
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective total synthesis of parnafungin A1 and 10a-epi-hirtusneanine†
Jiawei
Sun
a,
Wei
Gu
a,
He
Yang
*a and
Wenjun
Tang
 *ab
*ab
aState Key Laboratory of Bio-Organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, 345 Ling Ling Rd, Shanghai 200032, China
bSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
First published on 30th June 2021
Abstract
The first and enantioselective total synthesis of the heterodimeric biaryl antifungal natural product parnafungin A1 as well as complex biaryl tetrahydroxanthone 10a-epi-hirtusneanine is accomplished, by employing cross-coupling through the benzoxaborole strategy to construct their sterically hindered biaryl cores. Besides the powerful Suzuki–Miyaura cross-coupling, the synthesis of parnafungin A1 also features a highly diastereoselective oxa-Michael addition to construct a tetrahydroxanthone skeleton, and an effective Zn-mediated reductive cyclization-Mitsunobu sequence to furnish the isoxazolidinone structure. Key innovations in total synthesis of 10a-epi-hirtusneanine include the employment of DTBS protection for functional group manipulation on the tetrahydroxanthone skeleton, stereoselective methylations, and complete reversal of the stereochemistry of the C5-hydroxy group using oxidation/Evans–Saksena reduction, as well as the strategy of preparing both complex tetrahydroxanthone monomers from the same chiral intermediate 25.
Introduction
Dimeric tetrahydroxanthones exist widely in fungi and bacteria as secondary metabolites with rich pharmacological properties.1,2 In particular, biaryl dimeric tetrahydroxanthones have gained significant interest recently, not only because of their intriguing antibacterial and anticancer activities, but also owing to the synthetic challenges posed by their structures. Recent work from the groups of Bräse,3–7,21,22 Nicolaou,8 Tietze,9–11,18,19 Porco,12,15–17 Kumamoto,13 and Gao14,20et al. has shown significant progress in the synthesis of tetrahydroxanthone monomers3–14 or homodimeric tetrahydroxanthones.15–22 To the best of our knowledge, heterodimeric biaryl tetrahydroxanthones have rarely been synthesized, largely due to the synthetic challenges in assembling their sterically hindered biaryls.Parnafungin A1 (1), A2 (2), B1 (3), and B2 (4) are both structurally and biologically interesting heterodimeric tetrahydroxanthones isolated by a Merck team in 2008 from the lichenicolous fungi Fusarium larvarum as an equilibrating mixture of four interconverting species23 (Fig. 1A). Biologically, parnafungins specifically inhibit the activity of fungal polyadenosine polymerase (PAP) and have shown a broad spectrum of antifungal activities.24–27 Structurally, besides the interconversion between four species of parnafungins, the uniqueness of parnafungins also resides in the presence of an unprecedented isoxazolidinone unit, which is responsible for their broad spectrum of antifungal activity and selectivity. Under basic or neutral conditions, the cleavage of the sensitive isoxazolidinone can take place readily, leading to the inactive, ring-opened phenathridine isomers.
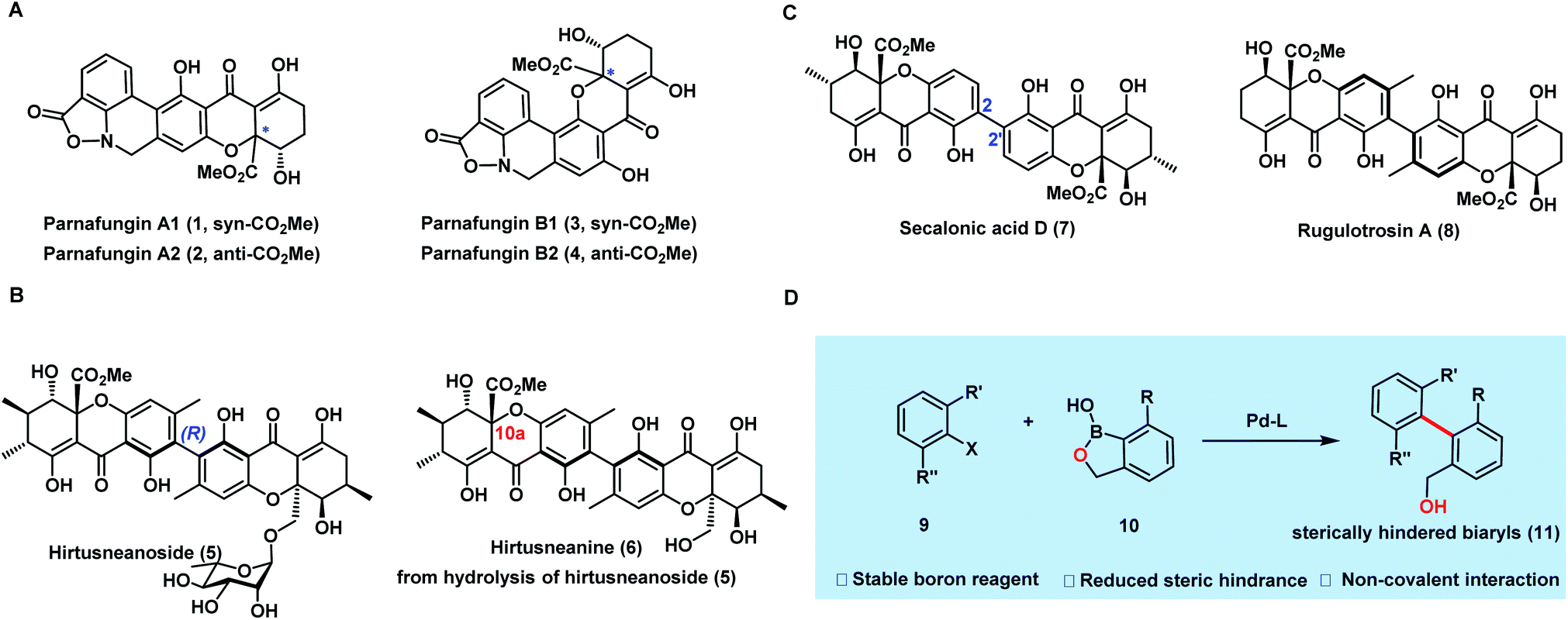 | ||
| Fig. 1 Dimeric tetrahydroxanthones and the cross-coupling strategy. (A) Parnafungins A1, A2, B1, and B2. (B) Hirtusneanoside and hirtusneanine. (C) Symmetrical biaryl dimeric tetrahydroxanthones. (D) Biaryl construction by cross-coupling through the benzoxaborole strategy. | ||
The significant antifungal activities as well as intriguing structure of parnafungins have attracted attention from a number of synthetic groups.28–33 In 2010, Zhou and Snider28–30 described the synthesis of hexacyclic parnafungin A and C models and further revealed the sensitivity of the isoxazolidinone structure. Later, Tietze,31 Gao,32 and Porco33 groups also reported their efforts on this elusive molecule in related dissertations, but with no success in completing the synthesis. It appears that “the ready isomerization of parnafungins and their propensity to rapidly decompose to phenathridines under neutral or basic conditions make these natural products especially challenging synthetic targets.1”
Hirtusneanoside (5) is an L-rhamnose-glycosylated biaryl heterodimeric tetrahydroxanthone (Fig. 1B), first isolated from the lichen Usnea hirta in 2007 by Rezanka and Sigler.34 Biologically, it inhibits the growth of Staphylococcus aureus and Bacillus subtilis at the nanomolar level (LD50 = 3.4 nM and LD50 = 14.0 nM, respectively), but is inactive against Gram-negative bacteria and yeast. This heterodimer bears a unique chemical structure which contains a rare L-rhamnopyranoside and an additional peripheral methyl group compared to secalonic acid D,15 and rugulotrosin A17 (Fig. 1C). The biosynthetic origin of this methyl group is yet to be elucidated, and the rotation along the biaryl axis is restricted by the bulky ortho-substituents, forming an axial chirality similarly to rugulotrosin A. Since its isolation, no synthetic studies have been reported towards hirtusneanoside (5) or its aglycon, hirtusneanine (6). We envision that the most challenging tasks in synthesizing 5 and 6 are the construction of complex tetrahydroxanthone monomers with up to four contiguous stereocenters and the installation of the sterically hindered axially chiral tetra-ortho-substituted biaryl unit.
Considering the synthetic challenges within the structures of both parnafungins and hirtusneanine, we believe that the paramount goal in our synthetic studies toward these elusive heterodimeric biaryl tetrahydroxanthones is to develop an efficient cross-coupling strategy for constructing the sterically hindered biaryls of these structures. Because of its excellent stability, less steric hindrance, and good ability in providing non-covalent interactions, benzoxaborole is recognized as one of most ideal and successful cross-coupling partners for the construction of sterically hindered biaryls35 (Fig. 1D). Herein, we report the employment of cross-coupling through the benzoxaborole strategy in accomplishing the first and enantioselective synthesis of the parnafungin A1 and 10a-epi-hirtusneanine.
Results and discussion
(a) Total synthesis of the parnafungin A1 (1)
Our retrosynthetic analysis of the parnafungin A1 (1) is depicted in Scheme 1A. Structure 12, the methylated form of parnafungin A1, proved to be stable and was characterized by X-ray crystallography.23 Because of its sensitive nature, the isoxazolidinone structure was planned to be installed at a late stage through the reduction of the nitro group followed by ring-closure. The formation of the biaryl linkage proved to be a challenge since a previous attempt on Suzuki–Miyaura cross-coupling between boronic ester 15 and an aryl halide containing the tetrahydroxanthone moiety failed to obtain the desired coupling product,32 possibly due to the issue of severe protodeboronation. We envisioned that benzoxaborole 16 would be more resistent to protodeboronation and suitable for the key cross-coupling.35 And benzoxaborole 16 could be derived from tetrahydroxanthone monomer 17. Although several effective strategies are developed by Bräse,3–7 Nicolaou,8 Tietze,9–11 Porco,12 Kumamoto13 and Gao14 groups in the preparation of the chiral tetrahydroxanthone structure, a highly enantioselective and diastereoselective version would be highly desirable. We anticipated that 17 could be efficiently synthesized from 18 and a known chiral intermediate 19 (ref. 36) through an oxa-Michael addition.8 Besides serving as a protecting group, the acetonide moiety in the structure of 19 was anticipated to be an anchor to forge a highly diastereoselective oxa-Michael addition.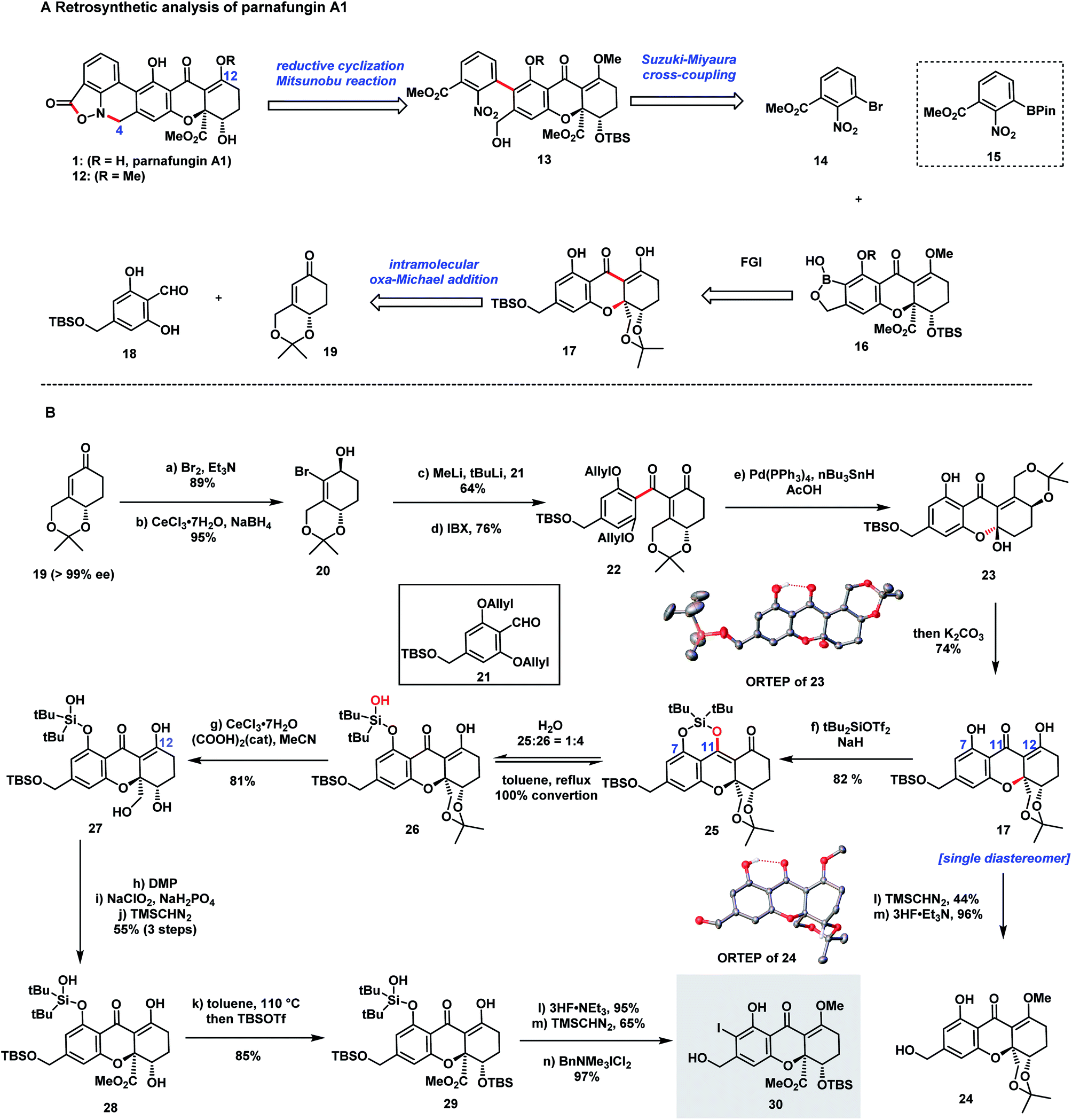 | ||
| Scheme 1 (A) Retrosynthetic analysis of parnafungin A1. (B) Synthesis of tetrahydroxanthone 30. | ||
Our synthesis commenced with the preparation of tetrahydroxanthone monomer 30 from known compound 19 (Scheme 1B). Bromination of 19 and a subsequent Luche reduction afforded 20, which was subjected to Li–Br exchange, treatment with aldehyde 21, and IBX oxidation providing diketone 22. Next, a Pd-catalyzed deallylation-intramolecular oxa-Michael addition cascade was explored to form 17. Surprisingly, instead of forming 17, a hemiketal 23 resulting from intramolecular 1,2-addition was isolated, whose structure was confirmed by X-ray crystallography. To our delight, 23 was smoothly converted to 17 as a single diastereomer under basic conditions (K2CO3/MeOH) and its stereochemistry (15R,15aR) was confirmed by 24 which was prepared from the methylation of 17 followed by desilylation. The perfect diastereoselectivity of the oxa-Michael reaction, likely due to the rigid conformation of the bicyclic ring system, was in contrast to the unsatisfactory diastereoselectivities (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 2:1 dr) observed in Nicolaou's report.8
:2 to 2:1 dr) observed in Nicolaou's report.8
We next turned our attention to furnish the ester moiety of the tetrahydroxanthone structure (Scheme 1B). Because of the labile nature of the tetrahydroxanthone once the ester was incorporated, the choice of the protecting group for the following manipulation was essential. We chose di-tert-butylsilylene (DTBS) protection for diol 17. The selective installation of the DTBS group at both C7-hydroxyl and C11-enol hydroxyl groups was successfully accomplished under conditions of DTBS(OTf)2/NaH to form 25. Owning to the labile nature of the vinylogous silyl ether, the ring opening of the DTBS protection occurred readily to give a mixture of 26 and 25 at 4:1 ratio. Interestingly, 26 was transformed back to 25 by heating in toluene with azeotropical removal of water. Next, selective removal of the acetonide protection was accomplished by treatment of 26 with CeCl3·7H2O and (COOH)2 (5 mol%),37 affording 27 in 81% yield. Subsequent Dess–Martin oxidation, Pinnick oxidation, and methylation with TMSCHN2 delivered ester 28 in 55% overall yield. Treatment of 28 in toluene at 110 °C resulted in DTBS ring closure, followed by selective TBS protection of the hydroxyl group adjacent to the ester group gave 29 in 85% yield. Selective desilylation, methylation with TMSCHN2, and ortho-iodination with BnMe3NICl2 successfully led to 30.
We then looked into the key Suzuki–Miyaura cross-coupling for the formation of biaryl moiety. Initial experiments on sterically hindered cross-coupling between aryl iodide 30 and boronic ester 15 proved to be futile and significant formation of protodeboronation and deiodination side-products was observed. We envisioned that benzoxaborole 31 would be stable and more resistant to protodeboronation. Thus, Miyaura borylation of 30 to form 31 was studied. As depicted in Scheme 2, Pd(dppf)Cl2 (Scheme 2, Table 1, entry 1) and Pd(PPh3)2Cl2 (Scheme 2, Table 1, entry 2), commonly used catalysts for borylation led to unsatisfactory yields (25% and 28% respectively). Encouragingly, SPhos (Scheme 2, Table 1, entry 3) provided an improved yield (62%). The use of tetrahydroxydiboron (Scheme 2, Table 1, entry 4) did not provide any activities. Surprisingly, the addition of water turned out to be crucial for the transformation and AntPhos, a prominent ligand for both Miyaura borylation and Suzuki coupling with sterically hindered substrates,38–41 providing the best yield for this transformation (67% yield) (Scheme 2, Table 1, entry 5 and 6). With 31 in hand, the Pd-catalyzed cross-coupling between 31 and 14 was carried out in THF/H2O with DIPEA as the base (Scheme 2, Table 2). AntPhos proved to be the most prominent ligand, providing coupling product 32 as an atropisomeric mixture (1.5:1 dr) in 63% yield with protodeboronation as the major side-product (Scheme 2, Table 2, entry 1). SPhos was similarly effective (Scheme 2, Table 2, entry 2). A series of P,P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ligands42,43 were also applicable and L2 delivered a moderate yield (53%) (Scheme 2, Table 2, entry 3–6).
O ligands42,43 were also applicable and L2 delivered a moderate yield (53%) (Scheme 2, Table 2, entry 3–6).
 | ||
| Scheme 2 (A) Studies of the Miyaura borylation (Table 1); studies of the Suzuki–Miyaura cross-coupling reaction (Table 2). (B) Total synthesis of parnafungin A1. | ||
The stage was set for generating the sensitive isoxazolidinone structure (Scheme 2B). Reported methods such as Zn reduction and reduction with RANEY® Ni/H2 (ref. 44) failed to provide the desired product. When the reductive cyclization was performed with Zn and using acetic acid as an additive,29 the target product 33 was successfully formed as an inseparable mixture (1.3:1) from both atropisomers of 33. Next, ring closure of 33 to form 34 was studied. Extensive experiments revealed that this transformation was successfully accomplished under Mitsunobu conditions (DIAD/PPh3) and 34 was formed in an excellent yield (80%). Finally, a global deprotection of 34 provided parnafungin A1 (1) in 66% yield along with the methylated form of parnafungin A1 (12) in 18% yield. Thus, we accomplished for the first time the total synthesis of the parnafungin A1. Because the interconversion of parnafungins proceeded slowly in CDCl3 solution, the pure spectra of parnafungin A1 (1) were acquired successfully for the first time.
Experiments revealed that the interconversion between parnafungins A1, A2, B1, and B2 took place more facilely in DMSO and an instant conversion from parnafungin A1 (1) to B1 (3) was observed. The formation of parnafungins A2 (2) and B2 (4) occurred after several hours. The NMR spectra of the interconverting mixture of the four parnafungin A1, A2, B1, and B2 in DMSO (the equilibrium ratio 4:1:5:1) was consistent with the reported data.23 Thus, we accomplished the total synthesis of the parnafungins for the first time.
(b) Total synthesis of 10a-epi-hirtusneanine
The successful diastereoselective synthesis of tetrahydroxanthone monomers such as 25 and the development of a reliable cross-coupling for biaryl formation in complex structure synthesis prompted us to explore the synthesis of hirtusneanine (6) (Scheme 3A). The absolute configuration of hirtusneanine was assigned as 6 in Rezanka and Sigler's original reports based on spectroscopic data and chemical degradation. Nevertheless, no original NMR spectra of hirtusneanoside (5) and hirtusneanine (6) could be provided by either the authors or the journal office, which significantly increases the difficulties of their total syntheses. As we meticulously compared the 1H NMR data of both hirtusneanoside (5) and hirtusneanine (6) provided by Rezanka and Sigler,34 with the reported 2,2′-linked tetrahydroxanthones dimers [secalonic acid D (H5, dd, J = 11.3, 1.6 Hz),15 rugulotrosin A (H5, dd, J = 12.3, 5.0 Hz),17 ascherxanthone A (H5, d, J = 10.3 Hz)20], the large H-5/H-6 coupling constant (d, J = 9.5 Hz) indicated that H-5 of hirtusneanine (6) should occupy an axial position and, thus, be in the syn-position (and not in the anti-position) with respect to the methyl carboxylate moiety at C10a (Scheme 3C).Taken together, we speculated that 10a-epi-hirtusneanine (36) should be the feasible structure that would match the reported NMR data. Therefore, our attention was concentrated on the total synthesis of 10a-epi-hirtusneanine (36), which could be the real natural product. Our synthetic plan toward 10a-epi-hirtusneanine (36) is outlined in Scheme 3B. We anticipated that the tetra-ortho-substituted biaryl could be formed from the coupling between benzoxaborole 39 and iodide 38. We expected that the presence of the benzyl hydroxyl group could be crucial for the synthesis of benzoxaborole 39 as well as the subsequent cross-coupling. More importantly, both fragments (benzoxaborole 39 and iodide 38) could be derived from a single intermediate 25.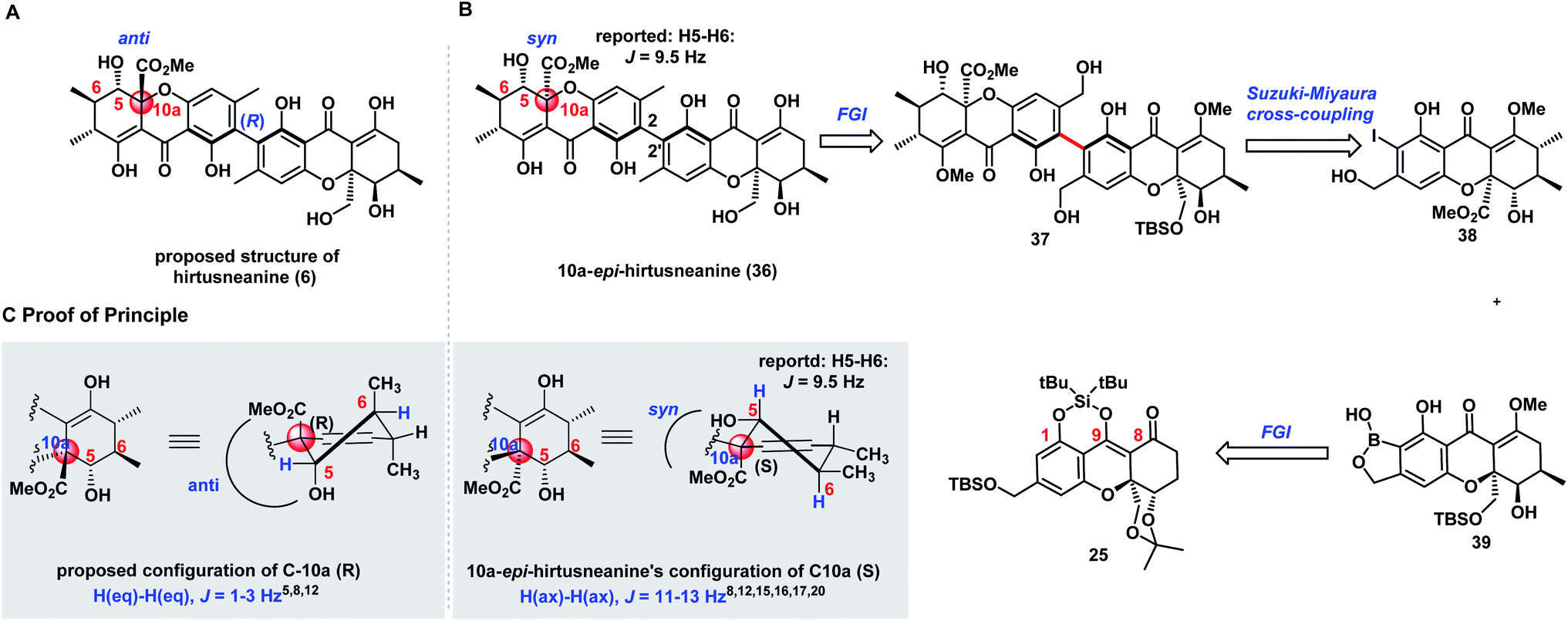 | ||
| Scheme 3 Comparison of the NMR coupling constants of H5/H6 and retrosynthetic analysis of 10a-epi-hirtusneanine (36). (A) Proposed structure of hirtusneanine (6). (B) 10a-epi-Hirtusneanine and retrosynthetic analysis. (C) Proof of principle. | ||
Thus, our first task was to synthesize iodide 38 from 25 with the expectation that the formation of cyclic DTBS ether within 25 would enable the keto form at C8 and be beneficial to subsequent methylations. Compound 25 was treated under dehydrogenation conditions in order to produce dihydroxanthone 40 (Scheme 4). The conditions of Saegusa oxidation and Nicolaou dehydrogenation led to severe decompositions, while the employment of Mukaiyama's protocol (N-tert-butylphenylsulfinimidoyl chloride)45 and Grieco elimination46 gave product 40 in 40% and 57% yield, respectively. Direct exposure of 40 to dimethylcopperlithium (3.0 equiv.) furnished the desired Michael addition product 41 as a single diastereoisomer in 85% yield. The perfect diastereoselectivity could be attributed to the steric bulk of acetonide at the si face. Installation of the second methyl group provided 42 which was elaborated to 45 through the well-developed five-step sequence in parnafungin syntheses: (1) removal of the acetonide protection with CeCl3·7H2O and (COOH)2 (cat); (2) selective oxidation of primary alcohol; (3) Pinnick oxidation; (4) methylation of the resulting acid with TMSCHN2; (5) removal of silyl groups with 3HF·Et3N. The relative stereochemistry of 45 was confirmed by its X-ray structure. As shown in Scheme 4, H-5 of compound 45 was at axial orientation, and the coupling constants (H-5/H6, J = 11.1 Hz) in DMSO-d6 were consistent with the H-5/H-6 coupling constant (9.5 Hz) reported by Rezanka and Sigler, indicating the correctness of our proposal on 10a-epi-hirtusneanine. Selective methylation of 45 with TMSCHN2 (74% yield) and ortho-iodination with BnMe3NICl2 afforded iodide 38 in 92% yield.
 | ||
| Scheme 4 Synthesis of iodide 38 and benzoxaborole 39. | ||
We then turned our attention to construct benzoxaborole 39 (Scheme 4). Different from iodide 38, the structure of benzoxaborole 39 has R configuration at the C5 position. Our immediate mission was to inverse the configuration of the C5–OH functionality of the mono-methylated tetrahydroxanthone intermediate 41. Attempts of employing the Mitsunobu protocol failed to provide the desired outcome. We then explored the strategy of the hydroxyl-directed Evans–Saksena reduction.47 Removal of both acetonide and TBS protection with HCl/MeOH (93% yield), followed by selective protection of 46 with TBSCl (99% yield), a subsequent Dess–Martin oxidation to form the ketone functionality (85% yield), and finally selective TBS deprotection to release the primary hydroxyl group (90% yield), afforded ketone 47. It was noteworthy that ketone 47 could undergo aromatization easily under acidic conditions. Fortunately, the mild conditions [CeCl3·7H2O/(COOH)2] previously employed for acetonide deprotection also applied effectively for TBS deprotection, therefore minimizing the formation of side-products. Pleasingly, ketone 47 facilitated Evans–Saksena reduction47 smoothly to give alcohol 48 with an inversed R configuration at the C5 position. Treatment of alcohol 48 with TBSCl followed by selective removal of the silyl protecting groups at both phenol and benzyl alcohol positions with HF·Py provided compound 50 in 81% yield, along with a fully-deprotected product 49 in 11% yield. Further transformation of 50 to iodide 51 was achieved through selective methylation with TMSCHN2 (65% yield) and ortho-iodination with BnMe3NICl2 (92% yield). A palladium-catalyzed Miyaura borylation of iodide 51 affected by Pd-AntPhos successfully furnished benzoxaborole 39.
With iodide 38 and benzoxaborole 39 in hand, we came to the stage of the key sterically hindered Suzuki–Miyaura cross-coupling (see Table S1†). Use of the reaction conditions (Pd-AntPhos, DIPEA) applied in the synthesis of parnafungins did not provide any desired coupling product. Realizing that the steric hindrance of this cross-coupling is even greater than those reported for rugulotrosin A17 and ascherxanthone A,20 we further screened various commercially available ligands and found that SPhos was one among the most effective ligands. With Pd(OAc)2/SPhos as the catalyst, the desired tetra-ortho-substituted biaryl 52 was isolated in 43% yield and an atropodiastereomerically pure form (Scheme 5).
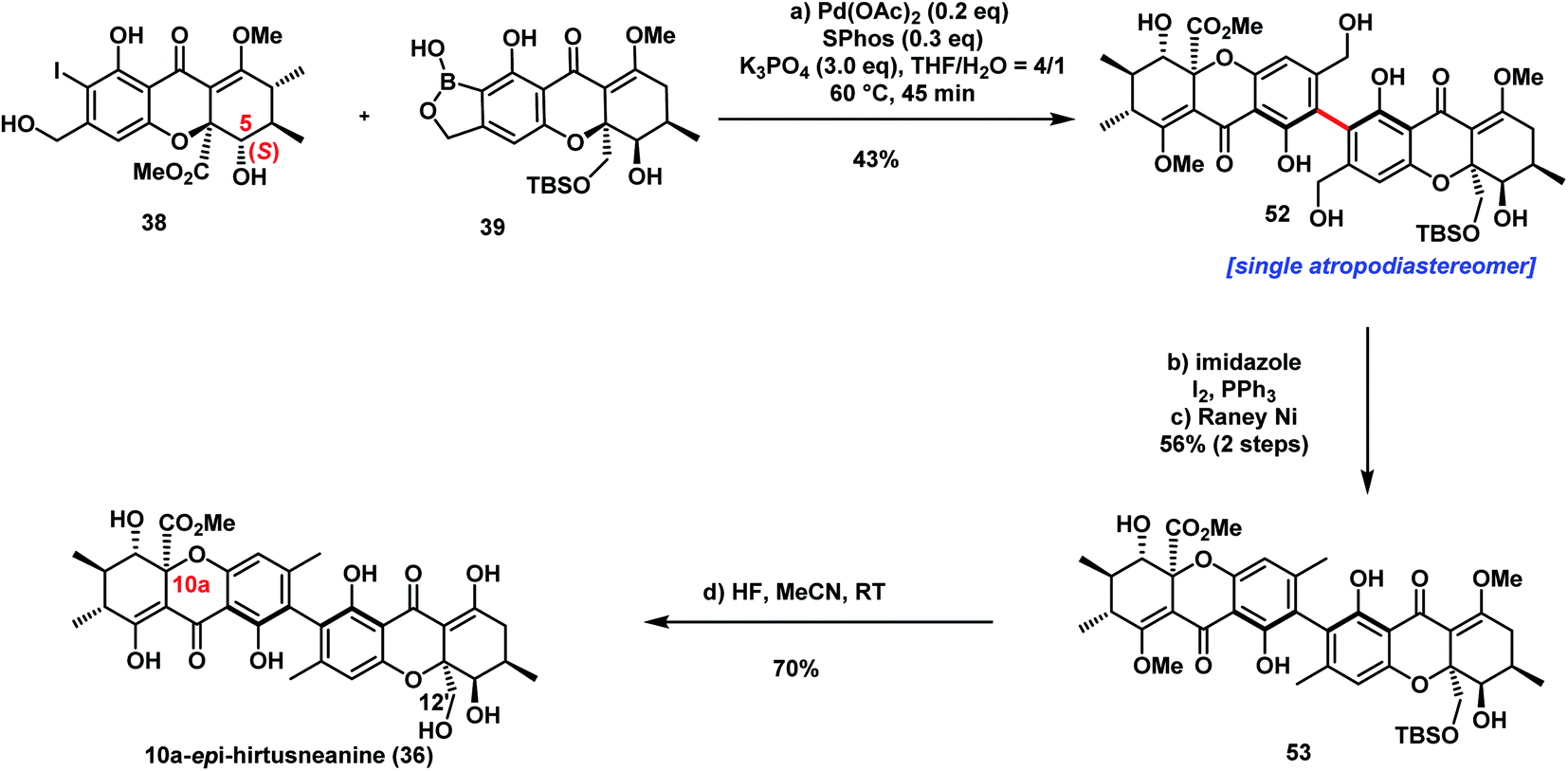 | ||
| Scheme 5 Synthesis of 10a-epi-hirtusneanine (36). | ||
With the heterodimeric biaryl tetrahydroxanthone skeleton 52 successfully assembled, the final task of the total synthesis was the deoxygenation of the two primary alcohols as well as the deprotection of TBSO- and MeO-functionalities. Treatment of 52 with iodine and triphenylphosphine led to the formation of iodide, which underwent dehalogenation with RANEY® nickel to form compound 53. The CD spectroscopic data of compound 53 revealed the R configuration of the axial chirality. Finally, a global deprotection of the protecting group with HF (aq) accomplished the total synthesis of 10a-epi-hirtusneanine 36. Unfortunately, our acquired spectroscopic data of 36 did not match those reported by Rezanka and Sigler.34 Although the H-5/H-6 coupling constant (11.1 Hz) of 10a-epi-hirtusneanine was close to the reported data of hirtusneanine (9.5 Hz), marked differences of chemical shifts were observed for enolic and phenolic protons, as well as C(12′)–H (see ESI Table 13†). It was unfortunate that the authors could not provide original NMR spectra of hirtusneanine or hirtusneanoside for further comparison. Efforts are currently in progress to elucidate the real structure of hirtusneanine or hirtusneanoside.
Conclusions
In summary, we have accomplished the first and enantioselective total synthesis of the heterodimeric biaryl antifungal natural product parnafungin A1 as well as complex biaryl tetrahydroxanthone 10a-epi-hirtusneanine, using cross-coupling through the benzoxaborole strategy to construct their key sterically hindered biaryl cores. Besides the powerful Suzuki–Miyaura cross-coupling, the synthesis of parnafungin A1 also features a highly diastereoselective oxa-Michael addition to construct the tetrahydroxanthone skeleton, and an effective Zn-mediated reductive cyclization-Mitsunobu sequence to furnish the isoxazolidinone structure. Key innovations in total synthesis of 10a-epi-hirtusneanine include the employment of DTBS protection for functional group manipulation on the tetrahydroxanthone skeleton, the stereoselective methylations, complete reversal of the stereochemistry of the C5-hydroxy group using oxidation/Evans–Saksena reduction, as well as the strategy of preparing both complex tetrahydroxanthone monomers from the same chiral intermediate 25. We strongly believe that the strategy of cross-coupling through benzoxaborole for sterically hindered biaryl cross-coupling should be applicable to the total syntheses of a number of heterodimeric biaryl tetrahydroxanthones.Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files, including experimental details, characterization data, and 1H and 13C NMR spectra of all new compounds.Author contributions
W. T. designed the research and experiments. J. S., H. Y. and W. G. conducted the synthetic work. W. T. and H. Y. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Strategic Priority Research Program of the Chinese Academy of Sciences XDB20000000, CAS (QYZDY-SSW-SLH029), NSFC (21725205, 21432007, 21572246, and 22001112), STCSM-18520712200, and the K.C. Wong Education Foundation.Notes and references
- K.-S. Masters and S. Bräse, Chem. Rev., 2012, 112, 3717–3776 CrossRef CAS PubMed.
- T. Wezeman, S. Bräse and K.-S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC.
- B. Lesch and S. Bräse, Angew. Chem., Int. Ed., 2004, 43, 115–118 CrossRef PubMed.
- C. F. Nising, U. K. Ohnemüller and S. Bräse, Angew. Chem., Int. Ed., 2006, 45, 307–309 CrossRef CAS PubMed.
- E. M. C. Gérard and S. Bräse, Chem.–Eur. J., 2008, 14, 8086–8089 CrossRef PubMed.
- M. C. Bröhmer, E. Bourcet, M. Nieger and S. Bräse, Chem.–Eur. J., 2011, 17, 13706–13711 CrossRef PubMed.
- A. C. Meister, A. Encinas, H. Sahin, E. M. Singer, C. F. Nising, M. Nieger and S. Bräse, Eur. J. Org. Chem., 2014, 2014, 4861–4875 CrossRef CAS.
- K. C. Nicolaou and A. Li, Angew. Chem., Int. Ed., 2008, 47, 6579–6582 CrossRef CAS.
- L. F. Tietze, S. Jackenkroll, C. Raith, D. A. Spiegl, J. R. Reiner and M. C. Ochoa Campos, Chem.–Eur. J., 2013, 19, 4876–4882 CrossRef CAS PubMed.
- L. F. Tietze, L. Ma, J. R. Reiner, S. Jackenkroll and S. Heidemann, Chem.–Eur. J., 2013, 19, 8610–8614 CrossRef CAS.
- L. F. Tietze, S. Jackenkroll, J. Hierold, L. Ma and B. Waldecker, Chem.–Eur. J., 2014, 20, 8628–8635 CrossRef CAS PubMed.
- T. Qin, R. P. Johnson and J. A. Porco Jr, J. Am. Chem. Soc., 2011, 133, 1714–1717 CrossRef CAS PubMed.
- K. Adachi, S. Hasegawa, K. Katakawa and T. Kumamoto, Tetrahedron Lett., 2017, 58, 4479–4482 CrossRef CAS.
- J. Chen, Y. Li, Z. Xiao, H. He and S. Gao, Org. Lett., 2020, 22, 1485–1489 CrossRef CAS PubMed.
- T. Qin and J. A. Porco Jr, Angew. Chem., Int. Ed., 2014, 53, 3107–3110 CrossRef CAS PubMed.
- T. Qin, T. Iwata, T. T. Ransom, J. A. Beutler and J. A. Porco Jr, J. Am. Chem. Soc., 2015, 137, 15225–15233 CrossRef CAS PubMed.
- T. Qin, S. L. Skraba-Joiner, Z. G. Khalil, R. P. Johnson, R. J. Capon and J. A. Porco, Nat. Chem., 2015, 7, 234–240 CrossRef CAS PubMed.
- D. Ganapathy, J. R. Reiner, L. E. Löffler, L. Ma, B. Gnanaprakasam, B. Niepoetter, I. Koehne and L. F. Tietze, Chem.–Eur. J., 2015, 21, 16807–16810 CrossRef CAS.
- D. Ganapathy, J. R. Reiner, G. Valdomir, S. Senthilkumar and L. F. Tietze, Chem.–Eur. J., 2017, 23, 2299–2302 CrossRef CAS PubMed.
- Z. Xiao, Y. Li and S. Gao, Org. Lett., 2017, 19, 1834–1837 CrossRef CAS PubMed.
- S. Lindner, M. Nieger and S. Braese, Adv. Synth. Catal., 2015, 357, 3303–3308 CrossRef CAS.
- L. Geiger, M. Nieger and S. Bräse, Adv. Synth. Catal., 2017, 359, 3421–3427 CrossRef CAS.
- C. A. Parish, S. K. Smith, K. Calati, D. Zink, K. Wilson, T. Roemer, B. Jiang, D. Xu, G. Bills and G. Platas, J. Am. Chem. Soc., 2008, 130, 7060–7066 CrossRef CAS PubMed.
- D. Overy, K. Calati, J. N. Kahn, M.-J. Hsu, J. Martín, J. Collado, T. Roemer, G. Harris and C. A. Parish, Bioorg. Med. Chem. Lett., 2009, 19, 1224–1227 CrossRef CAS PubMed.
- B. Jiang, D. Xu, J. Allocco, C. Parish, J. Davison, K. Veillette, S. Sillaots, W. Hu, R. Rodriguez-Suarez and S. Trosok, Chem. Biol., 2008, 15, 363–374 CrossRef CAS PubMed.
- G. F. Bills, G. Platas, D. P. Overy, J. Collado, A. Fillola, M. R. Jimenez, J. Martin, A. G. del Val, F. Vicente and J. R. Tormo, Mycologia, 2009, 101, 449–472 CrossRef CAS PubMed.
- G. C. Adam, C. A. Parish, D. Wisniewski, J. Meng, M. Liu, K. Calati, B. D. Stein, J. Athanasopoulos, P. Liberator and T. Roemer, J. Am. Chem. Soc., 2008, 130, 16704–16710 CrossRef CAS PubMed.
- Q. Zhou and B. B. Snider, J. Org. Chem., 2010, 75, 8224–8233 CrossRef CAS PubMed.
- Q. Zhou and B. B. Snider, Org. Lett., 2009, 11, 2936–2939 CrossRef CAS PubMed.
- Q. Zhou, PhD thesis, Brandeis University, 2011.
- S. Heidemann, PhD thesis, Georg-August University of Göttingen, 2016.
- Y. Li, MSc thesis, East China Normal University, 2017.
- X. Wu, PhD thesis, Boston University, 2020.
- T. Rezanka and K. Sigler, J. Nat. Prod., 2007, 70, 1487–1491 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, M. K. Cyrański, A. Żubrowska and A. Sporzyński, J. Organomet. Chem., 2009, 694, 3533–3541 CrossRef.
- E. Corey and H. Kigoshi, Tetrahedron Lett., 1991, 32, 5025–5028 CrossRef CAS.
- X. Xiao and D. Bai, Synlett, 2001, 535–537 CrossRef CAS.
- W. Tang, A. G. Capacci, X. Wei, W. Li, A. White, N. D. Patel, J. Savoie, J. J. Gao, S. Rodriguez and B. Qu, Angew. Chem., Int. Ed., 2010, 49, 5879–5883 CrossRef CAS PubMed.
- G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570–573 CrossRef CAS PubMed.
- H. Yang, J. Sun, W. Gu and W. Tang, J. Am. Chem. Soc., 2020, 142, 8036–8043 CrossRef CAS PubMed.
- G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101–1112 CrossRef CAS PubMed.
- C. Li, T. Chen, B. Li, G. Xiao and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3792–3796 CrossRef CAS PubMed.
- B. Li, T. Li, M. A. Aliyu, Z. H. Li and W. Tang, Angew. Chem., Int. Ed., 2019, 131, 11477–11481 Search PubMed.
- W. Wierenga, B. Evans and G. Zurenko, J. Med. Chem., 1984, 27, 1212–1215 CrossRef CAS PubMed.
- T. Mukaiyama, J.-i. Matsuo and H. Kitagawa, Chem. Lett., 2000, 29, 1250–1251 CrossRef.
- H. J. Reich, J. M. Renga and I. L. Reich, J. Org. Chem., 1974, 39, 2133–2135 CrossRef CAS.
- D. Evans, K. Chapman and E. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2026687, 2026689 and 2068312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02919c |
| This journal is © The Royal Society of Chemistry 2021 |