
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple valence electron detachment following Auger decay of inner-shell vacancies in gas-phase DNA
Wen
Li
a,
Oksana
Kavatsyuk
b,
Wessel
Douma
a,
Xin
Wang
a,
Ronnie
Hoekstra
a,
Dennis
Mayer
c,
Matthew S.
Robinson
 cd,
Markus
Gühr
c,
Mathieu
Lalande
e,
Marwa
Abdelmouleh
e,
Michal
Ryszka
e,
Jean Christophe
Poully
e and
Thomas
Schlathölter
*a
cd,
Markus
Gühr
c,
Mathieu
Lalande
e,
Marwa
Abdelmouleh
e,
Michal
Ryszka
e,
Jean Christophe
Poully
e and
Thomas
Schlathölter
*a
aUniversity of Groningen, Zernike Institute for Advanced Materials, Nijenborgh 4, 9747AG Groningen, Netherlands. E-mail: t.a.schlatholter@rug.nl
bUniversity College Groningen, Hoendiep 23/24, 9718BG Groningen, Netherlands
cUniversität Potsdam, Institut für Physik und Astronomie, 14476 Potsdam, Germany
dCentre for Free Electron Lasers (CFEL), DESY, Notkestraße 85, 22607 Hamburg, Germany
eCIMAP UMR 6252 (CEA/CNRS/ENSICAEN/Université de Caen Normandie), Boulevard Becquerel, 14070 Caen Cedex 5, France
First published on 10th September 2021
Abstract
We have studied soft X-ray photoabsorption in the doubly deprotonated gas-phase oligonucleotide [dTGGGGT–2H]2−. The dominating decay mechanism of the X-ray induced inner shell vacancy was found to be Auger decay with detachment of at least three electrons, leading to charge reversal of the anionic precursor and the formation of positively charged photofragment ions. The same process is observed in heavy ion (12 MeV C4+) collisions with [dTGGGGT–2H]2− where inner shell vacancies are generated as well, but with smaller probability. Auger decay of a single K-vacancy in DNA, followed by detachment of three or more low energy electrons instead of a single high energy electron has profound implications for DNA damage and damage modelling. The production of three low kinetic energy electrons with short mean free path instead of one high kinetic energy electron with long mean free path implies that electron-induced DNA damage will be much more localized around the initial K-shell vacancy. The fragmentation channels, triggered by triple electron detachment Auger decay are predominantly related to protonated guanine base loss and even loss of protonated guanine dimers is tentatively observed. The fragmentation is not a consequence of the initial K-shell vacancy but purely due to multiple detachment of valence electrons, as a very similar positive ion fragmentation pattern is observed in femtosecond laser-induced dissociation experiments.
Introduction
The production of inner-shell vacancies in biomolecular systems, in particular DNA, is one of the first steps in many types of biological radiation action.1–3 Therapeutic X-ray photons and electrons, protons and heavy ions at clinically relevant kinetic energies are all likely to create inner-shell holes but also natural nuclear decay processes can do so.2,4–6 Their large internal excitation energy renders such vacancies of key radiobiological importance.7 In light atoms, which make up biological tissue, the excitation energy is released mostly in (cascades of) radiationless Auger transitions in which the excess energy from the de-excitation process is transferred to a second electron that is emitted from the atom. The emission of these secondary electrons is considered responsible for a large part of the biological radiation damage.3 From the perspective of molecular damage, it makes a difference if one high-energy secondary electron is emitted or whether the Auger process is accompanied by many-electron detachment. In the latter case the molecular damage and break-up is expected to be more localized and prominent. Here we address the outstanding question how (un-)likely it is that inner-shell Auger decay in a single, large biomolecule is accompanied by many-electron detachment.In radiotherapy of deep-seated tumors, one uses hard X-rays of MeV photon energies,8 protons of hundreds of MeV or swift heavy ions of GeV energies.9 For such high-energy photons and particles cross sections for the creation of inner-shell vacancies in a single molecule are very small and their beams are technically difficult to handle.10 Therefore, for the efficient production of inner-shell vacancies in light atoms such as C, N, and O we use soft X-rays. The energies of soft X rays are tuned on and over the respective K-shell absorption edges.11
For investigation of the direct molecular response of DNA to inner-shell excitation and ionization without effects of the environment gas-phase studies are have proven very useful.12 Only in the gas-phase, it is directly possible to measure interaction products such as low energy electrons which are extremely short-lived in solution. Gas-phase studies allow to distinguish genuine molecular properties from effects that are due to the chemical environment and last but not least gas-phase studies are ideally suited for comparison to quantum chemical calculations. It has been shown that standard crossed molecular beam techniques in which the molecules are evaporated from an oven cannot be used for molecules, much larger than the nucleobases.13
Bari et al.14 and Milosavljevic et al.15 independently developed tandem-mass-spectrometry approaches tailored to the investigation of radiation action on complex gas-phase biomolecular systems. Here electro-sprayed biomolecular ions are mass-over-charge selected and accumulated in a radiofrequency ion trap where they can be exposed to photons or ions. The technique was quickly successfully applied to soft X-ray interaction with gas-phase protonated peptides16 and proteins.17
As DNA in biological systems is typically negatively charged, it is essential for a biologically relevant study to focus on DNA in a deprotonated state. Studies in negative ions are technically more challenging, in particular for singly or doubly deprotonated molecules, because (many-) electron detachment can lead to neutrals which are lost from the ion trap or to oppositely charged positive fragments. Many radiofrequency ion traps with linear geometries are not well-suited for simultaneous trapping of positive and negative ions. Our home-built system is based on the classical Paul-trap design allowing ion trapping independent of the sign of the electric charge.
In this study, we first present direct evidence for the X-ray induced generation of positive fragment ions from deprotonated gas-phase DNA. Multiple electron detachment Auger decay of the carbon 1s vacancy will be discussed as the underlying process. We will then present evidence for the same process in the therapeutically relevant collisions of 12 MeV C4+ ions on deprotonated DNA, in which carbon 1s vacancies are induced as well. In a last step, we will demonstrate that even though the carbon 1s vacancy is the initial step of the multiple ionization process, the subsequent fragmentation dynamics can also be triggered by multiple valence ionization, for instance by infrared multiphoton absorption in intense femtosecond laser pulses.
Methods
All experiments in this study were performed using a home-built tandem mass spectrometer, designed for interfacing with synchrotron or accelerator beamlines. The system has been described in detail before.14,18,19 For the soft X-ray experiments, we used the U49-2_PGM-1 soft X-ray beamline at BESSY II synchrotron at Helmholtz–Zentrum Berlin20 (Germany) with photon energy at the C K-edge 280–310 eV (typically 4.0 × 1013 photons per s−1 for a 600 μm slitwidth). The C4+ experiments were performed with the IRRSUD heavy ion beamline at the Grand Accélérateur National d’Ions Lourds (GANIL), in Caen (France). The C4+ ions at a total kinetic energy of 11.76 MeV (typically 1.4 × 1011 ions per s) passed a 2 mm diaphragm right before the ion trap.The IR fs-laser experiments were performed using the Potsdam Coherent Astrella Ti:Sapphire femtosecond laser system (center wavelength: 800 nm, repetition rate: 1 kHz, measured pulse length: 47 fs). A lens was used to focus the beam to a measured spot size of 103.5 μm at the laser–sample interaction point.
Briefly, oligonucleotide anions (synthesized at LGC Biosearch technologies, Risskov, Denmark) were generated from a 40μM solution (20/80 (v:v) water/methanol, UHPLC grade chemicals, Sigma-Aldrich) in an electrospray ionization (ESI) source. The anions were then phase-space compressed into a well-defined beam using an RF ion funnel. The ions were accumulated in an RF octopole ion trap/guide. Bunches of ions were extracted into a RF quadrupole mass filter to select the anions [dTGGGGT–2H]2− (m/z = 931 Da, see Fig. 4 and 1 for the schematic structure) and [dTTGGGT–2H]2− (m/z = 918 Da). The mass-selected ions were accumulated in an RF Paul trap and cooled by He buffer gas collisions. We present [dTGGGGT–2H]2− data for the soft X-ray and MeV ion studies but [dTTGGGT–2H]2− data for the IR fs-laser experiments. In the IR case, the recorded [dTGGGGT–2H]2− dataset was smaller, but featured almost identical mass spectra.
 | ||
| Fig. 1 Chemical structure of the dTGGGGT oligonucleotide. | ||
In the trap, the oligonucleotide anions where exposed to soft X-rays photons at the C K-edge (texposure ∼ 1s), to 11.76 MeV C4+ ions (texposure ∼ 1s), or IR fs laser pulses (texposure can be varied, typically below 1s). The trap content was then extracted into a time-of-flight (TOF) mass spectrometer. The sign of the extraction voltage was chosen in order to extract either negative or positive ions. A few hundred of individual TOF spectra were averaged to obtain a mass spectrum of sufficient signal to noise ratio. The typical acquisition time for single mass spectra presented in this work was between 15 and 60 minutes. It is important to realize that exposure of doubly deprotonated oligonucleotides to soft X-rays, MeV ions and IR fs laser pulses can also lead to formation of neutral precursor molecules or neutral fragments. Neutrals are always lost from the ion-trap and therefore inaccessible in our experiment. Furthermore, both trapping efficiency and detection efficiency depend strongly on the m/q of an ion. This, together with the fact that multiple charge fragments can be produced from a single precursor ion, makes it impossible to directly compare the amount of precursor loss with the total yield of positive and negative interaction products. It is however possible to compare the relevance of neutralization for the soft X-rays, MeV ions and IR fs pulses.
Results and discussion
In 2013, Gonzalez-Magaña and co-workers performed a pioneering study of a doubly protonated oligonucleotide (dGCAT, where G, C, A and T are guanine, cytosine, adenine and thymine).21 Mostly protonated and radical nucleobase cations as well as a prominent deoxyribose fragment were detected for X-ray photoabsorption at the various 1s absorption edges. Glycosidic bond cleavage was identified as a key mechanism and the radiation sensitivity of deoxyribose was confirmed. In biological systems and at physiological pH, the phosphate groups are deprotonated and thus negatively charged. Biologically relevant gas-phase studies can take this into account by using deprotonated oligonucleotides.Fig. 2a and b shows two [dTGGGGT–2H]2− photofragmentation spectra obtained at the C K-edge. The photon energies are EX-ray = 292 eV (in protonated peptides and proteins, this energy is within the carbon 1s excitation range;22,23 for deprotonated biomolecules it is most likely in the transition region between excitation and ionization) and EX-ray = 300 eV (this energy is deep in the ionization range).
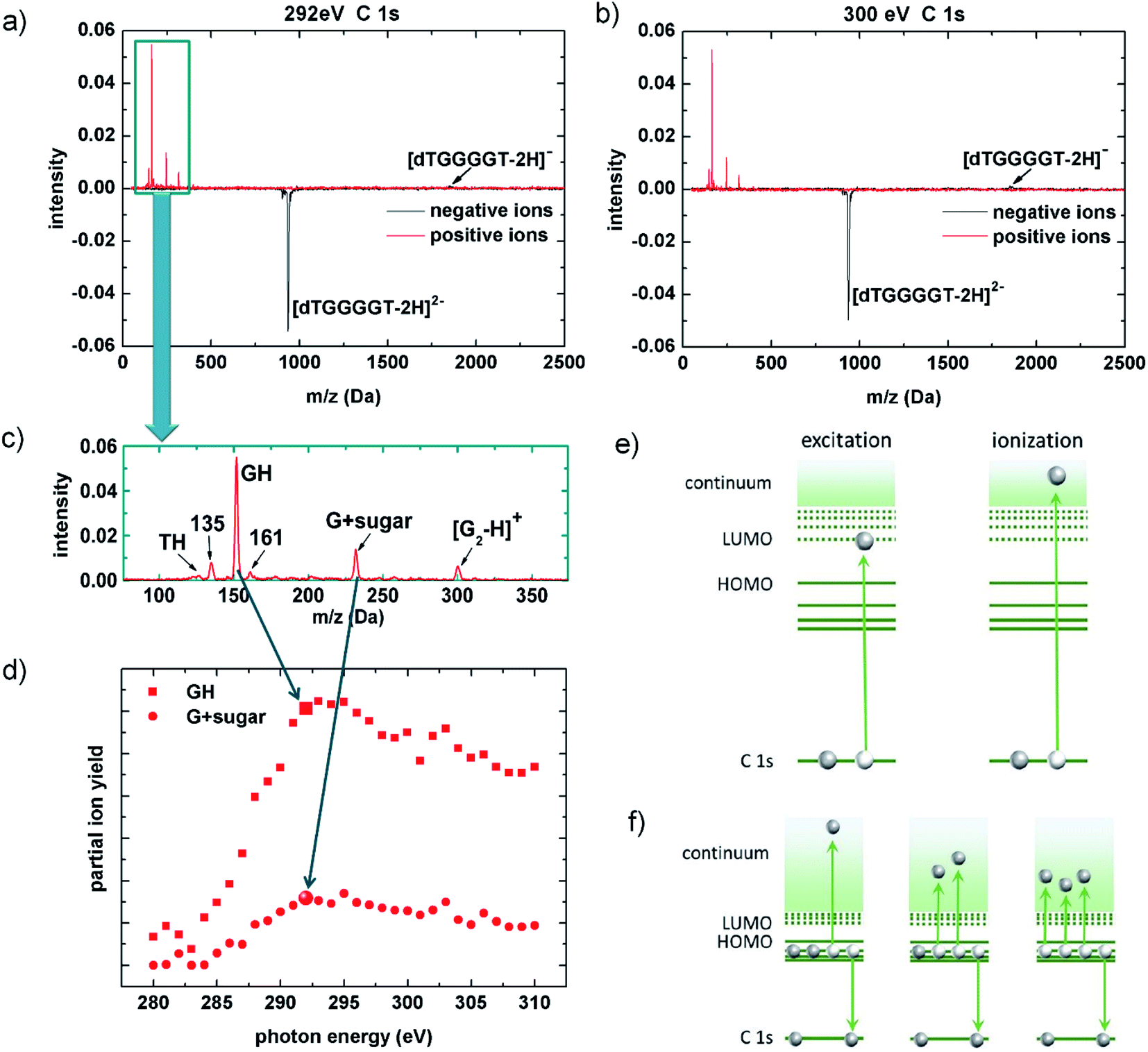 | ||
| Fig. 2 Top panel: X-ray photoabsorption mass spectra of [dTGGGGT–2H]2− anions (m/z = 930.6) for EX-ray = 292 eV (a) and 300 eV (b) (black: negative ions, red: positive ions). (c) Zoom into the cationic fragment region 75–375 Da. (d) Relative total photoabsorption cross sections as a function of photon energy for two fragments. (e) Schematics for C 1s excitation and ionization. (f) Sketch for single, double and triple valence electron detachment, associated to an Auger decay. | ||
The spectra show the net-effect of the photoabsorption on the ion-trap content. Loss of the precursor manifests as a peak of negative intensity and photoproducts (anions or cations) appear as positive peaks. The yields are normalized to the initial trap content and to the number of photons to which the trap content has been exposed. For both photon energies, non-dissociative detachment of one electron forming [dTGGGGT–2H]1− is observed as a very weak feature as compared to the depletion of the [dTGGGGT–2H]2− precursor from the trap. Even if detection efficiency decreases with m/z, this effect cannot compensate for the lack of signal attributed to [dTGGGGT–2H]1−. The absence of negatively charged fragments is different from what we observed at lower vacuum ultraviolet (VUV) energies, where only valence shell vacancies (that, in a radiobiological context, can be considered as oxidative damage) are induced:18 in deprotonated G-rich oligonucleotide anions, the photon-induced holes were found to migrate towards an energetically favorable G-rich site where they can weaken bonds and ultimately lead to bond cleavages and formation of anionic fragments.18
So, what is happening with the photoexcited/photoionized oligonucleotides in the soft X-ray case? As mentioned before, a straightforward explanation would be neutralization leading to loss from the ion trap. A second possibility is the formation of positive ions. K-shell ionization-induced production of positive ions from anionic precursors has only been observed for atoms and small molecules before.24 For fullerene anions, multiple electron detachment upon single VUV photon absorption has been reported.25 The red curves in Fig. 2a and b indeed show evidence for very efficient production of small cationic fragments with masses below 500 Da. No cationic fragments with m/q exceeding the range of 300 are observed and in particular, no non-dissociative multiple (three or more) electron detachment that would lead to positively charged dTGGGGT moieties is observed. This implies that positive ion formation due to X-ray induced multiple electron detachment induces extensive fragmentation and is thus closely related to X-ray induced DNA damage. This fact is even more clear, when zooming in on the small positive fragments (see Fig. 2c): the most intense fragment GH+ (152 Da) is due to glycosidic bond cleavage accompanied by double H/proton transfer, a very common process also observed in photofragmentation26 but also in collision-induced dissociation27 of protonated oligonucleotides. The peak at 135 Da is most likely formed by NH3 loss from GH+, which is a common fragmentation channel in protonated guanine.28,29 And the strong peak at 232 Da can be assigned to a cyclic nucleoside complex containing the guanine moiety and the five-membered sugar ring30 (G + sugar). One additional strong fragment that is not commonly observed is found at 301 Da. This fragment could be assigned to a (G2–H)+-dimer, formed by double glycosidic bond scission accompanied by single H transfer.
To demonstrate that positive fragment formation is due to inner-shell excitation/ionization rather than (non-resonant) valence ionization, we choose a spectroscopic approach: by recording the partial ion yields from the peak integrals of selected photoproducts in the mass spectra in Fig. 2c as a function of EX-ray, the [dTGGGGT–2H]2− near-edge X-ray absorption mass spectrometry (NEXAMS) spectra for GH+ and [G + sugar]+ shown in Fig. 2d have been obtained. In qualitative agreement with our results for smaller, protonated oligonucleotides,26 the spectra feature a strong increase between 285 eV and 292 eV that is due to resonant C 1s excitation into unoccupied molecular orbitals, mostly of π* character (see Fig. 2e, left). At higher photon energy, direct C 1s ionization sets in (see Fig. 2e, right). In the ionization continuum between 295 eV and 300 eV the ion yields show the expected slow decrease. The transition from 1s excitation to 1s ionization implies the removal of an additional electron in the ionization case. With the presented data, we are not able to quantify the influence of this effect.
Inner shell ionization clearly is not a requirement for positive ion formation, as positive nucleobase ion formation is observed already in the excitation regime. This rules out direct photodouble detachment31 and even single detachment as an underlying process. The induction of a K-shell vacancy is sufficient for positive fragment ion formation, and as a consequence, Auger de-excitation (see Fig. 2f) must be an important step. The Feifel group has systematically studied double Auger decay, i.e., Auger process in which two valence electrons are emitted, for a series of small molecules.32 The percentage of double Auger processes systematically increases with the number of valence electrons available in the vicinity of the core vacancy, i.e. with the density of valence orbitals (from 6.3% in CH4 to 29% in CCl4 for a carbon 1s hole).33 If we extrapolate this trend, double Auger decay could be a dominant channel in large molecules such as oligonucleotides. However, only triple electron detachment can explain the observed positive ion formation from a doubly negative precursor. For small neutral molecules such as CO2, about 1% of the total Auger process are due to triple Auger decay for a C 1s vacancy, with the percentage again increasing with valence state density.34 In negative ions, multiple electron detachment due to K-shell vacancy followed by Auger decay has until now only been studied for atoms and small molecules (i.e., C-35 and B2-/B3-36). Multiple detachment is generally much stronger in negative than in neutral systems. In a very recent study on C-, it was for instance found that upon excitation of the lowest energy resonance, triple detachment was 5% and quadruple detachment was about 0.1% of the double detachment cross sections.37 Triple electron detachment in a C 1s decay process could thus realistically be explained by a multiple electron detachment Auger decay, but it remains remarkable that this channel is so strong in our system under study.
Inner shell vacancies can also be produced in proton and heavy ion therapy. It is thus straightforward to investigate the response of the (dTGGGGT–2H)2− upon interactions with a therapeutically relevant ion beam. Carbon ion beams for cancer therapy typically have energies in the GeV range. This energy is however chosen such that the ions are slowed down to the 10 MeV range at the tumor location. At these kinetic energies, energy deposition per unit length is maximum (Bragg-peak),38 which is why we have chosen a 11.76 MeV C4+ beam for our study. MeV ion collisions predominantly lead to emission of valence electrons, and only collisions with very small impact parameters lead to inner shell ionization, as recently shown for 42 MeV C6+ collisions with the gas-phase adenine nucleobase, where only about 4% of the total ionization cross section where due to K shell ionization.39–42
A mass spectrum obtained after 11.76 MeV C4+ collisions with (dTGGGGT–2H)2− is shown in Fig. 3a. The negative ion spectrum is very similar to the soft X-ray case, with strong loss of the (dTGGGGT–2H)2− precursor, that is not compensated by the small yield of (dTGGGGT–2H)−. Non-dissociative electron detachment is relatively stronger as compared to the soft X-ray case, but it remains a minor channel also for the MeV ion case. The positive ion spectrum again shows clear peaks in the low mass region. However, relative to the precursor loss, these peaks are much weaker as compared to the soft X-ray case, in agreement with the very low probability for 1s ionization in MeV ion collisions. The comparable yields for non-dissociative detachment and the significantly lower yield of positive fragments implies that in the MeV ion case, a larger fraction of interaction products has to be neutral and remains inaccessible in our study. It is very likely that these additional neutral product are due to double valence ionization rather than K-shell ionization. Fig. 3b shows a zoom in the low mass region of the positive ion spectrum. Essentially the same fragments as those formed upon soft X-ray absorption (Fig. 2c) are observed (the additional peak for C4H5N4 – a guanine fragment43 lacked in the soft X-ray mass spectrum because of a slightly higher low-mass cutoff of the ion trap). In summary, the relatively small cross section for K-shell ionization in MeV ion collisions leads to a lower yield of positive fragments, but the 1s ionized oligonucleotides appear to decay into the same fragmentation pattern as observed for soft X-ray absorption. There might be a contribution from the much more likely MeV ion induced multiple valence ionization, but it is difficult to disentangle these two channels.
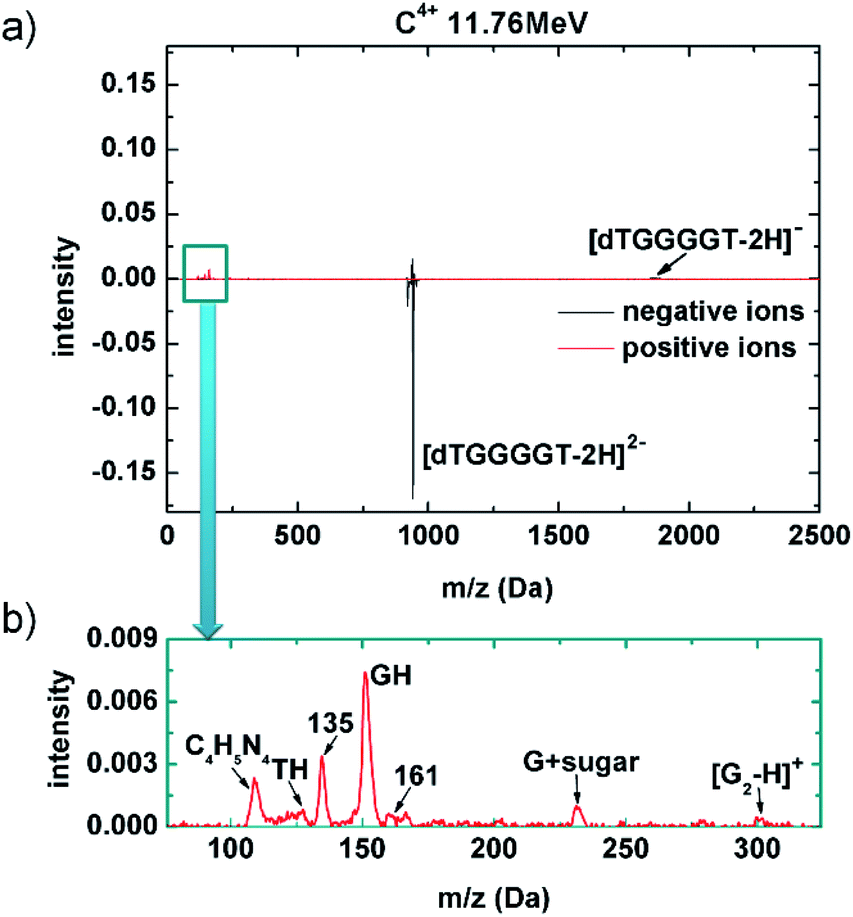 | ||
| Fig. 3 Mass spectra for 10 MeV C4+ collisions with (dTGGGGT–2H)2−. (a) Both negative (black) and positive (red) ion data are displayed. (b) Zoom into the region of small positive fragments. | ||
In a last step, we demonstrate that the K-shell vacancy is only required to facilitate multiple electron detachment in Auger decay, whereas the observed actual fragmentation (e.g., glycosidic bond cleavage) is due to valence ionization, only. We have performed complementary femtosecond laser-induced ionization/dissociation (fs-LID) experiments on (dTTGGGT–2H)2−. In fs-LID, molecules are exposed to near infrared pulses of several 10 fs duration and with peak powers of the order of 1013 W cm−2. We have employed pulses of length τ = 47 fs, photon energy Ephoton = 1.55 eV and peak power between 9.6 × 1012 W cm−2 and 8.1 × 1013 W cm−2. The ionization of molecular anions in femtosecond laser pulses has only been studied for a few small molecules such as SF6 before.44 Detachment of the weakly bound electrons from a doubly negative precursor requires less energy, and therefore much lower laser intensities, than positive ion formation from a neutral or cationic molecule.45
Daly et al. have recently performed a ns-pulse UV-vis spectroscopic study on deprotonated gas phase oligonucleotide 6mers in different charge states.45 For [dGGGGGG–2H]2−, they observed a gas-phase absorption spectrum very close to the one observed in solution, with a pronounced band 1 centered at about 4.9 eV and band 2 centered at about 6.4 eV. The energetic position of these maxima is only weakly dependent on nucleobase composition and deprotonation state and consists of a multitude of excited states. Besides radiative decay back to the ground-state, excitation into one of these bands can lead to the two different processes sketched in Fig. 4. In both cases, the excited oligo can decay back to the ground state via internal conversion (IC). The vibrational excitation that results from a single excitation process is insufficient to induce fragmentation of an oligonucleotide 6-mer.45 However, according to Daly et al.'s study,45 the sum of adiabatic detachment energy (ADE) and repulsive coulomb barrier (RCB) is just below the first absorption band (e.g. 4.1 eV for [dGGGTTT–2H]2−), opening up an efficient electron detachment channel. Interestingly, this channel appears to be inefficient for the case of excitation into band 2, leaving IC as the dominant channel.
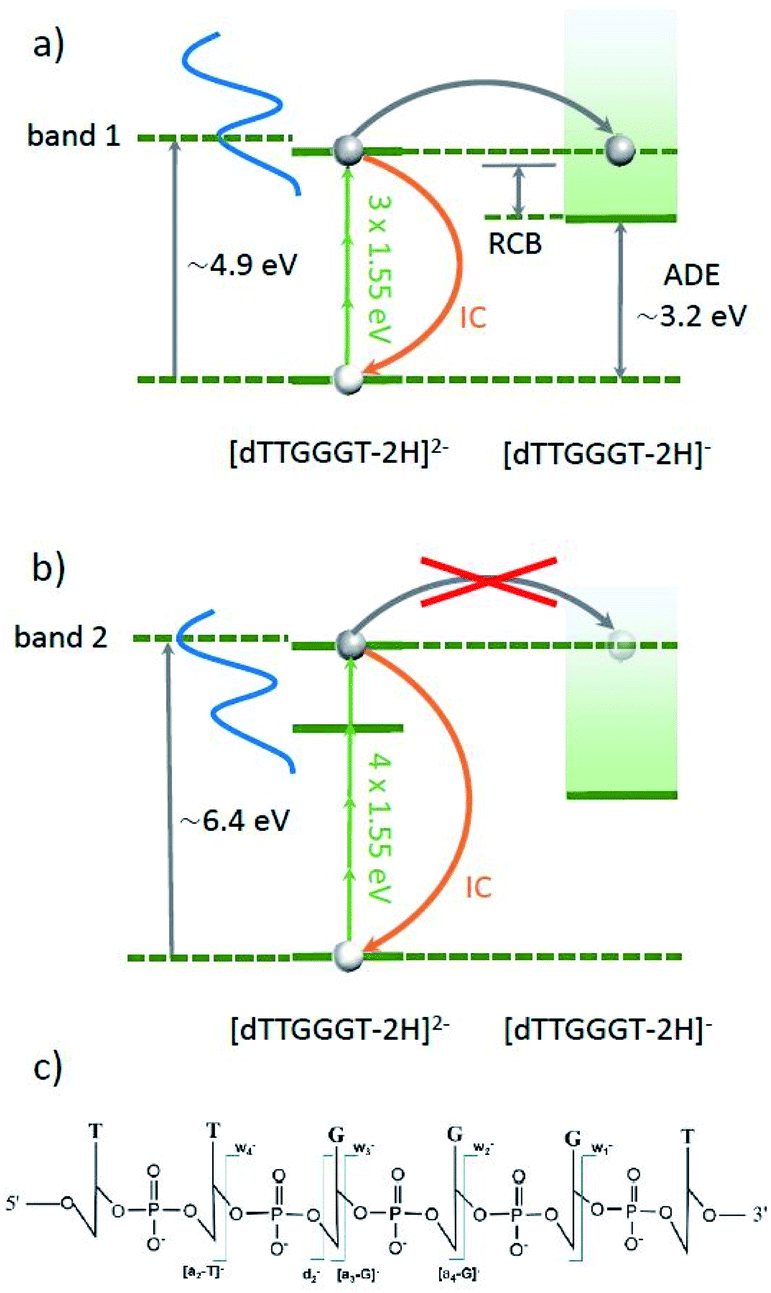 | ||
| Fig. 4 Energy schemes for resonant (3 IR-photon) excitation to band 1 (a) and for resonant (4 IR-photon) excitation to band 2 (b) of [dTTGGGT–2H]2−. In both cases, the oligonucleotide can decay radiatively back to the ground state (not shown) or the excitation energy is transferred into vibrational energy by internal conversion (IC). In both cases, electron detachment into [dTTGGGT–2H]1− is energetically allowed. For band 1, this channel is expected to be strong whereas for band two, electron detachment is very weak.45 Panel (c) shows the schematic structure of the oligonucleotide with the observed cleavage sites. | ||
In our experiment, band 1 can be reached by resonant absorption of 3 IR photons (3 × 1.55 eV ≈ 4.65 eV) and band 2 can be reached by resonant absorption of 4 IR photons (4 × 1.55 eV ≈ 6.2 eV). Variation of the fs-laser intensity allows for variation of the probability for multiphoton absorption. In line with the expectations, below a threshold peak power (here about 1013 W cm−2), the laser beam has no effect on the trapped [dTTGGGT–2H]2− anions. In the threshold region, where we expect 3 photons to be absorbed, only electron detachment into [dTTGGGT–2H]1− is observed (see Fig. 5c, 1st panel). It is likely, that many molecules decay to the precursor ground state by IC but the excitation energy is insufficient for fragmentation.
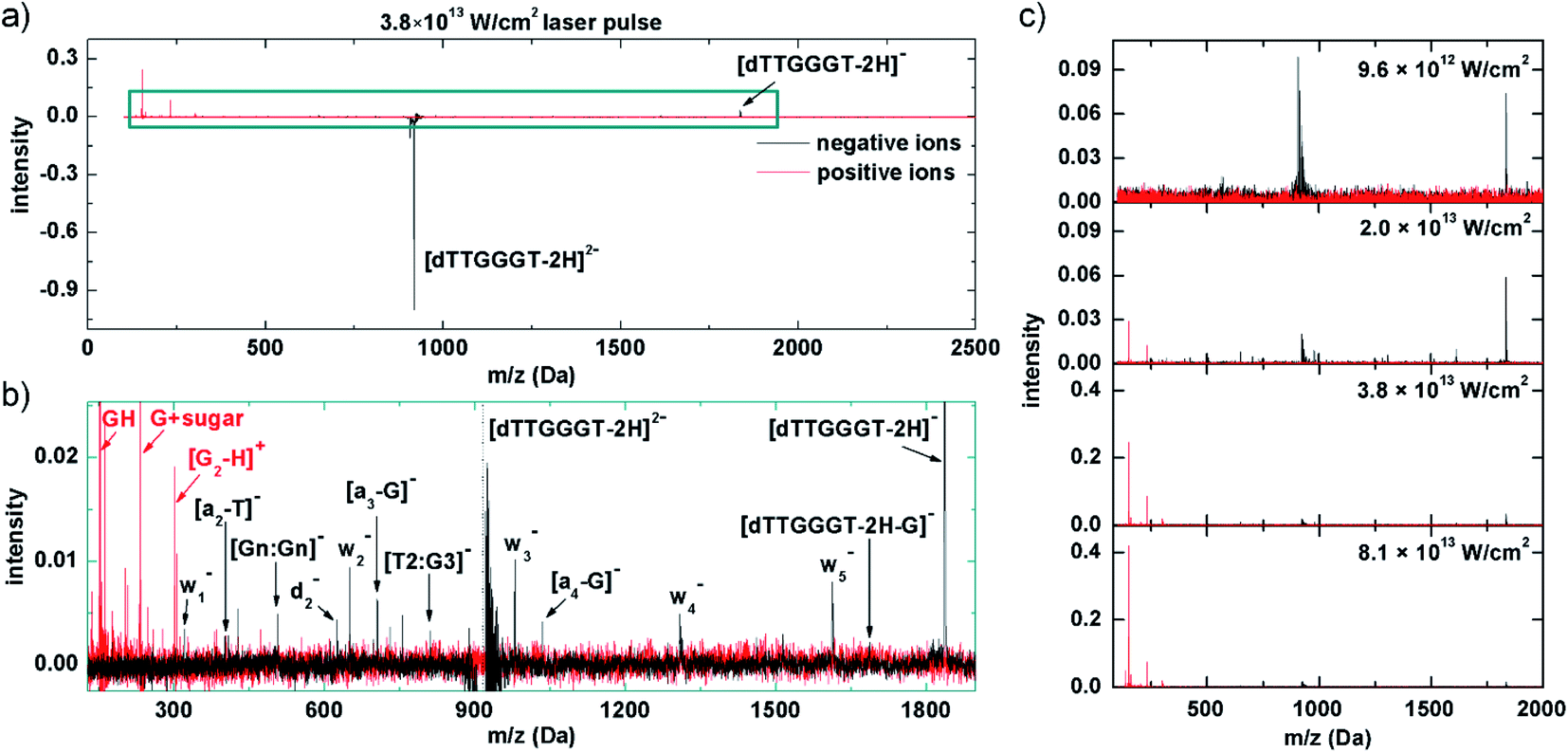 | ||
| Fig. 5 Mass spectra of the fs-LID products from (dTTGGGT–2H)2− for 47 fs 800 nm laser pulses. (a) Data for 3.8 × 1013 W cm−2 pulses (black: negative ions, red: positive ions); (b) zoom into the fragment region; (c) spectral evolution as a function of pulse energy; the strong positive peak observed for 9.6 × 1012 W cm−2 is an artifact at the m/q of the precursor ion. | ||
Doubling the laser power to 2 × 1013 W cm−2 leads to the mass spectrum in the 2nd panel of Fig. 5c. The electron detachment channel is still dominant, but a number of negative fragment ions appear which could be explained by increased vibrational energy in the system after IC from additional photon absorption (i.e. 3+). It is well established that moderately deprotonated oligonucleotides predominantly fragment by loss of a base B (here: G or T) followed by scission of the corresponding sugar 3′ C–O bond (see Fig. 5c).46 This process leads to wn− and (a–B)− and both types of fragments are abundant in the mass spectrum (see Fig. 5b for a zoom into a mass spectrum recorded at higher laser power, the negative-ion fragments are qualitatively similar at different laser powers). A similar pattern has also been observed in VUV photoionization of similar oligonucleotides.18. [Gn:Gn]− and [T2:G3]− are internal fragments.
Interestingly, also positive fragments are already formed at 2 × 1013 W cm−2. At this laser power, detachment of multiple electrons and sufficient energy transfer into vibrational modes via IC into the same molecule is not yet a likely combination of processes to happen. An alternative scenario would be a nucleobase-centered fs absorption process in which an electron is detached and the glycosidic bond is broken before the base can be neutralized. As a result, a positively charged nucleobase as well as 2 negative w- and (A–B)− fragments could be formed. To our knowledge, such a process has not been observed before, though.
Another doubling of the laser power to 3.8 × 1013 W cm−2 (3rd panel of Fig. 5a and c) leads to a moderate reduction in intensity for both, non-dissociative detachment and formation of negative fragments. The most dramatic change is observed for the positive fragment yields which increase in a non-linear fashion by almost one order of magnitude. The positive ion yield now clearly exceeds the negative ion yield, implying multiple electron detachment as an underlying process. Depending on the vertical detachment energy of the system, the Keldish parameter47 could be below a value of 1 and tunneling ionization of outer valence electrons (in addition to (resonant-enhanced) multi-photon ionization processes) could become possible.48 Tunneling ionization has occasionally been used in mass spectrometry before, in particular for protonated peptides,49,50 as no chromophores are required and new, non-ergodic fragmentation channels are known to open up. In any case, electron tunneling is strongly sensitive to electron binding energies which are dependent on detachment site and charge state. A thorough investigation of the respective dynamics is beyond the scope of this article, but it is clear that the mass spectrum results from removal of multiple valence electrons together with substantial electronic (and ultimately vibrational) excitation of the system. As observed for soft X-ray absorption and MeV ion impact, the positive ion spectrum is dominated by GH+, (G + sugar)+ and the tentative (G2–H)+ fragment (see Fig. 5b).
A further doubling of the laser power to 8.1 × 1013 W cm−2 leads to the positive and negative ion mass spectra in the 4th panel of Fig. 5c. Negative ion yields further decrease and the positive fragment ion yields approximately double with respect to the 3.8 × 1013 W cm−2 case. The positive ion fragmentation pattern very much resembles the one for the soft X-ray case (Fig. 2a). This confirms the proposed scenario in which valence ionization is responsible for the positive fragment formation.
It is important to note that for this spectrum the laser power is well in the tunneling-regime and the increase in positive ion yields has already leveled off. From this we conclude that the formation of neutral photoproducts is probably negligible but certainly not dominating. As the spectrum is very similar to Fig. 2a, the same is probably true for the soft X-ray case and only for MeV ions, where positive ion formation is relatively weaker, neutralization may play a bigger role.
Note that in the fs-laser experiments, different power regimes can contribute to the same mass spectrum; e.g. electron detachment is observed at both 9.6 × 1012 and 2.0 × 1013 W cm−2, and positive ions are observed at 2.0 × 1012 and 3.8 × 1012 W cm−2. This is because the beam has a Gaussian intensity profile: precursor ions are exposed to the nominal intensity solely at the center of the beam. Oligonucleotides can be multiply ionized and positive fragments form. Away from the center, intensities are lower and single electron detachment may still dominate. At the moment, we take care to mention that these proposed mechanisms are unfortunately still somewhat speculative due to lack of similar experiments in the literature. Despite basing our proposed mechanisms on similar power ranges scans and results performed by Daly et al., we do note that there are differences between the two experiments in terms of the photon energy used (IR vs. UV) and the duration of the laser pulses used (fs vs. ns), which will massively affect which states are populated, which mechanisms are involved as well as the timescales in which ionisation/dissociation occurs. More work is therefore needed to elucidate how exactly (1) photon energy, (2) pulse duration and (3) laser intensity all affect the dissociation of these systems in to both positive and negative fragments. The preliminary observations presented in this article are simply the first step into these new studies.
Conclusion
We have shown evidence for strong three (or more) electron detachment in the Auger decay of a K-shell vacancy in gas-phase deprotonated oligonucleotides. This process is characteristic for soft X-ray absorption around the carbon K-edge, but can also be observed in heavy ion induced DNA damage where inner shell vacancies are generated as well. The DNA damage processes that are triggered by triple electron detachment Auger decay are predominantly related to glycosidic bond cleavage accompanied by H transfer (base loss). An indication for loss of protonated guanine dimers is observed as well. The actual damage mechanism clearly is not related to the initial K-level vacancy but due to the induced multiple detachment of valence electrons, as a very similar positive ion fragmentation pattern is observed in fs-LID experiments.Triple electron detachment Auger decay of K-shell vacancies in DNA has profound consequences for DNA damage and for damage modeling. A single Auger electron emitted after decay of a carbon 1s vacancy at about 300 eV has a penetration range of about 10 nm whereas triple electron detachment decay would imply substantially smaller electron energies, with smaller penetration depths (∼3–4 nm for electron energies below 100 eV).51 As a result, damage due to secondary electrons from inner shell vacancy decay in DNA very likely is much more localized than previously thought. The localized damage of three or more low energy electrons emitted after the decay of a single K-shell vacancy in DNA could be sufficient to induce a double strand break or possibly even a cluster of DNA damage.52
In future experiments, we plan to measure the energy distribution of the emitted electrons and to investigate the influence of charge state, length and sequence of the deprotonated oligonucleotide on positive ion formation. This knowledge is crucial to understand how far the results of our gas-phase studies can be transferred to the (biologically relevant) liquid phase. Of particular interest would be the investigation of nanosolvated systems, i.e. deprotonated gas-phase oligonucleotides with attached water molecules. Furthermore, in particular electron spectroscopy will give much deeper insight into the actual Auger decay processes associated with individual excited states, the precise number of electrons emitted and possibly answer the question whether the soft-X-ray excited 1s electron participates actively in the subsequent Auger decay or whether it is merely a spectator.
Data availability
The data for Fig. 2, 3 and 5 can be found in the ESI.Author contributions
According to CASRAI CRediT (contribution role taxonomy). (1) Conceptualization; Wen Li, Thomas Schlathölter. (2) Data curation; Thomas Schlathölter. (3) Formal analysis; Wen Li, Xin Wang, Thomas Schlathölter. (4) Funding acquisition; Thomas Schlathölter, Ronnie Hoekstra, Markus Gühr, Jean Christophe Poully. (5) Investigation; Wen Li, Wessel Douma, Xin Wang, Dennis Mayer, Matthew S. Robinson, Markus Gühr, Mathieu Lalande, Marwa Abdelmouleh, Michal Ryszka, Jean Christophe Poully, Thomas Schlathölter. (6) Methodology; Thomas Schlathölter, Markus Gühr, Jean Christophe Poully. (7) Project administration; Wen Li, Thomas Schlathölter. (8) Resources; Thomas Schlathölter, Jean Christophe Poully, Ronnie Hoekstra, Markus Gühr. (9) Software; Thomas Schlathölter. (10) Supervision; Thomas Schlathölter, Markus Gühr, Jean Christophe Poully. (11) Validation; Wen Li, Matthew S. Robinson, Thomas Schlathölter. (12) Visualization; Wen Li, Oksana Kavatsyuk, Thomas Schlathölter. (13) Writing – original draft; Wen Li, Oksana Kavatyuk, Thomas Schlathölter. (14) Writing – review & editing; Wen Li, Oksana Kavatsyuk, Matthew S. Robinson, Ronnie Hoekstra, J. C. Poully, Thomas Schlathölter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank HZB for the allocation of synchrotron radiation beamtime. The research leading to this result has been supported by the project CALIPSOplus under the grant agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. W. L. and X. W. acknowledge support by the Chinese Scholarship Council (CSC). The authors would like to acknowledge the contribution of the EU COST action MDGAS (CA18212). We are grateful to the GANIL and CIRIL staff for the support during beamtimes and to Violaine Vizcaino for help with the IRRSUD experiments. D. M., M. S. R. and M. G. acknowledge a Lichtenberg Professur from the Volkswagen Foundation. M. G. acknowledges funding of the laser system via a DFG Großgerät INST 336/112-1 FUGG.Notes and references
- R. W. Howell, Advancements in the use of Auger electrons in science and medicine during the period 2015–2019, Int. J. Radiat. Biol., 2020, 1–26 CrossRef PubMed.
- P. Bernhardt, W. Friedland and H. G. Paretzke, The role of atomic inner shell relaxations for photon-induced DNA damage, Radiat. Environ. Biophys., 2004, 43, 77–84 CrossRef CAS PubMed.
- A. Yokoya and T. Ito, Photon-induced Auger effect in biological systems: a review, Int. J. Radiat. Biol., 2017, 93, 743–756 CrossRef CAS PubMed.
- J. D. Garcia, R. J. Fortner and T. M. Kavanagh, Inner-Shell Vacancy Production in Ion-Atom Collisions, Rev. Mod. Phys., 1973, 45, 111–177 CrossRef CAS.
- V. L. Jacobs and B. F. Rozsnyai, Multiple ionization and x-ray line emission resulting from inner-shell electron ionization, Phys. Rev. A, 1986, 34, 216–226 CrossRef CAS PubMed.
- J. D. Garcia, Inner-Shell Ionizations by Proton Impact, Phys. Rev. A, 1970, 1, 280–285 CrossRef.
- B. Fayard, A. Touati, F. Abel, M. A. Herve du Penhoat, I. Despiney-Bailly, F. Gobert, M. Ricoul, A. L'Hoir, M. F. Politis, M. A. Hill, D. L. Stevens, L. Sabatier, E. Sage, D. T. Goodhead and A. Chetioui, Cell Inactivation and Double-Strand Breaks: The Role of Core Ionizations, as Probed by Ultrasoft X Rays, Radiat. Res., 2002, 157, 128–140 CrossRef CAS.
- C. H. Rim, H. J. Yim, S. Park and J. Seong, Recent clinical applications of external beam radiotherapy for hepatocellular carcinoma according to guidelines, major trials and meta-analyses, J. Med. Imaging Radiat. Oncol., 2019, 63, 812–821 CrossRef PubMed.
- T. Rackwitz and J. Debus, Clinical applications of proton and carbon ion therapy, Semin. Oncol., 2019, 46, 226–232 CrossRef CAS PubMed.
- B. Tenorio, C. M. T. Nunes, M. A. C. Nascimento, A. B. Rocha and S. Coriani, Molecular inner-shell photoabsorption/photoionization cross sections at core-valence-separated coupled cluster level: Theory and examples, J. Chem. Phys., 2019, 150, 224104 CrossRef PubMed.
- S. Bari, L. Inhester, K. Schubert, K. Mertens, J. O. Schunck, S. Dörner, S. Deinert, L. Schwob, S. Schippers, A. Müller, S. Klumpp and M. Martins, Inner-shell X-ray absorption spectra of the cationic series NHy+ (y = 0-3), Phys. Chem. Chem. Phys., 2019, 21, 1655–16514 RSC.
- H. Zettergren, A. Domaracka, T. Schlathölter, P. Bolognesi, S. Díaz-Tendero, M. Łabuda, S. Tosic, S. Maclot, P. Johnsson, A. Steber, D. Tikhonov, M. C. Castrovilli, L. Avaldi, S. Bari, A. R. Milosavljević, A. Palacios, S. Faraji, D. G. Piekarski, P. Rousseau, D. Ascenzi, C. Romanzin, E. Erdmann, M. Alcamí, J. Kopyra, P. Limão-Vieira, J. Kočišek, J. Fedor, S. Albertini, M. Gatchell, H. Cederquist, H. T. Schmidt, E. Gruber, L. H. Andersen, O. Heber, Y. Toker, K. Hansen, J. A. Noble, C. Jouvet, C. Kjær, S. B. Nielsen, E. Carrascosa, J. Bull, A. Candian and A. Petrignani, Roadmap on dynamics of molecules and clusters in the gas phase, Eur. Phys. J. D, 2021, 75, 152 CrossRef CAS.
- K. Ueda, E. Sokell, S. Schippers, F. Aumayr, H. Sadeghpour, J. Burgdörfer, C. Lemell, X. Tong, T. Pfeifer, F. Calegari, A. Palacios, F. Martin, P. Corkum, G. Sansone, E. V. Gryzlova, A. Grum-Grzhimailo, M. N. Piancastelli, P. M. Weber, T. Steinle, K. Amini, J. Biegert, N. Berrah, E. Kukk, R. Santra, A. Müller, D. Dowek, R. R. Lucchese, C. W. McCurdy, P. Bolognesi, L. Avaldi, T. Jahnke, M. S. Schöffler, R. Dörner, Y. Mairesse, L. Nahon, O. Smirnova, T. Schlathölter, E. E. B. Campbell, J. Rost, M. Meyer and K. A. Tanaka, Roadmap on photonic, electronic and atomic collision physics: I. Light–matter interaction, J. Phys. B: At., Mol. Opt. Phys., 2019, 52, 171001 CrossRef CAS.
- S. Bari, O. Gonzalez-Magaña, G. Reitsma, J. Werner, S. Schippers, R. Hoekstra and T. Schlathölter, Photodissociation of protonated leucine-enkephalin in the VUV range of 8–40 eV, J. Chem. Phys., 2011, 134, 024314 CrossRef CAS PubMed.
- A. R. Milosavljević, C. Nicolas, J. Lemaire, C. Dehon, R. Thissen, J. Bizau, M. Réfrégiers, L. Nahon and A. Giuliani, Photoionization of a protein isolated in vacuo, Phys. Chem. Chem. Phys., 2011, 13, 15432–15436 RSC.
- O. González-Magaña, G. Reitsma, M. Tiemens, L. Boschman, R. Hoekstra and T. Schlathölter, Near-Edge X-ray Absorption Mass Spectrometry of a Gas-Phase Peptide, J. Phys. Chem. A, 2012, 116, 10745–10751 CrossRef.
- A. R. Milosavljević, F. Canon, C. Nicolas, C. Miron, L. Nahon and A. Giuliani, Gas-Phase Protein Inner-Shell Spectroscopy by Coupling an Ion Trap with a Soft X-ray Beamline, J. Phys. Chem. Lett., 2012, 3, 1191–1196 CrossRef.
- W. Li, E. Mjekiqi, W. Douma, X. Wang, O. Kavatsyuk, R. Hoekstra, J. Poully and T. Schlathölter, Hole Migration in Telomere-Based Oligonucleotide Anions and G-Quadruplexes, Chem.–Eur. J., 2019, 25, 16114–16119 CrossRef CAS PubMed.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Near edge X-ray absorption mass spectrometry of gas phase proteins: the influence of protein size, Phys. Chem. Chem. Phys., 2016, 18, 26213–26223 RSC.
- T. Kachel, The plane grating monochromator beamline U49-2 PGM-1 at BESSY II, Journal of Large-Scale Research Facilities, 2016, 2, A72 CrossRef.
- O. Gonzalez-Magana, M. Tiemens, G. Reitsma, L. Boschman, M. Door, S. Bari, P. O. Lahaie, J. R. Wagner, M. A. Huels, R. Hoekstra and T. Schlathölter, Fragmentation of protonated oligonucleotides by energetic photons and Cq+ ions, Phys. Rev. A, 2013, 87, 032702 CrossRef.
- A. R. Milosavljević, F. Canon, C. Nicolas, C. Miron, L. Nahon and A. Giuliani, Gas-Phase Protein Inner-Shell Spectroscopy by Coupling an Ion Trap with a Soft X-ray Beamline, J. Phys. Chem. Lett., 2012, 3, 1191–1196 CrossRef PubMed.
- S. Bari, D. Egorov, T. L. Jansen, R. Boll, R. Hoekstra, S. Techert, V. Zamudio-Bayer, C. Bülow, R. Lindblad and G. Leistner, Soft X-ray Spectroscopy as a Probe for Gas-Phase Protein Structure: Electron Impact Ionization from Within, Chem.–Eur. J., 2018, 24, 7631–7636 CrossRef CAS PubMed.
- N. Berrah, J. D. Bozek, G. Akerman, B. Rude, G. Turri, H. Zhou and S. T. K. Manson, K-shell photodetachment of He- : experiment and theory, Phys. Rev. Lett., 2002, 88, 093001 CrossRef CAS PubMed.
- R. C. Bilodeau, N. D. Gibson, C. W. Walter, D. Esteves-Macaluso, S. Schippers, A. Müller, R. A. Phaneuf, A. Aguilar, M. Hoener, J. M. Rost and N. Berrah, Single-Photon Multiple Detachment in Fullerene Negative Ions: Absolute Ionization Cross Sections and the Role of the Extra Electron, Phys. Rev. Lett., 2013, 111, 043003 CrossRef CAS PubMed.
- X. Wang, S. Rathnachalam, K. Bijlsma, W. Li, R. Hoekstra, M. Kubin, M. Timm, B. Von Issendorff, V. Zamudio-Bayer, J. T. Lau, S. Faraji and T. Schlathölter, Site-selective soft X-ray absorption as a tool to study protonation and electronic structure of gas-phase DNA, Phys. Chem. Chem. Phys., 2021, 20, 11900–11906 RSC.
- J. Wu and S. A. McLuckey, Gas-phase fragmentation of oligonucleotide ions, Int. J. Mass Spectrom., 2004, 237, 197–241 CrossRef CAS.
- L. Sadr-Arani, P. Mignon, H. Chermette, H. Abdoul-Carime, B. Farizon and M. Farizon, Fragmentation mechanisms of cytosine, adenine and guanine ionized bases, Phys. Chem. Chem. Phys., 2015, 17, 11813–11826 RSC.
- J. M. Gregson and J. A. McCloskey, Collision-induced dissociation of protonated guanine, Int. J. Mass Spectrom. Ion Processes, 1997, 165, 475–485 CrossRef.
- J. Ni, M. A. Mathews and J. A. McCloskey, Collision-induced Dissociation of Polyprotonated Oligonucleotides Produced by Electrospray Ionization, Rapid Commun. Mass Spectrom., 1997, 11, 535–540 CrossRef CAS PubMed.
- A. Mueller, A. Borovik Jr, S. Bari, T. Buhr, K. Holste, M. Martins, A. Perry-Saßmannshausen, R. A. Phaneuf, S. Reinwardt and S. Ricz, Near-K-edge double and triple detachment of the F− negative ion: observation of direct two-electron ejection by a single photon, Phys. Rev. Lett., 2018, 120, 133202 CrossRef CAS PubMed.
- A. H. Roos, J. Eland, J. Andersson, S. Zagorodskikh, R. Singh, R. J. Squibb and R. Feifel, Relative extent of double and single Auger decay in molecules containing C, N and O atoms, Phys. Chem. Chem. Phys., 2016, 18, 25705–25710 RSC.
- A. H. Roos, J. H. D. Eland, J. Andersson, R. J. Squibb, D. Koulentianos, O. Talaee and R. R. Feifel, Abundance of molecular triple ionization by double Auger decay, Sci. Rep., 2018, 8, 16405 CrossRef PubMed.
- A. H. Roos, J. Eland, J. Andersson, M. Wallner, R. J. Squibb and R. Feifel, Relative extent of triple Auger decay in CO and CO2, Phys. Chem. Chem. Phys., 2019, 21, 9889–9894 RSC.
- N. D. Gibson, C. W. Walter, O. Zatsarinny, T. W. Gorczyca, G. D. Ackerman, J. D. Bozek, M. Martins, B. M. McLaughlin and N. Berrah, K-shell photodetachment from C−: Experiment and theory, Phys. Rev. A, 2003, 67, 030703 CrossRef.
- N. Berrah, R. C. Bilodeau, J. D. Bozek, I. Dumitriu, D. Toffoli and R. R. Lucchese, Shape resonances in K-shell photodetachment of small size-selected clusters: Experiment and theory, Phys. Rev. A, 2007, 76, 042709 CrossRef.
- A. Perry-Sassmannshausen, T. Buhr, A. Borovik Jr, M. Martins, S. Reinwardt, S. Ricz, S. O. Stock, F. Trinter, A. Müller and S. Fritzsche, Multiple photodetachment of carbon anions via single and double core-hole creation, Phys. Rev. Lett., 2020, 124, 083203 CrossRef CAS PubMed.
- R. N. Kjellberg and M. Abe, Stereotactic Bragg Peak Proton Beam Therapy, in Modern Stereotactic Neurosurgery, ed. L. D. Lunsford, Springer US, Boston, MA, 1988, pp. 463–470 Search PubMed.
- S. Bhattacharjee, C. Bagdia, M. R. Chowdhury, A. Mandal, J. M. Monti, R. D. Rivarola and L. C. Tribedi, Bare-carbon-ion-impact electron emission from adenine molecules: Differential and total cross-section measurements, Phys. Rev. A, 2019, 100, 012703 CrossRef CAS.
- S. Bhattacharjee, A. Mandal, M. R. Chowdhury, C. Bagdia, J. M. Monti, R. D. Rivarola and L. C. Tribedi, Electron emission in ionization of adenine molecule induced by 5 MeV/u bare C ions, Eur. Phys. J. D, 2020, 74, 1–5 CrossRef.
- C. Bagdia, S. Bhattacharjee, M. R. Chowdhury, A. Mandal, G. Lapicki and L. C. Tribedi, 1s–1s electron transfer in collisions of fast C and O ions with adenine, Nucl. Instrum. Methods Phys. Res., Sect. B, 2020, 462, 68–74 CrossRef CAS.
- C. Bagdia, S. Bhattacharjee, M. Roychowdhury, A. Mandal, G. Lapicki and L. C. Tribedi, K–K electron capture from adenine and CO2 molecule by fast carbon ions using KLL-Auger electron technique, X-Ray Spectrom., 2020, 49, 160–164 CrossRef CAS.
- J. Liu, S. Cao, B. Jia, D. Wei, X. Liao, J. Lu and Y. Zhao, A theoretical and mass spectrometry study of the novel mechanism of N-glycosidic bond cleavage in nucleoside, Int. J. Mass Spectrom., 2009, 282, 1–5 CrossRef CAS.
- Y. Albeck, D. M. Kandhasamy and D. Strasser, Multiple Detachment of the SF6–Molecular Anion with Shaped Intense Laser Pulses, J. Phys. Chem. A, 2014, 118, 388–395 CrossRef CAS PubMed.
- S. Daly, M. Porrini, F. Rosu and V. Gabelica, Electronic spectroscopy of isolated DNA polyanions, Faraday Discuss., 2019, 217, 361–382 RSC.
- J. Wu and S. A. McLuckey, Gas-phase fragmentation of oligonucleotide ions, Int. J. Mass Spectrom., 2004, 237, 197–241 CrossRef CAS.
- L. V. Keldysh, Diagram technique for nonequilibrium processes, J. Exp. Theor. Phys., 1965, 20, 1018–1026 Search PubMed.
- C. L. Kalcic, G. E. Reid, V. V. Lozovoy and M. Dantus, Mechanism Elucidation for Nonstochastic Femtosecond Laser-Induced Ionization/Dissociation: From Amino Acids to Peptides, J. Phys. Chem. A, 2012, 116, 2764–2774 CrossRef CAS PubMed.
- C. L. Kalcic, T. C. Gunaratne, A. D. Jones, M. Dantus and G. E. Reid, Femtosecond Laser-Induced Ionization/Dissociation of Protonated Peptides, J. Am. Chem. Soc., 2009, 131, 940–942 CrossRef CAS PubMed.
- G. Reitsma, O. Gonzalez-Magaña, O. Versolato, M. Door, R. Hoekstra, E. Suraud, B. Fischer, N. Camus, M. Kremer, R. Moshammer and T. Schlathölter, Femtosecond laser induced ionization and dissociation of gas-phase protonated leucine enkephalin, Int. J. Mass Spectrom., 2014, 365, 365–371 CrossRef.
- I. Kyriakou, M. Šefl, V. Nourry and S. Incerti, The impact of new Geant4-DNA cross section models on electron track structure simulations in liquid water, J. Appl. Phys., 2016, 119, 194902 CrossRef.
- E. Surdutovich and A. V. Solov’yov, Double strand breaks in DNA resulting from double-electron-emission events, Eur. Phys. J. D, 2012, 66, 206 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |