
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a carborane-substituted bis(phosphanido) cobaltate(I), ligand substitution, and unusual P4 fragmentation†
Peter
Coburger‡
 ab,
Julia
Leitl
a,
Daniel J.
Scott
a,
Gabriele
Hierlmeier
a,
Ilya G.
Shenderovich
c,
Evamarie
Hey-Hawkins
*b and
Robert
Wolf
*a
ab,
Julia
Leitl
a,
Daniel J.
Scott
a,
Gabriele
Hierlmeier
a,
Ilya G.
Shenderovich
c,
Evamarie
Hey-Hawkins
*b and
Robert
Wolf
*a
aInstitute of Inorganic Chemistry, Universität Regensburg, 93040 Regensburg, Germany. E-mail: Robert.Wolf@chemie.uni-regensburg.de
bInstitute of Inorganic Chemistry, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Web: https://anorganik.chemie.uni-leipzig.de/anorganik/ak-hey-hawkins/
cInstitute of Organic Chemistry, Universität Regensburg, 93040 Regensburg, Germany
First published on 7th July 2021
Abstract
Oxidative addition of the P–P single bond of an ortho-carborane-derived 1,2-diphosphetane (1,2-C2(PMes)2B10H10) (Mes = 2,4,6-Me3C6H2) to cobalt(−I) and nickel(0) sources affords the first heteroleptic complexes of a carborane-bridged bis(phosphanido) ligand. The complexes also incorporate labile ligands suitable for further functionalisation. Thus, the cobalt(I) complex [K([18]crown-6)][Co{1,2-(PMes)2C2B10H10}(cod)] (cod = 1,5-cyclooctadiene) bearing a labile cyclooctadiene ligand undergoes facile ligand exchange reactions with isonitriles and tert-butyl phosphaalkyne with retention of the bis(phosphanido) ligand. However, in the reaction with one equivalent of P4, the electron-rich bis(phosphanido) moiety abstracts a single phosphorus atom with formation of a new P3 chain, while the remaining three P atoms derived from P4 form an η3-coordinating cyclo-P3 ligand. In contrast, when the same reaction is performed with two equivalents of the cobalt(I) complex, a dinuclear product is formed which features an unusual P4 chain in its molecular structure.
Introduction
Terminal phosphanides, PR2−, are remarkable ligands for transition metal coordination chemistry due to their flexible and unusual reaction behaviour. Even when acting as simple ‘spectator’ ligands, their properties as very strong σ and π donors can dramatically influence the electronic structure and hence the reactivity at the metal.1 In addition, the presence of sterically and electronically accessible lone pairs of electrons leads to highly reactive metal–phosphorus bonds,2–19 allowing for additional, ‘non-innocent’ reactivity toward substrates including Brønsted acids,4,10–12,17 electrophiles,4,7,10,17 and unsaturated π systems.19 Moreover, terminal phosphanido complexes can serve as metalloligands for heterobimetallic complexes.5,7,10 These exciting properties make transition metal phosphanido complexes attractive targets for study, and they have been exploited for a number of applications including palladium-catalysed Suzuki cross-couplings, where bimetallic phosphanido complexes of ruthenium and palladium show excellent catalytic performance.2,3 Terminal phosphanido complexes have also been utilised in catalytic hydrophosphination and dehydrocoupling reactions.6,8,11,13,16While monodentate phosphanido ligands have been quite extensively explored, complexes with chelating, terminal bis(phosphanido) ligands are comparably understudied. In addition to the improved stability and rigidity that are inherent to the use of chelates, such ligands are expected to lead to further enhancements in reactivity due to the presence of two non-innocent nucleophilic sites in close proximity, as well as the cumulative electronic effects of the two donor moieties on the transition metal centre. Pertinent examples of 3d metal complexes of such species are shown in Fig. 1.20–24 They have been synthesised in three different ways: (a) salt metathesis of alkali metal bis(phosphanides) (I and II),20,22 (b) oxidative addition of a P–P bond to a transition metal centre (III and IV),21,23 and (c) elimination of phenyl groups from precursor diphenylphosphinoarene ligands (V).24
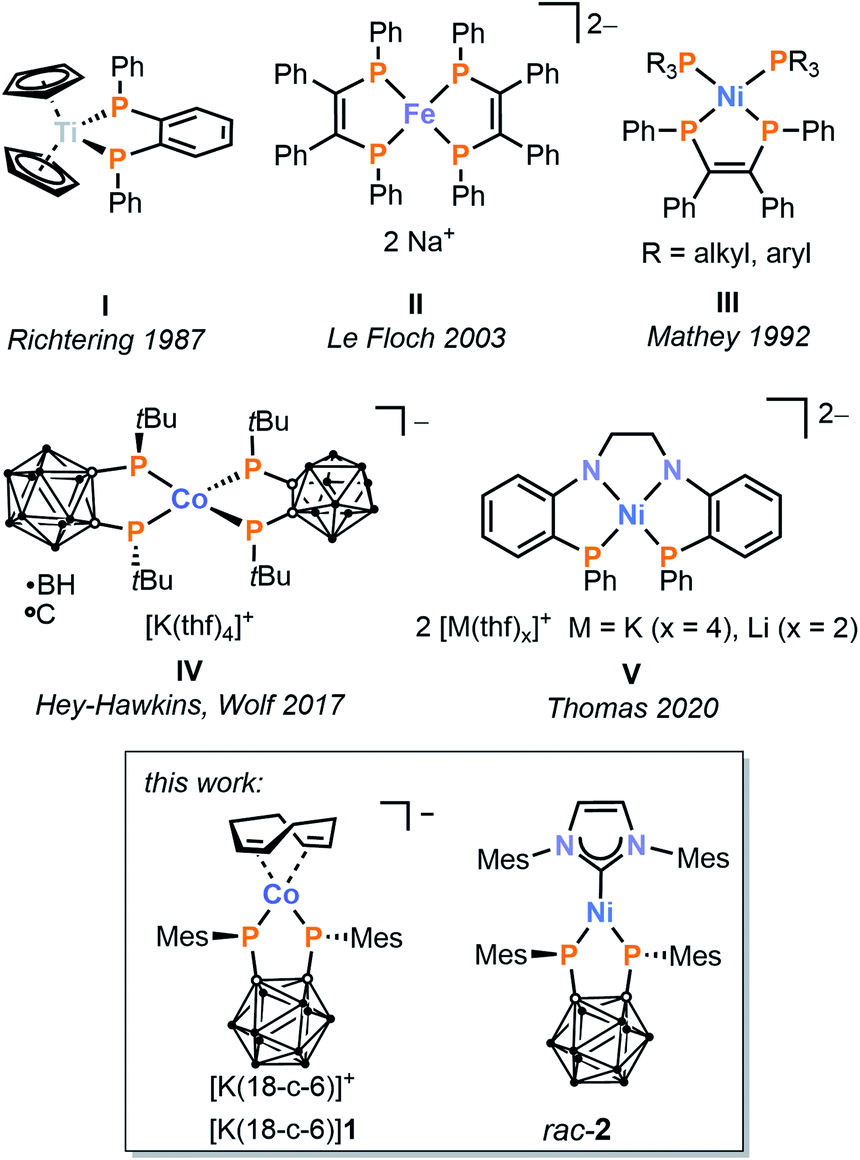 | ||
| Fig. 1 Examples of first-row transition metal complexes with chelating terminal bis(phosphanido) ligands. | ||
In our own contribution to this area, we recently reported the synthesis of anionic cobalt(III) complex IV by reaction of an ortho-carborane-based diphosphetane with the anionic cobalt(−I) complex [Co(cod)2]− (cod = 1,5-cyclooctadiene).23 Oxidative addition of the P–P single bonds from two equivalents of the diphosphetane to the cobalt(−I) centre affords two bis(phosphanido) moieties, which coordinate as chelate ligands at a single cobalt atom. Remarkably, the strong σ and π donation of the P donor atoms results in a low-spin (singlet) electronic ground state despite the (pseudo)tetrahedral coordination geometry.
Although this is an impressive demonstration of the powerful effect of the carborane-bridged bis(phosphanido) ligand, the further reactivity of the coordinatively saturated, heteroleptic complex IV is unfortunately limited. We therefore sought to test the generality of our synthetic approach to obtain new, low-valent 3d metal complexes, and particularly heteroleptic examples that retain vacant coordination sites and/or labile ligands suitable for further functionalisation.
Reported herein are the results of this investigation, including the preparation of complexes of Co ([rac-1]−) and Ni (rac-2), each incorporating a single bis(phosphanido) moiety alongside potentially labile ligands. While the insolubility and instability of the nickel compound 2 precluded follow-up chemistry, further studies have revealed a versatile reactivity of the cobalt complex [K(18-c-6)(thf)][rac-1] (18-c-6 = [18]crown-6) toward various substrates, resulting in a range of derivatives ([rac-6]− to [rac-9]2−), which have been structurally and spectroscopically characterised. These include a remarkable complex ([meso-8]−) incorporating both cyclo-P3 and carborane-bridged triphospholanide ligands, resulting from a well-behaved fragmentation of white phosphorus (P4).
Results and discussion
Synthesis of heteroleptic complexes [K(18-c-6)(thf)][rac-1] and rac-2
Previously we found that reactions of a tert-butyl-substituted ortho-carborane-based diphosphetane with the cobalt(−I) sources [Co(cod)2]− and [Co(anthracene)2]− in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio afford the homoleptic complex IV shown in Fig. 1, which features two bis(phosphanido) ligands.23
:1 stoichiometric ratio afford the homoleptic complex IV shown in Fig. 1, which features two bis(phosphanido) ligands.23
When the same reaction is performed in a 1:1 stoichiometric ratio, complex IV is again observed as the only product by 31P{1H} NMR spectroscopy, while formation of a heteroleptic 1:1 complex with one bis(phosphanido) ligand coordinating at cobalt(I) was not detectable. In light of these results and anticipating that the desired monoligation should be favoured by an increase in steric bulk, attention was therefore shifted to preparation of a larger, mesityl-substituted diphosphetane, which was achieved via the two-step reaction sequence shown in Scheme 1. This synthetic route was adapted from the preparation of the original tert-butyl-substituted diphosphetane.25 Deprotonation of the parent ortho-carborane 3 using nBuLi and subsequent in situ treatment of the product with dibromo(mesityl)phosphane26 affords the brominated bis(phosphane) 4 as a mixture of stereoisomers.27 Direct treatment of this crude material with potassium graphite (two equivalents) in diethyl ether straightforwardly affords diphosphetane rac-5 in modest overall yield.§ The proposed diphosphetane structure in rac-5 was confirmed by multinuclear NMR spectroscopy and elemental analysis (see the ESI†).
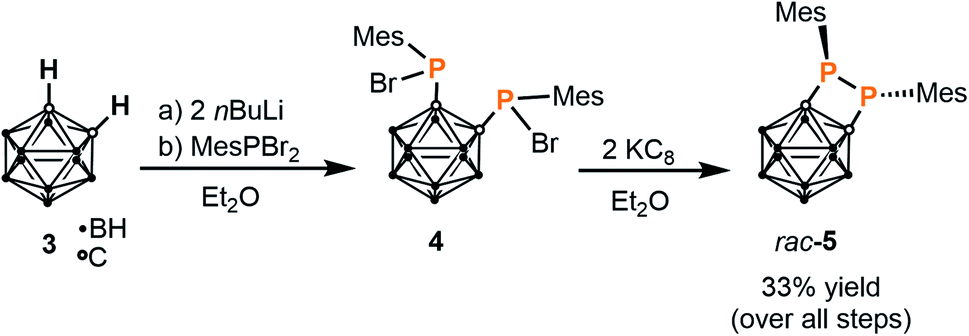 | ||
| Scheme 1 Synthesis of mesityl-substituted diphosphetane rac-5. | ||
With the desired precursor rac-5 in hand, attention was shifted to the preparation of the target metal complexes. Gratifyingly, treatment of rac-5 with [K(thf)0.2][Co(cod)2] in a diethyl ether/n-hexane mixture (2:5 v/v) cleanly affords a single reaction product, exhibiting a singlet in the 31P{1H} NMR spectrum at 101.3 ppm. This chemical shift is upfield compared to that observed for IV (201.8 ppm in THF-d8), suggesting that it does not correspond to a homoleptic 2:1 complex. Addition of [18]crown-6 to the crude reaction mixture affords the crown ether adduct [K(18-c-6)(thf)][rac-1]. This species can be isolated as dark orange crystals in good yield (67%) by crystallisation from THF/n-hexane (Scheme 2). The formulation of the anion [rac-1]− as [Co{1,2-(PMes)2C2B10H10}(cod)]− is supported by elemental analysis as well as multinuclear NMR spectroscopy. The 1H NMR spectrum in THF-d8 shows a single set of mesityl resonances and also demonstrates the presence of a coordinated cod ligand, with olefinic protons that are significantly shielded compared to the free ligand (2.93 ppm vs. 5.58 ppm) as a result of metal–ligand π backbonding. This chemical shift is similar to that observed in [K(thf)2][Co(BIAN)(cod)] (BIAN = 1,2-bis(2,6-diisopropylphenylimino)acenaphthene) (2.91 ppm).28 Two broad resonances are observed in the 11B{1H} NMR spectrum at −9.4 and −7.9 ppm, consistent with previous observations of the carborane-bridged bis(phosphanido) ligand in IV,23,29 while the carborane carbon atoms are detected at ca. 92 ppm in the 13C{1H} NMR spectrum.
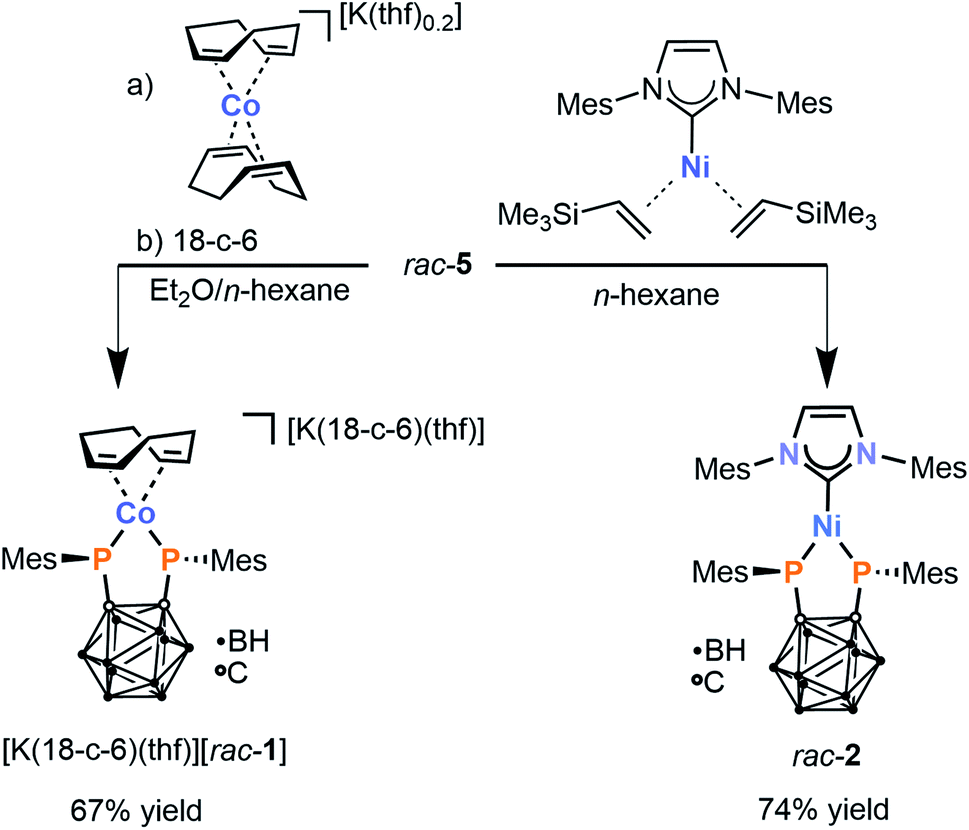 | ||
| Scheme 2 Synthesis of heteroleptic 3d metal complexes [K(18-c-6)(thf)][rac-1] and rac-2via oxidative addition of diphosphetane rac-5 (18-c-6 = [18]crown-6). | ||
The singlet in the 31P{1H} NMR spectrum of [K(18-c-6)][rac-1] is significantly broadened (νFWHM = 140 Hz), consistent with quadrupolar relaxation induced by the 59Co nucleus (100% natural abundance, I = 7/2). Further, conclusive proof of the structure of the anion [rac-1]− was obtained by X-ray crystallographic analysis of single crystals of the salt [K(dme)4][rac-1] (dme = 1,2-dimethoxyethane; vide infra).
Having succeeded in the isolation of the desired heteroleptic Co complex, an analogous Ni complex was also targeted. Indeed, a comparable reaction of diphosphetane rac-5 with the nickel(0) source [Ni(IMes)(η2-H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiMe3)2] (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene) in n-hexane furnished a single product rac-2 in good isolated yield (74%, Scheme 2).
CHSiMe3)2] (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene) in n-hexane furnished a single product rac-2 in good isolated yield (74%, Scheme 2).
Purple, crystalline rac-2 is poorly soluble in hydrocarbons and aromatic solvents. In polar solvents such as THF it is slightly soluble, but decomposes over the course of several minutes, as is evident from a colour change from purple to light yellow and the observation of several new, unidentified resonances in the 31P{1H} NMR spectrum (see the ESI† for details). While this prevented characterisation by conventional solution-phase NMR spectroscopy, rac-2 can nevertheless confidently be formulated as the neutral complex [Ni{1,2-(PMes)2C2B10H10}(IMes)] – the first example of a Ni complex incorporating a carborane-substituted bis(phosphanido) ligand – on the basis of elemental analysis, X-ray crystallography (vide infra), and solid-state 31P{1H} MAS NMR spectroscopy (see the ESI, Fig. S9†). Line fitting of the NMR spectrum reveals the three components of the chemical shift tensor in rac-2, δ11 = 424 ppm; δ22 = 33 ppm; δ33 = −67 ppm. This corresponds to an isotropic 31P NMR shift of 130 ppm, which is similar to the chemical shift of complex [K(18-c-6)(thf)][rac-1] (δ = 100.3 ppm) observed in solution.
Reactivity studies of complex [K(18-c-6)(thf)][rac-1]
While attempts to study the reactivity of Ni complex rac-2 have been hampered by its limited solubility and stability in common solvents, the Co complex [K(18-c-6)(thf)] [rac-1] is fully amenable to further investigation. Crucially, the anion [rac-1]− features a potentially labile 1,5-cyclooctadiene ligand, which could provide a valuable handle for further reactivity and thus allow the targeted synthesis of new and more elaborate bis(phosphanido) complexes.In order to demonstrate this potential for ligand substitution, we began by examining the reaction of [K(18-c-6)(thf)][rac-1] with two equivalents of cyclohexyl isonitrile (CyNC) as shown in Scheme 3. Satisfyingly, this reaction cleanly affords the 16 VE isonitrile complex [K(18-c-6)(dme)][Co{1,2-(PMes)2C2B10H10}(CyNC)2] ([K(18-c-6)(dme)][rac-6]), which can be isolated as red crystals in good yield (55%) after work-up and recrystallisation from dme/n-hexane. Like its precursor [K(18-c-6)(thf)][rac-1], the composition and structure of the product [K(18-c-6)(dme)][rac-6] could be confirmed by elemental analysis, multinuclear NMR spectroscopy (which shows comparable resonances for the bis(phosphanido) ligand, alongside those expected for the CyNC moieties), and X-ray crystallography (vide infra). The presence of the CyNC ligands was further confirmed by the IR spectrum of solid [K(18-c-6)(dme)][rac-6], which shows two strong CN vibrations at 2029 and 1938 cm−1. In comparison, the free isonitrile shows a CN stretching vibration at 2136 cm−1. The substantial changes of the CN stretching frequency of 198 cm−1 in case of the most intense vibration suggests that the bis(phosphanido) ligand in [rac-6]− acts as an exceptional σ-donor.¶ In fact, the observed CN stretching frequencies are considerably lower than typically found in mixed cobalt(I)-isonitrile complexes with phosphane ligands.30–32 However, in those complexes the cobalt atom is usually coordinated by three to four isonitrile ligands which hampers the comparability to [K(18-c-6)(dme)][rac-6].
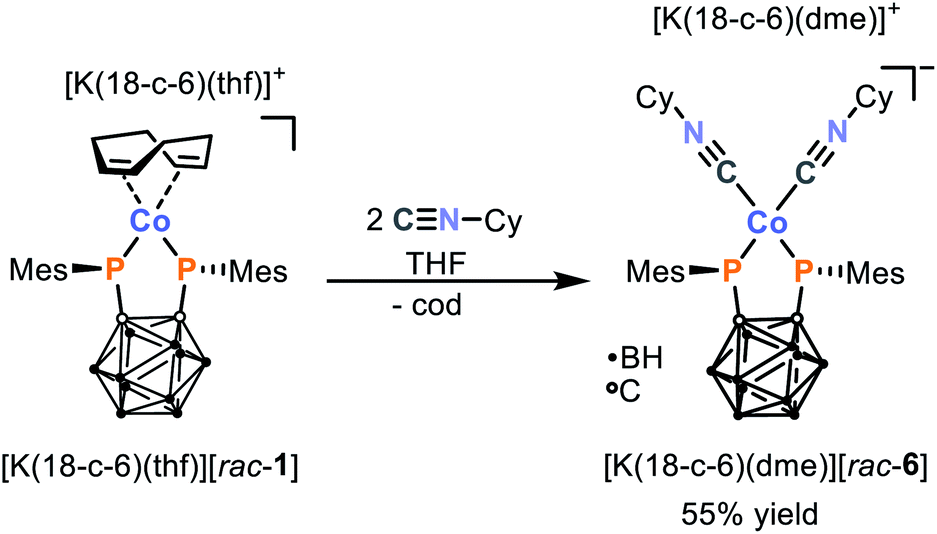 | ||
| Scheme 3 Synthesis of isonitrile complex [K(18-c-6)(dme)][rac-6] via ligand exchange of [K(18-c-6)(thf)][rac-1] (18-c-6 = [18]crown-6). | ||
As the simple ligand substitution reaction between [K(18-c-6)(thf)][rac-1] and CyNC was successful, more elaborate transformations were pursued. We have previously highlighted how the dimerisation of phosphaalkynes in the coordination sphere of low-valent Co complexes can be used to access coordinatively and chemically versatile complexes of 1,3-diphosphabutadienes.33–35 Thus, the reaction between [K(18-c-6)(thf)][rac-1] and tert-butyl phosphaalkyne, tBuCP, was investigated. As hoped, this leads to the clean and exclusive formation of the 16 VE complex [K(18-c-6)][Co(η4-1,3-tBu2C2P2){1,2-(PMes)2C2B10H10}] ([K(18-c-6)][rac-7]) which can be isolated in 65% yield after washing of the crude product with diethyl ether (Scheme 4). The complex anion [rac-7]− contains both the chelating bis(phosphanido) ligand and an η4-coordinating 1,3-diphosphacyclobutadiene (tBu2C2P2) ligand, as was confirmed by a combination of elemental analysis, NMR spectroscopy, and X-ray crystallography.
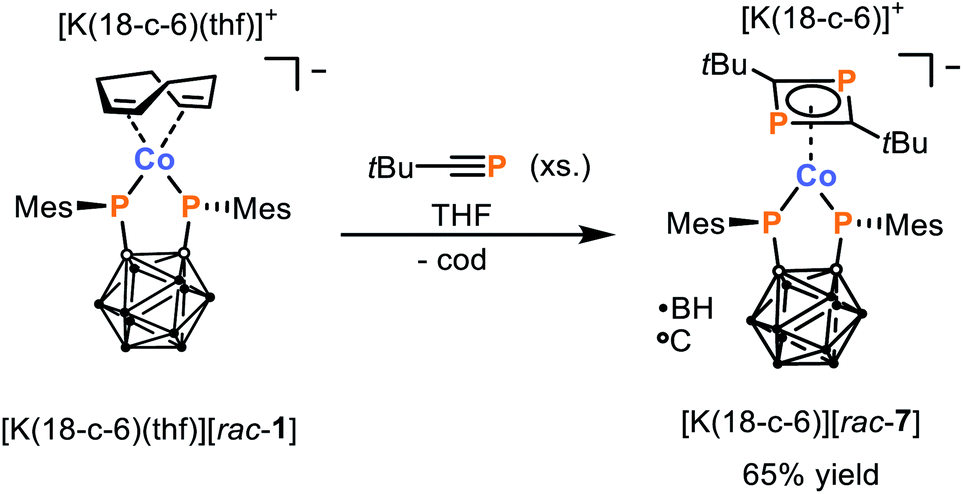 | ||
| Scheme 4 Synthesis of 1,3-diphosphabutadiene complex [K(18-c-6)][rac-7] via reaction of tBuCP with [K(18-c-6)(thf)][rac-1]. | ||
Finally, we also studied the reactivity of [K(18-c-6)(thf)][rac-1] toward white phosphorus (P4). The reactivity of P4 toward transition metal complexes has been a topic of intense interest in recent years due to a strong desire to discover alternatives to the wasteful and indirect methods currently used to transform P4 industrially.36–38 Notably, while this allotrope is well known for its high reactivity, the development of selective reactions of P4 is typically very challenging. Nevertheless, the 1:1 reaction between [K(18-c-6)(thf)][rac-1] and P4 results in formation of a single major product, namely the 18 VE cobalt(I) complex [K(18-c-6)][meso-8], which can be isolated from the reaction mixture in 49% yield (Scheme 5).||
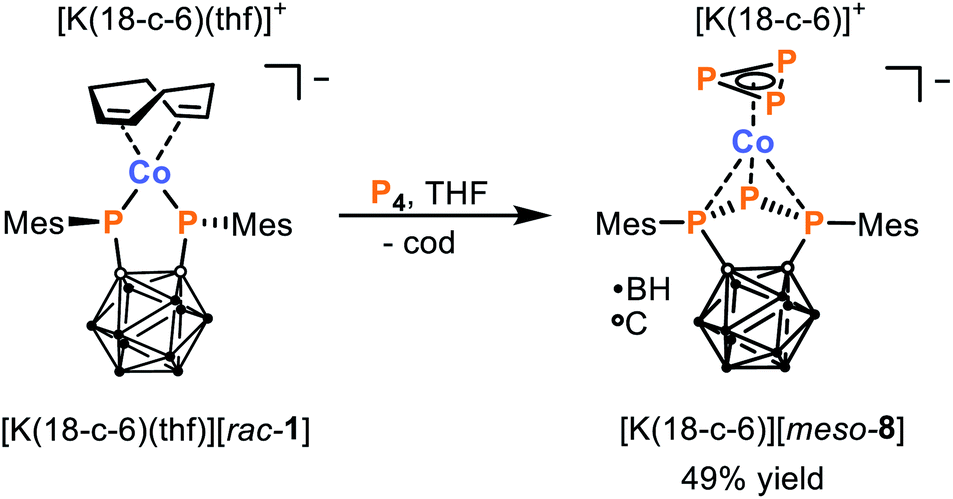 | ||
| Scheme 5 Fragmentation of P4via reaction with [K(18-c-6)(thf)][rac-1] to generate [K(18-c-6)][meso-8]. | ||
Based on the reactivity of [K(18-c-6)(thf)][rac-1] toward CyNC and tBuCP, it had been anticipated that the reaction with P4 would most likely lead to substitution of cod by a cyclo-P4 ligand, as has been observed in previous reactions of P4 with complexes of the type [L2Co(cod)]− (L2 = chelating bidentate ligand).28,39 Instead, X-ray crystallographic analysis (vide infra) revealed the unexpected fragmentation of P4 shown in Scheme 5. The anion [meso-8]− represents a rare anionic cyclo-P3 cobalt complex, the only other reported examples are [nBu4N][(PHDI)Co(η3-P3)(CN)] (PHDI = bis(2,6-diisopropylphenyl)phenanthrene-9,10-diimine), [Cat][Cp′′′Co(η3-P3)] ([Cat] = [K(thf)0.7]+ and [(TMC)2P]+, TMC = 1,3,4,5-tetramethyl-imidazolin-2-ylidene, Cp′′′ = 1,2,4-tri-tert-butyl-cyclopentadienide).39–41 Furthermore, instead of the expected dianionic bis(phosphanido) ligand, [meso-8]− features a monoanionic triphospholanido ligand, in an unprecedented η3-coordination mode.
Elemental analysis and NMR spectroscopic investigations are both consistent with the structure revealed crystallographically. The 11B{1H} NMR spectrum of [K(18-c-6)][meso-8] shows a similar pattern to those previously reported for ortho-carborane-substituted triphospholanes, with three broad resonances centred at −12.5, −8.6 and −2.3 ppm.42,43 The presence of the mesityl substituents is evidenced by singlets in the 1H NMR spectrum for the ortho-methyl groups (2.80 and 2.82 ppm), para-methyl groups (2.24 ppm), and aromatic hydrogen atoms (6.83 and 6.87 ppm), with the number of resonances indicating hindered rotation about the P–Caryl bond. The 31P{1H} NMR spectrum is likewise in agreement with the solid-state molecular structure, showing a doublet at 30.0 ppm and triplet at −250.7 ppm (1JPP = 325 Hz) for the triphospholanido ligand, and a characteristically upfield singlet for the cyclo-P3− ligand at −250.9 ppm.39,40
The structure of [meso-8]−, and its divergence from those observed previously for products derived from related [L2Co(cod)]−, again highlights the non-innocence and dramatic impact of the bis(phosphanido) ligand on the reactivity. Overall, it can be seen that the P4 molecule is formally split into P3− and P+ moieties, which are ultimately bound to the metal centre and bis(phosphanido) ligand, respectively. Intuitively, it seems likely that this fragmentation would proceed via initial coordination of P4 to the complex anion [rac-1]−, followed by intramolecular abstraction of a P+ fragment by the nucleophilic phosphanido moieties. Related transition-metal mediated [3 + 1] fragmentations of P4 have rarely been reported in the literature.40,44–46 To the best of our knowledge, our system is the first which incorporates both the resulting P3 and P1 fragment in the same product.
To provide deeper insight, a combined DFT and DLPNO-CCSD(T) study was conducted (see the ESI† for details), and the calculated mechanism is shown in Fig. 2. In the first step, the cod ligand of [rac-1]− is replaced with P4, leading to the formation of complex Int1 which contains a side-on coordinated P4 moiety where one P–P bond is significantly weakened (2.740 Å). In the next step – which is slightly exothermic with respect to Int1 (ΔE = −3.8 kcal mol−1) – removal of a P+ atom from the P4 moiety is achieved through attack by a single phosphanido moiety. This step proceeds via transition state TS1 to produce a second intermediate Int2 containing the thus formed P3− ligand, with the abstracted P atom bridging one of the original Co–P bonds.
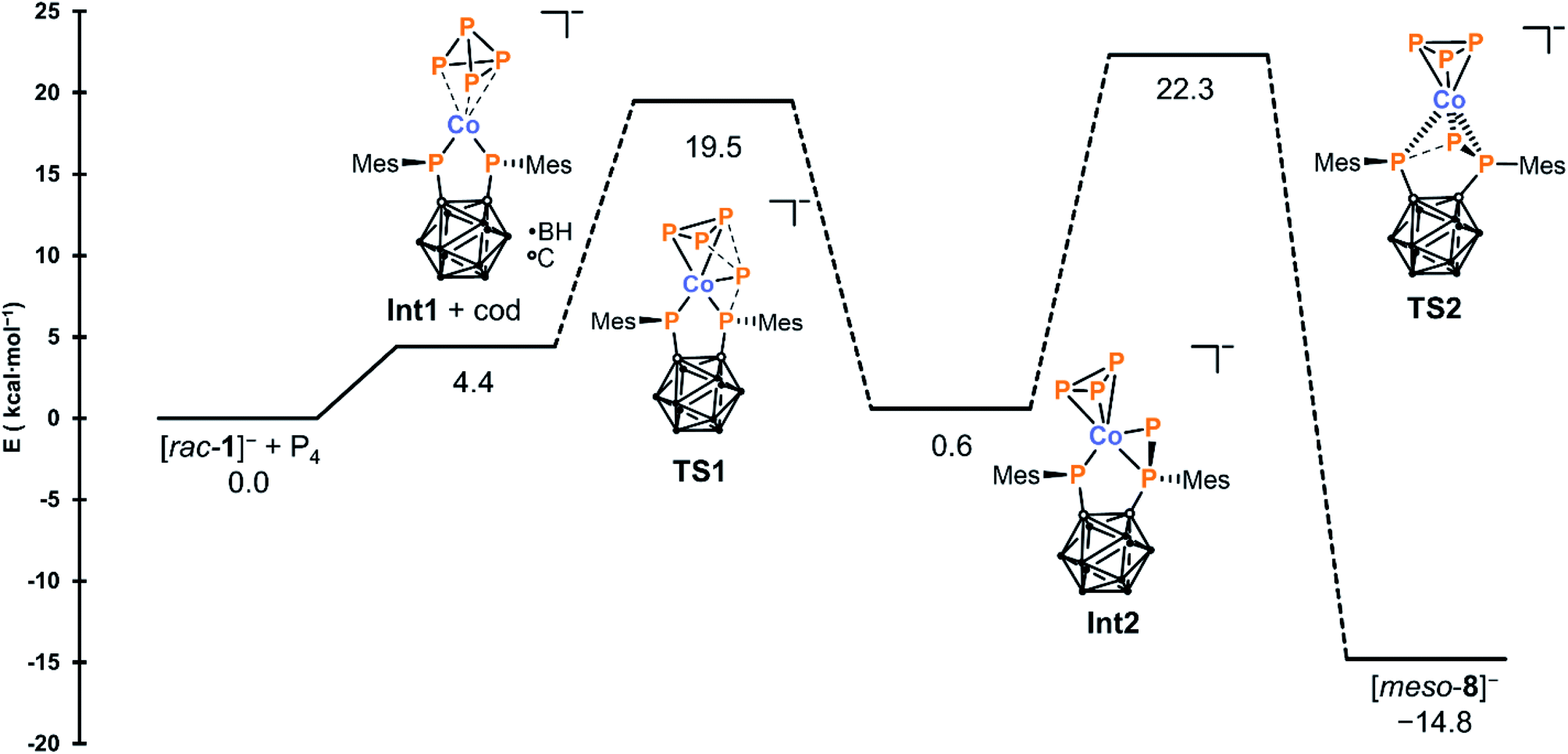 | ||
| Fig. 2 Calculated mechanism for the formation of [meso-8]− from [rac-1]− and P4. Energies were obtained at the DLPNO-CCSD(T)/CBS level. | ||
In the final, rate-determining step, interaction of this P atom with the second phosphanido moiety results in formation of the triphospholanido moiety, forming the final product rac-8−via transition state TS2 with a total activation barrier of 22.3 kcal mol−1. In the calculated mechanism, all activation barriers are easily accessible at 298 K and none of the intermediates is a thermodynamic sink. This is in agreement with reaction monitoring by 31P{1H} NMR spectroscopy, where only the direct formation of [meso-8]− without intermediates is observed (see the ESI, Fig. S26†).
In the above reaction, the complex [K(18-c-6)][meso-8] is consistently isolated containing ca. 5 to 10% of a minor impurity. This side product can be synthesised cleanly and deliberately by treating an in situ prepared solution of [rac-1]− in THF with just half an equivalent of P4, and subsequent addition of [18]crown-6. Washing of the crude product with diethyl ether affords the homobimetallic complex [K(18-c-6)]2[rac-9] as a purple solid in 21% yield (Scheme 6). X-ray crystallographic analysis (vide infra) revealed the structure of the complex dianion [rac-9]2−, which features a bridging, open-chain μ-η4:η4-P42− ligand that can be described as a cis-1,3-tetraphosphabutadienediide. Complexes of this ligand are extremely scarce, and to the best of our knowledge only three other examples are known, namely [K(dme)4][{(BIAN)Co}2(μ-η4:η4-P4)], [{Cp′′Fe}2(μ-η4:η4-P4)] (Cp′′ = 1,3-di-tert-butyl-cyclopentadienide) and [{Cp′′′Fe}2(μ-η4:η4-P4)].28,47,48
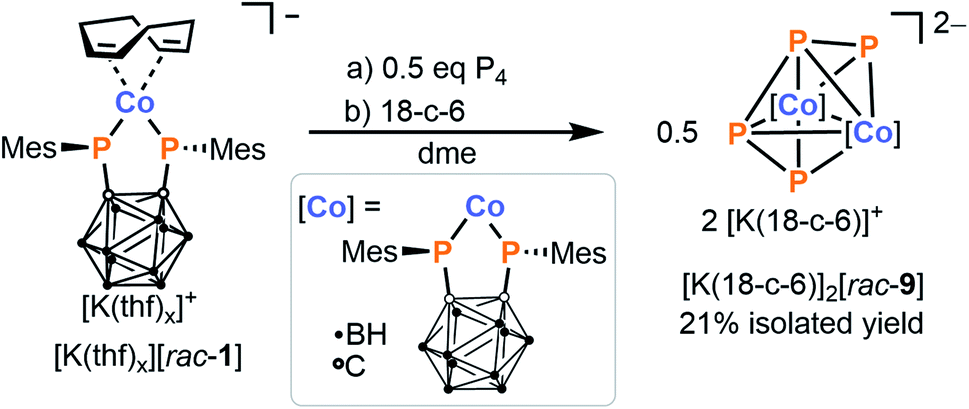 | ||
| Scheme 6 Synthesis of homodinuclear complex [K(18-c-6)]2[rac-9] via reaction of P4 with 2 equivalents of [K(thf)x][rac-1]. | ||
The 11B{1H} NMR spectrum of [K(18-c-6)]2[rac-9] shows a single broad resonance at −7.1 ppm for the carborane ligand backbone, while the 1H NMR spectrum shows the expected ortho-Me, para-Me and aromatic resonances for the inequivalent mesityl groups. At 298 K, the 31P{1H} NMR spectrum in THF-d8 solution consists of three broad resonances corresponding to the P42− ligand (P3: 423.6 ppm, P4, P5: −42.0 ppm, P6: 223.1 ppm; vide infra for detailed structural discussion, atom labels according to Fig. 3) and two singlets (P1: 58.0 ppm, P2: 119.2 ppm) corresponding to the two chemically inequivalent phosphorus nuclei within each bis(phosphanido) ligand. These assignments have been made by reference to DFT-calculated 31P NMR shifts in [rac-9]2− (vide infra). The number of signals excludes the presence of a fast fluxional process that would render the atoms of the P42− chain equivalent on the NMR timescale at room temperature, in contrast to the iron complexes [{Cp′′Fe}2(μ-η4:η4-P4)] and [{Cp′′′Fe}2(μ-η4:η4-P4)] (for each of which only one singlet was observed at 165 ppm and 91.0 ppm, respectively). Nevertheless, variable-temperature NMR studies do suggest some limited fluxionality, and at 233 K the resonances of the P42− moiety are split further, with four distinct signals (P3: 418.8 ppm, P4: −49.1, P5: −42.3 ppm, P6: 216.5 ppm) and resolved 1JPP couplings (1JP3–P4 = 379 Hz, 1JP4–P5 = 217 Hz, 1JP5–P6 = 478 Hz).
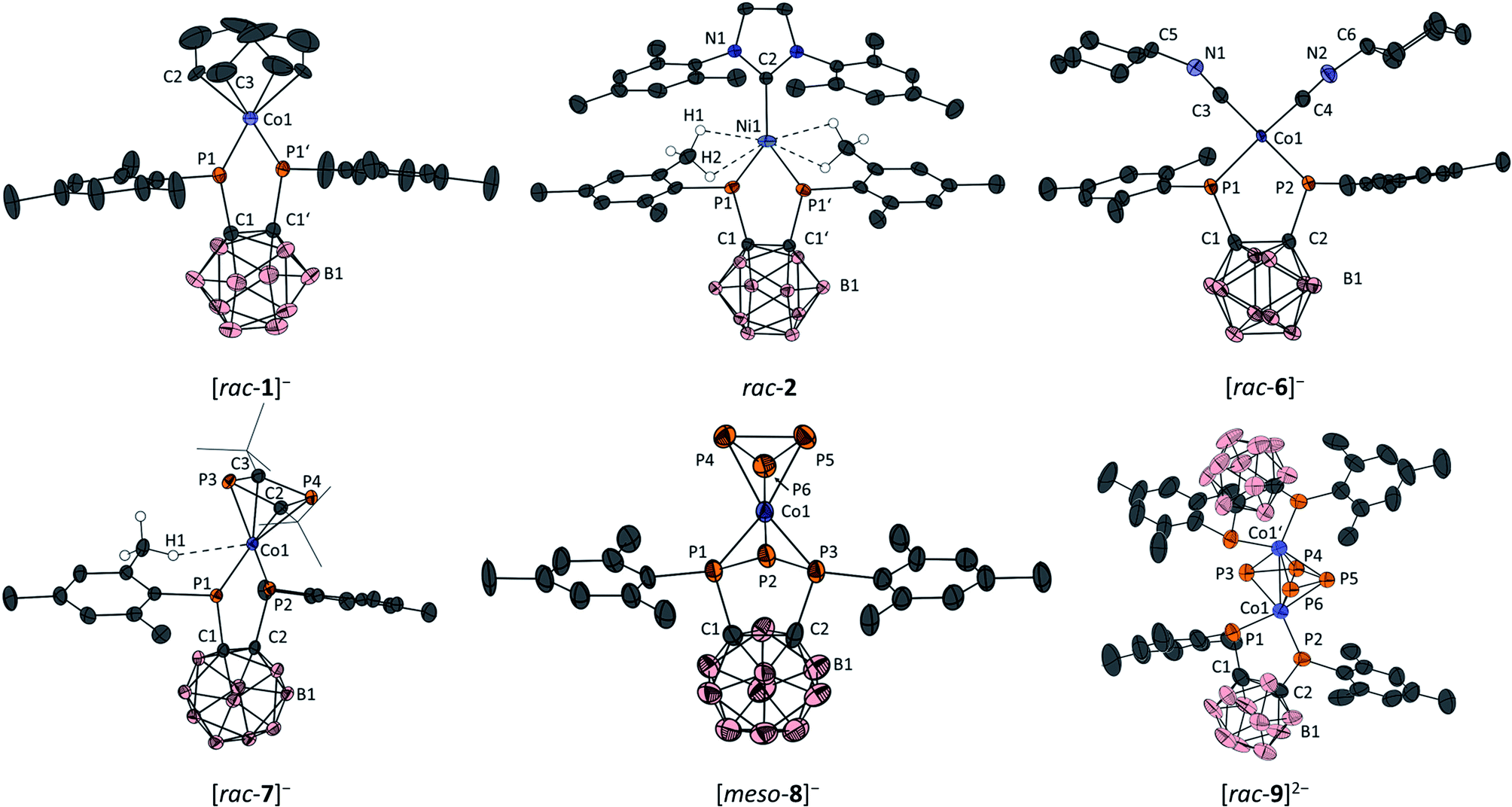 | ||
| Fig. 3 Molecular structures of [K(dme)4][rac-1] (top left), rac-2 (top centre), [K(18-c-6)(dme)][rac-6] (top right), [K(dme)4][rac-7] (bottom left), [K(18-c-6)(dme)2][meso-8] (bottom centre) and [K(dme)][K(dme)2][rac-9] (bottom right) with ellipsoids at the 50% probability level. Only the anions are shown; the counterions are omitted for clarity. For all compounds, hydrogen atoms, except those interacting with Co1 in [rac-7]− and with Ni1 in 2, are omitted for clarity. Additionally, the tert-butyl groups in 7− are drawn as wireframes for clarity. For [rac-9]2−, only one of two crystallographically independent anions in the asymmetric unit is shown; structural parameters of the second molecule are given in the ESI.† Symmetry-generated atoms are marked with a prime symbol (′). Selected bond lengths [Å] and angles [°] of [K(dme)4][rac-1]: C1–C1′ 1.657(5), Co1–P1 2.1857(8), Co1–C2 2.041(7), Co1–C3 2.073(6), C2–C3 1.343(9), P1–Co1–P1′ 88.84(2), ∑P1 339.8(3) (sum of bond angles around P1); rac-2: C1–C1′ 1.650(2), Ni1–P1 2.1264(4), Ni1–C2 2.041(7), Co1–C3 1.962(2), Ni1–H1 2.56, Ni1–H2 2.88, P1–Ni1–P1′ 91.35(1), ∑P1 323.2(2). [K(18-c-6)(dme)][rac-6]: C1–C2 1.669(2), Co1–P1 2.1874(6), Co1–P2 2.1570(6), Co1–C3 1.784(2), Co1–C4 1.784(2), C3–N1 1.183(2), C4–N2 1.174(3), P1–Co1–P2 91.47(2), C3–N1–C5 153.6(2), C4–N2–C6 153.3(2), ∑P1 328.5(2), ∑P2 337.8(2); [K(dme)4][rac-7]: C1–C2 1.672(2), Co1–P1 2.2371(4), Co1–P2 2.0871(5), Co1–C2 2.049(2), Co1–C3 2.099(1), Co1–P3 2.2750(5), Co1–P4 2.2454(5), Co1–H1 2.64, C2–P3 1.800(1), C2–P4 1.805(2), P1–Co1–P2 92.29(2), ∑P1 325.3(2), ∑P2 351.1(2); [K(18-c-6)(dme)2][meso-8]: C1–C2 1.65(1), Co1–P1 2.136(2), Co1–P2 2.382(2), Co1–P3 2.141(2), Co1–P4 2.283(4), Co1–P5 2.276(2), Co1–P6 2.267(2), P1–P2 2.175(3), P4–P5 2.142(3), P5–P6 2.164(3), P1–P2–P3 78.0(1); [K(dme)x]2[rac-9]: C1–C2 1.685(9), Co1–P1 2.246(2), Co1–P2 2.170(2), Co1–P3 2.293(2), Co1–P4 2.333(2), Co1–P5 2.340(2), Co1–P6 2.183(2), Co1–Co1′ 2.651(1), P3–P4 2.095(3), P4–P5 2.336(3), P5–P6 2.112(3), P1–Co1–P2 92.63(7), ∑P1 323.5(8), ∑P2 340.7(7). | ||
Time-resolved 31P{1H} NMR spectroscopic monitoring confirmed that formation of [rac-9]2− occurs via the reaction of [meso-8]− with a second equivalent of cobaltate [rac-1]−. Interestingly, when pre-formed [K(18-c-6)][rac-1] is used instead of in situ generated, crown ether-free [rac-1]−, the reaction proceeds considerably more slowly, indicating that the potassium counterion might play a significant role in facilitating the formation of [rac-9]2− (see the ESI, Fig. S27†).
Structural characterisation of rac-2 and complex anions [rac-1]−, [rac-6]−, [rac-7]−, [meso-8]− and [rac-9]2−
Crystals suitable for X-ray diffraction experiments could be obtained for all complexes described in the present study (see Fig. 3 for depictions and selected bond lengths and angles). Three complexes ([rac-1]−, [rac-7]− and [rac-9]2−) were crystallised from dme solutions without any 18-c-6 ligand. Of these, [rac-1]− and [rac-7]− were crystallised with the [K(dme)4]+ counterion. In the case of [rac-9]2−, two molecules of the complex dianion were contained in the asymmetric unit. Due to severe disorder and the low quality of the crystals, only the counterions [K(dme)n]+ (n = 1, 2) coordinating to one of the dianions in the asymmetric unit could be modelled properly. The counterions of the second dianion were removed from the unit cell using the SQUEEZE procedure of the PLATON program package,49 and the obtained bond lengths and angles should therefore be analysed with care. Nevertheless, the identity of [rac-9]2− can be unambiguously derived from the X-ray data. The other complexes were obtained as [K(18-c-6)(dme)]+ ([rac-6]−) or [K(18-c-6)(dme)2]+ ([meso-8]−) salts. Also, the anions [rac-1]−, [rac-6]− and [rac-9]2− are located on symmetry elements in the solid state.In all complexes, the C–C bond within the ortho-carborane backbone is in the typical range for carborane-substituted bis(phosphane) complexes.27,50 In the case of the complex anions [rac-1]−, [rac-6]− and [rac-9]2−, the Co–P bonds formed by the bis(phosphanido) ligand are in the range of single bonds,51–59 and significantly elongated compared to IV (2.1437(5) Å), consistent with the lower oxidation state of the metal (vide infra). For these complexes, the sums of bond angles around the phosphorus atoms range from 328.5(2)° ([rac-6]−) to 340.7(7)° ([rac-9]2−), showing significant pyramidalisation. Thus, these structural data suggest the presence of a dominant σ–donor interaction between the phosphanido moieties and the cobalt centres, with limited contributions from PCo π bonding. This is confirmed by quantum chemical calculations (vide infra).
For [rac-1]−, a distorted square-planar environment is observed (τ4 = 0.22, where a value of 0 corresponds to an ideal square-planar structure, while a value of 1 indicates a perfectly tetrahedral structure).60 The η4-coordinating cod ligand shows similar structural parameters to those found in the related cobalt(I) complex [K(thf)2][Co(BIAN)(cod)].28
In rac-2, the nickel atom adopts a perfectly trigonal planar environment (sum of bond angles around Ni: 360.00(2)°), and the Ni1–P1 bond (2.1264(4) Å) is shorter than observed for the related bis(phosphanido) nickel complex II (Ni–P 2.164(1) and 2.173(1) Å, Fig. 1). Despite this, the sums of bond angles around the phosphorus atoms in rac-2 (323.2(2)°) still indicate trigonal pyramidal geometries, which suggests that the bis(phosphanido) ligand again acts mainly as a σ donor. Another notable feature of the structure is a collection of short contacts between the nickel centre and several hydrogen atoms of the closest methyl groups of the mesityl substituents in the bis(phosphanido) ligand. The observed Ni⋯H distances range from 2.56 to 2.88 Å; such values indicate anagostic interactions, which are likely attributable to the comparatively low coordination number of the nickel atom.61
The solid state molecular structure of anion [rac-6]− confirms the substitution of cod by two isonitrile ligands. The cobalt atom adopts a highly distorted square planar environment close to a seesaw geometry (τ4 = 0.37). The two Co–P bond lengths (Co1–P1 2.1874(6) Å, Co1–P2 2.1570(6) Å) and the respective sums of angles around the phosphorus atoms (∑P1: 328.5(2)°, ∑P2: 337.8(2)°) are significantly different, presumably due to the interaction of the [K(18-c-6)(dme)]+ counterion with the cyclohexyl moiety of one of the isonitrile ligands which leads to a deviation from the expected ideal C2 symmetry of [rac-6]−. Significant backbonding from the cobalt atom to π* orbitals of the isonitrile ligands can be deduced from the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–C bond angles in the solid state which deviate significantly from 180° (C3–N1–C5: 153.6(2)°, C4–N2–C6: 153.3(2)°).
N–C bond angles in the solid state which deviate significantly from 180° (C3–N1–C5: 153.6(2)°, C4–N2–C6: 153.3(2)°).
The structure of [rac-7]− shows an anagostic61 interaction between H1 (part of an ortho-methyl group on the mesityl substituent attached to P1) and the cobalt atom (Co1⋯H1: 2.64 Å, Fig. 3). This leads to a rather large deviation from the expected C2 symmetry, with the tBu2C2P2 ring notably tilted away from H1. In addition, the two phosphorus atoms of the bis(phosphanido) ligand in [rac-7]− show distinctly different coordination environments. While the Co1–P1 bond length (2.2371(4) Å) and ∑P1 (325.3(2)°) indicate a Co–P single bond and a trigonal pyramidal environment around P1, the respective data for P2 indicate a CoP double bond and a trigonal planar environment (Co1–P2: 2.0871(5) Å, ∑P2: 351.1(2)°). Both this increased bond order and the anagostic interaction with H1 can be attributed to the more electron-poor and hence electrophilic Co atom in [rac-7]− than in [rac-1]− or [rac-6]− (vide infra for further discussion of electronic structures). It should be noted, however, that these features do not appear to be retained in solution, as evidenced by 31P{1H} NMR spectra of isolated [K(18-c-6)][rac-7] (vide supra; see also the ESI, Fig. S28 and S29,† for variable-temperature studies) which show only a single resonance for the bis(phosphanido) ligand with a similar chemical shift to those observed for [K(18-c-6)(thf)][rac-1] and [K(18-c-6)(dme)][rac-6]. The Co–C, Co–P and P–C bond lengths within the Co(η4-1,3-tBu2C2P2) fragment of [rac-7]− are all similar to those found in related complexes such as [Cp′′′Co(η4-1,3-tBu2C2P2)]62 and [K(18-c-6)(thf)2][Co(η4-1,3-tBu2C2P2)2].33
In the solid state, the cobalt atom in [meso-8]− adopts a distorted trigonal antiprismatic environment, similar to that found in the neutral complex [(triphos)Co(η3-P3)] (triphos = 1,1,1-tris(diphenylphosphinomethyl)ethane).63 While the Co–P1 and Co–P3 bonds (to the mesityl-substituted P atoms of the triphospholanido ligand) are in the range of Co–P single bonds (2.136(2) and 2.141(2) Å), the Co–P2 bond length (to the central unsubstituted P atom of the triphospholanido ligand) is considerably larger (2.382(2) Å), suggesting a rather weak interaction. Within the Co(cyclo-P3) fragment, the P–P bond lengths (2.142(3)–2.164(3) Å), as well as the Co–P bond lengths (2.267(2)–2.283(4) Å), are similar to those found in [nBu4N][(PHDI)Co(η3-P3)(CN)]39 and [(TMC)2P][Cp′′′Co(η3-P3)].40
The dianion [rac-9]2− consists of two neutral [CoII{1,2-(PMes)2C2B10H10}] fragments bridged by the cis-tetraphosphabutadienediide ligand. The dianion has a mirror plane formed by the planar P42− ligand. The phosphorus atoms within the bis(phosphanido) ligands are chemically inequivalent as indicated by their significantly different Co–P bond lengths (Co1–P1 2.246(2) Å, Co1–P2 2.170(2) Å) and sum of angles around the phosphorus atoms (∑P1: 323.5(8)°, ∑P2 340.7(7)°). The P–P bond lengths (2.095(3)–2.336(3) Å) as well as the Co–P bond lengths (2.183(2)–2.340(2) Å) within the Co2P4 fragment are comparable to those found in [K(dme)4][{(BIAN)Co}2(μ-η4:η4-P4)].28 In contrast, the Co–Co distance in [rac-9]2− (2.651(1) Å) is indicative of a Co–Co single bond and is notably shorter than in the aforementioned BIAN complex (2.8455(5) Å), although a similar metal–metal distance was found in [{Cp′′′Fe}2(μ-η4:η4-P4)] (2.6430(8) Å).47
Quantum chemical and electronic structure analysis of rac-2 and complex anions [rac-1]−, [rac-6]−, [rac-7]−, [meso-8]− and [rac-9]2−
Cobalt complexes [K(18-c-6)(thf)][rac-1], [K(18-c-6)(dme)][rac-6], [K(18-c-6)][rac-7], [K(18-c-6)] [meso-8] and [K(18-c-6)]2[rac-9] were further characterised by UV-vis spectroscopy in THF to provide experimental insight into their electronic structures. Gratifyingly, nickel complex rac-2 was sufficiently soluble and stable in THF to allow for the measurement of UV-vis spectra as well (see the ESI, Fig. S39–S50†). To complement these experimental studies, TDDFT calculations at the ωB97X-D3/def2-TZVP CPCM(THF) level were also conducted, to give further insight into the relevant electronic transitions. As is typically observed for range-separated functionals, the calculated spectra are blue-shifted with respect to the experimental spectra.Nevertheless, the overall shape of the spectra is very well reproduced (see the ESI, Fig. S39–S50†), and the calculated difference densities were thus used to assign the nature of the observed transitions. A summary of the UV-vis data is given in Table 1, and it can be seen that the spectra are dominated by charge-transfer transitions. It is noteworthy that [K(18-c-6)(thf)][rac-1], [K(18-c-6)(dme)][rac-6], and [K(18-c-6)][rac-7] share similar metal-to-ligand charge-transfer (MLCT) transitions from 3d electrons at cobalt into π* orbitals at their ligands as their lowest-energy transitions. Here, the wavelength λmax of these transitions increases in the order [K(18-c-6)(dme)][rac-6] < [K(18-c-6)(thf)][rac-1] < [K(18-c-6)][rac-7], which neatly coincides with the order of the π-accepting abilities of the ligands in these complexes (vide infra), since more strongly accepting ligands require less energy for an MLCT transition.
| λ max [nm] | ε [103 L mol−1 cm−1] | Transition type | |
|---|---|---|---|
| [rac-1]− | 337, 435, 768 | 13.3, 6.2, 1.9 | MLCT, LMCT, MLCT |
| rac-2 | 271, 340, 424, 527, 898 | 8.8, 9.7, 1.7, 1.9, 0.7 | MLCT, MLCT, LMCT/d–d LMCT/d–d, d–d |
| [rac-6]− | 277, 342, 490, 655 | 11.6, 8.6, 3.4, 0.7 | MLCT/n–π*, MLCT, MLCT, MLCT |
| [rac-7]− | 354, 404, 513, 810 | 11.5, 6.3, 4.3, 2.3 | n–π*, LMCT, d–d, MLCT |
| [meso-8]− | 287, 337, 398 | 16.3, 18.3, 6.8 | MLCT, MLCT, d–d/LMCT |
| [rac-9]2− | 357, 533 | 20.1, 14.4 | n–π*, LMCT/n–π* |
CASSCF calculations were also performed for the cobaltate anions [rac-1]−, [rac-6]−, [rac-7]−, [meso-8]− and the nickel complex rac-2 to gain a deeper insight into their electronic structures. Due to its size and the presence of two metal centres, complex dianion [rac-9]2− was instead analysed by broken-symmetry DFT (BS-DFT) calculations. The CASSCF calculations for the complexes [rac-1]−, rac-2, [rac-6]−, [rac-7]− and [meso-8]− revealed only moderate multireference character (for depiction of the CASSCF orbitals, see the ESI, Fig. S51–S55†) with contributions of the ground-state configuration function to the overall electronic structure of between 85% ([rac-7]−) and 88% ([rac-6]−). As a result, DFT-based energy decomposition methods such as EDA-NOCV (energy decomposition analysis with natural orbitals of chemical valence)64 could be used to quantify the interactions between the cobalt atom and the ligands in the cobalt complexes [rac-1]−, [rac-6]−, [rac-7]− and [meso-8]− (Table 2).
| Fragments | E int | E L→M | E M→L | |
|---|---|---|---|---|
| [rac-1]− | [Co{1,2-(PMes)2C2B10H10}]−, cod | 190 | 29b | 150 |
| [rac-6]− | [Co{1,2-(PMes)2C2B10H10}]−, 2 CyNC | 162 | 52a | 93 |
| [rac-7]− | [Co{1,2-(PMes)2C2B10H10}]−, tBu2C2P2 | 214 | 28b | 161 |
| [meso-8]− | [Co{1,2-(P3Mes2)C2B10H10}], P3− | 175 | 66b | 82 |
For the four cobalt complexes, the interactions between the metal centres and the ligands were found to be highly covalent in nature. Thus, assignments of oxidation states are somewhat ambiguous. Still, in the case of [rac-1]− and [rac-6]−, the CASSCF calculations indicate the expected presence of a 3d8 CoI atom coordinated by a σ-donating bis(phosphanido) ligand. Furthermore, the EDA-NOCV calculations show that the cod ligand in 1− acts as a weakly σ-donating (29 kcal mol−1) and strongly π-accepting (150 kcal mol−1) ligand (see the ESI† and Table 1). As expected, the isonitrile ligands in [rac-6]− act as comparatively stronger σ-donating (52 kcal mol−1) but weaker π-accepting (93 kcal mol−1) ligands.
In contrast to [rac-1]− and [rac-6]−, the oxidation state of the cobalt atom in [rac-7]− cannot be derived as straightforwardly from the CASSCF calculations. For example, one orbital pair describing the Co–((η4-1,3-tBu2C2P2) π interactions is depicted in Fig. 4. Here, the bonding orbital (183) shows almost equal contributions from cobalt and the diphosphacyclobutadiene ligand (44% Co, 41% tBu2C2P2). Based on these data, the electronic structure of [rac-7]− can be described as intermediate between two extreme cases: a cobalt(III) complex with a strong π-donating diphosphacyclobutadienediide ligand (case i) or a cobalt(I) complex with a strong π-accepting diphosphacyclobutadiene ligand (case ii). The same ambiguity was also found previously for the homoleptic complex [K(18-c-6)(thf)2][Co(η4-1,3-tBu2C2P2)2].34 However, similarly to [rac-1]− and [rac-6]−, [rac-7]− shows an MLCT band as the lowest absorption in its UV-vis spectrum (vide supra) which seems unlikely to occur in case i. Thus, the spectroscopic data suggest that the electronic structure of [rac-7]− is probably closer to case ii. For that case, EDA-NOCV calculations indeed show that the π-accepting properties of the tBu2C2P2 ligand dominate and account for 75% (160 kcal mol−1) of the total interaction between the cobalt atom and the tBu2C2P2 ligand.
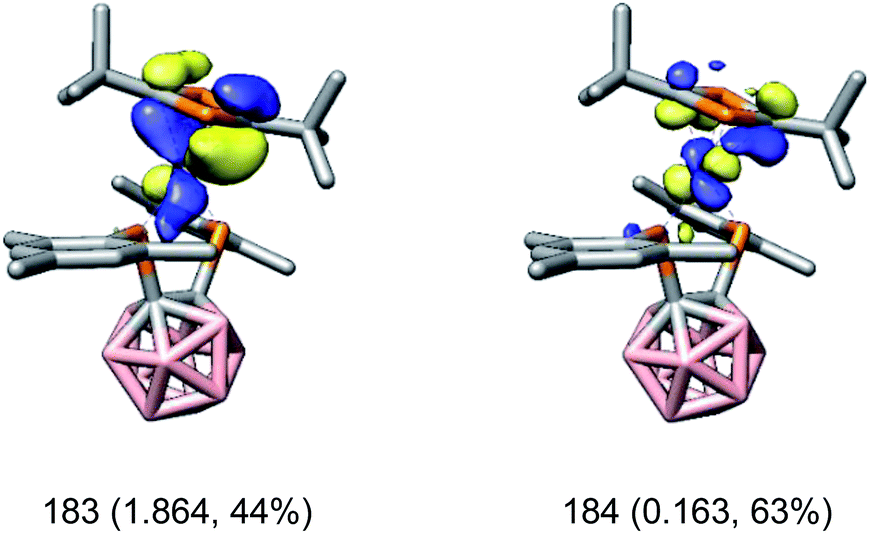 | ||
| Fig. 4 CASSCF natural orbitals of [rac-7]− describing the π interaction between the cobalt atom and the η4-1,3-tBu2C2P2 ligand (183: bonding; 184: antibonding). Orbital occupations and contributions of 3d orbitals on Co are given in parentheses. Surface isovalue = 0.05. | ||
Analysis of the wave function of [meso-8]− obtained from the CASSCF calculation confirms σ-bonding interactions between Co1 and P2 (the central P atom of the triphospholanido ligand) despite the relatively long distance between these atoms observed crystallographically (vide supra; see the ESI† for full details). In addition, the CASSCF calculations show that the cyclo-P3− ligand functions as both a strong π donor and a strong π acceptor. These findings are supported by the EDA-NOCV calculations which show that π donation accounts for 38% and backdonation accounts for 47% of the total interaction between cobalt and the cyclo-P3 ligand (Table 2).
For dianion [rac-9]2−, the BS-DFT calculations revealed that two low-spin 3d7 cobalt(II) atoms are antiferromagnetically coupled via P3 (one of the terminal P atoms of the P42− chain, J = −1837 cm−1; see Fig. 5 for a depiction of the spin density). The solution NMR data offer indirect proof of this antiferromagnetic coupling between the cobalt(II) atoms, since the calculated 31P NMR shifts are only in reasonable agreement with the experimental data when a broken-symmetry wave function is utilised. In particular, the shift of P3 showed an exceptionally large deviation from the experimental data when a closed-shell singlet wave function was used (218 vs. 419 ppm, see the ESI†). Notably, no direct Co–Co bonds were found, but bonding interactions between the cobalt atoms do occur via three-centre two-electron bonds involving Co1, Co1′ and P3 or P6, respectively (see the ESI†). Furthermore, in agreement with classical electron-counting rules, the Co2P4 fragment in [rac-9]2− contains 14 cluster electrons. Thus, [rac-9]2− obeys the 2n + 2 (n = number of cluster vertices) criterion to be classified as a closo cluster. The shape of the Co2P4 fragment – a distorted bicapped tetrahedron or capped trigonal bipyramid (Scheme 6) – differs from the octahedral shape predicted by the Wade–Mingos rules. However, such deviations are common, especially in mixed transition metal–main group element clusters.65
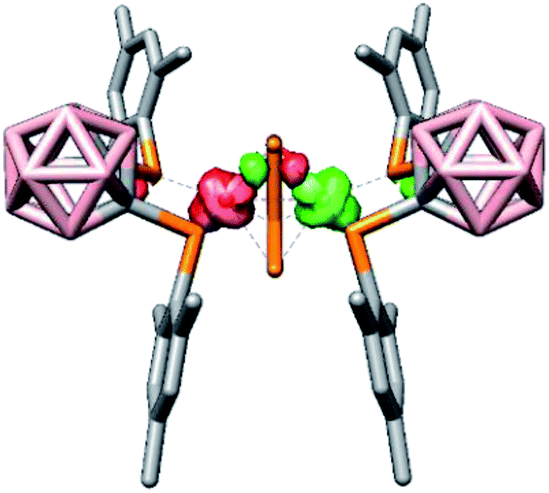 | ||
| Fig. 5 Spin density plot of the dianion [rac-9]2− obtained from a BS-DFT calculation. | ||
For nickel complex rac-2, the CASSCF calculations indicate the absence of π bonding between the bis(phosphanido) ligand and the 3d8 nickel(II) atom, thus supporting the conclusions drawn from the analysis of the solid-state molecular structure (see the ESI, Fig. S52†).
Redox properties of complexes [K(18-c-6)(thf)][rac-1], [K(18-c-6)(dme)][rac-6], [K(18-c-6)][rac-7], [K(18-c-6)][meso-8] and [K(18-c-6)]2[rac-9]
The redox properties of the cobalt complexes were investigated by cyclic voltammetry (CV) in THF/[nBu4N](PF6). The results are summarised for a scan rate of 0.20 V s−1 in Table 3 (all referenced against FcH/FcH+ (FcH = [Cp2Fe])). All complexes show one oxidation event in the range of −1.50 to −0.10 V, with the exception of [rac-9]2− which shows four oxidation events (see the ESI† for CV data of the complexes in a larger range). Cobalt complex [rac-1]− shows a quasi-reversible oxidation at E1/2 = −1.13 V which is considerably higher than that observed in the related cobalt(I) complex [K(thf)2][Co(BIAN)(cod)] (E1/2 = −1.72 V), presumably due to the strong inductive electron-withdrawing effect of the carborane backbone. When comparing the complexes [rac-1]−, [rac-6]− and [rac-7]−, their oxidation potentials increase in the order [rac-6]− < [rac-1]− < [rac-7]−. This trend nicely follows the order of the π-accepting properties of the ligands in these complexes (isonitrile < cod < 1,3-diphosphacyclobutadiene, see Table 3). Among the cobalt complexes, [meso-8]− is oxidised at the highest potential (E = −0.79 V). The bimetallic complex [rac-9]2− shows a particularly rich redox chemistry. Here, an irreversible oxidation at E = −0.97 V is followed by three quasi-reversible oxidations at E1/2 = −0.79, −0.56 and −0.18 V.| [rac-1]− | [rac-6]− | [rac-7]− | [meso-8]− | [rac-9]2− | |
|---|---|---|---|---|---|
| E ox1/2 | −1.13 | −1.48 | −0.81 | −0.79* | −0.97*, −0.79, −0.56, −0.18 |
Conclusions
Through rational ligand modification we have been able to prepare the first examples of heteroleptic transition metal complexes incorporating powerful electron-donating carborane-bridged bis(phosphanido) ligands. These complexes have been obtained through simple oxidation reactions of convenient, low-valent Co and Ni sources with a carborane-bridged diphosphetane. For the resulting Co complex [K(18-c-6)(thf)][rac-1] we have shown that the presence of a labile cod ligand allows for extensive further reactivity, which can be used to prepare various other bis(phosphanido)-containing products. These reactions include not only simple ligand substitutions, but also well-defined, inner-sphere small molecule aggregation and degradation. Particularly dramatically, the highly nucleophilic bis(phosphanido) ligand in [K(18-c-6)(thf)][rac-1] enables an unusual activation of white phosphorus to yield a remarkable triphospholanido complex [K(18-c-6)][meso-8], in an unprecedented inner-sphere fragmentation of the P4 molecule into P+ and P3− motifs, the latter resulting in a very rare cyclo-P3− ligand. The rather facile synthetic access to [K(18-c-6)][meso-8] renders this complex a promising candidate for further experimental studies with the aim of liberating and/or functionalising the cyclo-P3− fragment. These studies are ongoing in our laboratories.Data availability
The data supporting the findings of this study are available within the paper and its ESI† files. Crystallographic have been deposited at the CCDC under 2086046 ([K(dme)4][rac-1]), 2086047 (rac-2), 2086048 ([K(18-c-6)(dme)][rac-6]), 2086049 ([K(dme)4][rac-7]), 2086050 [K(18-c-6)(dme)2][meso-8] and 2086051 ([K(dme)][K(dme)2][rac-9]).Author contributions
E. H.-H., R. W. and P. C. conceived the project. P. C. performed the preparative experiments and theoretical investigations and analysed the data. J. L. recorded and analysed cyclic voltammograms und UV-vis spectra. G. H. contributed to the synthetic investigations. I. G. S recorded and analysed solid-state NMR spectra. P. C. and D. J. S. wrote the manuscript with input from R. W. and E. H.-H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Karolina Trabitsch for experimental assistance. Financial support by the European Research Council (ERC CoG 772299) is gratefully acknowledged.Notes and references
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606–626 CrossRef CAS.
- J. G. Planas and J. A. Gladysz, Inorg. Chem., 2002, 41, 6947–6949 CrossRef CAS PubMed.
- J. Giner Planas, F. Hampel and J. A. Gladysz, Chem.–Eur. J., 2005, 11, 1402–1416 CrossRef PubMed.
- E. J. Derrah, D. A. Pantazis, R. McDonald and L. Rosenberg, Organometallics, 2007, 26, 1473–1482 CrossRef CAS.
- P. Mastrorilli, Eur. J. Inorg. Chem., 2008, 4835–4850 CrossRef CAS.
- R. Waterman, Dalton Trans., 2009, 18–26 RSC.
- V. Nesterov, G. Schnakenburg, A. Espinosa and R. Streubel, Inorg. Chem., 2012, 51, 12343–12349 CrossRef CAS PubMed.
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC.
- L. Rosenberg, ACS Catal., 2013, 3, 2845–2855 CrossRef CAS.
- Y. Gloaguen, W. Jacobs, B. de Bruin, M. Lutz and J. I. van der Vlugt, Inorg. Chem., 2013, 52, 1682–1684 CrossRef CAS PubMed.
- P. E. Sues, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 4746–4760 CrossRef CAS PubMed.
- R. Waterman, D. J. Mindiola, C. R. Clough and G. L. Hillhouse, Inorg. Chim. Acta, 2014, 422, 57–64 CrossRef CAS.
- A. M. Geer, Á. L. Serrano, B. de Bruin, M. A. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2015, 54, 472–475 CAS.
- E. C. Y. Tam, D. C. Apperley, J. D. Smith, M. P. Coles and J. R. Fulton, Inorg. Chem., 2017, 56, 14831–14841 CrossRef CAS PubMed.
- D. Wang, Q. Chen, X. Leng and L. Deng, Inorg. Chem., 2018, 57, 15600–15609 CrossRef CAS.
- K. Kaniewska, A. Dragulescu-Andrasi, Ł. Ponikiewski, J. Pikies, S. A. Stoian and R. Grubba, Eur. J. Inorg. Chem., 2018, 4298–4308 CrossRef CAS.
- A. Schmer, N. Volk, A. E. Ferao and R. Streubel, Dalton Trans., 2018, 48, 339–345 RSC.
- V. Varela-Izquierdo, A. M. Geer, B. de Bruin, J. A. López, M. A. Ciriano and C. Tejel, Chem.–Eur. J., 2019, 25, 15915–15928 CrossRef CAS PubMed.
- J. Yang, S. Langis-Barsetti, H. C. Parkin, R. McDonald and L. Rosenberg, Organometallics, 2019, 38, 3257–3266 CrossRef CAS.
- H. Köpf and V. Richtering, J. Organomet. Chem., 1988, 346, 355–360 CrossRef.
- G. Sillett, L. Ricard, C. Patois and F. Mathey, J. Am. Chem. Soc., 1992, 114, 9453–9457 CrossRef CAS.
- A. Moores, N. Mézailles, L. Ricard, F. Mathey and P. L. Floch, Chem. Commun., 2003, 1914–1915 RSC.
- P. Coburger, S. Demeshko, C. Rödl, E. Hey-Hawkins and R. Wolf, Angew. Chem., Int. Ed., 2017, 56, 15871–15875 CrossRef CAS PubMed.
- K. Lee, C. E. Moore and C. M. Thomas, Organometallics, 2020, 39, 2053–2056 CrossRef CAS.
- A. Kreienbrink, M. B. Sárosi, E. G. Rys, P. Lönnecke and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2011, 50, 4701–4703 CrossRef CAS PubMed.
- F. Nief and F. Mathey, Tetrahedron, 1991, 47, 6673–6680 CrossRef CAS.
- A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS.
- S. Pelties, T. Maier, D. Herrmann, B. de Bruin, C. Rebreyend, S. Gärtner, I. G. Shenderovich and R. Wolf, Chem.–Eur. J., 2017, 23, 6094–6102 CrossRef CAS.
- P. Coburger, S. Demeshko, C. Rödl, E. Hey-Hawkins and R. Wolf, Angew. Chem., 2017, 129, 16087–16091 CrossRef.
- I. Beaumont and A. H. Wright, J. Organomet. Chem., 1992, 425, C11–C14 CrossRef CAS.
- J. W. Dart, M. K. Lloyd, R. Mason, J. A. McCleverty and J. Williams, J. Chem. Soc., Dalton Trans., 1973, 1747–1751 RSC.
- C. A. L. Becker, M. A. Salam Biswas and J. C. Cooper, Inorg. Chim. Acta, 1991, 188, 191–194 CrossRef CAS.
- R. Wolf, A. W. Ehlers, J. C. Slootweg, M. Lutz, D. Gudat, M. Hunger, A. L. Spek and K. Lammertsma, Angew. Chem., Int. Ed., 2008, 47, 4584–4587 CrossRef CAS.
- R. Wolf, A. W. Ehlers, M. M. Khusniyarov, F. Hartl, B. de Bruin, G. J. Long, F. Grandjean, F. M. Schappacher, R. Pöttgen, J. C. Slootweg, M. Lutz, A. L. Spek and K. Lammertsma, Chem.–Eur. J., 2010, 16, 14322–14334 CrossRef CAS.
- C. Rödl, K. Schwedtmann, J. J. Weigand and R. Wolf, Chem.–Eur. J., 2019, 25, 6180–6188 CrossRef PubMed.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- C. M. Hoidn, D. J. Scott and R. Wolf, Chem.–Eur. J., 2021, 27, 1886–1902 CrossRef CAS.
- L. Giusti, V. R. Landaeta, M. Vanni, J. A. Kelly, R. Wolf and M. Caporali, Coord. Chem. Rev., 2021, 441, 213927 CrossRef CAS.
- C. M. Hoidn, T. M. Maier, K. Trabitsch, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 18931–18936 CrossRef CAS PubMed.
- M. Piesch, S. Reichl, M. Seidl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2019, 58, 16563–16568 CrossRef CAS PubMed.
- M. Piesch, S. Reichl, M. Seidl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2021, 60, 15101–15108 CrossRef CAS.
- A. Kreienbrink, S. Heinicke, T. T. Duong Pham, R. Frank, P. Lönnecke and E. Hey-Hawkins, Chem.–Eur. J., 2014, 20, 1434–1439 CrossRef CAS PubMed.
- P. Coburger, H. Grützmacher and E. Hey-Hawkins, Chem. Commun., 2019, 55, 3187–3190 RSC.
- M. Peruzzini, J. A. Ramirez and F. Vizza, Angew. Chem., Int. Ed., 1998, 37, 2255–2257 CrossRef CAS.
- S. Du, J. Yin, Y. Chi, L. Xu and W.-X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 15886–15890 CrossRef CAS PubMed.
- R. Grünbauer, G. Balázs and M. Scheer, Chem.–Eur. J., 2020, 26, 11722–11726 CrossRef.
- M. D. Walter, J. Grunenberg and P. S. White, Chem. Sci., 2011, 2, 2120–2130 RSC.
- O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110–4112 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- I. B. Sivaev, M. Y. Stogniy and V. I. Bregadze, Coord. Chem. Rev., 2021, 436, 213795 CrossRef CAS.
- D. M. Jenkins, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 11238–11239 CrossRef CAS PubMed.
- D. M. Jenkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 7148–7165 CrossRef CAS PubMed.
- R. G. Hayter, J. Am. Chem. Soc., 1964, 86, 823–828 CrossRef CAS.
- H. Werner, W. Hofmann, R. Zolk, L. F. Dahl, J. Kocal and A. Kühn, J. Organomet. Chem., 1985, 289, 173–188 CrossRef CAS.
- H.-F. Klein, M. Gaß, U. Zucha and B. Eisenmann, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 927–932 CrossRef CAS.
- B. Pan, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2011, 30, 5560–5563 CrossRef CAS.
- A. N. Kornev, V. V. Sushev, Y. S. Panova, N. V. Belina, O. V. Lukoyanova, G. K. Fukin, S. Y. Ketkov, G. A. Abakumov, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2012, 51, 874–881 CrossRef CAS PubMed.
- R. Beck and H.-F. Klein, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m604 CrossRef CAS PubMed.
- M. W. Bezpalko, A. M. Poitras, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2017, 56, 503–510 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- E.-M. Rummel, M. Eckhardt, M. Bodensteiner, E. V. Peresypkina, W. Kremer, C. Gröger and M. Scheer, Eur. J. Inorg. Chem., 2014, 1625–1637 CrossRef CAS.
- M. Di Vaira, L. Sacconi and P. Stoppioni, J. Organomet. Chem., 1983, 250, 183–195 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- F. Z. Trodi, G. Lucas, M. Bencharif, J.-F. Halet, S. Kahlal and J.-Y. Saillard, J. Cluster Sci., 2007, 18, 729–740 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, detailed spectral and crystallographic characterisation, DFT calculations, electrochemical data, catalysis plots. See DOI: 10.1039/d1sc02948g |
| ‡ Present address: Dr. P. Coburger, Laboratory of Inorganic Chemistry, ETH Zürich, Vladimir-Prelog-Weg 1-5/10, CH-8093 Zürich, Switzerland. |
| § Reduction to generate product rac-5 is significantly slower than the analogous reaction to generate the tert-butyl-substituted analogue investigated previously, which is consistent with the increased steric bulk of the mesityl groups. |
| ¶ Similar to carbonyl ligands, the lowering of the CN-stretching vibration in isonitrile ligands is caused by the partial occupation of the respective π* antibonding orbitals as a consequence of backbonding from the metal centre. |
| || We wish to note that rac-5 does not react with P4 in THF at room temperature according to 31P NMR studies. |
| This journal is © The Royal Society of Chemistry 2021 |