
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a photochemical cascade process by flow-based interception of isomerising alkenes†
Mara
Di Filippo
 a,
Cristina
Trujillo
b,
Goar
Sánchez-Sanz
ac,
Andrei S.
Batsanov
d and
Marcus
Baumann
*a
a,
Cristina
Trujillo
b,
Goar
Sánchez-Sanz
ac,
Andrei S.
Batsanov
d and
Marcus
Baumann
*a
aSchool of Chemistry, University College Dublin, Science Centre South, D04 N2E2, Dublin, Ireland. E-mail: marcus.baumann@ucd.ie
bTrinity Biomedical Sciences Institute, School of Chemistry, The University of Dublin, Trinity College, Dublin, Ireland
cIrish Centre for High-End Computing (ICHEC), Grand Canal Quay, Dublin 2, D02 HP83, Ireland
dDepartment of Chemistry, Durham University, DH1 3LE, South Road, Durham, UK
First published on 5th July 2021
Abstract
Herein we report the discovery of a new photochemical cascade process through a flow-based strategy for intercepting diradicals generated from simple alkenes. This continuous process delivers a series of unprecedented polycyclic reaction products. Exploring the scope of this novel process revealed that this approach is general and affords a variety of structurally complex reaction products in high yields (up to 81%), short reaction times (7 min) and high throughputs (up to 5.5 mmol h−1). A mechanistic rationale is presented that is supported by computations as well as isolation of key intermediates whose identity is confirmed by X-ray crystallography. The presented photochemical cascade process demonstrates the discovery of new chemical reactivity and complex chemical scaffolds by continuously generating and intercepting high-energy intermediates in a highly practical manner.
Introduction
Recent years have witnessed a revival of photochemical transformations, and particularly photocatalysed reactions, where transition-metal catalysts and organic photocatalysts facilitate the construction of important chemical bonds via mild and orthogonal reaction conditions.1–6 The transformative power of modern photocatalysis and its potential impact on sustainability7 are evident in the number of recent reports detailing selective and highly efficient protocols that exploit visible light, operate at ambient temperature and are devoid of stoichiometric reagents or additives.Importantly, this has enabled the application of photochemical processes not only in academia but also in industry8 to bring about the functionalisation of valuable building blocks through previously elusive methods. Key to this development is thereby not only the development of photocatalysis as a field, but also the availability of cheap and monochromatic light emitting diodes (LEDs) as attractive light sources,9 and lastly the advent of new reactor technology to provide ready-to-use and standardised set-ups in batch10 and continuous flow mode.11
Whilst the use of blue light (ca. 450 nm, ca. 64 kcal mol−1) is commonly favoured in photoredox applications due to the tolerance of highly functionalised substrates, UV light still plays an important role in photochemistry. This can be seen in [2 + 2]-photocycloaddition processes of readily available alkenes affording important cyclobutanes as well as various photochemical rearrangements.3 Modern high-power LEDs emitting in the UV-A region (310–400 nm, ca. 71–92 kcal mol−1) are particularly interesting in view of replacing classical medium-pressure mercury lamps that do not provide monochromatic light, generate significant amounts of heat, and often require filters to block undesired wavelengths that would result in side-product formation.
Recent work by Noël and co-workers demonstrated the power of photocatalysed HAT processes using UV-A emitting LEDs to bring about the effective C–H functionalisation of unreactive alkanes such as methane, ethane and propane in a continuous manner.12 Additional studies by Booker-Milburn,13 Cochran14 and others15 clearly showcase the value of flow-based photochemistry exploiting uniform and selective irradiation processes to generate diverse targets in a scalable and effective manner.
Our own laboratory recently exploited a high-power LED lamp (tuneable 50–100 W) emitting at 365 nm to realise the clean generation of a set of quinolines16 (Scheme 1, top). In this application the direct irradiation of chalcones triggered isomerisation of the alkene and subsequent cyclocondensation. The replacement of a traditional medium-pressure mercury lamp with this high-power UV-A LED thereby minimised side-product formation, which increased reaction yields by 10–25% and furthermore enabled the telescoped hydrogenation of the quinolines to furnish a set of tetrahydroquinolines including the antimalarial alkaloid galipinine.
 | ||
| Scheme 1 Prior work and approach to the new photocascade process reported herein. | ||
Results and discussion
Taking inspiration from this quinoline forming photo-process we embarked on a study exploiting analogous chalcones17 bearing an alkyne group in place of the previous amine (Scheme 1, bottom). As the quinoline forming process involved the breaking the C–C π-bond and the isomerisation of the alkene prior to cyclocondensation, we proposed that such putative diradical intermediates (e.g.6) could be trapped by an adjacent alkyne moiety to facilitate the formation of a new C–C bond as well as a new ring system. By deviating from this simple alkene isomerisation path, we anticipated to open new avenues towards creating structural complexity whilst discovering new chemical reactivities. To test this hypothesis, the required scaffold (4) was readily prepared from 2-iodo benzaldehydes (5) via a Sonogashira cross-coupling followed by an aldol condensation process under alkaline conditions (see ESI† for full details). This straightforward route created a small set of substrates providing variation on the chalcone scaffold including three points of diversity (R1–3, Scheme 1, bottom).To study the fate of these entities under standardised photochemical conditions a Vapourtec E-series flow reactor (10 mL FEP coil, 1/16′′ i.d.) was exploited in combination with a high-power LED lamp emitting light at a wavelength of 365 nm (see ESI† for full details). This reactor set-up was advantageous as the LED power can be regulated between 50–100 W, while the flow processing allows for short and uniform light pathlength, narrow residence time distribution and thus excellent reproducibility. Initial efforts exploited piperonal-derived substrate 4a possessing a maximum absorbance (λmax) at 375 nm which closely matched the emission of the LED light source. Using the described set-up, a solution of 4a (50 mM in MeCN) was processed through the LED reactor set-up operating at 100 W with a residence time of 20 minutes. Pleasingly, TLC analysis indicated full conversion of 4a and formation of a major new product. Optimisation of this flow process quickly indicated that clean formation of this new product can be achieved using only 70 W lamp power, in combination with a shorter residence time of 7 minutes at a concentration of 80 mM while regulating the reactor temperature to 25–30 °C (Table 1). These findings indicate that shorter residence times minimise over-irradiation (i.e. secondary photo-reactions) through improved spatiotemporal control in the flow reactor, which renders the desired product in higher yields.
|
|
||||
|---|---|---|---|---|
| Entry | Concentration [mM] | Lamp power [W] | t Res [min] | Isolated yield |
| 1 | 50 | 100 | 20 | 56 |
| 2 | 50 | 70 | 15 | 60 |
| 3 | 50 | 70 | 10 | 67 |
| 4 | 50 | 70 | 7 | 75 |
| 5 | 80 | 70 | 7 | 81 |

This new product was subsequently purified by silica gel chromatography rendering a beige solid that was then studied by various spectroscopic techniques to establish its chemical structure. 1H-NMR spectroscopy highlighted that a single entity had been obtained whereby the presence of diastereotopic methylene protons indicated a chiral, albeit racemic structure. Most notably, a set of four resonances was observed up-field of the methylendioxybenzene unit (5.8–6.8 ppm, see Fig. 1).
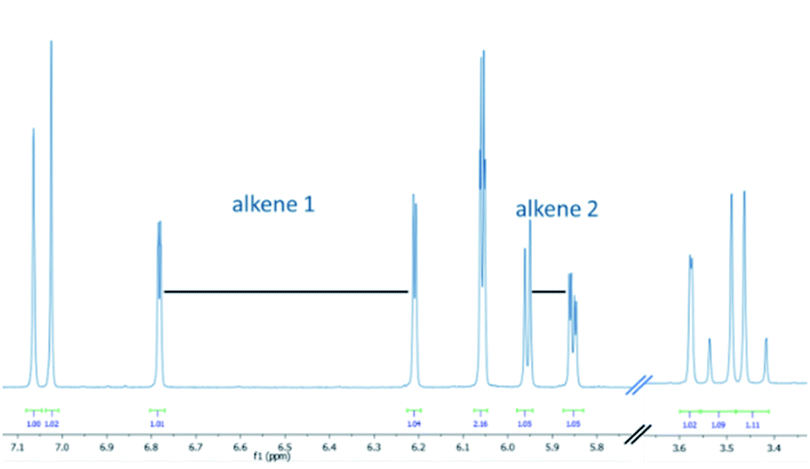 | ||
| Fig. 1 Partial 1H-NMR spectrum of the photoproduct (7a, CDCl3, 500 MHz). | ||
These four resonances integrated for one proton each and were part of two separate alkenes. Furthermore, a doublet with a coupling constant of 2.3 Hz was observed at 3.6 ppm (1 H), as well as an AB quartet at 3.5 ppm (2 × 1 H). This unexpected finding, together with the absence of the enone protons, pointed towards a set of secondary transformations that involved not only the alkene and alkyne moieties, but also the phenyl ring adjacent to the carbonyl in 4a. Analysis of 13C-NMR data along with various 2D NMR data corroborated this analysis suggesting that an unexpected and likely unprecedented photochemical cascade had taken place that transformed a benzene ring into two separate alkene moieties. High-resolution mass spectrometry confirmed a formula of the base peak of C22H21O3 (M + H+) which is identical to that of the substrate. UV-Vis spectroscopy revealed a diminished maximum absorbance of 348 nm indicating a less conjugated system compared to the substrate (Δλmax = 27 nm). While IR spectroscopy additionally indicated the presence of functionalities consistent with a conjugated ketone (1674 cm−1) as well as alkenes and alkanes, it was not possible to unambiguously establish the connectivity of this new structure.
To resolve this situation, the synthesis of this new photoproduct was scaled in flow mode to provide larger quantities for crystallisation and subsequent X-ray diffraction analysis. Gratifyingly, operating the flow process under the previously established conditions for 1 h reproduced the outcome of the initial experiments and rendered a total of 1.85 g of this product after purification. This not only equates to a high productivity of 5.5 mmol h−1, but moreover demonstrates the straightforward scalability of continuous photochemical processes.18 Attempts to crystallise this material were soon met with success and led to identifying the molecular structure based on single crystal X-ray diffraction. Fig. 2 shows this structure,19 which confirmed the prior spectroscopic data and highlighted the formation of a new fused pentacyclic ring system (excluding the methylenedioxy ring).
 | ||
| Fig. 2 Molecular structure of photo-cascade product 7a (CCDC-2072295). | ||
The structure of this unique architecture is characterised by a cyclopentenone with fused cyclopentene and cyclobutene rings that are joint by a methine carbon and two adjacent quaternary carbon atoms (C6 and C7, Fig. 2). The cyclobutene ring and butyl chain display a cis relationship imparting a distinct three-dimensional shape to this structure. It is noted that this tricyclic subunit of 7a occurs in the natural product tricycloclavulone20 (8, Fig. 3), though published studies describing the construction of this scaffold unsurprisingly require significantly more steps compared to our new photochemical cascade.
 | ||
| Fig. 3 Structural comparison between 7a and tricycloclavulone 8. | ||
Having demonstrated proof of principle for this new photochemical cascade we next wished to study the scope of this process. To this end we modified the scaffold of substrate 4 incorporating different appendages on both aryl moieties and the alkyne. Pleasingly the reaction tolerated different groups on the initial 2-iodo benzaldehyde unit, including 3,4-(methylene)dioxy, 3,4-dimethoxy and even unsubstituted systems (R1 = H, Fig. 4). A distinct blue-shift of the maximum absorbance (λmax) for substrates 4h–j to ca. 310 nm was observed, however a shoulder reaching as far as 360 nm appears to provide sufficient overlap with light emitted from the UV-LED. It was furthermore established that variations on the alkyne including phenyl and alkyl groups are well tolerated. However, placing a cyclopropyl ring next to the alkyne did not provide the desired product (7g), which is consistent with the involvement of nearby radicals.21 Lastly, introduction of substituents on the benzoyl moiety is possible providing the desired products (7d, 7j) with three contiguous quaternary centres. In all these cases the desired products were isolated in high chemical yields. It was furthermore established that the cascade products possess significantly lowered λmax values compared to the substrates (300–345 nm) which is consistent with the apparent loss of conjugation.
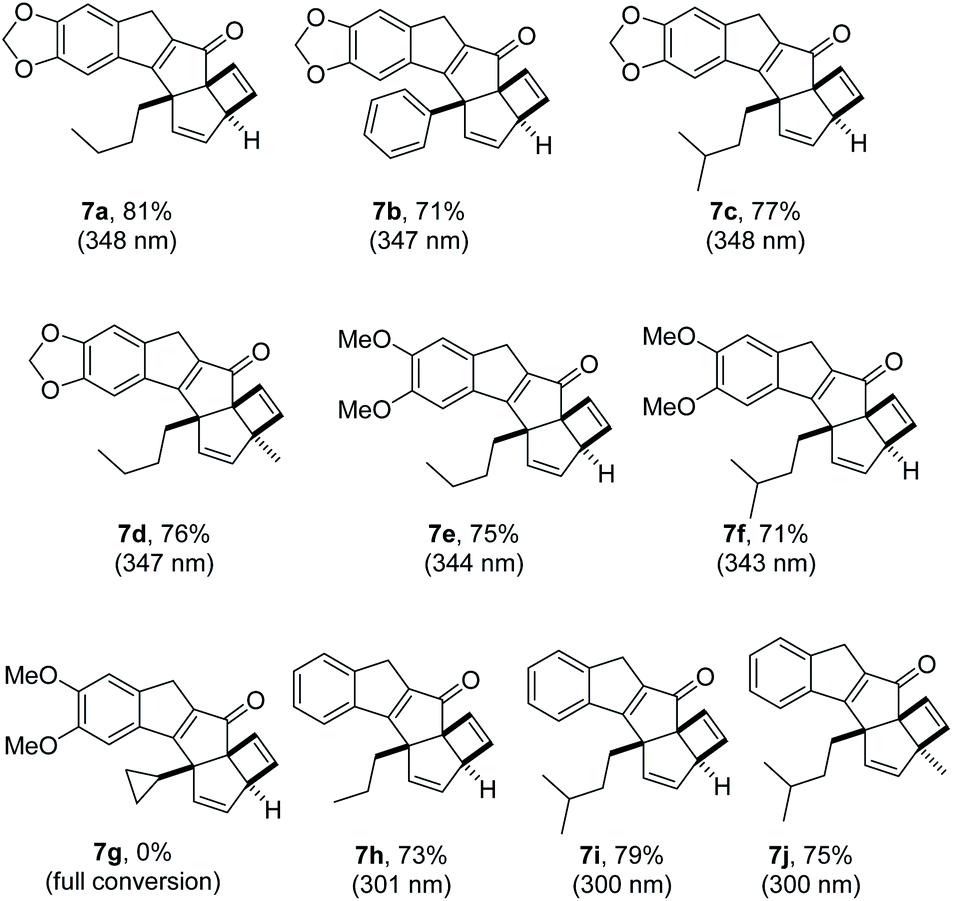 | ||
| Fig. 4 Structures and λmax values of photo-cascade products 7a–j. | ||
The next part of our study concerned probing the mechanism by which these unique photocascade products are generated. Specifically, this entailed the fate of the aryl ring that is transformed into the unusual bicyclo[3.2.0]hepta-2,6-diene scaffold. As we initially hypothesised that this ring system may have arisen from a cycloheptatriene by a photochemical 4π-electrocyclisation process, we attempted to experimentally find proof for this proposal. We therefore wished to again exploit the benefits of continuous flow processing allowing for fine tuning of reaction conditions based on superb spatiotemporal control.22
Therefore, the flow process was repeated for the photo-cascade of 4a, however, utilising conditions that would lead to incomplete conversion of the substrate. An experiment was thus run in which a more concentrated solution of 4a (100 mM MeCN) was processed with only 5 minute residence time at 50 W lamp power (Scheme 2).
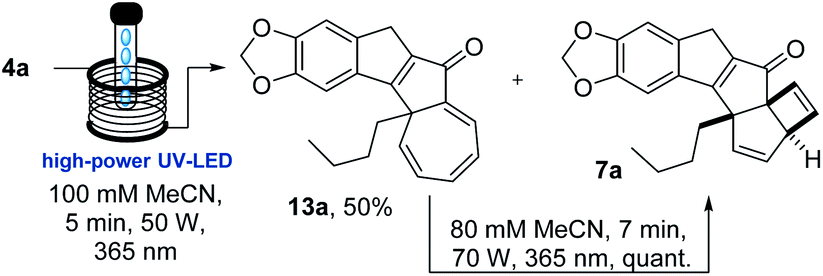 | ||
| Scheme 2 Flow synthesis of 13a and its conversion into 7a. | ||
Analysing the 1H-NMR spectrum of the crude product, we were pleased to identify a new species (13a, ca. 65%) besides the previous photo-cascade product (7a, ca. 35%). Purification by chromatography rendered this new product as a yellow solid (λmax = 383 nm) in 50% isolated yield. HR-MS analysis confirmed that it was an isomer of 7a (molecular formula: C22H20O3). In addition, the 1H-NMR spectrum of this compound clearly showed that the resonance for the methine proton at 3.6 ppm was absent, while a total of five olefinic resonances were visible (5.9–7.1 ppm; Fig. 5). COSY and NOESY experiments subsequently revealed that these five protons are part of the same spin system, which together with all other data suggested this species to be the cycloheptatriene intermediate 13a.
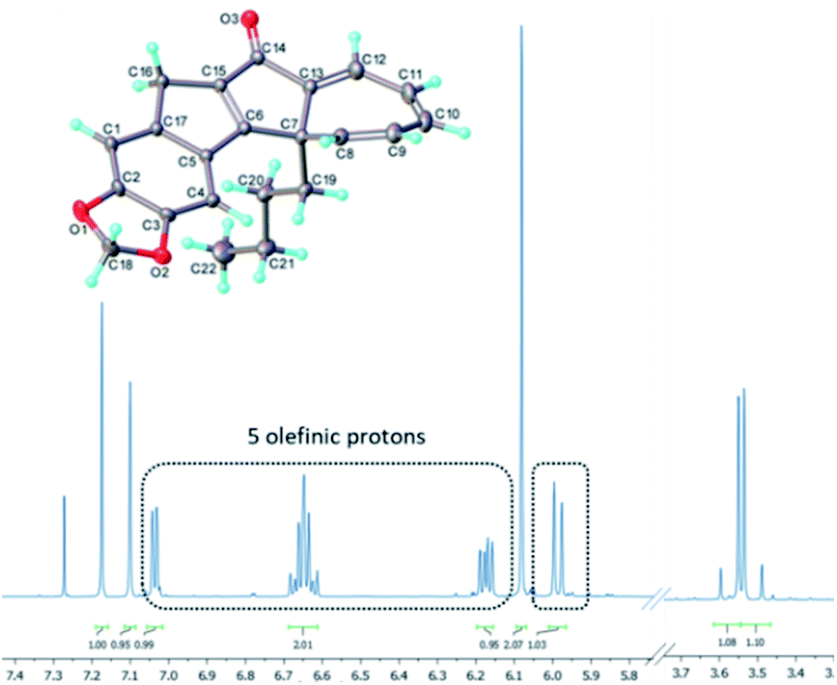 | ||
| Fig. 5 Molecular structure (CCDC-2072296) and partial 1H-NMR spectrum of cycloheptatriene 13a (CDCl3, 500 MHz). | ||
The molecular structure of 13a was subsequently secured through single crystal X-ray diffraction analysis19 and confirmed both our initial hypothesis and structural assignment.
Considerable amounts of 13a (ca. 250 mg) were soon prepared allowing for a final test to prove its role as a late-stage intermediate. Pleasingly, subjecting a solution of 13a (80 mM MeCN) to the regular photoreaction conditions (70 W, 7 minutes residence time, Scheme 3) afforded 7a as sole product in 97% purity (by 1H-NMR).
 | ||
| Scheme 3 Mechanistic rationale for continuous photocascade process. | ||
To shed some light on the mechanistic pathway operational for this photo-cascade process we furthermore performed DFT calculations as implemented in Gaussian 16.23 Specifically, calculations at the M062X24/6-31+G(d,p)25 level of theory in a solvent model SMD (acetonitrile) at 298 K were used and subsequently refined for single point energies using M062X/6-311+G(d,p)25 (acetonitrile). These calculations suggest a first step in which the alkene π-bond initially undergoes homolysis to a diradical species (int2) that is intercepted by the alkyne through a 5-exo-dig cyclisation step (Fig. 6).26 The resulting intermediate (int3) undergoes H-atom translocation to render a fully substituted cyclic enone with an adjacent carbene (int4).27 This carbene species then adds across the adjacent benzene ring to render a norcaradiene ring system.28 These calculations show the possibility of both a concerted (in the singlet state) and stepwise (via switch to triplet intermediate) pathway for this process. Subsequent 6π-electrocyclisation occurs in a conrotatory sense under photochemical conditions to furnish cycloheptatriene 13a, which upon disrotatory 4π-electrocyclisation yields the scaffold of 7a.
 | ||
| Fig. 6 Free-energy profile for the studied reaction of substrate 4a; S and T refer to singlet and triplet states, respectively. | ||
As depicted in Fig. 6, the cycloheptatriene species (int6) is thermodynamically more stable than the final cascade product, which is in good agreement with observations that solutions of this material (e.g.13a) are stable for several weeks under laboratory conditions. However, the high-power UV-LED lamp provides enough energy to overcome this barrier towards the final products. Diminished absorbance of the photoproduct 7a is the likely cause for the observed irreversibility of the final step in this cascade rendering the mechanism as shown in Scheme 3.
Armed with this mechanistic understanding we embarked on a final extension to probe the effect of further substituents in the para position of the aryl enone fragment. Both electron donating (R = OMe) and electron-withdrawing (R = F, CN, CF3) groups were chosen to complement the previously studied cases (R = H, Me) where the substrates uniformly featured the piperonyl moiety as well as a propyl chain introduced with the alkyne. Using the para-fluoro substrate quickly established the formation of the anticipated polycyclic scaffold 7 (Fig. 7) in good yield. However, when trialling either the methoxy derivative, or the electron-deficient nitrile and trifluoromethyl variants, the corresponding cycloheptatriene products 13 were obtained as principal products. These materials were found to absorb light in the UV-A region which may lead to secondary photoreactions and likely accounts for the modest yields (see ESI† for details). The results imply that the electronic effect of the para substituent is not decisive in determining the reaction outcome. To understand these results, we turned again to computations at the same level of theory as before which highlighted the interplay between the energy barrier (13 → 7) as well as the relative stability of the ultimate product with regards to the cycloheptatriene species. In the case of the p-methoxy system (13l), a relatively low energy barrier together with the reversibility of this reaction favours the more stable 7-membered ring as the product. On the other hand, the energy barrier for both the trifluoromethyl and nitrile system is excessively high thus precluding the final step to occur efficiently. Conversely, in case of the para-substituent being a proton or methyl group (e.g.7a, 7d) the energy barrier is surmountable under the reaction conditions whilst a significantly lower absorbance of the product impedes the reverse reaction.
 | ||
| Fig. 7 Correlation between energy barrier and product formation (a yield for butyl side chain; b attainable in 50% yield, see Scheme 2). | ||
Conclusions
In summary, the discovery of a novel photochemical cyclisation cascade is reported rendering complex three-dimensional structures in good to excellent yields. Flow processing in combination with a tunable high-power LED light source emitting at 365 nm was exploited to intercept in situ generated diradicals and rapidly generate a small selection of photoproducts with throughputs up to 5.55 mmol h−1. Besides, modifications to the optimised conditions enabled the generation and spectroscopic characterisation of an advanced cycloheptatriene intermediate that supports the computed mechanism. Continuous flow synthesis facilitated the discovery of this new cascade process, as well as the efficient and scalable production of additional members of this unprecedented class of compounds. Further efforts towards expanding the scope and applicability of this unique light-driven cascade process are underway in our laboratories.Data availability
Data available in the SI includes spectroscopic and computational data.Author contributions
The work was conceptualised by MB and MDF. Experimentation was performed by MDF and MB. Computations were performed by CT and GSS. X-ray analysis was undertaken by ASB. The first draft of the manuscript was prepared by MB and the final draft was edited by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the School of Chemistry (UCD) for providing a Research Demonstratorship (to MDF). This publication has emanated from research supported by Science Foundation Ireland (SFI 18/SIRG/5517; 18/RI/5702; 12/RC2275_P2). We are grateful to the Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities.Notes and references
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 803 CrossRef PubMed.
- For selected examples from industry-led groups, please see: (a) R. Grainger, T. D. Heighten, S. V. Ley, F. Lima and C. N. Johnson, Chem. Sci., 2019, 10, 2264 RSC; (b) D. M. Schultz, F. Levesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274 CrossRef CAS; (c) I. Abdiaj and J. Alcazar, Bioorg. Med. Chem., 2017, 25, 6190 CrossRef CAS PubMed; (d) H. E. Bonfield, K. Mercer, A. Diaz-Rodriguez, G. C. Cook, B. S. J. McKay, P. Slade, G. M. Taylor, W. X. Ooi, J. D. Williams, J. P. M. Roberts, J. A. Murphy, L. Schmermund, W. Kroutil, T. Mielke, J. Cartwright, G. Grogan and L. J. Edwards, ChemPhotoChem, 2020, 4, 45 CrossRef CAS.
- M. Sender and D. Ziegenbalg, Chem. Ing. Tech., 2017, 89, 1159 CrossRef CAS.
- C. C. Le, M. K. Wismer, Z.-C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647 CrossRef CAS PubMed.
- (a) C. Sambiago and T. Noël, Trends Chem., 2020, 2, 92 CrossRef; (b) M. Di Filippo, C. Bracken and M. Baumann, Molecules, 2020, 25, 356 CrossRef CAS PubMed; (c) T. H. Rehm, ChemPhotoChem, 2020, 4, 235 CrossRef CAS; (d) J. D. Williams and C. O. Kappe, Curr. Opin. Green Sustain. Chem., 2020, 25, 100351 CrossRef.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuno, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92 CrossRef CAS PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskil, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem.–Eur. J., 2014, 20, 15226 CrossRef CAS PubMed.
- J. E. Cochran and N. Waal, Org. Process Res. Dev., 2016, 20, 1533 CrossRef CAS.
- (a) F. Levesque, M. J. Di Maso, K. Narsihan, M. K. Wismer and J. R. Naber, Org. Process Res. Dev., 2020, 24, 2935 CrossRef CAS; (b) A. Steiner, P. M. C. Roth, F. J. Strauss, G. Gauron, G. Tekautz, M. Winter, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2020, 24, 2208 CrossRef CAS; (c) J. D. Williams, M. Nakano, R. Gerardy, J. A. Rincon, O. de Frutos, C. Mateos, J.-C. M. Monbaliu and C. O. Kappe, Org. Process Res. Dev., 2019, 23, 78 CrossRef CAS.
- M. Di Filippo and M. Baumann, Eur. J. Org. Chem., 2020, 39, 6199 CrossRef.
- (a) D. I. Schuster, in The photochemistry of enones, ed. S. Patai and Z. Rappoport, John Wiley & Sons Ltd., 1989, vol. 2, pp. 623–756 Search PubMed; (b) P. E. Eaton, Acc. Chem. Res., 1968, 1, 50 CrossRef CAS; (c) M. Sisa, S. L. Bonnet, D. Ferreira and J. H. Van der Westhuizen, Molecules, 2010, 15, 5196 CrossRef CAS.
- (a) K. Donnelly and M. Baumann, J. Flow Chem., 2021 DOI:10.1007/s41981-021-00168-z , in print; (b) E. Kayahan, M. Jacobs, L. Braeken, L. C. J. Thomassen, S. Kuhn, T. van Gerven and M. E. Leblebici, Beilstein J. Org. Chem., 2020, 16, 2484 CrossRef CAS PubMed.
- The crystal structures of compounds 7a and 13a were deposited with the Cambridge Crystallographic Database as CCDC-2072295 and 2072296, respectively.
- (a) M. Iwashima, I. Terada, K. Okamoto and K. Iguchi, J. Org. Chem., 2002, 67, 2977 CrossRef CAS PubMed; (b) H. Ito, M. Hasegawa, Y. Takenaka, T. Kobayashi and K. Iguchi, J. Am. Chem. Soc., 2004, 126, 4520 CrossRef CAS PubMed; (c) M. Harmata and S. Wacharasindhu, Org. Lett., 2005, 7, 2563 CrossRef CAS PubMed.
- Elongation of the C–C bond within the cyclopropane ring was observed through computation, see ESI† for full details.
- J. Yoshida, H. Kim and A. Nagaki, J. Flow Chem., 2017, 7, 60 CrossRef CAS and references therein..
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, in Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- Related diradical species have been reported in thermal alkyne-based cyclisation cascades; please see: S. Saaby, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2005, 3, 3365 RSC.
- Tautomerisation of int3 to an indene diradical (int3′, ΔG = 17.6 kcal mol−1) is also conceivable based on computations, followed by a 1,5-H shift yielding int4:
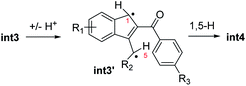 .
. - (a) R. Hoffmann, Tetrahedron Lett., 1970, 33, 2907 CrossRef; (b) G. Maier, Angew. Chem., Int. Ed., 1967, 6, 402 CrossRef CAS; (c) O. A. McNamara and A. R. Maguire, Tetrahedron, 2011, 67, 9 CrossRef CAS; (d) Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814 CrossRef CAS.

Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2072295 and 2072296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02879k |
| This journal is © The Royal Society of Chemistry 2021 |