DOI:
10.1039/D1SC02947A
(Edge Article)
Chem. Sci., 2021,
12, 10279-10289
On 1,3-phosphaazaallenes and their diverse reactivity†
Received
31st May 2021
, Accepted 30th June 2021
First published on 30th June 2021
Abstract
1,3-Phosphaazaallenes are heteroallenes of the type RP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNR′ and little is known about their reactivity. In here we describe the straightforward synthesis of ArPCNR (Ar = Mes*, 2,4,6-tBu-C6H2; MesTer, 2.6-(2,4,6-Me3C6H2)–C6H3; DipTer, 2.6-(2,6-iPr2C6H2)–C6H3; R = tBu; Xyl, 2,6-Me2C6H3) starting from phospha-Wittig reagents ArPPMe3 and isonitriles CNR. It is further shown that ArPCNtBu are thermally labile with respect to the loss of iso-butene and it is shown that the cyanophosphines ArP(H)CN are synthetically feasible and form the corresponding phosphanitrilium borates with B(C6F5)3, whereas deprotonation of DipTerP(H)CN was shown to give an isolable cyanidophosphide. Lastly, the reactivity of ArPCNR towards Pier's borane was investigated, showing hydroboration of the CN bond in Mes*PCNtBu to give a hetero-butadiene, while with DipTerPCNXyl the formation of the Lewis acid–base adduct with a B–P linkage was observed.
CNR′ and little is known about their reactivity. In here we describe the straightforward synthesis of ArPCNR (Ar = Mes*, 2,4,6-tBu-C6H2; MesTer, 2.6-(2,4,6-Me3C6H2)–C6H3; DipTer, 2.6-(2,6-iPr2C6H2)–C6H3; R = tBu; Xyl, 2,6-Me2C6H3) starting from phospha-Wittig reagents ArPPMe3 and isonitriles CNR. It is further shown that ArPCNtBu are thermally labile with respect to the loss of iso-butene and it is shown that the cyanophosphines ArP(H)CN are synthetically feasible and form the corresponding phosphanitrilium borates with B(C6F5)3, whereas deprotonation of DipTerP(H)CN was shown to give an isolable cyanidophosphide. Lastly, the reactivity of ArPCNR towards Pier's borane was investigated, showing hydroboration of the CN bond in Mes*PCNtBu to give a hetero-butadiene, while with DipTerPCNXyl the formation of the Lewis acid–base adduct with a B–P linkage was observed.
Introduction
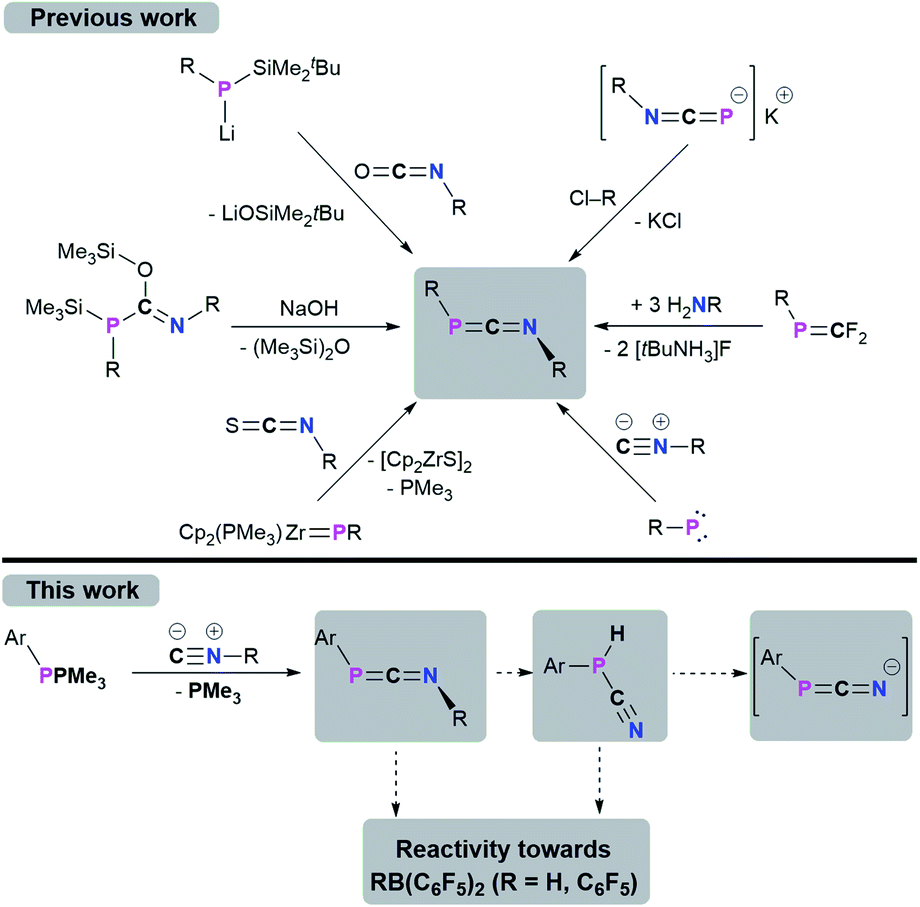
1,3-Phosphaazallenes (RPCNR) are a heteroallene subclass. The first derivative tBuPCNtBu was obtained by combining tBuP(SiMe3)C(OSiMe3)NtBu with NaOH under the release of hexamethyldisiloxane.1,2 Although known for almost 40 years, 1,3-phosphaazaallenes have been scarcely investigated, especially when compared to the “lighter” carbodiimides and other heteroallene analogues. Another synthetic route was disclosed by Yoshifuji,3,4 and Appel,5 who reacted Mes*P(Li)SiMe2tBu (Mes* = 2,4,6-tBu3C6H2) with isocyanates in a Peterson-type reaction to give Mes*PCNR (R = Ph, nPr, tBu). In 2000, Zhou and co-workers expanded this series to include Mes*PCN(4-ClC6H4).6 Mes*PCN(4-ClC6H4) and Mes*PCNPh4 are the only structurally characterized 1,3-phosphaazaallenes bearing classic organic substituents. Sterically demanding groups on the P atom suppress dimerization to the corresponding 1,3-diphosphetanes, which can only be reconverted to the 1,3-phosphaazallenes by flash vacuum pyrolysis.2,7 Even Mes*PCNPh slowly dimerizes in solution, whereas in the presence of catalytic amounts of Pd(PPh3)4 the unsymmetric four-membered heterocycle is obtained.8 Derivatives with a bulky cyclopropen-1-yl substituent at the phosphorus were synthesized by Regitz et al.9
In 1991 Grobe and co-workers demonstrated that the metastable (F3C)PCNtBu (can be handled at −40 °C) is feasible by reacting the phosphaalkene precursor (F3C)PCF2 with three equivalents of H2NtBu.10 Instead of dimerizing at higher temperatures, (F3C)PCNtBu decomposes to give fluorinated cyclophosphanes (PCF3)n and the isocyanide CNtBu. Stephan et al. showed that the zirconocene phosphinidene Cp2(PMe3)ZrPMes* reacts with an isothiocyanate in a [2 + 2] cycloaddition/cycloreversion sequence to yield Mes*PCNPh and [Cp2ZrS]2.11 That 1,3-phosphaazaallenes can function as ligands for transition metals was established by Streubel and Jones.12 Photochemical ring opening in the presence of an isoscyanide of a W(CO)5-stabilized 2H-azaphosphirene resulted in the formation of an 1,3-phosphaazaallene with the P atom remaining coordinated to W(CO)5. A transient terminal phosphinidene complex is assumed to react in a 1,1-addition with the respective isocyanide. The motif to generate 1,3-phosphaazaallenes directly in the coordination sphere of a transition metal by reactions of metal-bound phosphinidenes with isocyanides is more common in the literature.13 A trimethylstannyl substitution at the phosphorus centre in 1,3-phosphaazaallenes was achieved by reacting the potassium 1,3-azaphosphaallenide K[iPrNCP] with ClSnMe3 in a salt metathesis reaction, thus revealing another access to this substance class.14 The most recent examples of 1,3-phosphaazaallenes were synthesized by Bertrand et al. in coupling reactions of (phosphino)phosphinidenes with isocyanides.15 Moreover, Scheschkewitz et al. showed that a phosphasilene with a mobile NMe2-functionality on the phosphorus atom undergoes an NMe2-shift in the reaction with CNtBu to give a P-silyl-substituted 1,3-phosphaazaallene.16 The synthetic protocols towards 1,3-phosphaazaallenes are summarized in Scheme 1. Even though 1,3-phosphaazaallenes are without a doubt an interesting class of compounds, it is surprising that their general reactivity has not been studied in detail.
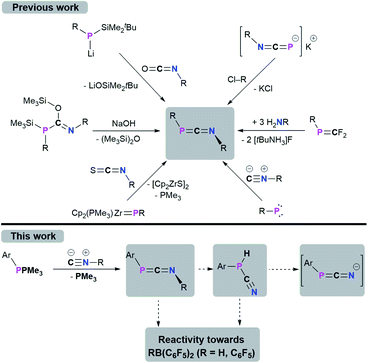 |
| | Scheme 1 Syntheses of 1,3-phosphaazaallenes and scope of this work. | |
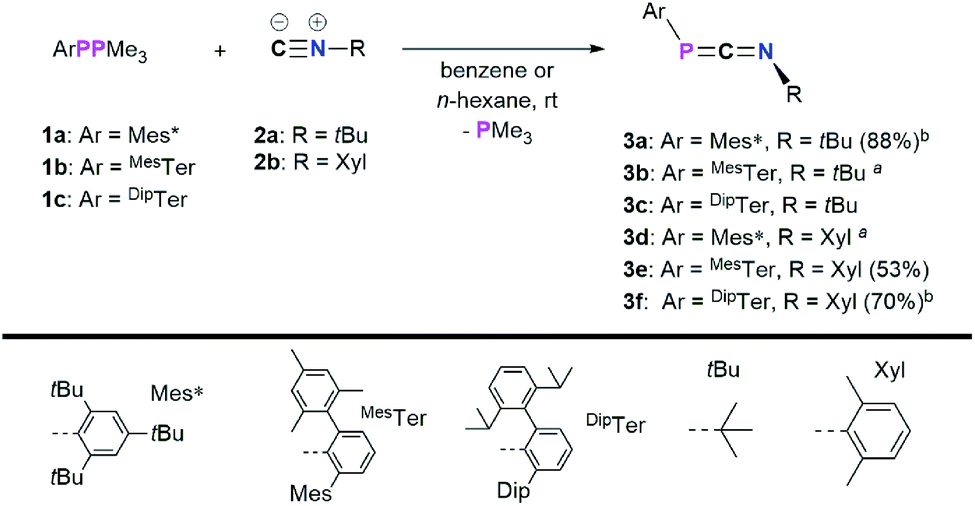
Recently, we have revisited the chemistry of phosphanylidene phosphoranes, so-called phospha-Wittig reagents, ArPPMe3 (1a–c) (1a: Ar = Mes*; 1b: Ar = 2,6-(2,4,6-Me3C6H2)–C6H3, MesTer, 1c: Ar = 2.6-(2,6-iPr2C6H3)–C6H3, DipTer).17 We successfully used them as phosphinidene transfer reagents in reactions with N-heterocyclic carbenes (NHCs) or N-heterocyclic olefins (NHOs),17 towards Al(I) species to give phosphaalumenes,18 and with Cp2Ti(C2(SiMe3)2) to afford terminal titanium phosphinidene complexes, respectively.19 In this contribution, the reactivity of the phospha-Wittig reagents 1a–c towards isocyanides is presented (Scheme 1, bottom), giving a series of 1,3-phosphaazaallenes. The tBu-substituted 1,3-phosphaazaallenes can be converted into primary cyanophosphines, which in one case can be transformed to the corresponding cyanophosphides. Finally, the reactivity of 1,3-phosphaazaallenes and primary cyanophosphines towards the perfluorinated arylboranes RB(C6F5)2 (R = H, C6F5) is illustrated.
Results and discussion
In a first series of experiments Mes*PPMe3 (1a)14 was dissolved in C6D6, and an excess of CNtBu (2a) was added (Scheme 2).20 Within 16 h at room temperature, two new signals were observed in the 31P{1H} NMR spectrum at −62.6 and −103.8 ppm, respectively, along with mostly unreacted starting materials. Heating to 60 °C for 24 h resulted in the consumption of 1a and one equivalent of CNtBu to give the targeted 1,3-phosphaazaallene Mes*PCNtBu (3a, δ31P{1H} = −103.8 ppm) upon PMe3 release. This is in contrast to the reaction of the phosphaketene [sP]PCO ([sP] = (H2CNDip)2P) with CN–Ad (Ad = adamantyl), which did not result in a CO for isonitrile substitution, but in the formation of a P2C2 heterocycle through attack of CN–Ad on the PCO carbon atom.21 However, sequential treatment of [sP]PCO with PPh3 and CNAd was shown to afford the corresponding heteroallenes [sP]PCNAd.22 Phosphaketenes are related to the 1,3-phosphaazaallenes through isolobal CO for CNR replacement.23 To further investigate the scope of this reaction, 1a–c were each reacted with both 2a and CNXyl (Xyl = 2,6-Me2C6H3, 2b) (Scheme 2). It was found that the desired 1,3-phosphaazaallene formation is generally faster at higher temperatures but accompanied by diphosphene ArPPAr formation unless DipTerPPMe3 (1c) is used. It is worthy to note that 3b and 3d could only be obtained as mixtures with the corresponding diphosphenes MesTerPPMesTer,24 and Mes*PPMes*,25 respectively, even if an excess of isocyanide was employed (Fig. S8 and S18†). Theoretical investigations at the PBE0-D3/def2SVP//DNLPO-CCSD(T)/def2TZVP level of theory revealed that the formation of 3a–f are exergonic (Fig. S86†).26,27 However, diphosphene formation from ArPPMe3 through recombination of ArP upon PMe3 release, has been calculated to be even more exergonic at the same level of theory, therefore explaining that formation of diphosphenes cannot be completely suppressed (Fig. S18†). Using a thermal approach, a diphosphene–poly(phenylenevinylene) has been prepared from the corresponding phospha-Wittig monomers upon PMe3 release. The monomer showed the same 2,6-Mes2Ar structural motif as in phospha-Wittig reagent 1b.28 In case of 1a free phosphinidenes are unlikely, as cyclometalated species are not observed.29 The NMR data of 3a–f are in accordance to the previously reported data of 3a in CDCl3 (Table 1).4 The 31P{1H} NMR signals of 3a–3f range from −103.9 to −145.4 ppm and are generally shifted to higher field when the N-substituent is aromatic with the P-substituent being the same. This was corroborated by DFT calculations at the PBE0-D3/def2SVP level of theory, which gave δcalc(31P) values that are systematically at lower ppm values, though the experimentally observed trends are followed (Table 1).
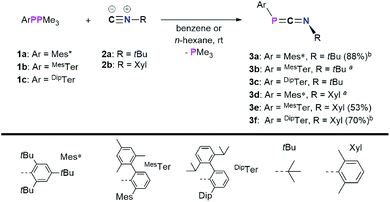 |
| | Scheme 2 Reactions of 1a–c with isocyanides to give 1,3-phosphaazaallenes 3a–f; a obtained as a mixture with the diphosphenes Mes*PPMes* and MesTerPPMesTer, respectively; b heated to 60° (3a) or 80 °C (3f). | |
Table 1 Characteristic 31P{1H} and 13C{1H} NMR data of 3a–f. Calculated 31P NMR shifts (PBE0-D3/def2SVP) are given in parentheses
| Compound |
δ
31P{1H}a (δcalc31P)a |
δ
13C{1H}(PCN)a |
1
J
P,C(PCN)a |
|
In C6D6 at room temperature; values given in ppm (δ) or Hz (J).
|
|
3a
|
−103.9 (−124.4) |
192.2 |
76.8 |
|
3b
|
−125.4 (−161.1) |
186.6 |
73.0 |
|
3c
|
−134.8 (−164.8) |
177.9 |
77.9 |
|
3d
|
−120.6 (−157.9) |
191.5 |
78.8 |
|
3e
|
−145.4 (−179.4) |
183.7 |
78.1 |
|
3f
|
−144.8 (−164.1) |
179.6 |
77.2 |
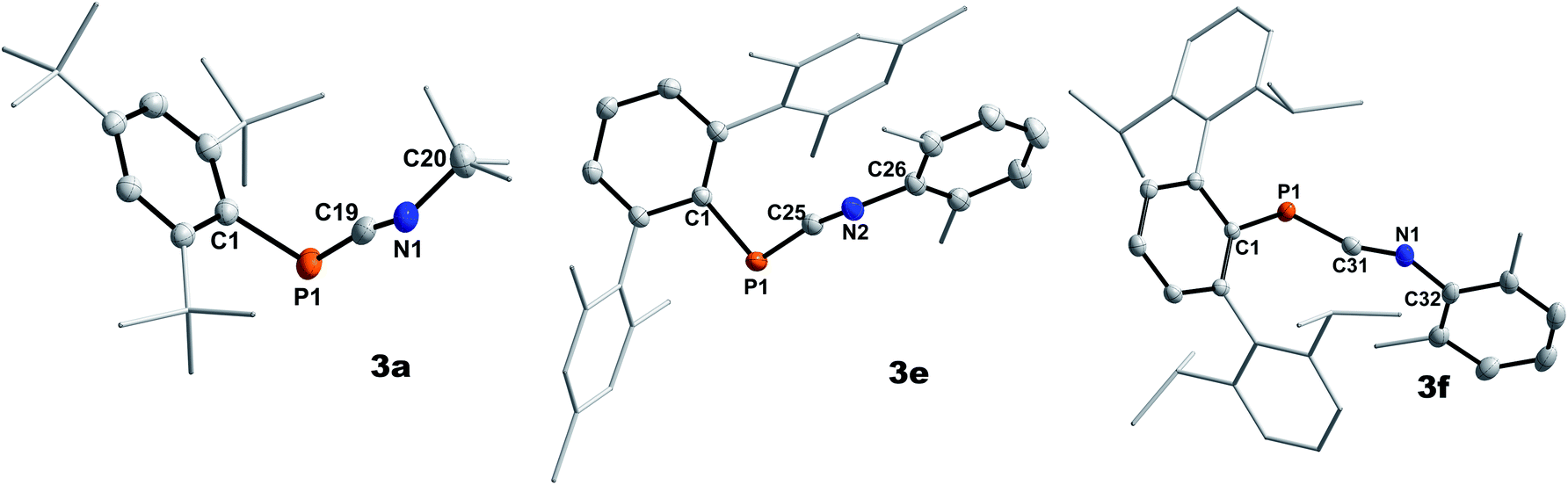
Highly characteristic for 3a–f are the 13C{1H}NMR signals of the two-coordinate carbon atoms of the PCN moieties, being significantly deshielded (δ13C{1H} = 177.9 to 192.2 ppm) and showing 1JP,C coupling constants of 73.0 to 78.1 Hz (Table 1). Additionally, the molecular structures of 3a, 3e, and 3f could be determined by single crystal X-ray diffraction (SC-XRD, Fig. 1, Table 2). The P–C bond lengths of 3a, 3e, and 3f of 1.6658(15) Å (3a) to 1.6785(12) Å (3f) are slightly elongated compared to I (1.651 Å) and II (1.642(5) Å), respectively, but are shorter than the sum of the covalent double bond radii (Σrcov(PC) = 1.69 Å).30 The N–C bond lengths of 3a, 3e, and 3f (1.2009(15) Å to 1.2037(19) Å) are in the expected range for heteroallenes (cf. XylNCNXyl d(C–N) 1.197(2), 1.206(3) Å).31,32 Noteworthy, the P–C–N angles deviate from linearity (as expected for sp-hybridized carbon atoms) but are in good agreement to previously structurally characterized 1,3-phosphaazaallenes (Table 2).
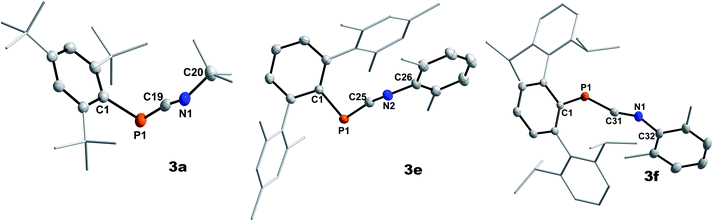 |
| | Fig. 1 Molecular structures of 3a (left), 3e (middle), and 3f (right). Hydrogen omitted and parts of the molecule rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Structural parameters are summarized in Table 2. | |
Table 2 Selected bond lengths [Å] and angles [°] of 3a, 3e and 3f (literature known species I and II for comparison)
| Compound |
P–C |
N–C |
P–C–N |
C–P–C |
C–N–C |
|
3a
|
1.6690(15) |
1.2034(18) |
170.38(12) |
97.92(6) |
130.02(13) |
|
3e
|
1.6658(15) |
1.2037(19) |
167.14(13) |
103.06(7) |
139.44(15) |
|
3f
|
1.6785(12) |
1.2009(15) |
160.00(10) |
107.53(5) |
143.24(12) |
| Mes*PCNPh (I)4 |
1.651 |
1.209 |
171.1 |
99.2 |
130.5 |
| Mes*PCN(p-ClC6H4) (II)6 |
1.642(5) |
1.214(6) |
170.8(4) |
99.8(2) |
128.3(4) |
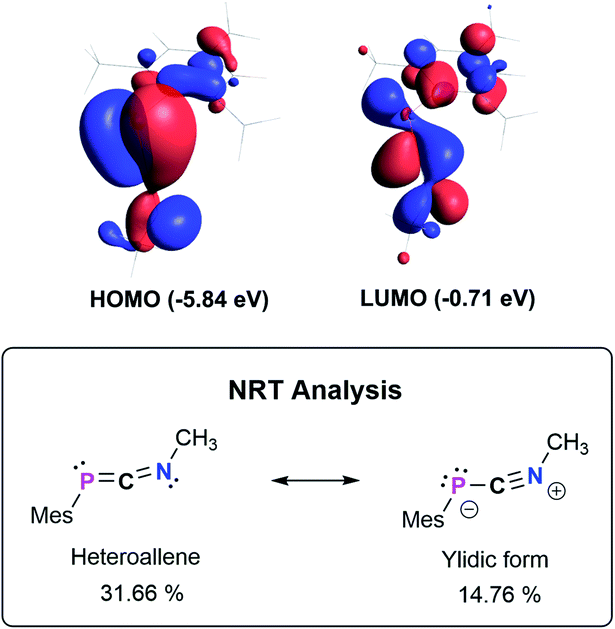
The bonding in 3a–f was studied using the truncated model compound MesPCNMe on the PBE0-D3/def2SVP level of theory. Inspection of the Kohn–Sham orbitals revealed a HOMO best described as a polarized P–C π-bond, while the LUMO shows major contribution from the C–N π* orbital interacting with a s-type lone pair on phosphorus (Fig. 2, top). With an energetically high lying HOMO the 1,3-phosphaazaallenes might be potentially oxidized to give the corresponding radical cation, as was recently shown for vinyl-substituted diphosphenes.33 CV studies on 3a show an irreversible oxidation event at E1/2 = 0.38 V vs. Fc/Fc+ (Fig. S82–S84†), and the corresponding radical cation might be synthetically feasible. We next evaluated the NPA (Natural Population Analysis) charges indicating a minimal charge transfer from the MesP-fragment to the CNMe moiety by −0.196e, with a positive partial charge on P of 0.37e and 0.07e on the two-coordinate C atom. Natural Bond Orbital (NBO) analysis supports the description as an heteroallene, with a LP of electrons on P and polarized σ- and π-PCNMe (WBI 1.64) and PCNMe (WBI 2.05) double bonds, respectively. In agreement with the KS-orbitals the π-component is polarized towards the P atom (58.3% P, 41.7% C), whereas the σ-component is inversely polarized (34.5% P, 65.5% C). Analysis of the second order perturbation of the Fock matrix revealed delocalization of the lone pair of electrons (LP) on P into the CN π*-orbital resulting in a stabilization energy of 12.7 kcal mol−1. Natural resonance theory analysis (NRT) revealed two leading resonance structures, with the 1,3-phosphaazaallene being the dominant form (31.7%) and an ylidic formulation with a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond and thus two LPs on P (14.8%) (Fig. 2, bottom).
N triple bond and thus two LPs on P (14.8%) (Fig. 2, bottom).
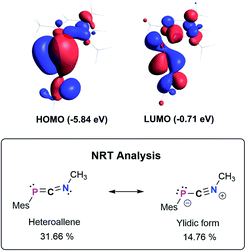 |
| | Fig. 2 Selected Kohn–Sham orbitals of the truncated model compound MesPCNMe (PBE0-D3/def2SVP) and leading resonance structures according to NRT analysis. | |
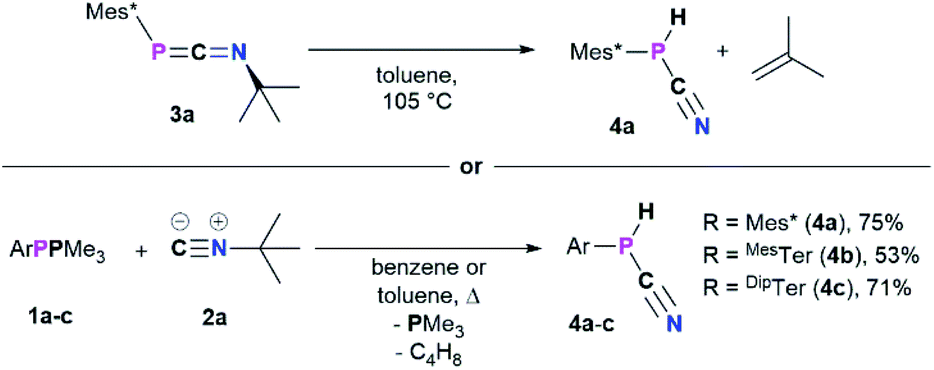
Formation of cyanophosphines starting from 3a–c
When a solution of 3a was heated to 105 °C in toluene-d8 a new species with a 31P{1H} NMR chemical shift of −105.6 ppm (cf.3aδ31P{1H} = −103.9 ppm) was observed along with minimal amounts of the diphosphene Mes*PPMes* (δ31P{1H} = 493.2 ppm). This transformation is accompanied by the formation of iso-butene, as evident from two signals in the 1H NMR spectrum in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at 1.60 (triplet) and 4.75 (heptet) ppm, respectively. Finally, the multinuclear and multidimensional NMR data clearly showed that the new compound is the cyanophosphine Mes*P(H)CN (4a) (Scheme 3). Isobutene elimination and formal HCN transfer has been previously observed with disilynes,34 whereas with boracummulenes and transient borylenes CN− transfer was observed, with formation of a mixture of iso-butane and -butene.35,36 Streubel and co-workers showed that the η1-1,3-phosphaazaallene complex (Me3Si)2HC–P(W(CO)5)CNtBu undergoes thermal loss of iso-butene to give the corresponding cyanophosphine tungsten complex.12 The thermodynamic feasibility of this transformation was elucidated at the PBE0-D3/def2SVP//DNLPO-CCSD(T)/def2TZVP level of theory, showing that the formation of 4a is exergonic by −36.93 kJ mol−1, whereas the dimerization of 3a to give Mes*PPMes* and CNtBu is less favored (ΔRG = −5.68 kJ mol−1). A scan of the potential energy surface revealed that the H-shift from the tBu-group to P occurs intramolecularly via a six-membered transition state (Δ#G = 156.1 kJ mol−1), resulting in P–H bond formation as the C(sp3)–H and N–CtBu bonds are cleaved (Fig. S87†). This rather high energy barrier is in line with prolonged heating of the reaction mixture at 105 °C. An alternative radical pathway through N–CtBu bond homolysis and formation of a tBu˙ radical was excluded as this would result in disproportionation and a mixture of iso-butene and iso-butane. The intermediate formation of free phosphinidenes is also unlikely, as this would give rise to cyclo-metalated species in case of Mes* and DipTer, which were not observed by NMR spectroscopy. Alternatively, 4a can be prepared directly in one pot starting from 1a and 2a (Scheme 3, bottom).16 Following this route 4a was isolated as a colourless solid in good yields of 75%. Given the PH functionality, the 1H NMR spectrum shows a characteristic doublet at 5.57 ppm with a 1JP,H coupling constant of 252.3 Hz, which is corroborated by the 31P NMR spectrum. To further elaborate the scope of this reaction, the analogous MesTerP(H)CN (4b) and DipTerP(H)CN (4c) derivatives were synthesized, and their characteristic NMR data is shown in Table 3. Surprisingly, in the IR spectrum of 4a and 4c no characteristic CN band is detected, in agreement with frequency analyses at the PBE0-D3/def2SVP level of theory. The presence of a P–H moiety was corroborated by a band at 2411 and 2310 cm−1 for 4a and 4c, respectively. Cyanophosphines of the general type RP(H)CN (R = alkyl, aryl) have long remained elusive and were either found to be unstable,37 or to be stabilized by coordination to a transition metal.38
:1 ratio at 1.60 (triplet) and 4.75 (heptet) ppm, respectively. Finally, the multinuclear and multidimensional NMR data clearly showed that the new compound is the cyanophosphine Mes*P(H)CN (4a) (Scheme 3). Isobutene elimination and formal HCN transfer has been previously observed with disilynes,34 whereas with boracummulenes and transient borylenes CN− transfer was observed, with formation of a mixture of iso-butane and -butene.35,36 Streubel and co-workers showed that the η1-1,3-phosphaazaallene complex (Me3Si)2HC–P(W(CO)5)CNtBu undergoes thermal loss of iso-butene to give the corresponding cyanophosphine tungsten complex.12 The thermodynamic feasibility of this transformation was elucidated at the PBE0-D3/def2SVP//DNLPO-CCSD(T)/def2TZVP level of theory, showing that the formation of 4a is exergonic by −36.93 kJ mol−1, whereas the dimerization of 3a to give Mes*PPMes* and CNtBu is less favored (ΔRG = −5.68 kJ mol−1). A scan of the potential energy surface revealed that the H-shift from the tBu-group to P occurs intramolecularly via a six-membered transition state (Δ#G = 156.1 kJ mol−1), resulting in P–H bond formation as the C(sp3)–H and N–CtBu bonds are cleaved (Fig. S87†). This rather high energy barrier is in line with prolonged heating of the reaction mixture at 105 °C. An alternative radical pathway through N–CtBu bond homolysis and formation of a tBu˙ radical was excluded as this would result in disproportionation and a mixture of iso-butene and iso-butane. The intermediate formation of free phosphinidenes is also unlikely, as this would give rise to cyclo-metalated species in case of Mes* and DipTer, which were not observed by NMR spectroscopy. Alternatively, 4a can be prepared directly in one pot starting from 1a and 2a (Scheme 3, bottom).16 Following this route 4a was isolated as a colourless solid in good yields of 75%. Given the PH functionality, the 1H NMR spectrum shows a characteristic doublet at 5.57 ppm with a 1JP,H coupling constant of 252.3 Hz, which is corroborated by the 31P NMR spectrum. To further elaborate the scope of this reaction, the analogous MesTerP(H)CN (4b) and DipTerP(H)CN (4c) derivatives were synthesized, and their characteristic NMR data is shown in Table 3. Surprisingly, in the IR spectrum of 4a and 4c no characteristic CN band is detected, in agreement with frequency analyses at the PBE0-D3/def2SVP level of theory. The presence of a P–H moiety was corroborated by a band at 2411 and 2310 cm−1 for 4a and 4c, respectively. Cyanophosphines of the general type RP(H)CN (R = alkyl, aryl) have long remained elusive and were either found to be unstable,37 or to be stabilized by coordination to a transition metal.38
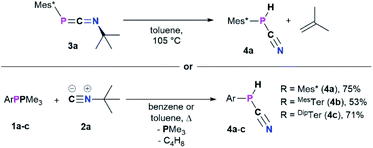 |
| | Scheme 3 Formation of the cyanophosphines 4a–c from 3a–c (top) or directly from 1a–c and tBuNC (2a). | |
Table 3 Characteristic 31P{1H}5.61 and 13C{1H} NMR data of 4a–c. Calculated 31P NMR shifts (PBE0-D3/def2SVP) are given in parentheses
| Compounda |
δ
1H (PH) |
δ
31P (δcalc31P) |
1
J
P,H (PH) |
δ
13C (CN) |
1
J
P,C (CN) |
|
In C6D6 at room temperature; values in ppm (δ) or Hz (J).
previously reported NMR data for 4a was collected in CD2Cl2 [values given in brackets].39
|
|
4a
|
5.57 [5.95]b |
−105.4 [-101.6]b (−139.1) |
252.3 [249.7]b |
120.8 [121.2]b |
76.3 [74.4]b |
|
4b
|
4.38 |
−120.6 (−154.9) |
244.8 |
116.7 |
76.7 |
|
4c
|
4.35 |
−120.4 (−154.6) |
247.2 |
116.6 |
75.3 |
|
5a
|
5.61 |
−99.2 () |
260.0 |
121.0 |
106.8 |
|
5b
|
4.61 |
−115.1 |
250.9 |
— |
— |
|
5c
|
5.03 |
−108.3 |
256.1 |
— |
— |
| MeP(H)CN39 |
4.15 |
−119.9 |
227.5 |
119.6 |
70.9 |
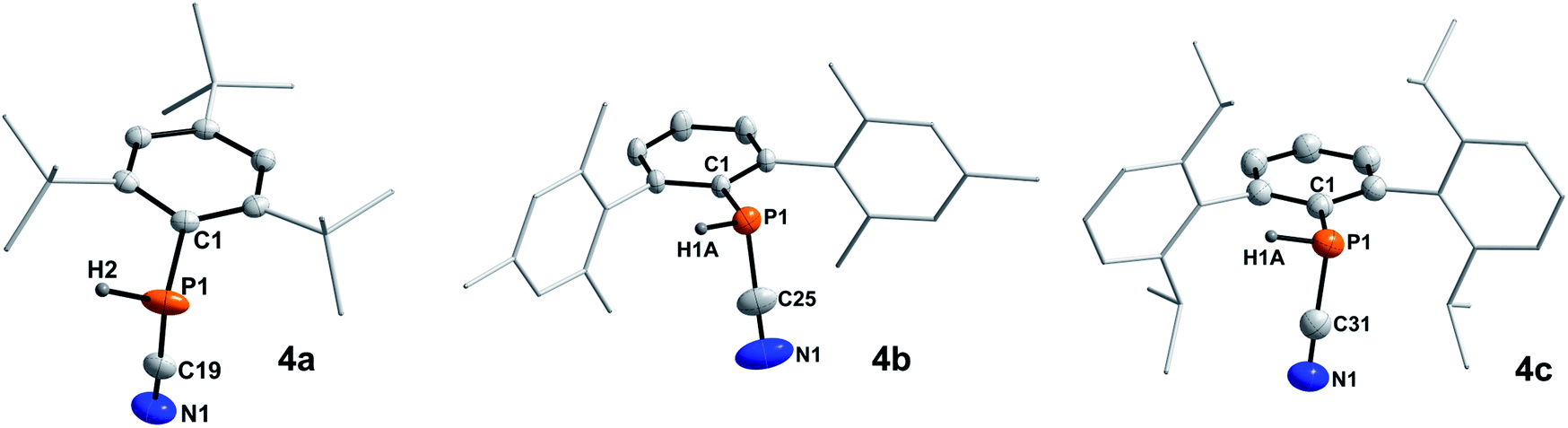
In 2001 the reaction of dicyanophosphines (RP(CN)2) with equimolar amounts of Schwartz's reagent ([Cp2Zr(H)Cl]n) was shown to afford the methyl, tert-butyl, and Mes* derivatives, respectively.39 However, structural data of this compound class is missing in the literature and the molecular structures of 4a–c could be determined by SC-XRD (Fig. 3, Table 4).The C–N bond lengths in 4a–c average 1.146 Å and indicate triple bonds (Σrcov(CN) = 1.14 Å),30 in agreement with the formulation as cyanophosphines. The average P–C bond length of 1.791 Å is shorter than the respective single bond covalent radii (Σrcov(P–C) = 1.86 Å),30 with a nearly linear arrangement of the P–C–N unit (>176°). Similar bond lengths were reported for Mes*P(CN)2 (P–Cavg 1.80 Å, N–Cavg 1.14 Å).39 NBO analyses of 4a–c at the PBE0-D3/def2SVP//PBE0/def2SVP level of theory support the notation as cyanophosphines with CN triple bonds (WBI CN 4a 2.88, 4b 2.87, 4c 2.87), a polar Pδ+–Cδ−CN single bond and a LP on P, which is minimally delocalized into two π* orbitals of the CN group with a stabilization energy of ca. 12 kcal mol−1.17
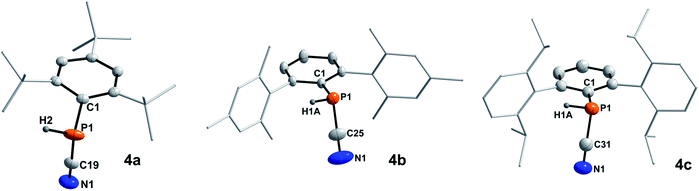 |
| | Fig. 3 Molecular structures of 4a (left), 4b (middle), and 4c (right). Hydrogen atoms (except on P1) omitted and parts of the molecules rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Structural parameters are summarized in Table 4. | |
Table 4 Selected bond lengths [Å] and angles [°] of 4a–c and 5a
| Compound |
P–C |
N–C |
P–C–N |
C–P–C |
|
4a
|
1.796(3) |
1.143(4) |
176.4(3) |
97.49(11) |
|
4b
|
1.7853(18) |
1.148(2) |
177.41(16) |
100.31(6) |
|
4c
|
1.793(2) |
1.146(3) |
177.5(2) |
99.32(9) |
|
5a
|
1.799(3) |
1.136(3) |
175.7(2) |
97.71(11) |
Reactivity of cyanophosphines towards B(C6F5)3
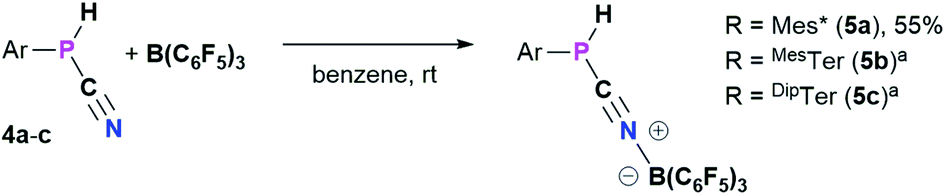
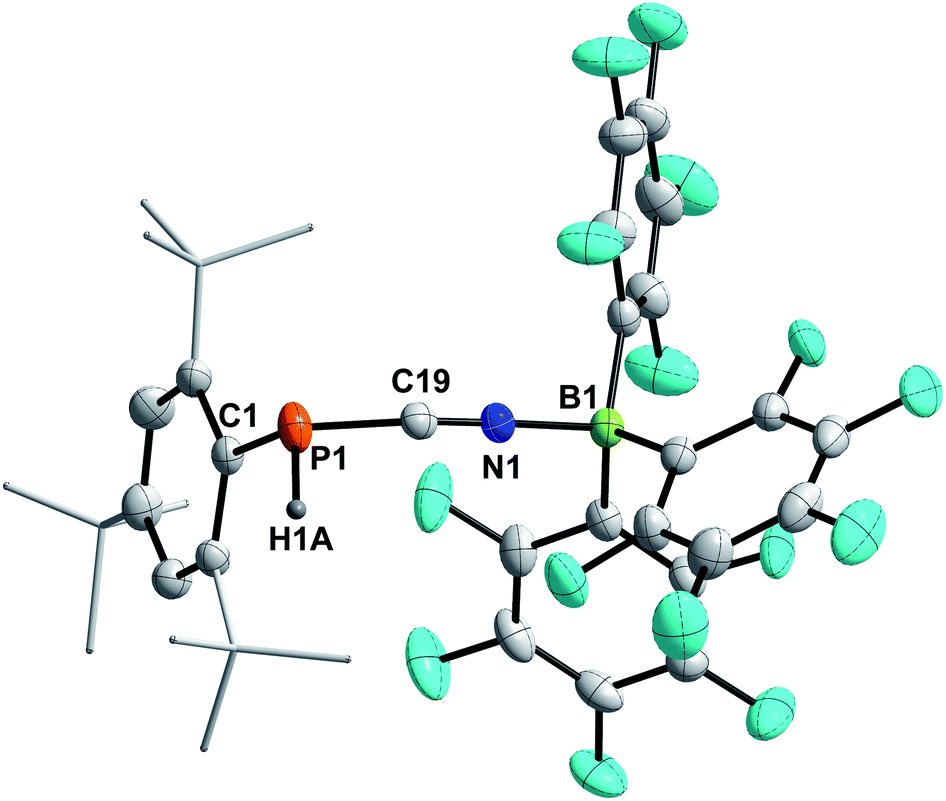
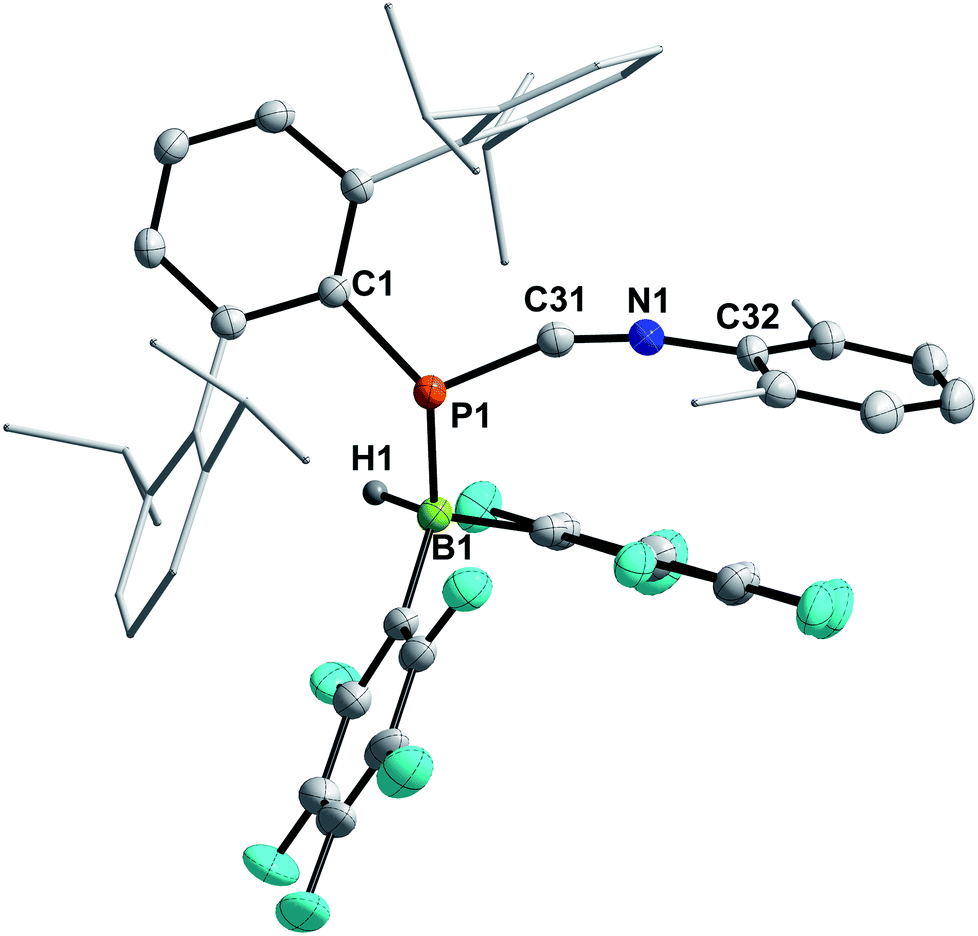
4a–c possess two potential binding sites for Lewis acids, the LPs on P and N, even though steric congestion should render the phosphorus rather inaccessible. By reacting 4a–c with B(C6F5)3, the first examples of the corresponding phosphanitrilium borates RP(H)CNB(C6F5)3 (R = Mes* (5a), R = MesTer (5b), R = DipTer (5c)) were prepared (Scheme 4). The reactions were performed on NMR scale and 5a was exemplarily isolated as a colourless solid in a moderate yield of 55%. The coordination to the borane moiety results in a minimal deshielding of the PH unit accompanied by a slight increase of the 1JP,H coupling constant in both the 1H and 31P NMR spectra (δ(1H) = 5.61 ppm, δ(31P) = −99.2 ppm, 1JP,H = 260.0 Hz; δcalc(31P) = −140.9 ppm), respectively, when compared to the starting material 4a (δ(1H) = 5.57 ppm, δ(31P) = −105.4 ppm, 1JP,H = 252.3 Hz). The signals of the CN group are unaltered (5a: δ(13C{1H}) = 121.0 ppm, c.f.4a: δ13C{1H} = 120.8 Hz), while the 1JP,C coupling constant increases significantly to 1JP,C = 106.8 Hz (cf.4a1JP,C = 76.3 Hz). Interestingly, in the IR spectrum the CN stretch is now visible as a weak band at 2265 cm−1. The 11B{1H} NMR resonance at −10.3 ppm is consistent with tetra-substituted boron atoms bearing perfluorinated aryl groups (cf. [K(18-crown-6)][SCNB(C6F5)3]: δ(11B{1H}) = −12.4 ppm).40 The three C6F5 groups are equivalent as verified by the respective 19F NMR spectrum (δ(19F{1H}4 = −133.9 (meta), −155.8 (para), and −163.4 (ortho) ppm; Δ(δ)19Fm,p = 7.6 Hz), which is in agreement with other nitrilium borates with a heteroatom at the carbon atom of the CN triple bond (cf. PhSCNB(C6F5)3: δ(19F{1H}) = −134.0 (meta), −155.7 (para), and −163.3 (ortho) ppm; Δ(δ)19Fm,p = 7.6 Hz).41 The molecular structure of 5a (Fig. 4) confirms the four-coordinate boron atom and the BNCP axis is in a nearly linear arrangement (P1–C19–N1 175.7(2)°, C19–N1–B1 178.3(2)°). The N1–C19 bond length of 1.136(3) Å is still diagnostic of a triple bond (cf.4a 1.143(4) Å; Σrcov(CN) = 1.14 Å).30 The newly formed N1–B1 bond (1.584(3) Å) is in the same range as reported for PhSCNB(C6F5)3 (1.5829(10) Å)41 and slightly shorter when compared to classic nitrile–B(C6F5)3 adducts (cf. MeCNB(C6F5)3 1.616(3) Å).42
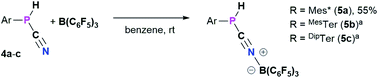 |
| | Scheme 4 Synthesis of phosphanitriliumborates ArP(H)CNB(C6F5)35a–c. a NMR reactions, products were not isolated. | |
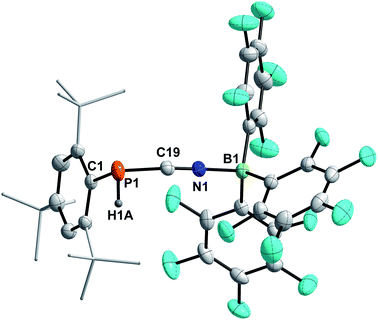 |
| | Fig. 4 Molecular structure of 5a. Hydrogen atoms (except H1) omitted and tBu-groups on Mes* rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): P1–C19 1.799(3), N1–C19 1.136(3), N1–B1 1.584(3), P1–C19–N1 175.7(2), C19–N1–B1 178.3(2), C19–P1–C1 97.71(11). | |
Attempted syntheses of cyanophosphides
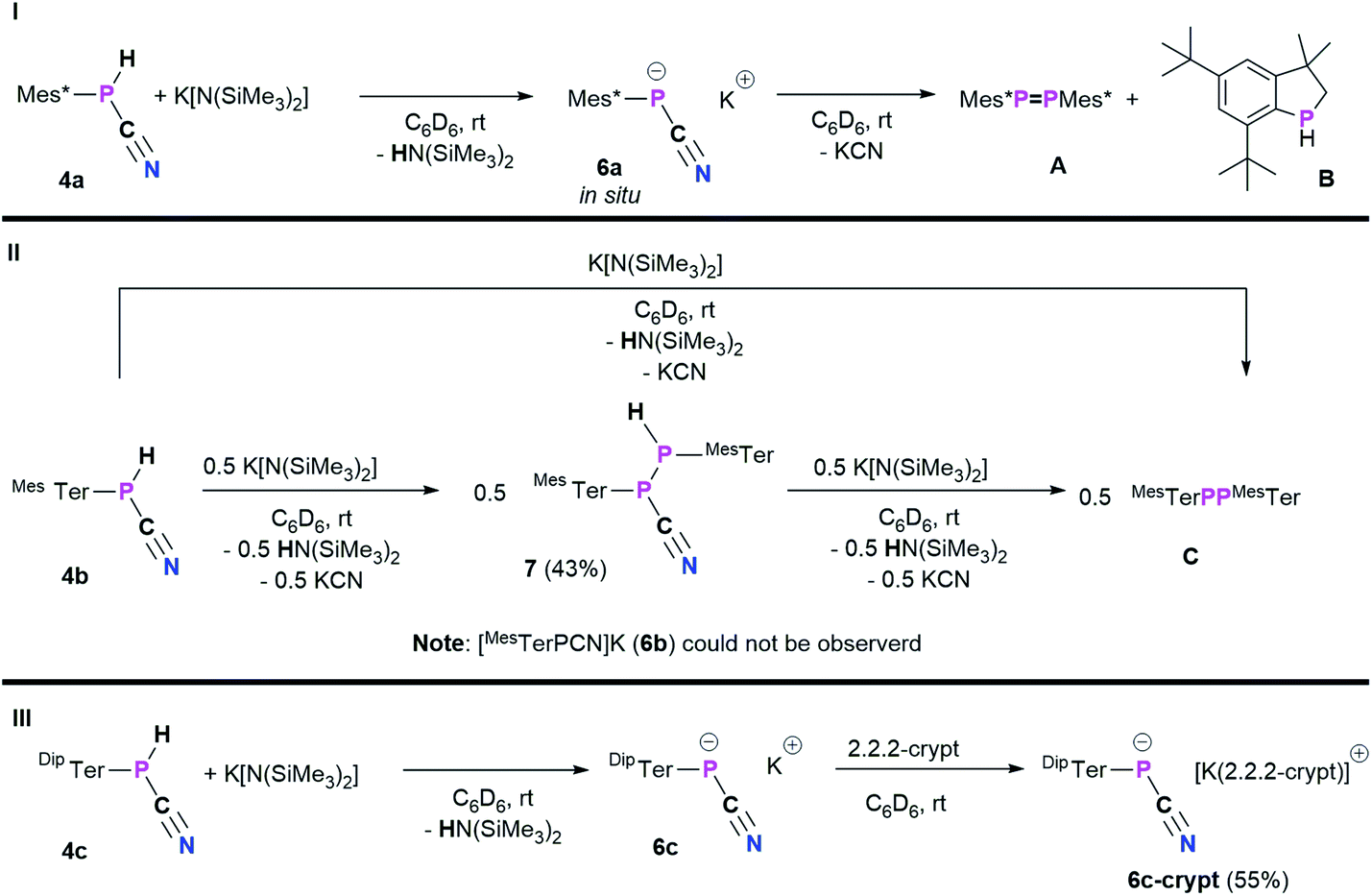
With 4a–c in hand, we envisioned to synthesize the corresponding cyanophosphides [RPCN]− through simple deprotonation of 4a–c. The first dicyanophosphides [P(CN)2]M were isolated by Schmidtpeter et al. through reductive decyanation of P(CN)3,43 and alternative synthetic strategies have surfaced since this initial report.44–46 Schmidtpeter and co-workers then synthesized [PhPCN]M (M+ = Na, K, [(Ph3P)2N]) by reacting Ph5P5 with the corresponding cyanides as equilibrium mixtures, which is shifted to [PhPCN]M when using weakly-coordinating cations.47–49 Recently, Grützmacher et al. introduced alkali phosphanyl cyanophospides [(NHP)PCN]M (NHP = N-heterocyclic phosphenium, M = Na, K) as versatile PCN building blocks, by an oxygen for nitrogen exchange from phosphanyl phosphaketenes of the general type (NHP)PCO with (M(NSiMe3)2) (M = alkali metal) and concomitant formation of O(SiMe3)2.50 Wolf, Weigand and co-workers observed the formation of the phosphanyl-substituted cyanophosphides ([R2PPCN]−; R = Ph, Cy, tBu, N(iPr)2) as counter-anions for anionic cyclo-triphosphido cobalt complexes.51 Inspired by these results, the potential of 4a–c being deprotonated by K[N(SiMe3)2] (KHMDS) was investigated (Scheme 5).
 |
| | Scheme 5 Reactivity of 4a–c towards KHMDS: (I) in situ synthesis of [Mes*PCN]K (6a) and decomposition towards phosphaindane A, diphosphene B and KCN; (II) synthesis of MesTerP(H)P(CN)MesTer (7) or diphosphene C dependent on the used stoichiometry; (III) synthesis of [DipTerPCN]K (6c) and [DipTerPCN][K(2.2.2-crypt)] (6c-crypt). | |
As a starting point, the cyanophosphine Mes*P(H)CN (4a) and KHMDS were combined on an NMR scale, accompanied by a color change from colorless to yellow and formation of a colorless precipitate. 1H and 31P{1H} NMR spectra were immediately recorded and show that the main species at this point showed a 31P{1H} NMR signal at −146.2 ppm, which according to 1H NMR spectroscopy does not bear a P–H function and HN(SiMe3)2 (HMDS, δ(1H) = 0.10 ppm) was observed as well.20 This indicated successful deprotonation to give [Mes*PCN]K (6a).
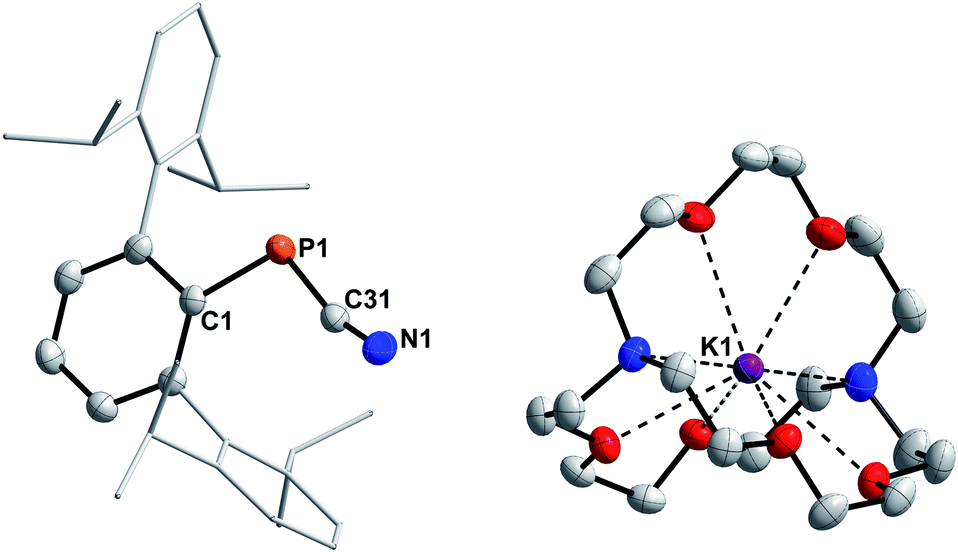
Nevertheless, 6a is unstable at room temperature and after 16 h at room temperature, the 31P{1H} NMR data revealed three signals at 493.2 (Mes*PPMes*, A),25 −79.7, and −146.2 ppm, respectively.20 The signal at −79.7 ppm is now the main species and was assigned to the known 3,3-dimethyl-5,7-di-tert-butylphosphaindane (B).52 Unfortunately, up to now all attempts to isolate, crystallize or trap 6a have not been successful and only crystals of A and B could be obtained. From a mechanistic point of view, we assume that deprotonation of 4a by KHMDS leads to the formation of HMDS and 6a, the latter then eliminates KCN to give a reactive phosphinidene intermediate capable of both dimerization to give A and capable of insertion of the phosphinidene fragment into one methyl group of one tert-butyl group of the Mes* substituent to yield B.52 Burg and Slota noted that the stability of species of the type RPHX is greatly enhanced by the steric profile of the substituent R.53 Therefore, the terphenyl-based cyanophosphines 4b and 4c were expected to make the anions isolable. The reaction of MesTerP(H)CN (4b) and KHMDS resulted in an immediate color change of the reaction mixture from colorless to yellow and precipitation of a colorless solid. Interestingly, the clean formation of MesTerPPMesTer (C) (δ(31P{1H}) = 492.5 ppm) and HMDS were observed even when the reaction mixture is directly analyzed by NMR spectroscopy after reacting both substrates.20 To get information whether any other phosphorus containing species can be observed (e.g. intermediate formation of a phosphinidene which dimerizes to C), 4b and 0.5 eq. of KHMDS were combined and the solution was directly analyzed by NMR spectroscopy. Intriguingly, two doublet signals were observed in the 31P{1H} NMR spectrum at −78.2 and −82.6 ppm with a coupling constant of 1JP,P = 326.6 Hz. The corresponding 31P NMR spectrum revealed the existence of two doublets of doublets with additional coupling constants of 38.3 Hz and 216.3 Hz, respectively. In addition, a highly diagnostic doublet of doublet signal in the 1H NMR spectrum at 3.97 ppm confirms that the above mentioned coupling constants correspond to 1JP,H and 2JP,H coupling, thus the obtained molecule bears a unique P(H)–P moiety.20 In accordance with the NMR data and high-resolution mass spectrometry, SC-XRD verified the formation of the diphosphane MesTerP(H)P(CN)MesTer (7, Fig. S1†). Treatment of 7 with additional amounts of KHMDS then resulted in the clean conversion to give diphosphene C as shown by 31P{1H} NMR spectroscopy. It is worth mentioning, that the reaction of 4a with half an equivalent of KHMDS only leads to the described concomitant formation of 6a, A, B, KCN, and HMDS with parts of 4a remaining unreacted. Finally, the even bulkier cyanophosphine DipTerP(H)CN (4c) was reacted with equimolar amounts of KHMDS, giving an immediate color change to yellow. A significantly shielded signal in the 31P{1H} NMR spectrum at −142.0 ppm (c.f. in situ prepared 6a: δ(31P{1H}) = −146.2 ppm; [(NHP)PCN]M: δ(31P{1H}) = −124 to −84 ppm50) indicated the formation of the corresponding cyanophosphide [DipTerPCN]K (6c). 6c proved to be stable in C6D6 solution for at least one week at room temperature. Subsequently, the potassium cation could be sequestered by adding 2.2.2-cryptand to quantitatively give the ion separated salt [DipTerPCN][K(2.2.2-crypt)] (6c-crypt). The ion separation leads to the expected low-field shift in the 31P{1H} NMR of approximately 20 ppm so that a signal at −120.7 ppm is detected. In addition, the molecular structure of 6c-crypt was determined by SC-XRD (Fig. 5).
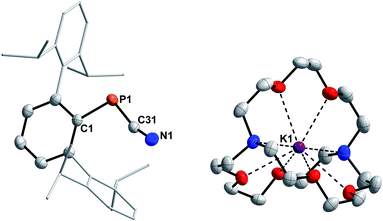 |
| | Fig. 5 Molecular structure of 6c-crypt. Hydrogen atoms omitted and Dip-groups rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): P1–C31 1.7680(14), N1–C31 1.1585(19), P1–C31–N1 165.45(12), C1–P1–C31 106.73(6). | |
The structural parameters of the P(−)CN unit indicate that the negative charge is mainly located at the phosphorus, with a N1–C31 bond length of 1.1585(19) Å (Σrcov(CN) = 1.14 Å,30cf. Ph3PC(H)CN 1.158(3) Å).54 This is minimally longer than in starting material 4a (1.146(3) Å), whereas the P1–C31 bond length is slightly shortened (1.7680(14) Å; c.f.4a: 1.793(2) Å). Therefore a major contribution from the resonance structure R–P(−)–CN and a minor contribution from the resonance structure R–PCN(−) is reasonable, and is further supported by the C–N stretching frequency of 2053 cm−1. The only other structurally characterized cyanophosphides bear phosphorus based substituents at the phosphorus atom of the PCN moiety but show nearly identical bond lengths across the PCN axis (c.f. [iPr2PPCN]−: P–C 1.763(1) Å, N–C 1.160(2) Å;51 [(NHP)PCN]−: P–Cavg. 1.75 Å, N–Cavg 1.16 Å).50 Whereas for the previously described cyanophosphides nearly linear arrangements of the PCN moieties are observed (N–C–P > 177°),50,51 the P1–C31–N1 bond angle of 165.45(12)° deviates significantly from linearity which might be caused by steric repulsion of the sterically demanding DipTer group.
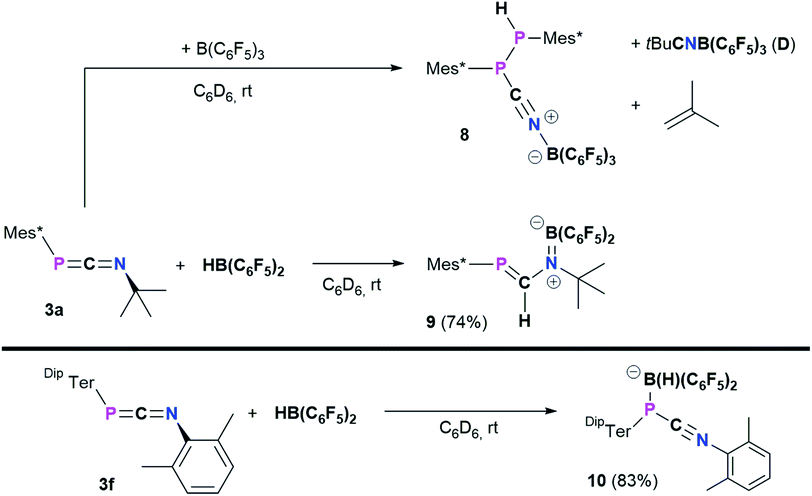
Reactivity of selected 1,3-phosphaazaallenes towards B(C6F5)3 and Pier's borane HB(C6F5)2
Heteroallenes like carbodiimides, isocyanates, and isothiocyanates have shown a diverse reactivity towards the perfluorinated boranes B(C6F5)3 and HB(C6F5)2 (Pier's borane), ranging from the development of new heterocycles to the formation of classic and frustrated Lewis pairs (FLPs) and 1,2-hydroboration reactions.41,55–58
Treatment of 3a with B(C6F5)3 in toluene afforded a colorless suspension (Scheme 6, top). After stirring for 16 h and subsequent workup,20 the isolated colorless solid was hardly soluble in aromatic hydrocarbons and started to polymerize tetrahydrofuran within minutes. From a saturated C6D6 solution sufficient 1H, 11B{H}, 19F{1H}, and 31P{1H} data was obtained and the 31P{1H} NMR spectrum showed two characteristic signals at −46.8 and −53.3 ppm, respectively with a characteristic 1JP,P = 247.5 Hz coupling constant, reminiscent of MesTerP(H)P(CN)MesTer (7) (c.f. δ(31P{1H}) = −78.2 and −82.6 ppm, 1JP,P = 326.6 Hz). The existence of a P(H)–P moiety was supported by the 1H and 31P NMR data, which show that the signal at −53.3 ppm is a doublet of doublets with 1JP,H = 224.0 Hz, which is further corroborated by a doublet signal with the same 1JP,H coupling constant in the 1H NMR spectrum at 5.44 ppm. The reaction is accompanied by significant amounts of byproducts as evident from two signals in the 11B{1H} NMR spectrum at −7.9 (significantly broadened) and −20.7 ppm, respectively. Similarly, the 19F{1H} NMR spectrum shows a total of nine signals. Moreover, iso-butene was identified as byproduct (δ(1H) = 1.60 and 4.74 ppm, Fig. S67†), similarly to the synthesis of the cyanophosphines 4a–c.
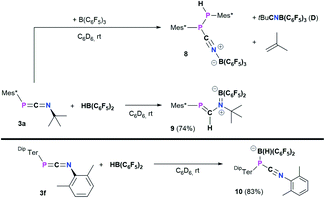 |
| | Scheme 6 Reactivity of 3a towards B(C6F5)3 and HB(C6F5)2, and reactivity of 3f towards HB(C6F5)2 to give 10. | |
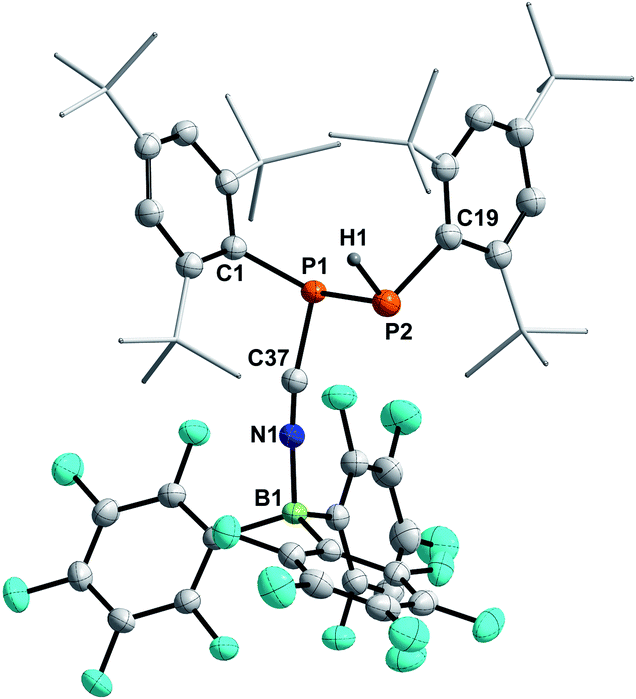
Crystallization attempts gave two types of colorless crystals, and SC-XRD confirmed that indeed the diphosphane Mes*P(H)P(CNB(C6F5)3)Mes* (8, Fig. 6) was formed alongside the literature known nitrile–borane adduct tBuCNB(C6F5)3 (D) (Scheme 6, top).42 It is worth mentioning, that all attempts to isolate 8 in pure fashion failed up to now, which is attributed to quite similar solubilities of 8 and D. In 8 the newly formed P1–P2 and N1–B1 bond lengths of 2.2464(8) Å and 1.572(3) Å are in good accordance with the formulation as single bonds (Σrcov(P–P) = 2.22 Å; Σrcov(N–B) = 1.56 Å).30 The N1–C37 bond length of 1.140(3) Å is a typical carbon nitrogen triple bond (Σrcov(CN) = 1.14 Å),30 and the P1,C37,N1,B1 axis is minimally bent (e.g. C37–N1–B1 174.9(2)°).
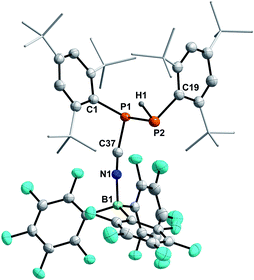 |
| | Fig. 6 Molecular structure of 8. Hydrogen atoms (except H1) omitted and tBu-groups on Mes* rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): P1–P2 2.2464(8), N1–B1 1.572(3), P1–C37 1.784(2), P1–C1 1.844(2), P2–C19 1.852(2), N1–C37 1.140(3); P1–C37–N1 165.4(2), C37–N1–B1 174.9(2). | |
All these metrics agree with phosphanitrilium borate 5a (Fig. 4). It is noteworthy that the phosphaketene [sP]PCO reacted with B(C6F5)3 to give a zwitterionic diphosphirenium with a P2C three-membered ring with an exocyclic C–O–B(C6F5)3 moiety.21 We continued to investigate the reactivity of 3a towards Pier's borane (HB(C6F5)2) to check its potential for hydroboration chemistry.59
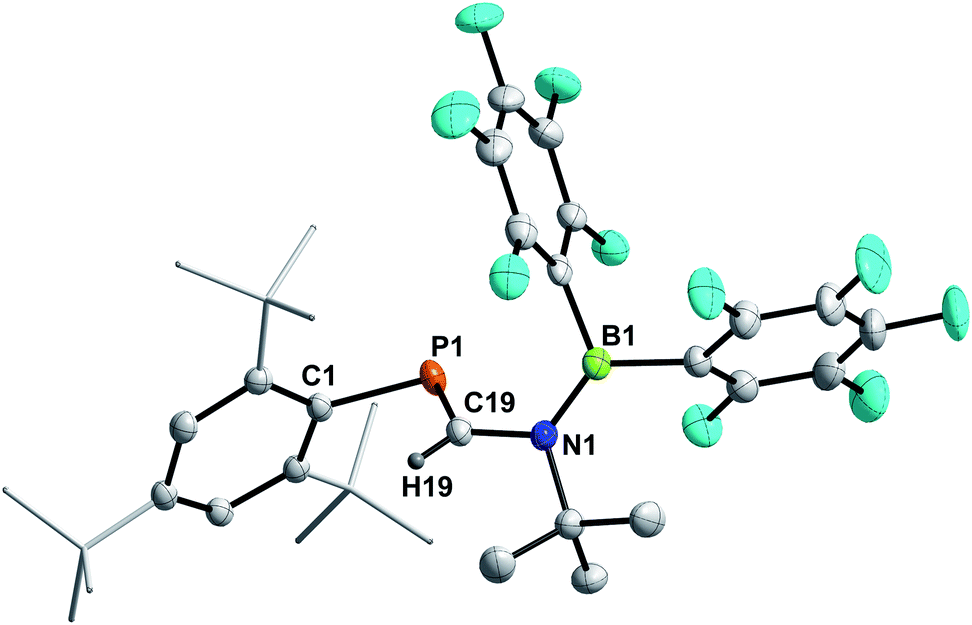
The reaction of 3a and HB(C6F5)2 yielded a yellow solid after workup (isolated yield 74%, Scheme 6, middle). Single crystals grown from layering a saturated C6D6 solution with n-hexane revealed the product to be Mes*PC(H)N(tBu)B(C6F5)2 (9, Fig. 7), showing that 1,2-hydroboration across the CN bond of 3a had occurred (Scheme 6, middle). Remarkably, the molecular structure of 9 reveals a novel heterodiene (PC–N(+)B(−)) structural motif. Both, the P1–C19 and N1–B1 bond lengths of 1.6751(13) Å and 1.3995(18) Å are best described as double bonds, respectively, which is also illustrated in the KS orbitals (PBE0-D3/def2-SVP, Fig. S91†). The HOMO is best described as the P–C and B–N π-bonds, respectively, with one node. The LUMO has π* character for P–C and B–N bonds resulting in two nodes and with π-character between C and N, as expected for a heterodiene.60 The nature of BN multiple bonds has been in the focus of recent computational studies,61,62 and NBO results for 9 show a σ (N 79.5, B 20.5%) and π (N 86.1, B13.9%) NBO, which are mainly formed by the natural atomic hybrid orbitals located on N. This agrees well with the values obtained for 9,10-diimino-9,10-dihydro-9,10-diboraanthracene.62,63 Topological analysis of the electron density using the QT-AIM approach revealed an electron density (ρ(3,−1) [e bohr−3]) of 0.198 at the BN bond critical point (BCP), as well as an electron density Laplacian (∇2 [e bohr−5]) of 0.651, which corresponds nicely with the aforementioned diminodiboraanthracene.20,62 In addition, the sum of angles around C19, N1, and B1 all add up to over 359.8°, in line with sp2-hybridization. The solution NMR spectra of 9 are indicative that this diene structure sustains in solution, with one resonance in the 11B NMR spectrum at 36.4 ppm, indicating a tri-coordinated boron atom (cf. (C6F5)2BNMe2 33.7 ppm).64 Given the double bond character of the BN bond, two distinct C6F5 groups are detected giving two sets of signals in the 19F NMR spectrum. Highly diagnostic is the 1H NMR chemical shift of the PC(H) proton at 7.80 ppm as a doublet with a 2JP,H coupling constant of 18.5 Hz (cf. Mes*PC(H)N(SiMe3)2 (ref. 65) δ(1H) = 8.24 (d, 2JP,H = 16.8 Hz)). The aforementioned 1H NMR signal, the deshielded 31P{1H} NMR signal at 228.5 ppm and the 13C{1H} NMR signal of the PC(H) functionality (δ13C{1H} = 177.5 ppm, 1JP,C = 37.3 Hz) clearly indicate a phosphaalkene (cf. 2,6-(Mes*PC(H))2(NC5H5) δ(31P) = 249.1 ppm).66 Interestingly, carbodiimides react with Pier's Borane to the corresponding four-membered boron amidinates.55 Similar four-membered heterocycles are formed when isothiocyanates are treated with HB(C6F5)2.41
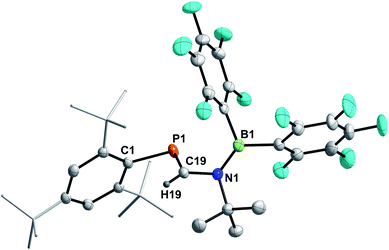 |
| | Fig. 7 Molecular structure of 9. Hydrogen (except H19) omitted and tBu-groups on Mes* rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): P1–C19 1.6751(13), N1–C19 1.4278(15), N1–B1 1.3995(18); C1–P1–C19 98.93(6), P1–C19–N1 123.69(9), C19–N1–B1 120.15(10). | |
Finally, the influence of the substitution pattern at both the phosphorus and nitrogen atoms was investigated exemplarily by reacting the 1,3-phosphaazaallene 3f (bearing DipTer and Xyl substituents) with HB(C6F5)2 (Scheme 6, bottom). One singlet signal in the 11B{1H} NMR spectrum at −19.3 ppm, indicated a four-coordinate boron atom. In contrast to 9, only three signals are observed in the 19F{1H} NMR spectrum and the 31P{1H} signal is observed at −83.1 ppm, over 300 ppm shifted towards higher field when compared to 9. These data together with the data obtained by SC-XRD showed that instead of 1,2-hydroboration, the Lewis acid base adduct DipTerP(HB(C6F5)2)CNXyl (10) with a newly formed P–B bond was obtained (Fig. 8). The C1–N1 bond length of 1.161(3) Å is shortened by approximately 0.04 Å when compared to the starting material 3f (c.f. 1.2009(15) Å) and is now close to a carbon nitrogen triple bond (Σrcov(CN) = 1.14 Å).25 Accordingly, the C31–N1–C32 bond angle increases to 165.1(3)° (c.f. 143.24(12)° (3f)). The P1–C31 bond length of 1.754(3) Å also increases compared to 3f (1.6785(12) Å) indicative of a single bond (c.f.DipTerP(H)CN 4c 1.793(2) Å). The P1–B1 bond length of 2.060(3) Å is 0.1 Å longer than the respective sum of the covalent radii (Σrcov(P–B) = 1.96 Å)25 and corresponds with dative bonding as further ascertained by a low WBI for the P–B bond of 0.85.
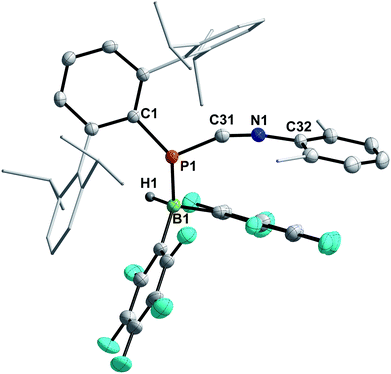 |
| | Fig. 8 Molecular structure of 10. Hydrogen (except H1) omitted and Dip and Xyl groups rendered as wireframe for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): P1–C31 1.754(3), N1–C31 1.161(3), P1–B1 2.060(3); P1–C31–N1 157.7(2). | |
Conclusions
Phospha-Wittig reagents have been shown to react with isocyanides to give 1,3-phosphaazaallenes 3a–f. In case of the CNtBu-derivatives these were further transformed in the corresponding cyanophosphines 4a–c. CNtBu acts in this case as a disguised HCN transfer reagent. This allowed the structural characterization of this underrepresented class of phosphines and deprotonation yielded in one case a rare example of an aryl-substituted cyanophosphide anion. Moreover, 3a was shown to be 1,2-hydroborylated along the CN bond to give the unique heterodiene 9 with alternating PC and BN double bonds. Studies using cyanophosphide 6c as a ligand and exploiting heterodiene 9 in FLP-type chemistry are currently underway.
Data availability
All experimental, crystallographic and computational data are provided in the ESI.
Author contributions
M. F. discovered and optimized the formation of 1,3-phosphaazaallenes, studied the scope and studied the reactivity of compounds 3a–f. M. F. prepared the experimental part and the first draft of the manuscript. C. H.-J. designed the overall research, supervised the work, carried out the computational work, contributed to IR analysis, finalized the manuscript, proofread the experimental part and coordinated the overall project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. H.-J. thanks Prof. M. Beller for his continuous support and the Boehringer Ingelheim Stiftung (BIS) is acknowledged for an Exploration Grant. We thank our technical and analytical staff for assistance, especially Dr Anke Spannenberg for her support regarding X-ray analysis. J.-E. Siewert is gratefully acknowledged for assisting with CV measurements. We wish to thank the ITMZ at the University of Rostock for access to the Cluster Computer and especially Malte Willert for technical support and Dr Jonas Bresien for helpful discussions.
Notes and references
- O. I. Kolodiazhnyi, Tetrahedron Lett., 1982, 23, 4933–4936 CrossRef CAS.
- O. I. Kolodiazhnyi, Phosphorus Sulfur Relat. Elem., 1983, 18, 39–42 CrossRef CAS.
- M. Yoshifuji, K. Toyota, K. Shibayama and N. Inamoto, Tetrahedron Lett., 1984, 25, 1809–1812 CrossRef CAS.
- M. Yoshifuji, T. Niitsu, K. Toyota, N. Inamoto, K. Hirotsu, Y. Odagaki, T. Higuchi and S. Nagase, Polyhedron, 1988, 7, 2213–2216 CrossRef CAS.
- R. Appel and C. Behnke, Z. Anorg. Allg. Chem., 1987, 555, 23–35 CrossRef CAS.
- X.-G. Zhou, L.-B. Zhang, R.-F. Cai, Q.-J. Wu, L.-H. Weng and Z.-E. Huang, J. Organomet. Chem., 2000, 604, 260–266 CrossRef CAS.
- C. Wentrup, H. Briehl, G. Becker, G. Uhl, H. J. Wessely, A. Maquestiau and R. Flammang, J. Am. Chem. Soc., 1983, 105, 7194–7195 CrossRef CAS.
- M.-A. David, J. B. Alexander, D. S. Glueck, G. P. A. Yap, L. M. Liable-Sands and A. L. Rheingold, Organometallics, 1997, 16, 378–383 CrossRef CAS.
- T. Wegmann, M. Hafner and M. Regitz, Chem. Ber., 1993, 126, 2525–2530 CrossRef.
- J. Grobe, D. Le Van and T. Großpietsch, Z. Naturforsch. B, 1991, 46, 978–984 CrossRef CAS.
- T. L. Breen and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 11914–11921 CrossRef CAS.
- E. Ionescu, G. von Frantzius, P. G. Jones and R. Streubel, Organometallics, 2005, 24, 2237–2240 CrossRef CAS.
- F. Basuli, L. A. Watson, J. C. Huffman and D. J. Mindiola, Dalton Trans., 2003, 4228–4229 RSC.
- G. Becker, H. Brombach, S. T. Horner, E. Niecke, W. Schwarz, R. Streubel and E.-U. Würthwein, Inorg. Chem., 2005, 44, 3080–3086 CrossRef CAS PubMed.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- P. Willmes, M. J. Cowley, M. Hartmann, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 2216–2220 CrossRef CAS.
- P. Gupta, J.-E. Siewert, T. Wellnitz, M. Fischer, W. Baumann, T. Beweries and C. Hering-Junghans, Dalton Trans., 2021, 50, 1838–1844 RSC.
- M. Fischer, S. Nees, T. Kupfer, J. T. Goettel, H. Braunschweig and C. Hering-Junghans, J. Am. Chem. Soc., 2021, 143, 4106–4111 CrossRef CAS PubMed.
- M. Fischer, F. Reiß and C. Hering-Junghans, Chem. Commun., 2021, 57, 5626–5629 RSC.
- Experimental and computational details, and details on the X-ray diffraction studies are included in the ESI.† CCDC 2086496–2086506 contain the X-Ray data..
- M. M. Hansmann, D. A. Ruiz, L. L. Liu, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 3720–3725 RSC.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138(49), 15885–15888 CrossRef CAS PubMed.
- J. Escudié, H. Ranaivonjatovo and L. Rigon, Chem. Rev., 2000, 100, 3639–3696 CrossRef PubMed.
- E. Urnéžius and J. D. Protasiewicz, Main Group Chem., 1996, 1, 369–372 CrossRef.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2004, 126, 2268–2269 CrossRef CAS PubMed.
- S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
-
M. B. Smith and J. March, Localized Chemical Bonding, In March's Advanced Organic Chemistry, ed. M. B. Smith and J. March, 2006, pp. 3–31 Search PubMed.
- T. Peddarao, A. Baishya, M. K. Barman, A. Kumar and S. Nembenna, New J. Chem., 2016, 40, 7627–7636 RSC.
- M. K. Sharma, D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem. Commun., 2019, 55, 10408–10411 RSC.
- K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2012, 134, 2954–2957 CrossRef CAS PubMed.
- J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469–4473 CrossRef PubMed.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed.
- R. C. Dobbie, P. D. Gosling and B. P. Straughan, J. Chem. Soc., Dalton Trans., 1975, 2368–2373 RSC.
- C. Schulten, G. von Frantzius, G. Schnakenburg and R. Streubel, Heteroat. Chem., 2011, 22, 275–286 CrossRef CAS.
- A. Maraval, A. Igau, C. Lepetit, A. Chrostowska, J.-M. Sotiropoulos, G. Pfister-Guillouzo, B. Donnadieu and J. P. Majoral, Organometallics, 2001, 20, 25–34 CrossRef CAS.
- I. C. Vei, S. I. Pascu, M. L. H. Green, J. C. Green, R. E. Schilling, G. D. W. Anderson and L. H. Rees, Dalton Trans., 2003, 2550–2557 RSC.
- M. Fischer and M. Schmidtmann, Chem. Commun., 2020, 56, 6205–6208 RSC.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- A. Schmidpeter and F. Zwaschka, Angew. Chem., Int. Ed. Engl., 1977, 16, 704–705 CrossRef.
- A. Schmidpeter, G. Burget, F. Zwaschka and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1985, 527, 17–32 CrossRef CAS.
- A. Schmidpeter, F. Zwaschka and W. S. Sheldrick, Chem. Ber., 1985, 118, 1078–1085 CrossRef CAS.
- J. F. Binder, S. C. Kosnik, P. B. J. St Onge and C. L. B. Macdonald, Chem.–Eur. J., 2018, 24, 14644–14648 CrossRef CAS PubMed.
- A. Schmidpeter, K.-H. Zirzow, G. Burget, G. Huttner and I. Jibril, Chem. Ber., 1984, 117, 1695–1706 CrossRef CAS.
- A. Schmidpeter and G. Burget, Z. Naturforsch. B, 1985, 40, 1306–1313 CrossRef.
- R. M. K. Deng and K. B. Dillon, J. Chem. Soc., Chem. Commun., 1981, 1170–1171 RSC.
- Z. Li, J. E. Borger, F. Müller, J. R. Harmer, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2019, 58, 11429–11433 CrossRef CAS PubMed.
- C. M. Hoidn, T. M. Maier, K. Trabitsch, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 18931–18936 CrossRef CAS PubMed.
- A. H. Cowley, F. Gabbai, R. Schluter and D. Atwood, J. Am. Chem. Soc., 1992, 114, 3142–3144 CrossRef CAS.
- A. B. Burg and P. J. Slota, J. Am. Chem. Soc., 1958, 80, 1107–1109 CrossRef CAS.
- C. Schwarz, L. T. Scharf, T. Scherpf, J. Weismann and V. H. Gessner, Chem.–Eur. J., 2019, 25, 2793–2802 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS PubMed.
- M. H. Holthausen, M. Colussi and D. W. Stephan, Chem.–Eur. J., 2015, 21, 2193–2199 CrossRef CAS PubMed.
- A. C. McQuilken, Q. M. Dao, A. J. P. Cardenas, J. A. Bertke, S. Grimme and T. H. Warren, Angew. Chem., Int. Ed., 2016, 55, 14335–14339 CrossRef CAS PubMed.
- M. Mehta and J. M. Goicoechea, Chem. Commun., 2019, 55, 6918–6921 RSC.
- E. A. Patrick and W. E. Piers, Chem. Commun., 2020, 56, 841–853 RSC.
- G. Desimoni and G. Tacconi, Chem. Rev., 1975, 75, 651–692 CrossRef CAS.
- S. Berski, Z. Latajka and A. J. Gordon, New J. Chem., 2011, 35, 89–96 RSC.
- G. Mierzwa, A. J. Gordon and S. Berski, J. Mol. Model., 2020, 26, 136 CrossRef CAS PubMed.
- P. Müller, S. Huck, H. Köppel, H. Pritzkow and W. Siebert, Z. Naturforsch., B: Chem. Sci., 1995, 50, 1476–1484 CrossRef.
- D. Winkelhaus, Y. V. Vishnevskiy, R. J. F. Berger, H.-G. Stammler, B. Neumann and N. W. Mitzel, Z. Anorg. Allg. Chem., 2013, 639, 2086–2095 CrossRef CAS.
- M. Song, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Chem.–Asian J., 2007, 2, 904–908 CrossRef CAS PubMed.
- Y.-H. Chang, Y. Nakajima and F. Ozawa, Organometallics, 2013, 32, 2210–2215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, crystallographic, and computational details. CCDC 2086496–2086506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02947a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Christian
Hering-Junghans
and
Christian
Hering-Junghans