-chiral and racemic dibenzyl sulfoxides (Scheme 3).34 A survey of a range of different chiral carboxylic acid ligands and achiral Cp ligands revealed that N-Piv-Me-Pro-OH and Cp*tBu were the optimal ligands for this transformation, providing an efficient and straightforward way to construct sulfur chiral centers. A variety of dibenzyl sulfoxides and dioxazolones were compatible with this process, giving access to a variety of highly functionalized sulfoxide compounds with synthetically attractive amide substitution groups in good yields with excellent enantioselectivities via desymmetrization (Scheme 3a) and parallel kinetic resolution (PKR, Scheme 3b). Moreover, the flexible derivatization of the amidated sulfoxide was elaborated, providing various types of chiral sulfoxide scaffolds that could be potentially useful in asymmetric catalysis as chiral bidentate and tridentate ligands.
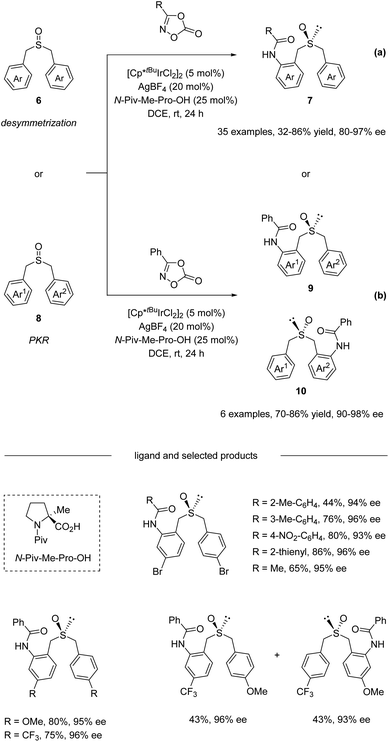 |
| | Scheme 3 Dual-ligand-enabled Ir(III)-catalyzed enantioselective C–H amidation of dibenzyl sulfoxides. (a) Desymmetrization pathway. (b) Parallel kinetic resolution pathway. | |
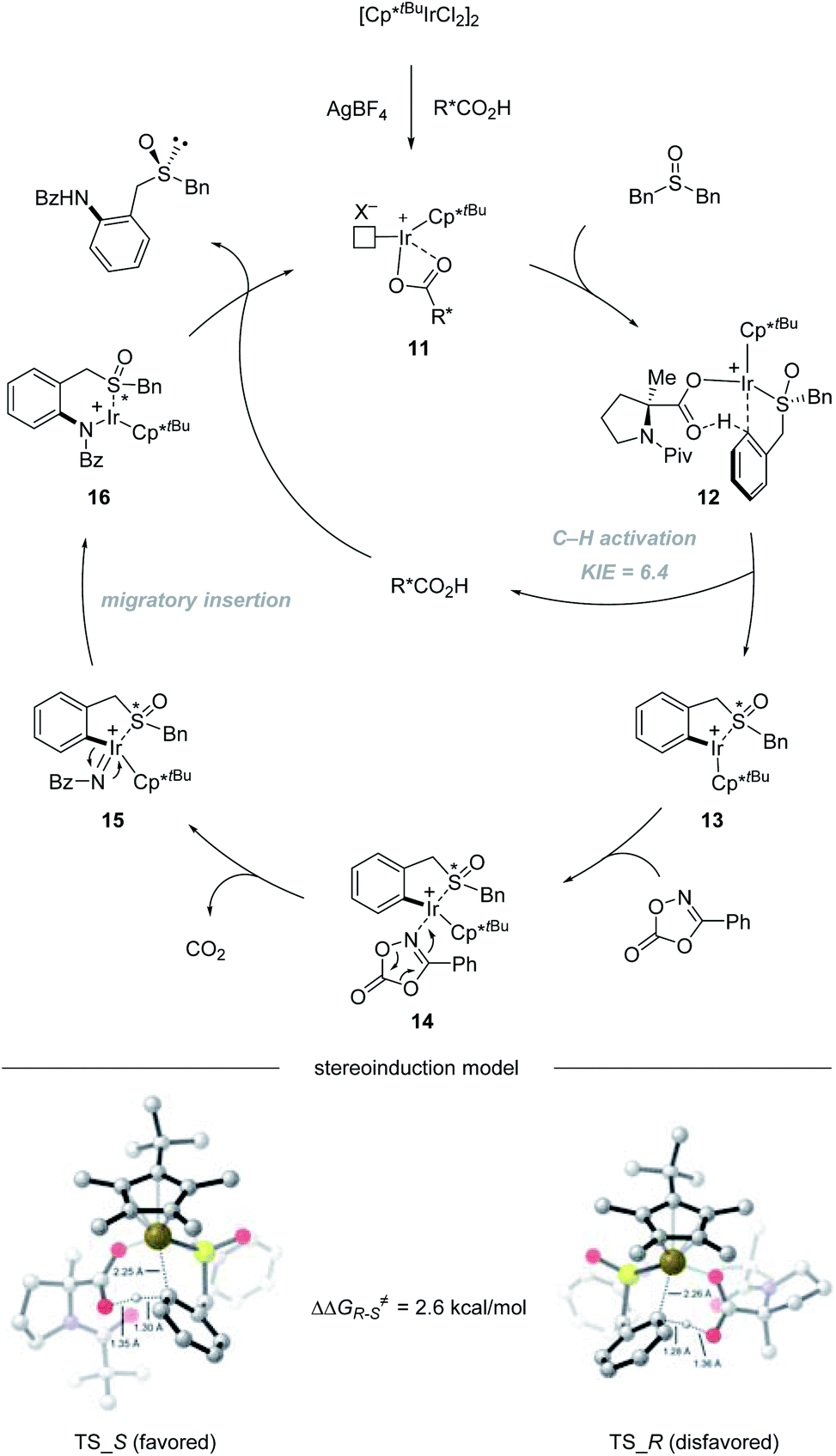
The catalytic cycle starts with the coordination of active species 11 with dibenzyl sulfoxide, giving intermediate 12. Then enantioselective C–H bond activation occurs as the rate-determining step (RDS) with a 6.4 KIE value via concerted metalation/deprotonation (CMD) assisted by chiral carboxylic acid, providing (S)-iridacycle 13. Preliminary DFT calculations reveal that TS_S is 2.6 kcal mol−1 lower in energy than TS_R. The synergistic use of the Cp*tBu ligand with the (Piv)-protected methyl substituted tertiary proline ligand creates a compact chiral transition state, which allows the perfect discrimination of the diastereotopic benzyl groups. Then, the coordination of dioxazolone with intermediate 13 generates intermediate 14, which can subsequently undergo CO2 extrusion (intermediate 14 to 15) and migratory insertion furnishes the desired amide bond (intermediate 15 to 16). Finally, protonolysis produces the amide product and regenerates 11, closing the catalytic cycle (Scheme 4).
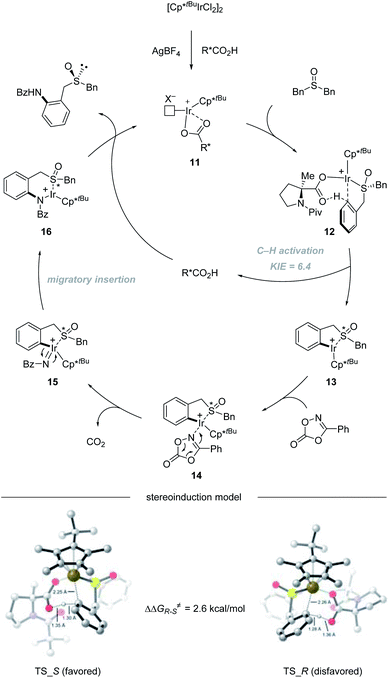 |
| | Scheme 4 Mechanism of Ir(III)-catalyzed enantioselective C–H amidation of dibenzyl sulfoxides. | |
2.2 Sulfoximine-directed enantioselective C–H functionalization
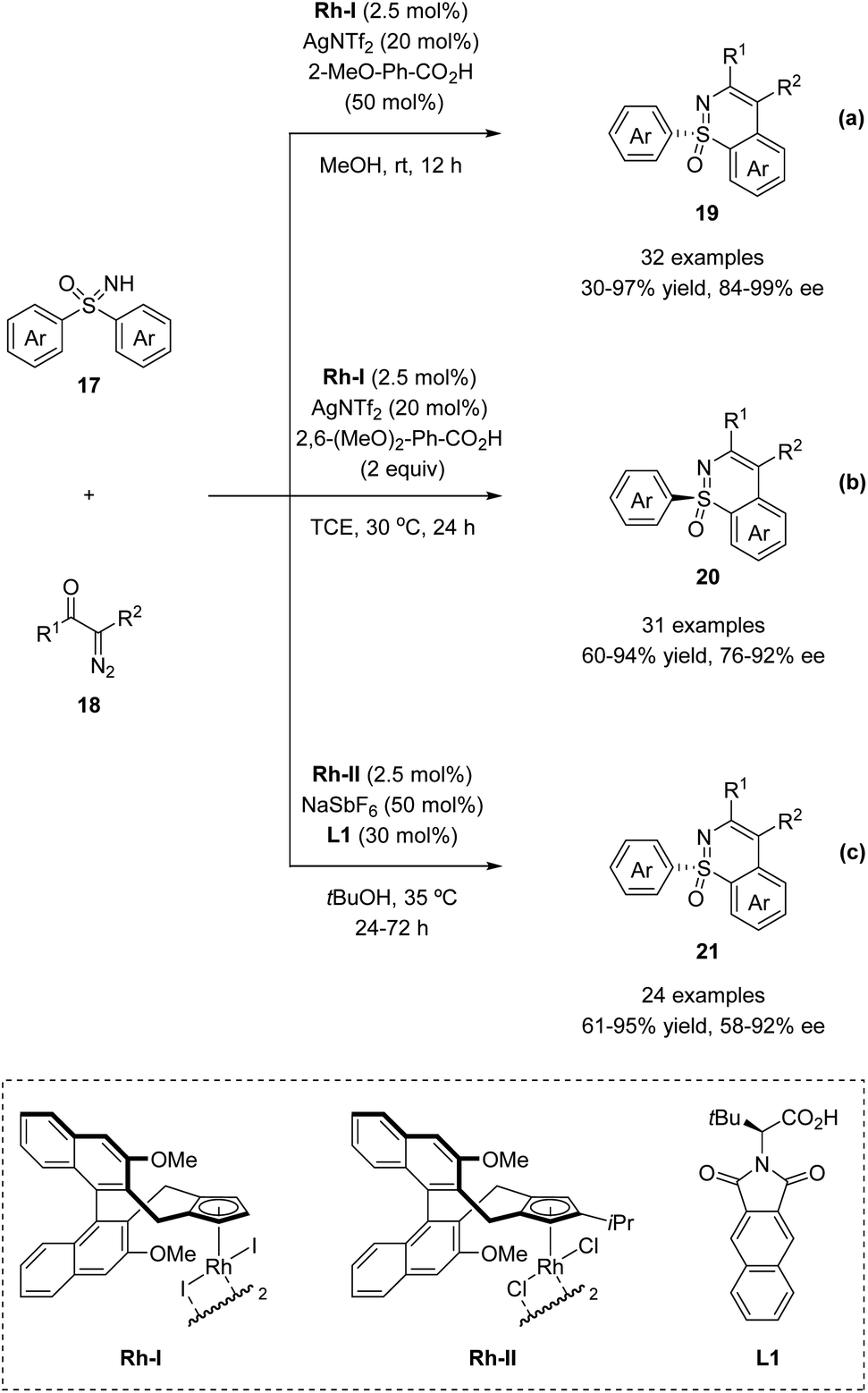
In 2018, the Li group and Cramer group independently reported the desymmetrization of diaryl sulfoximines under Rh(III)-catalyzed enantioselective annulative C–H functionalization with diazo compounds (Scheme 5).35,36
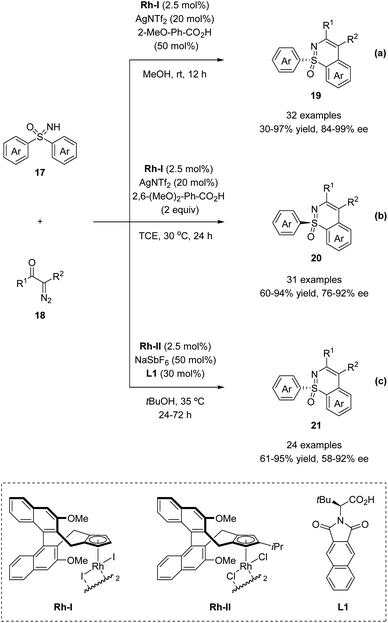 |
| | Scheme 5 CpxRh(III)-catalyzed enantioselective annulative C–H functionalization of diaryl sulfoximines. (a and b) Li's work toward (R) and (S)-products by altering carboxylic acid and solvent. (c) Cramer's work toward (R)-products. | |
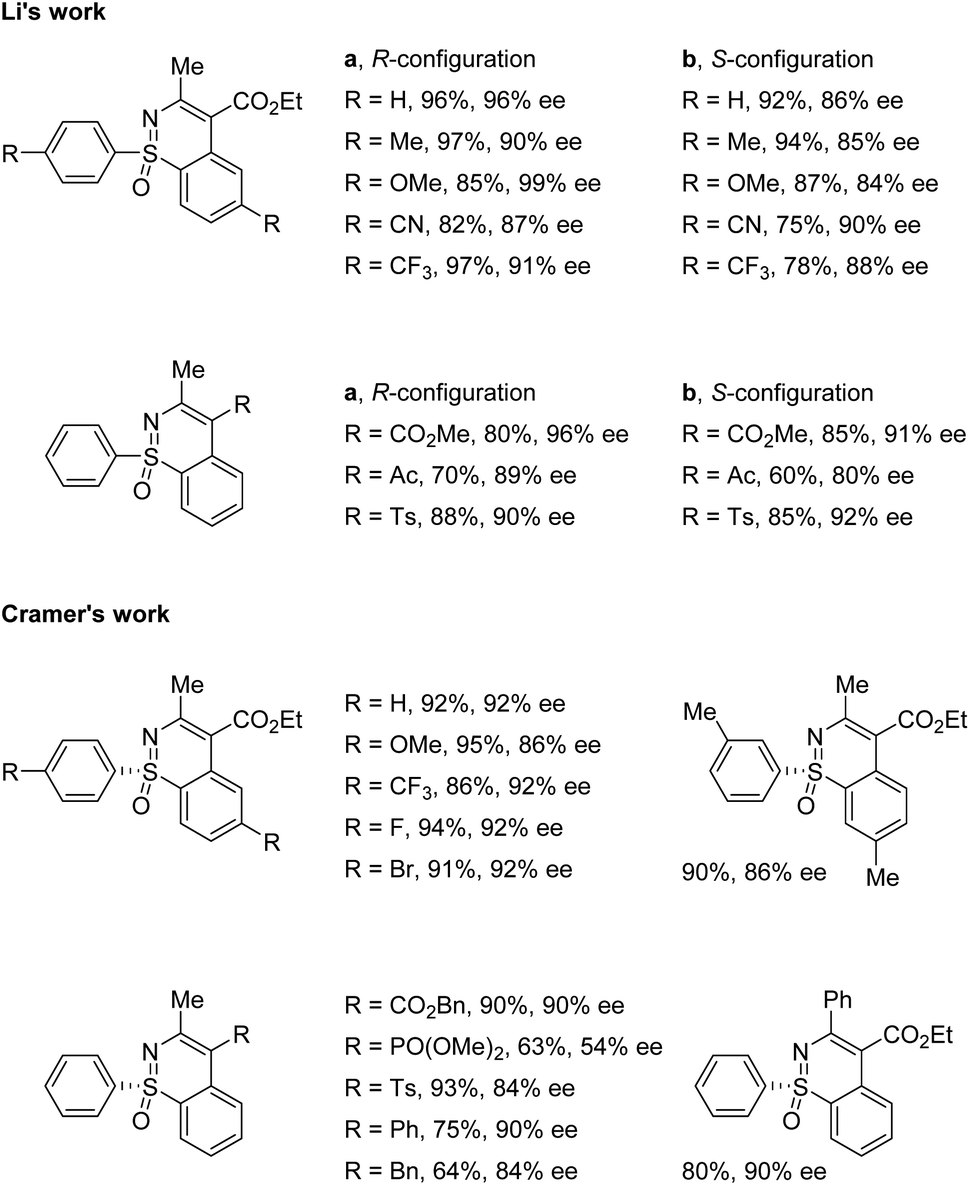
Li and co-workers applied the chiral CpxRh(III) catalyst Rh-I in combination with differently substituted achiral benzoic acid derivatives, achieving an enantiodivergent desymmetrization of sulfoximines 17. The use of Rh-I with 2-methoxybenzoic acid gave benzothiazines (R)-19 in high yields with good enantioselectivities (Scheme 5a). A broad scope of sulfoximines bearing various electron-donating, electron-withdrawing, and halogen substituted groups were coupled with diazo reagents successfully (Scheme 6). Interestingly, an optimal inversion of enantioselectivity was achieved when 2,6-dimethoxybenzoic acid was applied in the less polar solvent TCE (1,1,2,2-tetrachloroethane), and the opposite enantiomers (S)-20 could be obtained with moderate to good enantioselectivities (Scheme 5b).
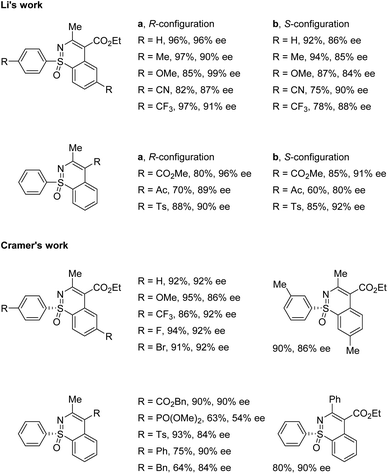 |
| | Scheme 6 Selected substrate scope. | |
Meanwhile, Cramer and co-workers applied their trisubstituted chiral Cpx ligand Rh-II in combination with chiral tert-leucine-derived ligand L1, also obtaining the annulative C–H functionalization products (R)-21 in high yields with good enantioselectivities (Scheme 5c). A variety of diaryl sulfoximines with different substitution patterns and a series of diazo compounds proved to be capable reaction partners for this transformation (Scheme 6).
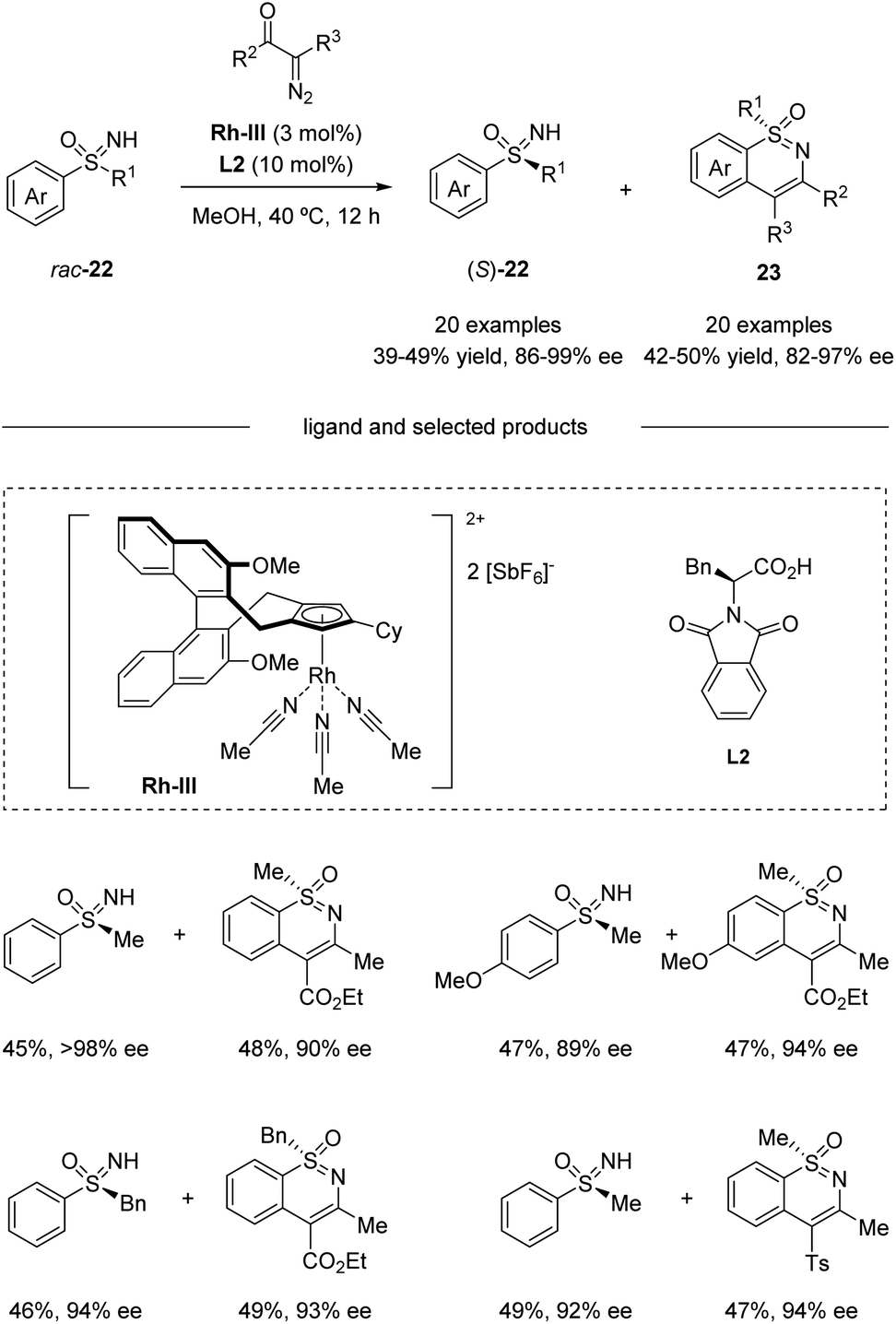
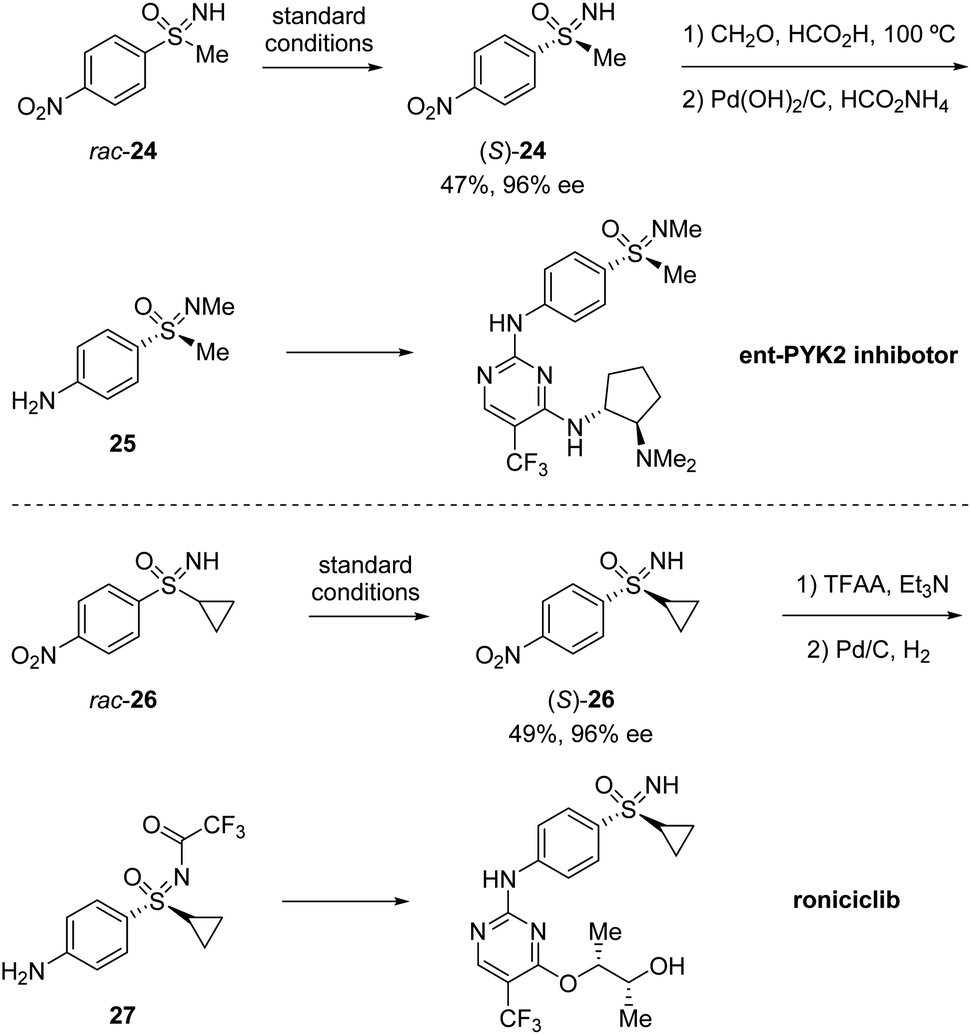
Based on their previous work, Cramer et al. further developed a kinetic resolution of alkyl aryl sulfoximines via cationic CpxRh(III)-catalyzed annulative C–H functionalization (Scheme 7).37 By using chiral Rh-III and phenylalanine-derived ligand L2, they realized the conversion of racemic alkyl aryl substituted sulfoximines 22 to cyclic benzothiazines 23 and maintained enantioenriched starting materials (S)-22 with high enantioselectivities. A variety of different aryl and alkyl substituted sulfoximines 22 and various diazo interceptors were well tolerated in the process. Additionally, the derivatization of the enantioenriched starting materials gave the precursors of bio-active molecules PYK2 inhibitor and roniciclib (Scheme 8).38,39
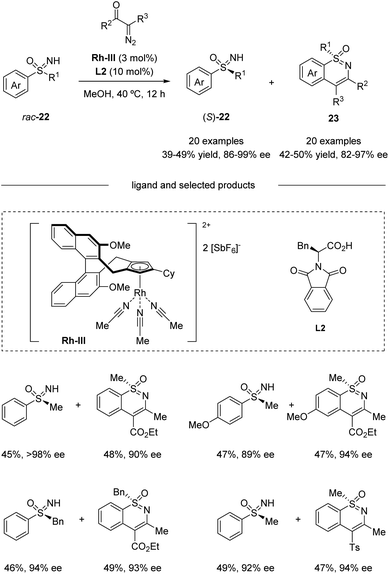 |
| | Scheme 7 Kinetic resolution of alkyl aryl sulfoximines via CpxRh(III)-catalyzed annulative C–H functionalization. | |
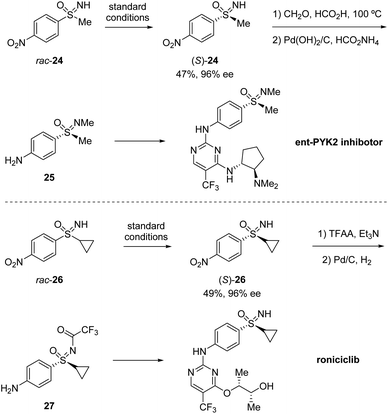 |
| | Scheme 8 Application of the kinetic resolution for bioactive compounds. | |
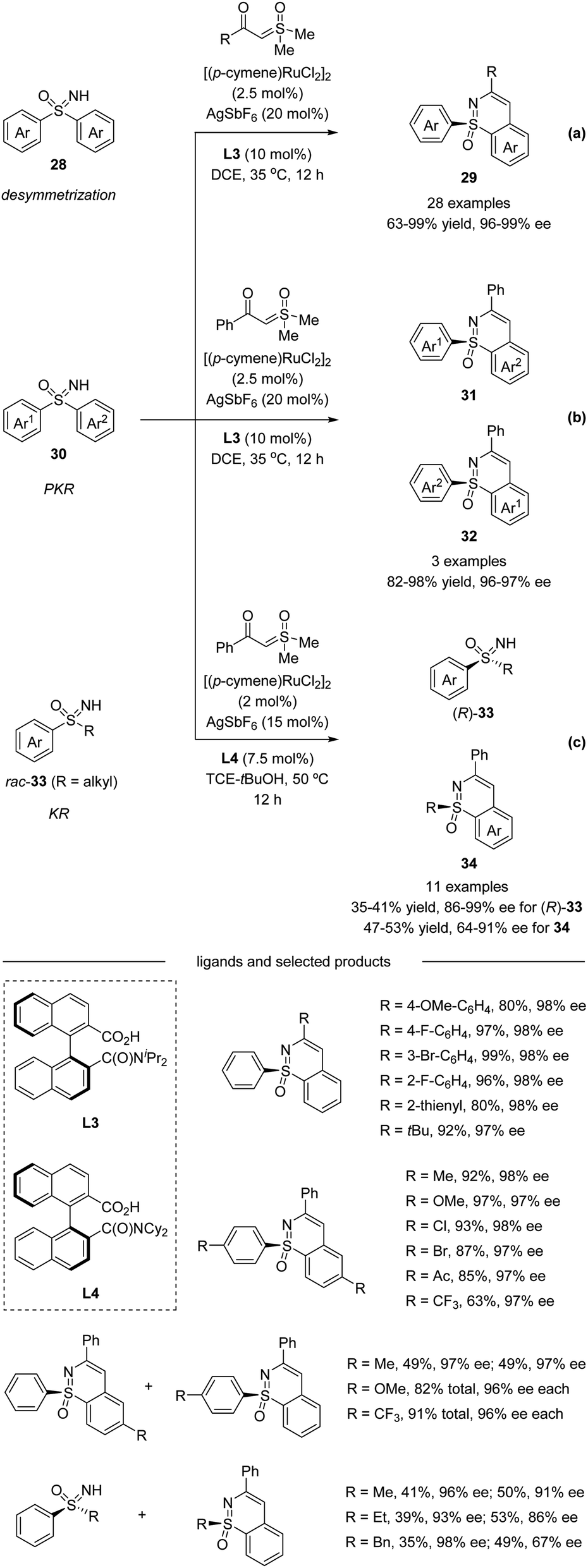
Very recently, Shi et al. described a Ru(II)-catalyzed enantioselective C–H activation/annulation of sulfoximines with α-carbonyl sulfoxonium ylides using C1-symmetric binaphthyl monocarboxylic acids as chiral ligands via desymmetrization (Scheme 9a), parallel kinetic resolution (PKR, Scheme 9b), and kinetic resolution (KR, Scheme 9c).40 A wide range of sulfoximines bearing various electron-donating groups and electron-withdrawing groups worked well with sulfoxonium ylides affording high yields with good to excellent enantioselectivities. α-Substituted benzoyl sulfoxonium ylides bearing electron-donating groups or halide substituents at the para, meta, and ortho positions of the aryl ring were all well-tolerated. α-Fatty acyl sulfoxonium ylides were also compatible with this transformation. Moreover, chiral sulfoximines obtained from kinetic resolution could be reduced stereospecifically to chiral sulfoxides. The inhibitors of human CYP24 hydroxylase could also be accessed from enantioenriched sulfoximines.
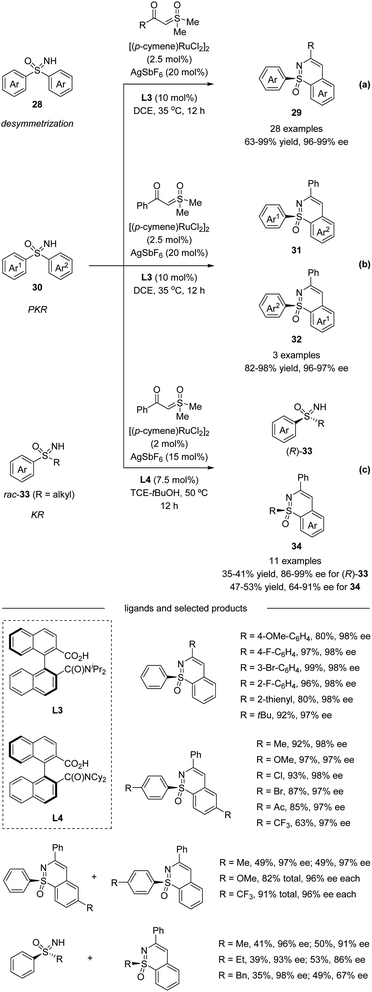 |
| | Scheme 9 Ru(II)-catalyzed enantioselective annulative C–H functionalization of diaryl sulfoximines and alkyl aryl sulfoximines. (a) Desymmetrization pathway. (b) Parallel kinetic resolution pathway. (c) Kinetic resolution pathway. | |
In terms of mechanism, the above sulfoximine directed enantioselective C–H functionalization reactions are proposed to begin with the formation of catalytically reactive species A. Then sulfoximine directed enantioselective C–H activation via concerted metalation/deprotonation (CMD) pathways gives metallacycle D. After that, carbene formation followed by migratory insertion of the M–aryl bond into the resulting carbene species leads to C–C bond formation. Finally, protonolysis and off-cycle condensation afford the annulated product H (Scheme 10).
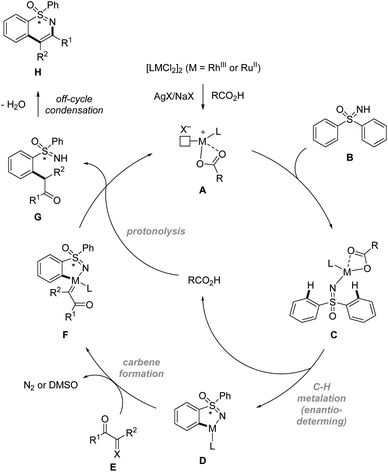 |
| | Scheme 10 Proposed mechanism of sulfoximine directed enantioselective C–H functionalization. | |
3. Utilization of sulfur stereocenters in asymmetric C–H functionalization
Sulfoxides are classical functional groups for directing the stoichiometric metalation41–45 and functionalization of C–H bonds. In recent years, a renaissance period in the application of sulfoxides arises from their versatility in directing C–H functionalization due to their unique reactivity. Furthermore, chiral sulfoxides have been considered as ideal ligand candidates for transition-metal-catalyzed asymmetric reactions due to their ready advantages of easy synthesis, stability, and special stereogenic control.
3.1 Chiral sulfoxide-directed diastereoselective C–H functionalization
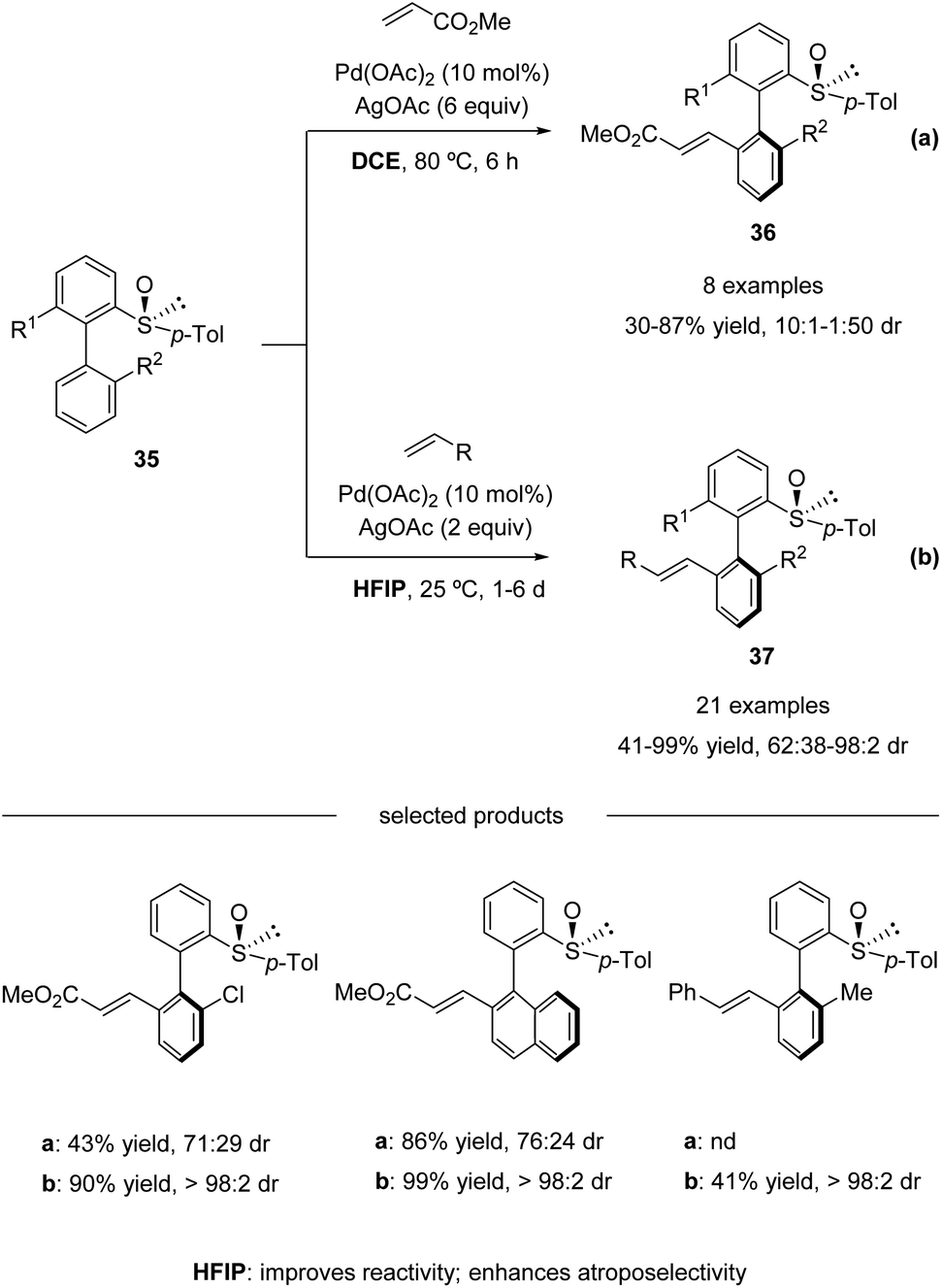
In 2013, Colobert and co-workers reported the first atropo-diastereoselective method for the synthesis of axially chiral biaryl scaffolds via C–H olefination using a chiral sulfoxide both as the directing group enabling the regioselective activation of a C–H bond and as the chiral auxiliary generating an asymmetric environment in the coordination sphere of the metal complex (Scheme 11a).46
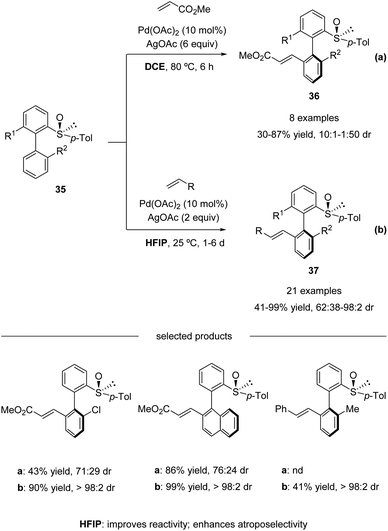 |
| | Scheme 11 Pd(II)-catalyzed sulfoxide-directed atropo-diastereoselective C–H olefination of biphenyls. (a) DCE as the solvent. (b) HFIP as the solvent. | |
Electron-deficient acrylates serve as efficient coupling partners, giving access to ortho-alkenylated products 36 in moderate to good yields and atropo-diastereoselectivities (Scheme 11a). However, a relatively high reaction temperature (80 °C), a large excess of silver-based oxidant (6 equiv. of AgOAc), and activated olefin coupling partners were needed in this process. To address these issues, they revisited their pioneering work and found that the use of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as the solvent enhanced the reactivity significantly. The amount of oxidant could be reduced, and the reactions could run at ambient temperature, albeit with a long reaction time. Both acrylates and styrenes worked properly in the solvent of HFIP. Under the update conditions, high yields with excellent atropo-diastereoselectivities were obtained (Scheme 11b).47
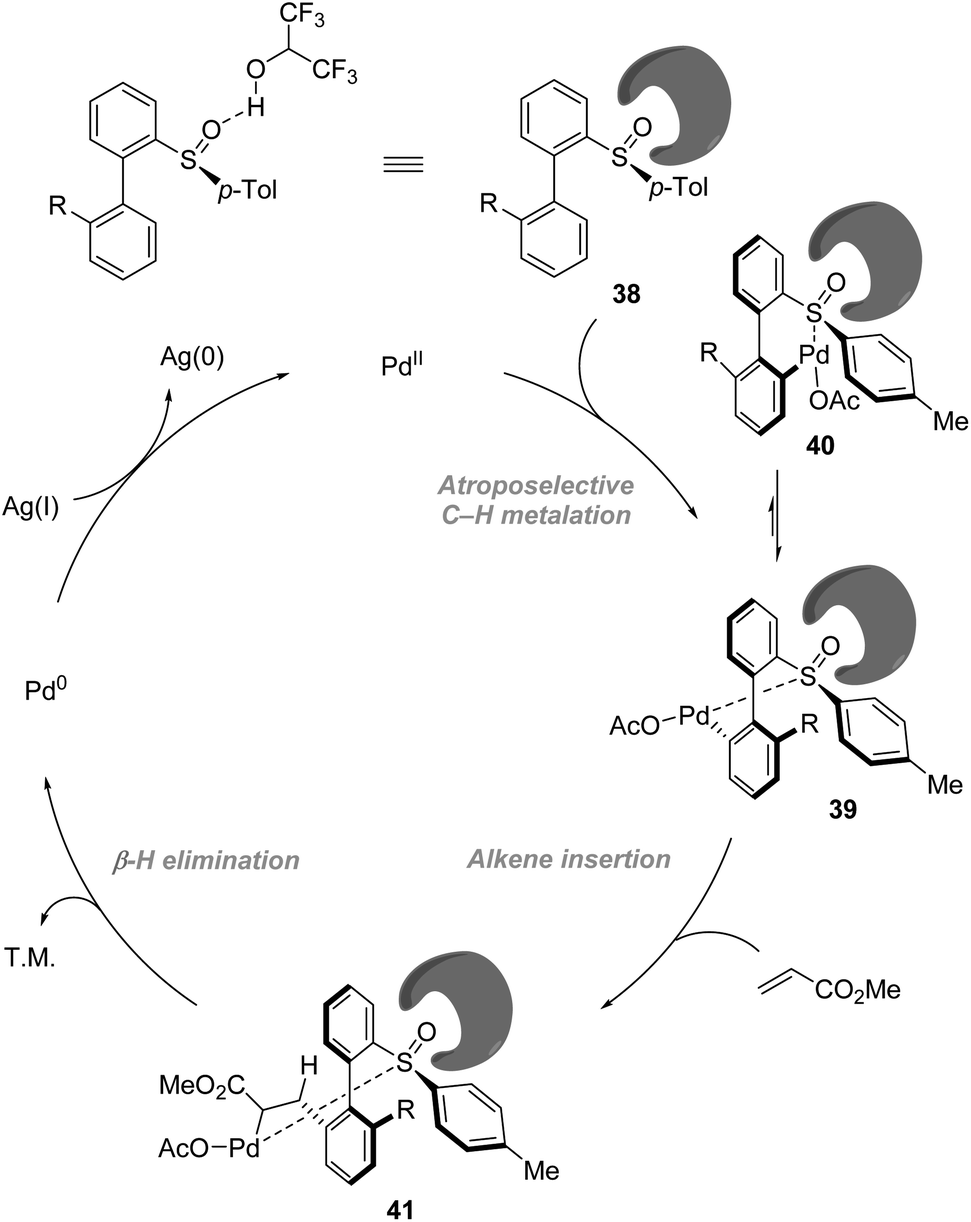
On the basis of NMR and IR studies, Colobert and co-workers showed that the hydrogen-bond donor solvent HFIP might coordinate to the oxygen of the sulfoxide group, affecting the electronics of the directing sulfoxide group and enhancing the rate determining C–H activation step (KIE = 2.2 in DCE vs. KIE = 5.9 in HFIP). This sulfoxide–HFIP interaction plays a key role in achieving high atropo-diastereoselectivity, which may be ascribed to the increased effective size of the sulfoxide directing group when coordinated to HFIP. The catalytic cycle starts with the coordination between Pd(OAc)2 and HFIP coordinated biarylsulfoxide substrate 38. Then irreversible, rate-determining C–H bond cleavage occurs, affording the favourable atropisomeric palladacyclic intermediate 39 with a decreased steric hindrance. Then, the insertion of the olefin coupling partner into the C–Pd bond, followed by β-H elimination, delivers an alkenylated product as a single atropisomer. The catalytic cycle is completed by the reoxidation of palladium(0) to palladium(II) by the silver salt (Scheme 12).
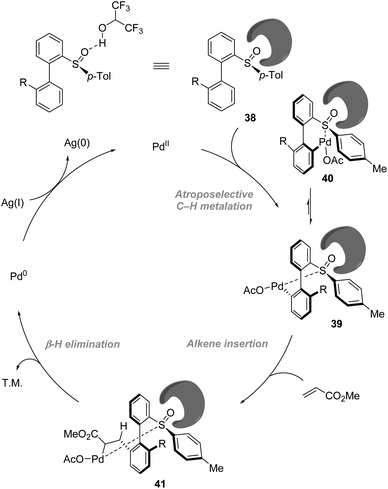 |
| | Scheme 12 Role of HFIP in the improvement of reactivity and enhancement of atropo-diastereoselectivity. | |
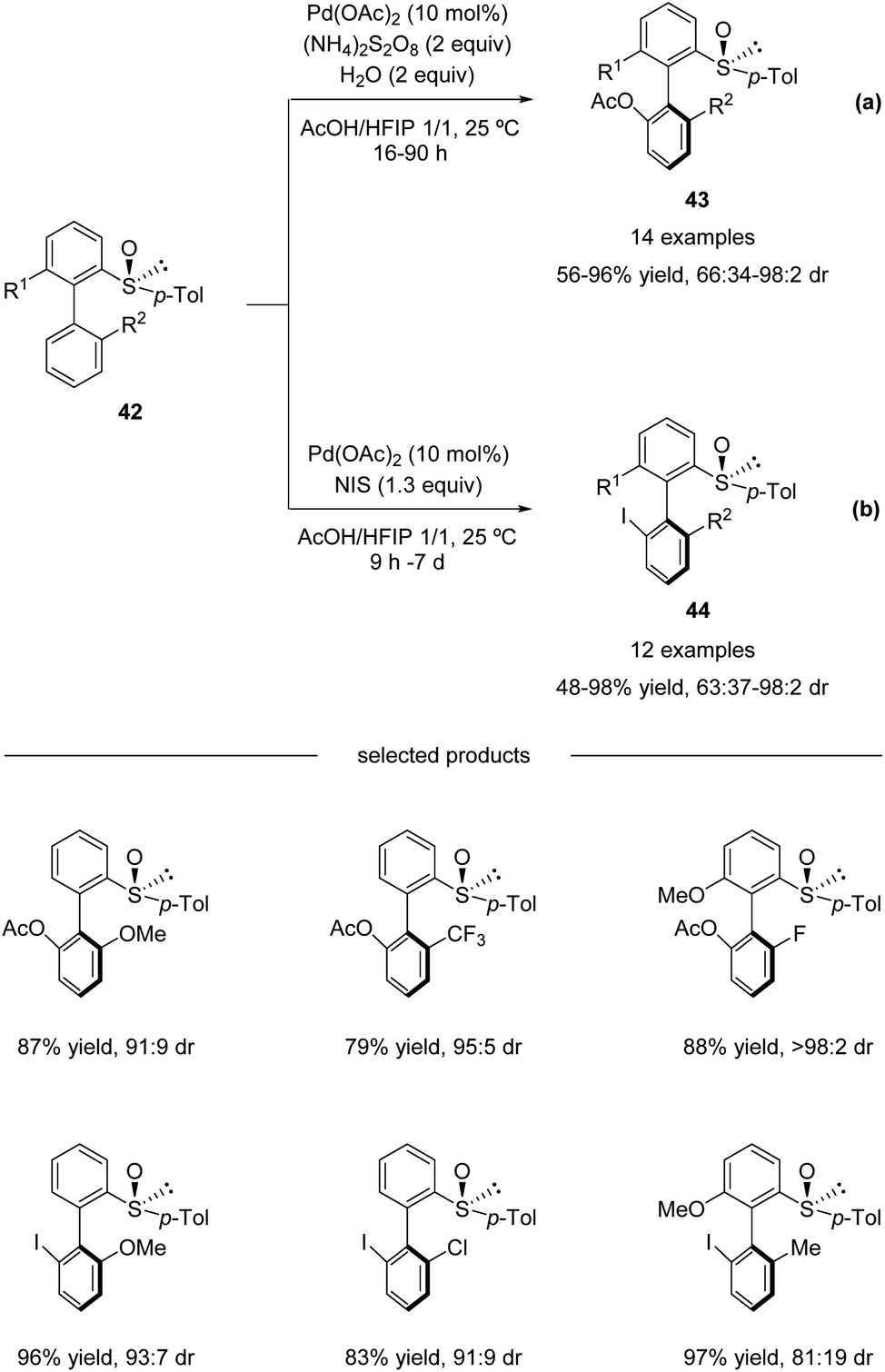
In continuation of their studies, Colobert and co-workers developed an atropo-diastereoselective C–H acetoxylation reaction (Scheme 13a).48 The use of acetic acid as a co-solvent in the presence of a catalytic amount of Pd(OAc)2 and 2 equivalents of ammonium persulfate as an oxidant permitted selective C–H oxidation to give the corresponding atropo-enriched acetate-substituted biaryls 43. This acetoxylation reaction is extremely robust, no precautions to avoid air or moisture are required, and the addition of a small amount of water is even beneficial. Under the optimized reaction conditions, a variety of substituted biaryl sulfoxides 42 underwent atroposelective acetoxylation with remarkable efficiency and stereoselectivity. Both electron-donating and electron-withdrawing substituents could be installed at either the 2′-position or the 6-position, and the corresponding C–O coupling products 43 were isolated in 56–96% yields with 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :34 to > 98:2 diastereoselectivities.
:34 to > 98:2 diastereoselectivities.
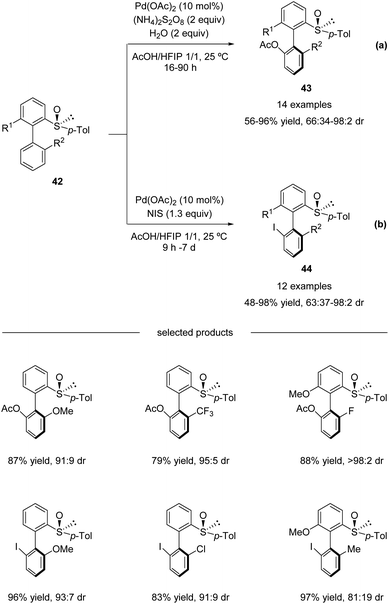 |
| | Scheme 13 Pd(II)-catalyzed sulfoxide-directed atropo-diastereoselective C–H acetoxylation and halogenation of biphenyls. (a) C–H acetoxylation. (b) C–H iodination. | |
Moreover, this methodology could also be efficiently used for the construction of halogenated atropo-pure products 44 (Scheme 13b). A simple replacement of the (NH4)2S2O8 oxidant by N-iodosuccinimide (NIS; 1.3 equiv.) led to a complete switch in the reactivity of the catalytic system, which allowed a smooth, mild, and highly diastereoselective C–I coupling reaction.
They undertook several mechanistic studies. First, a kinetic isotope effect of 1.09 was observed through two parallel reactions, verifying that C–H activation was not the rate-determining step (Scheme 14a). Second, when 6′-D-45 was reacted with Pd(OAc)2 in HFIP/AcOH, a significant D/H scrambling was observed, indicating that the C–H activation step was reversible (Scheme 14b). These results indicate that the overall outcome of this transformation is probably controlled by the kinetics of isomerization of the palladacycle and reductive elimination. They proposed that the steric hindrance around the biaryl axis was high enough to prevent atropo-epimerization (rotation around the biaryl axis) of the substrate 48. Then, the two diastereomers reacted with Pd(OAc)2 rapidly to generate the corresponding atropo-stereogenic palladacyclic intermediates Int-A and Int-B. The formation of such pallada-bridged cyclic species might increase the angle between the two aromatic units, thereby lowering the rotation barrier in this intermediate compared with that in the corresponding substrate, making rotation around the Ar–Ar bond possible. As the steric hindrance of such an intermediate was reduced when palladation occurred on the opposite side of the p-tolyl substituent of the sulfoxide moiety, the disfavored palladacycle Int-A underwent rapid atropo-epimerization to form Int-B. In this scenario, the reductive elimination from Int-A is unfavored and sufficiently slow to enable isomerization. Finally, reductive elimination from Int-B provided the product with high diastereo-selectivity (Scheme 14c).
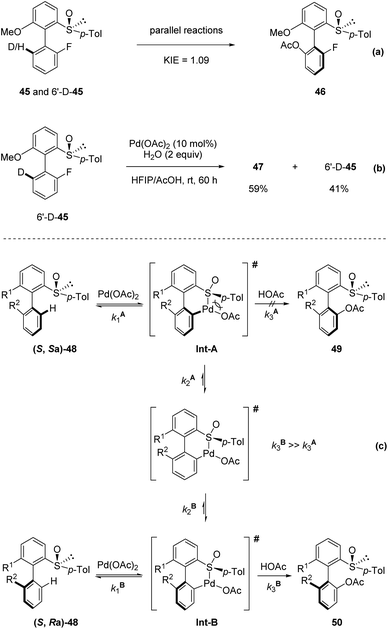 |
| | Scheme 14 Mechanistic studies and proposed mechanism. (a) KIE experiment. (b) D/H scrambling experiment. (c) Proposed mechanism. | |
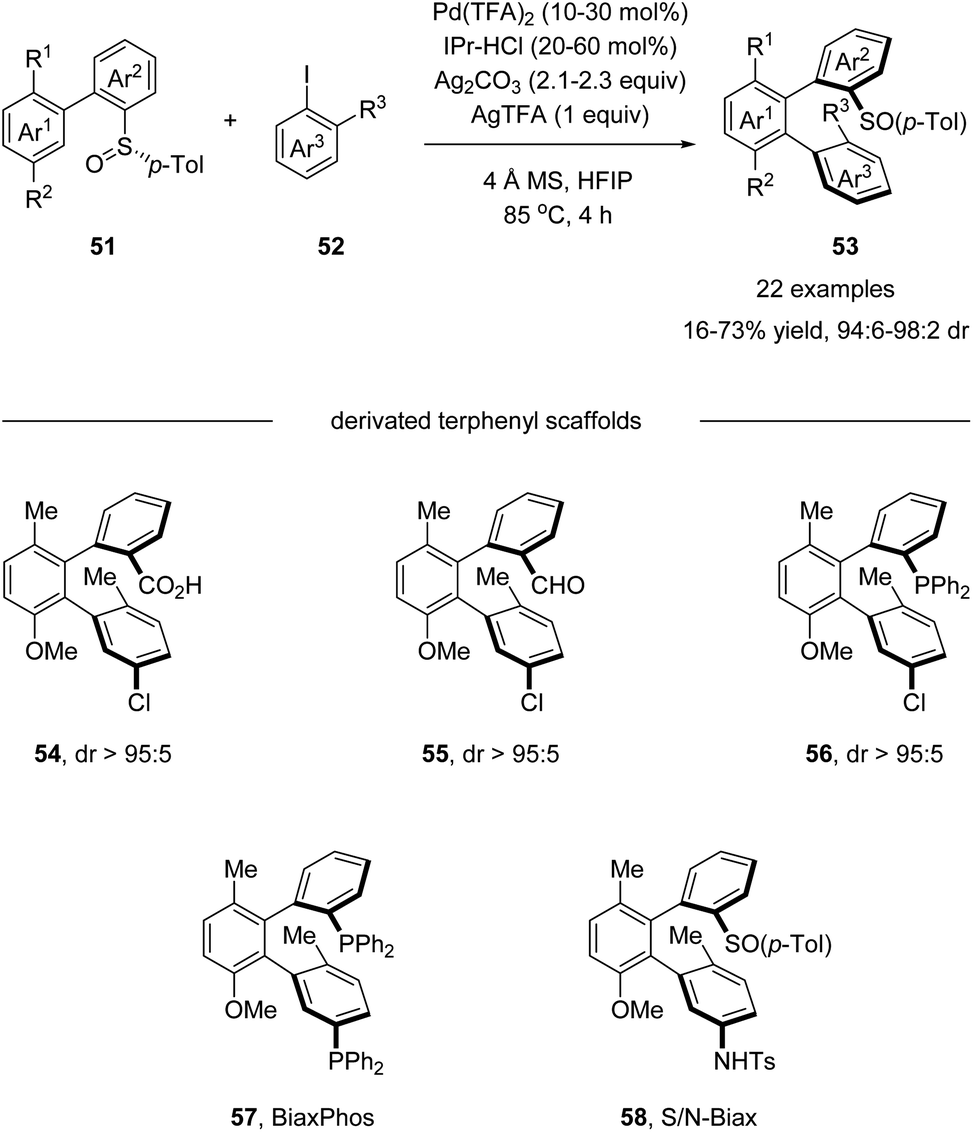
Later in 2018, Wencel-Delord and Colobert developed an elegant atropo-diastereoselective arylation reaction, affording terphenyls with two atropisomeric axes (Scheme 15).49 In this reaction, the sulfoxide moiety served as both the directing group and the chiral auxiliary, allowing the control of double chiral axes with excellent stereoselectivity in a single transformation without any other chiral ligands. Further elaboration of the arylation products generated a series of chiral terphenyl scaffolds, including diphosphine BiaxPhos 57 and S/N-Biax 58, which was successfully applied in the asymmetric hydrogenation of methyl (Z)-α-acetamidocinnamate and asymmetric 1,2-addition of Et2Zn to benzaldehyde.
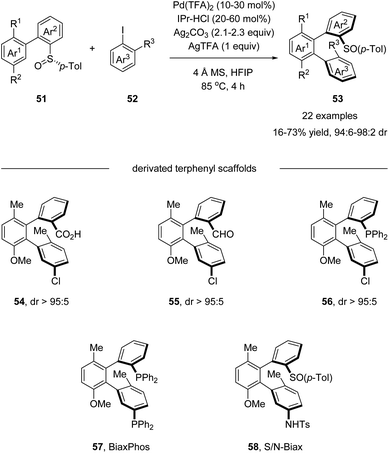 |
| | Scheme 15 Pd(II)-catalyzed sulfoxide-directed atropo-diastereoselective C–H arylation for the synthesis of terphenyls. | |
Combining D/H scrambling and KIE experiments, they proposed that the first stereogenic axis was established via a dynamic kinetic resolution (DKR) process. The diastereomer (Sa,S)-59 reacted irreversibly with the NHC–Pd catalyst, whereby the interactions between the p-Tol moiety and the NHC ligand are minimized. In contrast, the palladation of (Ra,S)-59 requires the accommodation of the bulky ligand and p-Tol moiety in the same plane, enforcing the isomerization of (Ra,S)-59 to (Sa,S)-59, forming the dominant palladacycle 61. Next, in the induction of the second stereogenic axis, the steric hindrance between the p-Tol moiety and the ortho substituent of the Ar–I coupling partner is the key element, making 63 the favored oxidative addition intermediate. Finally, reductive elimination from this sterically less congested Pd(IV) intermediate afforded the desired terphenyl product 64 (Scheme 16).
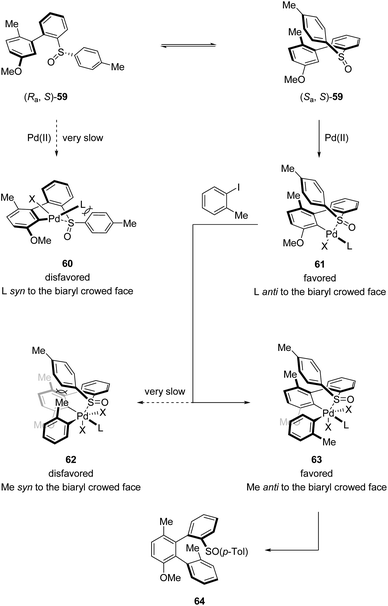 |
| | Scheme 16 Possible mechanism for stereoinduction. | |
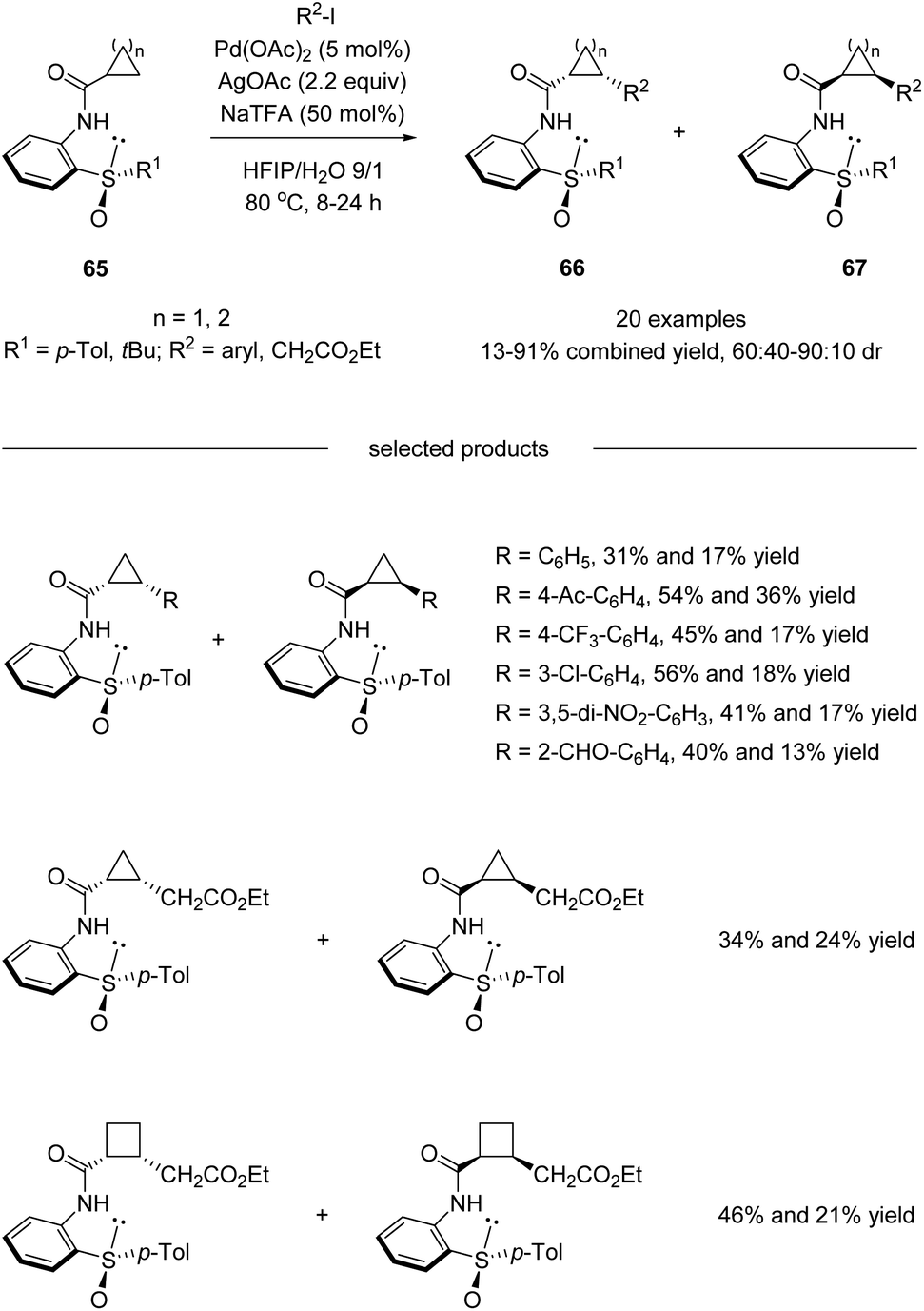
Besides diastereoselective C(sp2)–H functionalization mentioned above, more challenging asymmetric C(sp3)–H activation using sulfoxide as the directing chiral auxiliary has also been realized. In 2016, Wencel-Delord, Colobert and co-workers developed a chiral bidentate aniline sulfoxide-based directing group, which allowed a Pd(II)-catalyzed diastereoselective functionalization of the aliphatic C–H bond of cyclopropane and cyclobutane carboxylic acid derivatives (Scheme 17).50 Both arylation and more challenging alkylation reactions have been investigated, and moderate to good levels of diastereoselectivities could be obtained. It is worth mentioning that the APS (2-(p-tolylsulfinyl)aniline) directing group could be cleaved and recovered conveniently after the diastereoselective C(sp3)–H functionalization without the loss of optical purity.
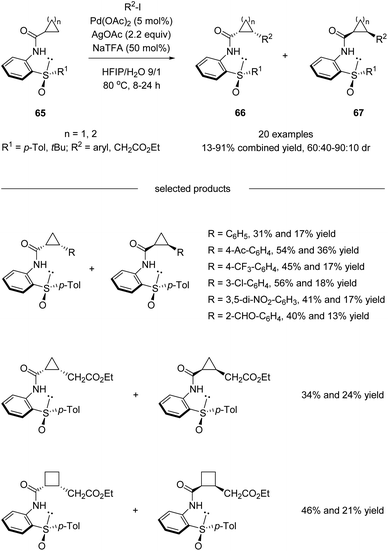 |
| | Scheme 17 Chiral APS as a recyclable auxiliary for asymmetric C(sp3)–H activation. | |
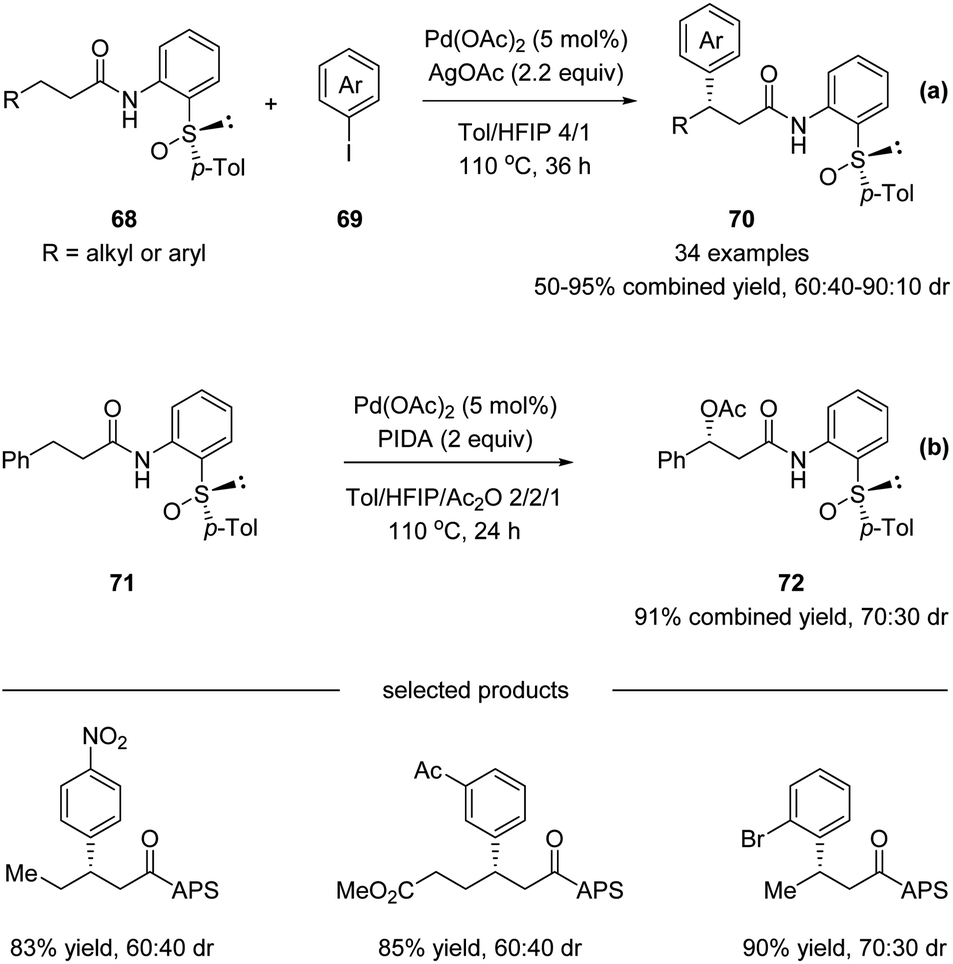
Later in 2017, Wencel-Delord, Colobert and co-workers reported another application of the APS auxiliary for a Pd(II)-catalyzed diastereoselective C(sp3)–H arylation of aliphatic amides 68 (Scheme 18a).51 A wide scope of aryl iodide coupling partners was tolerated for this diastereoselective functionalization of linear aliphatic chains. Moreover, a diastereoselective acetoxylation could also be conducted efficiently, delivering the desired C–O coupling products 72 in high yields yet with moderate dr (Scheme 18b). It is noteworthy that when using a simple amide substrate 73, a sequential C(sp3)–H arylation could be performed. One-pot, two-step diarylation of 73 afforded difunctionalized products 75 with two same aryl groups (Scheme 19a) and 78 with two different aryl groups (Scheme 19b).
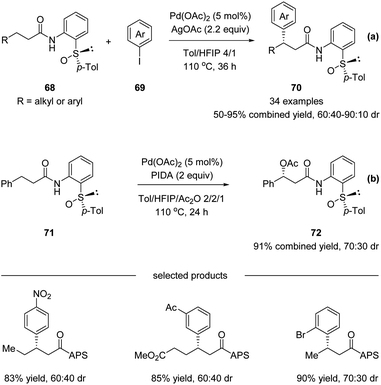 |
| | Scheme 18 Stereoselective APS-directed Pd(II)-catalyzed C–H arylation and acetoxylation of aliphatic amides. (a) C–H arylation. (b) C–H acetoxylation. | |
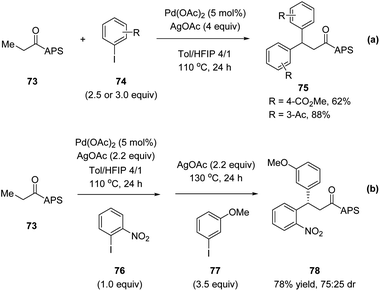 |
| | Scheme 19 Stereoselective APS-directed Pd(II)-catalyzed double C–H arylation. (a) Two same aryl groups. (b) Two different aryl groups. | |
3.2 Chiral sulfoxide ligand enabled enantioselective C–H functionalization
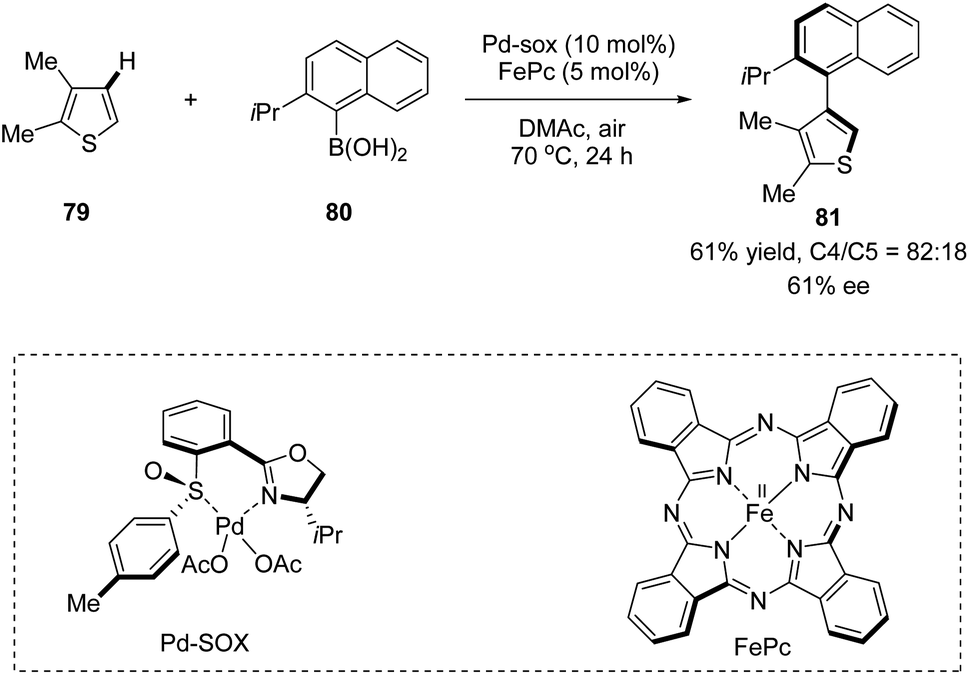
In 2013, Yamaguchi, Itami, and co-workers applied a chiral sulfoxide–oxazoline ligand in a Pd(II)/Fe(II) catalyzed C–H arylation reaction (Scheme 20).52 This is the very first example of the construction of axial chirality using sulfoxide–oxazoline as the chiral ligand. The cross-coupling of 2,3-dimethylthiophene 79 with (2-isopropylnaphthalen-1-yl)-boronic acid 80 in the presence of 10 mol% Pd–SOX and 5 mol% FePc as catalysts in dimethylacetamide (DMAc) at 70 °C under air gave the direct C–H arylation product 81 in 61% yield, with 82:18 rr and 61% ee.
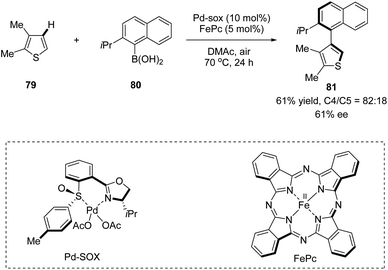 |
| | Scheme 20 Sulfoxide–oxazoline ligand enabled atroposelective aerobic oxidative C–H arylation. | |
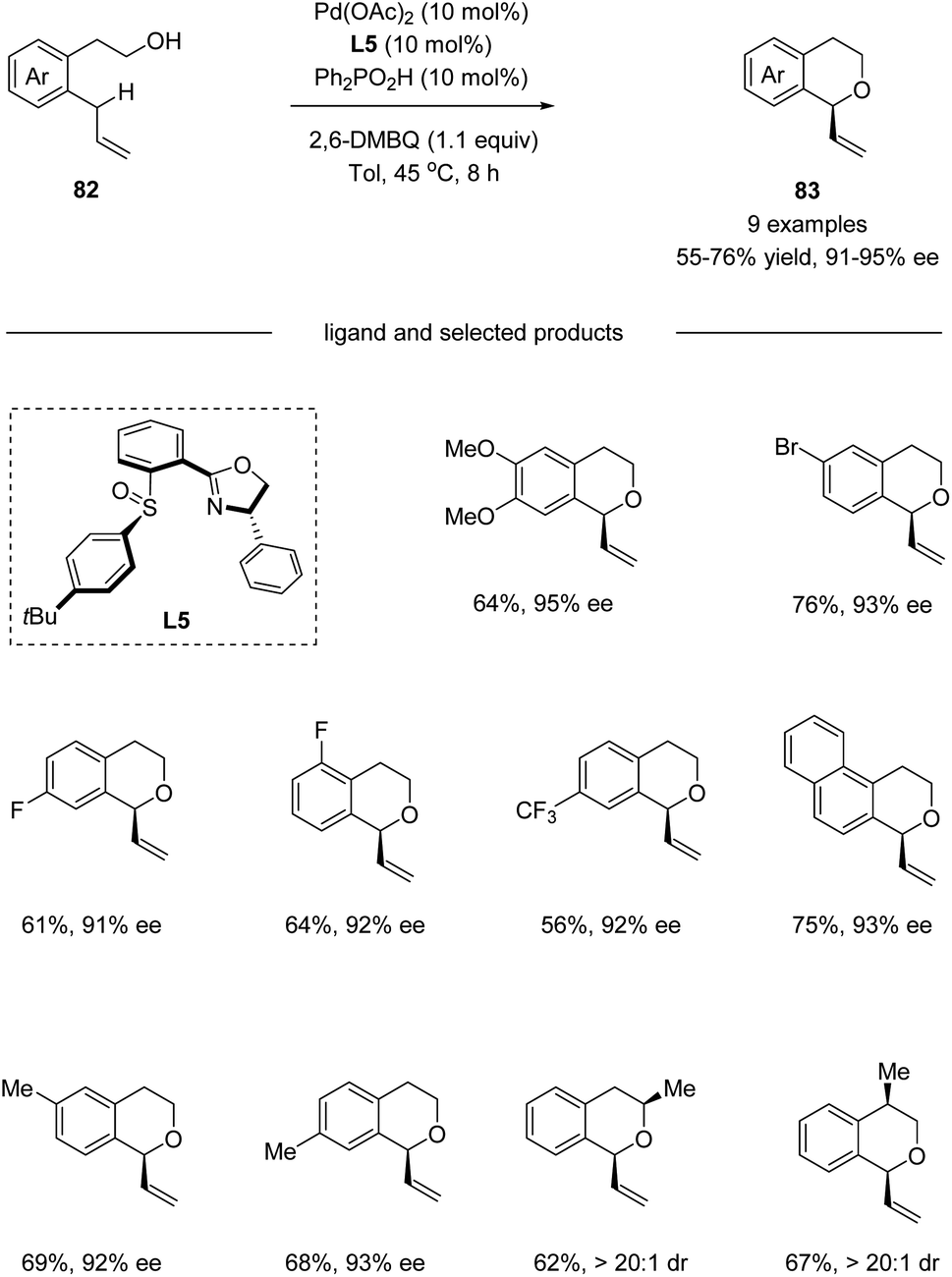
In 2016, by using chiral sulfoxide–oxazoline L5 as the ligand, White and co-workers developed a palladium(II)-catalyzed enantioselective intramolecular allylic C–H oxidation of prochiral C–H bonds (Scheme 21).53 Under optimized conditions, a series of chiral isochromans 83 could be obtained in good yields with excellent enantioselectivities. Taking advantage of this process, bio-active (S)-PNU-109291 and (S)-sonepiperazole were smoothly synthesized (Scheme 22).
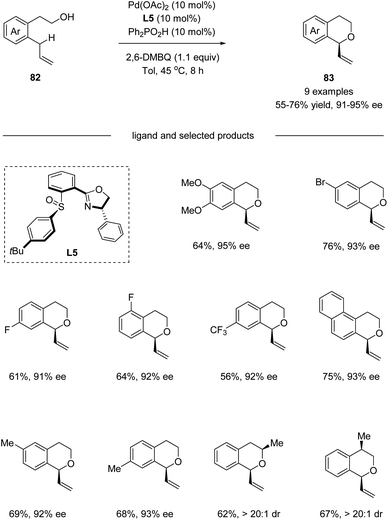 |
| | Scheme 21 Pd(II)/chiral sulfoxide catalyzed enantioselective allylic C–H oxidation. | |
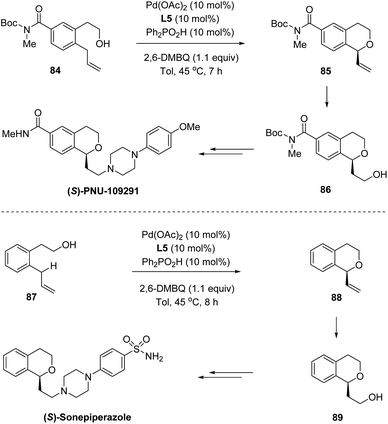 |
| | Scheme 22 Vinylisochromans as versatile chiral intermediates. | |
Further in 2018, White and co-workers reported the development of Pd(II)/cis-aryl sulfoxide–oxazoline (cis-ArSOX) catalysts for asymmetric allylic C–H alkylation of terminal olefins with a variety of synthetically versatile nucleophiles (Scheme 23).54 The modular, tunable, and oxidatively stable ArSOX scaffold is key to the broad substrate scope and high enantioselectivity (37 examples, 79–95% ee).
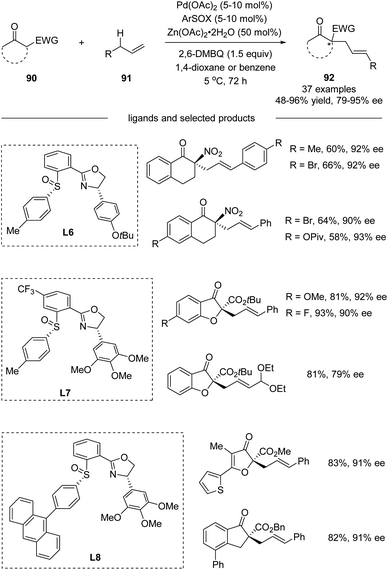 |
| | Scheme 23 Asymmetric intermolecular allylic C–H alkylation via Pd(II)/cis-ArSOX catalysis. | |
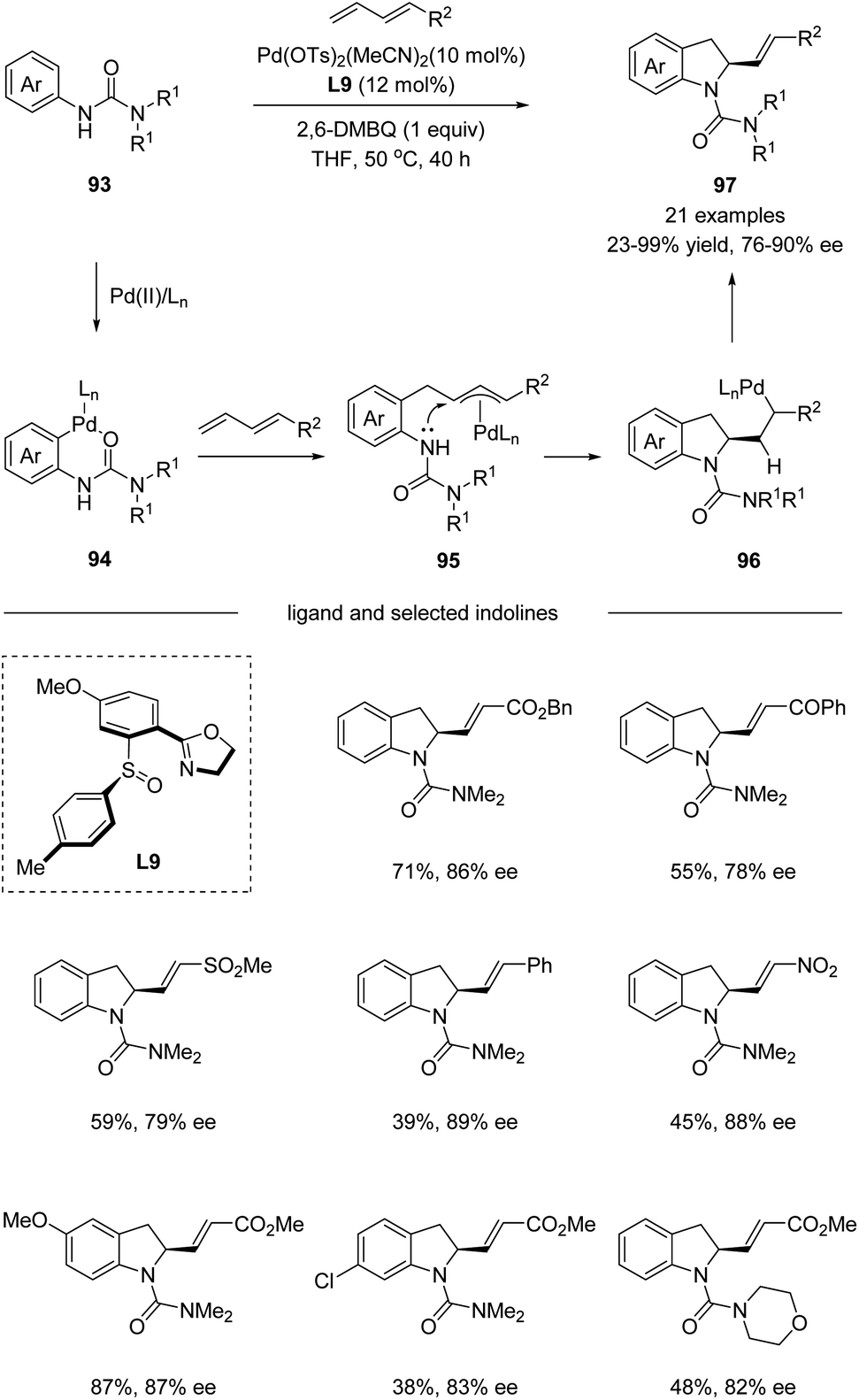
In 2017, Han and co-workers utilized chiral sulfoxide–oxazoline ligand (SOX) in a palladium-catalyzed cascade sp2 C–H activation/intramolecular asymmetric allylation reaction (Scheme 24).55 The process underwent a directing group assisted sp2 C–H palladation, followed by alkene insertion and asymmetric intramolecular allylation via an electrophilic palladation pathway. It is worth mentioning that the chiral sulfoxide–oxazoline ligand L9 bearing a single chiral center on the sulfur was identified as the optimal ligand for the reaction. A wide range of terminal substituted 1,3-dienes bearing ester, acyl, sulfonyl, and nitro groups reacted smoothly with a series of aryl ureas 93, affording the corresponding indoline products 97 with excellent E/Z selectivity and high enantioselectivities.
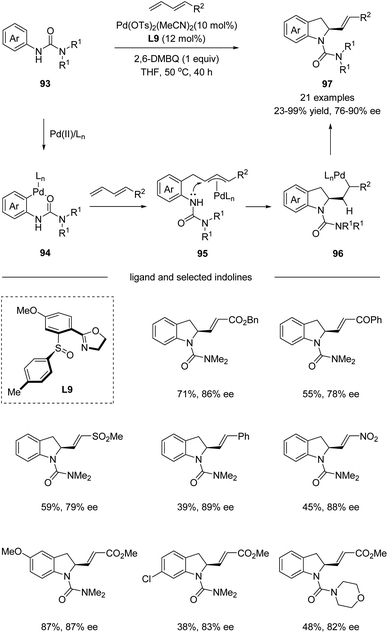 |
| | Scheme 24 Pd(II)-catalyzed cascade sp2 C–H activation/intramolecular asymmetric allylation. | |
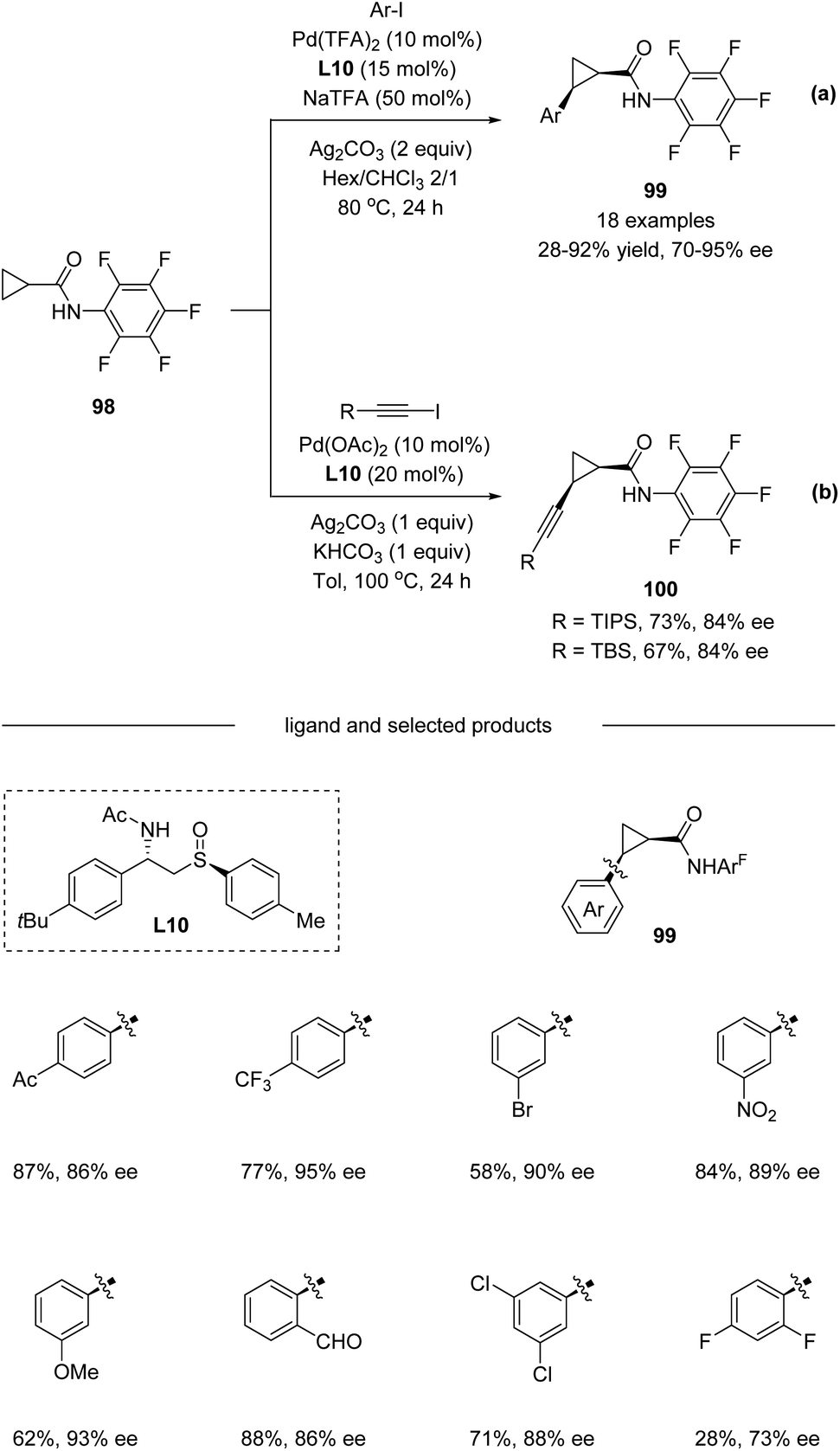
In 2019, Wencel-Delord, Colobert and co-workers developed a new family of chiral sulfoxide–amide ligands L10 for a Pd(II)-catalyzed asymmetric C(sp3)–H arylation of cyclopropanes (Scheme 25a).56 A variety of Ar–I bearing electron-donating substituents like OMe and electron-withdrawing substituents such as CF3, CO2Me, and Ac were all compatible in the reaction, affording corresponding products 99 with up to 95% ee. Moreover, direct alkynylation could also be performed with an enantioselectivity of up to 84% ee. Nevertheless, the substrate scope for alkynylation is limited in TIPS and TBS protected alkynyl iodide (Scheme 25b).
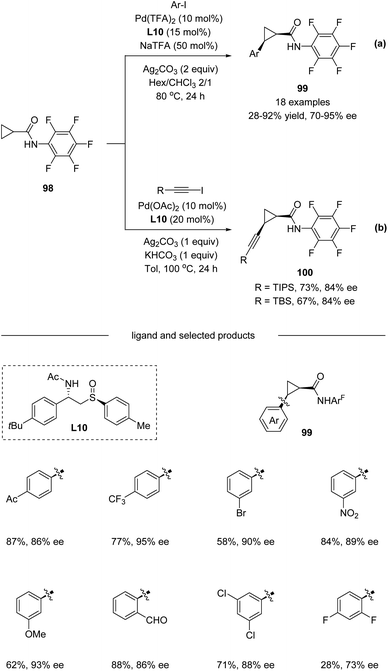 |
| | Scheme 25 Pd(II)-catalyzed C(sp3)−H arylation and alkynylation enable by chiral sulfoxide–amide ligands. (a) C–H arylation. (b) C–H alkynylation. | |
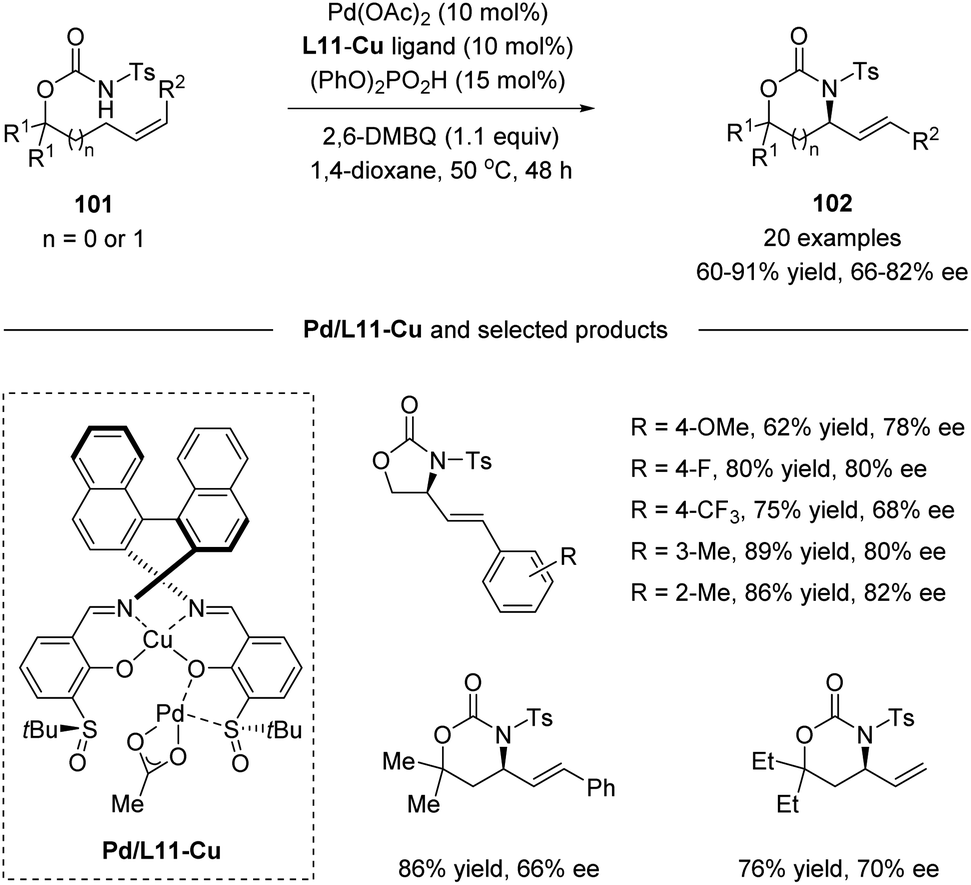
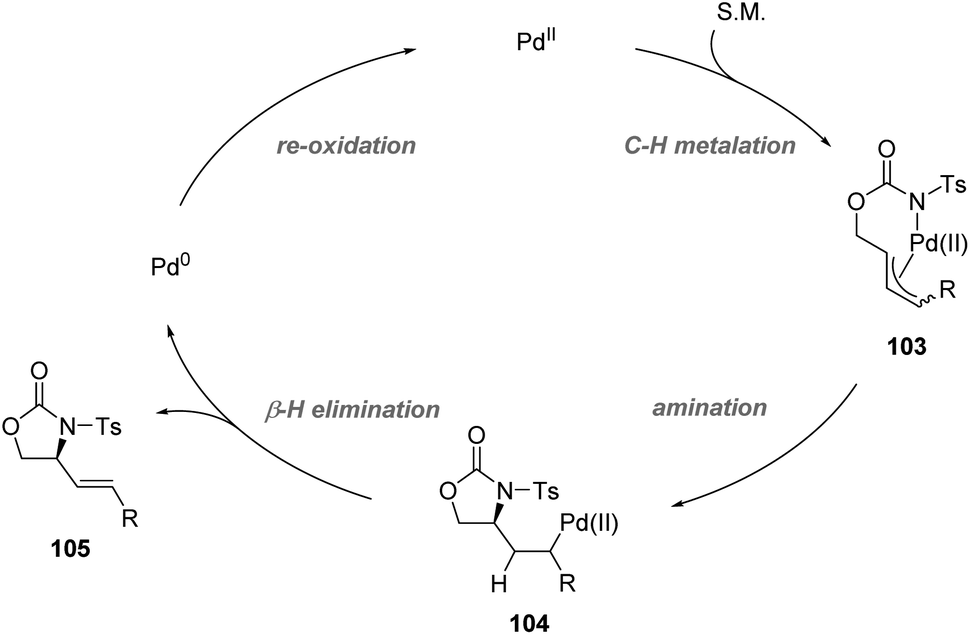
Recently, Yoshino, Matsunaga and co-workers demonstrated the synthetic utility of Cu-containing Schiff base/sulfoxide ligands for a Pd(II)-catalyzed asymmetric allylic C–H amination reaction (Scheme 26).57 The combination of the Schiff base-Cu(II)/sulfoxide with Pd(OAc)2 provided intramolecular C–H amination products with up to 82% ee. DFT calculations suggested that the chelation of the palladium complex by the sulfoxide and phenoxide lone pair is energetically favored, delivering the best outcome of enantioselectivity. Both internal and terminal alkenes were applicable in the transformation. The reaction is proposed to start with an allylic C–H activation, generating a Pd(II)-allyl intermediate 103. Then asymmetric amination followed by β-hydride elimination afforded the chiral product and Pd(0). Finally, the re-generation of Pd(II) finished the catalytic cycle (Scheme 27).
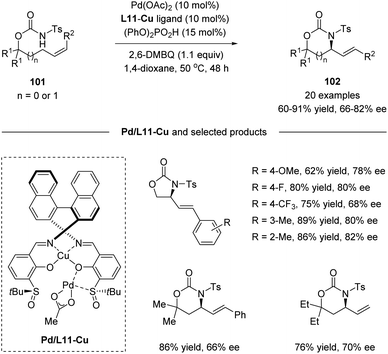 |
| | Scheme 26 Cu-containing Schiff base/sulfoxide ligands for a Pd(II)-catalyzed asymmetric allylic C–H amination. | |
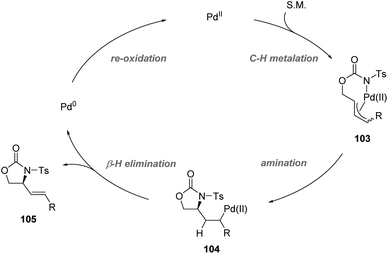 |
| | Scheme 27 Possible mechanism. | |
4. Conclusions
The past decade has witnessed the rapid development of the field of enantioselective C–H functionalization, and this transformation has gradually become a powerful tool for the synthesis of valuable chiral molecules. In particular, the use of sulfur-based directing groups has delivered a variety of new stereoselective C–H transformations via various activation modes, which have been incorporated into synthetic routes to many chiral organosulfur compounds. This perspective summarizes recent progress in the generation and utilization of sulfur stereogenic centers in asymmetric C–H transformations, including the construction of sulfur stereogenic centers of sulfoxide and sulfoximines via enantioselective C–H functionalization based on desymmetrization and (parallel) kinetic resolution strategies and chiral sulfoxide directing group or chiral sulfoxide ligand enabled stereoselective C–H functionalization.
Currently, the generation of sulfur stereocenters via C–H functionalization relies on desymmetrization and kinetic resolution pathways. New strategies are in high demand. The utilization of chiral sulfoxides as directing groups and ligands for stereoselective C–H functionalization has witnessed enormous growth, which allow the synthesis of optically active products with high yields and enantioselectivities. However, the scope of reactions using such directing groups and ligands remains limited, and the development of new types of ligands and reactions will be the topic of this area in the future. We believe that accessing and utilizing sulfur stereocenters via asymmetric C–H functionalization represents a continuously expanding field with exciting chances to develop new transformations with broad application potential.
Author contributions
C. H. and W. L. convinced the topic and assembled all the sections. W. L., J. K. and C. H. wrote and edited the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the start-up fund from the Southern University of Science and Technology, Shenzhen Science and Technology Innovation Committee (JCYJ20190809142809370), and Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002).
Notes and references
-
Handbook of C–H Transformations: Applications in Organic Synthesis, ed. G. Dyker, vol. 2, 2005 Search PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed.
- T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759 CrossRef CAS PubMed.
- J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS.
- J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- R. Bentley, Chem. Soc. Rev., 2005, 34, 609–624 RSC.
- P. Lindberg, A. Braendstroem, B. Wallmark, H. Mattsson, L. Rikner and K. J. Hoffmann, Med. Res. Rev., 1990, 10, 1–54 CrossRef CAS PubMed.
- I. Agranat and H. Caner, Drug Discovery Today, 1999, 4, 313–321 CrossRef CAS PubMed.
- A. R. Maguire, S. Papot, A. Ford, S. Touhey, R. O'Connor and M. Clynes, Synlett, 2001, 1, 41–44 CrossRef.
- A. Osorio-Lozada, T. Prisinzano and H. F. Olivo, Tetrahedron: Asymmetry, 2004, 15, 3811–3815 CrossRef CAS.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- Q. Tang, X. Yin, R. R. Kuchukulla, Q. Zeng and R. R. Kuchukulla, Chem. Rec., 2021, 21, 893–905 CrossRef CAS PubMed.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307–4359 RSC.
- N. Sundaravelu, S. Sangeetha and G. Sekar, Org. Biomol. Chem., 2021, 19, 1459–1482 RSC.
- M. Carmen Carreno, G. Hernandez-Torres, M. Ribagorda and A. Urbano, Chem. Commun., 2009, 41, 6129–6144 RSC.
- G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC.
- B. M. Trost and M. Rao, Angew. Chem., Int. Ed., 2015, 54, 5026–5043 CrossRef CAS PubMed.
- S. Otocka, M. Kwiatkowska, L. Madalinska and P. Kielbasinski, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed.
- T. Jia, M. Wang and J. Liao, Top. Curr. Chem., 2019, 377, 1–29 CrossRef CAS PubMed.
- I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef CAS PubMed.
- E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2010, 110, 4303–4356 CrossRef CAS PubMed.
- A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed.
- C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- K.-X. Tang, C.-M. Wang, T.-H. Gao, L. Chen, L. Fan and L.-P. Sun, Adv. Synth. Catal., 2019, 361, 26–38 CrossRef CAS.
- Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129–5133 CrossRef CAS PubMed.
- W. Liu, W. Yang, J. Zhu, Y. Guo, N. Wang, J. Ke, P. Yu and C. He, ACS Catal., 2020, 10, 7207–7215 CrossRef CAS.
- B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534–15538 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 15539–15543 CrossRef CAS PubMed.
- M. Brauns and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8902–8906 CrossRef CAS PubMed.
- D. P. Walker, M. P. Zawistoski, M. A. McGlynn, J.-C. Li, D. W. Kung, P. C. Bonnette, A. Baumann, L. Buckbinder, J. A. Houser, J. Boer, A. Mistry, S. Han, L. Xing and A. Guzman-Perez, Bioorg. Med. Chem. Lett., 2009, 19, 3253–3258 CrossRef CAS PubMed.
- M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed.
- T. Zhou, P.-F. Qian, J.-Y. Li, Y.-B. Zhou, H.-C. Li, H.-Y. Chen and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 6810–6816 CrossRef CAS PubMed.
- S. Ogawa and N. Furukawa, J. Org. Chem., 1991, 56, 5723–5726 CrossRef CAS.
- C. Quesnelle, T. Iihama, T. Aubert, H. Perrier and V. Snieckus, Tetrahedron Lett., 1992, 33, 2625–2628 CrossRef CAS.
- B. Ferber and H. B. Kagan, Adv. Synth. Catal., 2007, 349, 493–507 CrossRef CAS.
- J. L. G. Ruano and A. M. M. Castro, Heteroat. Chem., 2007, 18, 537–548 CrossRef CAS.
-
Organosulfur Chemistry in Asymmetric Synthesis, ed. T. Toru and C. Bolm, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS.
- Q. Dherbassy, G. Schwertz, M. Chesse, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2016, 22, 1735–1743 CrossRef CAS PubMed.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed.
- Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef CAS PubMed.
- S. Jerhaoui, F. Chahdoura, C. Rose, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2016, 22, 17397–17406 CrossRef CAS PubMed.
- S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2017, 23, 15594–15600 CrossRef CAS PubMed.
- K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 RSC.
- S. E. Ammann, W. Liu and M. C. White, Angew. Chem., Int. Ed., 2016, 55, 9571–9575 CrossRef CAS PubMed.
- W. Liu, S. Z. Ali, S. E. Ammann and M. C. White, J. Am. Chem. Soc., 2018, 140, 10658–10662 CrossRef CAS PubMed.
- S.-S. Chen, M.-S. Wu and Z.-Y. Han, Angew. Chem., Int. Ed., 2017, 56, 6641–6645 CrossRef CAS.
- S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, ACS Catal., 2019, 9, 2532–2542 CrossRef CAS.
- Y. Bunno, Y. Tsukimawashi, M. Kojima, T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 2663–2668 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*