
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed 1,1-alkynylbromination of alkenes with alkynyl bromides†
Yusuke
Ano
 *ab,
Natsuki
Kawai
a and
Naoto
Chatani
a
*ab,
Natsuki
Kawai
a and
Naoto
Chatani
a
aDepartment of Applied Chemistry, Faculty of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: ano@chem.eng.osaka-u.ac.jp
bCenter for Atomic and Molecular Technologies, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 14th August 2021
Abstract
The palladium-catalyzed 1,1-alkynylbromination of terminal alkenes with a silyl-protected alkynyl bromide is reported. The method tolerates a diverse range of alkenes including vinylarenes, acrylates, and even electronically unbiased alkene derivatives to afford propargylic bromides regioselectively. Mechanistic studies and DFT calculations indicate that the 1,1-alkynylbromination reaction proceeds via the migration of the Pd center followed by the formation of a π-allenyl Pd intermediate, leading to the stereoselective reductive elimination of the C(sp3)–Br bond at the propargylic positon.
Introduction
Development of new methodologies for the highly efficient, selective preparation of complex compounds is an important issue in current organic synthesis.1 Catalytic difunctionalization of alkenes has progressed to open new avenues for the introduction of diverse functionalities into easily accessible chemical feedstock.2 Given that halogenated organic compounds are valuable materials and have desirable properties,3 the transition-metal-catalyzed carbohalogenation of alkenes represents a potent approach for preparing multiply substituted alkyl halides.4 A traditional and reliable method for this type of catalytic carbohalogenation is the use of aryl metal reagents in combination with halogenating reagents (Scheme 1a). In 1968, Heck reported on the Pd-catalyzed 1,2-arylhalogenation of alkenes using aryl mercury reagents in the presence of copper halides.5 Yoshida subsequently reported on a preliminary example of the Pd-catalyzed 1,1-arylchlorination of terminal alkenes using aryl tributylstannanes and CuCl2.6 Following these prior reports, Sanford developed a general protocol for the Pd-catalyzed 1,1- and 1,2-arylhalogenation of terminal alkenes with aryl tributylstannanes, in which the regioselectivities can be controlled by fine-tuning the halogenation reagents, the reaction conditions, and the alkene substituents.7 The use of aryl boronic acids with electrophilic fluorination reagents has also been reported for the Pd-catalyzed 1,1-arylfluorination of alkenes,8 and this method is applicable to 1,2-arylfluorination in combination with directing groups.9 Mechanistic studies on the reported 1,1-arylhalogenations suggest that a β-hydride elimination/reinsertion process was involved and that the resulting π-benzyl Pd intermediate facilitated a 1,1-selective reaction.7b,8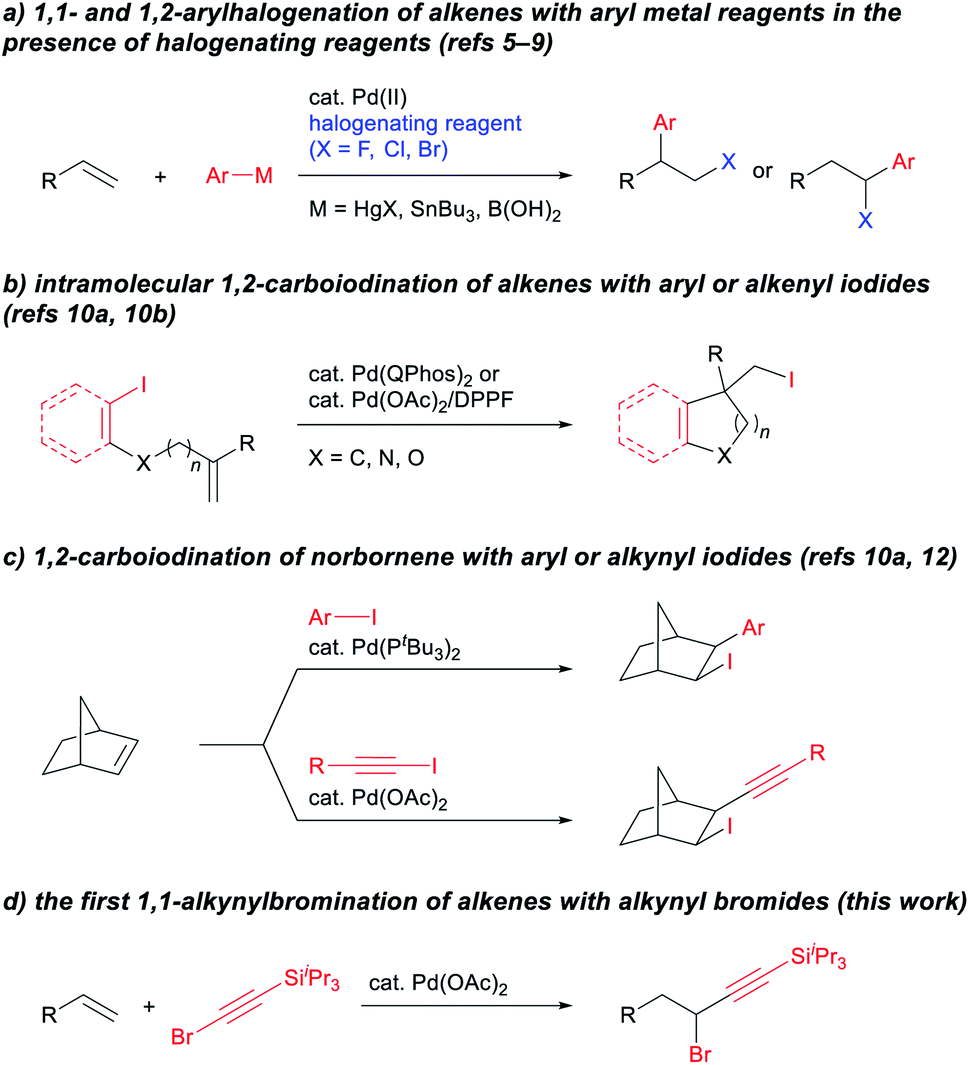 | ||
| Scheme 1 Palladium-catalyzed carbohalogenation of alkenes. | ||
In contrast, carbohalogenation reactions that involve the direct addition of a carbon–halogen bond to an alkene is a more atom-economical approach to providing alkyl halides. In 2011, Lautens10a and Tong10b independently reported on the Pd(0)-catalyzed intramolecular 1,2-addition of C(sp2)–I bonds to alkenes using QPhos or DPPF as ligands (Scheme 1b). Their reports triggered the development of the catalytic intramolecular 1,2-addition of C(sp2)–I bonds to alkenes utilizing not only Pd10 but also Ni11 catalysts. A key to the success of these reactions is the introduction of a substituent on the internal carbon atom of the alkene, which inhibits the undesirable β-hydride elimination process by forming a quaternary carbon center. Norbornene can be employed for Pd-catalyzed intermolecular 1,2-carboiodination reactions using aryl iodides10a or alkynyl iodides12 (Scheme 1c). The Pd13- and Ni14-catalyzed carbohalogenation of internal alkynes has also recently been developed using aryl halides. To the best of our knowledge, however, the catalytic 1,1-addition of organohalides to alkenes has not been reported so far. Moreover, except for the C–I bond, the addition of a C–halogen bond to alkenes continues to be a challenging issue in the Pd-catalyzed reaction.10i,15,16 Herein, we report on the first example of the Pd-catalyzed 1,1-alkynylbromination of terminal alkenes with alkynyl bromides (Scheme 1d).
Results and discussion
We initially investigated the Pd-catalyzed alkynylbromination of 2-vinylnaphthalene (1a) with alkynyl bromide 2 as a model reaction. To our delight, the 1,1-alkynylbromination product 3a was obtained in 71% NMR yield and 61% isolated yield when 1a (0.3 mmol) was reacted with 2 (1.2 equiv.) in the presence of Pd(OAc)2 (10 mol%) in toluene (1 mL) at 75 °C for 20 h under air (Scheme 2). It should be noted that the 1,2-alkynylbromination product of 1a was not observed, while a Mizoroki–Heck type reaction of 1a with 2 took place to give the alkynylalkene 4a in 9% NMR yield as a E/Z mixture (E/Z = 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Extensive optimization studies revealed that neither the addition of ligands, bases, and acids nor the use of other solvents was effective for improving the yield of 3a.17 In a gram–scale reaction using 1.16 g (7.5 mmol) of 1a under the optimal reaction conditions, 1.68 g of 3a was produced in 54% yield. Removal of the triisopropylsilyl group from 3a was also successful using TBAF/AcOH in THF at 0 °C, giving the corresponding terminal alkyne 5a in 81% yield (Scheme 3).
:1). Extensive optimization studies revealed that neither the addition of ligands, bases, and acids nor the use of other solvents was effective for improving the yield of 3a.17 In a gram–scale reaction using 1.16 g (7.5 mmol) of 1a under the optimal reaction conditions, 1.68 g of 3a was produced in 54% yield. Removal of the triisopropylsilyl group from 3a was also successful using TBAF/AcOH in THF at 0 °C, giving the corresponding terminal alkyne 5a in 81% yield (Scheme 3).
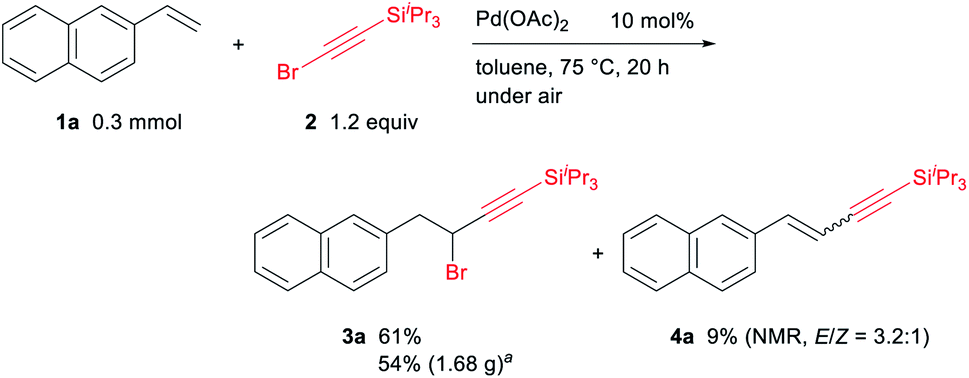 | ||
| Scheme 2 Optimal reaction conditions for the Pd-catalyzed 1,1-alkynylbrommination of 1a with 2. a1.16 g (7.5 mmol) of 1a was used. | ||
 | ||
| Scheme 3 Removal of triisopropylsilyl group. | ||
Having established the optimal reaction conditions, we explored the scope of vinylarenes toward the Pd-catalyzed 1,1-alkynylbromination (Scheme 4a). This reaction tolerates a wide variety of functional groups and substitution patterns on the aromatic ring. For example, both electron-withdrawing (1d–g) and electron-donating (1c, 1k, 1n, 1o) groups were compatible for this catalytic 1,1-alkynylbromination, giving the corresponding propargylic bromides in good yields. The reaction of styrenes bearing halogen (1h–j) or boronate (1m) groups was also successful with these functionalities remaining intact, which would be useful for further functionalization by cross-coupling protocols. In contrast, the reaction of the electron-rich 1l and 1q was impeded by competitive oligomerization under the optimal conditions, leading to decreased yields. Sterically demanding vinylarenes such as 1p and 1r were applicable to this reaction, while a longer time was required for the 1,1-alkynylbromination of 9-vinylphenanthrene (1s) to reach completion. The 1,1-alkynylbromination of vinylheteroarenes 1t and 1u proceeded slowly along with their partial decomposition, resulting in only moderate yields. It should be noted that this protocol could also be applied to vinylarenes derived from biologically active compounds or pharmaceuticals. For example, the estrone (3v), indomethacin (3w), ezetimibe (3x), and fenofibrate (3y) that contained a propargylic bromide core could be easily synthesized.
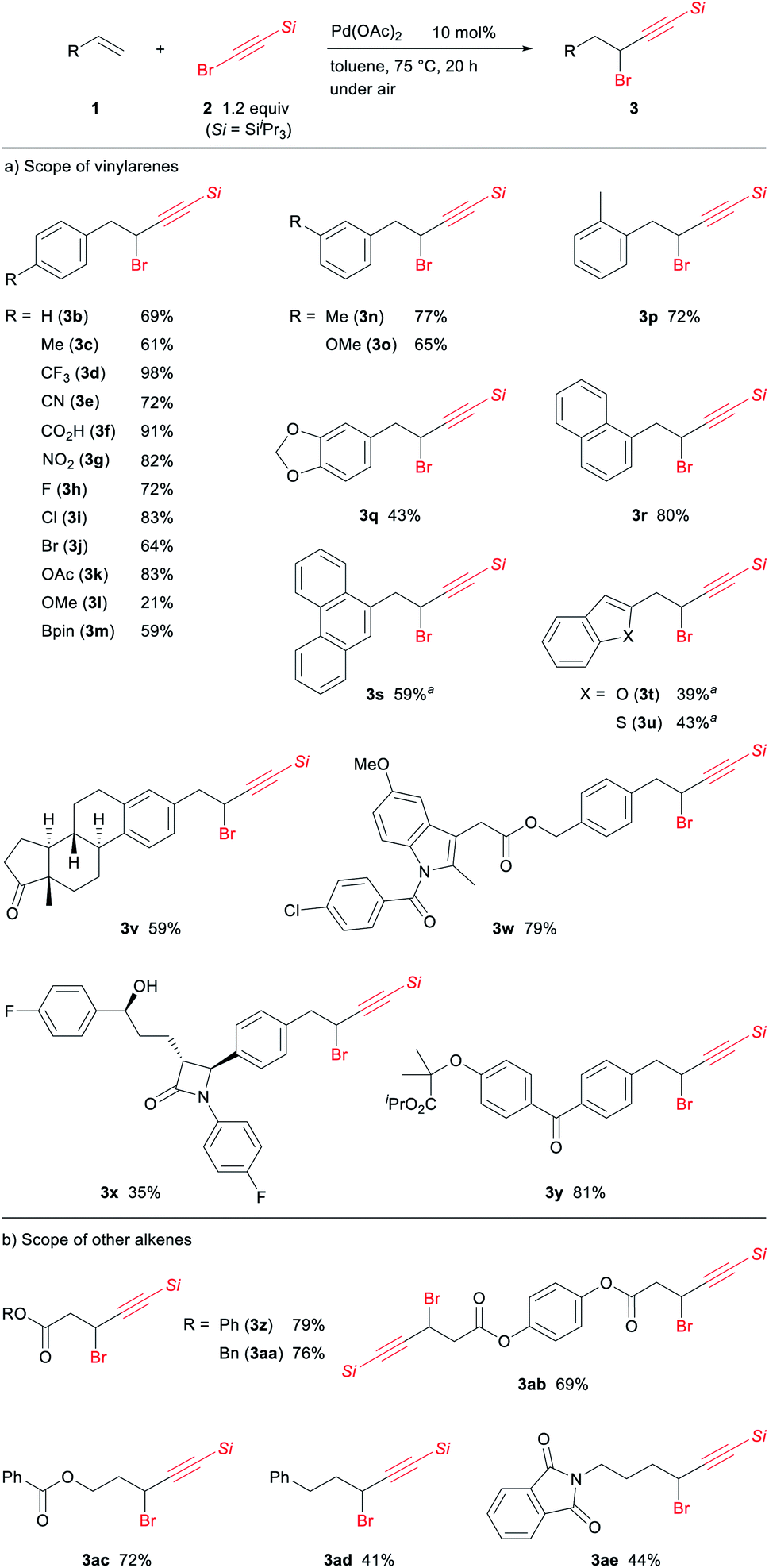 | ||
| Scheme 4 Scope of the reaction. Reaction conditions: alkene (1, 0.3 mmol), alkynyl bromide (2, 0.36 mmol), Pd(OAc)2 (0.03 mmol), toluene (1 mL), 75 °C, 20 h, under air. aRun for 48 h. | ||
We next evaluated the reactivity of other alkenes under the optimal conditions (Scheme 4b). Acrylates such as 1z and 1aa could be employed for this 1,1-alkynylbromination protocol, and bis-1,1-alkynylbromination of 1ab was also successful, giving 3ab as the sole product in good yield. To our surprise, the reaction of allyl benzoate 1ac afforded 3ac without the elimination of the benzoate moiety.18 Electronically unbiased alkenes such as 1ad and 1ae could be used in the 1,1-alkynylbromination, but the yields of 3ad and 3ae remained moderate due to the competitive alkene isomerization. Unfortunately, internal alkenes were unreactive under the optimal conditions.
Monitoring the progress of the 1,1-alkynylbromination of 1a by 1H NMR revealed that there was an induction period at the initial stage of the reaction under the optimal conditions, whereas no induction period was involved when the reaction was conducted with Pd2(dba)3·CHCl3 instead of Pd(OAc)2 (see the ESI†). This result indicated that a Pd(0) species, generated through the stoichiometric Wacker-type oxidation of 1a with Pd(OAc)2,19,20 was likely an active species.
A plausible reaction pathway is illustrated in Scheme 5. To the Pd(0) species generated by reduction of Pd(OAc)2, the oxidative addition of the alkynyl bromide 2 takes place to afford the alkynylpalladium bromide A. Coordination of alkene 1 to A forms B, and the migratory insertion of 1 into the Pd–C(sp) bond of B gives intermediate C. A subsequent β-hydride elimination followed by reinsertion of the alkynylalkene into Pd–H bond leads to the migration of a Pd(II) center to form intermediate E. Finally, reductive elimination to form a C(sp3)–Br bond gives the 1,1-alkynylbromination product 3 along with the generation of Pd(0).
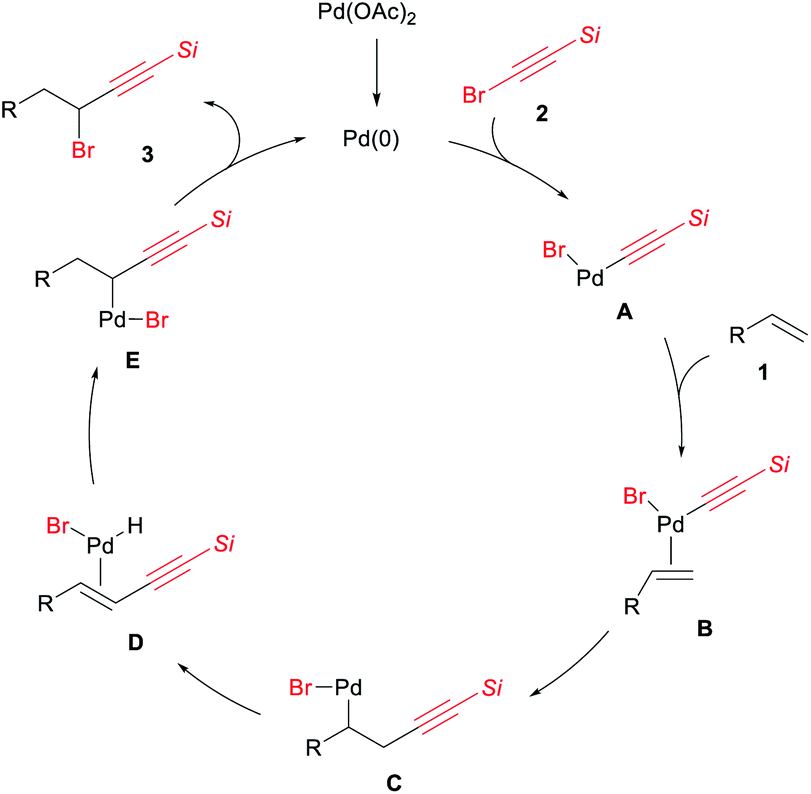 | ||
| Scheme 5 A plausible mechanism (Si = SiiPr3). | ||
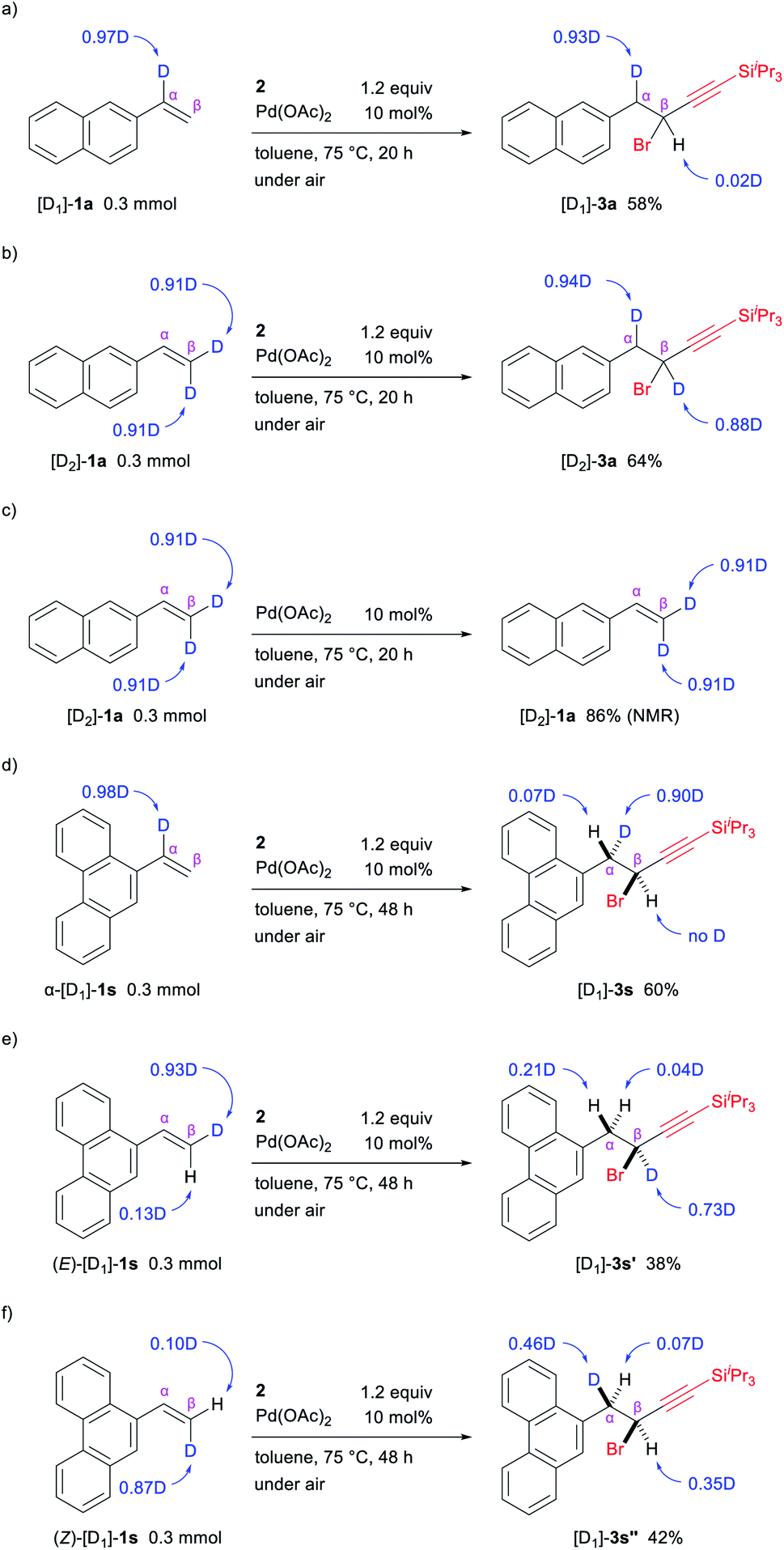
To gain further insights into the mechanism for this reaction, the 1,1-alkynylbromination of deuterated 2-vinylnaphthalenes was examined under the optimal conditions. The reaction of [D1]-1a (0.97D at the α-carbon) with 2 gave [D1]-3a in 58% yield with the deuterium incorporation at the α-carbon being retained (Scheme 6a). On the other hand, when [D2]-1a bearing two deuterium atoms at the β-carbon (both are 0.91D incorporation) was used, the reaction proceeded without the loss of deuterium atoms and afforded [D2]-3a in which one deuterium atom remained at the β-carbon (0.88D) and the other was observed to be located on the α-carbon (0.94D) (Scheme 6b). In addition, no loss of deuterium atoms was observed when the reaction of [D2]-1a was carried out in the absence of 2 (Scheme 6c). These results indicate that a deuterium atom at the β-carbon of the alkene migrated to the α-carbon during the reaction, which was consistent with a mechanism involving β-hydride elimination followed by reinsertion (C → D → E, in Scheme 5).21
 | ||
| Scheme 6 Deuterium-labelling experiments. | ||
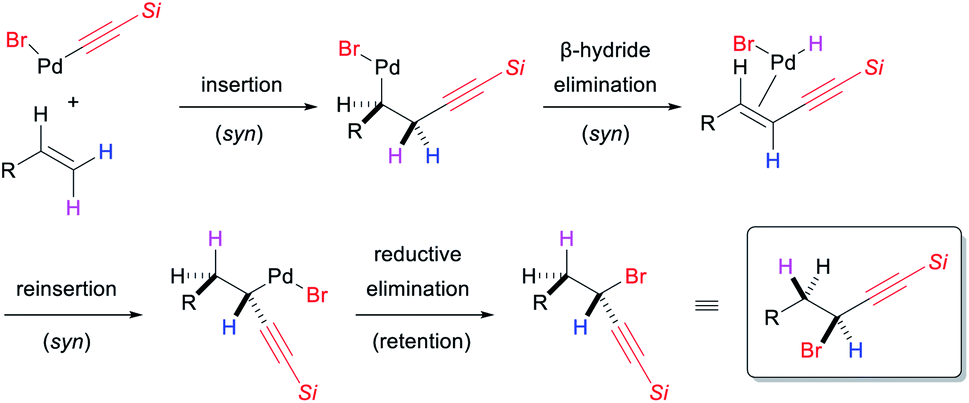
Interest in the stereochemical course of the reaction prompted us to perform some deuterium-labelling experiments using 1s, in which the 1,1-alkynylbromination product 3s has diastereotopic benzylic protons that can be distinguished by 1H NMR analysis. Similar to the reaction of [D1]-1a, the reaction of α-[D1]-1s (0.98D at the α-carbon) with 2 proceeded while the deuterium incorporation at the α-carbon being retained, giving [D1]-3s in 60% yield (Scheme 6d). The configuration between this deuterium atom and the hydrogen atom at the β-carbon in [D1]-3s was assigned as syn based on 1H NMR analysis. Furthermore, the reaction of (E)-[D1]-1s (0.93D and 0.13D at the E- and Z-position, respectively) afforded [D1]-3s′ in which the deuterium was mainly at the β-carbon (0.73D), while a partial incorporation of deuterium at the α-carbon (0.21D) also occurred in an anti-configuration with respect to the deuterium atom at the β-carbon (Scheme 6e). This result suggests that the major reaction pathway involves the cleavage of the C–H bond at the Z-position to the 9-phenanthrenyl group by β-hydride elimination. In the light of the syn-stereochemical course for the migratory insertion of alkenes to Pd–C(sp) bond22 and β-hydride elimination/reinsertion,23 the findings for these deuterium-labelling experiments suggest that the reductive elimination of the C(sp3)–Br bond likely proceeds with retention of configuration, as shown in Scheme 7. We also examined the 1,1-alkynylbromination of (Z)-[D1]-1s (0.10D and 0.87D at E- and Z-position, respectively) with the expectation that the reaction would proceed via the migration of the deuterium atom from the β-carbon to the α-carbon. However, this reaction provided [D1]-3s′′ with deuterium scrambling (Scheme 6f), indicating that another reaction pathway appears to be involved as a minor pathway in this 1,1-alkynylbromination.24
 | ||
| Scheme 7 A plausible stereochemistry of the reaction (Si = SiiPr3). | ||
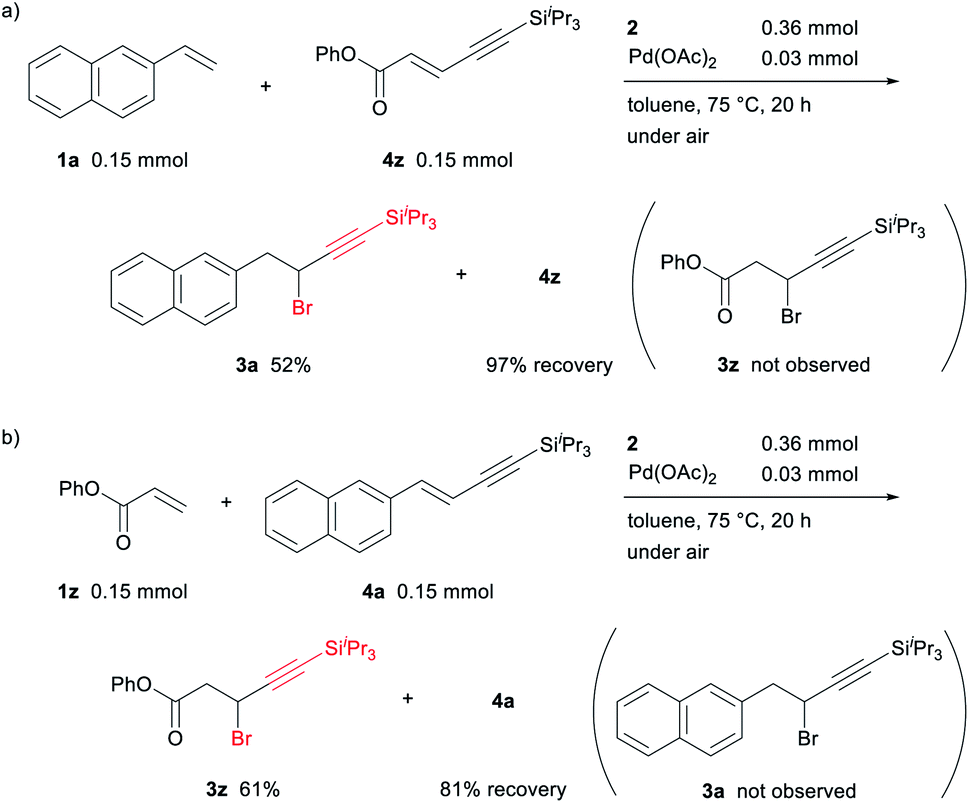
A crossover experiment using an equimolar mixture of 1a and the alkynylalkene 4z, provided by Mizoroki–Heck type reaction of 1z with 2, afforded 3a in 52% yield along with 97% of 4z being recovered (Scheme 8a). Similarly, using an equimolar mixture of 1z and alkynylalkene 4a in the reaction resulted in the formation of 3z as the sole 1,1-alkynylbromination product with 81% of 4a being recovered (Scheme 8b). These results indicate that the coordinating alkynylalkene 4 in intermediate D is reinserted into the Pd–H bond without exchanging a free alkynylalkene in the catalytic cycle (Scheme 5).
 | ||
| Scheme 8 Crossover experiments. | ||
In an attempt to shed light on the unusual regioselectivity of this reaction, we calculated the Gibbs free energy profiles for the 1,1-alkynylbromination of 1b with the alkynyl bromide 2′ at the SMD (toluene) M06L/def2TZVP//B3LYP/6-31G(d)–LANL2DZ (for Pd and Br) level of theory.25,26 The calculated Gibbs free energy profiles are shown in Fig. 1a, where a Pd(0)/2′ complex INT0 was set as the relative zero point. The initial oxidative addition of 2′ on the Pd(0) center of INT1 proceeds viaTS1-2 with an energy barrier of 24.9 kcal mol−1, providing the three-coordinated intermediate INT2. In TS1-2, the lengths of the cleaving C–Br bond and the forming Pd–Br bond are 2.23 and 2.70 Å, respectively (Fig. 1b). The coordination of 1b to INT2 gives INT3 along with an energy release of 8.7 kcal mol−1, and the following migratory insertion of 1b into the Pd–C bond viaTS3-4 requires an activation barrier of 12.9 kcal mol−1, giving the benzyl Pd intermediate INT4. The computed structure of TS3-4 shows that the lengths of the forming C–C bond and the breaking Pd–C bond are 2.06 and 2.00 Å, respectively. The subsequent C–C bond rotation in INT4 gives INT5 along with the dissociation of the alkyne moiety from the Pd center. The migration of Pd from the benzylic position (INT5) to the propargylic position (INT 8) is initiated by a β-hydride elimination viaTS5-6 with an activation barrier of 14.3 kcal mol−1 to give INT6. The computational results showed that TS7-8 for the migratory reinsertion of the coordinating alkynylalkene into the Pd–H bond requires higher activation energy than that for TS5-6 (ΔΔG‡ = 2.3 kcal mol−1) and the overall energy barrier for the Pd migration step is 16.6 kcal mol−1. The σ-propargyl Pd intermediate INT8 can be converted into the π-allenyl Pd intermediate INT9 with an exothermal energy of 6.9 kcal mol−1. It should be noted that the π-allenyl Pd intermediate INT9 was found to be 6.7 kcal mol−1 more thermodynamically stable than the σ-benzyl Pd intermediate INT5.
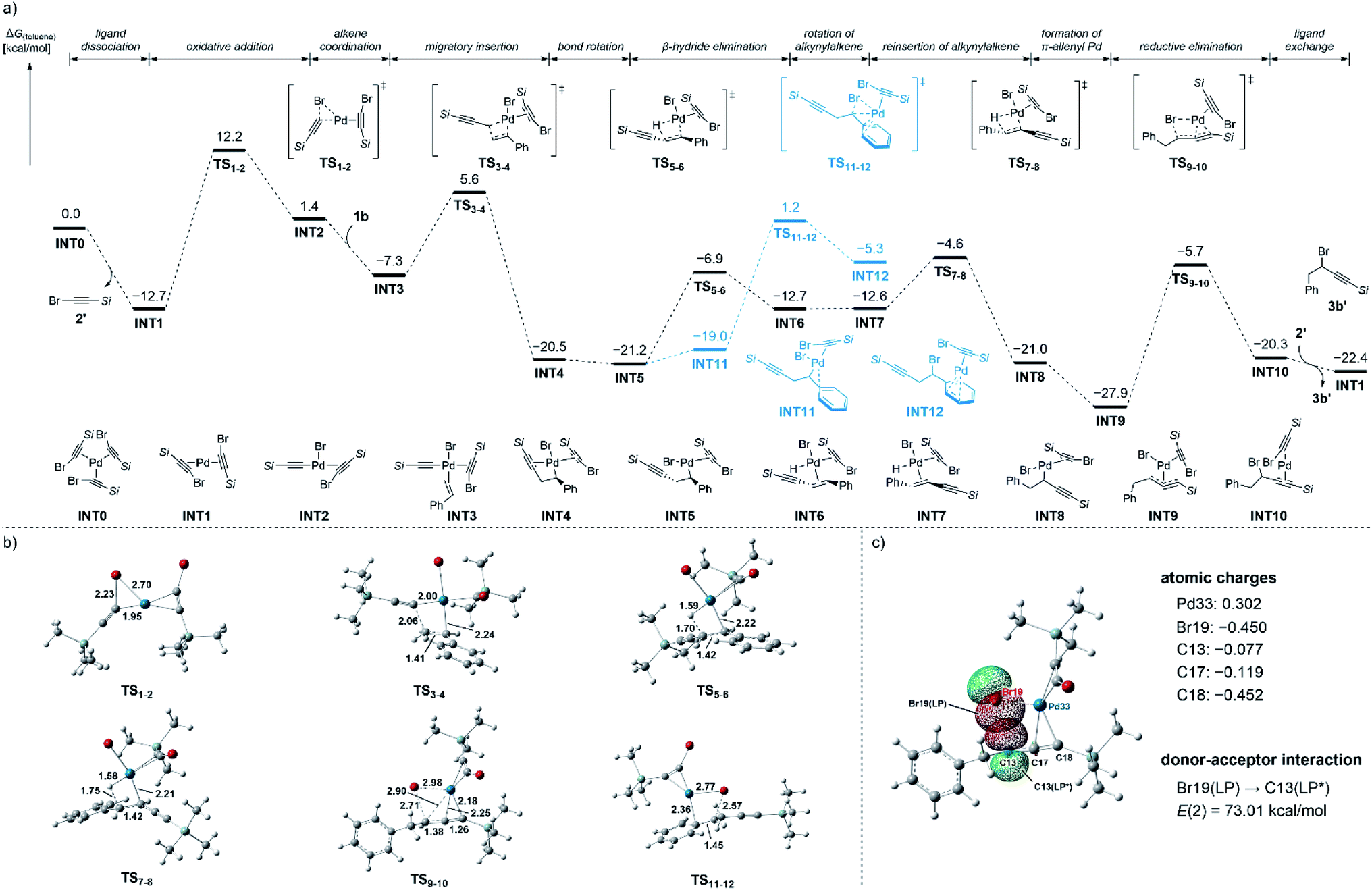 | ||
| Fig. 1 (a) Gibbs free energy profiles for the Pd-catalyzed 1,1-alkynylbromination of 1b with 2′ (Si = SiMe3). (b) Optimized structures of TSs. (c) Natural population analysis of TS9-10. | ||
The reductive elimination proceeds from INT9viaTS9-10 with an activation barrier of 22.2 kcal mol−1, and INT10 is formed with retention of configuration at the propargylic carbon atom,27 which is consistent with the observations from our deuterium-labelling experiments (Scheme 6). Structural information regarding TS9-10 shows that the length of the cleaving Pd–Br bond is 2.98 Å and that of the forming C–Br bond is 2.71 Å, and the propargylic carbon is a distance of 2.90 Å from the Pd center. The geometry of TS9-10 suggests that the reductive elimination likely proceeds via the release of the bromine ligand from Pd center prior to the formation of the C–Br bond. This mechanism is in agreement with the reverse process of the oxidative addition of propargylic halides to zero-valent group 10 metal complexes.28Fig. 1c shows the result of the natural population analysis (NPA) of TS9-10, where the values of the natural charges for Pd33 and Br19 atoms were found to be 0.302 and −0.450, respectively, while the negative charge on the allenyl moiety was distributed on C18 (C13, −0.077; C17, −0.119; C18, −0.452). Moreover, the natural bond orbital (NBO) analysis29 of TS9-10 revealed that the donor–acceptor interaction from the p-orbital of the Br19 atom to the p-orbital of the C13 atom was large, which also supports the above mechanism. Finally, ligand exchange provides 3b′ and INT1 along with an exothermal energy of 2.1 kcal mol−1. The calculations also revealed that the reductive elimination of C(benzyl)–Br bond from INT11 occurs viaTS11-12, the free energy of which is higher than that of TS7-8 and TS9-10 (ΔΔG‡ = 5.8 and 6.9 kcal mol−1, respectively), indicating that the formation of 1,2-alkynylbromination product is a kinetically unfavourable process.
Conclusions
In summary, we report on the first Pd-catalyzed 1,1-alkynylbromination of terminal alkenes using a silyl-protected alkynyl bromide as an alkynylbromination reagent. A variety of alkenes including vinylarenes, acrylates, and electronically unbiased alkenes were found to be applicable to this protocol thus providing direct access to functionalized propargylic bromides. Deuterium-labelling experiments revealed that the migration of a Pd center is involved in the formation of the 1,1-alkynylbromination product and suggest that the reductive elimination proceeds with the retention of configuration. Computational studies also support that the conclusion that the reductive elimination occurs from a π-allenyl Pd intermediate and that the C–Br bond is formed in a stereoretentive fashion at the propargylic carbon atom. Further mechanistic studies are currently underway in our group.Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
Y. A. conceived the study, carried out the computations, and wrote the manuscript. N. K. performed the experiments and analyzed the data. N. C. discussed the results with Y. A. and N. K. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grant-in-Aid for Early-Career Scientists (18K14218 to Y. A.) and Grant-in-Aid for Specially Promoted Research (17H06091 to N. C.) from JSPS. We thank the Analytical Instrumentation Facility, Graduate School of Engineering, Osaka University, for their assistance with HRMS.Notes and references
- (a) B. M. Trost, Science, 1983, 219, 245–250 CrossRef CAS PubMed; (b) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed; (c) Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 CrossRef CAS PubMed; (d) J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CrossRef CAS.
- For recent reviews on catalytic difunctionalization of alkenes, see: (a) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem. – Asian J., 2018, 13, 2277–2291 CrossRef CAS; (b) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990–8003 CrossRef CAS; (c) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC; (d) R. K. Dhungana, R. R. Sapkota, D. Niroula and R. Giri, Chem. Sci., 2020, 11, 9757–9774 RSC.
- For selected reviews on halogenated bioactive compounds and natural products, see: (a) C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed; (b) P. M. Pauletti, L. S. Cintra, C. G. Braguine, A. A. da Silva Filho, M. L. A. e. Silva, W. R. Cunha and A. H. Januário, Mar. Drugs, 2010, 8, 1526–1549 CrossRef CAS; (c) G. W. Gribble, Environ. Chem., 2015, 12, 396–405 CrossRef CAS; (d) G. W. Gribble, Mar. Drugs, 2015, 13, 4044–4136 CrossRef CAS PubMed; (e) W.-J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 CrossRef CAS.
- For recent reviews on the formation of carbon–halogen bonds, see: (a) X. Jiang, H. Liu and Z. Gu, Asian J. Org. Chem., 2012, 1, 16–24 CrossRef CAS; (b) C. Chen and X. Tong, Org. Chem. Front., 2014, 1, 439–446 RSC; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed; (d) D. A. Petrone, C. M. Le, S. G. Newman and M. Lautens, in New Trends in Cross-Coupling: Theory and Applications, ed. T. J. Colacot, The Royal Society of Chemistry, Cambridge, 2015, pp. 276–321 Search PubMed; (e) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS; (f) D. J. Jones, M. Lautens and G. P. McGlacken, Nat. Catal., 2019, 2, 843–851 CrossRef CAS. See also recent reviews on catalytic carbohalogenation of carbon–carbon multiple bonds: (g) D. Bag, S. Mahajan and S. D. Sawant, Adv. Synth. Catal., 2020, 362, 3948–3970 CrossRef CAS; (h) D. Bag, H. Kour and S. D. Sawant, Org. Biomol. Chem., 2020, 18, 8278–8293 RSC.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5538–5542 CrossRef CAS.
- (a) Y. Tamaru, M. Hojo, H. Higashimura and Z.-I. Yoshida, Angew. Chem., Int. Ed. Engl., 1986, 25, 735–737 CrossRef; (b) Y. Tamaru, M. Hojo, S. Kawamura and Z.-I. Yoshida, J. Org. Chem., 1986, 51, 4089–4090 CrossRef CAS. A related reaction was also reported, see: (c) J. P. Parrish, Y. C. Jung, S. I. Shin and K. W. Jung, J. Org. Chem., 2002, 67, 7127–7130 CrossRef CAS PubMed.
- (a) D. Kalyani and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 2150–2151 CrossRef CAS PubMed; (b) D. Kalyani, A. D. Satterfield and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 8419–8427 CrossRef CAS PubMed.
- (a) Y. He, Z. Yang, R. T. Thornbury and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 12207–12210 CrossRef CAS PubMed; (b) J. Miró, C. del Pozo, F. D. Toste and S. Fustero, Angew. Chem., Int. Ed., 2016, 55, 9045–9049 CrossRef.
- (a) E. P. A. Talbot, T. A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101–4104 CrossRef CAS; Recently, Pd-catalyzed 1,2-arylfluorination of internal enamides was reported: (b) Y. Xi, C. Wang, Q. Zhang, J. Qu and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 2699–2703 CrossRef CAS.
- For selected examples of Pd-catalyzed 1,2-addition of C(sp2)–I bonds to alkenes, see: (a) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778–1780 CrossRef CAS; (b) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187–6193 CrossRef CAS; (c) S. G. Newman, J. K. Howell, N. Nicolaus and M. Lautens, J. Am. Chem. Soc., 2011, 133, 14916–14919 CrossRef CAS PubMed; (d) X. Jia, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 9870–9872 CrossRef CAS; (e) D. A. Petrone, H. A. Malik, A. Clemenceau and M. Lautens, Org. Lett., 2012, 14, 4806–4809 CrossRef CAS PubMed; (f) D. A. Petrone, M. Lischka and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 10635–10638 CrossRef CAS; (g) D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 7908–7912 CrossRef CAS; (h) Z.-M. Zhang, B. Xu, L. Wu, L. Zhou, D. Ji, Y. Liu, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 8110–8115 CrossRef CAS; (i) X. Chen, J. Zhao, M. Dong, N. Yang, J. Wang, Y. Zhang, K. Liu and X. Tong, J. Am. Chem. Soc., 2021, 143, 1924–1931 CrossRef CAS PubMed.
- For selected examples of Ni-catalyzed 1,2-addition of C(sp2)–I bonds to alkenes, see: (a) H. Yoon, A. D. Marchese and M. Lautens, J. Am. Chem. Soc., 2018, 140, 10950–10954 CrossRef CAS PubMed; (b) A. D. Marchese, F. Lind, Á. E. Mahon, H. Yoon and M. Lautens, Angew. Chem., Int. Ed., 2019, 58, 5095–5099 CrossRef CAS; (c) A. D. Marchese, L. Kersting and M. Lautens, Org. Lett., 2019, 21, 7163–7168 CrossRef CAS PubMed; (d) A. D. Marchese, T. Adrianov, M. F. Köllen, B. Mirabi and M. Lautens, ACS Catal., 2021, 11, 925–931 CrossRef CAS.
- H. Liu, C. Chen, L. Wang and X. Tong, Org. Lett., 2011, 13, 5072–5075 CrossRef CAS.
- (a) C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 254–257 CrossRef CAS; (b) Y. H. Lee and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 6444–6448 CrossRef CAS PubMed.
- (a) T. Takahashi, D. Kuroda, T. Kuwano, Y. Yoshida, T. Kurahashi and S. Matsubara, Chem. Commun., 2018, 54, 12750–12753 RSC; (b) T. Takahashi, T. Kurahashi and S. Matsubara, ACS Catal., 2020, 10, 3773–3777 CrossRef CAS.
- Pd-catalyzed 1,3-alkynylbromination of norbornene with alkynyl bromides, see: Y. Li, X. Liu, H. Jiang, B. Liu, Z. Chen and P. Zhou, Angew. Chem., Int. Ed., 2011, 50, 6341–6345 CrossRef CAS PubMed.
- Au-catalyzed 1,2-alkynylhalogenation of alkenes with alkynyl halides, see: (a) M. Kreuzahler and G. Haberhauer, J. Org. Chem., 2019, 84, 8210–8224 CrossRef CAS PubMed; (b) M. E. de Orbe, M. Zanini, O. Quinonero and A. M. Echavarren, ACS Catal., 2019, 9, 7817–7822 CrossRef CAS; (c) P. D. García-Fernández, C. Izquierdo, J. Iglesias-Sigüenza, E. Díez, R. Fernández and J. M. Lassaletta, Chem.–Eur. J., 2020, 26, 629–633 CrossRef PubMed.
- The reaction of 1a with tBuMe2Si-protected alkynyl bromide gave the corresponding 1,1-alkynylbromination product in 51% NMR yield. The use of iPr3Si-protected alkynyl chloride instead of 2 were less effective for the reaction with 1a (14% NMR yield). See also the ESI† for details.
- J. Le Bras and J. Muzart, Tetrahedron, 2012, 68, 10065–10113 CrossRef CAS.
- J. Tsuji, Synthesis, 1984, 369–384 CrossRef CAS.
- When the reaction of 1a with 2 was performed, the formation of 2-acetylnaphthalene was observed by 1H NMR analysis of the crude reaction mixture (2% NMR yield).
- For a related discussion on the Pd-catalyzed 1,1-difunctionalization of terminal alkenes, see: (a) K. B. Urkalan and M. S. Sigman, Angew. Chem., Int. Ed., 2009, 48, 3146–3149 CrossRef CAS PubMed; (b) V. Saini, L. Liao, Q. Wang, R. Jana and M. S. Sigman, Org. Lett., 2013, 15, 5008–5011 CrossRef CAS PubMed; (c) M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688–12695 CrossRef CAS PubMed.
- A. Tenaglia, K. Le Jeune, L. Giordano and G. Buono, Org. Lett., 2011, 13, 636–639 CrossRef CAS PubMed.
- R. F. Heck, J. Am. Chem. Soc., 1969, 91, 6707–6714 CrossRef CAS.
- This result was different from our expectation based on the reaction mechanism shown in Scheme 7. See Scheme S1 in the ESI† for the detailed discussion.
- I. Kalvet, K. J. Bonney and F. Schoenebeck, J. Org. Chem., 2014, 79, 12041–12046 CrossRef CAS.
- DFT calculations indicated that the Pd catalyst bearing 2′ as a ligand provided an energetically favorable reaction pathway in comparison with that containing 1b as a ligand. See the ESI† for details.
- Despite many attempts, we have failed to find a TS for the reductive elimination with inversion of the configuration at the propargylic carbon atom at this stage.
- (a) T. Nishida, S. Ogoshi, K. Tsutsumi, Y. Fukunishi and H. Kurosawa, Organometallics, 2000, 19, 4488–4491 CrossRef CAS. Stoichiometric study on the reactivity of π-allenyl Pd complex was also reported: (b) K. Tsutsumi, S. Ogoshi, S. Nishiguchi and H. Kurosawa, J. Am. Chem. Soc., 1998, 120, 1938–1939 CrossRef CAS.
- (a) E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold, NBO Version 3.1 Search PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02873a |
| This journal is © The Royal Society of Chemistry 2021 |