
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of crystal packing in semiconducting spin-crossover materials with fractionally charged TCNQδ− anions (0 < δ < 1)†
Ökten
Üngör
 a,
Eun Sang
Choi
b and
Michael
Shatruk
*ab
a,
Eun Sang
Choi
b and
Michael
Shatruk
*ab
aDepartment of Chemistry and Biochemistry, Florida State University, 95 Chieftan Way, Tallahassee, FL 32306, USA. E-mail: shatruk@chem.fsu.edu
bNational High Magnetic Field Laboratory, 1800 E Paul Dirac Dr, Tallahassee, FL 32310, USA
First published on 5th July 2021
Abstract
Co-crystallization of the prominent Fe(II) spin-crossover (SCO) cation, [Fe(3-bpp)2]2+ (3-bpp = 2,6-bis(pyrazol-3-yl)pyridine), with a fractionally charged TCNQδ− radical anion has afforded a hybrid complex [Fe(3-bpp)2](TCNQ)3·5MeCN (1·5MeCN, where δ = −0.67). The partially desolvated material shows semiconducting behavior, with the room temperature conductivity σRT = 3.1 × 10−3 S cm−1, and weak modulation of conducting properties in the region of the spin transition. The complete desolvation, however, results in the loss of hysteretic behavior and a very gradual SCO that spans the temperature range of 200 K. A related complex with integer-charged TCNQ− anions, [Fe(3-bpp)2](TCNQ)2·3MeCN (2·3MeCN), readily loses the interstitial solvent to afford desolvated complex 2 that undergoes an abrupt and hysteretic spin transition centered at 106 K, with an 11 K thermal hysteresis. Complex 2 also exhibits a temperature-induced excited spin-state trapping (TIESST) effect, upon which a metastable high-spin state is trapped by flash-cooling from room temperature to 10 K. Heating above 85 K restores the ground-state low-spin configuration. An approach to improve the structural stability of such complexes is demonstrated by using a related ligand 2,6-bis(benzimidazol-2′-yl)pyridine (bzimpy) to obtain [Fe(bzimpy)2](TCNQ)6·2Me2CO (4) and [Fe(bzimpy)2](TCNQ)5·5MeCN (5), both of which exist as LS complexes up to 400 K and exhibit semiconducting behavior, with σRT = 9.1 × 10−2 S cm−1 and 1.8 × 10−3 S cm−1, respectively.
Introduction
Transition metal complexes that exhibit spin crossover (SCO), arguably, offer one of the most appealing examples of molecular bistability, which manifests itself as dramatic changes in structural, optical, magnetic, and dielectric properties of the SCO material.1,2 The large disparity in properties between the high-spin (HS) and low-spin (LS) electronic configurations3 offers a convenient parameter space to manipulate the bistable behavior by variations in temperature,4–7 pressure,8–13 or light irradiation.14–20In recent years, SCO building blocks have been actively explored in the pursuit of multifunctional molecule-based materials that would combine spin-state switching with other useful functional properties, such as conductivity, magnetic ordering, or luminescence.21–24 For example, the approach that combines SCO metal ions with organic π-donors or π-acceptors, known to form conducting charge transfer salts,25 has been recognized as a viable design strategy toward hybrid SCO conductors. The main goal of these efforts is to achieve coupling between spin-state switching and electrical transport. One can envision two approaches to designing such materials. In the first strategy, the unit that provides conducting pathways is attached to the metal complex directly, as part of the ligand structure. This approach has been actively explored by a number of groups by using ligands functionalized with tetrathiafulvalene (TTF) or its derivatives,26–30 although partial oxidation of the TTF moieties that leads to appreciable conductivity of the resulting hybrid material was reported only in two of such cases.31,32 Alternatively, the SCO metal complex can be co-crystallized with organic radical anions of required charge. In our opinion, this approach is more promising, as it offers a greater flexibility in the choice of building blocks, essentially allowing modular design of functional materials. Additionally, it provides more degrees of freedom for combining molecular fragments to achieve the optimal crystal packing.
Initially, the design of hybrid SCO conductors of the second type focused mainly on combining cationic Fe(III) complexes with metal–dithiolene anions.33–37 Recently, however, there has been an increased interest in the co-crystallization of fractionally charged anions of 7,7,8,8-tetracyanoquinodimethane (TCNQ) with other transition metal ions for the synthesis of such hybrid materials. In this vein, our and other groups reported the first examples of TCNQ-containing SCO semiconductors. The first of these reports employed a cationic Fe(II) complex that afforded a complete albeit gradual SCO as well as a light-induced excited spin-state trapping (LIESST) effect at lower temperatures.38 Two other reports included co-crystallization of Co(II) complexes with fractionally charged TCNQδ− anions; in both cases, the SCO was very gradual, with the midpoint of the LS ↔ HS conversion (T1/2) seen above 350 K.39,40 Shvachko et al. reported an Fe(II)–TCNQ complex with an onset of SCO above room temperature and an even higher T1/2 value, estimated as 445 K.41 A recently reported Mn(III) complex has offered the first example of a hysteretic spin transition (T1/2,↓ = 73 K upon cooling and T1/2,↑ = 123 K upon heating) in a complex with fractionally charged TCNQδ− anions, albeit the measured conductivity was rather low, perhaps, due to sequestration of the anions into isolated columns separated by cationic complexes.42
Despite these encouraging reports, it is surprising that Fe(II) complexes, which constitute more than 90% of known SCO compounds,1 are largely underused in the search for new SCO conductors. We hypothesized that, to achieve the complete SCO within an accessible temperature range, one needs to begin with a well-defined SCO cation that has been shown to form complexes with abrupt spin transitions. In this vein, we turned our attention to the well-studied tridentate ligand, 2,6-bis(pyrazol-3-yl)pyridine (3-bpp). Complexes of the homoleptic [Fe(3-bpp)2]2+ cation have been shown to afford abrupt spin transitions that are highly sensitive to the nature of anion and degree of solvation.43,44 This sensitivity is explained by the tendency of 3-bpp to couple to counter ions and interstitial solvent molecules through hydrogen-bonding interactions, thus offering more interesting opportunities for materials design, as compared to the related ligand, 2,6-bis(pyrazol-1-yl)pyridine (1-bpp).43 Herein, we report the first observation of a hysteretic SCO in an Fe(II) complex co-crystallized with fractionally charged TCNQδ− (0 < δ < 1) radical anions. We demonstrate that the properties of such solids can be strongly dependent on interstitial solvent molecules. In this vein, we also show that the stability of single crystals of such materials can be improved by modifying the 3-bpp ligand to achieve more extensive intermolecular contacts between the Fe(II) cationic complexes. To that end, a related tridentate ligand, 2,6-bis(benzimidazol-2′-yl)pyridine (bzimpy), previously shown to form SCO complexes with the Fe(II) ion,45 has been used to obtain conducting Fe(II)–TCNQ complexes that are more stable towards the loss of interstitial solvent. Finally, we discuss pathways to improve the design of molecule-based conductors with spin-state switching behavior.
Results and discussion
Synthesis
The synthetic pathways to all complexes discussed in this work are summarized in Scheme 1. The SCO complex [Fe(3-bpp)2](TCNQ)3·5MeCN (1·5MeCN), containing TCNQδ− anions with an average charge of −2/3, was obtained as dark-green plate-shaped crystals by slow diffusion of solutions of [Fe(3-bpp)2](BF4)2 and Et3NH(TCNQ)2 in acetonitrile (MeCN). The thermal gravimetric analysis (TGA) of this complex revealed a 4.7% mass loss at 100 °C (Fig. S1a†), which can be attributed to the loss of ∼1.5 MeCN molecule per formula unit (f.u.). A gradual decrease in the mass of the sample was observed immediately upon heating, suggesting that the solvent is partially lost from the crystals even at room temperature (r.t.). Heating above 200 °C led to multi-step decomposition of the complex. When 1·5MeCN was kept under vacuum (∼10−1 mbar) for 12 h, the resulting sample showed a TGA curve without any mass loss up to 200 °C (Fig. S1a,† inset), suggesting that evacuation led to the complete loss of interstitial solvent and the formation of [Fe(3-bpp)2](TCNQ)3 (1).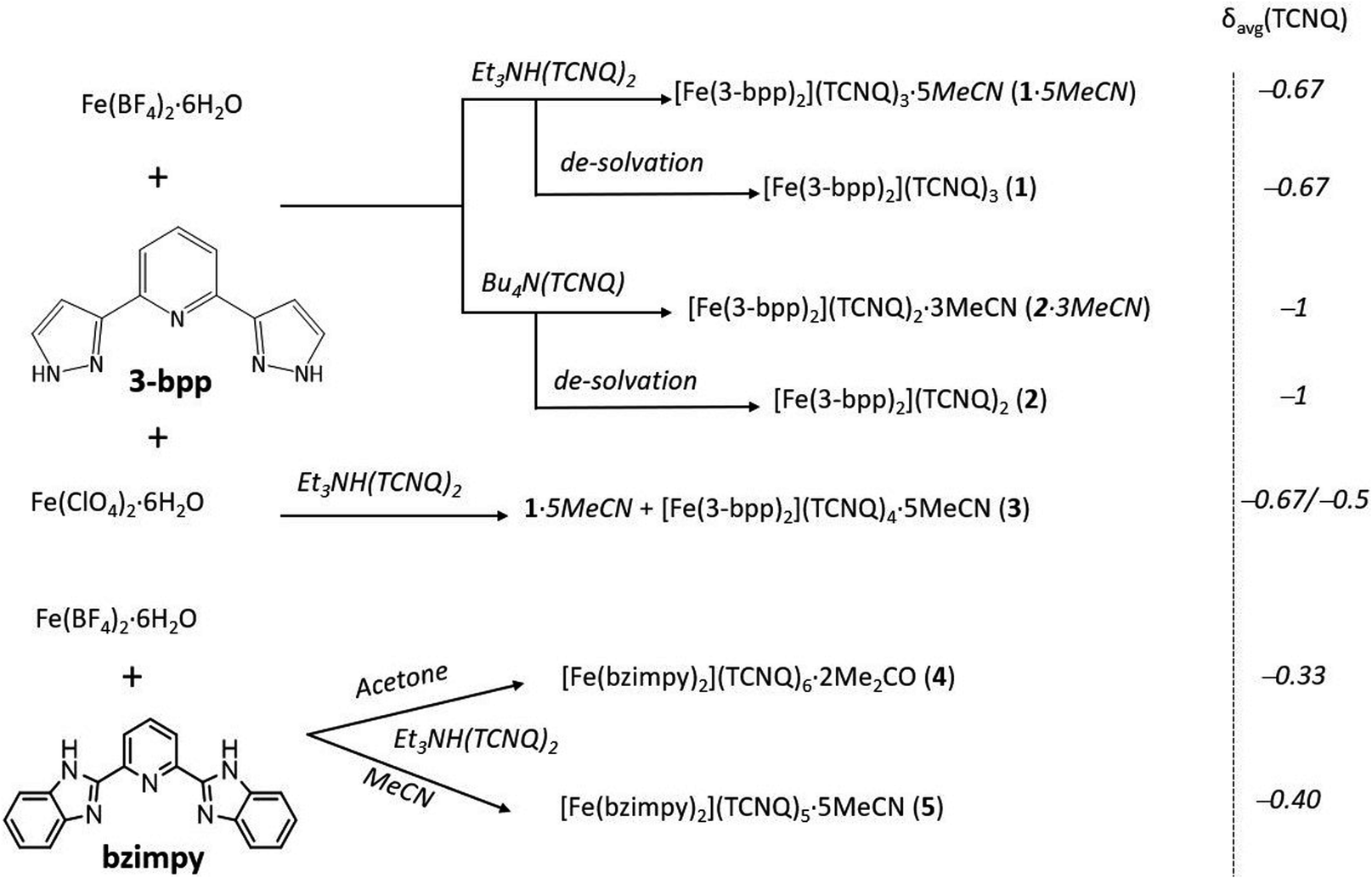 | ||
| Scheme 1 The synthesis of complexes containing [Fe(3-bpp)2]2+ or [Fe(bzimpy)2]2+ cations and TCNQδ− anions. | ||
As a reference material, we synthesized a complex with integer-charged TCNQ− anions, [Fe(3-bpp)2](TCNQ)2·3MeCN (2·3MeCN). This complex was crystallized in a manner similar to that described for 1·5MeCN, but with the use of Bu4N(TCNQ) as a precursor. TGA revealed that 2·3MeCN was more thermally stable than 1·5MeCN and the solvent loss occurred in a single step (Fig. S1b†). At 100 °C, the mass loss was 13.6%, corresponding to the loss of three MeCN molecules per f.u. Drying under vacuum (∼10−1 mbar) for 12 h or heating to 120 °C for 1 h afforded a completely desolvated complex, [Fe(3-bpp)2](TCNQ)2 (2), as judged by the TGA curve of the resulting sample (Fig. S1b,† inset)To assess the generality of the reaction, the synthesis of 1·5MeCN was repeated by reacting Et3NH(TCNQ)2 with another Fe(II) precursor, i.e., Fe(ClO4)2·6H2O. This reaction produced complex 1·5MeCN, but also led to a minor impurity of a different complex, [Fe(3-bpp)2](TCNQ)3.5·6MeCN (3), in the form of thin plates that were easily distinguishable from the crystals of 1·5MeCN. Attempts to synthesize and isolate complex 3 in a bulk amount were unsuccessful. Given its low yield, poorly crystallinity, and the lack of its formation in the reactions performed with Fe(BF4)2·6H2O, complex 3, most likely, represents a kinetic byproduct.
To achieve better crystal packing and increase the stability of crystals toward the solvent loss (see the Crystal structure section below), we extended this study to include a tridentate mer-coordinating ligand, 2,6-bis(benzimidazol-2′-yl)pyridine (bzimpy), which should offer better intermolecular interactions than 3-bpp due to the more extended aromatic system. The reaction between [Fe(bzimpy)2](BF4)2 and Et3NH(TCNQ)2 in acetone (Me2CO) yielded [Fe(bzimpy)2](TCNQ)6·2Me2CO (4) that contains TCNQδ− anions with an average charge of −1/3. The same reaction carried out in MeCN yielded a complex [Fe(bzimpy)2](TCNQ)5·5MeCN (5) with an average charge equal to –2/5 per TCNQδ− anion. The dark, block-like single crystals of 4 and the needle-like single crystals of 5 appear to be more stable when exposed to air and do not lose the interstitial solvent as quickly as the complexes with 3-bpp. TGA of 4 revealed a 1.6% mass decrease by 123 °C, which corresponds to the loss of the residual interstitial solvent (Fig. S2a†). Indeed, as shown by the elemental analysis, drying the sample under vacuum at room temperature leads to the complete loss of interstitial solvent. TGA of 5 revealed a 5.7% mass decrease by 110 °C (Fig. S2b†), which corresponds to the loss of two MeCN molecule per f.u. A gradual decrease in the mass of the sample was observed upon heating, accompanied by two-step decomposition of 5. The de-solvated sample of 5 is thermally stable up to 200 °C.
Crystal structure
The crystal structures of 1·5MeCN and 2·3MeCN were determined at 90 and 250 K (Table S1†). Complex 1·5MeCN crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) at both temperatures. The asymmetric unit includes a [Fe(3-bpp)2]2+ cation, three TCNQδ− anions (hereafter denoted as TCNQA, TCNQB, and TCNQC), and five interstitial MeCN molecules (Fig. S3a†).
at both temperatures. The asymmetric unit includes a [Fe(3-bpp)2]2+ cation, three TCNQδ− anions (hereafter denoted as TCNQA, TCNQB, and TCNQC), and five interstitial MeCN molecules (Fig. S3a†).
Each FeII center exhibits a distorted octahedral coordination furnished by six N-donor atoms from two mer-coordinating 3-bpp ligands. The average Fe–N bond length is 2.194(1) Å at 90 K and 2.187(2) Å at 250 K. The octahedral distortion parameter ∑90, defined as the sum of absolute deviations of the twelve cis-N–Fe–N bond angles from the ideal octahedral value of 90°, also shows only minor changes between 90 and 250 K (Table 1). These data indicate that the FeII center in the structure of 1·5MeCN remains in the HS state at both temperatures.
| 1 ·5MeCN | 2 ·3MeCN | 2 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a The fraction of the HS state was determined by comparing to the Fe–N distances in the LS and HS structures of the reference SCO complex, [Fe(3-bpp)2](NCS)2, for which d(Fe–N)avg = 1.961 Å in the LS state and 2.162 Å in the HS state.47 | |||||||||
| Temperature, K | 90 | 250 | 90 | 250 | 90 | 100 | 250 | 250 | 250 |
| d(Fe–N)avg, Å | 2.194(1) | 2.187(2) | 2.107(3) | 2.167(2) | 2.080(1) | 2.090(1) | 2.167(1) | 1.956(1) | 1.953(2) |
| ∑90, ° | 168.3(4) | 163.7(1) | 149.1(2) | 144.5(4) | 128.4(2) | 134.9(1) | 145.0(1) | 87.3(3) | 89.6(2) |
| FeII spin state | HS | HS | ∼50% HSa | HS | ∼40% HS | ∼45% HS | HS | LS | LS |
The crystal packing of 1·5MeCN reveals alternating layers of the [Fe(3-bpp)2]2+ cations and TCNQδ− anions (Fig. 1a). In the cationic layers, the [Fe(3-bpp)2]2+ complexes form chains parallel to the a axis (Fig. 1b). The cohesion between the cations within these chains is provided by π–π interactions between the pyrazolyl rings of the 3-bpp ligands, with a rather short interplanar distance of 3.24 Å. On the other hand, the separation between the chains is substantial (Fig. 1a); the intrachain Fe–Fe distance is 7.85 Å while the interchain Fe–Fe distance is 13.81 Å. Such packing leads to large channels filled with the solvent molecules (Fig. S3b†) that are easily lost upon heating or evacuation of the sample (Fig. S1a†). The loss of solvent results in disintegration of crystals of 1·5MeCN. Nevertheless, powder X-ray diffraction on the dried sample of 1 reveals that the crystallinity is preserved (Fig. S4†), although the diffraction pattern becomes substantially different from the one calculated from the crystal structure of 1·5MeCN.
 | ||
| Fig. 1 The crystal structure of 1·5MeCN viewed in the bc plane (a) and in the ac plane (b). The TCNQA, TCNQB, and TCNQC units are shown in green, orange, and red, respectively. The dashed red ovals emphasize the repeating sequence in panel (a) and the π–π interactions between the cations in panel (b), where the cationic chains are highlighted with pale-blue shadows. The color scheme for other atoms: Fe = orange, N = blue, C = gray. The H atoms and interstitial solvent molecules are omitted for clarity. | ||
Two different types of anionic layers alternate in the structure. One type of layers contains one-dimensional stacks of TCNQδ− anions that propagate along the b axis (Fig. 1a). These stacks are formed by TCNQA and TCNQB units with a repeating sequence [ABBA]. The TCNQA–TCNQB overlap is of the ring-over-ring type, whereas the TCNQA–TCNQA and TCNQB–TCNQB overlaps are of the ring-over-external-bond type (Fig. S5a†).46 At 90 K, the interplanar separations between the TCNQ units in these tetrads are 3.162, 3.108, and 3.125 Å, with a distance of 3.415 Å between the neighboring tetrads. At 250 K, these distances increase to 3.250, 3.198, 3.200, and 3.440 Å, respectively, due to the thermal expansion of the lattice.
The second type of anionic layers consists of stacks of dimers built of TCNQC units and extending along the a direction of the lattice, perpendicular to the stacks built of the TCNQA and TCNQB units (Fig. 1). The intra-dimer TCNQC–TCNQC overlap is of the ring-over-external-bond type (Fig. S5b†). Both the intra- and inter-dimer interplanar separations increase with temperature, from 3.076 and 3.349 Å at 90 K to 3.124 and 3.510 Å at 250 K, respectively.
The electroneutrality of 1·5MeCN requires that the total charge on the three TCNQδ− anions be equal to −2. The approximate charge on each TCNQ unit can be calculated from the equation −δ = −41.67[c/(b + d)] + 19.83,48 where the letters represent the bond lengths in the TCNQ structure (Table S3†). Using this equation, the charge on the dimer-forming TCNQC anion is equal to −1.01, in agreement with the tendency of TCNQ− monoanions to dimerize via the strong π–π interaction in the solid state.49 The charges on TCNQA and TCNQB are −0.58 and −0.41, suggesting an even charge distribution along the 1D stack, which is expected to afford good conducting properties (to be discussed in a later section).
Complex 2·3MeCN crystallizes in the monoclinic space group P21/n at both 90 and 250 K. The asymmetric unit is composed of a [Fe(3-bpp)2]2+ cation, two TCNQ− anions, and three interstitial MeCN molecules (Fig. S6†). Each FeII center shows a distorted octahedral coordination similar to that observed in 1·5MeCN. At 250 K, the average Fe–N bond length is 2.167(2) Å, which corresponds to the HS FeII ion. At 90 K, however, the average Fe–N bond length decreases to 2.107(3) Å, suggesting a partial conversion to the LS state.
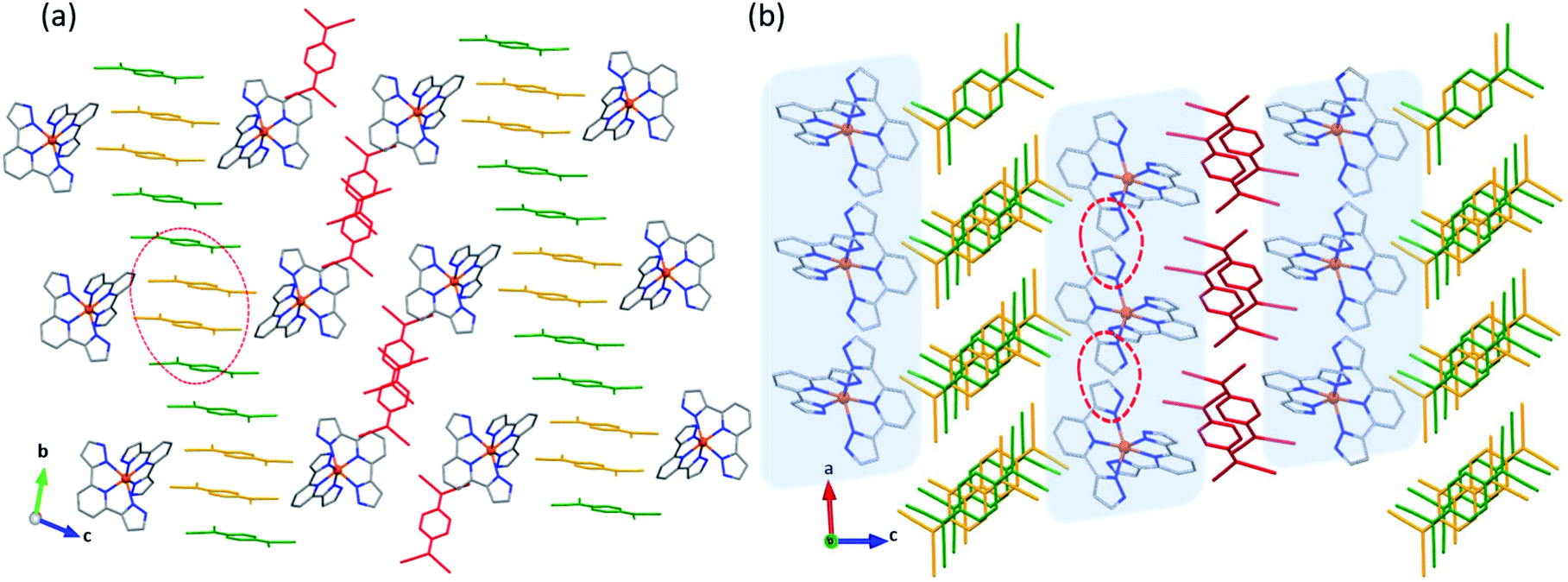
Similar to 1·5MeCN, the crystal packing of 2·3MeCN reveals cationic and anionic layers. The cations are arranged in chains assembled via π–π and σ–π interactions between the pyrazolyl rings of 3-bpp (Fig. 2a). At 250 K, the intrachain Fe–Fe separation is 8.42 Å while the interchain separations are 9.41 and 11.20 Å. The difference between the intra- and interchain Fe–Fe distances is not as drastic as observed in 1·5MeCN. As a result, the interstitial solvent mainly resides in the anionic layer, separating (TCNQ)22− dimers that stack parallel to the a axis. The overlap of the TCNQ units in the dimer is of the ring-over-ring type. The interplanar π–π separation within the dimer increases from 3.056 Å at 90 K to 3.160 Å at 250 K, while the inter-dimer distance changes from 6.87 Å to 6.97 Å, respectively.
 | ||
| Fig. 2 The crystal structures of 2·3MeCN (a) and 2 (b) viewed in the ac plane. The interstitial solvent molecules in panel (a) are emphasized with purple color. The color scheme for other atoms: Fe = orange, N = blue, C = gray. The H atoms are omitted for clarity. The dashed blue shadowed circles indicate the (TCNQ)22− dimers. The inter-dimer distance and the distance between the pyrazolyl rings of the adjacent cations at 250 K are shown with black arrows (the distances are given in Å). | ||
In contrast to 1·5MeCN, complex 2·3MeCN does not lose the interstitial solvent as readily, but the solvent can be completely removed by exposing the complex to vacuum for 12 h or maintaining it under inert atmosphere at 120 °C for 1 h. Remarkably, despite the removal of three solvent molecules per f.u., the crystal lattice is preserved, and the X-ray analysis of 2 could be performed on the same crystals that were used for the structure determination of 2·3MeCN.
Complex 2 crystallizes in the space group P21/n at both 90 and 250 K. The asymmetric unit includes one [Fe(3-bpp)2]2+ cation and two TCNQ− anions. The average Fe–N bond lengths are 2.167(2) Å at 250 K and 2.080(1) Å at 90 K, indicating a partial HS → LS conversion upon cooling. (The reasons for the incomplete SCO will be discussed in the Magnetic Properties section.) The view of the crystal packing of 2 shown in Fig. 2b reveals that the crystal lattice of 2·3MeCN remains essentially preserved upon the solvent loss. The loss of interstitial solvent results in a large decrease in the unit cell volume (15% at 250 K), which translates into smaller intrachain (8.64 Å) and interchain (9.30 and 10.06 Å) Fe–Fe distances in the cationic layer and a closer separation between the (TCNQ)22− dimers (6.63 Å) and within the dimers (3.079 Å). The obvious densification of the crystal structure (Fig. S7†) leads to stronger interactions between the SCO cations, causing a substantial difference in the magnetic behavior of the solvated and desolvated forms of 2, as will be shown below.
Complex 3 was obtained as a kinetic byproduct during crystallization of 1·5MeCN using the Fe(ClO4)2·6H2O precursor. The crystal structure was determined at 100 K. The poor quality of X-ray diffraction achieved with the crystals of 3 necessitated the use of the solvent-mask (SQUEEZE) procedure.50 Even after eliminating the contribution from the disordered solvent to the X-ray diffraction intensities, the quality of the crystal structure refinement remained low (Table S2†). Nevertheless, the structural features important for this work could be reliably identified, and we discuss them here.
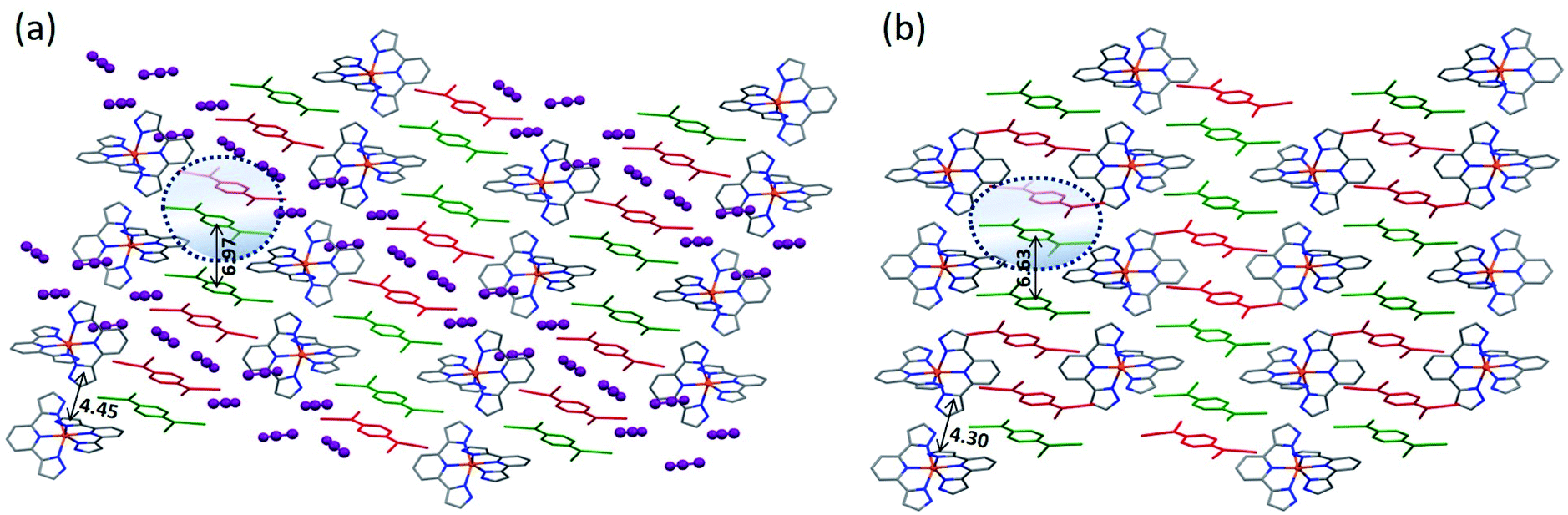
Complex 3 crystallizes in the space group C2/c, with the asymmetric unit containing one [Fe(3-bpp)2]2+ cation, three and a half TCNQδ− anions, and six disordered MeCN molecules, as determined through the solvent-mask procedure (Fig. S8†). The average Fe–N bond distance is 1.945(5) Å, indicating the LS state of the Fe(II) ion. Unfortunately, due to poor quality of the crystals of 3, we were unable to obtain the crystal structure at higher temperatures to evaluate the possibility of SCO.
Similar to the other structures described above, the crystal structure of 3 reveals alternating layers of cations and anions (Fig. 3). The anionic layers contain 1D stacks of TCNQδ− along the b axis. The average charge per TCNQ is −0.5, but the analysis of charges on crystallographically distinct TCNQ units could not be performed due to poor accuracy of the bond lengths. The poor crystallinity of this complex and low quality of its X-ray diffraction pattern are in agreement with the suggested kinetic nature of this product.
 | ||
| Fig. 3 The crystal packing of 3 viewed down the c axis. The crystallographically non-inequivalent TCNQ units are shown in different colors. The color scheme for other atoms: Fe = orange, N = blue, C = gray. The H atoms are omitted for clarity. The solvent molecules were not refined. | ||
Given the easy loss of interstitial solvent by complex 1·5MeCN, which leads to disintegration of its crystals, we sought to devise an approach to improve the structural stability of such materials. The use of the bzimpy ligand with the more extended aromatic system afforded complexes 4 and 5, both of which showed much higher stability in the single-crystal form. Interestingly, the use of different crystallization solvents causes changes in the stoichiometry and the crystal packing of 4 and 5.
The crystal structures of 4 and 5 were determined at 250 K. Both compounds crystallize in the space group P. In each structure, the FeII ion is octahedrally coordinated by six N-donor atoms provided by two mer-coordinating bzimpy ligands. The average Fe–N bond length of 1.954(2) Å for 4 and 1.953(2) for 5 indicates that both complexes are in the LS state at 250 K. Similar to the structures of 1·5MeCN and 2·3MeCN, the crystal packing of 4 and 5 is built of alternating layers of [Fe(bzimpy)2]2+ cations and TCNQδ− anions (Fig. 4).
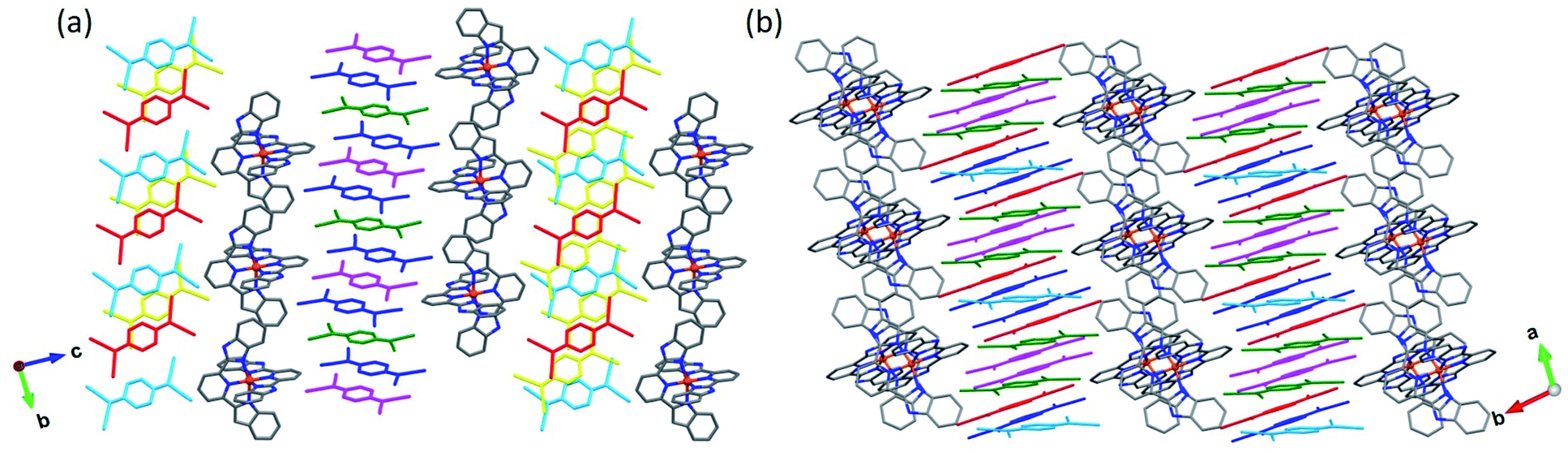 | ||
| Fig. 4 The crystal packing of 4 (a) and 5 (b). The distinct TCNQ anions are highlighted with different colors. The color scheme for other atoms: Fe = orange, N = blue, C = gray. The interstitial solvent molecules and H atoms are omitted for clarity. | ||
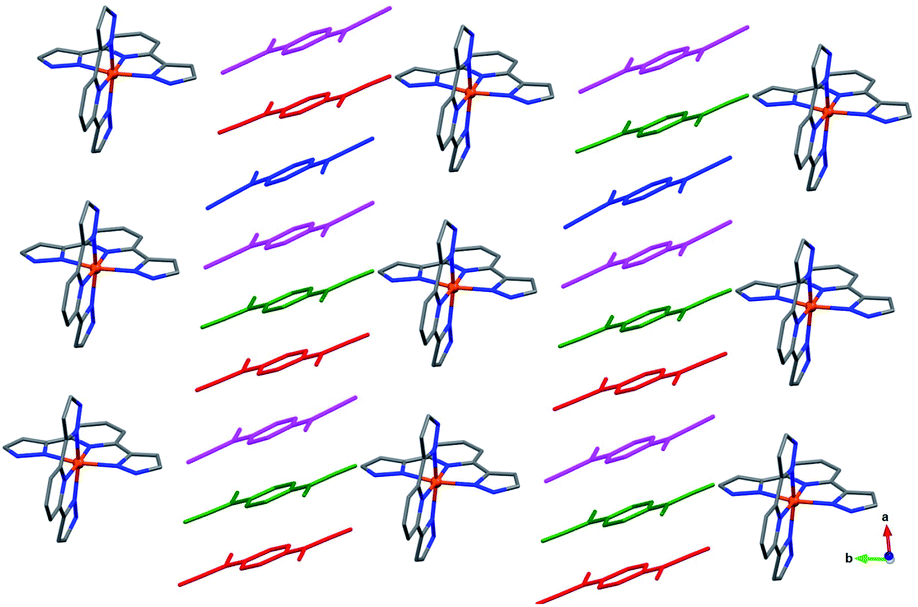
The asymmetric unit of 4 contains one [Fe(bzimpy)2]2+ cation, six TCNQδ− anions (hereafter denoted as A, B, C, A′, B′, and C′), and two Me2CO molecules (Fig. S9†). The change in the molecular structure of the ligand has a profound impact on the packing of cations. In contrast to the structures observed for the complexes with [Fe(3-bpp)2]2+, in which cations tend to arrange in chains, in the structure of 4 the [Fe(bzimpy)2]2+ cations form an efficiently packed 2D array. Each cation exhibits extensive interactions with neighboring cations, with the interplanar (π–π) and carbon-to-plane (σ–π) distances shown in Fig. 5a.
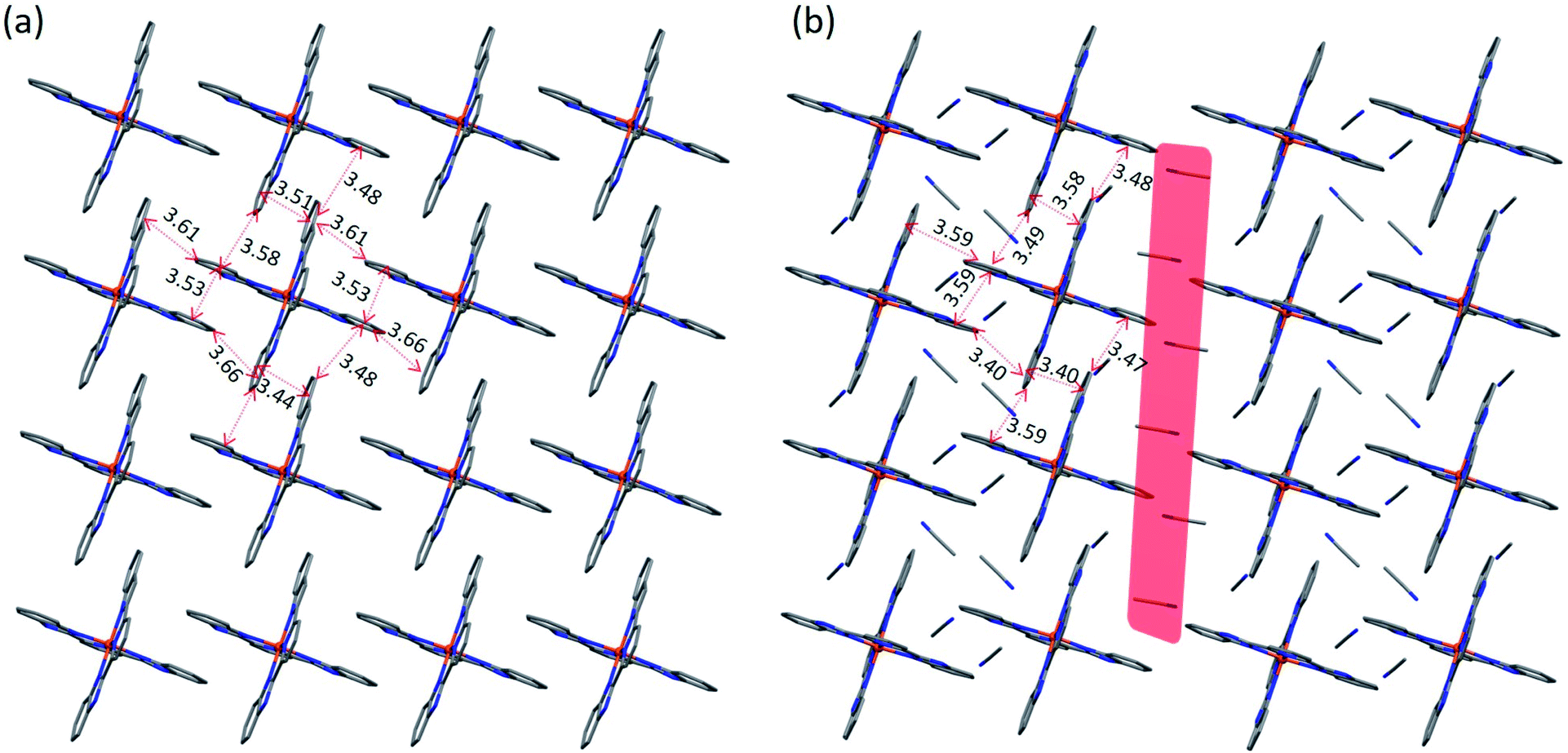 | ||
| Fig. 5 The cationic layers in crystal structures of 4 (a) and 5 (b). The π–π and σ–π intermolecular interactions (in Å) are indicated with red arrows for one of the cations in each panel. The solvent layer that separates the double chains of cations is highlighted with red in panel (b). The color scheme: Fe = orange, N = blue, C = gray. The H atoms are omitted for clarity. | ||
Another interesting feature of the crystal structure of 4 is that the direction of the TCNQ stacks alternates in every other anionic layer (Fig. 4a). The first type of stacks consists of TCNQA, TCNQB, and TCNQC units that form tetrads with a repeating sequence [ABCA], with the interplanar distances of 3.142, 3.481, and 3.481 Å within the tetrad and 3.142 Å between the tetrads. The second type of stacks contains tetrads of TCNQA′, TCNQB′, and TCNQC′ units arranged in the repeating sequence [A′B′C′B′], with the interplanar distances of 3.167, 3.411, 3.411, and 3.167 Å.
The electroneutrality requirement gives the average charge of −1/3 per TCNQδ− anion in 4. The calculated charges reveal a more non-uniform charge distribution (Table S3†). The charges on the TCNQA, TCNQB, and TCNQC anions are equal to −0.70, −0.28, and −0.21, respectively, while the charges on the TCNQA′, TCNQB′, and TCNQC′ anions are −0.47, −0.36, and 0.02, respectively.
The asymmetric unit of 5 includes one [Fe(bzimpy)2]2+ cation, five crystallographically unique TCNQ anions (denoted as A, B, C, A′, and B′), and five MeCN molecules (Fig. S10†). The arrangement of cations shows a similar topology to that observed in structure 4, but the higher solvent content in 5 causes partial disruption of the π–π and σ–π interactions between the cations, making their packing to appear as double chains rather than uniform layers (Fig. 5b). The change in the crystal packing was also reflected in the crystal shape; the crystals of 4 formed as thick plates while the crystals of 5 appeared as long flat needles.
Similar to 4, the direction of TCNQ stacks in 5 alternates in the adjacent anionic layer (Fig. 4b). The stacks of first type consist of the TCNQA, TCNQB, and TCNQC anions that form triads with the repeating sequence [ABC] and interplanar distances of 3.359 and 3.346 Å within the triad and 3.790 Å between the triads. The second type of stacks is formed by triads of TCNQA′ and TCNQB′ anions, with the repeating sequence [A′B′B′] and the interplanar distances of 3.054, 3.354, and 3.410 Å.
According to the stoichiometry of 5, the average charge per TCNQδ− anion is −0.4. The charges calculated for the individual TCNQA, TCNQB, TCNQC, TCNQA′, and TCNQB′ anions are −0.40, −0.62, −0.82, −0.16, and −0.01, respectively. Thus, similar to the crystal structure of 4, the anionic charge distribution in the structure of 5 is rather non-uniform. Moreover, the TCNQA′ and TCNQB′ units are nearly neutral, which suggests that one half of the TCNQ stacks in 5 should not be providing effective charge-transport pathways.
Overall, both the crystal packing and stoichiometry of these FeII–TCNQ hybrid structures show very strong dependence on the size of the cationic complex and the content of interstitial solvent. The improvement in the cationic packing observed for the [Fe(bzimpy)2]2+-containing structures suggests that an optimization of these structures should be possible, to improve their stability and potential coupling between the structural, magnetic, and conducting properties.
Magnetic properties
Magnetic susceptibility (χ) was measured as a function of temperature (T) for polycrystalline samples of all complexes shown in Scheme 1, except for 3, which was obtained only in trace amounts. In addition, the magnetic properties of 1·5MeCN were also measured on a fresh sample covered with mother liquid and sealed in NMR tubes, to prevent any loss of interstitial solvent due to the slight vacuum present in the magnetometer sample chamber.The sample of 1·5MeCN under mother liquid showed a very gradual an incomplete SCO upon cooling from 300 to 5 K (the gray curve in Fig. 6). The χT value decreases from ∼3.0 emu K mol−1 at 300 K to a plateau of ∼2.5 emu K mol−1 at 50 K, suggesting that only ∼15% of the sample undergoes the temperature-driven HS → LS conversion.
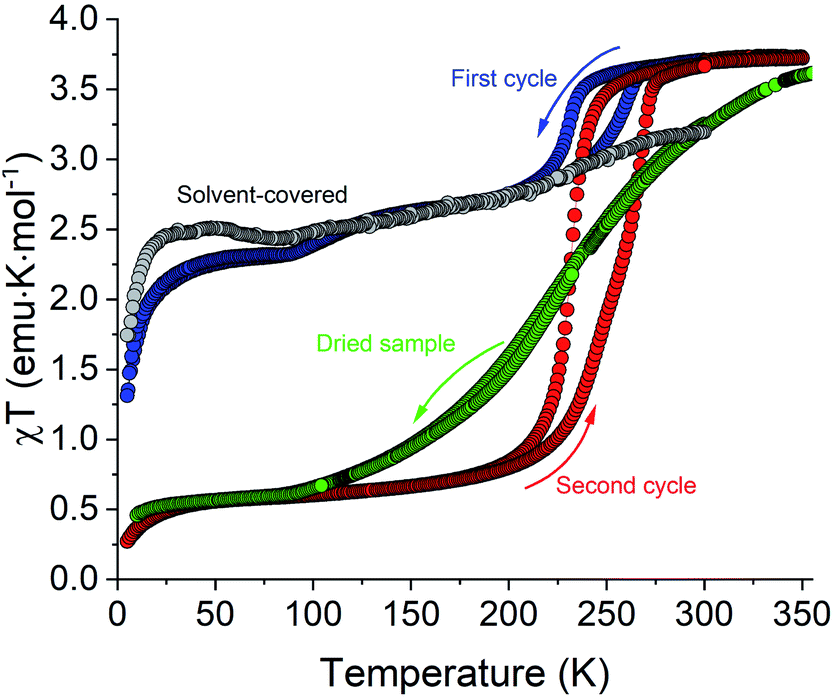 | ||
| Fig. 6 The temperature dependence of χT for complex 1·5MeCN covered with mother liquid (gray), filtered without prolonged drying (blue and red, corresponding to the first and second cooling–heating cycles, respectively), and dried under vacuum for 12 h (green). The rate of cooling and heating was 1 K min−1. | ||
Different magnetic behavior was observed for the sample of 1·5MeCN that was filtered and loaded in a polycarbonate capsule for magnetic measurements. This sample also showed the decrease in χT as the temperature was lowered from 350 K (blue curve in Fig. 6), but this decrease proceeded in two steps. First, the χT decreased from a high-temperature plateau of ∼3.7 emu K mol−1, corresponding to a fully populated HS state of the Fe(II) ion, to a plateau of ∼2.7 emu K mol−1 at ∼180 K, and then a more gradual decrease to a plateau of ∼2.3 emu K mol−1 at ∼70 K was observed, suggesting ∼40% HS → LS conversion. (The decrease in the χT value below 25 K is due to zero-field splitting effects.) Upon heating the sample back to 350 K, a thermal hysteresis was detected for the higher-temperature SCO stage, with the midpoints at T1/2,↓ = 230 K and T1/2,↑ = 261 K in the cooling and heating modes, respectively. In the second cooling–heating cycle, however, a single SCO step was observed, corresponding to a complete and hysteretic spin transition with T1/2,↓ = 231 K and T1/2,↑ = 269 K (the red curve in Fig. 6). The χT value decreased to a plateau of ∼0.5 emu K mol−1 in the low-temperature region, suggesting that either a small amount (∼10%) of the HS species still remains in the sample after the transition or a minor paramagnetic impurity is present.
The complete spin transition observed in the second cooling–heating cycle nearly coincided with the smaller hysteretic event seen in the first cycle (the blue curve). Thus, the behavior observed in the first cycle appears to be an additive result of the χT curve recorded for the sample covered with mother liquid and the χT curve recorded in the second cycle. Evidently, the behavior observed in the first cycle stems from the partial transformation of the fully solvated sample (1·5MeCN) to the sample with a lower solvent content, which subsequently exhibits the complete and hysteretic SCO. To test this hypothesis, a sample of 1·5MeCN was introduced in the magnetometer chamber and kept for 1 h at 300 K. The subsequent measurement revealed that this sample showed the same complete hysteretic SCO (Fig. S11†) as observed in the second cooling–heating cycle above.
To establish the extent of solvent loss that afforded the observation of the hysteretic spin transition, the sample obtained after the second cooling–heating cycle was extracted from the magnetometer chamber and subjected to TGA from r.t. to 473 K at the heating rate of 2 K min−1. The sample showed a solvent loss step of ∼4.1% by mass, estimated from the inflection point at 408 K (Fig. S12†), which corresponds to the loss of ∼1 MeCN molecule per f.u., assuming that a fully desolvated sample is obtained above this temperature. Despite its promising magnetic properties, the partially desolvated sample, which we henceforth denote as “1·MeCN”, has proven to be elusive. While the complete hysteretic SCO could be consistently achieved by exposing the fully solvated sample to the slight vacuum of the magnetometer chamber, the complete drying of 1·5MeCN by exposure to vacuum (∼10−1 mbar) at room temperature led to sample 1 that showed a complete but very gradual SCO spanning the temperature range of ∼200 K, with T1/2 ≈ 223 K (the green curve in Fig. 6).
The only case of a hysteretic SCO in a structure with fractionally charged TCNQδ− anions was reported in a MnIII complex, [Mn(5-Cl-sal-N-1,5,8,12)]TCNQ1.5·2MeCN (5-Cl-sal-N-1,5,8,12 = N,N′-bis(3-(2-oxy-5-chlorobenzylideneamino)propyl)-ethylenediamine), which showed a spin transition centered at 98 K, with a 50 K hysteresis.42 The partially desolvated 1·5MeCN provides the first example of such behavior in a structure assembled of FeII SCO cations and TCNQδ− anions. Complexes [Fe(tpma)(xbim)](X) (TCNQ)1.5·DMF (X = ClO4−, BF4−; tpma = tris(2-pyridylmethyl)amine, xbim = 1,1′-(α,α′-o-xylyl)-2,2′-biimidazole) also showed complete but gradual SCO, without thermal hysteresis.51
It is well established that the changes in the interstitial solvent content can lead to dramatic alteration of SCO behavior. For example, such changes were observed in the complexes of the [Fe(3-bpp)2]2+ cations with innocent counter ions, such as ClO4−, BF4−, or OTf−.44,52,53 The strong influence of the solvent content on the magnetic properties of such complexes is explained by the high sensitivity of SCO to structural distortions caused by changes in the intermolecular interactions.47 Thus, the changes in the SCO behavior upon progressive desolvation of 1·5MeCN can be explained by gradual modification of the crystal packing. The initial partial desolvation stabilizes the LS state and improves the cooperativity of intermolecular interactions, due to the closer approach of the SCO cations, thus promoting the hysteretic SCO. On the other hand, the complete desolvation, most likely, causes an increased structural disorder that leads to the very gradual SCO.
In the same vein, the solvent impact on magnetic properties was observed for 2·3MeCN. The solvated complex showed χT value of 3.50 emu K mol−1 at 300 K, which corresponds to the HS state of the FeII ion. As the temperature was lowered, the χT value remained nearly constant but decreased quickly below 150 K (the purple curve in Fig. 7a). A plateau of 1.75 emu K mol−1 reached at 90 K suggests that the low-temperature phase contains approximately equal amounts of the HS and LS FeII ions. This finding is in excellent agreement with the fraction of the HS state found from the average Fe–N bond length determined by X-ray crystallography (Table S1†).
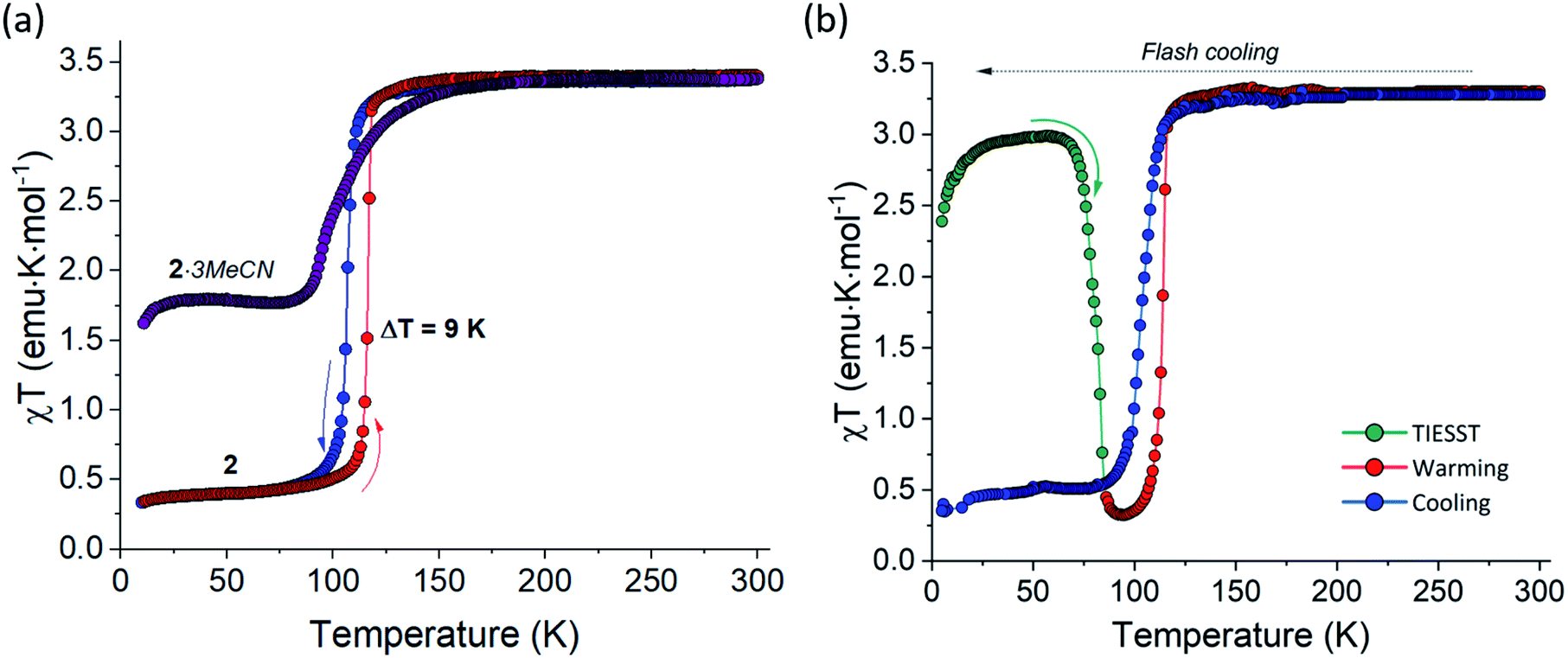 | ||
| Fig. 7 (a) The temperature dependence of χT for 2·3MeCN (purple) and 2 measured in cooling (blue) and warming (red) modes at 1 K min−1. (b) The temperature dependence of χT for 2 measured by flash-cooling the sample from room temperature to 10 K (indicated with the gray horizontal arrow), warming at 0.3 K min−1 (first teal, then red data points), and cooling at 1 K min−1 (blue data points). The low-temperature part of the curve measured in the warming mode is shown with teal color to emphasize the TIESST effect. See the main text for the more detailed explanation of these measurements. | ||
As shown by the TGA results and X-ray crystallography, 2·3MeCN can be completely desolvated without the loss of crystallinity. So obtained desolvated sample 2 exhibits an abrupt and hysteretic temperature-driven spin transition with T1/2,↓ = 106 K and T1/2,↑ = 117 K in the consecutive cooling and heating cycles (blue and red curves, respectively, in Fig. 7a). Thus, the loss of the interstitial solvent leads to both complete and much more abrupt SCO as compared to the behavior observed for 2·3MeCN. Such change is well explained by the closer approach and stronger interactions between the SCO cations in the crystal structure of 2 (Fig. S7†).
While the hysteretic SCO in 2 was observed under the cooling and heating rate of 1 or 2 K min−1, flash-cooling the sample of 2 to 10 K in the magnetometer chamber resulted in quenching of the metastable HS state. This phenomenon is commonly known as the temperature-induced excited spin-state trapping (TIESST).54,55 Warming the sample from 10 K at 0.3 K min−1 revealed that the trapping of the HS state was nearly quantitative; the χT plateau of 3.00 emu K mol−1 observed between 25 and 70 K (the green curve in Fig. 7b) indicates the fraction of the HS state equal to ∼85%. The efficient TIESST stems from the relatively low temperature of the thermal spin transition observed in 2, a pre-requisite noted for other complexes with such behavior.56,57 The trapped HS state remained stable at lower temperatures, but decayed rapidly to the ground LS state when the temperature exceeded 75 K. The characteristic relaxation temperature, TTIESST = 85 K, was determined from the minimum of the d(χT)/dT dependence. This TTIESST value is not as high as those reported for some other SCO complexes of the FeII ion,58–63 but this is the first reported case of the TIESST behavior in hybrid TCNQ-containing SCO materials. Further heating to 300 K followed by cooling to 5 K, both at 2 K min−1, resulted in the thermal spin transition with hysteresis (the red and blue curves, respectively, in Fig. 7b).
Materials with abrupt thermal spin transitions and TIESST almost always exhibit the LIESST effect,64 which proceeds via spin-allowed photoexcitation from the ground LS state followed by intersystem crossing to the HS state. If the irradiation is performed at sufficiently low temperature, the metastable HS state can become trapped, similar to the TIEEST effect. We were unable to observe the LIESST behavior for either 2·3MeCN or 2. A possible reason for the lack of LIESST might be the very dark color of the materials that causes rapid absorption of light provided by a moderate-power diode laser, thus leading to a low penetration depth. In the nearest future, we will attempt to repeat these experiments with a higher-power laser irradiation.
Despite the lack of the LIESST behavior, the X-ray crystal structure determination suggests that complex 2 might show X-ray induced excited spin state trapping (XIESST). Indeed, while the magnetic data demonstrated unequivocally the complete HS → LS transition for this complex below 100 K, the crystal structure determination at 90 and 100 K afforded average Fe–N bond lengths indicative of a substantial fraction of the HS state (∼40% and ∼45%, respectively). It is well known that the LIESST relaxation temperature is increased if an SCO material is warmed under continuous laser irradiation.18,65 Given the high intensity of the microfocus Cu-Kα source used in the X-ray diffraction experiments and the high TTIESST (85 K) established for complex 2, we believe the reason for the large HS fraction observed in the crystal structures determined at 90 and 100 K is the X-ray induced excitation from the LS state to the metastable HS state. (Unfortunately, the diffraction data obtained with the less powerful Mo-Kα source were of insufficient intensity for the crystal structure determination.) This hypothesis can be confirmed by X-ray crystal structure analysis below TTIESST, but that temperature range was not accessible in our experiments. Nevertheless, the hypothesis is supported by the other reported precedents of the XIESST effect.66–68 We stress that, in the case of complex 2, such an effect is made possible by the high value of TTIESST (which should be similar to TXIESST) and the proximity of TTIESST to the thermally induced spin transition.
Complexes 4 and 5 showed diamagnetic behavior from 300 to 400 K (Fig. S13†), corresponding to the LS state of the FeII ion. A parent complex, [Fe(bzimpy)2](ClO4)2·0.25H2O, was reported to undergo a hysteretic spin transition with T1/2,↓ = 398 K and T1/2,↑ = 406 K, while [Fe(bzimpy)2](BPh4)2·4H2O showed a gradual SCO with T1/2,↑ = 310 K.45 The lack of SCO in complexes 4 and 5 might be explained by changes in the crystal packing that lead to the increased stability of the LS state. Further exploration of such complexes with bzimpy or related ligands appears to be a promising avenue for the preparation of stable crystalline phases combining SCO and conductivity.
Transport properties
The electrical resistance was measured on several crystals of 1·5MeCN, using a four-probe method. The current was applied along the longest axis of the single crystal, i.e., in the direction coinciding with the propagation of the TCNQ stacks in the crystal structure. To minimize the solvent loss during the measurements, half of the samples studied were covered with N-grease, while the other half was not covered with N-grease for the sake of comparison. Then the sample holder was moved into a cryostat and subjected to vacuum. Unfortunately, such process can still lead to the loss of interstitial solvent, despite the protection of the sample with grease, and, therefore, we refer to these samples as 1·xMeCN, to indicate the variability of the solvent content from one crystal to another.All crystals of 1·xMeCN showed increasing resistance (R) upon cooling (Fig. 8a), indicating semiconducting behavior. The typical room-temperature conductivity value (σRT) was equal to 3.1 × 10−3 S cm−1, falling within a common range for semiconducting materials. The conductivity value for the samples not covered with grease was found to be two orders of magnitude lower, which confirms the detrimental effect of solvent loss on the transport properties. The samples became very resistive below ∼140 K, causing the abundant noise in the low-temperature data, as the resistance became too high to be measured in the available dynamic range.
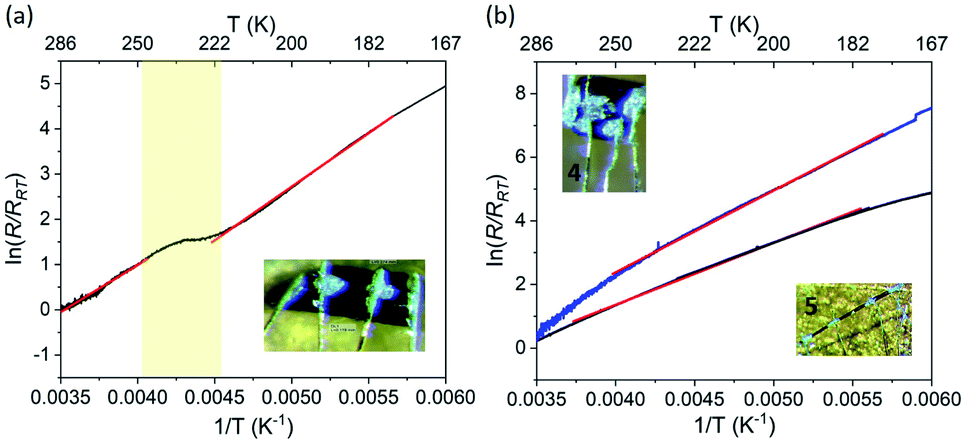 | ||
| Fig. 8 (a) The temperature dependence of the log of normalized electrical resistance measured on single crystals of 1·xMeCN (a) and 4 and 5 (b). The crystals used for measurements are shown in the insets of the corresponding panels. The solid red lines show the fit to the Arrhenius equation. The region of hysteretic SCO observed for 1·MeCN is highlighted with pale-yellow. | ||
The most striking feature in the resistance behavior of 1·xMeCN is the anomaly seen as an inflection point in the temperature range that coincides with the hysteretic SCO observed for the partially desolvated sample, 1·MeCN, by magnetic measurements (Fig. 6). Thus, we can conclude that the grease-protected sample should be similar in its composition to the sample obtained by the partial loss of the interstitial solvent in the magnetometer chamber. The linear regime of the normalized resistance, observed in the temperature regions above and below the inflection point (Fig. 8a), was fit to the Arrhenius equation, (R/RRT) = eEa/kT, yielding the activation energy Ea = 179(6) and 190(2) meV, respectively, for the HS and LS states of the sample. Such change in the activation energy across the SCO region was also observed previously and attributed to the modification of the interactions between the organic moieties caused by changes in the crystal packing due to SCO.31,38
In contrast to 1·xMeCN, complexes 2·3MeCN and 2 lack conductive pathways, since their structures show dimerization of integer-charged TCNQ− anions. Thus, these materials are expected to show high resistance, which we indeed observed experimentally. The studies performed on a single crystal of 2 revealed σRT = 4.3 × 10−6 S cm−1.
Despite the LS behavior of complexes 4 and 5, they are expected to show appreciable conductivity, based on the presence of stacks of fractionally charged TCNQδ− anions in their crystal structures. The measurements performed on single crystals of these materials revealed σRT = 9.1 × 10−2 S cm−1 and 1.8 × 10−3 S cm−1, respectively, which are of the same order of magnitude as the conductivity observed for 1·xMeCN. Samples 4 and 5 also showed semiconducting behaviour, with the activation energies of 191(4) and 172(3) meV, respectively, obtained by the Arrhenius fit to the linear region of the normalized resistance (Fig. 8b).
Concluding remarks
As described in the Introduction, a number of materials combining cationic SCO complexes with fractionally charged metal–dithiolene or TCNQ-based anions have been reported in the literature. This work, however, represents the first systematic attempt to assess the influence of the relative size and shape of the cation, the charge on the TCNQδ− anion, and the interstitial solvent content on the structural organization, stability, and magnetic properties of such complexes, as summarized in Table 2. For the sake of completeness and comparison, this table also lists the properties of all conducting SCO materials reported to date. Based on the observations made in this work, several generalizations can be made:| Complexa | T 1/2 (K) | σ RT (S cm−1) | −δavg per radical-anion | Reference |
|---|---|---|---|---|
| a Abbreviations for the radical anions: mnt = maleonitriledithiolate, dmit = 1,3-dithia-2-thione-4,5-dithiolate; the abbreviations and molecular structures of the blocking ligands used in specific complexes can be found in the corresponding references. b In the cases when SCO is incomplete, the residual percentage of the HS state observed below 50 K is indicated. For complex 1·5MeCN, the T1/2 value cannot be defined because the SCO is very gradual and the extent of the HS → LS conversion is only 20%. c The values were obtained by extrapolation. | ||||
| [FeII(3-bpp)2](TCNQ)3·5MeCN (1·5MeCN) | N/A (80% HS) | — | −0.67 | This work |
| [FeII(3-bpp)2](TCNQ)3·MeCN (1·MeCN) | 231–269 | 3.1 × 10−3 | −0.67 | This work |
| [FeII(3-bpp)2](TCNQ)3 (1) | 223 | — | −0.67 | This work |
| [FeII(3-bpp)2](TCNQ)2·3MeCN (2·3MeCN) | 100 (50% HS) | — | −1 | This work |
| [FeII(3-bpp)2](TCNQ)2 (2) | 106–117 | 4.3 × 10−6 | −1 | This work |
| [FeII(bzimpy)2](TCNQ)6·2Me2CO (4) | LS | 9.1 × 10−2 | −0.4 | This work |
| [FeII(bzimpy)2](TCNQ)5·5MeCN (5) | LS | 1.8 × 10−3 | −0.33 | This work |
| [FeII(tpma)(xbim)](TCNQ)1.5(ClO4)·DMF | 145 | 2.0 × 10−1 | −0.67 | 38 |
| [FeII(tpma)(xbim)](TCNQ)1.5(BF4)·DMF | 160 | n/a | −0.67 | 38 |
| [FeII(HC(pz)3)2](TCNQ)3 | 445c | 1.5 × 10−2 | −0.67 | 41 |
| [CoII(terpy)2](TCNQ)3·MeCN | >400c | 1.3 × 10−1 | −0.67 | 39 |
| [CoII(terpy)2](TCNQ)4·3DMF·0.5H2O | >400c | 3.3 × 10−4 | −0.5 | 40 |
| [MnIII(5-Cl-sal-N-1,5,8,12)]TCNQ1.5·2MeCN | 73/123 | 1.9 × 10−4 | −0.67 | 42 |
| [FeII(dppTTF)2][Ni(mnt)2]2(BF4)·PhCN | 240 | 2.6 × 10−3 | −0.5 | 31 |
| [FeIII(qnal)2][Pd(dmit)2]5·Me2CO | 220 | 1.6 × 10−2 | −0.2 | 36 |
| [FeIII(salEen)2]2[Ni(dmit)2]5·6MeCN | 220 | 1.2 × 10−1 | −0.4 | 35 |
| [FeIII(qsal)2][Ni(dmit)2]3·MeCN·H2O | 160 | 2.0 | −0.33 | 37 |
(1) The fractional charge on the TCNQδ− anions is paramount to promoting the formation of rather uniform 1D stacks of anions that provide efficient conducting pathways. Unfortunately, the scarcity of examples of such materials reported to date does not allow drawing any correlations between the average charge on the anion (−δ) and the conducting properties at this time.
(2) The anionic stacks consistently pack in layers that alternate with layers of SCO cations. Such structural organization is observed in nearly all structures of transition metal cations co-crystallized with fractionally charged TCNQδ− anions.
(3) Depending on the size of the ligands, the packing of the cations can be rather loose, as shown by the examples of hybrid structures formed with the [Fe(3-bpp)2]2+ cations in this work. As a result, a large amount of interstitial solvent can be present in the cationic layer.
(4) The incorporation of the interstitial solvent molecules disrupts interactions between the cations, lowering the cooperativity of the SCO. A more subtle affect is the influence of varying crystal-packing forces on the distortion of coordination environment around the transition metal ion. It is well established, in general, that such changes in cooperativity and coordination geometry can lead to strong variation in the character and temperature of SCO.
(5) The packing of cations can be optimized by changing the size of the ligand, to enhance the intermolecular contacts and expel the interstitial solvent from cationic layers. The effectiveness of such a strategy has been demonstrated with the [Fe(bzimpy)2]2+ cations, which form cationic layers with much more extensive π–π and σ–π interactions, thus increasing the stability of the crystals. If SCO within the accessible temperature range can be achieved with such structure of the cation, we predict that an abrupt and hysteretic spin transition will be observed.
These general principles provide guidance to improving the design of such hybrid structures and achieving more stable conducting materials with abrupt spin transitions. We would like to emphasize, once again, the importance of the complete structural characterization to improve the understanding of the structure–property relationships in such materials. To date, the scope of structurally characterized SCO complexes with appreciable conductivity remains rather limited. Our future efforts will focus on modifying the ligand field strength of bzimpy by minor substitutions into the aromatic rings, with the goal to preserve the efficient crystal packing of cations, shift the spin transition to lower temperatures, and achieve stronger coupling between SCO and conducting properties.
Materials and methods
Starting materials
All reactions were performed under an inert N2 atmosphere using standard Schlenk techniques, unless stated otherwise. [Fe(H2O)6](BF4)2 (Sigma-Aldrich) and TCNQ (TCI) were used as received. Literature procedures were followed to synthesize 2,6-di-(1H-pyrazol-3-yl)pyridine (3-bpp),69 2,6-bis(benzimidazol-2′-yl)pyridine (bzimpy),70 Et3NH(TCNQ)2,71 and Bu4N(TCNQ).72 Anhydrous acetonitrile (MeCN) and diethyl ether (Et2O) were obtained as HPLC or ACS reagent grade solvents and further purified by passing through a system of double-drying columns packed with activated alumina and molecular sieves (Glass Contour Inc.). Other anhydrous solvents were purchased in SureSeal® bottles from Sigma-Aldrich and used as received. Elemental analysis was performed by Atlantic Microlab, Inc. (Norcross, GA, USA).Physical measurements
Thermal gravimetric analysis was performed on the TA instruments Q50 Thermogravimetric Analyzer, in the temperature range from 25 to 500 °C, at a heating rate of 2 or 5 °C min−1. Infrared (IR) spectra were acquired in the range of 4000–400 cm−1 using JASCO 6800 FT-IR spectrometer.[Fe(3-bpp)2](TCNQ)3·3MeCN (1·5MeCN)
To a solution of 38.3 mg (0.075 mmol) of Et3NH(TCNQ)2 in 5 mL of MeCN was slowly added a solution of 16.3 mg (0.025 mmol) of [Fe(3-bpp)2](BF4)2 in 2 mL of MeCN. The resulting bilayer solution was left undisturbed for 2 days at 7 °C, affording dark-green crystals that were recovered by filtration. Yield = 20.0 mg (66%). Keeping the complex under vacuum for 12 h afforded a desolvated phase, [Fe(3-bpp)2](TCNQ)3 (1). Elem. analysis: calcd (found) for FeC58N22H34O2 (1a·2H2O), %: C 61.82 (61.71), H 3.04 (3.23), N 27.34 (27.17). Elemental analysis shows that storing the complex in air leads to the loss of MeCN molecules and absorption of H2O molecules. IR (cm−1), ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N): 2173, 2205, 2258; ν(C
N): 2173, 2205, 2258; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N): 1510; ν(C–H): 828. TGA: calcd (found) mass loss, %: 4.75 (4.70) at 100 °C for the loss of 1.5MeCN per f.u.
N): 1510; ν(C–H): 828. TGA: calcd (found) mass loss, %: 4.75 (4.70) at 100 °C for the loss of 1.5MeCN per f.u.
[Fe(3-bpp)2](TCNQ)2·3MeCN (2·3MeCN)
To a solution of 22.3 mg (0.050 mmol) of Bu4N(TCNQ) in 4 mL of MeCN was slowly added a solution of 16.3 mg (0.025 mmol) of [Fe(3-bpp)2](BF4)2 in 2 mL of MeCN. The bilayer solution was left undisturbed for 2 days at 7 °C, affording dark-green crystals that were recovered by filtration. Yield = 16.2 mg (73%). Keeping the complex under vacuum for 12 h or heating at 127 °C for 1 h afforded a desolvated phase, [Fe(3-bpp)2](TCNQ)2 (2). Elem. analysis: calcd (found) for FeC46N18H26: (2·3MeCN), %: C 62.31 (62.08), H 2.96 (2.98), N 28.43 (28.29). IR (cm−1), ν(CN): 2150, 2183, 2267; ν(CN): 1503; ν(C–H): 840. TGA: calcd (found) mass loss, %: 12.2 (13.6) at 100 °C for the loss of 3MeCN per f.u.
[Fe(3-bpp)2](TCNQ)4·3MeCN (3)
To a solution of 22.3 mg (0.050 mmol) of Et3NH(TCNQ)2 in 4 mL of MeCN was slowly added a solution of 16.3 mg (0.025 mmol) of [Fe(3-bpp)2](ClO4)2 in 2 mL of MeCN. The resulting bilayer solution was left undisturbed for 2 days at 7 °C, affording a mixture of dark-green thin-plate-shaped crystals and thick-plate-like crystals that were recovered by filtration. The elemental analysis could not be performed because the crystals of 3 were obtained only as a minor impurity phase.[Fe(bzimpy)2](TCNQ)6·2Me2CO (4)
To a solution of 38.3 mg (0.075 mmol) of Et3NH(TCNQ)2 in 4 mL of Me2CO and 0.5 mL DMF was slowly added a solution of 28.5 mg (0.025 mmol) of [Fe(bzimpy)2](ClO4)2 in 2 mL of Me2CO. The resulting solution was left undisturbed for 4 days at 7 °C, affording black block-like crystals that were recovered by filtration. Yield = 25.1 mg (65%). Elem. analysis: calcd (found) for FeC110N34H50 (fully desolvated 4), %: C 69.4 (69.12), H 2.65 (2.90), N 25.02 (24.80). IR (cm−1), ν(CN): 2204, 2186, 2170; ν(CN): 1503; ν(C–H): 827. TGA: calcd (found) mass loss, %: 1.80 (1.60) at 123 °C for the loss of 0.5Me2CO per f.u.
[Fe(bzimpy)2](TCNQ)5·5MeCN (5)
To a solution of 38.3 mg (0.075 mmol) of Et3NH(TCNQ)2 in 5 mL of MeCN and was slowly added a solution of 28.5 mg (0.025 mmol) of [Fe(bzimpy)2](ClO4)2 in 3 mL of MeCN. The mixture was stirred for 10 min, then filtered. The resulting solution was left undisturbed for 4 days at 7 °C, affording dark needle-like crystals that were recovered by filtration. Yield = 19.2 mg (40%). Elem. analysis: calcd. (found) for FeC98N30H46 (fully desolvated 5), %: C 69.26 (68.85), H 2.73 (2.96), N 24.73 (24.01). IR (cm−1), ν(CN): 2202, 2195, 2154; ν(CN): 1518; ν(C–H): 829. TGA: calcd (found) mass loss, %: 5.14 (5.70) at 110 °C for the loss of 2MeCN per f.u.
Magnetic measurements
Magnetic properties were measured on polycrystalline samples, using a superconducting quantum interference device (SQUID) magnetometer MPMS-XL (Quantum Design). Magnetic susceptibility was measured in a direct-current applied magnetic field of 1000 Oe in the 5–400 K temperature range, at cooling and heating rates of 1 K min−1. The data were corrected for the diamagnetic contribution from the sample holder and for the intrinsic diamagnetism using tabulated constants.73Conductivity measurements
Electrical resistance was measured on selected single crystals, with representative sizes of 0.78 × 0.57 × 0.10 mm3 for 1, 0.52 × 0.37 × 0.10 mm3 for 2, 0.27 × 0.24 × 0.08 mm3 for 4, and 1.50 × 0.50 × 0.10 mm3 for 5. A four-probe measurement setup was used, with the current applied parallel to the longest direction of the crystal. The direct current was applied by means of a Keithley 6221 source while a Keithley 2182 nanovoltmeter or a Keithley 6517A electrometer was used to measure the voltage.X-ray crystallography
Single-crystal X-ray diffraction was performed on a Rigaku-Oxford Diffraction Synergy-S diffractometer equipped with a HyPix detector and a monochromated Mo-Kα radiation source (λ = 0.71073 Å). A chosen single crystal was suspended in Parabar® oil (Hampton Research) and mounted on a cryoloop, which was cooled to the desired temperature in an N2 cold stream. The data set was recorded as ω-scans at 0.3° step width and integrated with the CrysAlis software package, which was also used for space group determination.74 Empirical adsorption correction was applied based on spherical harmonics as implemented in the SCALE3 ABSPACK algorithm.75 The crystal structure solution and refinement were carried out with SHELX76 using the interface provided by Olex2.77 The final refinement was performed with anisotropic atomic displacement parameters for all non-hydrogen atoms, except for some strongly disordered solvent molecules, which were refined isotropically. All H atoms were placed in calculated positions and refined in the riding model. Full details of the crystal structure refinement and the final structural parameters have been deposited with the Cambridge Crystallographic Data Centre (CCDC). The CCDC registry numbers and a brief summary of data collection and refinement parameters are provided in Tables S1 and S2.†Data availability
Data reported in this article are available on demand by contacting the corresponding author.Author contributions
Ö. Ü. – methodology, investigation, formal analysis, validation, visualization, writing – original draft. E. S. C. – investigation, formal analysis, writing – review & editing. M. S. – conceptualization, methodology, formal analysis, validation, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (award CHE-1955754 to M. S.) The Rigaku Synergy-S single-crystal X-ray diffractometer used for crystallographic work was acquired through the NSF MRI program (award CHE-1828362). This research also used resources provided by the X-ray Crystallography Center (FSU075000XRAY) and the Materials Characterization Laboratory (FSU075000MAC) at the FSU Department of Chemistry and Biochemistry. A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by the NSF Cooperative Agreement No. DMR-1644779 and the State of Florida.Notes and references
- P. Gütlich and H. A. Goodwin, Spin crossover – an overall perspective, Top. Curr. Chem., 2004, 233, 1–47 CrossRef
.
-
K. S. Murray, in Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons Ltd., 2013, pp. 1–54 Search PubMed
- A. Hauser, Ligand field theoretical considerations, Top. Curr. Chem., 2004, 233, 49–58 CrossRef CAS
- S. Brooker, Spin crossover with thermal hysteresis: practicalities and lessons learnt, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC
- B. Weber, W. Bauer and J. Obel, An iron(II) spin-crossover complex with a 70 K wide thermal hysteresis loop, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS
- M. A. Halcrow, Spin-crossover compounds with wide thermal hysteresis, Chem. Lett., 2014, 43, 1178–1188 CrossRef CAS
- V. García-López, M. Palacios-Corella, S. Cardona-Serra, M. Clemente-León and E. Coronado, Spin-crossover iron(II) complex showing thermal hysteresis around room temperature with symmetry breaking and an unusually high T(LIESST) of 120 K, Chem. Commun., 2019, 55, 12227–12230 RSC
- H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnár, J.-F. Létard and A. Bousseksou, Antagonism between extreme negative linear compression and spin crossover in [Fe(dpp)2(NCS)2]·py, Angew. Chem., Int. Ed., 2012, 51, 3910–3914 CrossRef CAS PubMed
- D. Pinkowicz, M. Rams, M. Mišek, K. V. Kamenev, H. Tomkowiak, A. Katrusiak and B. Sieklucka, Enforcing multifunctionality: a pressure-induced spin-crossover photomagnet, J. Am. Chem. Soc., 2015, 137, 8795–8802 CrossRef CAS
- S. P. Vallone, A. N. Tantillo, A. M. dos Santos, J. J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Giant barocaloric effect at the spin crossover transition of a molecular crystal, Adv. Mater., 2019, 31, 1807334 CrossRef
- L. J. Kershaw Cook, F. L. Thorp-Greenwood, T. P. Comyn, O. Cespedes, G. Chastanet and M. A. Halcrow, Unexpected spin-crossover and a low-pressure phase change in an iron(II)/dipyrazolylpyridine complex exhibiting a high-spin Jahn–Teller distortion, Inorg. Chem., 2015, 54, 6319–6330 CrossRef CAS PubMed
- Y. Garcia, V. Ksenofontov, G. Levchenko, G. Schmitt and P. Gütlich, Pressure-induced high spin state in [Fe(btr)2(NCS)2]·H2O (btr = 4,4′-bis-1,2,4-triazole), J. Phys. Chem. B, 2000, 104, 5045–5048 CrossRef CAS
- G. A. Craig, J. S. Costa, O. Roubeau, S. J. Teat, H. J. Shepherd, M. Lopes, G. Molnár, A. Bousseksou and G. Aromí, High-temperature photo-induced switching and pressure-induced transition in a cooperative molecular spin-crossover material, Dalton Trans., 2014, 43, 729–737 RSC
- O. Sato, J. Tao and Y. Z. Zhang, Control of magnetic properties through external stimuli, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS
- S. Ohkoshi and H. Tokoro, Photomagnetism in cyano-bridged bimetal assemblies, Acc. Chem. Res., 2012, 45, 1749–1758 CrossRef CAS PubMed
- Y. Jiang, L. C. Liu, H. M. Mueller-Werkmeister, C. Lu, D. Zhang, R. L. Field, A. Sarracini, G. Moriena, E. Collet and R. J. D. Miller, Structural dynamics upon photoexcitation in a spin crossover crystal probed with femtosecond electron diffraction, Angew. Chem., Int. Ed., 2017, 56, 7130–7134 CrossRef CAS
- S. Marcén, L. Lecren, L. Capes, H. A. Goodwin and J. F. Létard, Critical temperature of the LIESST effect in a series of hydrated and anhydrous complex salts [Fe(bpp)2]X2, Chem. Phys. Lett., 2002, 358, 87–95 CrossRef
- J. F. Létard, Photomagnetism of iron(II) spin crossover complexes-the T(LIESST) approach, J. Mater. Chem., 2006, 16, 2550–2559 RSC
- S. Hayami, Z.-z. Gu, Y. Einaga, Y. Kobayasi, Y. Ishikawa, Y. Yamada, A. Fujishima and O. Sato, A novel LIESST iron(II) complex exhibiting a high relaxation temperature, Inorg. Chem., 2001, 40, 3240–3242 CrossRef CAS PubMed
- J. S. Costa, C. Balde, C. Carbonera, D. Denux, A. Wattiaux, C. Desplanches, J.-P. Ader, P. Gütlich and J.-F. Létard, Photomagnetic properties of an iron(II) low-spin complex with an unusually long-lived metastable LIESST state, Inorg. Chem., 2007, 46, 4114–4119 CrossRef PubMed
-
Multifunctional Molecular Materials, ed. L. Ouahab, Pan Stanford Publishing, Singapore, 2013 Search PubMed
-
O. Sato, Z.-Y. Li, Z.-S. Yao, S. Kang and S. Kanegawa, in Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons Ltd., 2013, pp. 303–319 Search PubMed
- A. B. Gaspar, V. Ksenofontov, M. Seredyuk and P. Guetlich, Multifunctionality in spin crossover materials, Coord. Chem. Rev., 2005, 249, 2661–2676 CrossRef CAS
- Y. S. Koo and J. R. Galan-Mascaros, Spin crossover probes confer multistability to organic conducting polymers, Adv. Mater., 2014, 26, 6785–6789 CrossRef CAS
-
Organic Conductors: Fundamentals and Applications, ed. J.-P. Farges, CRC Press, New York, 1994 Search PubMed
- S. Schönfeld, K. Dankhoff, D. Baabe, M.-K. Zaretzke, M. Bröring, K. Schötz, A. Köhler, G. Hörner and B. Weber, Iron(II) spin crossover complexes based on a redox active equatorial Schiff-base-like ligand, Inorg. Chem., 2020, 59, 8320–8333 CrossRef PubMed
- S. Tiaouinine, J. Flores Gonzalez, B. Lefeuvre, T. Guizouarn, M. Cordier, V. Dorcet, L. Kaboub, O. Cador and F. Pointillart, Spin crossover and field-induced single-molecule magnet behaviour in Co(II) complexes based on terpyridine with tetrathiafulvalene analogues, Eur. J. Inorg. Chem., 2021, 2021, 2374–2383 CrossRef
- A. Kawamura, J. Xie, J.-N. Boyn, K. A. Jesse, A. J. McNeece, E. A. Hill, K. A. Collins, J. A. Valdez-Moreira, A. S. Filatov, J. W. Kurutz, D. A. Mazziotti and J. S. Anderson, Reversible switching of organic diradical character via iron-based spin-crossover, J. Am. Chem. Soc., 2020, 142, 17670–17680 CrossRef CAS
- Y.-R. Qiu, L. Cui, P.-Y. Cai, F. Yu, M. Kurmoo, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Enhanced dielectricity coupled to spin-crossover in a one-dimensional polymer iron(II) incorporating tetrathiafulvalene, Chem. Sci., 2020, 11, 6229–6235 RSC
- F. Pointillart, X. Liu, M. Kepenekian, B. Le Guennic, S. Golhen, V. Dorcet, T. Roisnel, O. Cador, Z. You, J. Hauser, S. Decurtins, L. Ouahab and S. X. Liu, Thermal and near-infrared light induced spin crossover in a mononuclear iron(II) complex with a tetrathiafulvalene-fused dipyridophenazine ligand, Dalton Trans., 2016, 45, 11267–11271 RSC
- M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Spin-crossover behavior and electrical conduction property in iron(II) complexes with tetrathiafulvalene moieties, Dalton Trans., 2011, 40, 2154–2156 RSC
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J.-L. Zuo, Photo- and electronically switchable spin-crossover iron(II) metal–organic frameworks based on a tetrathiafulvalene ligand, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS PubMed
- S. Dorbes, L. Valade, J. A. Real and C. Faulmann, [Fe(sal2-trien)][Ni(dmit)2]: towards switchable spin crossover molecular conductors, Chem. Commun., 2005, 69–71 RSC
- C. Faulmann, S. Dorbes, B. Garreau de Bonneval, G. Molnar, A. Bousseksou, C. J. Gomez-Garcia, E. Coronado and L. Valade, Towards molecular conductors with a spin-crossover phenomenon: crystal structures, magnetic properties and Mössbauer spectra of [Fe(salten)Mepepy][M(dmit)2] complexes, Eur. J. Inorg. Chem., 2005, 3261–3270 CrossRef CAS
- C. Faulmann, K. Jacob, S. Dorbes, S. Lampert, I. Malfant, M. L. Doublet, L. Valade and J. A. Real, Electrical conductivity and spin crossover: a new achievement with a metal bis dithiolene complex, Inorg. Chem., 2007, 46, 8548–8559 CrossRef CAS PubMed
- K. Takahashi, H. B. Cui, Y. Okano, H. Kobayashi, H. Mori, H. Tajima, Y. Einaga and O. Sato, Evidence of the chemical uniaxial strain effect on electrical conductivity in the spin-crossover conducting molecular system: [FeIII(qnal)2][Pd(dmit)2]5·acetone, J. Am. Chem. Soc., 2008, 130, 6688–6689 CrossRef CAS PubMed
- K. Takahashi, H. B. Cui, Y. Okano, H. Kobayashi, Y. Einaga and O. Sato, Electrical conductivity modulation coupled to a high-spin-low-spin conversion in the molecular system [FeIII(qsal)2][Ni(dmit)2]3·CH3CN·H2O, Inorg. Chem., 2006, 45, 5739–5741 CrossRef CAS PubMed
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Photomagnetic response in highly conductive iron(II) spin-crossover complexes with TCNQ radicals, Angew. Chem., Int. Ed., 2015, 54, 823–827 CrossRef CAS
- X. Zhang, Z.-X. Wang, H. Xie, M.-X. Li, T. J. Woods and K. R. Dunbar, A cobalt(II) spin-crossover compound with partially charged TCNQ radicals and an anomalous conducting behavior, Chem. Sci., 2016, 7, 1569–1574 RSC
- J.-Y. Zhang, L.-J. Su, Q.-J. Guo and J. Tao, Semiconducting spin-crossover cobalt(II) compound with non-integer charge distribution among TCNQ radicals, Inorg. Chem. Commun., 2017, 82, 39–43 CrossRef CAS
- Y. N. Shvachko, D. V. Starichenko, A. V. Korolyov, E. B. Yagubskii, A. I. Kotov, L. I. Buravov, K. A. Lyssenko, V. N. Zverev, S. V. Simonov, L. V. Zorina, O. G. Shakirova and L. G. Lavrenova, The conducting spin-crossover compound combining Fe(II) cation complex with TCNQ in a fractional reduction state, Inorg. Chem., 2016, 55, 9121–9130 CrossRef CAS PubMed
- A. V. Kazakova, A. V. Tiunova, D. V. Korchagin, G. V. Shilov, E. B. Yagubskii, V. N. Zverev, S. C. Yang, J.-Y. Lin, J.-F. Lee, O. V. Maximova and A. N. Vasiliev, The first conducting spin-crossover compound combining a MnIII cation complex with electroactive TCNQ demonstrating an abrupt spin transition with a hysteresis of 50 K, Chem.–Eur. J., 2019, 25, 10204–10213 CrossRef CAS PubMed
- M. A. Halcrow, The synthesis and coordination chemistry of 2,6-bis(pyrazolyl)pyridines and related ligands – versatile terpyridine analogues, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS
- S. A. Barrett, C. A. Kilner and M. A. Halcrow, Spin-crossover in [Fe(3-bpp)2][BF4]2 in different solvents – a dramatic stabilisation of the low-spin state in water, Dalton Trans., 2011, 40, 12021–12024 RSC
- B. Papánková, M. Vrbová, R. Boča, P. Šimon, K. Falk, G. Miehe and H. Fuess, DSC monitoring of the spin crossover in Fe(II) complexes, J. Therm. Anal. Calorim., 2002, 67, 721–731 CrossRef
-
H. Endres, in Extended Linear Chain Compounds, ed. J. S. Miller, Plenum, 1983, vol. 3, pp. 263–317 Search PubMed
- M. A. Halcrow, I. Capel Berdiell, C. M. Pask and R. Kulmaczewski, Relationship between the molecular structure and switching temperature in a library of spin-crossover molecular materials, Inorg. Chem., 2019, 58, 9811–9821 CrossRef CAS
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and D. O. Cowan, Structure of the red, semiconducting form of 4,4′,5,5′-tetramethyl-Δ2,2′-bi-1,3-diselenole-7,7,8,8-tetracyano-p-quinodimethane, TMTSF-TCNQ, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1193–1199 CrossRef
- Ö. Üngör and M. Shatruk, Transition metal complexes with fractionally charged TCNQ radical anions as structural templates for multifunctional molecular conductors, Polyhedron, 2020, 177, 114254 CrossRef
- P. Van der Sluis and A. L. Spek, BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 194–201 CrossRef CAS
- H. V. Phan, P. Chakraborty, M. M. Chen, Y. M. Calm, K. Kovnir, L. K. Keniley, J. M. Hoyt, E. S. Knowles, C. Besnard, M. W. Meisel, A. Hauser, C. Achim and M. Shatruk, Heteroleptic FeII complexes of 2,2′-biimidazole and its alkylated derivatives: spin-crossover and photomagnetic behavior, Chem.–Eur. J., 2012, 18, 15805–15815 CrossRef CAS PubMed
- G. A. Craig, J. S. Costa, O. Roubeau, S. J. Teat and G. Aromí, Local coordination geometry and spin state in novel FeII complexes with 2,6-bis(pyrazol-3-yl)pyridine-type ligands as controlled by packing forces: structural correlations, Chem.–Eur. J., 2012, 18, 11703–11715 CrossRef CAS
- K. H. Sugiyarto, K. Weitzner, D. C. Craig and H. A. Goodwin, Structural, magnetic and Mössbauer studies of bis(2,6-bis(pyrazol-3-yl)pyridine)iron(II) triflate and its hydrates, Aust. J. Chem., 1997, 50, 869–874 CrossRef CAS
- N. Paradis, G. Chastanet and J.-F. Létard, When stable and metastable HS states meet in spin-crossover compounds, Eur. J. Inorg. Chem., 2012, 2012, 3618–3624 CrossRef CAS
- M. Marchivie, P. Guionneau, J. F. Létard, D. Chasseau and J. A. K. Howard, Thermal trapped iron(II) high spin state investigated by X-ray diffraction, J. Phys. Chem. Solids, 2004, 65, 17–23 CrossRef CAS
- O. Roubeau, A. F. Stassen, I. F. Gramage, E. Codjovi, J. Linarès, F. Varret, J. G. Haasnoot and J. Reedijk, Surprising features in old and new [Fe(alkyl-tetrazole)6] spin-crossover systems, Polyhedron, 2001, 20, 1709–1716 CrossRef CAS
- R. Hinek, P. Gutlich and A. Hauser, Cooperative effects in the [Fe(mtz)6](BF4)2 spin-crossover system: fine tuning the energy gap, Inorg. Chem., 1994, 33, 567–572 CrossRef CAS
- V. Gómez, C. Sáenz de Pipaón, P. Maldonado-Illescas, J. C. Waerenborgh, E. Martin, J. Benet-Buchholz and J. R. Galán-Mascarós, Easy excited-state trapping and record high TTIESST in a spin-crossover polyanionic FeII trimer, J. Am. Chem. Soc., 2015, 137, 11924–11927 CrossRef
- A. Moneo-Corcuera, D. Nieto-Castro, C. Sáenz de Pipaón, V. Gómez, P. Maldonado-Illescas and J. R. Galan-Mascaros, Tuning the spin crossover behavior of the polyanion [(H2O)6Fe3(μ-L)6]6−: the case of the cesium salt, Dalton Trans., 2018, 47, 11895–11902 RSC
- H. Wang, C. Sinito, A. Kaiba, J. S. Costa, C. Desplanches, P. Dagault, P. Guionneau, J.-F. Létard, P. Negrier and D. Mondieig, Unusual solvent dependence of a molecule-based FeII macrocyclic spin-crossover complex, Eur. J. Inorg. Chem., 2014, 2014, 4927–4933 CrossRef CAS
- V. A. Money, C. Carbonera, J. Elhaïk, M. A. Halcrow, J. A. K. Howard and J.-F. Létard, Interplay between kinetically slow thermal spin-crossover and metastable high-spin state relaxation in an iron(II) complex with similar T1/2 and T(LIESST), Chem.–Eur. J., 2007, 13, 5503–5514 CrossRef CAS
- N. Paradis, G. Chastanet, T. Palamarciuc, P. Rosa, F. Varret, K. Boukheddaden and J.-F. Létard, Detailed investigation of the interplay between the thermal decay of the low temperature metastable HS state and the thermal hysteresis of spin-crossover solids, J. Phys. Chem. C, 2015, 119, 20039–20050 CrossRef CAS
- J. S. Costa, P. Guionneau and J.-F. Létard, Photomagnetic properties of the [Fe(L222(N3O2))(CN)2]·H2O complex: a fascinating example of multi-metastability, J. Phys.: Conf. Ser., 2005, 21, 67–72 CrossRef CAS
- A. Hauser, Light-induced spin crossover and the high-spin → low-spin relaxation, Top. Curr. Chem., 2004, 234, 155–198 CrossRef CAS
- F. Varret, K. Boukheddaden, G. Chastanet, N. Paradis and J. F. Létard, When T(LIESST) meets thermal hysteresis - a theoretical approach, Eur. J. Inorg. Chem., 2013, 763–769 CrossRef CAS
- D. Collison, C. David Garner, C. M. McGrath, J. Frederick, W. Mosselmans, M. D. Roper, J. M. W. Seddon, E. Sinn and N. A. Young, Soft X-ray induced excited spin state trapping and soft X-ray photochemistry at the iron L2,3 edge in [Fe(phen)2(NCS)2] and [Fe(phen)2(NCSe)2] (phen = 1,10-phenanthroline),
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J. Chem. Soc. Dalton Trans., 1997, 4371–4376 RSC
J. Chem. Soc. Dalton Trans., 1997, 4371–4376 RSC - C. Wäckerlin, F. Donati, A. Singha, R. Baltic, S. Decurtins, S.-X. Liu, S. Rusponi and J. Dreiser, Excited spin-state trapping in spin crossover complexes on ferroelectric substrates, J. Phys. Chem. C, 2018, 122, 8202–8208 CrossRef
- P. Homenya, L. Heyer and F. Renz, Electromagnetic radiation as a spin crossover stimulus, Pure Appl. Chem., 2015, 87, 293–300 CAS
- P. Gamez, R. H. Steensma, W. L. Driessen and J. Reedijk, Copper(II) compounds of the planar-tridentate ligand 2,6-bis(pyrazol-3-yl)pyridine, Inorg. Chim. Acta, 2002, 333, 51–56 CrossRef CAS
- A. W. Addison, T. N. Rao and C. G. Wahlgren, Synthesis of some benzimidazole- and benzothiazole-derived ligand systems and their precursory diacids, J. Heterocycl. Chem., 1983, 20, 1481–1484 CrossRef CAS
- L. R. Melby, R. J. Harder, W. R. Hertler, W. Mahler, R. E. Benson and W. E. Mochel, Substituted quinodimethans. II. Anion-radical derivatives and complexes of 7,7,8,8-tetracyanoquinodimethan, J. Am. Chem. Soc., 1962, 84, 3374–3387 CrossRef CAS
- S. A. O'Kane, R. Clérac, H. Zhao, X. Ouyang, J. R. Galán-Mascarós, R. Heintz and K. R. Dunbar, New crystalline polymers of Ag(TCNQ) and Ag(TCNQF4): structures and magnetic properties, J. Solid State Chem., 2000, 152, 159–173 CrossRef
- G. A. Bain and J. F. Berry, Diamagnetic corrections and Pascal's constants, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS
-
CrysAlis, Oxford Diffraction Ltd., Abingdon, England, 2006 Search PubMed
-
SCALE3 ABSPACK - An Oxford Diffraction program (1.0.4, gui:1.0.3), Oxford Diffraction Ltd., Abingdon, England, 2005 Search PubMed
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2085553–2085560, 2085716 and 2085812. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02843j |
| This journal is © The Royal Society of Chemistry 2021 |