
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric synthesis of spirocyclobutyl oxindoles and beyond via [2+2] cycloaddition and sequential transformations†
Xia
Zhong
 ,
Jiuqi
Tan
,
Jianglin
Qiao
,
Yuqiao
Zhou
,
Cidan
Lv
,
Zhishan
Su
,
Shunxi
Dong
* and
Xiaoming
Feng
*
,
Jiuqi
Tan
,
Jianglin
Qiao
,
Yuqiao
Zhou
,
Cidan
Lv
,
Zhishan
Su
,
Shunxi
Dong
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: dongs@scu.edu.cn; xmfeng@scu.edu.cn; Web: http://www.scu.edu.cn/chem_asl/
First published on 22nd June 2021
Abstract
Efficient asymmetric synthesis of a collection of small molecules with structural diversity is highly important to drug discovery. Herein, three distinct types of chiral cyclic compounds were accessible by enantioselective catalysis and sequential transformations. Highly regio- and enantioselective [2+2] cycloaddition of (E)-alkenyloxindoles with the internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of N-allenamides was achieved with N,N′-dioxide/Ni(OTf)2 as the catalyst. Various optically active spirocyclobutyl oxindole derivatives were obtained under mild conditions. Moreover, formal [4+2] cycloaddition products occurring at the terminal CC bond of N-allenamides, dihydropyran-fused indoles, were afforded by a stereospecific sequential transformation with the assistance of a catalytic amount of Cu(OTf)2. In contrast, performing the conversion under air led to the formation of γ-lactones via the water-involved deprotection and rearrangement process. Experimental studies and DFT calculations were performed to probe the reaction mechanism.
C bond of N-allenamides was achieved with N,N′-dioxide/Ni(OTf)2 as the catalyst. Various optically active spirocyclobutyl oxindole derivatives were obtained under mild conditions. Moreover, formal [4+2] cycloaddition products occurring at the terminal CC bond of N-allenamides, dihydropyran-fused indoles, were afforded by a stereospecific sequential transformation with the assistance of a catalytic amount of Cu(OTf)2. In contrast, performing the conversion under air led to the formation of γ-lactones via the water-involved deprotection and rearrangement process. Experimental studies and DFT calculations were performed to probe the reaction mechanism.
Introduction
Diversity-oriented synthesis (DOS)1 aims to create a library of small molecules with structural complexity and diversity in an efficient manner. Over the past two decades, great endeavors have been devoted to developing new methods and strategies towards this goal.2 Sequential reactions of chiral molecules generated in situ from asymmetric catalysis greatly enriched the chemical space,3 but discovering such synthons and convenient conditions without erosion of stereoselectivity remains challenging. N-Allenamides have attracted considerable interest as a versatile reagent in organic synthesis.4 Structurally, the amide moiety in N-allenamides is critical to strike the right balance between their reactivity and stability. This special structure endows N-allenamides with unique reactivity in diverse transformations,4a,b,5 such as addition reactions, cycloadditions and so on. Interestingly, both internal and terminal CC bonds of N-allenamides have been disclosed in cycloaddition reactions4b–e,6 including asymmetric versions4d (Scheme 1a). The regioselectivity of allenes depends on the choice of the catalyst,7 substrate8 and the substituent on the amide group.9 These impressive features make N-allenamides a promising substrate in diversity-oriented synthesis.
 | ||
| Scheme 1 Asymmetric [2+2] cycloadditions of N-allenamides. | ||
Cyclobutane is present in many biologically active compounds,10 and is also an appealing building block in synthetic organic chemistry.11 Among a number of protocols for the synthesis of cyclobutanes,12 the [2+2] cycloaddition represents one of most powerful strategies due to its high efficiency and stereoselectivity.12d–f In this context, the [2+2] cycloadditions of olefins with either terminal or internal CC bonds of N-allenamides have been accomplished by using a chiral cationic Au(I) catalyst13 or Lewis acid catalyst14 (Scheme 1a). Considering the frequent occurrence of the spirocyclobutyl oxindole skeleton in bioactive molecules,12h–j,15 it is highly desirable to develop reliable access to spirocyclobutyl oxindole derivatives.16 In continuation with our interest in chiral N,N′-dioxide/metal Lewis acid catalysis17 and inspired by elegant studies5,13,14 on the N-allenamide chemistry, herein, we demonstrated that catalysts could promote the direct asymmetric [2+2] cycloaddition of (E)-alkenyloxindoles with N-allenamides for straightforward access to optically active spirocyclobutyl oxindole derivatives (Scheme 1b). Moreover, further diversified transformation18 of the internal CC bond-involved product catalyzed by a copper salt enabled the one-pot synthesis of tetradropyranoindole derivatives, which seem to be the formal [4+2] cycloaddition adducts of the terminal CC bond of N-allenamides. Further transformation in the presence of acid and water gave 4′-methylenedihydrofuranone-substituted oxindole derivatives.
Results and discussion
Initially, (E)-alkenyloxindole 1a and N-allenaminde 2a were selected as the model substrates. The key reaction condition optimization is listed in Table 1 (for more details, see the ESI†). In the preliminary screening, only tetradropyranoindole derivative 4aa, the formal [4+2] cycloaddition product19 reacting at the terminal CC bond of N-allenamide, was obtained in the presence of Ni(OTf)2 (entry 1, 13% yield). Interestingly, when the reaction was performed by the use of the chiral L3-PiPr2/NiII complex, the [2+2] cycloaddition reaction took place smoothly, affording the corresponding internal CC bond involved spirocyclobutyl oxindole 3aa as the major product with 91% ee (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a = 89:11, in total 58% yield; entry 2). It is worth mentioning that when excessive amounts of the metal salt existed in the catalytic system with the chiral ligand, it led to the exclusive isolation of the product 4aa in 39% yield and 60% ee (entry 3). The examination of ligands suggested that the length of the carbon tether has a significant effect on the reaction, and the longer linker of N,N′-dioxide resulted in better chemoselectivity and enantioselectivity (entries 2, 4 and 5, the ratio of 3aa to 4aa, from 60:40 to 97:3). Variation of the chiral backbone exhibited that (S)-2-pipecolic acid derived L4-PiPr2 gave better enantioselectivity, while L-ramipril derived L4-RaPr2 resulted in a higher yield and comparable ee value (entries 5–7). The level of enantioselectivity increased to 95% ee when L5-RaPr2 with a five-carbon tether was used as the ligand (entry 8). We also identified other parameters for this asymmetric [2+2] cycloaddition reaction (see ESI, Tables S1–S4† for details), and 94% yield and 95% ee were obtained when 1.2 equivalents of 2a were used at higher temperature and concentration (entry 9). It should be noted that the use of other ligands, such as BINOL L1 or oxazoline L2, only led to the formation of [4+2] cycloaddition product 4aa in moderate yield with a low ee value (entries 10 and 11). Noteworthily, the diastereoselectivity of [2+2] cycloaddition was very high as only one diastereomer was detected in all cases.
:4a = 89:11, in total 58% yield; entry 2). It is worth mentioning that when excessive amounts of the metal salt existed in the catalytic system with the chiral ligand, it led to the exclusive isolation of the product 4aa in 39% yield and 60% ee (entry 3). The examination of ligands suggested that the length of the carbon tether has a significant effect on the reaction, and the longer linker of N,N′-dioxide resulted in better chemoselectivity and enantioselectivity (entries 2, 4 and 5, the ratio of 3aa to 4aa, from 60:40 to 97:3). Variation of the chiral backbone exhibited that (S)-2-pipecolic acid derived L4-PiPr2 gave better enantioselectivity, while L-ramipril derived L4-RaPr2 resulted in a higher yield and comparable ee value (entries 5–7). The level of enantioselectivity increased to 95% ee when L5-RaPr2 with a five-carbon tether was used as the ligand (entry 8). We also identified other parameters for this asymmetric [2+2] cycloaddition reaction (see ESI, Tables S1–S4† for details), and 94% yield and 95% ee were obtained when 1.2 equivalents of 2a were used at higher temperature and concentration (entry 9). It should be noted that the use of other ligands, such as BINOL L1 or oxazoline L2, only led to the formation of [4+2] cycloaddition product 4aa in moderate yield with a low ee value (entries 10 and 11). Noteworthily, the diastereoselectivity of [2+2] cycloaddition was very high as only one diastereomer was detected in all cases.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Yield of (3aa + 4aa)b (%) |
3aa:4aac |
eec (%) | |
| 3aa | 4aa | ||||
|
a Unless otherwise noted, all reactions were carried out with 1a (0.05 mmol), 2a (1.0 equiv.), and ligand/Ni(OTf)2 (1:1, 10 mol%) in CH2Cl2 (0.1 M) at 30 °C for 16 h under N2.
b Isolated yield of 3aa and 4aa.
c Determined by HPLC on a chiral stationary phase.
d Ligand/Ni(OTf)2 (1:1.5).
e
2a (1.2 equiv.) in CH2Cl2 (0.2 M) at 35 °C. Ts = p-tosyl.
|
|||||
| 1 | w/o | 13 | 1:99 |
— | 0 |
| 2 | L3-PiPr2 | 58 | 89:11 |
91 | 51 |
| 3d | L3-PiPr2 | 39 | 1:99 |
— | 60 |
| 4 | L2-PiPr2 | 59 | 60:40 |
58 | 83 |
| 5 | L4-PiPr2 | 55 | 97:3 |
95 | — |
| 6 | L4-PrPr2 | 55 | 98:2 |
79 | — |
| 7 | L4-RaPr2 | 75 | 97:3 |
90 | — |
| 8 | L5-RaPr2 | 74 | 98:2 |
95 | — |
| 9e | L5-RaPr2 | 94 | 98:2 |
95 | — |
| 10 | L1 | 29 | 1:99 |
— | 0 |
| 11 | L2 | 33 | 1:99 |
— | −11 |

It was observed that the presence of the metal salt (Table 1, entry 1 and 3) led to the formation of 4aa. The control experiment showed that the isolated spirocyclobutyl oxindole 3aa could be transformed into 4aa in 37% yield accompanied by a tiny amount of the by-product by treatment with 5 mol% of Ni(OTf)2 after 16 h under an air atmosphere (See ESI, Table S5 and Fig. S4† for more details). In contrast, [4+2] product 4aa did not transform into the spirocyclobutyl oxindole 3aa at all under the same conditions. It was indicated that the [2+2] cycloaddition occurred initially, followed by an achiral Lewis acid-accelerated rearrangement to yield tetradropyranoindole 4aa. Further investigation of other Lewis acids for a one-pot sequential reaction (Table 2), such as, Ni(OTf)2, Mg(OTf)2, Zn(OTf)2 or Cu(OTf)2, manifested that Cu(OTf)2 performed better (Table 2, entries 1–4). When 4 Å MS was added as a water scavenger, the total yield of 4aa increased to 70% in CHCl3 (entry 5), and the formation of the byproduct was obviously reduced. During these procedures, the enantioselectivity of the product 4aa was high and 93% ee was obtained after Cu(OTf)2 promoted sequential transformation. The result of one-pot sequential reactions was nearly the same as the two-step transformation (entry 6 vs. entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Metal salt | Yieldb (%) |
3aa:4aac |
ee of 4aac (%) |
| a Unless otherwise noted, all reactions were initially carried out with 1a (0.05 mmol), 2a (1.2 equiv.), L5-RaPr2 (10 mol%) and Ni(OTf)2 (10 mol%) in CH2Cl2 (0.2 M) at 35 °C for 16 h. Then, the metal salt (5 mol%) in CH2Cl2 (0.1 M) was added and stirred at 30 °C for 16 h under air. b Isolated yield of 4aa based on 1a. c Determined by HPLC on a chiral stationary phase. d 4 Å MS (10 mg) in CHCl3 (0.05 M) was used for the second transformation for 3 h. e Isolated 3aa was treated with Cu(OTf)2 (5 mol%) and 4 Å MS (10 mg) in CHCl3 (0.05 M) at 30 °C for 4 h under N2. | ||||
| 1 | Ni(OTf)2 | 76 | 82:18 |
92 |
| 2 | Zn(OTf)2 | 54 | 6:94 |
91 |
| 3 | Mg(OTf)2 | 52 | 34:66 |
91 |
| 4 | Cu(OTf)2 | 49 | 1:99 |
93 |
| 5d | Cu(OTf)2 | 70 | 1:99 |
93 |
| 6e | Cu(OTf)2 | 70 | 1:99 |
93 |

With the optimized reaction conditions in hand, the substrate scopes to synthesize chiral spirocyclobutyl oxindoles via chiral nickel(II) complex catalyzed [2+2] cycloaddition, and to synthesize tetradropyranoindoles via one-pot sequential transformation were investigated (Table 3). The effects of the ester groups of (E)-alkenyloxindole were first evaluated, and both yields and ee values were elevated gradually with the increase of steric hindrance of the ester group (3aa–3ea). The substrates with different substituents on the indole ring at the C5, C6, and C7 positions took part in the reaction smoothly, providing the desired spirocyclobutyl oxindoles 3fa–3pa in 72–94% yield with 87–96% ee. Moreover, the C5,C6-difluoro substituted alkenyloxindole was also applicable, affording 3qa in 76% yield and 92% ee. Subsequently, we turned our attention to the scope of N-allenamindes 2. Various substituted N-allenamindes were suitable in the current system, giving the corresponding products in good yields and excellent enantioselectivities (3ab–3ag, 69–97% yield, and 93–96% ee). The absolute configuration of product 3aa was determined to be (1R,2R,4S) by X-ray crystallography analysis,20 and the others were assigned by comparing their CD spectra with that of 3aa (see the ESI† for details). In comparison, the trend of enantioselectivity of the products 4 was similar to that of the corresponding spirocyclobutyl oxindoles. As depicted in Table 3, (E)-alkenyloxindoles and N-allenamides transformed into the tetradropyranoindoles in 33–83% yields and 70–93% ee after one-pot sequential transformation. The absolute configuration of 4ka was assigned as (R,E) on the basis of the absolute configuration of 3ka and the relative configuration of racemic 4ka.20
|
a Unless otherwise noted, the [2+2] cycloaddition conditions were the same as in Table 1, entry 8; and the synthesis of product 4 followed the same procedure as in Table 2, entry 5. Isolated yields of 3 and 4 are based on 1. The ee value was determined by HPLC on a chiral stationary phase.
b Cu(OTf)2 (10 mol%).
c Cu(OTf)2 (7.5 mol%).
d
4fa:3fa = 19:1.
e
4la:3la = 9:1.
f Cu(OTf)2 (12.5 mol%).
|
|---|

|
The structure of byproduct 5aa accompanied by the formation of 4aa in the sequential reaction was determined to be a γ-lactone derivative by X-ray crystal diffraction analysis.20 We proposed that γ-lactone 5aa was probably generated via water-involved hydrolysis with concomitant deprotection of the N-Boc group, following rearrangement (mechanism explanation is shown in Fig. 2b, cycle B). Interestingly, treatment of spirocyclobutyl oxindole 3aa with Cu(OTf)2 under air afforded γ-lactone 5aa with sharply improved yield (Scheme 2a; 70%, 65:35 dr, 95% ee for each diastereomer). Moreover, exposure of product 4aa to trifluoroacetic acid (TFA) also led to the formation of 5aa in 59% yield, 65:35 dr and 95% ee for each diastereomer (Scheme 2a). Subsequently, the conversion of spirocyclobutyl oxindoles 3 to γ-lactone substituted oxindoles 5 was examined (Scheme 2b).21 Under the influence of Cu(OTf)2, [2+2] products 3 with different substituents transformed into the corresponding products 5 in moderate yields and diastereoselectivities (44–70% yield, 65:35–69:31 dr) without any loss of enantioselectivity. The poor diastereoselectivity is due to the epimerization balance at the C3-position of oxindoles, and stereochemistry at the γ-lactone structure was maintained.
 | ||
| Scheme 2 The preparation of 4′-methylenedihydrofuranone-substituted oxindoles 5 and dihydropyran-fused indole products 7. | ||
Alkoxyallenes 6 were turned out to be suitable reaction partners. In these cases, [4+2] products 7 were obtained directly without the detection of the [2+2] cycloaddition products in the reaction system (see ESI, Table S10† for details). As shown in Scheme 2c, representative examples of alkoxyallenes 6 were tested, and all the reactions proceeded well, providing the corresponding dihydropyran-fused indole products in moderate yields and excellent enantioselectivities (7aa–7ac, 48–60% yield, and 94–97% ee).
To show the potential synthetic utility of the current method, scale-up synthesis of 3aa, 4qa and 5ab was carried out. Under the optimized reaction conditions, 1a (2.0 mmol) reacted with 2a (2.4 mmol) smoothly, providing the spirocyclobutyl oxindole 3aa in 89% yield (1.12 g) with 90% ee (Scheme 3a). It should be noted that for the synthesis of dihydropyran-fused indole product 4qa, increasing the amount of Cu(OTf)2 (7.5 mol%) and 4 Å MS, and the reaction concentration (0.07 M) was necessary to get high yield (58% yield for two steps) in 88% ee by employing 1.60 mmol 1q (Scheme 3b). γ-Lactone 5ab was obtained in 63% yield (0.72 g), 66:34 dr and 94% ee for each diastereomer (Scheme 3c). Furthermore, hydrogenation of 3aa in the presence of Pd/C and H2 gave rise to densely substituted spirocyclobutyl oxindole 8 in 95% yield, 85:15 dr and 95% ee (Scheme 3d). Hydrogenation of 4aa resulted in C–O bond cleavage of the pyran ring, delivering the product 9 in 75% yield, 60:40 dr and 92/92% ee (Scheme 3e).
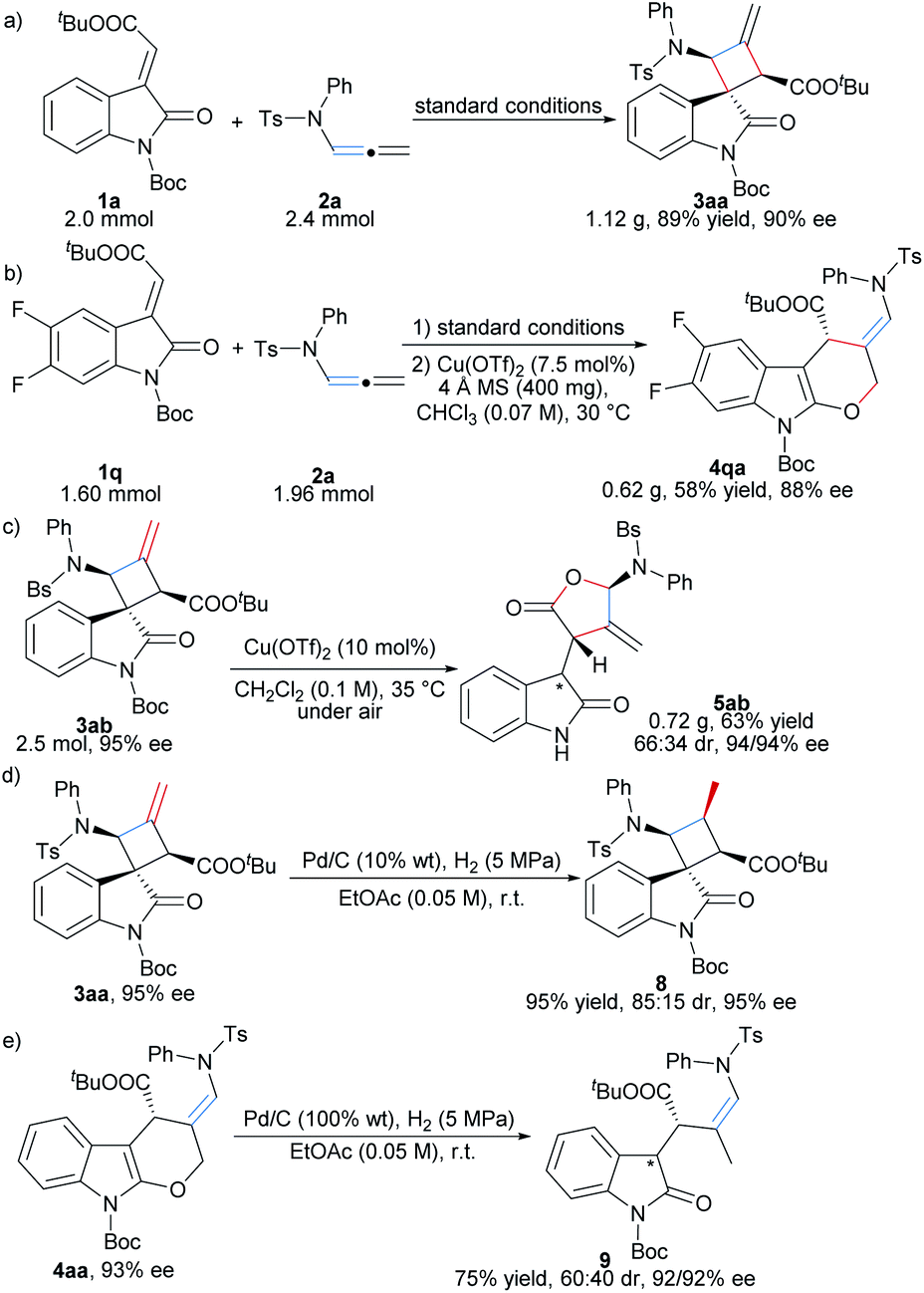 | ||
| Scheme 3 Scale-up reactions and further transformations of the products. | ||
To probe the mechanism for the transformation, the Cu(OTf)2-catalyzed isomerizations from 3aa to E-4aa or Z-4aa were studied at the M06-D3/6-31G(d,p) (SMD, dichloromethane) theoretical level and the energy profiles are shown in Fig. 1 (see ESI,† computational details part for more details). 3aa coordinated to Cu(OTf)2 in a bidentate fashion, forming an intermediate IM1. This process was exothermic by 25.5 kcal mol−1. Then, it underwent a ring-opening process via transition state TS1, generating intermediate I. Suffering from the steric repulsion from the OTf− anion in the catalyst, the relative Gibbs free energy of intermediate II with s-cis-unsaturated imine s-cis-II was slightly higher than that with the s-trans-one by 0.8 kcal mol−1. In the following step, the C–O bond in E-IM2 was constructed via transition state E-TS2, with ΔG≠ of 12.3 kcal mol−1. In contrast, the activation barrier associated with the formation of Z-IM2viaZ-TS2 was 14.3 kcal mol−1. In addition, Z-IM2 was less stable than E-IM2 by 5.0 kcal mol−1. These results indicated that E-4aa was predominantly formed in the presence of Cu(OTf)2. Although heating 3aa in DCE at 80 °C for 24 h led to the generation of E-4aa, the DFT studies indicated that the presence of Cu(OTf)2 could accelerate this conversion with low activation barriers (12.8 and 12.3 kcal mol−1), thus making the reaction possible at 30 °C (see ESI, Fig. S5† for more details).
 | ||
| Fig. 1 Energy profiles for the isomerization from 3aa to E-4aa or Z-4aa catalyzed by Cu(OTf)2. | ||
Based on the above analysis and the X-ray crystal structures of product 3aa and the L3-PiPr2/NiII complex,22 a possible working mode was proposed to elucidate the stereoselectivity of the [2+2] reaction. As shown in Fig. 2a, chiral N,N′-dioxide and alkenyloxindole 1a coordinated to NiII in tetradentate and bidentate fashions respectively to form a slightly distorted hexahedral complex. The Si-face of the substrate 1a was shielded by the substituted aniline group on the ligand. Consequently, N-allenamide 2a approached from its β-Re-face to form the zwitterionic intermediate, followed by cyclization from the Re-face of the imine moiety to afford [2+2] cycloaddition adduct (1R,2R,4S)-3aa.
 | ||
| Fig. 2 Proposed transition state model and mechanism. | ||
When Lewis acid Cu(OTf)2 comes into contact with [2+2] adduct 3aa, it will bond the N-Boc oxindole in a bidentate manner, leading to ring opening of the strained cyclobutyl structure to deliver the zwitterionic intermediate I (Fig. 1). Then the isomerizarion of intermediate I provided unsaturated imine s-trans-II, which was trapped by the intramolecular Lewis acid-bonded enol anion to generate the dihydropyran-fused indole product E-4aa. This reaction pathway was rationalized by DFT studies as well (see ESI, Fig. S11–S13† for more details). During the process, the stereocenter at the ester substituent is unaffected, and thus the enantioselectivity of 4aa is maintained. On the other hand, when 3aa was isolated and the second step was performed under an air atmosphere, after the formation of intermediate I, deprotection of the Boc group and hydrolysis of the ester unit successively take place under the influence of Lewis acid Cu(OTf)2,3c,23 affording the acid intermediate III. Subsequently, diastereoselective intramolecular Mannich-type cyclization and proton transfer occurs, yielding the lactone product 5aa with high enantioselectivity (Fig. 2b, cycle A). Moreover, treatment of product 4aa with TFA gave rise to intermediate V, which underwent ring-opening and proton transfer to produce 5aa (Fig. 2b cycle B, see ESI, Fig. S6† for more details).
Conclusions
In summary, we have successfully developed a highly enantioselective [2+2] cycloaddition reaction between the internal CC bond of N-allenamides and (E)-alkenyloxindole catalyzed by a chiral L5-RaPr2/Ni(OTf)2 catalyst. The appropriate steric and electronic properties of the catalyst enable the controllable formation of chiral spirocyclobutyl oxindole derivatives in good yields with high stereoselectivities, suppressing the rearrangement into thermodynamically stable formal [4+2] adducts. Nevertheless, diversity-oriented synthesis via sequential transformations was available, such as dihydropyran-fused indoles and lactones from chiral spirocyclobutyl oxindoles, which could be performed in a one-pot manner by the use of additional copper salt in the absence or presence of water. On the basis of control experiments and DFT studies, plausible transition state model and catalytic cycles were proposed to explain the origin of chem- and stereoselective control and transformation processes. The synthesis of three different kinds of enantioenriched cyclic compounds from the same starting materials and chiral catalysts makes the current methodology attractive.
Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, SFC spectra, CD spectra,computational methods and X-ray crystallographic data for 3aa, rac-4ka and rac-5aa are available in the ESI.Author contributions
X. Z. and J. L. Q. performed the experiments. J. Q. T. repeated data. C. D. L. and Z. S. S. analyzed the computational data. S. X. D. participated in structure characterization and discussion. Y. Q. Z. analyzed the X-ray diffraction crystal data. X. M. F. and X. H. L. supervised the project. X. M. F., X. H. L., S. X. D. and X. Z. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 21772127 and 21921002) for financial support.Notes and references
- (a) M. D. Bruke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed; (b) C. J. O' Connor, H. S. Beckmann and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444 RSC; (c) K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288 CrossRef CAS.
- (a) D. Tan, Nat. Chem. Biol., 2005, 1, 74 CrossRef CAS; (b) W. R. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
- For reviews, see: (a) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed; (b) G. Moura-Letts, C. M. DiBlasi, R. A. Bauer and D. S. Tan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6745 CrossRef CAS PubMed. For selected examples, see: (c) H. F. Zheng, Y. Wang, C. R. Xu, Q. Xiong, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327 CrossRef CAS; (d) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 142, 15975 CrossRef PubMed; (e) X. Lin, Y. Liu and C. Li, Chem.–Eur. J., 2020, 26, 14173 CrossRef CAS PubMed.
- For reviews, see: (a) L.-L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 CrossRef CAS; (b) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef CAS; (c) M. R. Fructos and A. Prieto, Tetrahedron, 2016, 72, 355 CrossRef CAS; (d) E. Manoni and M. Bandini, Eur. J. Org. Chem., 2016, 3135 CrossRef CAS; (e) C. Praveen, Catal. Rev., 2019, 61, 406 CrossRef CAS.
- (a) W. B. Dickinson and P. C. Lang, Tetrahedron Lett., 1967, 8, 3035 CrossRef; (b) S. Ma, in Modern Allene Chemistry, ed. A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, vol. 1–2, p. 595 Search PubMed.
- For selected examples, see: (a) W. Zhou, X.-X. Li, G.-H. Li, Y. Wu and Z. Chen, Chem. Commun., 2013, 49, 3552 RSC; (b) G.-H. Li, W. Zhou, X.-X. Li, Q.-W. Bi, Z. Wang, Z.-G. Zhao, W.-X. Hu and Z. Chen, Chem. Commun., 2013, 49, 4770 RSC; (c) S. Montserrat, H. Faustino, A. Lledós, J. L. Mascareñas, F. López and G. Ujaque, Chem.–Eur. J., 2013, 19, 15248 CrossRef CAS PubMed; (d) V. Pirovano, L. Decataldo, E. Rossi and R. Vicente, Chem. Commun., 2013, 49, 3594 RSC; (e) H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef CAS PubMed; (f) P. Bernal-Albert, H. Faustino, A. Gimeno, G. Asensio, J. L. Mascareñas and F. López, Org. Lett., 2014, 16, 6196 CrossRef CAS PubMed; (g) H. Faustino, I. Varela, J. L. Mascareñas and F. López, Chem. Sci., 2015, 6, 2903 RSC; (h) E. López, J. González and L. A. López, Adv. Synth. Catal., 2016, 358, 1428 CrossRef; (i) S. Peng, D. Ji and J. Sun, Chem. Commun., 2017, 53, 12770 RSC; (j) N. De, C. E. Song, D. H. Ryu and E. J. Yoo, Chem. Commun., 2018, 54, 6911 RSC; (k) D. C. Marcote, I. Varela, J. Fernández-Casado, J. L. Mascareñas and F. López, J. Am. Chem. Soc., 2018, 140, 16821 CrossRef CAS; (l) C. Wang, G. Xu, Y. Shao, S. Tang and J. Sun, Org. Lett., 2020, 22, 5990 CrossRef CAS PubMed.
- J. M. Wiest, M. L. Conner and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 15943 CrossRef CAS PubMed.
- S. Peng, S. Cao and J. Sun, Org. Lett., 2017, 19, 524 CrossRef CAS PubMed.
- M. Yang, L. Kang and S. He, Heterocycles, 2019, 98, 1725 CrossRef.
- (a) The Chemistry of Cyclobutanes, ed. Z. Rappoport and J. F. Liebman, Wiley, Chichester, 2005 Search PubMed; (b) A. Fernández-Tejada, F. Corzana, J. H. Busto, G. Jiménez-Osés, J. M. Peregrina and A. Avenoza, Chem.–Eur. J., 2008, 14, 7042 CrossRef PubMed; (c) K.-G. Wen, Y.-Y. Peng and X.-P. Zeng, Org. Chem. Front., 2020, 7, 2576 RSC; (d) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448 RSC.
- For selected reviews, see: (a) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485 CrossRef CAS PubMed; (b) E. Leemans, M. D'hooghe and N. De Kimpe, Chem. Rev., 2011, 111, 3268 CrossRef CAS PubMed; (c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS. For selected examples, see: (d) B.-S. Li, W.-X. Liu, Q.-W. Zhang, S.-H. Wang, F.-M. Zhang, S.-Y. Zhang, Y.-Q. Tu and X.-P. Cao, Chem.–Eur. J., 2013, 19, 5246 CrossRef CAS PubMed; (e) B.-M. Yang, P.-J. Cai, Y.-Q. Tu, Z.-X. Yu, Z.-M. Chen, S.-H. Wang, S.-H. Wang and F.-M. Zhang, J. Am. Chem. Soc., 2015, 137, 8344 CrossRef CAS PubMed; (f) W. Mazumdar, N. Jana, B. T. Thurman, D. J. Wink and T. G. Driver, J. Am. Chem. Soc., 2017, 139, 5031 CrossRef CAS PubMed; (g) J. Guo, X. Xu, Q. Xing, Z. Gao, J. Gou and B. Yu, Org. Lett., 2018, 20, 7410 CrossRef CAS PubMed; (h) M.-H. Xu, K.-L. Dai, Y.-Q. Tu, X.-M. Zhang, F.-M. Zhang and S.-H. Wang, Chem. Commun., 2018, 54, 7685 RSC; (i) F.-S. He, Y. Wu, X. Li, H. Xia and J. Wu, Org. Chem. Front., 2019, 6, 1873 RSC; (j) Y. Kim and D. Y. Kim, Bull. Korean Chem. Soc., 2021, 42, 510 CrossRef CAS; (k) H. Zheng, R. Wang, K. Wang, D. Wherritt, H. Arman and M. P. Doyle, Chem. Sci., 2021, 12, 4819 RSC.
- (a) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449 CrossRef CAS; (b) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559 CrossRef CAS; (c) J.-P. Deprés, P. Delair, J.-F. Poisson, A. Kanazawa and A. E. Greene, Acc. Chem. Res., 2016, 49, 252 CrossRef; (d) G. Otárola, J. J. Vaquero, E. Merino and M. A. Fernández-Rodríguez, Catalysts, 2020, 10, 1178 CrossRef; (e) F. Secci, A. Frongia and P. P. Piras, Molecules, 2013, 18, 15541 CrossRef; (f) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918 CrossRef CAS PubMed; (g) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed; (h) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946 RSC; (i) G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 RSC; (j) A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026 RSC.
- For selected examples of [2+2] cycloadditions accomplished using a chiral cationic Au(I) catalyst, see: (a) S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552 CrossRef PubMed; (b) M. Jia, M. Monari, Q.-Q. Yang and M. Bandini, Chem. Commun., 2015, 51, 2320 RSC; (c) Y. Wang, P. Zhang, Y. Liu, F. Xia and J. Zhang, Chem. Sci., 2015, 6, 5564 RSC; (d) H. Hu, Y. Wang, D. Qian, Z.-M. Zhang, L. Liu and J. Zhang, Org. Chem. Front., 2016, 3, 759 RSC.
- (a) R.-R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811 RSC; (b) X. Zhong, Q. Tang, P. F. Zhou, Z. W. Zhong, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Commun., 2018, 54, 10511 RSC; (c) W.-F. Zheng, G.-J. Sun, L. Chen and Q. Kang, Adv. Synth. Catal., 2018, 360, 1790 CrossRef CAS; (d) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef CAS.
- For selected examples, see: (a) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1 CAS; (b) A. Tahri, S. M. H. Vendeville, T. H. M. Jonckers, P. J.-M. B. Raboisson, L. Hu, S. D. Demin and L. P. Cooy-Mans, PCT Int. Appl., WO2014060411A, 2014 Search PubMed; (c) A. Fensome, R. Bender, J. Cohen, M. A. Collins, V. A. Mackner, L. L. Miller, J. W. Ullrich, R. Winneker, J. Wrobel, P. Zhang, Z. Zhang and Y. Zhu, Bioorg. Med. Chem. Lett., 2002, 12, 3487 CrossRef CAS; (d) M. Yoshikawa, H. Kamisaki, J. Kunitomo, H. Oki, H. Kokubo, A. Suzuki, T. Ikemoto, K. Nakashima, N. Kamiguchi, A. Harada, H. Kimura and T. Taniguchi, Bioorg. Med. Chem., 2015, 23, 7138 CrossRef CAS PubMed; (e) R. K. Ujjinamatada, S. Samajdar, C. Abbineni, S. Mukeherjee, T. Linnanen and G. Wohlfahrt, PCT Int. Appl., WO2016203112A1, 2016 Search PubMed.
- For selected asymmetric synthesis examples of spirocyclobutyl oxindoles, see: (a) X. Y. Hao, X. H. Liu, W. Li, F. Tan, Y. Y. Chu, X. H. Zhao, L. L. Lin and X. M. Feng, Org. Lett., 2014, 16, 134 CrossRef CAS; (b) H.-M. Zhang, Z.-H. Gao and S. Ye, Org. Lett., 2014, 16, 3079 CrossRef CAS; (c) L.-W. Qi, Y. Yang, Y.-Y. Gui, Y. Zhang, F. Chen, F. Tian, L. Peng and L.-X. Wang, Org. Lett., 2014, 16, 6436 CrossRef CAS; (d) K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685 CrossRef CAS PubMed; (e) S. S. Guo, P. Dong, Y. S. Chen, X. M. Feng and X. H. Liu, Angew. Chem., Int. Ed., 2018, 57, 16852 CrossRef CAS PubMed; (f) J.-H. Jin, J. Zhao, W.-L. Yang and W.-P. Deng, Adv. Synth. Catal., 2019, 361, 1592 CrossRef CAS.
- For reviews on chiral N,N'-dioxides, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (e) X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16604 CrossRef CAS PubMed; (f) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 CAS; (g) M.-Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969 CrossRef CAS.
- For selected reviews on cyclobutanes as 1,4-dipoles, see: (a) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC; (b) H.-U. Ressig and R. Zimmer, Angew. Chem., Int. Ed., 2015, 54, 5009 CrossRef; (c) N. Vemula and B. L. Pagenkopf, Org. Chem. Front., 2016, 3, 1205 RSC. For selected examples, see: (d) A. Levens, A. Ametovski and D. W. Lupton, Angew. Chem., Int. Ed., 2016, 55, 16136 CrossRef CAS PubMed; (e) L. K. B. Garve, A. Kreft, P. G. Jones and D. B. Werz, J. Org. Chem., 2017, 82, 9235 CrossRef CAS; (f) L.-W. Feng, H. Xiong, P. Wang, L. Wang and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 3055 CrossRef CAS PubMed; (g) J.-L. Hu, L. Zhou, L. Wang, Z. Xie and Y. Tang, Chin. J. Chem., 2018, 36, 47 CrossRef CAS; (h) D. Tong, J. Wu, N. Bazinski, D. Koo, N. Vemula and B. L. Pagenkopf, Chem.–Eur. J., 2019, 25, 15244 CrossRef CAS PubMed; (i) M. Yamazaki, T. Yoshimura and J. Matsuo, Tetrahedron Lett., 2021, 64, 151804 CrossRef CAS.
- When the racemic reaction (Table 1, entry 1) was carried out at 0 °C, the [2+2] cycoaddition product was detected. Thereofre, we deemed that the formal [4+2] adduct ws generated from the transfomation of the [2+2] cycoaddition product. See ESI, Table S1† entryt 15 for details..
- CCDC 2042864 (3aa), CCDC 2052253 (rac-4ka) and CCDC 2079315 (rac-5aa).†.
- Performing the reaction with a one-pot procedure resulted in 4 as the major product, See ESI, Table S5† for more details..
- K. Zheng, X. H. Liu, J. N. Zhao, Y. Yang, L. L. Lin and X. M. Feng, Chem. Commun., 2010, 46, 3771 RSC.
- For reviews of deprotection of Boc-protected amides using Lewis acids, see: (a) K. Jarowicki and P. Kocieński, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 1589 RSC; (b) G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199 CrossRef CAS PubMed. For selected examples, see: (c) J. A. Stafford, M. F. Brackeen, D. S. Karanewsky and N. L. Valvano, Tetrahedron Lett., 1993, 34, 7873 CrossRef CAS; (d) Z.-Y. Wei and E. E. Knaus, Tetrahedron Lett., 1994, 35, 847 CrossRef CAS; (e) H. Kotsuki, T. Ohishi, T. Araki and K. Arimura, Tetrahedron Lett., 1998, 39, 4869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR spectra, HPLC chromatograms, and CD spectra (PDF). X-ray crystallographic data for 3aa, rac-4ka and rac-5aa. CCDC 2042864, 2052253 and 2079315. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02681j |
| This journal is © The Royal Society of Chemistry 2021 |