
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/Cu-Catalyzed amide-enabled selectivity-reversed borocarbonylation of unactivated alkenes†
Fu-Peng
Wu
a and
Xiao-Feng
Wu
 *ab
*ab
aLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 30th June 2021
Abstract
The addition reaction between CuBpin and alkenes to give a terminal boron substituted intermediate is usually fast and facile. In this communication, a selectivity-reversed procedure has been designed and established. This selectivity-reversed borocarbonylation reaction is enabled by a cooperative action between palladium and copper catalysts and proceeds with complete regioselectivity. The key to the success of this transformation is the coordination of the amide group and slower CuBpin formation by using KHCO3 as the base. A wide range of β-boryl ketones were produced from terminal unactivated aliphatic alkenes and aryl iodides. Further synthetic transformations of the obtained β-boryl ketones have been developed as well.
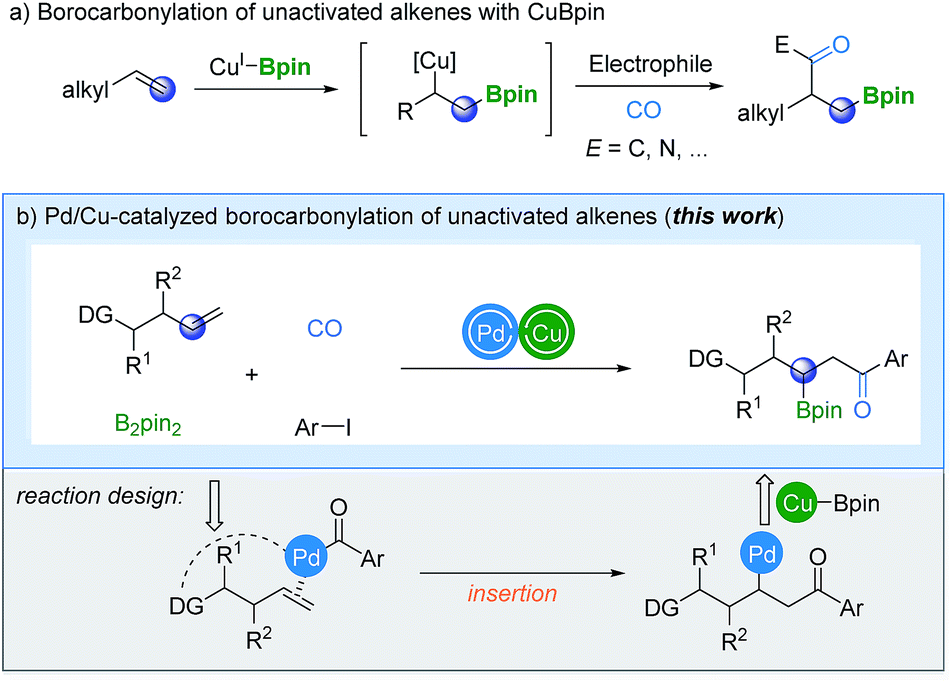
The catalytic borocarbonylation of alkenes represents a novel synthetic tool for the simultaneous installation of boron and carbonyl groups across alkenes, enabling rapid construction of molecules with high complexity from abundant alkenes. In particular, the obtained organoboron compounds are versatile synthetic intermediates that can be readily converted into a wide range of functional groups with complete stereospecificity.1 Consequently, several catalytic systems have been developed to diversify the molecular frameworks through carbonylative borofunctionalization.2 In general, carbonylative borofunctionalization of alkenes proceeds via an alkyl-copper intermediate, which was produced by the addition of CuBpin to the terminal position of the alkene starting material,3 followed by CO insertion and other related steps. A new C–B bond is formed at the terminal position of the alkene and a carbonyl group has been installed at the β-position simultaneously (Scheme 1a). However, in contrast to the progress in the borocarbonylation, a selectivity-reversed procedure (the boryl group is installed at the internal position) to give β-boryl ketone products is still unprecedented.
 | ||
| Scheme 1 Strategies for borocarbonylation of activated alkenes. | ||
Recently, several attractive strategies have emerged for the borofunctionalization of unactivated alkenes to give β-boryl products.4–7 In 2015, Fu, Xiao and their co-workers established a copper-catalyzed regiodivergent alkylboration of alkenes.4a In the same year, Miura and Hirano's group reported a copper-catalyzed aminoboration of terminal alkenes.4b In these two attractive procedures, the regioselectivity was controlled by the ligand applied. More recently, an intermolecular 1,2-alkylborylation of alkenes was described by Ito's research group.5 A radical-relay strategy was used to achieve the targeted regioselective addition. Furthermore, Engle and co-workers explored a palladium-catalyzed 1,2-carboboration and -silylation reaction of alkenes.6 Stereocontrol can be achieved in this new procedure with the assistance of a chiral auxiliary which is a coordinating group in this case.
Inspired by these pioneering studies, we assumed that if the reaction could be initiated by the insertion of an acylpalladium complex into alkenes, followed by transmetalation with CuBpin before reductive elimination, β-boryl ketones can finally be produced (Scheme 1b). However, due to the inherent reactivity of the palladium species toward alkenes, olefin substrates were usually restricted to styrenes and a large excess of them is typically required (>6 equivalents).8,9 Therefore, the critical part of the reaction design is to promote the reaction of the acylpalladium intermediate with alkenes faster than the insertion of CuBpin into olefins. One of the ideas is taking advantage of the coordinating group to transform the reaction from intermolecular to intramolecular. Among the developed directing groups,10 8-aminoquinoline (AQ) is interesting and has been relatively well studied by various groups in a number of novel transformations.11–13 Although the AQ directing group contains a NH group which can participate in intramolecular C–N bond formation,14 we believe that the selectivity-reversed borocarbonylation of alkenes can potentially be achieved through cooperative Pd/Cu catalysis. Then, valuable β-boryl ketones can be produced from readily available substrates directly and effectively.
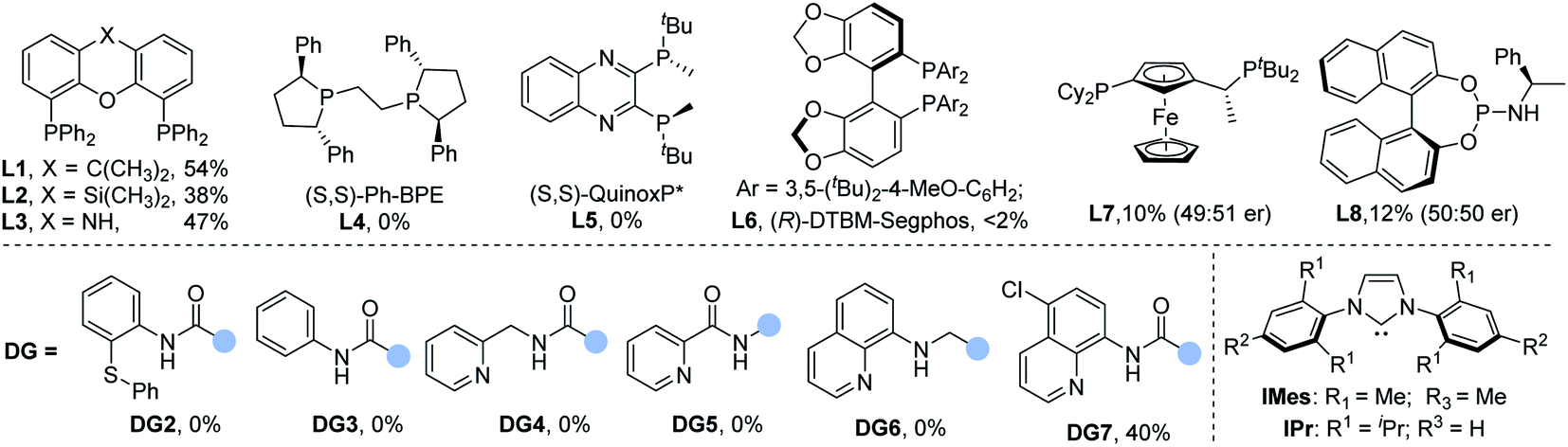
To test the viability of our design on selectivity-reversed borocarbonylation of alkenes, N-(quinolin-8-yl)pent-4-enamide (1a), iodobenzene (2a), and bis(pinacolato)diboron (B2pin2) were chosen as model substrates for systematic studies. As shown in Table 1, by using IMesCuCl and Pd(TFA)2 as the co-catalyst system, xantphos L1 as the ligand and K2CO3 as the base at 70 °C under 10 bar of CO for 18 h, the desired product 3a can be successfully obtained in 29% yield (Table 1, entry 1). In the reaction mixture, a small amount of N-phenyl-N-(quinolin-8-yl)pent-4-enamide can be detected due to copper promoted C–N bond coupling.15 In the testing of palladium precursors, allylpalladium chloride dimer proved to be the best palladium catalyst for this reaction, affording 3a in 41% yield (Table 1, entries 1–4). Subsequently, we evaluated the influence of copper precursors. The NHC ligand is proved to be unnecessary as copper iodide can give the target product in 50% yield (Table 1, entries 5–8). Various phosphine ligands were studied to examine ligand effects. Monodentate phosphine ligands failed to deliver the desired product 3a (for details, see the ESI†) and tend to generate the by-product β-aminoketone. Xantphos was found to be superior to the other tested bidentate ligands (Table 1, entries 9 and 10). Commonly used chiral ligands failed to exert any chiral induction (Table 1, entries 11–15). One possibility is that the AQ group was coordinated to palladium at the beginning of the reaction. In the testing of bases, to our delight, weaker base KHCO3 can provide 58% yield of the final product (Table 1, entries 16–19). Increasing the loading of xantphos to 10 mol% inhibited the reaction, indicating that xantphos only coordinates with palladium (Table 1, entry 20). Notably, other directing groups DG2–DG7 had no positive effect on the reaction outcome. Using N-phenyl-N-(quinolin-8-yl)pent-4-enamide as the substrate could not give any carbonylation product.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Cu | Base | Yield of 3a (%) |
| a All reactions were carried out on a 0.1 mmol scale with alkene (0.1 mmol) and aryl iodide (2.0 equiv.). Yields were determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard. b Isolated yield. c Xantphos (10 mol%). | |||||
| 1 | Pd(TFA)2 | L1 | IMesCuCl | K2CO3 | 29 |
| 2 | Pd(OAc)2 | L1 | IMesCuCl | K2CO3 | 34 |
| 3 | [Pd(η3-C3H5)Cl]2 | L1 | IMesCuCl | K2CO3 | 41 |
| 4 | [Pd(cinnamyl)Cl]2 | L1 | IMesCuCl | K2CO3 | 36 |
| 5 | [Pd(η3-C3H5)Cl]2 | L1 | IPrCuCl | K2CO3 | 0 |
| 6 | [Pd(η3-C3H5)Cl]2 | L1 | CuCl | K2CO3 | 33 |
| 7 | [Pd(η3-C3H5)Cl]2 | L1 | CuBr | K2CO3 | 41 |
| 8 | [Pd(η3-C3H5)Cl]2 | L1 | CuI | K2CO3 | 50 |
| 9 | [Pd(η3-C3H5)Cl]2 | L2 | CuI | K2CO3 | 38 |
| 10 | [Pd(η3-C3H5)Cl]2 | L3 | CuI | K2CO3 | 47 |
| 11 | [Pd(η3-C3H5)Cl]2 | L4 | CuI | K2CO3 | 0 |
| 12 | [Pd(η3-C3H5)Cl]2 | L5 | CuI | K2CO3 | 0 |
| 13 | [Pd(η3-C3H5)Cl]2 | L6 | CuI | K2CO3 | <2 |
| 14 | [Pd(η3-C3H5)Cl]2 | L7 | CuI | K2CO3 | 10 |
| 15 | [Pd(η3-C3H5)Cl]2 | L8 | CuI | K2CO3 | 12 |
| 16 | [Pd(η3-C3H5)Cl]2 | L1 | CuI | KHCO3 | 58 (51)b |
| 17 | [Pd(η3-C3H5)Cl]2 | L1 | CuI | K2HPO4 | 26 |
| 18 | [Pd(η3-C3H5)Cl]2 | L1 | CuI | NaHCO3 | 0 |
| 19 | [Pd(η3-C3H5)Cl]2 | L1 | CuI | NaOtBu | 11 |
| 20c | [Pd(η3-C3H5)Cl]2 | L1 | CuI | KHCO3 | <5 |
| 21 | [Pd(η3-C3H5)Cl]2 | L7 | CuI | KHCO3 | 40 |
|
|||||

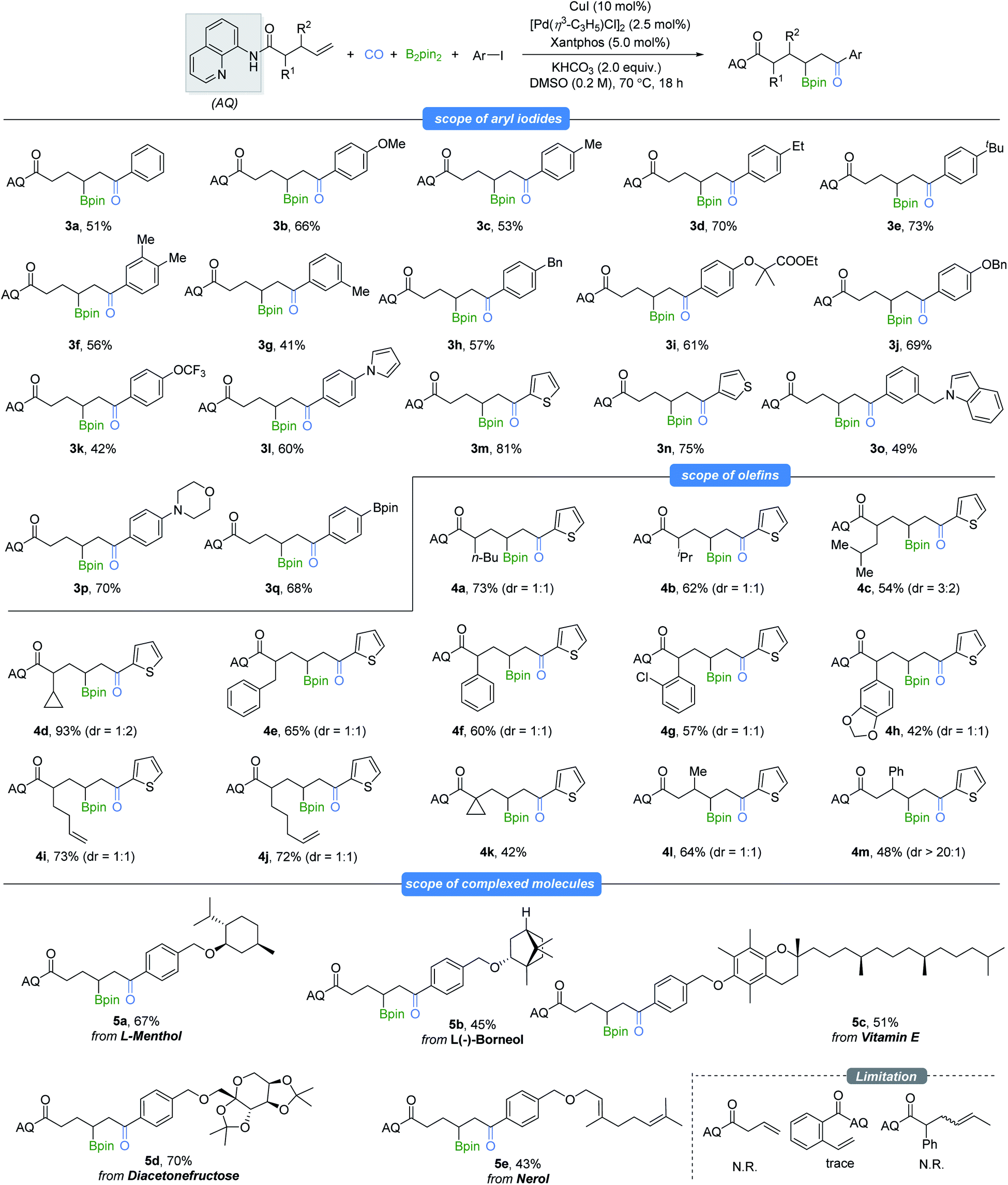
With the optimized reaction conditions in hand, we examined the scope of this selectivity-reversed borocarbonylation with various unactivated alkenes and aryl iodides toward the synthesis of β-boryl ketones (Table 2). Aryl iodides spanning a range of electronic properties were tested for this transformation (3a–3h). Functional groups such as ether (3i and 3j), pyrrole (3l), thiophene (3m and 3n), indole (3o), morpholine (3p), Bpin (3q) and highly lipophilic group OCF3 (3k) were all compatible with the reaction conditions to produce the target products in moderate to excellent yields. Bromobenzene and 4-bromobenzotrifluoride were also tested in place of iodobenzene, and no desired product could be detected even at increased reaction temperature. Phenyl triflate and alkenyl halides and triflate were tested with the model alkenes as well, and no reaction occurred. However, 87% yield of cinnamic acid was obtained when (E)-(2-bromovinyl)benzene was tested under our standard conditions. Subsequently, we evaluated a series of aliphatic alkenes in the reaction with 2-iodothiophene as a representative electrophile. When α-monosubstituted 4-enenoic amides with alkyl, cyclopropyl, benzyl or phenyl substitution were tested, the borocarbonylation reaction occurred smoothly and gave the desired product (4a–4h) in moderate to excellent yields, albeit with low diastereoselectivtiy. Notably, monocarbonylative coupling occurred selectively at the γ,δ-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond even when there are two CC bonds in the amide substrates (4i and 4j). In addition, sterically hindered 4-pentenoic amide was subjected to the optimized reaction conditions, and the corresponding product was formed in 42% yield (4k). Furthermore, mono-substitution at the β-position of 4-pentenoic amides could also be employed, affording the corresponding products in moderate yields (4l and 4m). Iodoarenes containing more complex substrates such as L-menthol, L-borneol, vitamin E, diacetonfructose and nerol were also competent substrates and gave moderate to good yields of the corresponding products. Finally, no desired product could be detected when 3-butenoic amide, 2-vinylbenzamide or internal alkene was tested under our standard conditions.
C bond even when there are two CC bonds in the amide substrates (4i and 4j). In addition, sterically hindered 4-pentenoic amide was subjected to the optimized reaction conditions, and the corresponding product was formed in 42% yield (4k). Furthermore, mono-substitution at the β-position of 4-pentenoic amides could also be employed, affording the corresponding products in moderate yields (4l and 4m). Iodoarenes containing more complex substrates such as L-menthol, L-borneol, vitamin E, diacetonfructose and nerol were also competent substrates and gave moderate to good yields of the corresponding products. Finally, no desired product could be detected when 3-butenoic amide, 2-vinylbenzamide or internal alkene was tested under our standard conditions.
| a All reactions were carried out on a 0.1 mmol scale. Alkenes (0.1 mmol), aryl iodides (2.0 equiv.), B2pin2 (1.5 equiv.), CuI (10 mol%), [Pd(η3-C3H5)Cl]2 (2.5 mol%), xantphos (5 mol%), KHCO3 (2.0 equiv.), CO (10 bar), and DMSO (0.2 M) were stirred at 70 °C for 18 h. The dr value given was determined by 1H NMR. |
|---|
|
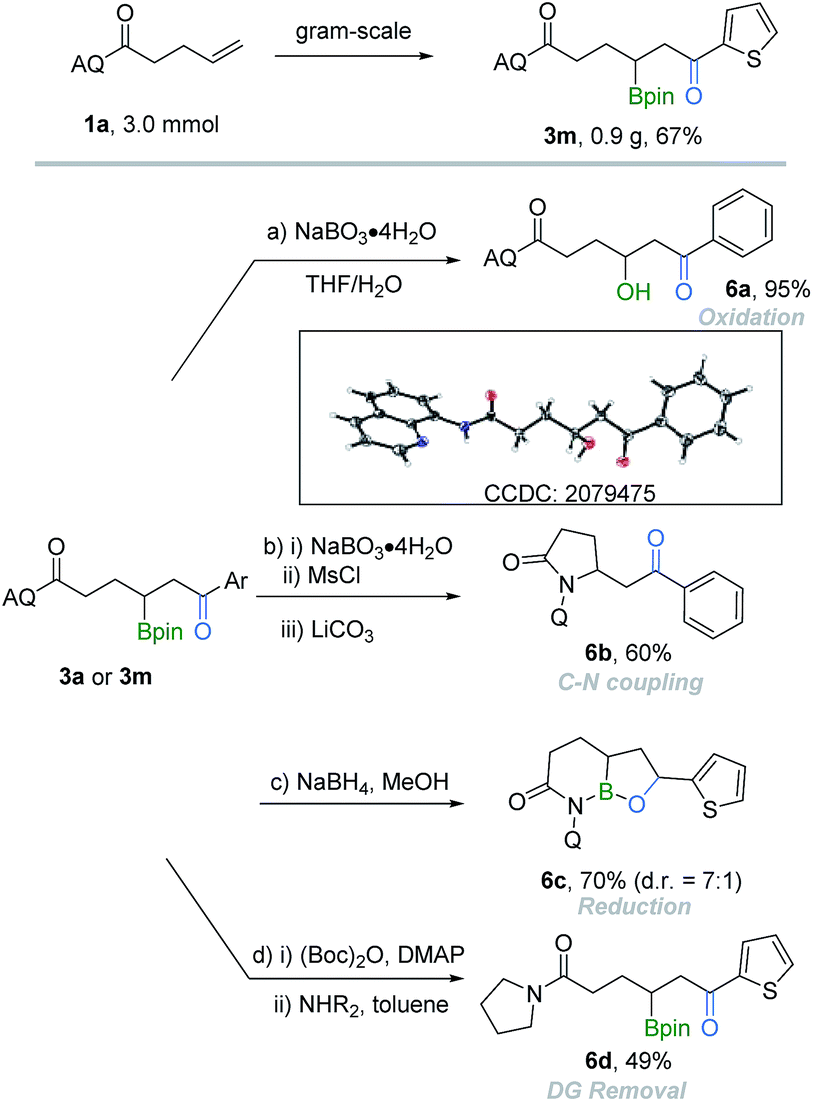
To demonstrate the synthetic utilities of the obtained borocarbonylation products, a series of further synthetic transformations of the β-boryl ketones were performed (Scheme 2). From a practical point of view, the reaction can be easily performed on the gram-scale and gave the target product 3m in 67% yield. β-Hydroxyl ketone 6a (CCDC: 2079475;† determined by X-ray crystallography and the ORTEP drawing with 50% thermal ellipsoids) was produced in 95% yield by oxidation of the parent β-boryl ketone 3a. Furthermore, the C–B bond can be easily converted into a C–N bond, affording β-aminoketone 6b in 60% yield. Upon the reduction reaction of 3m with NaBH4, the corresponding reduced oxaborole amide 6c could be isolated in 70% yield. Finally, a two-step transamination process was performed to remove the AQ group.16
 | ||
| Scheme 2 Diversification of β-boryl ketones. | ||
To gain some insight into the mechanism of this selectivity-reversed borocarbonylation of alkenes, several control experiments were performed. The target product 3a was not formed, instead byproduct 6b was obtained in 40% yield, in the case without xantphos. Possible explanations for this result are: (i) the bidentate directing group AQ increases the stability of Pd(II) species and promotes the carbonylation step; (ii) the role of xantphos is to coordinate to C(sp3)–Pd(II) species after its formation and inhibit the formation of the C–N bond to give byproduct 6b (Scheme 3a). In addition, copper and B2pin2 were proven to be important, and KHCO3 was essential for the carbonylation step (Scheme 3b). Analysis of the copper system in the absence of palladium and iodobenzene revealed that alkenes failed to undergo CuBpin insertion under this condition and no hydroboration products could be detected after work-up (Scheme 3c). Additionally, alkenes without the directing group were also tested under our standard conditions, and no reaction occurred.
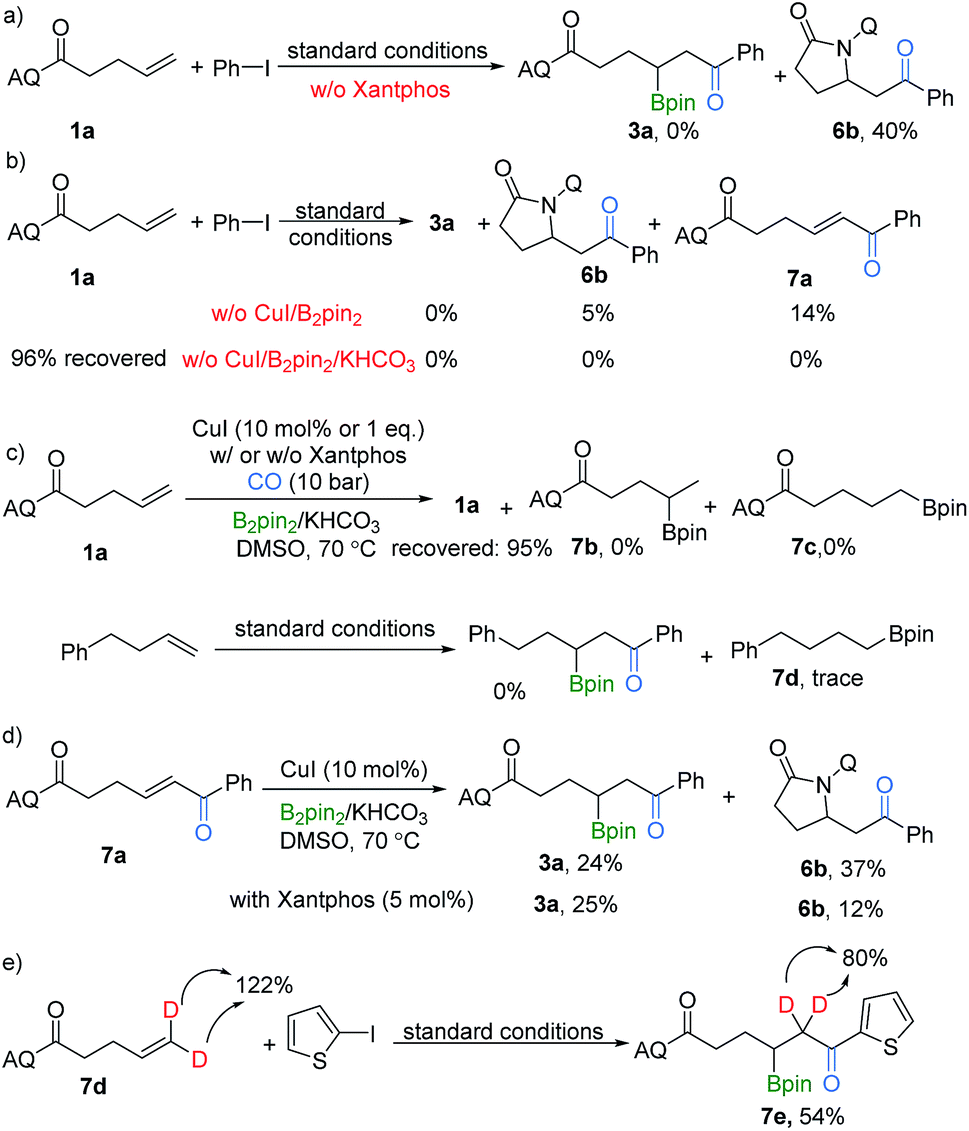 | ||
| Scheme 3 Control experiments. | ||
Although we did not observe compound 7a during our optimization and substrate scope processes, even after stopping the reaction after 8 hours, we tested the possibility that 7a might act as an intermediate. When 7a was subjected to this transformation, the product 3a was delivered in 24% yield and 6b was generated in 37% (Scheme 3d). No significant difference in the yield outcome was observed when xantphos was added. Additionally, in our deuterated substrate testing, the amount of the deuterated product obtained is lower than the theoretical value (Scheme 3e). Thus, a pathway of β-H elimination followed by hydroboration could be involved as well. However, we believe the direct reaction between palladium and copper intermediates is the main one for this procedure due to the proven importance of the AQ group and the known achievements of copper-catalyzed hydroboration of enones, even with enantioselective versions.17
On the basis of the above results and related literature studies,7,11–14 a possible reaction pathway is proposed (Scheme 4). Initially, the AQ directing group coordinates with Pd0, which produces the active AQ-Pd0 catalyst I. This is followed by oxidative addition to aryl iodides to generate PdII species II, and then by base promoted iodine dissociation to form complex III. After the CO insertion step, the acyl-PdII species IV coordinates with the alkene and undergoes migratory insertion to generate C(sp3)–PdII intermediate V, which is stabilized by the xantphos ligand and AQ directing group. Subsequently, C(sp3)–PdII complex V reacts especially with CuBpin to give the desired product β-boryl ketone and regenerate the Pd(0) complex. Finally, ligand exchange of Pd0Ln regenerates AQ-Pd0I for the next catalytic cycle. Additionally, another minor pathway that involves the carbonylative Heck reaction to give an enone derivative, followed by its hydroboration to give the final product could be included as well.
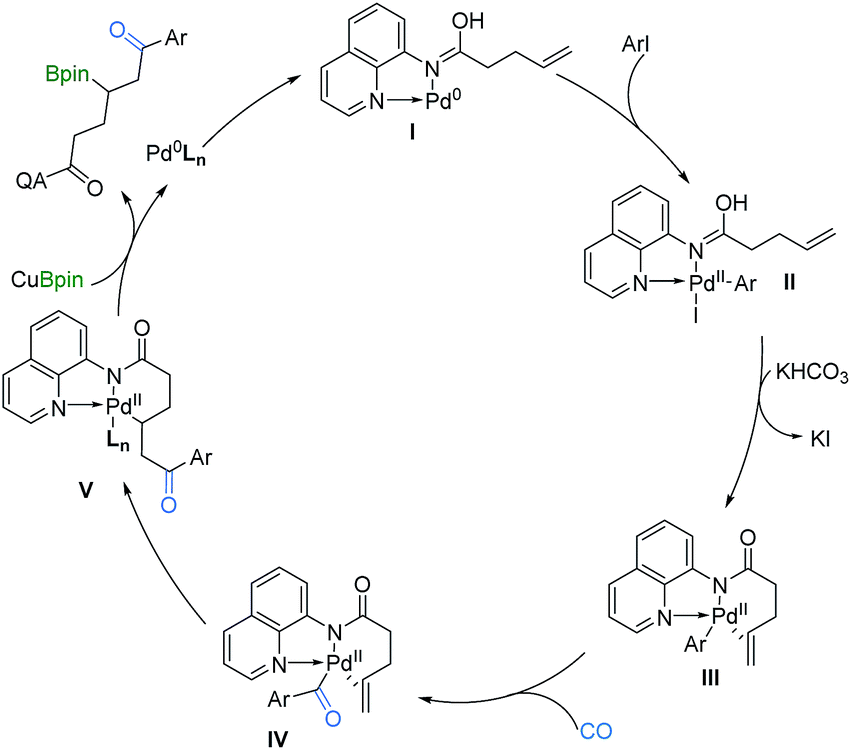 | ||
| Scheme 4 Proposed catalytic cycle. | ||
In summary, a novel Pd/Cu catalyzed amide-directed selectivity-reversed borocarbonylation for the selective synthesis of β-boryl ketones from terminal alkenes has been developed. Various aryl iodides and aliphatic alkenes were transformed into the desired β-boryl ketones in moderate to excellent yields. In this catalyst system, the assistance from the AQ directing group is essential for successful reaction design.
Author contributions
XFW supervised this project and revised the manuscirpt. FPW performed all the experiments and prepared this draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Chinese Scholarship Council for financial support. We also thank the analytical department of Leibniz Institute for Catalysis at the University of Rostock for their excellent analytical service. We also thank Dr Anke Spannenberg for the X-ray crystal structure analysis of compound 6a.Notes and references
- (a) D. G. Hall, Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications, in Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2006, pp. 1–99 Search PubMed; (b) E. C. Neeve, S. J. Geier, I. A. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS; (c) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- (a) A. Sawada, T. Fujihara and Y. Tsujia, Adv. Synth. Catal., 2018, 360, 2621–2625 CrossRef CAS; (b) L. J. Cheng and N. P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 10328–10332 CrossRef CAS; (c) T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS; (d) F.-P. Wu, Y. Yuan, C. Schunemann, P. C. J. Kamer and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 10451–10455 CrossRef CAS; (e) F.-P. Wu, J. Holz, Y. Yuan and X.-F. Wu, CCS Chem., 2020, 2, 2643–2654 Search PubMed; (f) F.-P. Wu, X. Luo, U. Radius, T. B. Marder and X.-F. Wu, J. Am. Chem. Soc., 2020, 142, 14074–14079 CrossRef CAS; (g) F.-Y. Yang, M. Shanmugasundaram, S.-Y. Chuang, P.-J. Ku, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2003, 125, 12576–12583 CrossRef CAS PubMed.
- (a) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS; (b) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, ACS Catal., 2020, 11, 1–18 CAS.
- (a) W. Su, T. J. Gong, X. Lu, M. Y. Xu, C. G. Yu, Z. Y. Xu, H. Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957–12961 CrossRef CAS; (b) R. Sakae, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 6460–6463 CrossRef CAS PubMed.
- S. Akiyama, N. Oyama, T. Endo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 5260–5268 CrossRef CAS PubMed.
- Z. Liu, J. Chen, H. X. Lu, X. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068–17073 CrossRef CAS PubMed.
- (a) Z. Liu, H.-Q. Ni, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 3223–3227 CrossRef CAS PubMed; (b) Z. Liu, X. Li, T. Zeng and K. M. Engle, ACS Catal., 2019, 9, 3260–3265 CrossRef CAS PubMed; (c) Z. Bai, S. Zheng, Z. Bai, F. Song, H. Wang, Q. Peng, G. Chen and G. He, ACS Catal., 2019, 9, 6502–6509 CrossRef CAS.
- Y. Yuan, F.-P. Wu, J.-X. Xu and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed.
- (a) X.-F. Wu, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 5284–5288 CrossRef CAS PubMed; (b) X.-F. Wu, H. Neumann, A. Spannenberg, T. Schulz, H. Jiao and M. Beller, J. Am. Chem. Soc., 2010, 132, 14596–14602 CrossRef CAS PubMed; (c) J. Schranck, X. F. Wu, H. Neumann and M. Beller, Chem.–Eur. J., 2012, 18, 4827–4831 CrossRef CAS PubMed; (d) X.-F. Wu, H. Jiao, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 726–733 CrossRef CAS; (e) H. Yin, D. U. Nielsen, M. K. Johansen, A. T. Lindhardt and T. Skrydstrup, ACS Catal., 2016, 6, 2982–2987 CrossRef CAS.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed; (b) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS PubMed; (c) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043–6046 CrossRef CAS PubMed; (d) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342–9345 CrossRef CAS PubMed.
- (a) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984–12986 CrossRef CAS; (b) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354–357 CrossRef CAS PubMed; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS PubMed; (d) Y. Aihara and N. Chatani, Chem. Sci., 2013, 4, 664–670 RSC; (e) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201–2208 RSC.
- For selected examples on using 8-aminoquinoline (AQ) as the directing group in organic transformations, see: (a) Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261–11270 CrossRef CAS; (b) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC; (c) M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805–5813 CrossRef CAS; (d) V. T. Tran, J. A. Gurak Jr, K. S. Yang and K. M. Engle, Nat. Chem., 2018, 10, 1126–1133 CrossRef CAS; (e) Z. Bai, Z. Bai, F. Song, H. Wang, G. Chen and G. He, ACS Catal., 2019, 10, 933–940 CrossRef; (f) J. Jeon, H. Ryu, C. Lee, D. Cho, M. H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 10048–10059 CrossRef CAS; (g) J. Jeon, C. Lee, H. Seo and S. Hong, J. Am. Chem. Soc., 2020, 142, 20470–20480 CrossRef CAS PubMed; (h) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS PubMed; (i) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457–5191 CrossRef CAS PubMed; (j) C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T.-P. Loh, J. Am. Chem. Soc., 2018, 140, 9332–9336 CrossRef CAS PubMed.
- (a) Z. Bai, H. Zhang, H. Wang, H. Yu, G. Chen and G. He, J. Am. Chem. Soc., 2021, 143, 1195–1202 CrossRef CAS PubMed; (b) J.-B. Peng, F.-P. Wu, D. Li, H.-Q. Geng, X. Qi, J. Ying and X.-F. Wu, ACS Catal., 2019, 9, 2977–2983 CrossRef CAS; (c) P. Shi, J. Wang, Z. Gan, J. Zhang, R. Zeng and Y. Zhao, Chem. Commun., 2019, 55, 10523–10526 RSC.
- G.-W. Zhang, A.-X. Zhou, W. He and X.-F. Xia, Synlett, 2018, 29, 2269–2274 CrossRef.
- (a) L. S. Fitzgerald and M. L. O'Duill, Chem.–Eur. J., 2021, 27, 8411–8436 CrossRef CAS PubMed; (b) O. Verho, M. P. Lati and M. Oschmann, J. Org. Chem., 2018, 8, 4464–4476 CrossRef PubMed.
- (a) J. A. Schiffner, K. Müther and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 1194–1196 CrossRef CAS PubMed; (b) E. Hartmann, D. J. Vyas and M. Oestreich, Chem. Commun., 2011, 47, 7917–7932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedures, analytical data, and NMR spectra. CCDC 2079475. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02785a |
| This journal is © The Royal Society of Chemistry 2021 |