
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IPr# – highly hindered, broadly applicable N-heterocyclic carbenes†
Qun
Zhao‡
a,
Guangrong
Meng‡
a,
Guangchen
Li‡
a,
Carol
Flach
 a,
Richard
Mendelsohn
a,
Roger
Lalancette
a,
Roman
Szostak
b and
Michal
Szostak
*a
a,
Richard
Mendelsohn
a,
Roger
Lalancette
a,
Roman
Szostak
b and
Michal
Szostak
*a
aDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, NJ 07102, USA. E-mail: michal.szostak@rutgers.edu
bDepartment of Chemistry, Wroclaw University, F. Joliot-Curie 14, Wroclaw 50-383, Poland
First published on 2nd July 2021
Abstract
IPr (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) represents the most important NHC (NHC = N-heterocyclic carbene) ligand throughout the field of homogeneous catalysis. Herein, we report the synthesis, catalytic activity, and full structural and electronic characterization of novel, sterically-bulky, easily-accessible NHC ligands based on the hash peralkylation concept, including IPr#, Np# and BIAN-IPr#. The new ligands have been commercialized in collaboration with Millipore Sigma: IPr#HCl, 915653; Np#HCl; 915912; BIAN-IPr#HCl, 916420, enabling broad access of the academic and industrial researchers to new ligands for reaction optimization and screening. In particular, the synthesis of IPr# hinges upon cost-effective, modular alkylation of aniline, an industrial chemical that is available in bulk. The generality of this approach in ligand design is demonstrated through facile synthesis of BIAN-IPr# and Np#, two ligands that differ in steric properties and N-wingtip arrangement. The broad activity in various cross-coupling reactions in an array of N–C, O–C, C–Cl, C–Br, C–S and C–H bond cross-couplings is demonstrated. The evaluation of steric, electron-donating and π-accepting properties as well as coordination chemistry to Au(I), Rh(I) and Pd(II) is presented. Given the tremendous importance of NHC ligands in homogenous catalysis, we expect that this new class of NHCs will find rapid and widespread application.
Introduction
N-Heterocyclic carbenes (NHCs) have emerged as tremendously valuable ligands in homogeneous catalysis.1,2 The broad application of NHC ligands is a consequence of strong σ-donation of the carbene center3 and variable steric bulk of wingtip groups4 that are often not easily attainable using other classes of ligands, including phosphines. Furthermore, through exploiting the flexible steric bulk of the wingtips, NHC ligands allow for kinetic stabilization of metals and intermediates at unusual oxidation states,5 while their well-defined topology has found widespread application in fine-tuning of reactivity at the metal center.6In this context, by far the most important NHC ligand in the field of homogeneous catalysis is bulky IPr (Fig. 1, 1) reported by Nolan and Arduengo in 1999.1,2 While its synthesis from Dipp (Dipp = 2,6-diisopropylaniline) is facile,7 the problem lies in the preparation of Dipp precursor. The preparation of Dipp is severely limited by challenges in controlling the alkylation selectivity, and the most common industrial route exploits lengthy and inflexible route through phenol alkylation.8
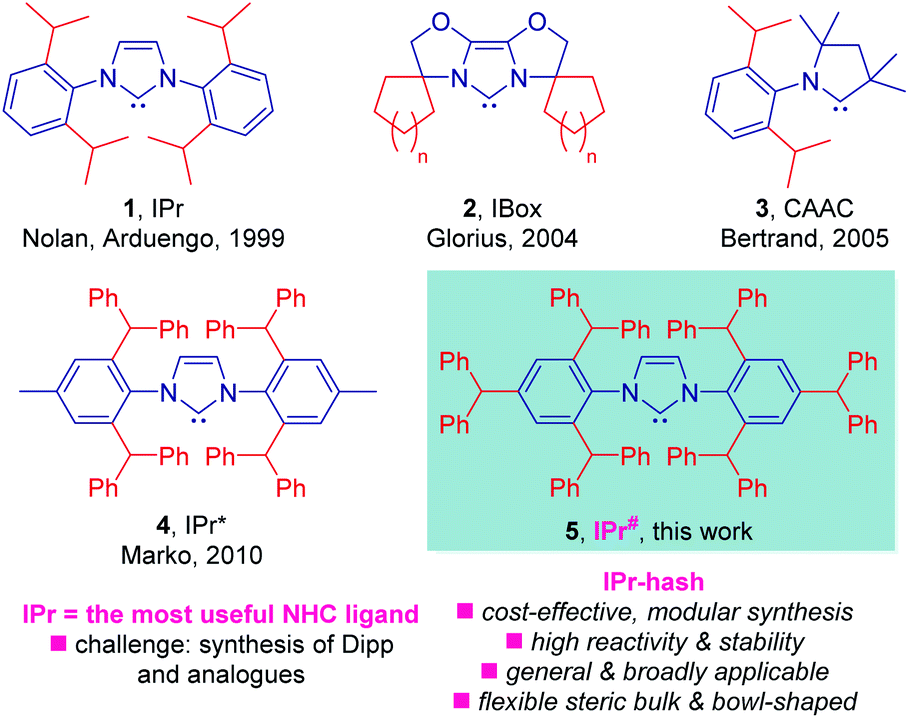 | ||
| Fig. 1 State-of-the-art of sterically-demanding N-heterocyclic carbenes in catalysis. Note that IPr-hash ligands are commercially available from Millipore Sigma (IPr#HCl, 915653; Np#HCl; 915912; BIAN-IPr#HCl, 916420). | ||
Taking inspiration from elegant studies in NHC design by Nolan,9 Glorius,10 Bertrand,11 Marko12 and others13 (Fig. 1), as well as following our interest in NHC catalysis14 and the synthesis of N-containing molecules,15 we sought to expand the concept of sterically-hindered, easily accessible NHC ligands. We considered that a bulky version of IPr, wherein the amine is prepared directly via modular alkylation of aniline, an industrial chemical that is available in bulk16 in a cost-effective manner should be accessible. Further, we hoped to demonstrate the generality of this approach through facile synthesis of NHC analogues with varying steric and electronic properties of the NHC topology as well as generality of the ligand design in cross-couplings by numerous bond breaking events.
Results and discussion
The synthesis of IPr# (Scheme 1, 5) was selected as our starting point. It should be noted that the direct three-fold peralkylation of aniline is significantly more challenging than the alkylation of para-blocked toluidine as a consequence of N-/C-alkyl migration.17 After experimentation, we were pleased to find that 2,4,6-tribenzhydrylaniline 7 could be prepared in 79% yield (93 g, 200 mmol scale) by adding HCl (1.0 equiv.) to a solution of aniline (1.0 equiv.), diphenylmethanol (3.5 equiv.) and ZnCl2 (0.5 equiv.) at 160 °C.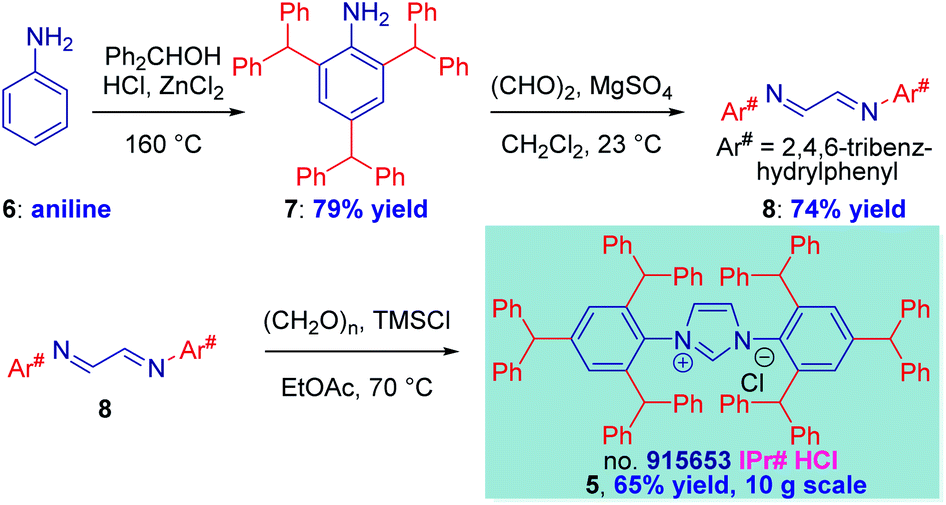 | ||
| Scheme 1 Synthesis of IPr# and the Diimine Precursors. Conditions: (a) 6 (1.0 equiv.), Ph2CHOH (3.5 equiv.), ZnCl2 (0.5 equiv.), HCl (36%, aq, 1.0 equiv.), 160 °C. (b) 7 (1.0 equiv.), (CHO)2 (40%, aq, 0.5 equiv.), MgSO4 (2.5 equiv.), 23 °C. (c) 8 (1.0 equiv.), (CH2O)n (1.1 equiv.), TMSCl (1.1 equiv.), EtOH, 70 °C. | ||
Routinely, 70–75% yields were obtained on 10–20 mmol scale. This represents a significant improvement over the previous Friedel–Crafts method involving addition of a mixture of ZnCl2 and HCl at 160 °C,12 which led to irreproducible results. We note that alkylation of aniline at para position occurs last; the N-alkylation products were not observed. The synthesis of diimine 8 was smoothly effected by reacting glyoxal (1.0 equiv.) with amine 7 (2.0 equiv.) and MgSO4 (2.5 equiv.) (70 g, 160 mmol scale). We found that the reaction was slower than in the synthesis of IPr*,12 suggesting more pronounced steric character. The optimum results were obtained by monitoring the reaction progress by 1H NMR, and driving the reaction to completion by slightly elevating the temperature (40 °C) or adding slight excess of glyoxal, if needed. The diimine was formed as exclusively the s-trans isomer. The cyclization step occurred smoothly upon exposing the diimine (1.0 equiv.) and paraformaldehyde (1.1 equiv.) to TMSCl (1.1 equiv.) in EtOAc at 70 °C (25 g, 40 mmol scale).18 We found that this procedure allows for much milder cyclization to 5 than the HCl/ZnCl2 combination,12 which again proved problematic for large-scale runs and was riddled with retro-Friedel–Crafts products. The TMSCl procedure could be conveniently followed by color change from yellow to light grey, indicating completion of the reaction.
Crucially, the entire sequence 6 → 5 is highly practical: (1) the three steps do not require purification of the intermediates, (2) the final product is obtained after facile work-up (filtration), (3) the procedure uses industrial chemicals available in bulk, (4) the sequence is routinely performed within two days.
With multigram access to IPr# secured, we next comprehensively evaluated steric and electronic properties of this novel NHC ligand. As shown in Scheme 2, the gold complex [Au(IPr#)Cl] (9) was prepared using the general method,19 while Rh(I) complexes, [Rh(IPr#)(CO)2Cl] (10) and [Rh(IPr#)(acac)CO] (11) were prepared after generating the free carbene in situ by deprotonation of IPr#HCl with a slight excess of either KHMDS or KOtBu. The Rh(I) complex (10) could be prepared either directly by using rhodium dicarbonyl chloro dimer [{Rh(CO)2(μ-Cl)}2] (path b) or in a mild two-step procedure via [Rh(IPr#)(cod)Cl]20 and the reaction with carbon monoxide (path c). We have also generated the selenium adduct [Se(IPr#)] (12) by adding the free carbene generated in situ to excess of selenium.21 The Pd(II) complex [Pd(IPr#)(cin)Cl] (13) was prepared by generating the free carbene in situ and coordinating to the palladium cinnamyl dimer [{Pd(cin)(μ-Cl)}2].22 Importantly, all complexes were found to be stable to air and moisture. Complexes 9 and 13 were fully characterized by X-ray crystallography (Fig. 2 and 3, vide infra).23
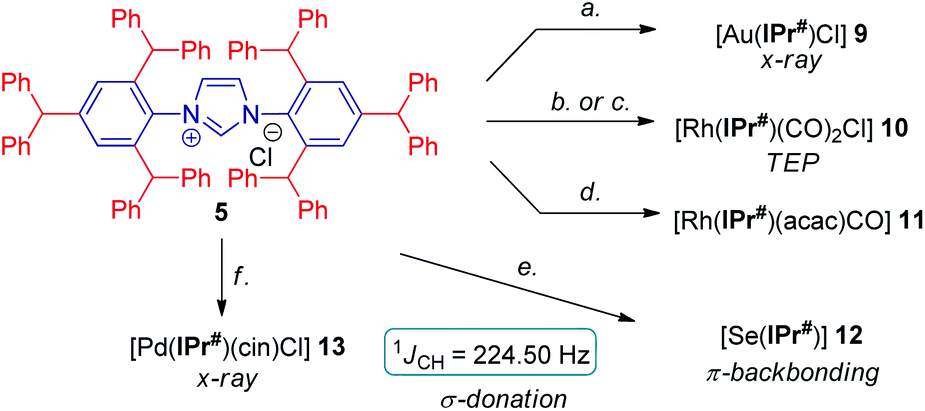 | ||
| Scheme 2 Synthesis of IPr# complexes. Conditions: (a) AuCl·Me2S (1.0 equiv.), K2CO3 (6.0 equiv.), acetone, 60 °C, 2 h, 90%. (b) KHMDS (1.8 equiv.), [Rh(CO)2Cl]2 (0.5 equiv.), toluene, 23 °C, 15 h, 88%. (c) [Rh(cod)Cl]2 (0.5 equiv.), K2CO3 (2.0 equiv.), acetone, 60 °C, 8 h, 71%, then CO, CH2Cl2, 23 °C, 15 h, 90%. (d) KOtBu (2.0 equiv.), [Rh(acac)(CO)2] (1.0 equiv.), THF, 23 °C, 15 h, 93%. (e) Se (3.0 equiv.), NaHMDS (1.2 equiv.), THF, −78 to 23 °C, 15 h, 95%. (f) KOtBu (1.1 equiv.), [Pd(cin)Cl]2 (0.45 equiv.), THF, 23 °C, 15 h, 89%. | ||
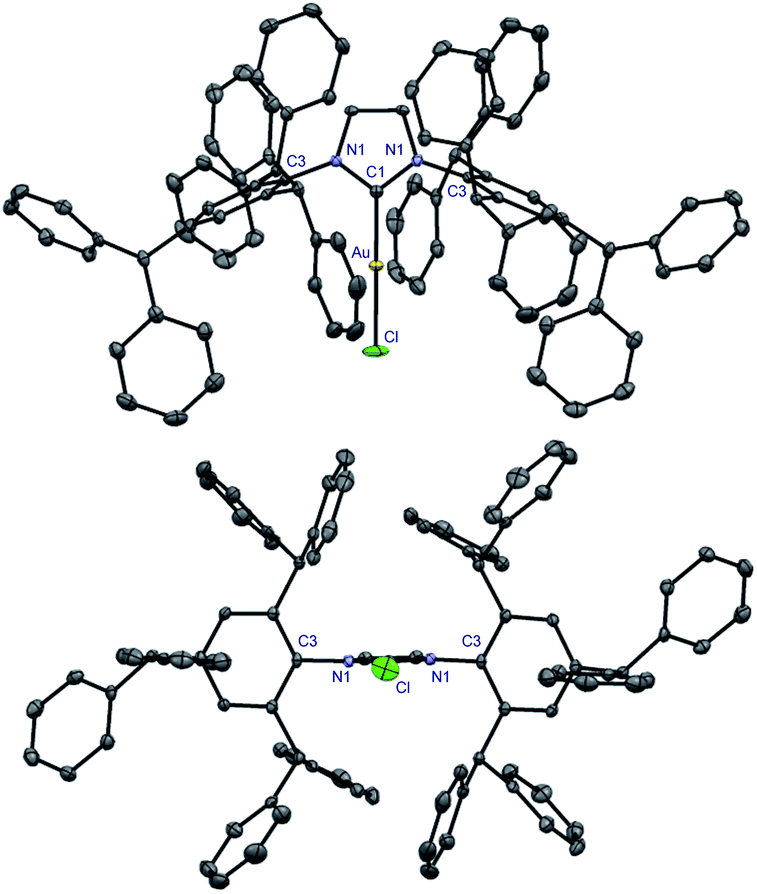 | ||
| Fig. 2 X-ray crystal structure of complex 9. Two views: front (top); side (bottom). Hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Au–C1, 1.972; Au–Cl, 2.2768; C1–N1, 1.356; C3–N1, 1.442(2); C1–Au–Cl, 180.0; N1–C1–N1, 104.9; C3–N1–C1, 122.7; N1–C1–Au, 127.5. Note the symmetry across the Cl–Au–C1 axis in 9. CCDC 2077050.† | ||
 | ||
| Fig. 3 X-ray crystal structure of complex 13. Two views: front (top); side (bottom). Hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Pd–C1, 2.044(4); Pd–Cl, 2.374(1); Pd–C49, 2.121(4); Pd–C50, 2.130(6); Pd–C51, 2.210(7); C1–N1, 1.364(5); C1–N2, 1.368(4); C4–N1, 1.452(4); C58–N2, 1.440(5); C1–Pd–C49, 103.2(2); C1–Pd–C50, 138.5(2); C1–Pd–C51, 169.4(2); C49–Pd–C51, 67.0(2); C1–Pd–Cl, 93.7(1); N1–C1–N2, 103.3(3); C4–N1–C1, 124.9(3); C58–N2–C1, 124.7(3). CCDC 2077053.† | ||
Extensive studies by Cavallo et al. demonstrated the % buried volume (%Vbur) and steric maps of model [Au(NHC)Cl] complexes as the best indication of quantifying the steric impact of NHC ligands.4,24 In our case, [Au(IPr#)Cl] is linear (C–Au–Cl, 180.0°; C–Au, 1.972 Å), making it a good model for evaluating %Vbur. Thus, with the (%Vbur) of 54.4%, [Au(IPr#)Cl] represents one of the bulkiest NHC ligands prepared to date (Table 1).1,2,4 This value compares well with the (%Vbur) of 50.4% determined for [Au(IPr*)Cl] (C–Au–Cl, 178.3°; C–Au, 1.987 Å),19 indicating a subtle but important effect of the para-diphenylmethyl substitution on the steric properties of the ligand.
A graphical representation of the steric mapping of the metal center in [Au(IPr#)Cl] is shown in Fig. 4A (vide infra).
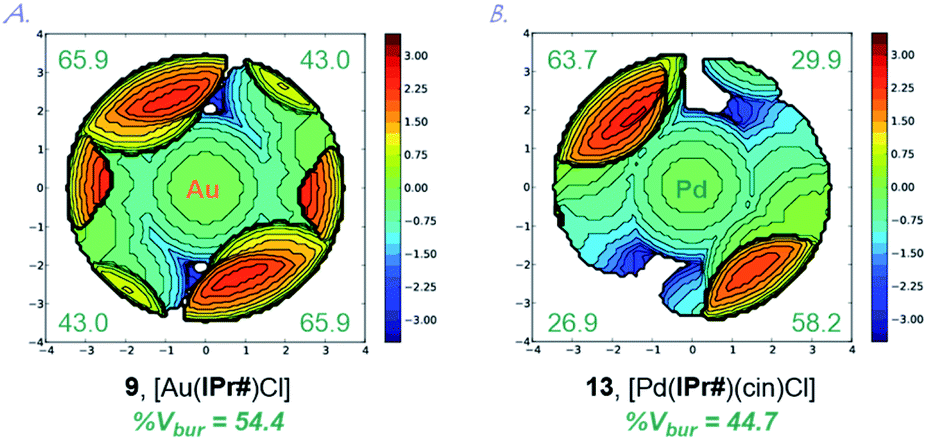 | ||
| Fig. 4 Topographical steric maps of [Au(IPr#)Cl] (9) and [Pd(IPr#)(cin)Cl] (13) showing %Vbur per quadrant. | ||
The Tolman electronic parameter (TEP) allows to evaluate electronic properties of NHC ligands.3 Thus, the CO stretching frequencies of [Rh(IPr#)(CO)2Cl] are νsym = 2079.5 cm−1 and νasym = 1999.5 cm−1 (CH2Cl2, 0.20 M),25 respectively, which corresponds to a TEP of 2051.8 cm−1 as a combined measure of the electronic properties of the ligand. These values match well with the IPr ligand (TEP of 2051.5 cm−1), IPr* (TEP of 2052.7 cm−1) and CAACCy (TEP of 2048.6 cm−1) and indicate one of the most donating 5-membered NHCs prepared to date.
Likewise, the use of selenourea adducts permits to evaluate π-backbonding from the 77Se NMR spectra.21 The δSe value of 108.11 ppm for [Se(IPr#)] (CDCl3) suggests that the expanded substitution leads to slightly higher π-accepting properties than IPr (δSe = 90 ppm), and IPr* (δSe = 106 ppm) and much lower than CAACMe2 (δSe = 492 ppm), as expected from the C-substitution.
Furthermore, one-bond CH J coupling constants obtained from 13C satellites of the 1H NMR spectrum26 provide good indication of σ-donating properties of an NHC ligand. The value of 224.50 Hz for IPr#HCl (CDCl3) is consistent with this ligand being as strongly σ-donating as IPr (1JCH = 223.70 Hz), but at the same time significantly more sterically-demanding and flexible. The chemical shift of the iminium proton in IPr#HCl was found at 12.6 ppm (CDCl3), which is significantly downfield compared with other imidazolium salts.27
The synthesis of [Rh(IPr#)(acac)CO] demonstrates that the extremely bulky IPr# is able to accommodate asymmetrical, κ2-O,O-bound ligands like acac to the metal center. Presumably, this leads to steric adjustment of the ligand topology to fit the Rh coordination plane.
This property is confirmed through the synthesis and full crystallographic characterization of [Pd(IPr#)(cin)Cl] (Fig. 3). Following the earlier reports,9,13a Pd(II) allyl-type complex (13) was selected as a model well-defined, air- and moisture-stable Pd(II)–NHC precatalyst to evaluate the performance of IPr# in cross-coupling reactions. The X-ray crystallographic analysis revealed the (%Vbur) of 44.7% with 63.7%, 29.9%, 58.2%, 26.9% for each quadrant (Fig. 4B). The values can be compared with the (%Vbur) of 54.4% for the linear [Au(IPr#)Cl] with 65.9%, 43.0%, 65.9%, 43.0% for each quadrant (Fig. 4A). Thus, the IPr# ligand is capable of both (1) adjusting the steric environment, and (2) asymmetrical twisting around the metal center, which furnishes important in catalysis differentiated quadrant distribution arising from the very bulky yet flexible ligand topology. The C–Pd, Pd–Cl, and Pd–C(Ph) bond lengths of 2.044 Å, 2.374 Å, and 2.210 Å in 13 are in the range for Pd(II)-allyl type complexes ([Pd(IPr)(cin)Cl], C–Pd, 2.027 Å; Pd–Cl, 2.357 Å; Pd–C(Ph), 2.245 Å).
It is further worth noting that diphenylmethyl substituents of the IPr# wingtips extend beyond the metal center in both 9 and 13, which might influence both (1) substrate approach, and (2) coordination of active intermediates formed during the catalysis. We hypothesize that the addition of a para-substituent has steric effect due to proximity to the metal center, while two additional effects are increase of donation and inhibition of N-wingtip rotation around the N–Ar axis.
The activity of IPr# was evaluated in various palladium-catalyzed cross-couplings (Scheme 3). As stated above, [Pd(IPr#)(cin)Cl] was selected due to the success of well-defined Pd(II)–NHCs supported by allyl-type throw-away ligands and the potential to tune the catalyst activity by allyl modifications.9,13a The results outlined in Scheme 3 indicate very high degree of generality of IPr#. As such, amide N–C(O) Suzuki cross-coupling (entry 1),14 ester C–O amidation (entry 2),28 amide N–C(O) transamidation (entry 3),29 C–Cl Suzuki cross-coupling (entry 4),30 C–Cl Buchwald–Hartwig amination (entry 5),31 C–Br Feringa coupling using both aryl- (entry 6)32 and challenging alkyllithium possessing β-hydrogens (entry 7),33 C–Cl ketone α-arylation (entry 8),34 C–S sulfur metathesis (entry 9)35 and C–H activation (entry 10)36 all proceeded in good to excellent yields. More importantly, these results demonstrate that the IPr# ligand is effective in an array of N–C, O–C, C–Cl, C–Br, C–S and C–H bond cross-couplings with various organometallics (B, Li, enolate, amine, sulfide) across some of the most broadly employed cross-couplings in industrial and academic settings.1,2,6a–c In view of its general reactivity across numerous bonds, IPr# might offer a significant potential for catalytic reaction development.
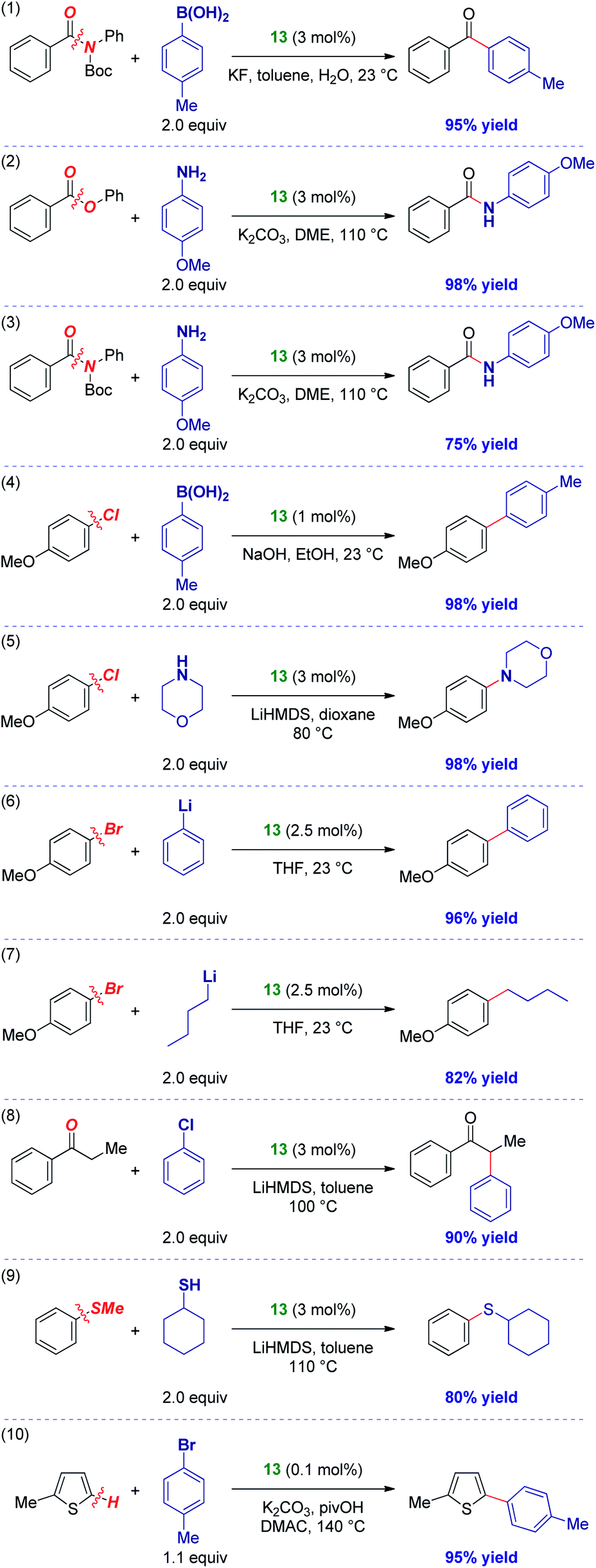 | ||
| Scheme 3 Activity of [Pd(IPr#)(cin)Cl] 13 in cross-coupling reactions. See ESI† for details. | ||
Generality and two further applications of the IPr# ligand design peralkylation concept are presented in Scheme 4. We selected the synthesis of BIAN-IPr# (14) and Np# (15) as two representative examples to showcase the synthetic potential of the “hash” NHC modular framework. Thus, BIAN (BIAN = bis(imino)acenaphthalene) ligands have emerged as powerful ligands in catalysis because of the structural rigidity of C–H bonds bringing the wingtip substituents closer to the metal center as well as redox-active properties, and strong σ-donating character of the carbene center.37 With the access to 2,4,6-tribenzhydrylaniline 7 in hand, the synthesis of BIAN-IPr# (14) proceeded uneventfully readily furnishing 1.5 gram of 14 using acenaphthoquinone (16) as the NHC precursor (see ESI†).
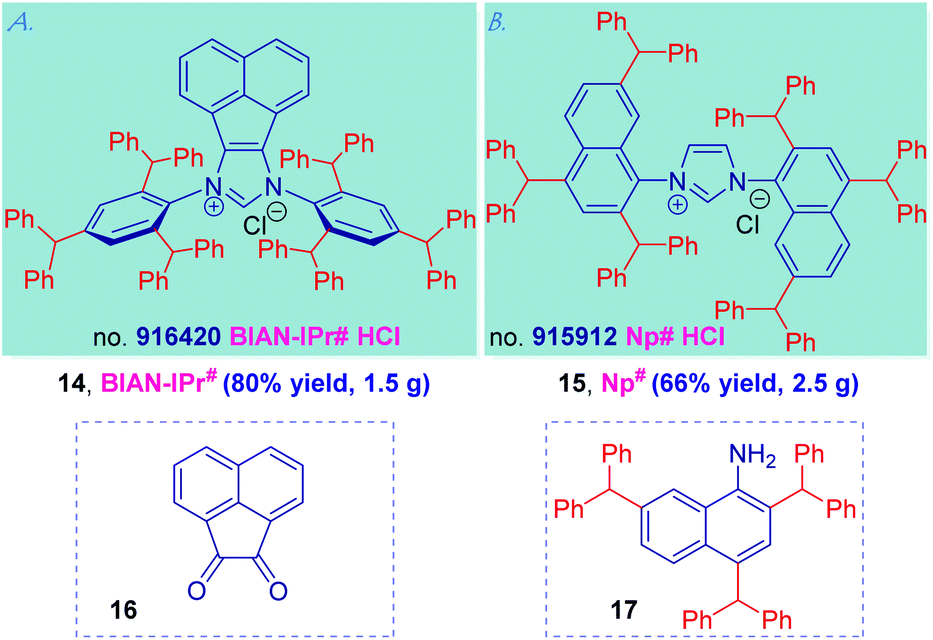 | ||
| Scheme 4 Generalization of IPr# NHCs: (a) BIAN-IPr# and (b) Np#. | ||
In the similar vein, the C2-symmetric imidazolin-2-ylidenes bearing substituted naphthyl N-wingtips reported by Dorta have emerged as the most active NHC ancillary ligands in Pd-catalyzed cross-coupling and Ru-metathesis;38 however, the synthesis of 2,7-substituted naphthyl wingtips has been a major limitation. Applying the concept described herein, the synthesis of Np# (15) exploiting the facile synthesis of 2,4,7-tribenzhydrylnaphthalen-1-amine (17) by Friedel–Crafts alkylation proceeded uneventfully and furnished 2.5 g of this sterically-differentiated NHC ligand (see ESI†). Thus, the use of “hash” concept permits a modular, rapid and cost-effective ligand assembly that might be applicable to both (1) various carbene classes (cf. BIAN-IPr#), and (2) diverse amines (cf. Np#). It is further worth noting that BIAN-IPr# is the first member of the intriguing family of BIAN ligands2m that has been commercialized (collaboration with Millipore Sigma, no. 916420, BIAN-IPr#HCl).39,40 Further applications of this concept are underway in our laboratory.
To further assess the effect of substitution on electronic properties of 5, HOMO and LUMO energy levels of IPr# and other NHCs were determined at the B3LYP 6-311++g(d,p) level (Table 2, Fig. 5 and ESI†).5a–c,11 Computation of HOMO and LUMO provides the most accurate estimation of nucleophilicity (more σ-donating, higher HOMO) and electrophilicity (more π-accepting, lower LUMO) of NHCs, however, the values for comparison must be available at the same level of theory.
| NHC | HOMO [eV] | LUMO [eV] |
|---|---|---|
| a See ESI for details. b rac-Np# (C2-symmetric), meso-Np# (CS-symmetric), −6.13 eV and −0.94 eV. rac-Np# is more stable than meso-Np# by 0.56 kcal mol−1 calculated at the B3LYP 6-311++g(d,p) level. | ||
| IPr# | −6.16 | −0.96 |
| BIAN-IPr# | −6.16 | −0.95 |
| Np# | −6.15b | −0.97b |
| IPr* | −6.12 | −0.90 |
| IPr | −6.01 | −0.48 |
| IMes | −5.90 | −0.33 |
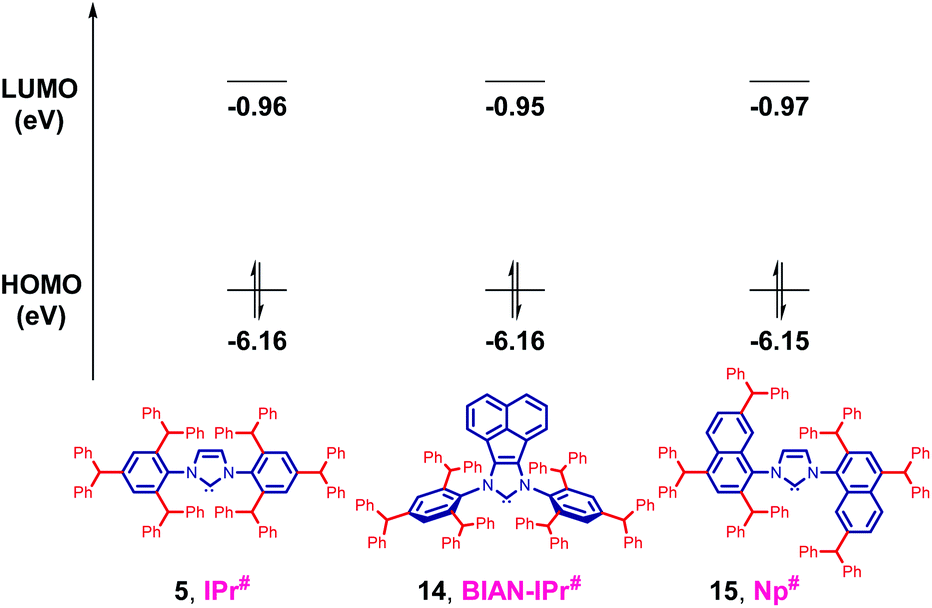 | ||
| Fig. 5 HOMO and LUMO energy levels (eV) calculated at B3LYP 6-311++g(d,p). See ESI† for details. | ||
The HOMO of IPr# (−6.16 eV) is comparable with IPr (−6.01 eV), which is a routine model for σ-donating NHCs. The more hindered acenaphthene ring of BIAN-IPr# results in a similar strongly nucleophilic (σ-donating) (HOMO−1, in-plane σ-orbital, −6.16 eV), and electrophilic (π-accepting) (LUMO+1 due to required symmetry, −0.95 eV) ligand. Replacement of the N-phenyl ring with N-naphthyl results in a C2-symmetric Np# ligand similar to classic NHCs (HOMO+2, in-plane σ-orbital, −6.15 eV), with similar electrophilicity (LUMO−2 due to required symmetry, −0.97 eV). Thus, it is evident that the strong σ-donation in combination with the differentiated steric impact renders the class of IPr# ligands well-suited for homogeneous catalysis.
Conclusions
In summary, we have developed a new class of extremely sterically-bulky, easily prepared NHC ligands by peralkylation concept. The parent ligand, IPr#, is readily accessible in three simple synthetic steps, exploiting the cost-effective, modular peralkylation of aniline, an industrial chemical that is available in bulk. Evaluation of structural and electronic properties has provided insight into the steric impact, σ-donation and π-accepting properties. The parent IPr# is one of the largest yet flexible NHC ligands developed to date, while at the same time one of the strongest σ-donors. Crucially, the potential to utilize the IPr# ligand in various Pd-catalyzed cross-coupling reactions by numerous bond breaking events has been demonstrated. The facile preparation of sterically-differentiated analogues BIAN-IPr# and Np# has been described, and highlights the capacity of this peralkylation design platform. The modularity of this approach makes is attractive for future development of NHC ligands. Given the tremendous importance of NHC ligands and the commercial availability of the developed ligands Millipore Sigma (IPr#HCl, 915653; Np#HCl; 915912; BIAN-IPr#HCl, 916420), we expect that this new class of NHCs will find rapid and widespread application.39,40Data availability
The datasets supporting this article have been uploaded as part of the supplementary material. Crystallographic data for 9 and 13 have been deposited at the CCDC under 2077050 and 2077053 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
Q. Z., G. M., G. L. and M. S. conceived the work. Q. Z., G. M., G. Z. performed the synthetic and characterization experiments and analyzed the data. C. F. and R. M. contributed to the IR studies. R. L. performed crystallographic studies. R. S. performed computational studies. M. S. supervised the project and wrote the manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): Rutgers University has filed patent(s) on ligands and precatalysts described in this manuscript (US 62/958565, Jan 8, 2020).Acknowledgements
We gratefully acknowledge Rutgers University (MS), the NIH (1R35GM133326, MS), the NSF (CAREER CHE-1650766, MS) and the ACS PRF (DNI-55549) for generous financial support. The Bruker 500 MHz spectrometer used in this study was supported by the NSF-MRI grant (CHE-1229030). Additional support was provided by the Rutgers Graduate School in the form of Dean's Dissertation Fellowship (GM). Supplement funding for this project was provided by the Rutgers University Newark Chancellor's Research Office. We thank the Wroclaw Center for Networking and Supercomputing (grant number WCSS159).Notes and references
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS ; for the original study by Arduengo, see:; (b) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef ; for a representative review, see:; (c) A. J. Arduengo III, Acc. Chem. Res., 1999, 32, 913–921 CrossRef . For the first study by Nolan, see: ; (d) J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9890–9899 Search PubMed.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) S. P. Nolan, N-Heterocyclic Carbenes, Wiley, 2014 Search PubMed; (c) S. Diez-Gonzalez, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, RSC, 2016 RSC; (d) H. V. Huynh, The Organometallic Chemistry of N-Heterocyclic Carbenes, Wiley, 2017 CrossRef; (e) C. S. J. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer, 2011 CrossRef; (f) E. A. B. Kantchev, C. J. O. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed; (g) W. A. Hermann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef; (h) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (i) G. Sipos and R. Dorta, Coord. Chem. Rev., 2018, 375, 13–68 CrossRef CAS; (j) M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC; (k) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed; (l) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (m) C. Chen, F. S. Liu and M. Szostak, Chem.–Eur. J., 2021, 27, 4478–4499 CrossRef CAS PubMed , for recent reviews on CCACs, see:; (n) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed; (o) J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714–1748 CrossRef CAS.
- (a) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (b) S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef CAS; (c) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS; (d) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) A. Gomez-Suarez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- (a) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS; (b) M. Melaimi, M. Soleihavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (d) J. Cheng, L. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed; (e) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362–3375 CrossRef CAS.
- For Pd-catalyzed cross-coupling reactions, see: (a) A. de Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley, 2014 CrossRef; (b) G. A. Molander, J. P. Wolfe and M. Larhed, Science of Synthesis: Cross-Coupling and Heck-Type Reactions, Thieme, 2013 Search PubMed; (c) T. J. Colacot, New Trends in Cross-Coupling: Theory and Applications, RSC, 2015, for Ru-catalyzed olefin metathesis, see Search PubMed; (d) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef CAS PubMed; (e) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed; (f) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9889–9890 CrossRef CAS.
- U. Kohler, F. F. Pape, M. Irgang, J. Wulff-Doring, M. Hesse and P. Polanek, Preparation of cyclic amines, US Pat., US5663438, 1995 Search PubMed.
- (a) O. Navarro, R. A. Kelly III and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 16194–16195 CrossRef CAS PubMed; (b) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS PubMed; (b) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed; (c) S. Würtz, C. Lohre, R. Fröhlich, K. Bergander and F. Glorius, J. Am. Chem. Soc., 2009, 131, 8344–8345 CrossRef PubMed.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172–6175 CrossRef CAS PubMed; (c) C. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed.
- (a) G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. H. Reek and I. E. Marko, Dalton Trans., 2010, 39, 1444–1446 RSC; (b) F. Izquierdo, S. Manzini and S. P. Nolan, Chem. Commun., 2014, 50, 14926–14937 RSC.
- (a) T. Scatollin and S. P. Nolan, Trends Chem., 2020, 2, 721–736 CrossRef; (b) Y. Wei, B. Rao, X. Cong and X. Zeng, J. Am. Chem. Soc., 2015, 137, 9250–9253 CrossRef CAS; (c) M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908–912 CrossRef CAS PubMed; (d) J. Diesel, A. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489–4493 CrossRef CAS PubMed; (e) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- (a) S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589–2599 CrossRef CAS PubMed; (b) P. Lei, G. Meng and M. Szostak, ACS Catal., 2017, 7, 1960–1965 CrossRef CAS; (c) P. Lei, G. Meng, S. Shi, Y. Ling, J. An, R. Szostak and M. Szostak, Chem. Sci., 2017, 8, 6525–6530 RSC.
- (a) G. Meng, S. Shi, R. Lalancette, R. Szostak and M. Szostak, J. Am. Chem. Soc., 2018, 140, 727–734 CrossRef CAS PubMed; (b) G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2015, 54, 14518–14522 CrossRef CAS PubMed; (c) F. Hu, R. Lalancette and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 5062–5066 CrossRef CAS PubMed.
- The current estimated value of aniline market is in excess of $15.66 billion, accessed on May 11, 2021, https://transparencymarketresearch.com/aniline-market.html.
- F. H. Howell, Alkylation and aralkylation of aromatic amines, US Pat., US4436936, 1984 Search PubMed.
- (a) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed; (b) D. Nelson, Eur. J. Inorg. Chem., 2015, 2012–2027 CrossRef CAS.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
- R. Savka and H. Plenio, Dalton Trans., 2015, 44, 891–893 RSC.
- (a) S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gomez-Suarez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC; (b) A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS , for a recent study on the Se scale, see:; (c) G. P. Junor, J. Lorkowski, C. M. Weinstein, R. Jazzar, C. Pietraszuk and G. Bertrand, Angew. Chem., Int. Ed., 2020, 59, 22028–22033 CrossRef CAS.
- A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem.–Eur. J., 2012, 18, 4517–4521 CrossRef CAS.
- Crystallographic data have been deposited with the Cambridge Crystallographic Data Center (CCDC 2077050, 2077053).
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- G. Meng, L. Kakalis, S. P. Nolan and M. Szostak, Tetrahedron Lett., 2019, 60, 378–381 CrossRef CAS.
- D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 CrossRef CAS.
- T. B. Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef.
- G. Meng, P. Lei and M. Szostak, Org. Lett., 2017, 19, 2158–2161 CrossRef CAS PubMed.
- M. T. Chen, D. A. Vicic, M. L. Turner and O. Navarro, Organometallics, 2011, 30, 5052–5056 CrossRef CAS.
- J. Huang, G. Grasa and S. P. Nolan, Org. Lett., 1999, 1, 1307–1309 CrossRef CAS.
- M. Giannerini, M. Fananas-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667–672 CrossRef CAS.
- E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef PubMed.
- M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053–4056 CrossRef CAS PubMed.
- Z. Lian, B. N. Bhwal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- A. Martin, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2012, 8, 1637–1643 CrossRef CAS PubMed.
- (a) D. A. Evans, I. Vargas-Baca and A. H. Cowley, J. Am. Chem. Soc., 2013, 135, 13939–13946 CrossRef CAS PubMed , for select recent applications, see:; (b) Y. Cai, X. T. Yang, S. Q. Zhang, F. Li, Y. Q. Li, L. X. Ruan, X. Hong and S. L. Shi, Angew. Chem., Int. Ed., 2018, 57, 1376–1380 CrossRef CAS PubMed; (c) See ref. 13d..
- (a) X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848–6858 CrossRef CAS PubMed; (b) D. Gallenkamp and A. Fürstner, J. Am. Chem. Soc., 2011, 133, 9232–9235 CrossRef CAS PubMed.
- M. Szostak, Q. Zhao, G. Meng and G. Li, Ligands for Transition Metal Catalysis, US Pat., US62/958,565, 2020 Search PubMed.
- The following ligands are available from Millipore Sigma: 915653, IPr#HCl; 915912, Np#HCl; 916420, BIAN-IPr#HCl, accessed on May 11, 2021. The commercial availability enables broad access of the academic and industrial researchers to the ligands described for reaction optimization and screening, www.sigmaaldrich.com/catalog/product/aldrich/915653.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and computational data. CCDC 2077050 and 2077053. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02619d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |