
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (+)-spiroindimicin A and congeners unveils their antiparasitic activity†
Zhen
Zhang
 a,
Sneha
Ray
a,
Leah
Imlay
a,
Lauren T.
Callaghan
ab,
Hanspeter
Niederstrasser
a,
Prema Latha
Mallipeddi
a,
Bruce A.
Posner
a,
Dawn M.
Wetzel
ab,
Margaret A.
Phillips
a and
Myles W.
Smith
*a
a,
Sneha
Ray
a,
Leah
Imlay
a,
Lauren T.
Callaghan
ab,
Hanspeter
Niederstrasser
a,
Prema Latha
Mallipeddi
a,
Bruce A.
Posner
a,
Dawn M.
Wetzel
ab,
Margaret A.
Phillips
a and
Myles W.
Smith
*a
aDepartment of Biochemistry, UT Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390, USA. E-mail: myles.smith@utsouthwestern.edu
bDepartment of Pediatrics, UT Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390, USA
First published on 30th June 2021
Abstract
The spiroindimicins are a unique class of chlorinated indole alkaloids characterized by three heteroaromatic rings structured around a congested spirocyclic stereocenter. Here, we report the first total synthesis of (+)-spiroindimicin A, which bears a challenging C-3′/C-5′′-linked spiroindolenine. We detail our initial efforts to effect a biomimetic oxidative spirocyclization from its proposed natural precursor, lynamicin D, and describe how these studies shaped our final abiotic 9-step solution to this complex alkaloid built around a key Pd-catalyzed asymmetric spirocyclization. Scalable access to spiroindimicins A, H, and their congeners has enabled discovery of their activity against several parasites relevant to human health, providing potential starting points for new therapeutics for the neglected tropical diseases leishmaniasis and African sleeping sickness.
Introduction
Dimeric tryptophan natural products represent an important class of compounds that has grown significantly in recent decades and contains several medicinally important members like rebeccamycin (1) and staurosporine (2) (Fig. 1).1 Among this broad class, the spiroindimicins constitute a unique subset of non-planar molecules isolated from marine Streptomycetes. The inaugural members of this family, spiroindimicins A–D (3–6), were reported by Zhang and coworkers in 2012, followed by two monochlorinated members, spiroindimicins E and F (7, 8), described by Luzhetskyy et al. in 2017.2a,b Two deschloro congeners, spiroindimicins G and H (9, 10), were also isolated by the Zhang group from a bacterial mutant with an inactivated halogenase gene.2c In the limited biological assays conducted thus far, the spiroindimicins displayed moderate cytotoxicity against several cancer cell lines (IC50 = 9–44 μM).2a,c | ||
| Fig. 1 Bioactive tryptophan dimers and the spiroindimicin family of alkaloids. | ||
Biosynthetically, the spiroindimicins are proposed to derive from the lynamicins, a previously isolated family of antibacterial alkaloids,3via a spirocyclization of C-3′ of one indole unit onto either C-5′′ or C-2′′ of the neighboring indole fragment (Fig. 2, top; spiroindimicin numbering, used throughout). This process transforms one indole into a spiroindolenine or -indoline and creates the congested C-3′ quaternary spirocenter. In line with this hypothesis, lynamicins A (13) and D (12) were co-isolated with 3–6, and further biosynthetic investigations by the Zhang group have shed light on their biogenesis as halogenated dimers of tryptophan and their viability as precursors to 3–6.4,2c At present, however, the enzyme(s) responsible for their oxidative spirocyclization remain unelucidated.
 | ||
| Fig. 2 Spiroindimicin biosynthesis from L-tryptophan and our synthetic approach to spiroindimicin A (3). | ||
In light of their appealing structures and preliminary bioactivities, it is unsurprising that the spiroindimicins have attracted interest from the synthetic community.5 One prior racemic synthesis of spiroindimicins B (4) and C (5) has been reported by Sperry and Blair (15–16 steps), centering upon early-stage construction of the spirocenter via an intramolecular Heck reaction, followed by stepwise introduction of the remaining heterocycles.6 To the best of our knowledge, no synthetic studies toward either of the more challenging C-3′/C-5′′-linked members, spiroindimicins A (3) and H (10), have been disclosed.
Herein, we describe the first total synthesis of (+)-spiroindimicin A (3) relying upon a short, gram-scale preparation of a triaryl precursor and its Pd-catalyzed asymmetric spirocyclization. We apply the developed strategy to the preparation of spiroindimicin H (10), lynamicins A and D (13, 12), and several structural analogues. Finally, with >100 mg of 3 in hand and a panel of congeners, we disclose their promising activity against the parasites Trypanosoma brucei, Plasmodium falciparum, and Leishmania amazonensis, causative agents of African trypanosomiasis (sleeping sickness), malaria, and leishmaniasis, respectively, diseases which constitute a serious and ongoing problem in the developing world.7
Results and discussion
The main challenge associated with total synthesis of 3–10 arises in constructing their core quaternary spirocenters, especially in an enantiocontrolled fashion.8 This challenge is amplified when targeting spiroindimicin A (3), as this entails linking C-3′ of one indole unit to the less reactive C-5′′ position of the other indole ring (C-4 in indole nomenclature); in the case of the 4–9 the nucleophilic C-2′′ carbon is joined to this position. Our approach to spiroindimicin A (3, SPM A) is outlined in Fig. 2 (bottom) and focused on two possible solutions to the challenging C-3′ spirocenter, namely a biomimetic final C-3′/C-5′′ spirocyclization (shown in blue) of a lynamicin D-type precursor (14), or a non-natural C-3′/C-4 spirocyclization (shown in red) of an ‘iso-lynamicin’-type compound (15). In both cases, the spirocyclization might be effected in either an oxidative sense (14, 15, X = H) or via a functional handle (X = I, Br, etc.). Control of the absolute stereochemistry in this key cyclization event remained a daunting prospect, however, given limited literature precedent. Precursors 14 and 15 should both be readily assembled via cross-coupling of appropriately functionalized heteroaryl fragments 16 and 17, themselves available via C–H functionalization of inexpensive indole and pyrrole starting materials.Our initial efforts focused on the biomimetic approach wherein oxidative spirocyclization of lynamicin D (12) might deliver either SPM A (3) directly, or possibly a spiroindolenine precursor to SPM D (6). For this purpose, we required a short and scalable synthesis of lynamicin D (12). 12 has been prepared once before in 6 steps (longest linear sequence) by Sarli and Nikolakaki utilizing a Suzuki coupling-based assembly of its triaryl moiety.9 Using their approach as inspiration, we were able to develop a shorter route to 12 leveraging the tools of C–H functionalization.10 Thus, we could prepare pyrrole dibromide 18via iron-catalyzed C–H methoxycarbonylation11 of commercial ester 17, followed by dibromination (Scheme 1).12 For the other partner, we could advance 5-chloroindole (19) to C-3 boronic ester 20 through an efficient Ir-catalyzed C–H borylation sequence.13 A high-yielding Suzuki coupling using Buchwald's SPhos ligand14 and removal of the Boc protecting groups delivered lynamicin D (12, 4 steps LLS). Using this scalable route we have been able to prepare over 1.7 g of 12, and additionally have achieved the first synthesis of lynamicin A (13) via a monohydrolysis/decarboxylation sequence.15
 | ||
| Scheme 1 Attempted biomimetic synthesis of spiroindimicin A (3) from lynamicin D (12). | ||
Unfortunately, despite extensive investigation we have been unable to achieve formation of C-5′′ or C-2′′-linked spiroindolenines from 12 under a range of oxidative conditions (reagent-based, electrochemical, photochemical; see ESI† for full details). Not surprisingly, 2′,2′′-linked indolocarbazole products such as 21 and 22 were often isolated.16 Similarly, efforts to utilize electronically differentiated monoprotected variants of 12 (e.g., 14, PG = Ts, Ac, Boc, cf.Fig. 2) or use the pyrrole ester/acid to direct C-5′′ functionalization also proved unsuccessful.
Given the challenge of achieving direct C-3′/C-5′′ oxidative spirocyclization, we planned to prepare an analogue with a suitable functional handle to allow for regioselective spirocyclization. While we initially targeted a C-5′′-functionalized variant of lynamicin D (e.g., 14, X = Br, I, cf.Fig. 2), preliminary efforts toward its assembly proved difficult. Our ultimate solution involved switching the order of bond formations to C-3′, where we first aimed to install the more challenging C-3′/C-5′′ bond in the form of an ‘iso-lynamicin’-type precursor (15, Fig. 2). For this purpose, we prepared 4-iodoindole 24 in 3 steps on multigram scale from 4-nitroindole (23) by improving a known sequence (Scheme 2A).17 The previously elusive C–C bond could then be formed via Suzuki coupling with boronic ester 20 in quantitative yield. Hereafter, indole C-3 iodination set the stage for a Stille coupling with pyrrole stannane 27, which was available from previously prepared 26via stannylation of a known18 monoiodide. The fragment coupling required significant optimization, however, with many common Stille conditions giving only low yields of the desired triaryl compound (not shown). Ultimately, we found that the use of Pd–NHC catalyst 2819 in the presence of Cs2CO3 and 4 Å molecular sieves gave the desired product in a serviceable but scalable 68% yield. A final iodination20 of the pyrrole ring and thermolytic Boc deprotection set the stage for the key spirocyclization, providing triaryl 30 which appears to exist as two separable atropisomers (dr ∼3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) that slowly interconvert at room temperature.
:1) that slowly interconvert at room temperature.
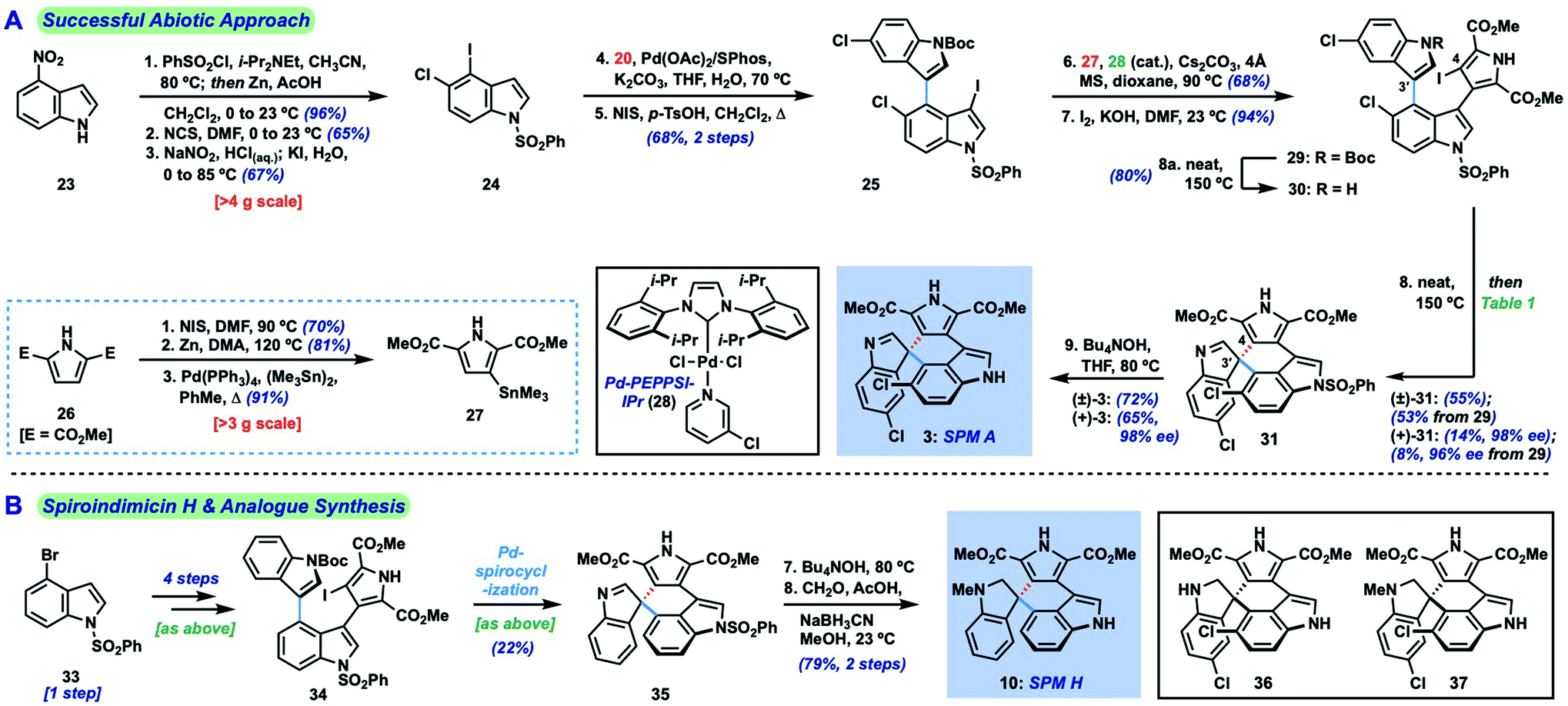 | ||
| Scheme 2 (A) Revised approach to spiroindimicin A (3) via Pd-catalyzed spirocyclization; (B) synthesis of spiroindimicin H (10) and potentially undiscovered spiroindimicins. | ||
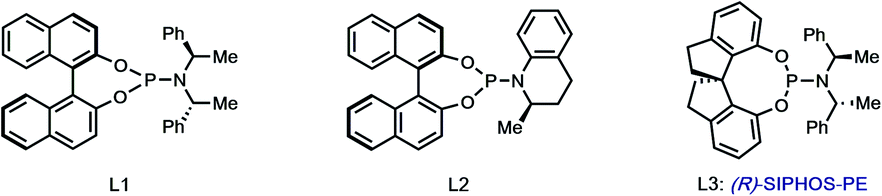
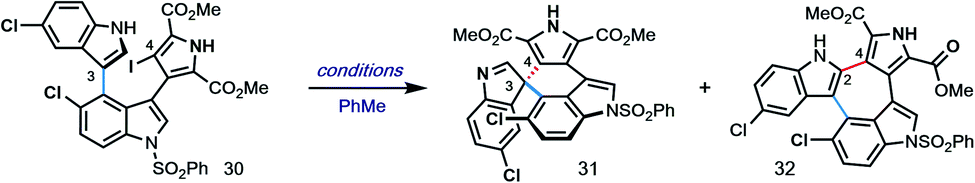
With hundred-milligram quantities of 30 in hand, we explored the racemic spirocyclization to 31 using Pd-catalyzed conditions developed by You as a starting point.21 Although their optimal conditions ([Pd(allyl)Cl]2/PPh3, K2CO3, PhMe, Δ) were unproductive, we did observe formation of C-2′-linked product 32 when employing Cs2CO3 as base in the presence of several phosphine ligands (Table 1; see ESI† for details). This 7-membered product appears to arise through direct C-2′ coupling rather than via C-3′ to C-2′ bond migration in desired spiroindolenine 31 based on control experiments with pure 31.22 Ultimately, we found that the ligand plays a crucial role in providing the desired connectivity, with NHC–Pd systems proving optimal: using Pd-PEPPSI-IPr (28)19 as catalyst under otherwise identical conditions provided protected SPM A (31) in 55% yield (Table 1, entry 1). After screening over 40 chiral ligands (see ESI† for full details), we discovered that the use of chiral phosphoramidites provided the best balance between enantioselectivity and selectivity for 31 over 32 (entries 2–4). With optimal phosphoramidite L3,23 enantioselectivity and especially yield were initially moderate (9%, 75% ee; entry 4) under our prior Cs2CO3 conditions. Ultimately, extensive investigations involving systematic variation of reaction parameters showed that the combination of both Cs2CO3 and Ag2CO3 as base (2.5 equiv. each) and lowering of the temperature to 70 °C could effect spirocyclization in 14% yield and an excellent 98% ee (entry 7; see ESI†). Here, the modest yield of 31 is due to competitive formation of 32, as well as protodeiodination of 30.24 Intriguingly, we also found that work-up conditions had an impact on the enantiopurity of isolated 31 (see ESI† for details). Despite the moderate efficiency, to the best of our knowledge, this is the first report of a highly enantioselective arylative indole to spiroindolenine transformation, and the first use of such a reaction – racemic or asymmetric – in natural product synthesis.21,25,26
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd/ligand | Base | T (°C) |
31:32b |
Yield (%)c | eed |
| a [Pd] = [Pd(allyl)Cl]2; standard conditions: Pd source (10 mol%), ligand (15 mol%), base (1.5 equiv.). b Determined by 1H NMR analysis of the crude reaction mixture. c Yield of isolated 31. d Determined by HPLC analysis. e [Pd]/ligand (30/45 mol%) prestirred in PhMe for 1 h. f 2.5 equiv. each. g [Pd]/ligand (40/60 mol%). | ||||||
| 1 | Pd-PEPPSI-IPr | Cs2CO3 | 115 | 1.8:1 |
55 | n/a |
| 2 | [Pd]/L1 | Cs2CO3 | 90 | 1:1.1 |
11 | −14 |
| 3 | [Pd]/L2 | Cs2CO3 | 90 | 1.7:1 |
14 | 4 |
| 4 | [Pd]/L3 | Cs2CO3 | 90 | 1:1.5 |
9 | 75 |
| 5 | [Pd]/L3e | Cs2CO3/Ag2CO3f | 105 | 1:2 |
8 | 83 |
| 6 | [Pd]/L3e | Cs2CO3/Ag2CO3f | 80 | 1:2.1 |
21 | 86 |
| 7 | [Pd]/L3g | Cs2CO3/Ag2CO3f | 70 | 1:2.2 |
14 | 98 |
| 8 | [Pd]/L1e | Cs2CO3/Ag2CO3f | 70 | 1:1.8 |
6 | −53 |
|
||||||
Additionally, we found that Boc deprotection and spirocyclization could be conducted as a one-pot procedure by simply subjecting the residue remaining after thermolysis to the spirocyclization conditions [(±)-31:53%; (+)-31:8%, 96% ee]. A final removal of the benzenesulfonyl group of 31 with Bu4NOH at 80 °C delivered spiroindimicin A [(±)-3: 72%; (+)-3: 65%], completing the first total synthesis of this target in 9 steps (longest linear sequence from commercial 4-nitroindole). Spectral data of our synthetic material matched those reported by Zhang and co-workers, and its chromatographic behavior was identical to an authentic sample (TLC; HPLC). The optical rotation was of the same sign and similar magnitude {[α]26D = +64.0 (c = 0.05, MeOH) for 98% ee; lit.: [α]20D = +46.49 (c = 0.15, MeOH)} confirming that we had prepared the natural enantiomer of 3. Overall, our synthetic efforts have yielded over 100 mg of 3 to date.
Utilizing our developed strategy, we have also been able to complete the first synthesis of spiroindimicin H [(±)-10, Scheme 2B] from 4-bromoindole (33). This material was similarly advanced to triaryl iodide 34, which could be spirocyclized to 35 under our Pd-PEPPSI-IPr-catalyzed conditions in 22% yield. High-yielding indole deprotection (89%) and reductive methylation (89%) of the indolenine then completed the synthesis of 10 in 8 steps overall. Moreover, the dihydrospiroindimicin A congeners 36 and 37, potentially as yet undiscovered natural products (cf.4–10), were prepared via similar indolenine reductions of spiroindimicin A (3).
Finally, with scalable access to spiroindimicin A (3) and a panel of related compounds, we have begun to explore their biological properties. Given that several tryptophan dimers, including staurosporine (2), have demonstrated antiparasitic activity,27 preliminary testing was conducted against the parasites Trypanosoma brucei, Plasmodium falciparum, and Leishmania amazonensis,7 revealing promising activity (Table 2). Specifically, SPM A (3) inhibits the growth of all three parasites (EC50 = 1.3–11 μM), with the potencies of natural (S)-3, ent-(R)-3, and racemic 3 being similar, suggesting a non-protein-based target. SPM H (10) and SPM A derivatives 36 and 37 are also active, demonstrating similar or slightly improved potencies in some cases. Lynamicin-type compounds showed activity, with 2′,2′′-linked indolocarbazole 21 displaying the highest potency against T. brucei (EC50 = 0.37 μM). Several compounds are also active against both multidrug-resistant (Dd2) and drug-sensitive (3D7) strains of P. falciparum. Importantly, in most cases the compounds did not display significant cytotoxicity against mammalian HepG2 and RAW 264.7 cells (a macrophage cell line) at 10 μM; when toxicity was observed, reasonable selectivity was maintained in several cases (e.g., for 21, HepG2 vs. T. brucei: selectivity index ∼12). The efficacy observed against T. brucei and L. amazonensis is noteworthy and comparable to that of existing therapeutics; for example, the approved leishmaniasis drug miltefosine displays an EC50 = 1.2 μM against L. amazonensis.28,29 Natural spiroindimicin A [(S)-3], in particular, may warrant further study against the neglected tropical disease leishmaniasis given its activity (EC50 = 1.3 μM) and lack of significant cytotoxicity in RAW cells.
| Compound | Antiparasitic activity | Selectivity | ||||
|---|---|---|---|---|---|---|
| T. brucei EC50 (μM) | P. falciparum 3D7 EC50 (μM) | P. falciparum Dd2 EC50 (μM) | L. amazonensis EC50 (μM) | RAW CC50 (μM) | HepG2 CC50 (μM) | |
| a Data represent the mean EC50 ± standard error for 3 biological replicates. EC50 calculations for each biological replicate were based on data from technical triplicates. n.t. = not tested. | ||||||
| (±)-3 | 7.5 ± 1.1 | 2.8 ± 0.49 | 4.2 ± 0.11 | 1.4 ± 0.35 | 5.5 ± 0.41 | 10 ± 1.2 |
| (S)-3 | 11 ± 1.2 | 3.9 ± 0.81 | 6.6 ± 0.12 | 1.3 ± 0.33 | >10 | >10 |
| (R)-3 | 11 ± 1.2 | 4.8 ± 1.2 | 7.1 ± 0.33 | 5.3 ± 1.1 | >10 | >10 |
| (±)-10 | 7.1 ± 1.2 | n.t. | n.t. | 4.5 ± 0.98 | 8.1 ± 0.38 | >10 |
| (±)-36 | 12 ± 1.1 | 4.4 ± 0.93 | 7.1 ± 0.83 | 6.3 ± 1.2 | 9.3 ± 0.67 | >10 |
| (±)-37 | 3.2 ± 0.64 | 3.7 ± 0.90 | 5.5 ± 0.51 | 6.0 ± 1.2 | >10 | >10 |
| 12 | 8.3 ± 1.0 | >10 | n.t. | >10 | >10 | >10 |
| 13 | 8.2 ± 0.45 | >10 | n.t. | 8.9 ± 0.9 | >10 | >10 |
| 21 | 0.37 ± 0.073 | 0.79 ± 0.11 | 1.0 ± 0.030 | 4.5 ± 0.26 | 3.4 ± 1.1 | 4.6 ± 1.1 |
Conclusions
In summary, we have reported the first total synthesis of (+)-spiroindimicin A (3). Our 9-step synthesis relies upon an efficient assembly of a triaryl scaffold with distinct connectivity to its natural precursor via cross-coupling, and a novel Pd-catalyzed asymmetric spirocyclization to construct the challenging C-3′/C-5′′-linked spiroindolenine in high enantiopurity. We have also prepared spiroindimicin H and lynamicins A and D in a concise fashion and tested the conversion of the latter to the spiroindimicins through biomimetic oxidative spirocyclization. Although unproductive, these studies did inform our ultimately successful approach to 3 using an alternate triaryl fragment. With meaningful quantities of spiroindimicin A (3) and its congeners now available, we have begun to explore their biological activity more broadly. Studies to date have unveiled promising antiparasitic activity that may provide a starting point for developing compounds to treat leishmaniasis and African trypanosomiasis.28,29Author contributions
Z. Z. and M. W. S. conceived and executed the synthetic studies. S. R., L. I., and L. T. C. conducted the biological investigations under the supervision of M. A. P. and D. M. W., with high-throughput assistance provided by H. N., P. L. M., and B. A. P. M. W. S composed the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by UT Southwestern through the W. W. Caruth Jr. Scholarship. Funding from the Welch Foundation to M. W. S. (I-2045), M. A. P. (I-1257), and D. M. W. (I-2086) is gratefully acknowledged. This work was also partially funded by the NIH grants, R01AI103947 (to M. A. P), R01AI146349 (to D. M. W.), and 1S10OD026758-01 (to B. A. P.), as well as by the Children's Clinical Research Advisory Committee Early Investigator Award, 2019 Harrington Scholar-Innovator Award, and 2020 UTSW Circle of Friends Pilot Synergy Grant (all to D. M. W.). We thank Dr Fan Xu for attempting electrochemical and photoredox spirocyclizations of 12 and its protected variants. We are grateful to Prof. Changseng Zhang, Dr Liang Ma, and Wenjun Zhang (South China University of Oceanology) for providing spectra and an authentic sample of SPM A, and Prof. Peter Kündig (Université de Genève) for providing chiral NHC precursors. We thank the Tambar, Ready, Qin, DeBrabander, Chen, and Falck groups (UT Southwestern) for generous access to equipment and chemicals, as well as helpful discussions, and Dr Feng Lin for assistance with NMR studies.Notes and references
- For reviews, see: (a) K. S. Ryan and C. L. Drennan, Chem. Biol., 2009, 16, 351 CrossRef CAS PubMed; (b) H. Nakano and S. Omura, J. Antibiot., 2009, 62, 17 CrossRef CAS PubMed.
- Spiroindimicin isolations: (a) W. Zhang, Z. Liu, S. Li, T. Yang, Q. Zhang, L. Ma, X. Tian, H. Zhang, C. Huang, S. Zhang, J. Ju, Y. Shen and C. Zhang, Org. Lett., 2012, 14, 3364 CrossRef CAS PubMed; (b) C. Paulus, Y. Rebets, B. Tokovenko, S. Nadmid, L. P. Terekhova, M. Myronovskyi, S. B. Zotchev, C. Rückert, S. Braig, S. Zahler, J. Kalinowski and A. Luzhetskyy, Sci. Rep., 2017, 7, 42382 CrossRef CAS PubMed; (c) Z. Liu, L. Ma, L. Zhang, W. Zhang, Y. Zhu, Y. Chen, W. Zhang and C. Zhang, Org. Biomol. Chem., 2019, 17, 1053 RSC.
- K. A. McArthur, S. S. Mitchell, G. Tsueng, A. Rheingold, J. D. White, J. Grodberg, K. S. Lam and B. C. M. Potts, J. Nat. Prod., 2008, 71(10), 1732 CrossRef CAS PubMed.
- L. Ma, W. Zhang, Y. Zhu, G. Zhang, H. Zhang, Q. Zhang, L. Zhang, C. Yuan and C. Zhang, Appl. Microbiol. Biotechnol., 2017, 101, 6123 CrossRef CAS PubMed.
- For methods targeting the spiroindimicins, see: (a) R. K. Nandi, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2016, 18, 1716 CrossRef CAS; (b) B. Singh, S. K. Bankar, K. Kumar and S. S. V. Ramasastry, Chem. Sci., 2020, 11, 4948 RSC.
- L. M. Blair and J. Sperry, Chem. Commun., 2016, 52, 800 RSC.
- For reviews, see: (a) M. C. Field, D. Horn, A. H. Fairlamb, M. A. Ferguson, D. W. Gray, K. D. Read, M. De Rycker, L. S. Torrie, P. G. Wyatt, S. Wyllie and I. H. Gilbert, Nat. Rev. Microbiol., 2017, 15, 217 CrossRef CAS; (b) M. A. Phillips, J. N. Burrows, C. Manyando, R. H. van Huijsduijnen, W. C. Van Voorhis and T. N. C. Wells, Nat. Rev. Dis. Primers, 2017, 3, 17050 CrossRef PubMed.
- For pertinent reviews, see: (a) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed; (b) C. Zheng and S.-L. You, Chem, 2016, 1, 830 CrossRef CAS; (c) C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276 RSC; (d) C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432 CrossRef CAS PubMed.
- I. Sigala, G. Ganidis, S. Thysiadis, A. L. Zografos, T. Giannakouros, V. Sarli and E. Nikolakaki, Bioorg. Med. Chem., 2017, 25, 1622 CrossRef CAS PubMed.
- For reviews, see: (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (c) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2018, 7, 1178 CrossRef CAS; (d) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925 RSC; (e) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767 CrossRef CAS.
- R. I. Khusnutdinov, A. R. Baiguzina, R. R. Mukminov, I. V. Akhmetov, I. M. Gubaidullin, S. I. Spivak and U. M. Dzhemilev, Russ. J. Org. Chem., 2010, 46, 1053 CrossRef CAS.
- A. Fürstner, A. Krause and O. R. Thiel, Tetrahedron, 2002, 58, 6373 CrossRef.
- C. C. C. Johansson Seechurn, V. Sivakumar, D. Satoskar and T. J. Colacot, Organometallics, 2014, 33, 3514 CrossRef CAS.
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871 CrossRef CAS PubMed.
- For a similar pyrrole dealkoxycarbonylations, see: (a) D. L. Boger and M. Patel, J. Org. Chem., 1988, 53, 1405 CrossRef CAS; (b) A. Rana, B. S. Kumar and P. K. Panda, Org. Lett., 2015, 17, 3030 CrossRef CAS.
- Attempts to transform indolocarbazole 21 to a 5-membered spirooxindole revelant to SPM D via treatment with oxidants delivered 22.
- D. Skolc, A. Ates, E. Jnoff and A. Valade, PCT Int. Appl. WO 2016055482 A1, April 14 2016.
- For the preparation of the monoiodide precursor to 27, see: K. Hasse, A. C. Willis and M. G. Banwell, Eur. J. Org. Chem., 2011, 88 CrossRef CAS.
- For reviews on Pd-PEPPSI precatalysts, see: (a) M. G. Organ, G. A. Chass, D.-C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2776 CrossRef CAS; (b) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS.
- Treatment of desiodo-30 with several oxidants to induce spirocyclization to 31 proved unsuccessful.
- K.-J. Wu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 3772 CrossRef CAS PubMed . These authors report a single example of enantioselective spirocyclization, proceeding in 61% ee with a diastereomer of L3 as ligand..
- Spiroindolenine 31 was stable to heating in toluene at 90 °C for 40 h, as well as in the presence of Cs2CO3 (90 °C, 16 h). Partial decomposition occurs in the presence of both base and Pd catalyst at this temperature (16 h), but no indole 32 is formed. 31 is stable to purification on silica gel.
- SIPHOS-PE ligand: W. Zhang, L.-X. Wang, W.-J. Shi and Q.-L. Zhou, J. Org. Chem., 2005, 70, 3734 CrossRef CAS.
- The corresponding bromo analogue of 30 proved less reactive in this transformation, requiring higher temperatures and leading to 31 in low enantioselectivity (see ESI† for details).
- For a recent report of a related asymmetric process incorporating an alkyne partner, see: (a) H. Chu, J. Cheng, J. Yang, Y.-L. Guo and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 21991 CrossRef CAS PubMed . For related enantioselective Pd-catalyzed cyclizations of phenols, see:; (b) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS PubMed; (c) R.-Q. Xu, Q. Gu, W.-T. Wu, Z.-A. Zhao and S.-L. You, J. Am. Chem. Soc., 2014, 136, 15469 CrossRef CAS PubMed; (d) K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033 CrossRef CAS; (e) G. Zhao, G. Xu, C. Qian and W. Tang, J. Am. Chem. Soc., 2017, 139, 3360 CrossRef CAS PubMed; (f) K. Du, H. Yang, P. Guo, L. Feng, G. Xu, Q. Zhou, L. W. Chung and W. Tang, Chem. Sci., 2017, 8, 6247 RSC . For organocatalyzed asymmetric indole C-3 arylation to indolenines, see:; (g) Y.-C. Zhang, J.-J. Zhao, F. Jiang, S.-B. Sun and F. Shi, Angew. Chem., Int. Ed., 2014, 53, 13912 CrossRef CAS PubMed; (h) Y. Wang, M. Sun, L. Yin and F. Shi, Adv. Synth. Catal., 2015, 357, 4031 CrossRef CAS.
- For other enantioselective indole to indolenine transformations, see: (a) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314 CrossRef CAS PubMed; (b) J. García-Fortanet, F. Kessler and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 6676 CrossRef PubMed; (c) Q.-F. Wu, H. He, W.-B. Liu and S.-L. You, J. Am. Chem. Soc., 2010, 132, 11418 CrossRef CAS PubMed; (d) Y. Liu and H. Du, Org. Lett., 2013, 15, 740 CrossRef CAS PubMed; (e) C. Romano, M. Jia, M. Monari, E. Manoni and M. Bandini, Angew. Chem., Int. Ed., 2014, 53, 13854 CrossRef CAS PubMed; (f) Z.-S. Liu, W.-K. Li, T.-R. Kang, L. He and Q.-Z. Liu, Org. Lett., 2015, 17, 150 CrossRef CAS PubMed; (g) M. J. James, J. D. Cuthbertson, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 7640 CrossRef CAS; (h) Y. Zhou, Z.-L. Xia, Q. Gu and S.-L. You, Org. Lett., 2017, 19, 762 CrossRef CAS; (i) V. Magné, A. Marinetti, V. Gandon, A. Voituriez and X. Guinchard, Adv. Synth. Catal., 2017, 359, 4036 CrossRef; (j) Y. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 15093 CrossRef CAS PubMed; (k) Z.-L. Xia, C. Zheng, S.-G. Wang and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 2653 CrossRef CAS PubMed; (l) R.-D. Gao, L. Ding, C. Zheng, L.-X. Dai and S.-L. You, Org. Lett., 2018, 20, 748 CrossRef CAS PubMed; (m) B. M. Trost, W.-J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710 CrossRef CAS; (n) L. Huang, Y. Cai, H.-J. Zhang, C. Zheng, L.-X. Dai and S.-L. You, CCS Chem, 2019, 1, 106 CAS; (o) A. Becker, C. P. Grugel and B. Breit, Org. Lett., 2021, 23, 3788 CrossRef CAS . For a Pd-catalyzed asymmetric enamine arylation to indolenines, see:; (p) R.-X. Liang, C. Zhong, Z.-H. Liu, M. Yang, H.-W. Tang, J.-F. Chen, Y.-F. Yang and Y.-X. Jia, ACS Catal., 2021, 11, 1827 CrossRef CAS.
- (a) M. V. Braga and W. de Souza, FEMS Microbiol. Lett., 2006, 256, 209–216 CrossRef CAS; (b) L. Cartuche, I. Sifaoui, A. López-Arencibia, C. J. Bethencourt-Estrella, D. San Nicolás-Hernández, J. Lorenzo-Morales, J. E. Piñero, A. R. Díaz-Marrero and J. J. Fernández, Biomolecules, 2020, 10, 657 CrossRef CAS PubMed.
- The clinical candidate acoziborole (currently in Phase III) has an EC50 value of 0.6 μM against T. brucei, see: (a) E. A. Dickie, F. Giordani, M. K. Gould, P. Mäser, C. Burri, J. C. Mottram, S. P. S. Rao and M. P. Barrett, Trop. Med. Infect. Dis., 2020, 5, 29 CrossRef PubMed. The bar for a clinical antimalarial candidate is significantly higher, typically EC50 ≤ 10 nM is desirable, see: (b) T. D. Ashton, S. M. Devine, J. J. Möhrle, B. Laleu, J. N. Burrows, S. A. Charman, D. J. Creek and B. E. Sleebs, J. Med. Chem., 2019, 62, 10526 CrossRef CAS PubMed.
- The approved leishmaniasis drug miltefosine has EC50 values of 6.83–10.12 μM and 1.2 μM against intracellular L. mexicana and extracellular L. amazonensis, respectively, see: (a) H. Sindermann, S. L. Croft, K. R. Engel, W. Bommer, H. J. Eibl, C. Unger and J. Engel, Med. Microbiol. Immunol., 2004, 193, 173 CrossRef CAS PubMed; (b) I. Ullah, S. Gahalawat, L. M. Booshehri, H. Niederstrasser, S. Majumdar, C. Leija, J. M. Bradford, B. Hu, J. M. Ready and D. M. Wetzel, ACS Infect. Dis., 2020, 6, 2057 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02838c |
| This journal is © The Royal Society of Chemistry 2021 |