
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive electrophilic C–H alkylation of quinolines by a reusable iridium nanocatalyst†‡
Rong
Xie
,
Wenhui
Mao
,
Huanhuan
Jia
,
Jialu
Sun
,
Guangpeng
Lu
,
Huanfeng
Jiang
 and
Min
Zhang
*
and
Min
Zhang
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, People's Republic of China. E-mail: minzhang@scut.edu.cn
First published on 27th September 2021
Abstract
The incorporation of a coupling step into the reduction of unsaturated systems offers a desirable way for diverse synthesis of functional molecules, but it remains to date a challenge due to the difficulty in controlling the chemoselectivity. Herein, by developing a new heterogeneous iridium catalyst composed of Ir-species (Irδ+) and N-doped SiO2/TiO2 support (Ir/N-SiO2/TiO2), we describe its application in reductive electrophilic mono and dialkylations of quinolines with various 2- or 4-functionalized aryl carbonyls or benzyl alcohols by utilizing renewable formic acid as the reductant. This catalytic transformation offers a practical platform for direct access to a vast range of alkyl THQs, proceeding with excellent step and atom-efficiency, good substrate scope and functional group tolerance, a reusable catalyst and abundantly available feedstocks, and generation of water and carbon dioxide as by-products. The work opens a door to further develop more useful organic transformations under heterogeneous reductive catalysis.
Introduction
Over the past decade, hydrogen transfer-mediated coupling reactions have emerged as appealing tools in the construction of valuable products,1 since there is no need for compressed H2 gas and elaborate experimental setups. For instance, except for the well-established reductive amination,2 the groups of Beller,3 Kempe,4 Kirchner,5 Liu6 and others7 have achieved the alkylation of amines and the α-site of carbonyl compounds with the hydrogen-borrowing strategy. Bruneau and co-workers have realized the β-alkylation of N-substituted cyclic amines.8 Li et al. demonstrated amine syntheses from abundantly available phenols.9 Recently, our group reported a direct reductive quinolyl β-C–H alkylation with aldehydes.10 In this process the heterogeneous Co-catalyst (CoOx/MSCC) effects the slow catalytic transfer hydrogenation (CTH) of the quinolines, thus favoring the coupling process (Scheme 1a; MSCC = multispherical cavity carbon). Despite these advances, the concept of merging the reduction of N-heteroarenes with a coupling step still encounters significant challenges, and one of the most important factors is that the N-heteroarenes tend to undergo multiple hydrogenations to form non-coupling cyclic amines.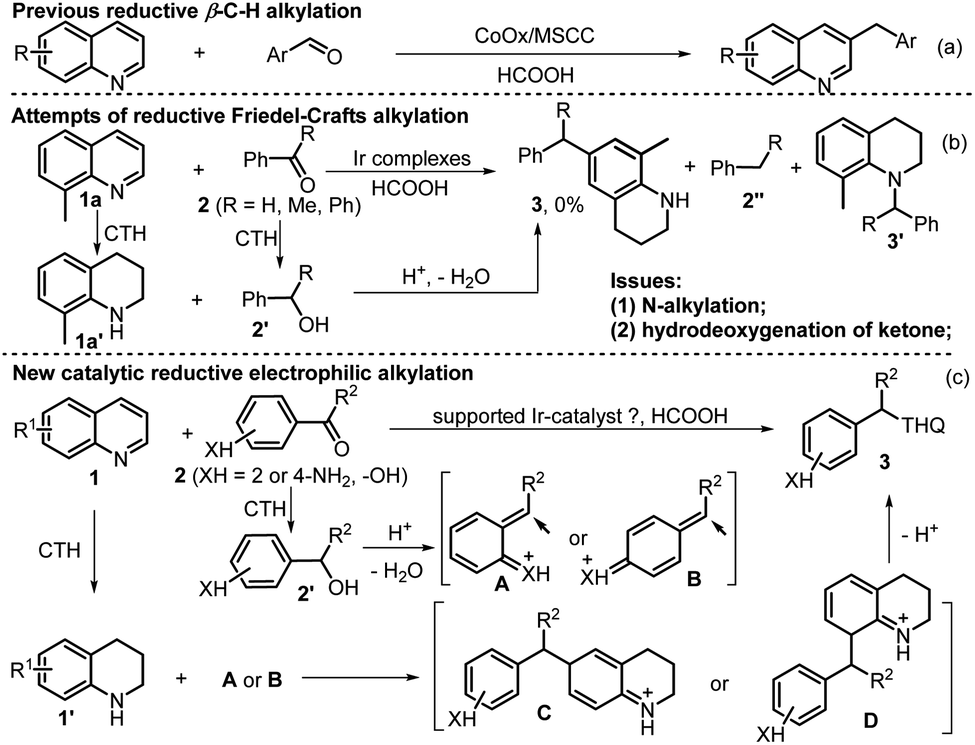 | ||
| Scheme 1 Reductive alkylation of quinolines. | ||
Tetrahydroquinolines (THQs) constitute a class of significantly important N-heterocycles, and they frequently occur in bioactive natural products, and are widely utilized as versatile building blocks for the preparation of numerous fine chemicals, pharmaceuticals, agrochemicals, and functional materials.11 Although the reduction of quinolines offers a useful way to directly access THQs,12 its synthetic diversity is easily restricted due to the limited availability of quinolines. Motivated by the quinolyl10 (Scheme 1a) and indoyl β-C–H alkylation13 and the excellent CTH capability of iridium,14 we initially envisioned a synthesis of alkyl THQs 3via reductive Friedel–Crafts alkylation of 8-methylquinoline 1a with different aryl carbonyls 2 (Scheme 1b). However, by utilizing renewable formic acid as a preferred reductant,15 the reaction with different iridium complexes only generated alkyl benzenes 2′′ and N-alkyl THQs 3′, deriving from the hydrodeoxygenation of 2′ and reductive amination of THQ 1a′ and 2, respectively. So, to achieve a selective synthesis of THQ 3, at least two challenging issues have to be addressed: (1) the in situ formed intermediates should favor the electrophilic coupling process, rather than the formation of by-products 2′′ and 3′. (2) It is preferable to develop a compatible and reusable Ir-catalyst due to the high cost of noble metal.
Regarding the above challenging issues, we believe that the development of a suitable supported Ir-catalyst and application of ortho and para effects of functional groups might offer a solution for a desired synthetic purpose. As shown in Scheme 1c, the introduction of a –OH or –NH2 group ortho or para to the carbonyl group of reactant 2 is able to afford more stable resonance form A or B of benzyl carbocation, deriving from CTH of 2 and acid-assisted dehydration of alcohol 2′, which is beneficial to the conjugate addition of THQ 1′ (see C or D) to afford product 3.
It is important to note that support dopants drastically affect the physicochemical properties of resulting supported catalysts including morphology, basicity and electrical conductivity.16 Moreover, introduction of two or more dopants may exhibit synergistic effects on catalytic performace.17 Attracted by these characteristics and the capability of silica for adjusting the micropore size of the supporting materials,18 we report herein the preparation and characterization of a new N-doped SiO2/TiO2-supported iridium nanocatalyst, and describe, for the first time, its application in reductive electrophilic alkylation of quinolines, which offers a platform to directly access a wide range of alkyl THQs from abundantly available quinolines and aryl carbonyls or benzyl alcohols.
Typically, the iridium heterogeneous catalyst was prepared through the following procedure: a mixture of IrCl3·3H2O and 1,10-phenanthroline in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio in EtOH was initially refluxed at 80 °C for 1 hour to form the coordinated Phen–Ir complex. Then, the silica was introduced by in situ hydrolysis of Si(OC2H5)4 (TEOS) with aqueous ammonia. After that, titanium dioxide powder (TiO2 P25) was added and the mixture was stirred at 80 °C for 4 h. The resulting composites were pyrolyzed under Ar flow at 500 °C for 3 h after removing the solution, which afforded the supported iridium material Ir/N-SiO2/TiO2 (see the ESI‡ for details).
:3 molar ratio in EtOH was initially refluxed at 80 °C for 1 hour to form the coordinated Phen–Ir complex. Then, the silica was introduced by in situ hydrolysis of Si(OC2H5)4 (TEOS) with aqueous ammonia. After that, titanium dioxide powder (TiO2 P25) was added and the mixture was stirred at 80 °C for 4 h. The resulting composites were pyrolyzed under Ar flow at 500 °C for 3 h after removing the solution, which afforded the supported iridium material Ir/N-SiO2/TiO2 (see the ESI‡ for details).
First, X-ray diffraction (XRD) was employed to test the constituents of the obtained iridium material. Except for the characteristic peaks of titanium dioxide (PDF no. 21-1276 and PDF no. 71-1166) and silicon dioxide (PDF no. 86-1563), no other significant peaks corresponding to the iridium species are found in the XRD pattern of Ir/N-SiO2/TiO2 (Fig. S1 in the ESI‡), suggesting that iridium species consist of a well-dispersed amorphous phase. The N2 adsorption–desorption experiment shows a typical IV isotherm, which indicates the existence of mesoporous structure (Fig. S2 in ESI‡). Further, the BET specific surface area is detected as 110.06 m2 g−1, and the corresponding pore size is approximately concentrated around 20 nm (Fig. S2 in ESI‡), which further supports the mesoporous structure of the iridium-based material.
Then, the morphology of the Ir/N-SiO2/TiO2 material was analyzed by TEM, HRTEM, and EDS techniques. The TEM and HRTEM images (Fig. 1a and b) do not show individual Ir clusters, illustrating that the iridium species are uniformly dispersed, which are in good accordance with the XRD results. In addition, the element mapping images (Fig. 1e and S3‡) also reveal that the elements of Ir, Ti, Si, N, C and O are highly overlapped and interconnected with each other, and the iridium species are embedded on the doped support. The initial coordination between the 1,10-phenanthroline (Phen) and iridium constitutes the key to prevent the iridium from agglomeration during pyrolysis process.
 | ||
| Fig. 1 (a and b) TEM, HRTEM images of Ir/N-SiO2/TiO2. (c) TEM images of the reused Ir/N-SiO2/TiO2 after ten runs. (d) STEM images of Ir/N-Si–TiO2 and (e) the corresponding elemental mapping of Ir, Si, N, Ti. | ||
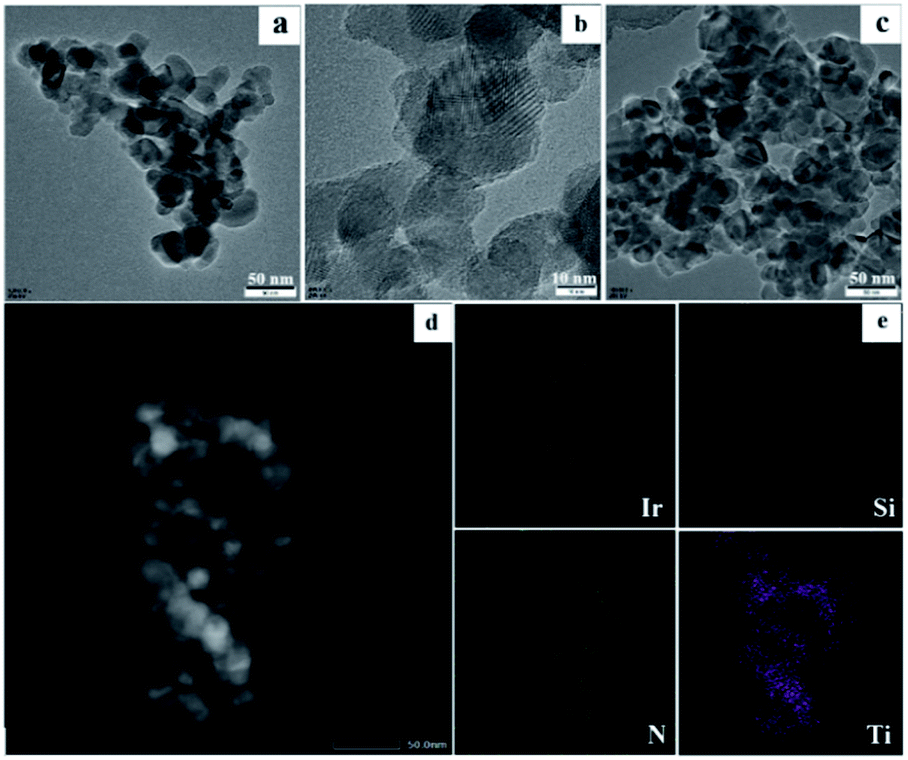
To further analyze the surface chemistry of the prepared iridium material, X-ray photoelectron spectroscopy (XPS) characterization was implemented. A range of elements corresponding to Ir, N, Si, C, O and Ti on the sample surface were detected as 0.56%, 1.68%, 11.4%, 30.93%, 43.48% and 11.95%, respectively, which are in accordance with the elemental mapping results (Fig. S3 in the ESI‡). In comparison with the initial N:Ir ratio of the starting materials (6:1), a 3:1 ratio of the N and Ir on the resulting catalyst surface indicates that three N-atoms are bound to one Ir-atom, whereas the unbound N-atoms are sparsely distributed on the material surface and could not be accurately detected by XPS.19 The N 1s XPS spectrum (Fig. S4b in the ESI‡) discloses almost one peak, corresponding to pyridinic N (399.9 eV),20 which derives from the Phen-ligand coordinated to iridium. The spectrum of Ir 4f (Fig. 2) shows four peaks, two typical characteristic peaks located at 62.4 eV and 65.4 eV are assigned to Irδ+ species,21 while the binding energy of 60.5 eV and 63.9 eV belong to the peaks of metallic Ir.21b,22
 | ||
| Fig. 2 Ir 4f XPS spectra of Ir/N-SiO2/TiO2. | ||
To investigate the catalytic performance of the developed nanomaterial, we chose the reaction of 8-methylquinoline 1a and salicylaldehyde 2a as a model system. After a systematic investigation of various reaction parameters, an optimal isolated yield of product 3aa was obtained when the reaction in mixed solution H2O/dioxane (8/1) was performed at 115 °C for 24 h in the presence of 0.65 mol% of catalyst using HCOOH as the reductant (standard condition, see Table S1 in ESI‡).
With the optimal reaction conditions available, we then set out to explore the universality of the developed chemistry. Initially, we tested the reductive electrophilic alkylation of quinoline with salicylaldehyde, but the reaction generated a mixture of C6- and C8-alkyalted THQs as well as C6,8-dialkylated THQ in 49, 8, and 14% yields, respectively. So, we employed 8-methylquinoline 1a to react with different kinds of salicylaldehydes (2a–2o, see Scheme S1 for their structures in ESI‡). As shown in Scheme 2, all the reactions proceeded smoothly and furnished C6-alkylated THQs in reasonable to excellent isolated yields (3aa–3ao). Various functional groups (–Me, –OMe, –OH, –NEt2, –CO2Me, allylic, halogens) were well tolerated in the transformation. The retention of these functionalities offers the potential for post-functionalization of the obtained alkyl THQs. Noteworthily, reactants 2 bearing electron-withdrawing groups (3aj–3am) afforded much higher product yields than those with electron-donating ones (3ae, 3ag, and 3ai). This phenomenon is attributed to electron-withdrawing groups favoring the formation of more reactive electrophilic intermediates (see Scheme 1c). In addition to salicylaldehydes and o-hydroxyaryl ketones (2p and 2q), 4-hydroxyaryl aldehydes (2r–2v) and 4-hydroxyaryl ketones (2w–2y) were also applicable for the reaction, delivering the desired products in reasonable to good yields (3ap–3ay).
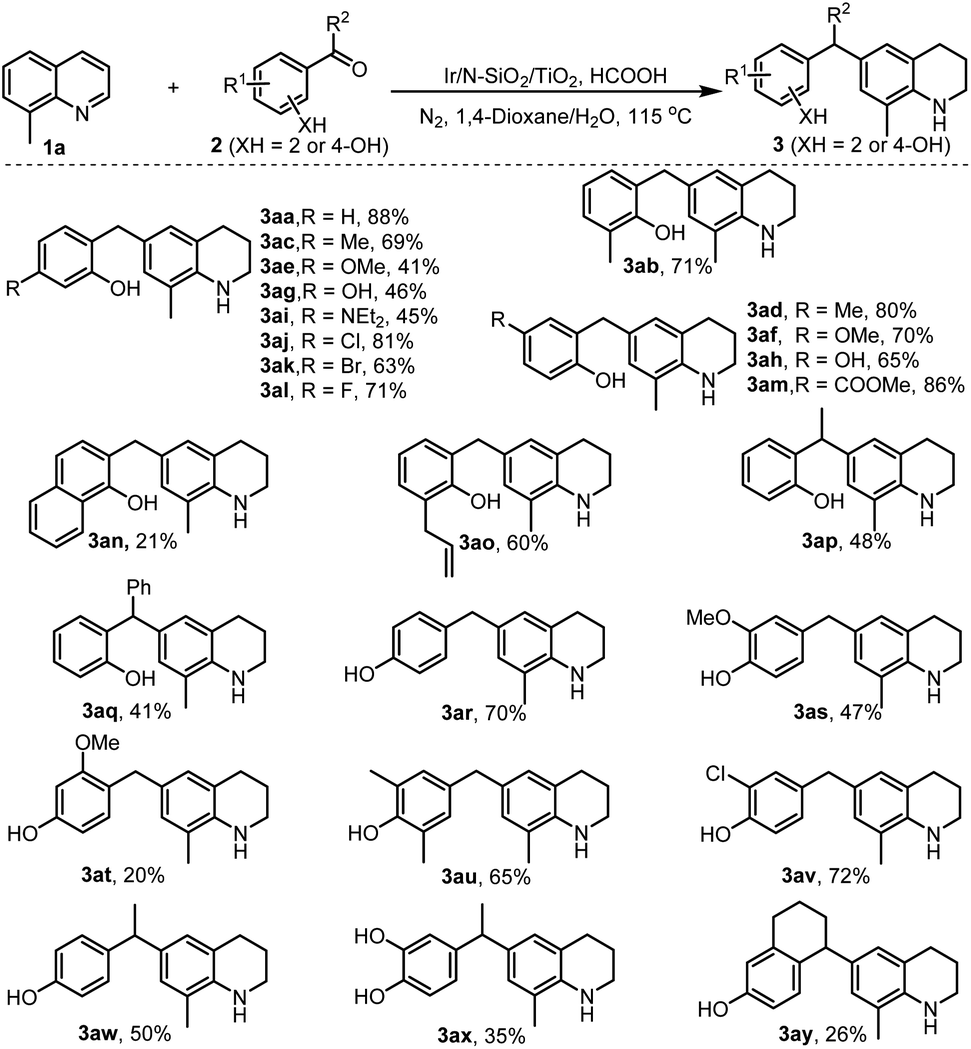 | ||
| Scheme 2 Variation of 2- and 4-hydroxyaryl ketones. | ||
Subsequently, we turned our attention to the variation of substituted quinolines 1 by employing salicylaldehyde 2a as a benchmark substrate (Scheme 3). First, various C8-substituted quinolines (1b–1k, see Scheme S1 for their structures in ESI‡) were tested. All the reactions smoothly afforded the C6-alkylated tetrahydroquinolines in moderate to excellent isolated yields (3ba–3ka). It was found that quinolines bearing an electron-donating group (3ba–3ca and 3ha–3ia) afforded the desired products in higher yields than those having an electron-withdrawing group (3da, and 3ja–3ka), which is assigned to the in situ formed THQs arising from catalytic transfer hydrogenation (CTH) of the electron-rich quinolines favoring the electrophilic coupling process (Scheme 1c). Gratifyingly, the reactions of C6-substituted quinolines (1l and 1m) were able to produce the C8-alkylated THQs, albeit the product yields were somewhat low (3la and 3ma). In addition to C6 and C8-substituted quinolines, C6 and C8 non-substituted quinolines (1n and 1r) were also amenable to the transformation. Interestingly, they resulted in two easily separable regioisiomers, and the 6-alkylated and 8-alkylated THQs were afforded as the major and minor products, respectively (3na-1 and 3na-2, 3ra-1 and 3ra-2). These examples show that the developed chemistry enables to afford C6- and C8-nonsubstituted alkyl THQs. Noteworthily, all the halogenated reactants (3aj–3al and 3da–3ga) in Schemes 2 and 3 did not undergo hydrodehalogenation, a side reaction occurring frequently in homogeneous reductive catalysis, showing that the developed heterogeneous catalyst merits good chemoselectivity in the current transformation.
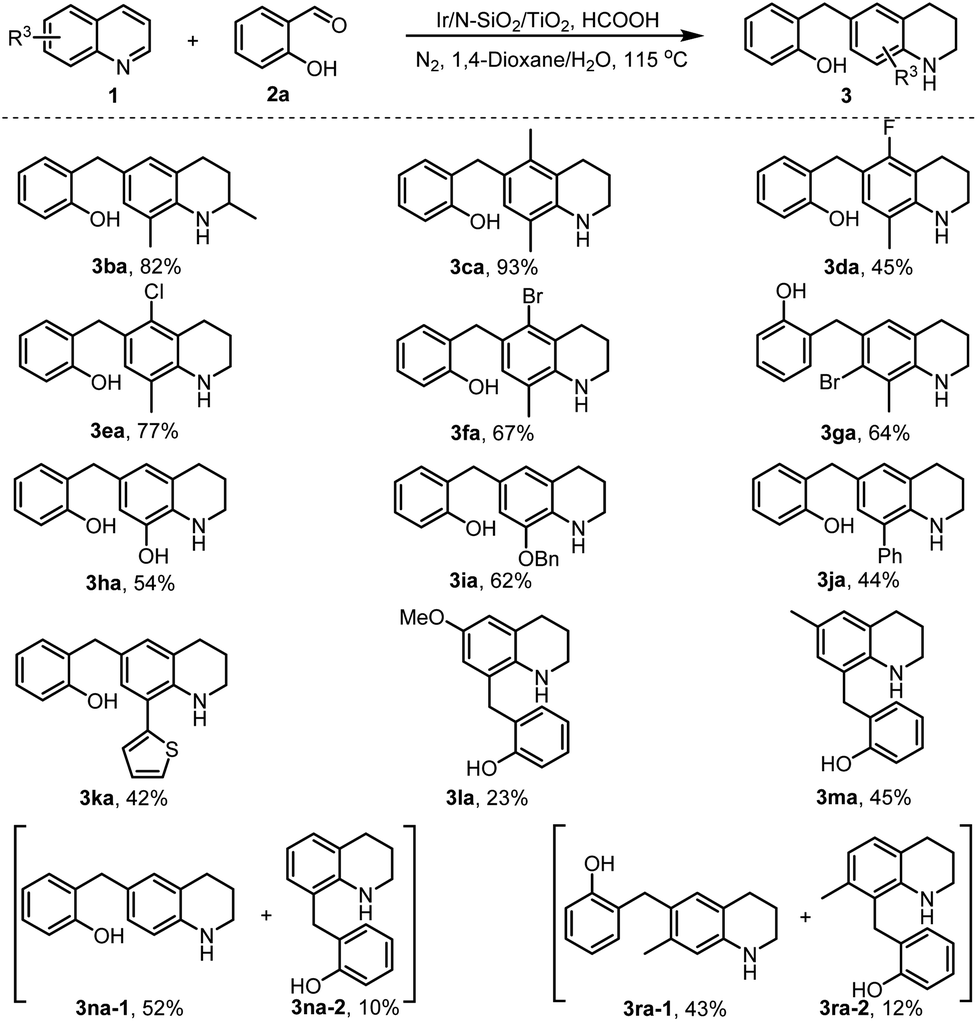 | ||
| Scheme 3 Monoalkylation of various quinolines. | ||
Encouraged by the C6 and C8-alkylations described in Schemes 2 and 3, we then tested the dialkylation of quinolyl C6 and C8-sites. Thus, various C6 and C8 non-substituted quinolines (1n–1s) were employed to couple with sufficient salicylaldehyde (2a). As shown in Scheme 4, all the reactions smoothly generated the desired dialkylated products (3) in moderate to excellent yields upon isolation (3naa–3saa). Interestingly, except for the quinolines, N-alkyl anilines were also applicable for the catalytic transformation. For instance, N,N-dimethylaniline (1t) and N-methylaniline (1u) directly coupled with salicylaldehyde 2a to afford the C2 and C4-dialkylated products (3taa and 3uaa). These two examples demonstrate the potential of the developed chemistry in further synthesis of functionalized anilines, a class of synthetically useful compounds applied for numerous purposes.
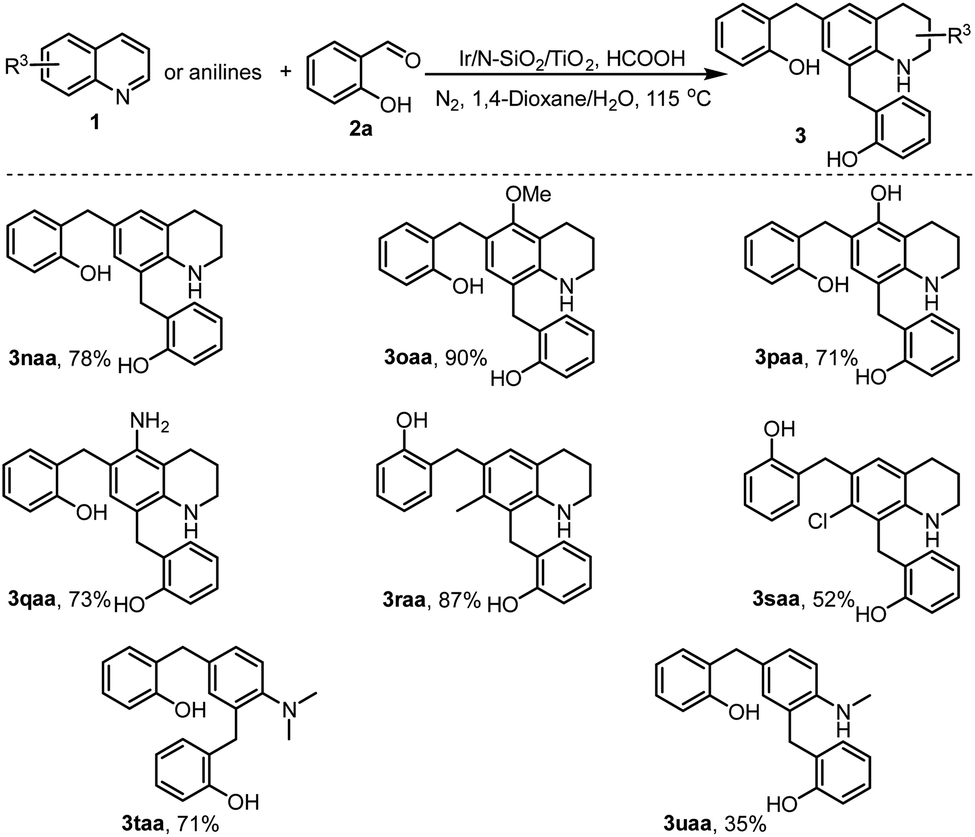 | ||
| Scheme 4 Dialkylation of various quinolines. | ||
Further, in consideration of the easy occurrence of cross-condensation between two molecules of 2-aminoaryl carbonyls, we therefore turned our attention to the transformation of 2-aminobenzyl alcohols 4 including one 2-aminoheteroaryl example (4k) with quinoline 1a. Gratifyingly, all the reactions efficiently afforded the desired 6-(2-aminobenzyl) THQs in reasonable to high isolated yields (Scheme 5, 5aa–5ak). Noteworthily, α-phenyl substituted alcohol 4c afforded relatively higher product yield (5ac) than other 2-aminobenzyl alcohols (5aa and 5ad–5ak), which is attributed to such a substrate (4c) favoring the formation of more stable electrophilic intermediate (Scheme 1c). Interestingly, 1,2,3,4-tetrahydroquinolin-4-ol 4l and 6-hydroxymethyl THQ 4m effectively coupled with quinoline 1a, affording the products featuring two tetrahydroquinolyl motifs in moderate yields (5al and 5am). 4-Hydroxymethyl aniline 4n also underwent efficient reductive alkylation to afford product 5an in 51% yield. Similar to the result described in Scheme 3, the reaction of C6-substituted quinoline 1m and alcohol 4b produced the C8-alkylated THQ 5mb in 32% yield. The C6 and C8 non-substituted quinolines (1n and 1o) smoothly coupled with amino alcohols (4b and 4e) to give two sets of separable regioisomers in reasonable yields (5nb-1 and 5nb-2, 5oe-1 and 5oe-2). It is important to note that all products in Scheme 5 possess two amino groups, which could serve as a class of valuable compounds for the preparation of functional polyamides.
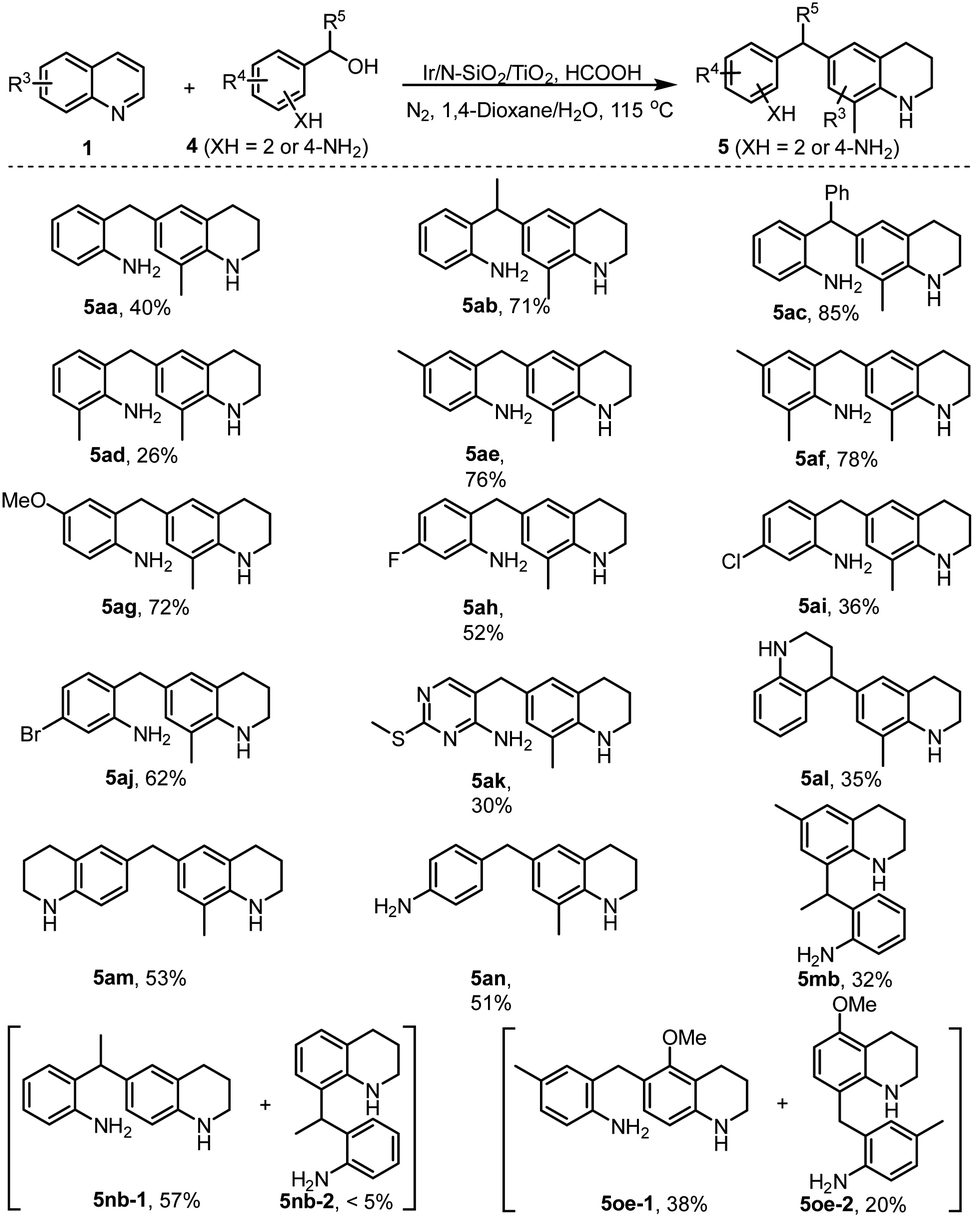 | ||
| Scheme 5 The application of 2- or 4-aminobenzyl alcohols. | ||
To examine the stability of the newly developed heterogeneous catalyst, we performed recycling experiments with the model reaction. As shown in Fig. 3, the catalytic activity maintained very well even after ten consecutive runs, albeit with a slight decline of product yields, which is assigned to the aggregation of some smaller nanoparticles and the mechanical abrasion during the reaction. Furthermore, the XRD spectrum, TEM images and Ir 4f XPS spectra (Fig. 1c, S1 and S4c in ESI‡) of the used Ir/N-SiO2/TiO2 material after 10 runs clearly demonstrate that its morphology is well retained, thus revealing that the catalyst has excellent stability.
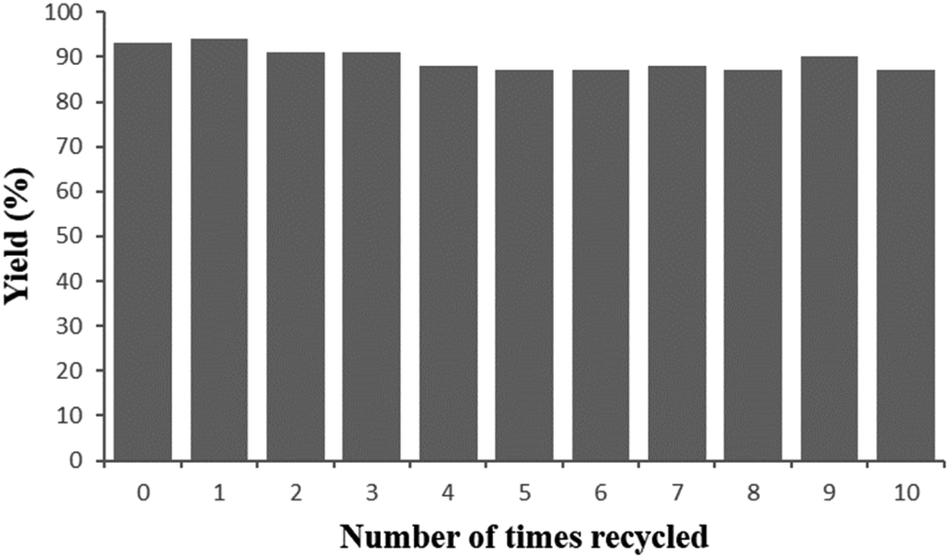 | ||
| Fig. 3 Reusability of the Ir/N-SiO2/TiO2 catalyst. | ||
To gain mechanistic insights into the reaction, several control experiments were performed. First, the model reaction was interrupted after 1 h, which resulted in product 3aa, THQ 1a′ and 2-hydroxybenzyl alcohol 2a′ in 2%, 80%, and 5% yields, respectively (Scheme 6a). Under the standard conditions, the reaction of THQ 1a′ with salicylaldehyde 2a or 2-(hydroxymethyl)phenol 2a′, or the coupling of quinoline 1a with 2a′ all generated product 3aa in excellent yields (Scheme 6b–d), which support that compounds 1a′ and 2a′ are key reaction intermediates. Further, by replacing aqueous HCOOH with DCOOD in D2O, the reaction of 8-methylquinoline 1a and salicylaldehyde 2a generated product 3aa-dn with different D-ratios at the α, β, γ and benzylic sites (Scheme 6e), indicating that HCO2H serves as the hydrogen donor, and there exists a hydrogenation of reactant 2a and a tautomerization during the formation of reactive electrophilic intermediates. Noteworthy, benzaldehyde or acetophenone failed to couple with 8-methylquinoline (1a) to give target products 3 (Scheme 6f). Therefore, all these results are in good agreement with the proposed reaction pathway in Scheme 1c. In addition, the poisoning experiment by addition of KSCN to the model reaction led to loss of catalyst activity in the formation of the target product 3aa,23 which reveals that the Irδ+ species serve as the catalytically active sites.
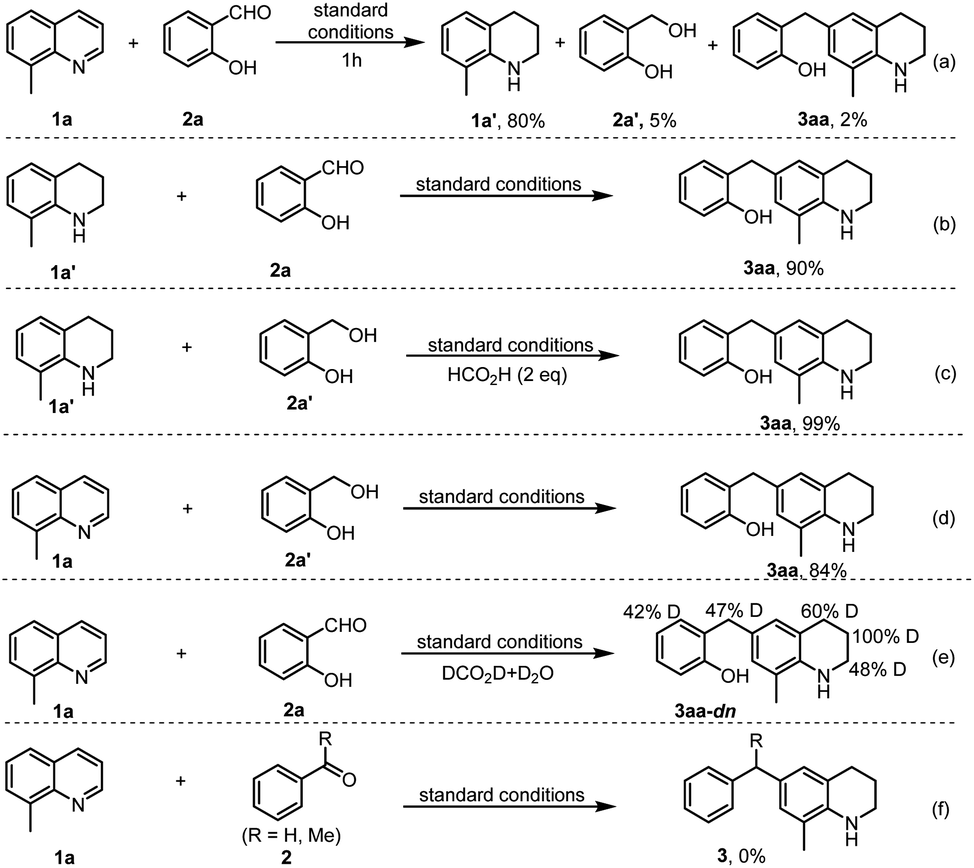 | ||
| Scheme 6 Control experiments. | ||
To explore the utility of the developed chemistry, further transformations of product 3aa were conducted. As shown in Scheme 7, a mixture of compound 3aa and t-BuONa in DMSO was stirred at 60 °C under O2 atmosphere for 6 h, which resulted in dehydroaromatization product 3aa-1 in 81% yield (Scheme 7a).24 In addition, treatment of 3aa with Tf2O and pyridine in dichloromethane solution at room temperature for 6 h initially produced a trifluoromethanesulfonylated product 3aa-2 in 88% yield,25 which then underwent palladium-catalysed Sonogashira cross-coupling with phenylacetylene, to afford product 3aa-3 in 93% yield (Scheme 7b). The coupling of compound 3aa with dihaloalkanes produced medium-sized ring products (3aa-4 and 3aa-5) in reasonable yields, which shows the potential of the obtained compounds in further construction of different cyclic products by mediating the chain-length of difunctionalized agents (Scheme 7c). Further, the synthetic method was successfully applied to reductive dialkylation of bisquinoline, affording the bistetrahydroquinoline 3–6 in 51% yield (Scheme 7d), a promising candidate for the development of optoelectronic materials and nitrogen-containing ligands.26 Finally, product 3–7, an analogue of therapeutical molecule used for the treatment of tauopathies,27 was prepared in high yield by hydrogen transfer-mediated annulation of C6-alkylated THQ 3aa with 1,3-propanediol (Scheme 7e).28
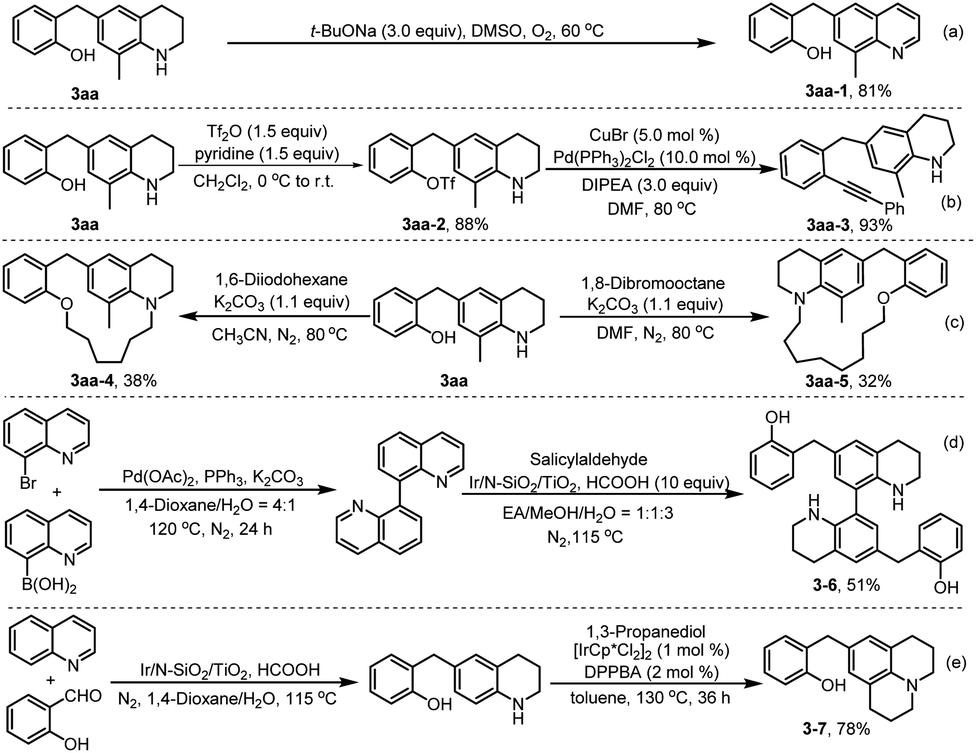 | ||
| Scheme 7 Synthetic applications. | ||
Conclusions
A heterogeneous nanocatalyst composed of iridium and nitrogen-doped SiO2/TiO2 support (Ir/N-SiO2/TiO2) is successfully applied to reductive electrophilic alkylation of quinolines with salicylaldehydes, 4-hydroxyaryl ketones, and 2- or 4-aminobenzylic alcohols. The catalytic transformation proceeds with good substrate and functional group compatibility, uses reusable catalyst and abundantly available feedstocks, and generates water and carbon dioxide as by-products, which offers a practical platform for direct access to a vast range of carbo-alkylated tetrahydroquinolines, a class of significantly important compounds employed frequently for the discovery and preparation of functional products. The strategy applying activity-tunable characteristic of supported heterogeneous catalysts in reductive coupling is anticipated to further develop more useful but challenging organic transformations.Data availability
The datasets supporting this article have been uploaded as part of the ESI.‡Author contributions
R. X. performed the experiments and the data analyses, and wrote the manuscript draft. M. Z. conceived the idea, revised the manuscript, and directed the project. W. M. provided some experimental assistance. H. J., J. S. and G. L. discussed and analysed the data of the catalyst. H. F. J. suggested the substrate scope and discussed the reaction mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21971071), the Natural Science Foundation of Guangdong Province (2021A1515010155), and Fundamental Research Funds for the Central Universities (2020ZYGXZR075) for financial support.Notes and references
- (a) W. Y. Ai, R. Zhong, X. F. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed; (b) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed; (c) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (d) X. W. Chen, H. Zhao, C. L. Chen, H. F. Jiang and M. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14232–14236 CrossRef CAS PubMed.
- Selected examples: (a) Y. Chen, Y. M. He, S. S. Zhang, T. T. Miao and Q. H. Fan, Angew. Chem., Int. Ed., 2019, 58, 3809–3813 CrossRef CAS PubMed; (b) Y. Chen, Y. X. Pan, Y. M. He and Q. H. Fan, Angew. Chem., Int. Ed., 2019, 58, 16831–16834 CrossRef CAS PubMed; (c) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- (a) S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (b) S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed.
- (a) F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS PubMed; (b) G. Y. Zhang, T. Irrgang, T. Dietel, F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 9131–9135 CrossRef CAS PubMed; (c) N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786–10789 CrossRef CAS PubMed; (d) S. Roesler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef CAS PubMed.
- (a) M. Mastalir, B. Stoeger, E. Pittenauer, M. Puchberger, G. Allmaier and K. Kirchner, Adv. Synth. Catal., 2016, 358, 3824–3831 CrossRef CAS; (b) M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed.
- (a) Y. Wang, Z. Shao, K. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2018, 57, 15143–15147 CrossRef CAS PubMed; (b) S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- Selected examples: (a) T. Liu, L. Wang, K. Wu and Z. Yu, ACS Catal., 2018, 8, 7201–7207 CrossRef CAS; (b) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS; (c) F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef CAS PubMed; (d) Z. J. Xu, X. L. Yu, X. X. Sang and D. W. Wang, Green Chem., 2018, 20, 2571–2577 RSC; (e) D. Wang, K. Zhao, C. Xu, H. Miao and Y. Ding, ACS Catal., 2014, 4, 3910–3918 CrossRef CAS.
- (a) K. Yuan, F. Jiang, Z. Sahli, M. Achard, T. Roisnel and C. Bruneau, Angew. Chem., Int. Ed., 2012, 51, 8876–8880 CrossRef CAS PubMed; (b) B. Sundararaju, M. Achard, G. V. M. Sharma and C. Bruneau, J. Am. Chem. Soc., 2011, 133, 10340–10343 CrossRef CAS PubMed; (c) Z. Sahli, B. Sundararaju, M. Achard and C. Bruneau, Green Chem., 2013, 15, 775–779 RSC.
- (a) Z. Qu, H. Zeng and C. Li, Acc. Chem. Res., 2020, 53, 2395–2413 CrossRef PubMed; (b) Z. Chen, H. Zeng, H. Gong, H. Wang and C.-J. Li, Chem. Sci., 2015, 6, 4174–4178 RSC; (c) Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C.-J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed.
- F. Xie, R. Xie, J. X. Zhang, H. F. Jiang, L. Du and M. Zhang, ACS Catal., 2017, 7, 4780–4785 CrossRef CAS.
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed; (b) R. Rahi, M. Fang, A. Ahmed and R. A. Sanchez-Delgado, Dalton Trans., 2012, 41, 14490–14497 RSC; (c) I. Muthukrishnan, V. Sridharan and J. C. Menendez, Chem. Rev., 2019, 119, 5057–5191 CrossRef CAS PubMed.
- (a) K. Murugesan, V. G. Chandrashekhar, C. Kreyenschulte, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2020, 59, 17408–17412 CrossRef CAS PubMed; (b) A. Vivancos, M. Beller and M. Albrecht, ACS Catal., 2018, 8, 17–21 CrossRef CAS; (c) D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6439–6450 RSC.
- (a) S. Ott, Science, 2011, 333, 1714–1715 CrossRef CAS PubMed; (b) R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang, X. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS; (c) R. Xie, G.-P. Lu, H.-F. Jiang and M. Zhang, J. Catal., 2020, 383, 239–243 CrossRef CAS; (d) R. Xie, F. Xie, C.-J. Zhou, H.-F. Jiang and M. Zhang, J. Catal., 2019, 377, 449–454 CrossRef CAS.
- (a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed; (b) S. Lu, C. Li, H. Li, Y. Zhao, Y. Gong, L. Niu, X. Liu and T. Wang, Appl. Surf. Sci., 2017, 392, 966–974 CrossRef CAS; (c) Q. Zhou, W. Ju, X. Su, Y. Yong, X. Li, Z. Fu and C. Wang, RSC Adv., 2017, 7, 43521–43530 RSC; (d) J. Wang, Z. Xu, Y. Gong, C. Han, H. Li and Y. Wang, ChemCatChem, 2014, 6, 1204–1209 CAS.
- (a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed; (b) S. Lu, C. Li, H. Li, Y. Zhao, Y. Gong, L. Niu, X. Liu and T. Wang, Appl. Surf. Sci., 2017, 392, 966–974 CrossRef CAS; (c) Q. Zhou, W. Ju, X. Su, Y. Yong, X. Li, Z. Fu and C. Wang, RSC Adv., 2017, 7, 43521–43530 RSC.
- (a) Z. Li, Y. Li, Y. Wang and X. Wang, Can. J. Phys., 2016, 94, 929–932 CAS; (b) J. A. Talla and A. A. Ghozlan, Chin. J. Phys., 2018, 56, 740–746 CrossRef CAS.
- (a) S. M. Sachau, M. Zaheer, A. Lale, M. Friedrich, C. E. Denner, U. B. Demirci, S. Bernard, G. Motz and R. Kempe, Chem. –Eur. J., 2016, 22, 15508–15512 CrossRef CAS PubMed; (b) J. Yoo, J.-I. Kim, H.-J. Cho, S.-S. Choo, S.-I. Kim, K. Lee, W. H. Shin, H.-S. Kim and J. W. Roh, Phys. Soc., 2018, 72, 775–779 CAS; (c) H. L. Jia, B. G. Ren, M. Li, X. J. Liu, J. X. Wu and X. Tan, Solid State Commun., 2018, 277, 45–49 CrossRef CAS.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhofer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Bruckner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS.
- (a) Y. Xing, Y. Yang, D. Li, M. Luo, N. Chen, Y. Ye, J. Qian, L. Li, D. Yang, F. Wu, R. Chen and S. Guo, Adv. Mater., 2018, 30, 1803124 CrossRef PubMed; (b) W. Cao, L. Lin, H. Qi, Q. He, Z. Wu, A. Wang, W. Luo and T. Zhang, J. Catal., 2019, 373, 161–172 CrossRef CAS.
- (a) S. J. Freakley, J. Ruiz-Esquius and D. J. Morgan, Surf. Interface Anal., 2017, 49, 794–799 CrossRef CAS; (b) M. Scohy, S. Abbou, V. Martin, B. Gilles, E. Sibert, L. Dubau and F. Maillard, ACS Catal., 2019, 9, 9859–9869 CrossRef CAS; (c) R. Jin, M. Peng, A. Li, Y. Deng, Z. Jia, F. Huang, Y. Ling, F. Yang, H. Fu, J. Xie, X. Han, D. Xiao, Z. Jiang, H. Liu and D. Ma, J. Am. Chem. Soc., 2019, 141, 18921–18925 CrossRef CAS PubMed; (d) Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Green Chem., 2020, 22, 6855–6861 RSC.
- (a) C. Tang, A. E. Surkus, F. Chen, M. M. Pohl, G. Agostini, M. Schneider, H. Junge and M. Beller, Angew.Chem. Int. Ed., 2017, 56, 16616–16620 CrossRef CAS PubMed; (b) Z. Ma, T. Song, Y. Yuan and Y. Yang, Chem. Sci., 2019, 10, 10283–10289 RSC; (c) H. Luo, L. Wang, S. Shang, G. Li, Y. Lv, S. Gao and W. Dai, Angew.Chem. Int. Ed., 2020, 59, 19268–19274 CrossRef CAS PubMed.
- R. Yang, S. Yue, W. Tan, Y. Xie and H. Cai, J. Org. Chem., 2020, 85, 7501–7509 CrossRef CAS PubMed.
- L. Cao, H. Zhao, Z. D. Tan, R. Q. Guan, H. F. Jiang and M. Zhang, Org. Lett., 2020, 22, 4781–4785 CrossRef CAS PubMed.
- (a) C. Wang, D. M. Flanigan, L. N. Zakharov and P. R. Blakemore, Org. Lett., 2011, 13, 4024–4027 CrossRef CAS PubMed; (b) T. P. Loh and J. Xiao, US Pat., 2012088916A1, 2012 Search PubMed.
- (a) S. Clunas, J. M. D. Strory, J. E. Rickard, D. Horsley, C. R. Harrington and C. M. Wischik, GB Pat., WO2010067078A2, 2010 Search PubMed; (b) M. A. Delong,K. A. Biedermann, D. L. Bissett, A. S. Boyer, S. L. Cohen and C. E. Snider, US Pat., WO2004037213A2, 2004 Search PubMed; (c) G. Q. Xu, Z. T. Feng, J. T. Xu, Z. Y. Wang, Y. Qin and P. F. Xu, Chem. –Eur. J., 2018, 24, 13778–13782 CrossRef CAS PubMed.
- A. Labed, F. Jiang, I. Labed, A. Lator, M. Peters, M. Achard, A. Kabouche, Z. Kabouche, G. V. M. Sharma and C. Bruneau, ChemCatChem, 2015, 7, 1090–1096 CrossRef CAS.
Footnotes |
| † This work is dedicated to Prof. Huanfeng Jiang on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all the compounds. See DOI: 10.1039/d1sc02967c |
| This journal is © The Royal Society of Chemistry 2021 |