
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational generation of lasso peptides based on biosynthetic gene mutations and site-selective chemical modifications†
Tan
Liu‡
a,
Xiaojie
Ma‡
a,
Jiahui
Yu
a,
Wensheng
Yang
b,
Guiyang
Wang
a,
Zhengdong
Wang
a,
Yuanjie
Ge
a,
Juan
Song
a,
Hua
Han
b,
Wen
Zhang
b,
Donghui
Yang
a,
Xuehui
Liu
*c and
Ming
Ma
 *a
*a
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China. E-mail: mma@bjmu.edu.cn
bSchool of Medicine, Tongji University, 1239 Siping Road, Shanghai 200092, China
cCAS Research Platform for Protein Sciences, Institute of Biophysics, Chinese Academy of Sciences, 15 Datun Road, Chao-yang District, Beijing 100101, China. E-mail: xhliu@ibp.ac.cn
First published on 10th August 2021
Abstract
Lasso peptides are a unique family of natural products whose structures feature a specific threaded fold, which confers these peptides the resistance to thermal and proteolytic degradation. This stability gives lasso peptides excellent pharmacokinetic properties, which together with their diverse reported bioactivities have garnered extensive attention because of their drug development potential. Notably, the threaded fold has proven quite inaccessible by chemical synthesis, which has hindered efficient generation of structurally diverse lasso peptides. We herein report the discovery of a new lasso peptide stlassin (1) by gene activation based on a Streptomyces heterologous expression system. Site-directed mutagenesis on the precursor peptide-encoding gene is carried out systematically, generating 17 stlassin derivatives (2–17 and 21) with residue-replacements at specific positions of 1. The solution NMR structures of 1, 3, 4, 14 and 16 are determined, supporting structural comparisons that ultimately enabled the rational production of disulfide bond-containing derivatives 18 and 19, whose structures do not belong to any of the four classes currently used to classify lasso peptides. Several site-selective chemical modifications are first applied on 16 and 21, efficiently generating new derivatives (20, 22–27) whose structures bear various decorations beyond the peptidyl monotonicity. The high production yields of these stlassin derivatives facilitate biological assays, which show that 1, 4, 16, 20, 21 and 24 possess antagonistic activities against the binding of lipopolysaccharides to toll-like receptor 4 (TLR4). These results demonstrate proof-of-concept for the combined mutational/chemical generation of lasso peptide libraries to support drug lead development.
Introduction
Lasso peptides are a unique class of ribosomally synthesized and post-translationally modified peptides (RiPPs); their structures are characterized by a specific threaded fold, in which the C-terminal amino acid residues thread through an N-terminal macrolactam ring in a structure resembling the eponymous “lasso”.1–4 Owing to this threaded fold, lasso peptides exhibit resistance to heat,5 denaturing agents,6 and proteases6 (with the notable exception of lasso peptide isopeptidases7–10). These thermal and proteolytic stabilities, viewed alongside their various reported biological activities (e.g., antibacterial, anticancer and anti-HIV activities),1,11 have garnered strong interest in pursuing lasso peptides as drug leads with desirable pharmacokinetic properties. For example, by introducing a tripeptide Arg-Gly-Asp (RGD) motif into the MccJ25 lasso peptide, this engineered MccJ25 derivative acquires potent binding affinity for integrins (with IC50 values in the nanomolar range) and can inhibit capillary formation of human umbilical vein endothelial cells (HUVECs). This MccJ25 derivative shows impressive proteolytic stability: more than half of it was still present in the serum after 30 h, whereas a linear peptide analogue was completely degraded after only 4 h.6However, different from many other peptides that can be prepared by solid-phase peptide synthesis (SPPS), lasso peptides have proven quite inaccessible by chemical synthesis. Nevertheless, the identification and engineering of lasso peptide biosynthetic pathways from native producer organisms—as well as their subsequent chemical modifications—should help to generate potentially many structurally diverse lasso peptides for basic studies and for drug development. Notably, genome mining studies have suggested that the number of candidate biosynthetic gene clusters (BGCs) far exceeds the number of lasso peptides structurally characterized to date (currently around 80 structures).12–14 As with the BGCs of many natural products, it can be challenging to culture the native producer cells; moreover, the lack of relevant biological understanding often prevents the transcriptional activation of a BGC in its native producer.
In lasso peptide biosynthesis, the precursor peptide A (consisting of a leader peptide and a core peptide) is first processed by a protease B, which cleaves off the leader peptide; in the biosynthesis of some lasso peptides from Actinobacteria and Firmicutes phyla, protease B is an enzyme complex comprising protein B1 (an example of the RiPP precursor peptide recognition element, RRE)14,15 and protein B2. After cleavage of the leader peptide, the so-called “core peptide” is the substrate for N-terminal macrolactam ring formation as catalyzed by a cyclase C enzyme, thereby generating the mature lasso peptide product (Fig. 1B). Although the precise catalytic mechanisms employed by the B/C proteins remain uncharacterized, there are reports indicating that they display some degree of substrate promiscuity.16–22 This presents an opportunity for using genetic engineering approaches to efficiently generate lasso peptide derivatives; that is, it should be possible to simply substitute amino acid residues of the core peptide, provided such substitutions can be tolerated by downstream processing B/C proteins.
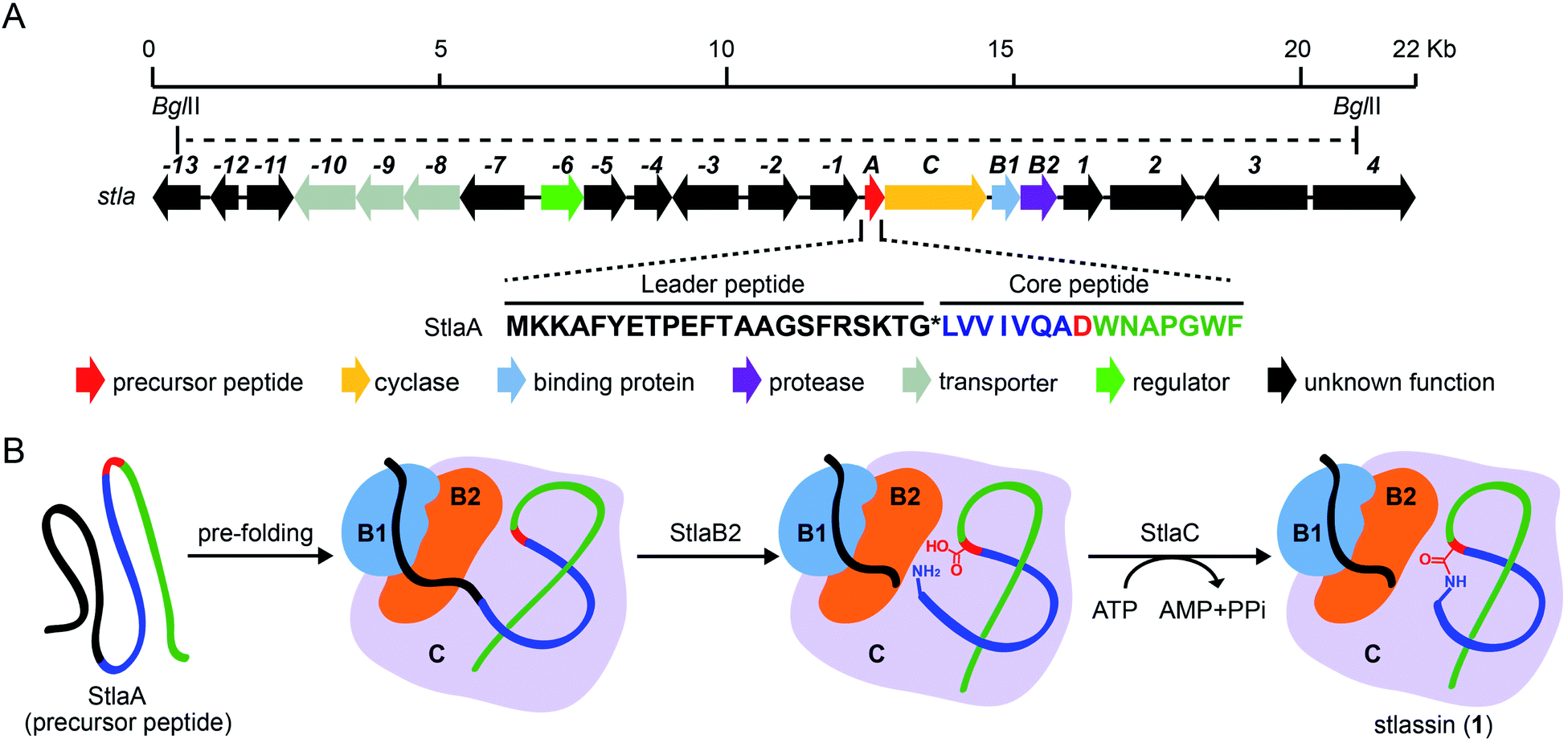 | ||
| Fig. 1 The biosynthetic gene cluster stla and the proposed biosynthesis of stlassin (1). (A) The biosynthetic gene cluster stla from the marine Streptomyces sp. PKU-MA01240. The amino acid sequence of the precursor peptide StlaA is displayed, with the leader peptide in black, the N-terminal macrolactam residues of the core peptide in blue (except the macrolactam-forming residue Asp8 in red), and the C-terminal residues of the core peptide in green. The functional annotations of numbered genes are shown in ESI Table S1.† The region between two BglII restriction sites indicates the DNA fragment used in the heterologous expression (some parts of genes −13 and 4 were not included). (B) The proposed biosynthetic pathway of 1, with StlaB1/B2/C together responsible for the pre-folding of the precursor peptide StlaA (leader peptide in black, and core peptide in blue, red and green), StlaB2 responsible for proteolysis of StlaA to cleave the leader peptide from the core peptide, and StlaC responsible for the formation of the N-terminal macrolactam ring from the core peptide to generate mature stlassin. | ||
Recalling the aforementioned problems when working with BGCs from native producers, it is unsurprising that such peptide-encoding gene mutations have been carried out using E. coli as a heterologous host. However, the production yields for lasso peptides (and engineered derivatives) are often very low in E. coli, limiting the amount of material available for biological activity assays and synthetic modification studies. Thus, and given the reports about the successful site-selective chemical modification of peptides and proteins under mild conditions (e.g., O-glycosylation of phenolic functionality and S-arylation of cysteine residues in aqueous solution at room temperature),23–25 we were interested in the idea of generating substantial structural diversity in lasso peptide derivatives by combining an efficient heterologous expression system with site-selective chemical modifications of biosynthetic products.
Pursuing this, we report here the discovery of a new class II lasso peptide (stlassin, 1) based on heterologous expression and activation of a BGC (stla) from the marine Streptomyces sp. PKU-MA01240. Using a Streptomyces heterologous expression system, we mutationally substituted each position of the core peptide StlaA and thereby simultaneously generated new lasso peptide derivatives (2–17 and 21), and identified key residues involved in the post-translational processing of stlassin peptides by the B/C enzymes of the stla BGC. Moreover, we determined solution NMR structures for 1, 3, 4, 14, and 16, which supported structural comparisons that ultimately enabled the rational design and successful production of new disulfide bond-containing derivatives 18 and 19, whose structures do not belong to any of the four classes currently used to classify lasso peptides.5,14 Our structural insights also supported the identification of modification-accessible residues (tyrosine or cysteine) at specific core peptide positions, and we successfully achieved site-selective chemical modifications at these sites, generating new derivatives (20 and 22–27) with O-glycosylation, S-glycosylation, S-arylation, disulfide bond formation, in addition to both the cleavage and formation of C–S bonds. Finally, we harnessed the high production yields from our heterologous production and chemical modification efforts and conducted biological activity assays: these revealed antagonistic activities of 1, 4, 16, 20, 21, and 24 against the binding of lipopolysaccharides (LPS) to toll-like receptor 4 (TLR4). Our study thus identifies an exciting class of lasso peptides and illustrates the powerful combination of biosynthetic gene-manipulations and chemical modifications for the generation of structurally diverse and bioactive lasso peptide molecules.
Results and discussion
Discovery of stlassin by heterologous expression of a lasso peptide biosynthetic gene cluster stla
As part of an ongoing project for natural product discovery from marine bacteria,26–29 we detected a candidate lasso peptide biosynthetic gene cluster stla (accession number: MZ182273) from the genome of a marine Streptomyces sp. PKU-MA01240. The stla gene cluster apparently encodes a precursor peptide StlaA, a RRE-containing protein StlaB1, a protease StlaB2, and a cyclase StlaC (Fig. 1A and Table S1†). Sequence analysis of StlaA indicated that the mature product may be a class II lasso peptide comprising 15 amino acid residues from the core peptide (LVVIVQADWNAPGWF), and an N-terminal macrolactam ring should be formed between the amino group of Leu1 and the side chain carboxyl group of Asp8 (Fig. 1).Given that no lasso peptide product was detected in cultures of S. sp. PKU-MA01240 assessed with HPLC or LC-MS, and considering that this strain does not readily accept exogenous plasmids, we explored the heterologous expression of the stla cluster in experimentally tenable Streptomyces hosts. Using RecET direct cloning and the Redαβ recombineering system,30,31 we constructed a plasmid (pMM2002) bearing the 20.3 Kb fragment covering the stla gene cluster (Fig. 1A and S2†). pMM2002 was integrated into S. coelicolor A3(2) and S. lividans K4-114,28 but no lasso peptide was detected by HPLC or LC-MS analyses of cultures grown using three different media (Fig. S3†), suggesting that the stla cluster may not be expressed in these heterologous hosts.
Seeking to activate the stla cluster, a constitutive promoter KasOp*, which has been used for the activation of other natural product biosynthetic gene clusters,32–34 was cloned into the upstream region of the stlaA locus, generating the plasmid pMM2004 (Fig. S4†). pMM2004 was integrated into S. coelicolor A3(2) and S. lividans K4-114 and HPLC analysis showed a product from both strains (∼80% amount in the supernatant and ∼20% amount in cells, Fig. S5†) cultured in all three tested media, with the highest yield detected for S. coelicolor A3(2) cultured in M2 medium (ESI and Fig. S3†). MS analysis of the product showed an [M–H]− ion at m/z 1694.8662 (Table S4 and Fig. S16†). This ion would be consistent with the aforementioned proposed product from the stla cluster. Subsequently, a 5 L fermentation and isolation using a combination of chromatographic methods yielded 51 mg of this compound (named stlassin, 1, Fig. 2). Currently, heterologous expression of actinobacterial lasso peptide clusters in E. coli is difficult. Given the increasing discovery of lasso peptides from Streptomyces,35–41 the Streptomyces expression system here could benefit the investigation of interesting clusters from this bacterial genus.
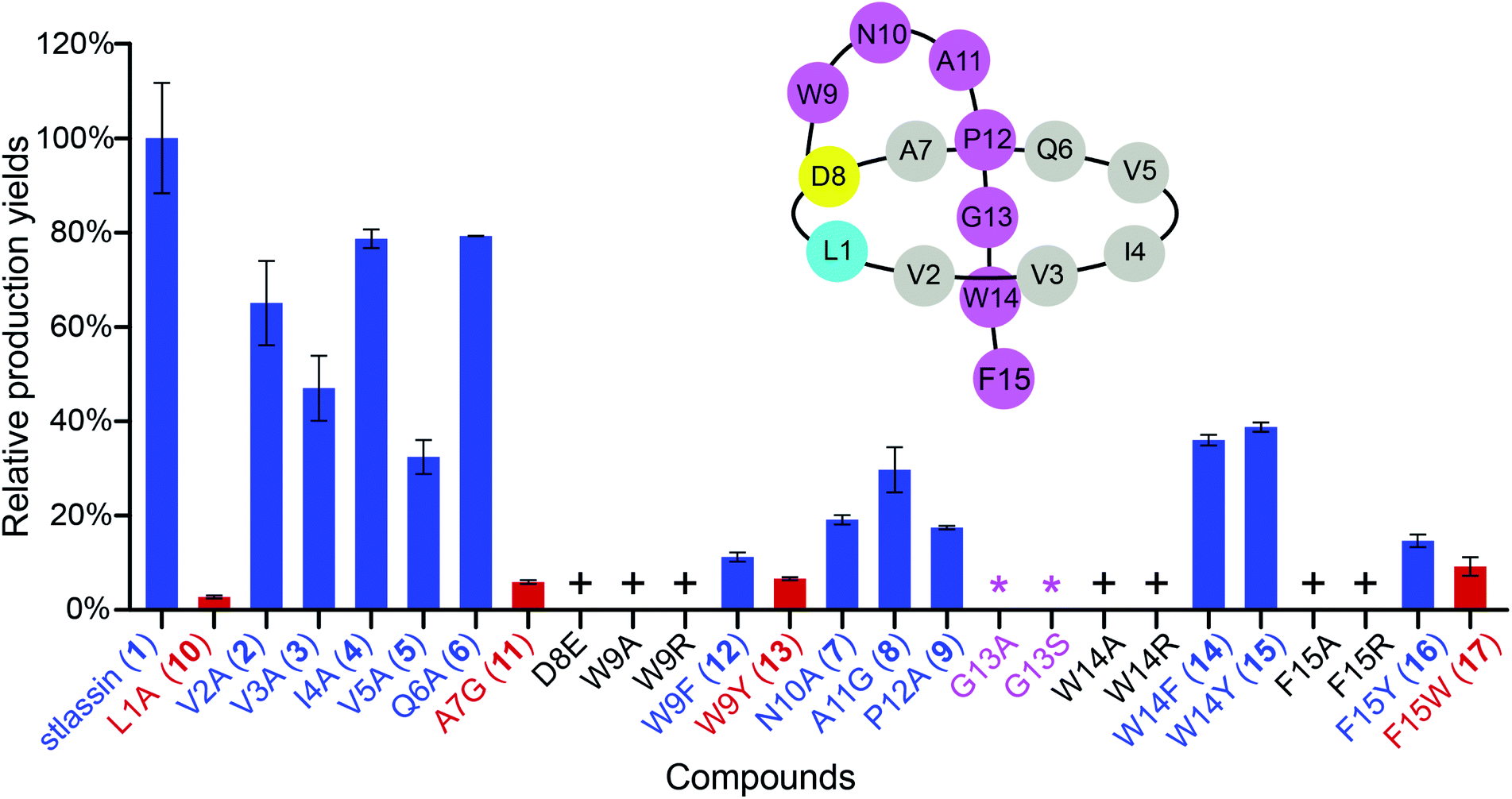 | ||
| Fig. 2 The relative production yields of stlassin derivatives from core peptide mutations to that of stlassin (1). The stlassin derivatives with relative production yields over 10% (based on HPLC peak area calculations) are shown in blue, and those below 10% are shown in red. “+” means that the production of stlassin derivatives can only be detected by mass, and “*” indicates no production even based on mass detection. A schematic presentation of stlassin (1) containing the 15 amino acid residues is also shown. | ||
The sufficient amount of 1 generated from the heterologous expression enabled us to determine its solution NMR structure. The structure of 1 shows a typical right-handed lasso fold. The N-terminal macrolactam ring is formed between Leu1 and Asp8, and the C-terminal residues thread through the macrolactam ring (Fig. 3A). Note that in so-called class II lasso peptides, a defining feature is the steric lock that keeps the C-terminal tail from escaping the confines of the macrolactam ring;5 this differs from the disulfide bond present in class I, III, and IV lasso peptides (Fig. 4B). The structure of 1 obeys the conserved feature in class II, with bulky residues including Trp14 and Phe15, which protrude in opposite directions under the macrolactam ring (Fig. 3A).
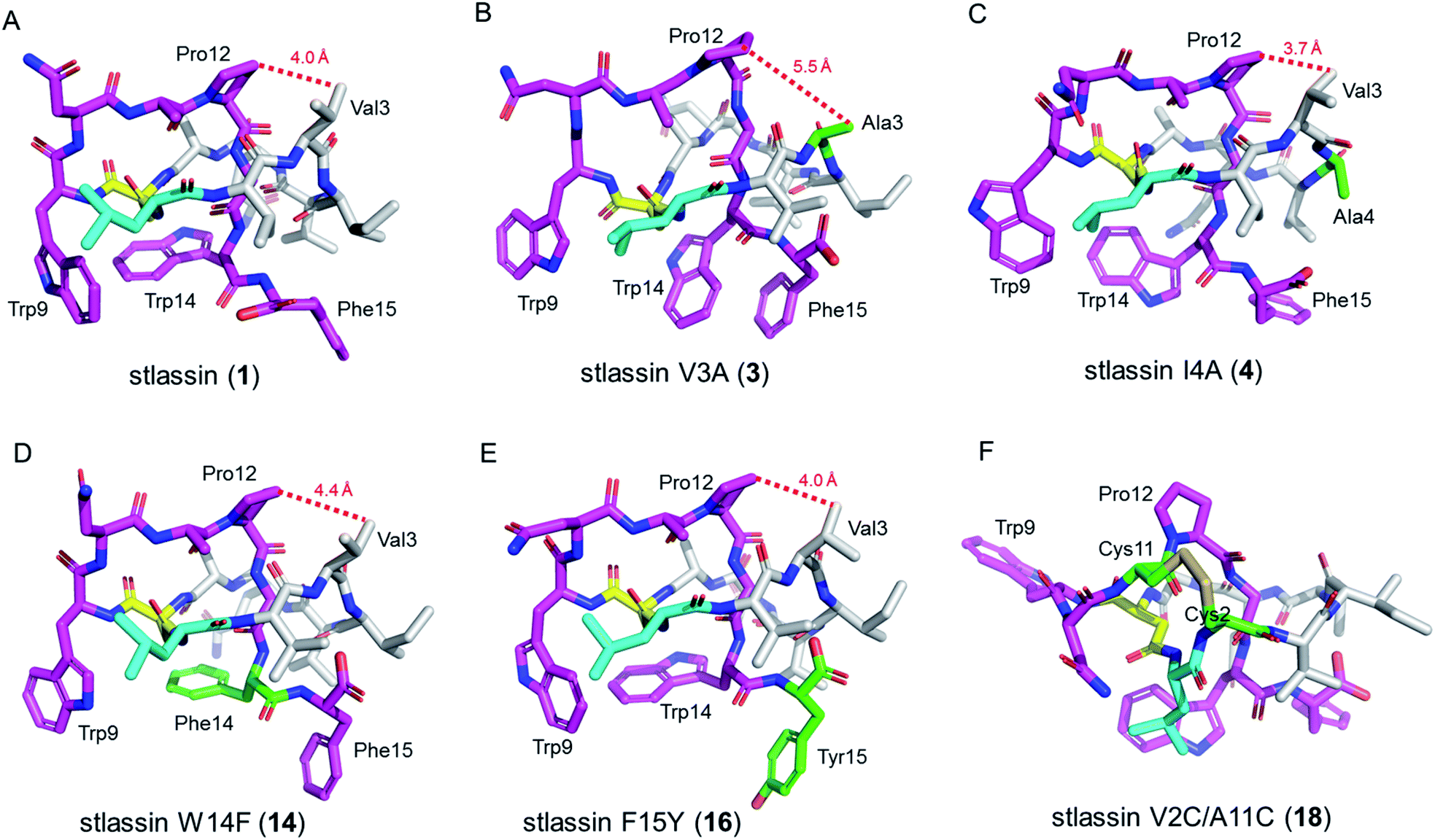 | ||
| Fig. 3 The solution NMR structures of stlassin (1) and its derivatives generated from mutations in the core peptide of StlaA. (A)–(F) The solution NMR structures of 1, 3, 4, 14, 16, and 18. The N-terminal macrolactam residues are shown in gray (except the macrolactam ring-forming Leu1 in cyan and Asp8 in yellow). The C-terminal residues are shown in magenta, and the residues introduced via mutations are shown in green. The distances between Val3 (or Ala3) and Pro12 measured from the NMR solution structures in (A)–(E) are indicated with red dashed lines. | ||
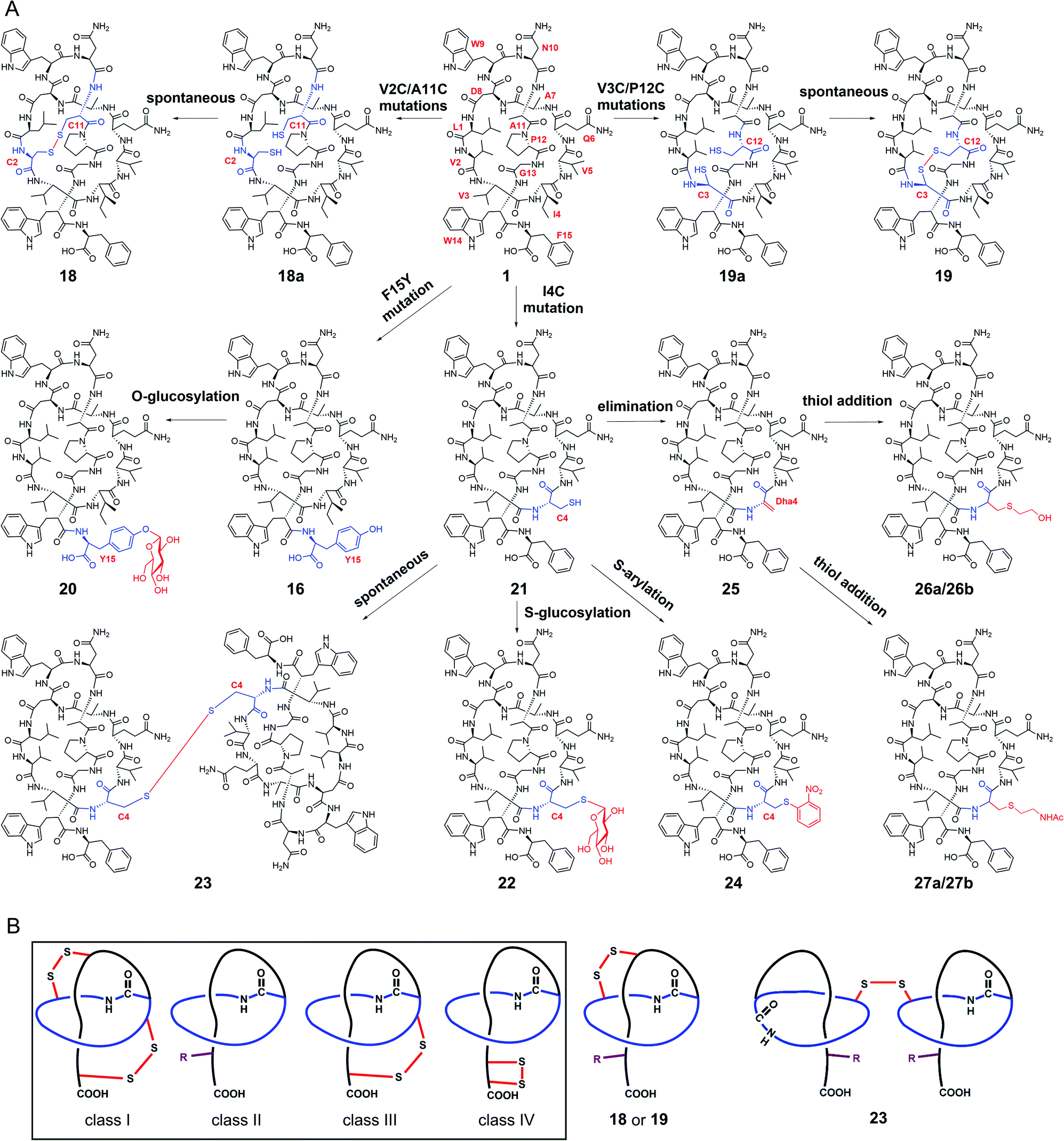 | ||
| Fig. 4 Generation of stlassins 18–27, and comparisons of 18, 19 and 23 with the four previously defined classes of lasso peptides. (A) Generation of 18–27 by site-directed mutagenesis of the core peptide of 1 and by site-selective chemical modifications under mild conditions. These chemical modifications efficiently generate new derivatives with O-glycosylation (20), S-glycosylation (22), intermolecular disulfide bond-bridged dimerization (23), S-arylation (24), cysteine sulfur elimination (25), and thiol-conjugate addition (26 and 27). (B) Comparisons of 18, 19 and 23 with the four previously defined classes of lasso peptides. 18, 19 and 23 are rationally designed to contain both the C-terminal steric lock (shown as “R”) and disulfide bond (not present in class II lasso peptides). The disulfide bonds in 18, 19 and 23 are generated at different positions from those in class I, III and IV lasso peptides. | ||
Generation of stlassin derivatives by single-mutations in the core peptide
We used the Redαβ recombineering system to explore the production of potential stlassin derivatives with a mutation strategy focused on 15 residues of the core peptide (ESI and Fig. S7†). These residues (LVVIVQADWNAPGWF) were first individually mutated to alanine residues (except Ala7, Asp8, and Ala11, which were mutated to Gly7, Glu8 and Gly11, respectively). HPLC and LC-MS analyses of extracts from the cells expressing the V2A, V3A, I4A, V5A, Q6A, N10A, A11G and P12A variants showed that they produced lasso peptide derivatives (2–9) at comparable yields to 1 in the control strain without mutation (Fig. 2 and S8†), suggesting that mutations at these positons can be tolerated by the downstream processing proteins StlaB1/B2/C.Cells expressing the L1A and A7G variants produced lasso peptide derivatives L1A (10) and A7G (11), which the HPLC data suggested to be produced at much reduced yields compared to 1. Even lower product yields were detected from cells expressing the D8E, W9A, and W9R variants (these putative products could only be detected by LC-MS, Fig. 2, S8 and S27†). Later experiments showed that cells expressing W9F and W9Y variants produced higher yields of products (named stlassins W9F (12) and W9Y (13)) compared to products from cells expressing the W9A and W9R variants. These results suggest that an aromatic side chain at this position may somehow support the post-translational processing of these peptides, or help maintain the threaded lasso products to avoid degradation before export out of cells.
No product was detected for cells expressing G13A or G13S variants (Fig. 2 and S8†). The solution NMR structure of 1 shows that the Gly13 residue is located exactly in the middle of the macrolactam ring (Fig. 3A). Thus, any residue with a side chain at this position may generate a steric effect, which may prevent proper pre-folding. Mutations of the W14 and F15 residues suggested that aromatic side chains may be required for the pre-folding step of stlassin production: briefly, only low-abundance products were detected from cells expressing the W14A and F15A variants, whereas cells with W14F, W14Y, F15Y and F15W produced lasso peptide derivatives named stlassins W14F (14), W14Y (15), F15Y (16) and F15W (17). Collectively, these mutational studies revealed a rich diversity of stlassin derivatives and highlighted individual residues of the core peptide that exert pronounced effects on the post-translational processing of the precursor peptide StlaA or the stability of the mature threaded lasso products.
Solution NMR structure comparisons and thermal stability evaluation of stlassin derivatives
We conducted large-scale fermentation and product isolation efforts with cells expressing the V3A, I4A, W14F, and F15Y variants (ESI†), and successfully obtained stlassin V3A (3), I4A (4), W14F (14) and F15Y (16), respectively. The solution NMR structures of 3, 4, 14 and 16 were determined and their structures were comparatively analyzed with 1.Our NMR data indicated similar right-handed lasso folds for 3, 4, 14, and 16 (Fig. 3B–E) to that in 1, and structural superimposition indicated that the five main chains overlap well (Fig. S12A†). In contrast, the side chains of Gln6, Trp9, Asn10, Trp14 (or Phe14), and Phe15 (or Tyr15) have distinct positions in these various structures (Fig. S12B†). Interestingly, among these stlassin derivative structures (except 3), the side chains of Trp9 and Trp14 (or Phe14) are invariably positioned facing each other (Fig. 3), which likely help keep the C-terminal tail from escaping the confines of the macrolactam ring.
The 20 solution structures with the lowest total energy in each of 1, 3, 4, 14 and 16 are generally the same, with Trp9, Trp14, and Phe15 (or Tyr15 in 16) displaying the highest extent of flexibility (Fig. S11†). We tested the thermal and proteolytic stabilities of 1, 3, 4, 14, and 16 by heating experiments and carboxypeptidase Y-treatment experiments,5,21,42–44 and found that all of these compounds are stable, with the exception of stlassin V3A (3) which could be converted to its unthreaded counterpart upon heating to 95 °C for 3 h (ESI†). In the solution NMR structures of 1, 4, 14 and 16, the side chains of Val3 and Pro12 are positioned facing each other (with their shortest distances ranging from 3.7 Å to 4.4 Å) (Fig. 3A and 3C–E). However, for 3 there was a larger distance (5.5 Å) between Ala3 and Pro12 (Fig. 3B). More NOESY correlations are observed between Val3 and Pro12 in each of 1, 4, 14 and 16, whereas only one NOESY correlation is observed between Ala3 and Pro12 in 3, due to the mutation from Val3 to a smaller side chain-containing Ala3 (Fig. S13†). The V3A mutation has additional structural consequences: Gly13 in 3 is not located in the middle of the macrolactam ring, and the indole ring of 3's Trp14 is not facing the indole ring of Trp9 any more. Ultimately, these differences suggest weaker steric locking in 3 than in 1, 4, 14 and 16. This is another example of a residue in the macrolactam ring contributing to thermal stability, which is additional to the case that a specific P8A mutation in lasso peptide caulosegnin II causes higher macrolactam ring flexibility and subsequent loss of thermal stability.45
Rational generation of disulfide bond-containing derivatives based on double mutations in the core peptide
We next explored the generation of additional lasso peptide derivatives that extend beyond the typical lasso peptide classification. That is, the lasso peptide family is traditionally divided into four classes based on the presence (class I/III/IV) or absence (class II) of disulfide bond(s). Class I peptides have two disulfide bonds: one between the macrolactam ring and the C-terminal loop, and another between the macrolactam ring and the C-terminal tail (the C-terminal region after threading through the macrolactam ring). Class II peptides lack any disulfide bonds. Class III peptides have one disulfide bond between the macrolactam ring and the C-terminal tail. Class IV peptides have one disulfide bond within their C-terminal tails (Fig. 4B).5,11,14 The disulfide bonds in class I, III, and IV peptides—together with C-terminal bulky residues—are known to promote the stability of the lasso fold.5,41,46,47 The stability of class II peptides results from steric locking.We generated double mutant StlaA variants by introducing two cysteine residues, seeking to generate derivatives of 1 having one disulfide bond positioned between the macrolactam ring and the C-terminal loop. If successful, this should generate a type of lasso fold that is outside the traditional four classes of lasso peptides. We excluded several residues including Asp8, Trp9, and Gly13 as potential cysteine residue replacement sites based on our results from the single mutant-variants. Given that the lengths of C–S and S–S bonds are about 1.82 Å and 2.07 Å, respectively, the distance between the two β-carbons of two cysteine residues in a disulfide bond should be smaller than 5.71 Å (1.82 Å × 2 + 2.07 Å).
We checked the solution NMR structures of 1, 3, 4, 14, and 16 and found four pairs of residues Leu1/Ala11, Val2/Ala11, Val3/Pro12, and Ala7/Pro12 whose β-carbons distances are smaller than 5.71 Å (Fig. S9†). We then generated double mutations of L1C/A11C, V2C/A11C, V3C/P12C and A7C/P12C, and fermented the strains bearing these mutations (MM20030-MM20033). HPLC and LC-MS analyses showed that cells expressing the V2C/A11C and V3C/P12C double cysteine mutant variants produced derivatives 18 and 19, with [M–2H]2− ions at m/z 863.8757 and 850.8723, respectively (Fig. S8, S34 and S35†). No products were detected in cells expressing the L1C/A11C and A7C/P12C variants (Fig. S8†), perhaps owing to the negative effects from the mutations of Leu1 and Ala7.
Large-scale fermentation and isolation of 18 supported solution NMR analysis: 18 has a right-handed lasso fold with a similar C-terminal steric lock to 1 (Fig. 3F). The disulfide bond between Cys2 and Cys11 causes distinct positions of most residues in 18 from those of 1, 3, 4, 14, and 15. The thermal and proteolytic stabilities of 18 were tested, showing similarly high stability to that of 1 (Fig. S6G†). The disulfide bond may decrease the flexibilities of both the macrolactam ring and C-terminal residues, thereby contributing to the thermal stability of 18. This is supported by the invariably positioned residues around the disulfide bonds in the 20 solution structures of 18 with the lowest total energy, particularly the C-terminal residues from Pro12 to the steric lock Trp14 (Fig. S11†).
Rational generation of stlassin derivatives using site-selective chemical modifications
The stlassin derivatives generated above are all based on core peptide-encoding gene mutations and the substrate promiscuities of the post-translation processing proteins StlaB1/B2/C. Notably, the absence of additional biosynthetic genes beyond StlaB1/B2/C in the stla cluster results in peptidyl monotonicity of the stlassin skeletons. We wanted to break through this structural limitation and generate additional derivatives, so we adopted a site-selective chemical modification approach. Some elegant recent work has demonstrated high-efficiency and selective glycosylation or arylation of tyrosine and cysteine residues of different peptides under mild reaction conditions.24,25 Starting from stlassin F15Y (16) with its C-terminal tyrosine residue, we incubated 16 with α-D-fluoroglucose and Ca(OH)2 at room temperature in water for 1 h,24 which yielded the glucosylation product stlassin F15Y-I (20) ([M–2H]2− ion at m/z 935.9551) (Fig. S36†) in a high yield (84%) (Fig. 4A and 5, lane III). The reaction of 1 with α-D-fluoroglucose under the same conditions produced no product (Fig. 5, lane I), thereby confirming the site-selective O-glucosylation of Tyr15 in the chemically modified product 20.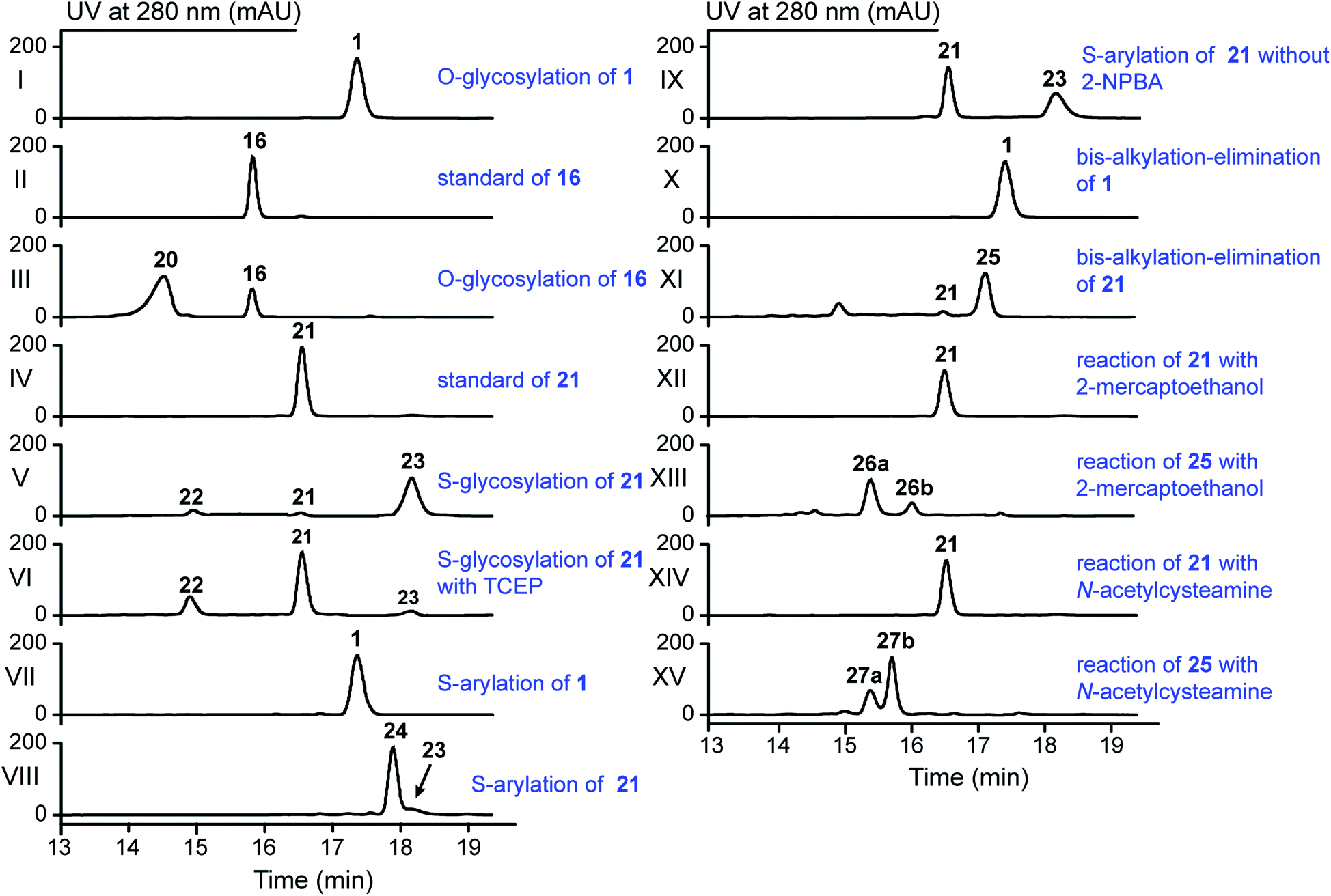 | ||
| Fig. 5 HPLC analysis of the reactions in the generation of 20 and 22–27 by the indicated site-selective chemical modifications (all achieved under mild conditions). | ||
Seeking to introduce a cysteine residue in the lasso peptide, we checked the solution NMR structures of 1, 3, 4, 14, 16, and 18, and selected Ile4 for mutation because (i) the I4A mutation gave a high yield of the product (4) and (ii) its position far from the C-terminal loop region may avoid potential steric effects. Cells expressing a I4C variant produced stlassin I4C (21) ([M–H]− ion at m/z 1684.7978) (Fig. S8 and S35†), which was isolated and incubated with α-D-fluoroglucose and Ca(OH)2 at room temperature in water for 1 h.24 We detected the glucosylation product stlassin I4C-I (22) ([M–H]− ion at m/z 1846.8480), but in a low yield (3%) (Fig. 4A and 5, lane V; Fig. S38†). Interestingly, a dimerization product stlassin I4C-II (23, [M–2H]2− ion at m/z 1683.7888) with an intermolecular disulfide bond was produced in this reaction, which suggests that the reaction conditions may provide an oxidative environment facilitating the disulfide bond formation (Fig. 4A and 5, lane V; Fig. S39†). To improve the glucosylation efficiency of 21, TCEP was added to prevent the dimerization, which led to an increased yield (24%) of 22 (Fig. 5, lane VI).
Beyond these glycosylation modifications, we also explored arylation of 21 by using the recently developed nickel(II)-promoted cysteine S-arylation reaction with 2-nitro-substituted arylboronic acid.25 Compound 21 was incubated with 2-nitrophenylboronic acid and Ni(OAc)2 in N-methylmorpholine (NMM) buffer at 37 °C for 30 min, generating the arylation product stlassin I4C-III (24, [M–H]− ion at m/z 1805.8148) in a high yield (93%) (Fig. 4A and 5, lane VIII; Fig. S40†). Note that a small amount of the aforementioned dimerization product 23 was also produced in this reaction (6%).
Previous studies have shown that some RiPPs such as lanthipeptides, thiopeptides, and goadsporins feature dehydroalanine (Dha) residue(s).48 Generation of Dha residues can result from the dehydration of serine by a dehydratase, but no dehydratase-encoding gene is present in lasso peptide biosynthetic gene clusters reported to date. However, it is possible to generate Dha residues through sulfur elimination from cysteine residues under mild conditions.49 Thus, we carried out the bis-alkylation–elimination reaction of 21 for the conversion of Cys4 to Dha4 using the reagent α,α′-dibromo-adipyl(bis)amide. When compound 21 was treated with α,α′-dibromo-adipyl(bis)amide and K2CO3 for 30 min at room temperature, followed by an additional 4 h at 37 °C, the Dha4-containing product stlassin I4C-IV (25, [M + H]+ ion at m/z 1652.8258) was generated in a high yield (91%) (Fig. 4A and 5, lane XI; Fig. S41†).
Dha residues are highly amenable to a diverse range of modifications, including phosphorylation, glycosylation, methylation, acetylation, and lipidation.49,50 We further modified 25 by Michael addition-like reactions with thiol molecules.50 The conjugate addition of 2-mercaptoethanol generated stlassins I4C-V (26a, [M + H]+ ion at m/z 1728.81) and I4C-VI (26b, [M + H]+ ion at m/z 1728.81) (Fig. 5, lane XIII and Fig. S42 and S43†), while conjugate addition of N-acetylcysteamine generated stlassins I4C-VII (27a, [M + H]+ ion at m/z 1771.89) and I4C-VIII (27b, [M + H]+ ion at m/z 1771.83) (Fig. 5, lane XV and Fig. S44 and S45†). Products 26a/26b and 27a/27b are hypothesized as two pairs of epimer products from the thiol additions, and similar epimer products from the addition of thiols to Dha have been mentioned in previous literature;49 these were generated in high yields at room temperature after only 2 h. The reactions of 21 with 2-mercaptoethanol or N-acetylcysteamine generated no products (Fig. 5, lanes XII and lane XIV), confirming the site-selective modifications on Dha4 in 26a, 26b, 27a, and 27b. Thus, along with the generation of 20 and 22–27, chemical modifications under mild conditions can be employed to expand the structural diversity beyond the limitation of lacking biosynthetic modifying genes.
Biological activity assays reveal antagonistic activities against the binding of LPS to TLR4
Lasso peptides exert various biological activities, including for example antibacterial effects.1,11 We found that 1 showed no antibacterial activities in assays against several Gram-positive and -negative strains; nor did it exert any obvious cytotoxicity against multiple human cancer cell lines (ESI†). However, we did find that 1 showed antagonistic activity against the binding of lipopolysaccharides (LPS) to toll-like receptor 4 (TLR4) in experiments based on biotin–streptavidin-based enzyme-linked immunosorbent assays (ELISA). For context, binding of LPS is known to promote dimerization of the TLR4–MD-2 (myeloid differentiation protein-2) complex, which subsequently recruits several signalling adaptor molecules and initiates a series of signaling cascades that result in the release of inflammatory mediators.51–53 Pharmacological regulation of TLR4 activation could be a beneficial strategy for preventing or treating inflammatory diseases.54To evaluate the antagonistic activities of other stlassin derivatives, we carried out large-scale fermentations of the corresponding mutant strains, followed by chemical modifications of the isolated products, to prepare for assays with 2–12, 14–16, 18–21, and 24. Our LPS antagonistic activity assays showed that stlassins I4A (4), I4C (21), I4C-III (24), F15Y (16), and F15Y-I (20) exerted comparable inhibitory activities to stlassin (1). The other stlassin derivatives showed no or less inhibitory activity (Fig. 6). Dose–response experiments were carried out for 1, 4, 21, 24, 16, and 20, showing increased antagonistic activities with increased concentrations of each compound (Fig. S15†). IC50 values were determined based on the dose–response curves (Fig. S15†), in which 16 (IC50 value of 0.273 ± 0.022 μM) and 20 (IC50 value of 0.501 ± 0.057 μM) gave smaller IC50 values than 1 (IC50 value of 0.753 ± 0.015 μM). The different inhibitions of these stlassin derivatives provide important information useful for the generation of more potent inhibitors, e.g. more mutations and chemical modifications on Ile4 and Phe15 positions in the future.
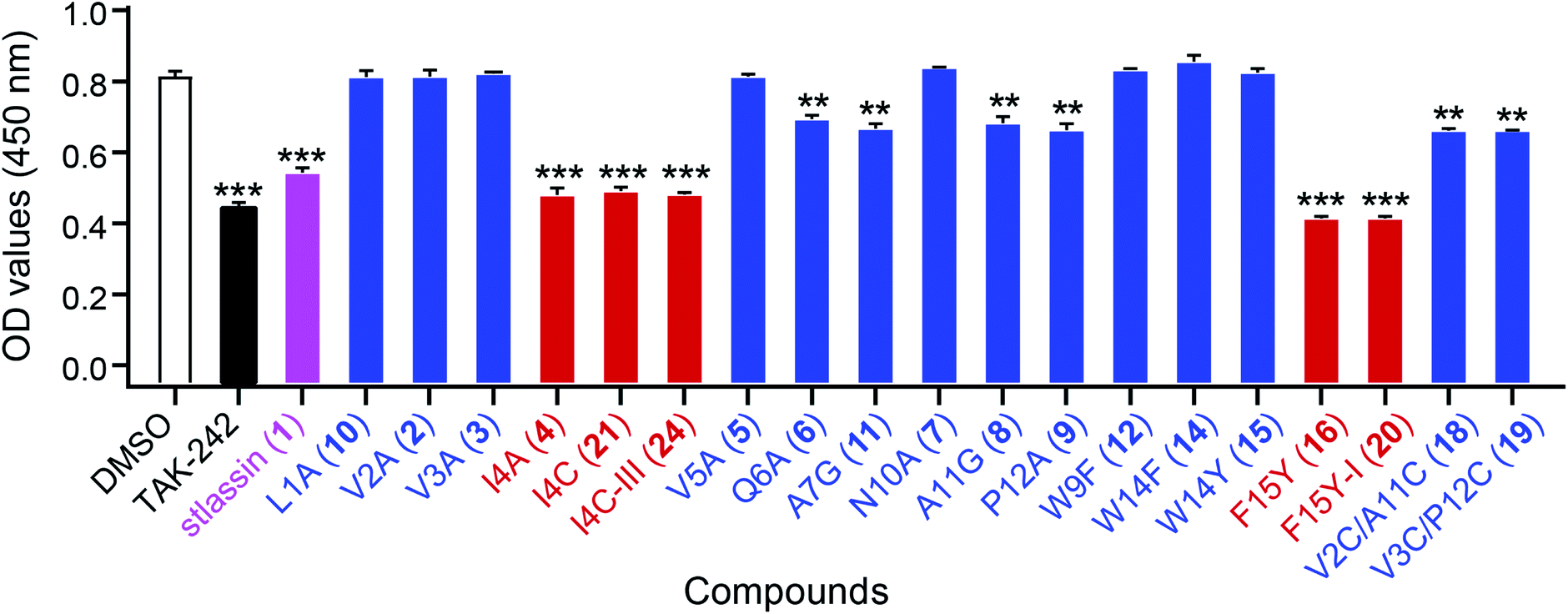 | ||
| Fig. 6 The antagonistic activities of stlassins against the binding of lipopolysaccharides (LPS) to toll-like receptor 4 (TLR4). The antagonistic activities were assessed as optical density (OD) values, measured at 450 nm using enzyme-linked immunosorbent assays (ELISA). Four parallel experiments were carried out for each sample, and the OD values and error bars were generated based on the calculation of mean ± standard deviation from the four parallel experiments. Stlassin (1) is shown in magenta; stlassins less potent than 1 are shown in blue; stlassins with comparable potency to 1 are shown in red. For these assays, DMSO was the negative control and the TLR4 inhibitor TAK-242 was the positive control. The statistical significances (versus DMSO treatments) were set at **P < 0.01 and ***P < 0.001, using one-way ANOVA with Dunnett's test. | ||
Conclusions
We successfully expressed the lasso peptide biosynthetic gene cluster stla from marine Streptomyces sp. PKU-MA01240 using a Streptomyces heterologous expression system, which produced the class II lasso peptide stlassin (1). To date, most mutational investigations of lasso peptides have been carried out in E. coli,16,42,55–59 with the single exception being a mutational analysis of lariatin A peptides in the Gram-positive bacteria Rhodococcus jostii.60 In the present study, our single-mutation analysis of the 15 positions of the core peptide revealed functional impacts of residues on stlassin peptide formation (including post-translational processing by StlaB1/B2/C), including Leu1, Asp8, Ala7, and Trp9 (related to the macrolactam ring formation), Gly13 (in the middle of the macrolactam ring), and Trp14 and Phe15 (related to the C-terminal steric lock formation).Mutations of Val2–Gln6 and Asn10–Pro12 in the core peptide were well tolerated and enabled production of structurally diverse products, as did substitution of Trp9, Trp14, and Phe15 with other aromatic residues. These results pave the way for carrying out more mutations at these positions, which could be an alternative way to random isolation from natural resources for obtaining new lasso peptides. Seventeen stlassin derivatives (2–17 and 21, Fig. S10†) were generated, and six solution NMR structures of stlassin derivatives (1, 3, 4, 14, 16 and 18) were determined. The solution NMR structural comparisons of 1, 3, 4, 14 and 16 revealed the structural differences related to the loss of thermal stability of 3. We harnessed this structural insight to support rational double mutations that enabled the production of 18 and 19, each of which contains both an intramolecular disulfide bond and a C-terminal steric lock; these structural features extend beyond the current classification of lasso peptides (Fig. 4B). Notably, our productions of 18 and 19 support that disulfide bonds in class I, III and IV lasso peptides may form spontaneously.
Moreover, we performed site-selective chemical modifications at specific positions in the stlassin peptides, generating 20 and 22–27. It should be possible to further expand the structural diversity of stlassin-type lasso peptides by for example introducing tyrosine, cysteine, or other residues into the core peptide to support chemical modifications. Finally, we found that stlassin derivatives with mutations and chemical modifications at the Ile4 and Phe15 positions (4, 16, 20, 21 and 24) showed antagonistic activity against the binding of LPS to TLR4. To the best of our understanding, no previous studies have reported this activity for lasso peptides. In sum, our application of biosynthetic gene mutations and site-selective chemical modifications sets the stage for constructing a unique lasso peptide library, which can support further efforts for bioactivity-guided structural optimization and anti-inflammatory agent development.
Data availability
All data are available in the manuscript and ESI.† The atomic coordinates of compounds 1, 3, 4, 14, 16, and 18 are accessible via PDB IDs of 7BZA, 7BZ8, 7BZ9, 6M19, 7BZ7, and 7CU6, respectively, at http://www.rcsb.org.Author contributions
Ming Ma for conceptualization; Tan Liu and Xiaojie Ma for major investigation; Wensheng Yang help perform the biological activity assays; Jiahui Yu, Guiyang Wang, Zhengdong Wang, Yuanjie Ge, Juan Song help the investigation; Xuehui Liu for providing resource for solution NMR structure determination and conceptualized discussion; Hua Han, Wen Zhang, Donghui Yang, Xuehui Liu and Ming Ma for formal analysis; Ming Ma for supervision and funding acquisition; Tan Liu and Xiaojie Ma for writing-original draft; Ming Ma for writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jun Li, Wen Ma and Yuan Wang of the State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, for LC-MS analysis. We thank Hailong Wang and Prof. Youming Zhang of Shandong University-Helmholtz Institute of Biotechnology, State Key Laboratory of Microbial Technology, Shandong University, for providing the RecET direct cloning and Redαβ recombineering system. This research was supported in part by the National Key Research and Development Program of China (2019YFC0312502), the National Natural Science Foundation of China (grant numbers 21877002, 81673332, 81991525, 22077007), the Key Project at Central Government Level: The Ability Establishment of Sustainable Use for Valuable Chinese Medicine Resources (2060302-1903-03), and the China Postdoctoral Science Foundation (2018M641123).References
- M. O. Maksimov, S. J. Pan and A. J. Link, Lasso peptides: structure, function, biosynthesis, and engineering, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- M. O. Maksimov and A. J. Link, Prospecting genomes for lasso peptides, J. Ind. Microbiol. Biotechnol., 2014, 41, 333–344 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. L. Xie and M. A. Marahiel, Lasso peptides: an intriguing class of bacterial natural products, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- J. D. Hegemann, Factors governing the thermal stability of lasso peptides, ChemBioChem, 2020, 21, 7–18 CrossRef CAS PubMed.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. L. Xie and M. A. Marahiel, Introducing lasso peptides as molecular scaffolds for drug design: engineering of an integrin antagonist, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS PubMed.
- M. O. Maksimov and A. J. Link, Discovery and characterization of an isopeptidase that linearizes lasso peptides, J. Am. Chem. Soc., 2013, 135, 12038–12047 CrossRef CAS PubMed.
- M. O. Maksimov, J. D. Koos, C. H. Zong, B. Lisko and A. J. Link, Elucidating the specificity determinants of the AtxE2 lasso peptide isopeptidase, J. Biol. Chem., 2015, 290, 30806–30812 CrossRef CAS PubMed.
- J. R. Chekan, J. D. Koos, C. H. Zong, M. O. Maksimov and A. J. Link, Structure of the lasso peptide isopeptidase identifies a topology for processing threaded substrates, J. Am. Chem. Soc., 2016, 138, 16452–16458 CrossRef CAS PubMed.
- C. D. Fage, J. D. Hegemann, A. J. Nebel, R. M. Steinbach, S. Z. Zhu, U. Linne, K. Harms, G. Bange and M. A. Marahiel, Structure and mechanism of the sphingopyxin I lasso peptide isopeptidase, Angew. Chem., Int. Ed., 2016, 55, 12717–12721 CrossRef CAS PubMed.
- C. Cheng and Z. C. Hua, Lasso Peptides: Heterologous production and potential medical application, Front. Bioeng. Biotechnol., 2020, 28, 571165 CrossRef PubMed.
- J. D. Hegemann, M. Zimmermann, S. Z. Zhu, D. Klug and M. A. Marahiel, Lasso peptides from proteobacteria: Genome mining employing heterologous expression and mass spectrometry, Biopolymers, 2013, 100, 527–542 CrossRef CAS PubMed.
- M. O. Maksimov, I. Pelczer and A. J. Link, Precursor-centric genome-mining approach for lasso peptide discovery, Proc. Natl. Acad. Sci., 2012, 109, 15223–15228 CrossRef CAS PubMed.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, A new genome-mining tool redefines the lasso peptide biosynthetic landscape, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, A prevalent peptide-binding domain guides ribosomal natural product biosynthesis, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS PubMed.
- O. Pavlova, J. Mukhopadhyay, E. Sineva, R. H. Ebright and K. Severinov, Systematic structure-activity analysis of microcin J25, J. Biol. Chem., 2008, 283, 25589–25595 CrossRef CAS PubMed.
- S. J. Pan and A. J. Link, Sequence diversity in the lasso peptide framework: Discovery of functional microcin J25 variants with multiple amino acid substitutions, J. Am. Chem. Soc., 2011, 133, 5016–5023 CrossRef CAS PubMed.
- J. D. Hegemann, C. J. Schwalen, D. A. Mitchell and W. A. van der Donk, Elucidation of the roles of conserved residues in the biosynthesis of the lasso peptide paeninodin, Chem. Commun., 2018, 54, 9007–9010 RSC.
- S. Duquesne, D. Destoumieux-Garzón, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Two enzymes catalyze the maturation of a lasso peptide in Escherichia coli, Chem. Biol., 2007, 14, 793–803 CrossRef CAS PubMed.
- K.-P. Yan, Y. Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, Dissecting the maturation steps of the lasso peptide microcin J25 in vitro, ChemBioChem, 2012, 13, 1046–1052 CrossRef CAS PubMed.
- A. J. DiCaprio, A. Firouzbakht, G. A. Hudson and D. A. Mitchell, Enzymatic reconstitution and biosynthetic investigation of the lasso peptide fusilassin, J. Am. Chem. Soc., 2019, 141, 290–297 CrossRef CAS PubMed.
- J. D. Koos and A. J. Link, Heterologous and in vitro reconstitution of fuscanodin, a lasso peptide from Thermobifida fusca, J. Am. Chem. Soc., 2018, 141, 928–935 CrossRef PubMed.
- O. Boutureira and G. J. Bernardes, Advances in chemical protein modification, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Rapid phenolic O-glycosylation of small molecules and complex unprotected peptides in aqueous solvent, Nat. Chem., 2018, 10, 644–652 CrossRef CAS.
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Rapid nickel(II)-promoted cysteine S-arylation with arylboronic acids, Chem. Commun., 2019, 55, 2841–2844 RSC.
- M. J. Zhou, F. W. Liu, X. Y. Yang, J. Jin, X. Dong, K. W. Zeng, D. Liu, Y. T. Zhang, M. Ma and D. H. Yang, Bacillibactin and bacillomycin analogues with cytotoxicities against human cancer cell lines from marine Bacillus sp. PKU-MA00093 and PKU-MA00092, Mar. Drugs, 2018, 16, 22 CrossRef PubMed.
- J. Jin, X. Y. Yang, T. Liu, H. Xiao, G. Y. Wang, M. J. Zhou, F. W. Liu, Y. T. Zhang, D. Liu, M. H. Chen, W. Cheng, D. H. Yang and M. Ma, Fluostatins M–Q featuring a 6-5-6-6 ring skeleton and high oxidized A-rings from Marine Streptomyces sp. PKU-MA00045, Mar. Drugs, 2018, 16, E87 CrossRef PubMed.
- T. Liu, J. Jin, X. Y. Yang, J. Song, J. H. Yu, T. T. Geng, Z. Y. Zhang, X. Y. Ma, G. Y. Wang, H. xiao, Y. J. Ge, X. X. Sun, B. Y. Xing, X. J. Ma, C. B. Chi, Y. Kuang, M. Ye, H. L. Wang, Y. M. Zhang, D. H. Yang and M. Ma, Discovery of a phenylamine-incorporated angucyclinone from marine Streptomyces sp. PKU-MA00218 and generation of derivatives with phenylamine analogues, Org. Lett., 2019, 21, 2813–2817 CrossRef CAS PubMed.
- Y. J. Ge, G. Y. Wang, J. Jin, T. Liu, X. Y. Ma, Z. Y. Zhang, T. T. Geng, J. Song, X. J. Ma, Y. T. Zhang, D. H. Yang and M. Ma, Discovery and biosynthesis of pepticinnamins G−M featuring three enzymes-catalyzed nonproteinogenic amino acid formation, J. Org. Chem., 2020, 85, 8673–8682 CrossRef CAS PubMed.
- H. L. Wang, Z. Li, R. N. Jia, Y. Hou, J. Yin, X. Y. Bian, A. Y. Li, R. Müller, A. F. Stewart, J. Fu and Y. M. Zhang, RecET direct cloning and Redαβ recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression, Nat. Protoc., 2016, 11, 1175–1190 CrossRef CAS.
- J. Fu, X. Y. Bian, S. B. Hu, H. L. Wang, F. Huang, P. M. Seibert, A. Plaza, L. Q. Xia, R. Müller, A. F. Stewart and Y. M. Zhang, Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting, Nat. Biotechnol., 2012, 30, 440–446 CrossRef CAS PubMed.
- W. S. Wang, X. Li, J. Wang, S. H. Xiang, X. Z. Feng and K. Q. Yang, An engineered strong promoter for Streptomycetes, Appl. Environ. Microbiol., 2013, 79, 4484–4492 CrossRef CAS PubMed.
- G. Y. Tan, K. H. Deng, X. H. Liu, H. Tao, Y. Y. Chang, J. Chen, K. Chen, Z. Sheng, Z. X. Deng and T. G. Liu, Heterologous biosynthesis of spinosad: an omics-guided large polyketide synthase gene cluster reconstitution in Streptomyces, ACS Synth. Biol., 2017, 6, 995–1005 CrossRef CAS PubMed.
- M. M. Zhang, F. T. Wong, Y. J. Wang, S. W. Luo, Y. H. Lim, E. Heng, W. L. Yeo, R. E. Cobb, B. Enghiad, E. L. Ang and H. M. Zhao, Crispr-Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters, Nat. Chem. Biol., 2017, 13, 607–609 CrossRef CAS PubMed.
- S. Kodani and K. Unno, How to harness biosynthetic gene clusters of lasso peptides, J. Ind. Microbiol. Biotechnol., 2020, 47, 703–714 CrossRef CAS PubMed.
- S. Um, Y. J. Kim, H. Kwon, H. Wen, S. H. Kim, H. C. Kwon, S. Park, J. Shin and D. C. Oh, Sungsanpin, a lasso peptide from a deep-Sea Streptomycete, J. Nat. Prod., 2013, 76, 873–879 CrossRef CAS PubMed.
- I. Kaweewan, H. Hemmi, H. Komaki, S. Harada and S. Kodani, Isolation and structure determination of a new lasso peptide specialicin based on genome mining, Bioorg. Med. Chem., 2018, 26, 6050–6055 CrossRef CAS PubMed.
- S. Son, M. Jang, B. Lee, Y. S. Hong, S. K. Ko, J. H. Jang and J. S. Ahn, Ulleungdin, a lasso peptide with cancer cell migration inhibitory activity discovered by the genome mining approach, J. Nat. Prod., 2018, 81, 2205–2211 CrossRef CAS PubMed.
- J. P. Gomez-Escribano, J. F. Castro, V. Razmilic, S. A. Jarmusch, G. Saalbach, R. Ebel, M. Jaspars, B. Andrews, J. A. Asenjo and M. J. Bibb, Heterologous expression of a cryptic gene cluster from Streptomyces leeuwenhoekii C34T Yields a Novel Lasso Peptide, Leepeptin, Appl. Environ. Microbiol., 2019, 85, e01752-19 CrossRef PubMed.
- C. Cortés-Albayay, S. A. Jarmusch, F. Trusch, R. Ebel, B. A. Andrews, M. Jaspars and J. A. Asenjo, Downsizing class II lasso peptides: genome mining-guided isolation of huascopeptin containing the first Gly1-Asp7 macrocycle, J. Org. Chem., 2020, 85, 1661–1667 CrossRef PubMed.
- Y. Y. Li, R. Ducasse, S. Zirah, A. Blond, C. Goulard, E. Lescop, C. Giraud, A. Hartke, E. Guittet, J. L. Pernodet and S. Rebuffat, Characterization of sviceucin from Streptomyces provides insight into enzyme exchangeability and disulfide bond formation in lasso peptides, ACS Chem. Biol., 2015, 10, 2641–2649 CrossRef CAS.
- J. D. Hegemann, M. Zimmermann, X. L. Xie and M. A. Marahiel, Caulosegnins I-III: a highly diverse group of lasso peptides derived from a single biosynthetic gene cluster, J. Am. Chem. Soc., 2013, 135, 210–222 CrossRef CAS.
- C. H. Zong, M. J. Wu, J. Z. Qin and A. J. Link, lasso peptide benenodin-1 is a thermally actuated [1]rotaxane switch, J. Am. Chem. Soc., 2017, 139, 10403–10409 CrossRef CAS PubMed.
- W. L. Cheung-Lee, M. E. Parry, A. J. Cartagena, S. A. Darst and A. J. Link, Discovery and structure of the antimicrobial lasso peptide citrocin, J. Biol. Chem., 2019, 294, 6822–6830 CrossRef CAS PubMed.
- J. D. Hegemann, C. D. Fage, S. Z. Zhu, K. Harms, F. S. Di Leva, E. Novellino, L. Marinelli and M. A. Marahiel, The ring residue proline 8 is crucial for the thermal stability of the lasso peptide caulosegnin II, Mol. BioSyst., 2016, 12, 1106–1109 RSC.
- W. L. Cheung-Lee, L. Cao and A. J. Link, Pandonodin: a proteobacterial lasso peptide with an exceptionally long C-terminal tail, ACS Chem. Biol., 2019, 14, 2783–2792 CrossRef CAS PubMed.
- K. Jeanne Dit Fouque, V. Bisram, J. D. Hegemann, S. Zirah, S. Rebuffat and F. Fernandez-Lima, Structural signatures of the class III lasso peptide BI-32169 and the branched-cyclic topoisomers using trapped ion mobility spectrometry-mass spectrometry and tandem mass spectrometry, Anal. Bioanal. Chem., 2019, 411, 6287–6296 CrossRef CAS PubMed.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H. G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J. K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, New developments in RiPP discovery, enzymology and engineering, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Methods for converting cysteine to dehydroalanine on peptides and proteins, Chem. Sci., 2011, 2, 1666–1676 RSC.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Conversion of cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications, Angew. Chem., Int. Ed., 2012, 51, 1835–1839 CrossRef CAS.
- U. Ohto, K. Fukase, K. Miyake and T. Shimizu, Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2, Proc. Natl. Acad. Sci., 2012, 109, 7421–7426 CrossRef CAS.
- T. Kawai and S. Akira, The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors, Nat. Immunol., 2010, 11, 373–384 CrossRef CAS PubMed.
- L. H. Guo and H. J. Schluesener, The innate immunity of the central nervous system in chronic pain: the role of Toll-like receptors, Cell. Mol. Life Sci., 2007, 64, 1128–1136 CrossRef CAS PubMed.
- J. E. Koo, Z. Y. Park, N. D. Kim and J. Y. Lee, Sulforaphane inhibits the engagement of LPS with TLR4/MD2 complex by preferential binding to Cys133 in MD2, Biochem. Biophys. Res. Commun., 2013, 434, 600–605 CrossRef CAS PubMed.
- M. Zimmermann, J. D. Hegemann, X. L. Xie and M. A. Marahiel, Characterization of caulonodin lasso peptides revealed unprecedented N-terminal residues and a precursor motif essential for peptide maturation, Chem. Sci., 2014, 5, 4032–4043 RSC.
- J. D. Hegemann, M. Zimmermann, S. Z. Zhu, H. Steuber, K. Harms, X. L. Xie and M. A. Marahiel, Xanthomonins I–III: a new class of lasso peptides with a seven residue macrolactam ring, Angew. Chem., Int. Ed., 2014, 53, 2230–2234 CrossRef CAS PubMed.
- M. Zimmermann, J. D. Hegemann, X. L. Xie and M. A. Marahiel, The astexin-1 lasso peptides: biosynthesis, stability, and structural studies, Chem. Biol., 2013, 20, 558–569 CrossRef CAS PubMed.
- T. A. Knappe, U. Linne, L. Robbel and M. A. Marahiel, Insights into the biosynthesis and stability of the lasso peptide capistruin, Chem. Biol., 2009, 16, 1290–1298 CrossRef CAS PubMed.
- H. Martin-Gomez, U. Linne, F. Albericio, J. Tulla-Puche and J. D. Hegemann, Investigation of the biosynthesis of the lasso peptide chaxapeptin using an E. coli-based production system, J. Nat. Prod., 2018, 81, 2050–2056 CrossRef CAS PubMed.
- J. Inokoshi, N. Koyama, M. Miyake, Y. Shimizu and H. Tomoda, Structure-activity analysis of gram-positive bacterium-producing lasso peptides with antimycobacterial activity, Sci. Rep., 2018, 6, 30375 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental and spectroscopic details. See DOI: 10.1039/d1sc02695j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |