
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in chemical conjugation strategies targeting native amino acids in proteins and their applications in antibody–drug conjugates
Min Sun
Kang
 ,
Theresa Wai See
Kong
,
Joycelyn Yi Xin
Khoo
and
Teck-Peng
Loh
*
,
Theresa Wai See
Kong
,
Joycelyn Yi Xin
Khoo
and
Teck-Peng
Loh
*
Division of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: esther.kang@ntu.edu.sg; teckpeng@ntu.edu.sg
First published on 6th October 2021
Abstract
Many fields in chemical biology and synthetic biology require effective bioconjugation methods to achieve their desired functions and activities. Among such biomolecule conjugates, antibody–drug conjugates (ADCs) need a linker that provides a stable linkage between cytotoxic drugs and antibodies, whilst conjugating in a biologically benign, fast and selective fashion. This review focuses on how the development of novel organic synthesis can solve the problems of traditional linker technology. The review shall introduce and analyse the current developments in the modification of native amino acids on peptides or proteins and their applicability to ADC linker. Thereafter, the review shall discuss in detail each endogenous amino acid's intrinsic reactivity and selectivity aspects, and address the research effort to construct an ADC using each conjugation method.
 Min Sun Kang | Min Sun Kang, born in South Korea, received her BSc (2018) in chemistry from Imperial College London. She then worked as an Investor Relations representative at SillaJen (South Korea). She is presently working as a research officer with Professor Teck Peng Loh. Her work focuses on protein functionalization, antibody–drug conjugate research and water-based synthetic methodology. |
 Theresa Wai See Kong | Kong Wai See, Theresa received her BSc in chemistry and biological chemistry from Nanyang Technological University, Singapore in 2021. |
 Joycelyn Yi Xin Khoo | Khoo Yi Xin, Joycelyn received her BSc in chemistry and biological chemistry from Nanyang Technological University, Singapore in 2021. She is currently pursuing her PhD at the same institution under the supervision of Professor Teck-Peng Loh. Her research interests are in the area of antibody–drug conjugates and the green synthesis of natural products. |
 Teck-Peng Loh | Teck-Peng Loh is a distinguished university professor of chemistry at Nanyang Technological University, Singapore. Under the tutelage of Professor E. J. Corey, he obtained his PhD (1994) from Harvard University. He has published 422 international refereed papers in reputable chemistry journals bearing high impact factor and 10 patents. He has been invited to give more than 100 lectures in many institutions in the world and plenary and keynote talks in conferences such as the ICOS, OMCOS, Asian European Symposium, etc. He has also served as chairman of many of these conferences. He has been awarded outstanding researcher awards from both National University of Singapore and Nanyang Technological University. In 2017 he received the Yoshida Prize (Japan) and the prestigious President's Science Award (individual) Singapore. He has been elected Fellow, Academia of Sciences, Singapore (2018) and Fellow of Academia of Sciences, Malaysia since 2010. His research work mainly focuses on the development of new synthetic methodology, green chemistry, and synthesis of natural and unnatural products. |
1. Introduction
1.1. Bioconjugation and its applications
Bioconjugation, combining biomolecules such as proteins or cells with exogenous moieties, leads to enhanced therapeutic efficacy and a wider range of functions.1 Among the conjugated molecules, one prominent example that is rising as a promising therapeutic modality in oncology is antibody–drug-conjugate (ADC) (Fig. 1). The ability of the antibody to deliver the payload to specific targets to kill the cancer cells with fewer side effects makes this class of compounds an attractive target for new therapeutics development. | ||
| Fig. 1 General structure of antibody–drug conjugate. | ||
With such exciting applications of bioconjugates, the development of a suitable linker, which connects biomolecules with payloads in a selective and efficient fashion, is increasingly garnering attention from the biomedical field. In this review, we will be focusing on the ADC chemical conjugation technology.
1.2. Antibody–drug-conjugate (ADC)
The synergy of a target-specific antibody and cytotoxic chemical led to the United States Food and Drug Administration (US FDA) approval of eleven ADCs, and 302 clinical studies, as of the second quartile of 2021. The first FDA-approved ADC is gemtuzumab ozogamicin (GO, Mylotarg™), developed by Pfizer to treat patients with acute myeloid leukemia (AML) who first relapsed.2 It is a conjugate of an engineered humanized monoclonal IgG4 antibody directed against the CD33 antigen present on leukemic myeloblasts, and a potent DNA-binding cytotoxic antibiotic, calicheamicin derivative (N-acetyl-γ-calicheamicin 1,2-dimethyl hydrazine dichloride).3 A bifunctional linker, 4-(4-acetylphenoxy)butanoic acid (AcBut) connects the CD33 targeting antibody and a calicheamicin derivative (Fig. 2).3 Pfizer conjugated the same AcBut linker-calicheamicin derivative payload to G544, an IgG4 isotype that specifically recognizes human CD22, to synthesize inotuzumab ozogamicin (InO, Besponsa™). It was approved in 2017 for the treatment of acute lymphoblastic leukemia (ALL).4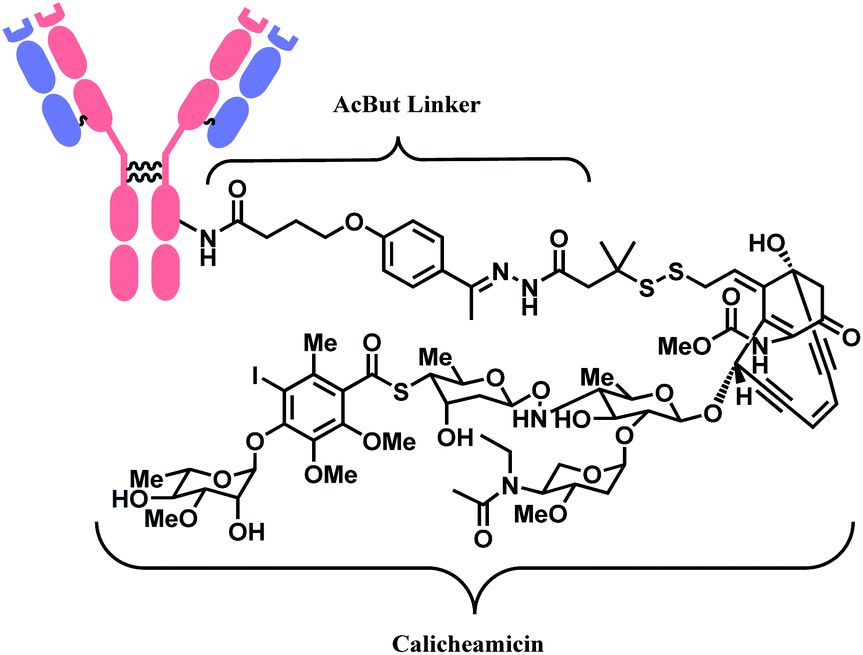 | ||
| Fig. 2 | Structure of Mylotarg™ (antibody: CD33-targeting IgG4 mAb) and Besponsa™ (antibody: CD22-targeting IgG4 mAb). | ||
In 2011, brentuximab vedotin (BV, Adcetris™), developed by Seattle Genetics, received a FDA approval to treat relapsed Hodgkin lymphoma (HL) and relapsed systemic anaplastic large-cell lymphoma (sALCL).5 BV is generated by conjugating synthetic antitubulin agent monomethyl auristatin E (MMAE) to CD30-targeting mAb, via a maleimidocapryl L-valine-citrulline p-aminobenzyl alcohol (mc-vc-PABC) linker (Fig. 3).6 Seattle Genetics conjugated the same linker-payload to Nectin-4 targeting mAb to produce enfortumab vedotin (EV, Padcev™),7 which is approved in 2019 to treat urothelial cancer.8 The MC-VC-PABC linker–MMAE payload is also employed by polatuzumab vedotin (PV, Polivy™), and it is conjugated to monoclonal antibody against CD79b, which is a B cell receptor component. PV is developed by Roche and it is approved for the third-line treatment of adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL).9
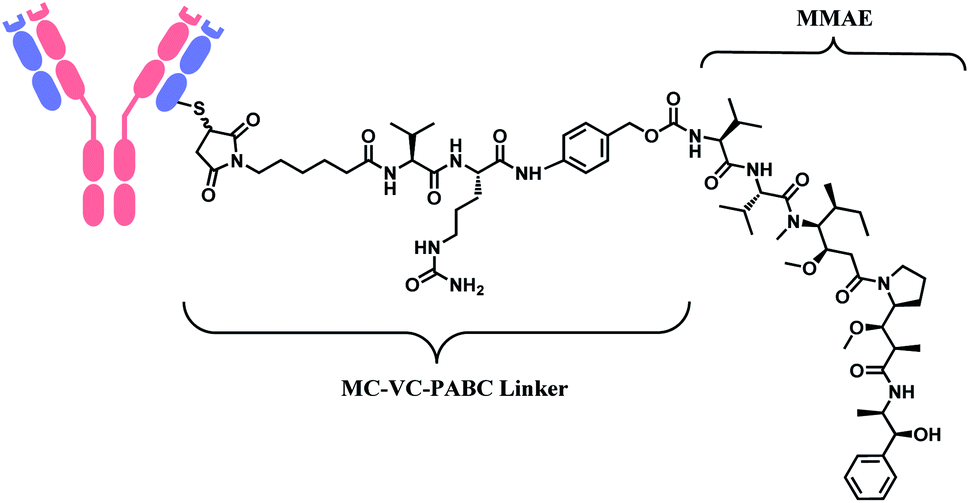 | ||
| Fig. 3 Structure of Adcetris™ (antibody: CD30-targeting IgG1 mAb), Padcev™ (antibody: Nectin-4-targeting IgG1 mAb), and Polivy™ (antibody: CD79b-targeting IgG1 mAb). | ||
In 2013, trastuzumab emtansine (T-DM1, Kadcyla™), developed by Roche, received a FDA approval to treat human epidermal growth factor receptor-2 (HER2) expressing metastatic breast cancer and to improve patient prognosis.10 T-DM1 conjugates HER2 targeting monoclonal IgG1 antibody trastuzumab with potent microtubule-disrupting agent DM1 via a linker, succinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (SMCC) (Fig. 4).11
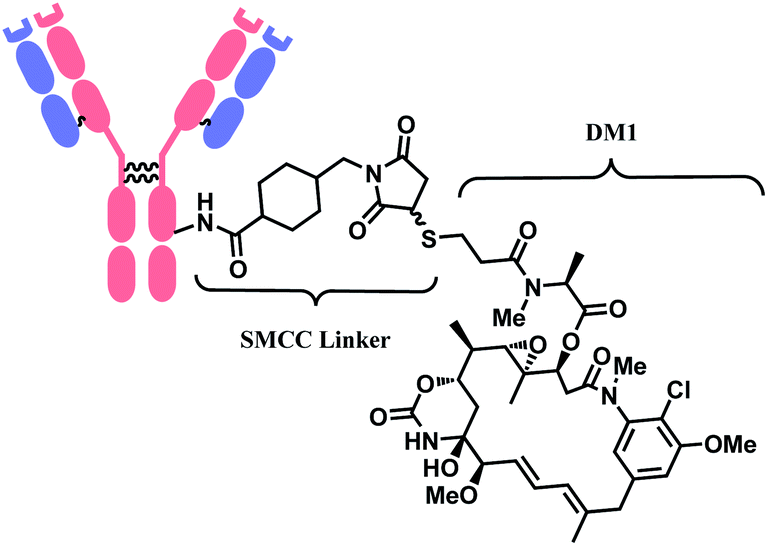 | ||
| Fig. 4 Structure of Kadcyla™ (antibody: trastuzumab). | ||
In 2019, trastuzumab deruxtecan (T-Dxd, Enhertu™) developed by AstraZeneca was approved as an unresectable or metastatic HER2+ breast cancer therapy.12 Trastuzumab and the payload, a camptothecin (CPT) derivative, were linked via MC-glycyl glycyl phenylalanyl glycine (GGFG)–aminomethyl (AM) linker. The CPT forms a stable complex with human DNA topoisomerase I (Topo I) and DNA, which induces DNA damage and cell apoptosis.13 Its structure is shown in Fig. 5.
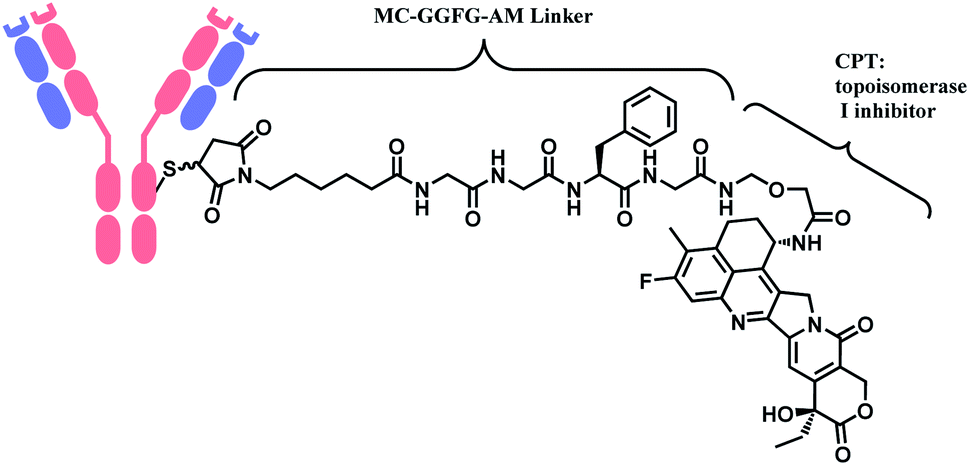 | ||
| Fig. 5 Structure of Enhertu™ (antibody: trastuzumab). | ||
In 2020, sacituzumab govitecan (SG, Trodelvy™), developed by Immunomedics was approved as a second-line therapy for triple-negative breast cancer.14 It is a conjugate of mAb targeting trophoblast cell-surface antigen 2 (TROP-2) and topoisomerase inhibitor, SN-38 (irinotecan metabolite). The linker is known as CL2A, containing polyethylene glycol (PEG) groups (Fig. 6).
 | ||
| Fig. 6 Structure of Trodelvy™ (antibody: TROP-2 targeting IgG1 mAb). | ||
Belantamab mafodotin (Belamaf, Blenrep™) is developed by GlaxoSmithKline, for the treatment of multiple myeloma. It couples an IgG1 mAb targeting B-cell maturation antigen (BCMA) and a microtubule-disrupting agent, monomethyl auristatin F (MMAF) via a MC linker (Fig. 7).15
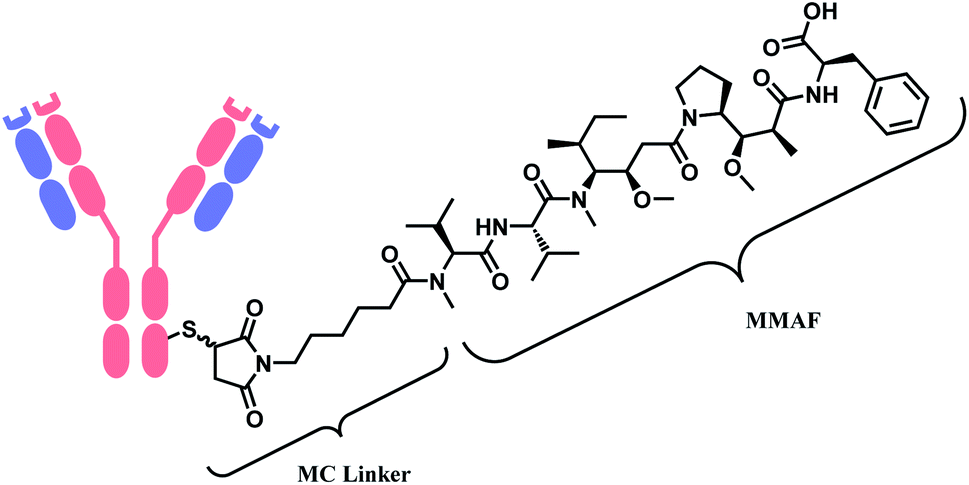 | ||
| Fig. 7 Structure of Blenrep™ (antibody: BCMA-targeting IgG1 mAb). | ||
In 2021, loncastuximab tesirine (LT, Zynlonta™), developed by ADC Therapeutics was approved as a treatment for relapsed/refractory diffuse large B cell lymphoma (DLBCL).16 A CD19-targeting mAb and a pyrrolobenzodiazepine (PBD) DNA-alkylating warhead are connected via the linker consisted of maleimide group that binds to the antibody, polyethylene glycols (PEGs), protease-cleavable valine–alanine (VA) and PABC (Fig. 8).
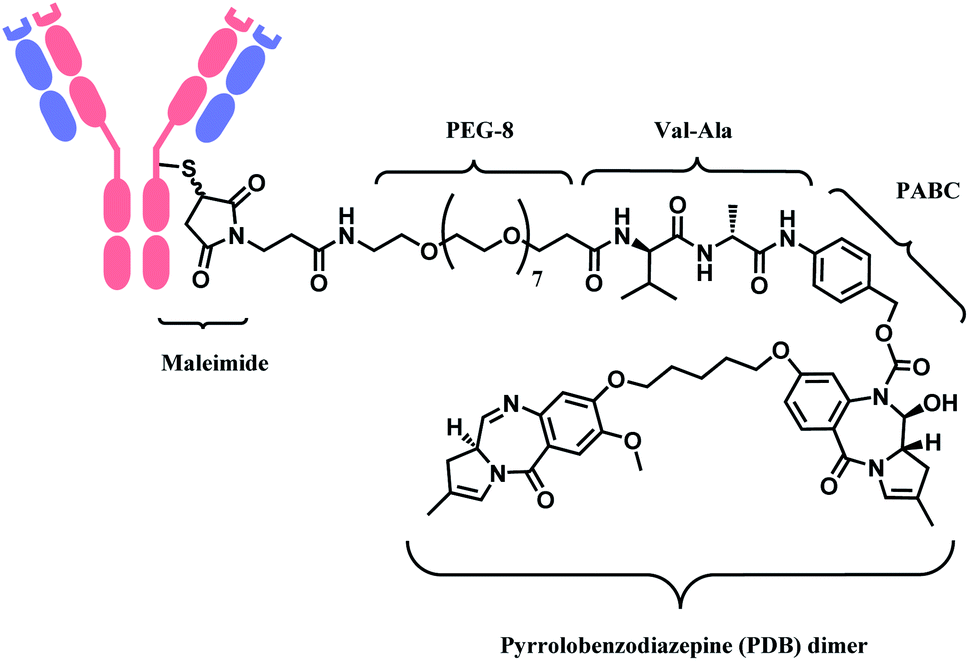 | ||
| Fig. 8 Structure of Zynlonta™ (antibody: CD19-targeting IgG1 mAb). | ||
Moxetumomab Pasudotox (MP, Lumoxiti™) is a conjugate of CD22-targeting mAb and a truncated form of Pseudomonas exotoxin (PE38).17 PE38 is connected to the VH chains of the mAb via a ASGGPE peptide bond, which is achieved by recombinant technology.18 MP was approved to treat hairy cell leukaemia.19
1.3. Benefits of ADC over antibody
Proteins alone serve as therapeutic agents, and they have increased dramatically in number and frequency of use since the introduction of the first recombinant protein therapeutic, human insulin, 25 years ago.20 Antibodies, especially, have become the predominant class of new drugs in chronic diseases, arthritis, and cancer.21 As oncological drugs, they trigger immune system response by exploiting antigenic differences between malignant cells and healthy cells.22 79 therapeutic monoclonal antibodies (mAbs) have been approved by the US FDA, including 30 mAbs in cancer.21 While they show significant advantages over traditional chemotherapies, a number of limitations can be addressed; (1) ineffectiveness in tumour cells with low expression of target antigens,23 (2) insufficient antitumor response after binding to antigen,24 and (3) significantly higher production cost compared to chemical drugs.25,26 Conjugating mAbs with cytotoxins has a potential to overcome such limitations and lead to great synergy. The following paragraphs introduce clinical or pre-clinical results of ADC which show such potential.In a phase III clinical trial on HER2 positive breast cancer which compared T-DM1 and trastuzumab head-to-head, T-DM1 was shown to significantly reduce the risk of invasive breast cancer recurrence or death from any cause by 50% compared to the antibody trastuzumab alone.27 A stable SMCC linker in the T-DM1 construct has enabled the incorporation of a highly potent drug DM1 (in vitro IC50 = 0.030 nM), which is unable to be safely dosed on its own, thereby achieving improved anti-cancer effects.11
On top of the enhanced therapeutic efficacy, ADC requires fewer doses as compared to monoclonal antibodies alone. The required dosage of T-DM1 is 3.6 mg per kilogram of body weight, whereas trastuzumab is administered 6 mg per kilogram of body weight.27 Another ADC of trastuzumab and cytotoxin, T-Dxd (Enhertu™) is administered 5.4 mg per kilogram of body weight.12 ADCs, with their chemical drugs, drive down the amount of expensive mAbs needed for the therapeutic dose.
The mechanism of ADC involves ‘bystander effect’, depending on the linker type,28 and it enables this mAb-based therapeutic to be efficacious in tumours with low or heterogenous expression of antigens as well. The bystander effect refers to the killing of proximal antigen-negative tissues by cytotoxic payloads which diffuse out from the cell.29 In the preclinical study, the ADC of anti-CanAg mAb and cytotoxin, DM1 or the DNA alkylator DC1, eradicated tumors containing both antigen-positive and antigen-negative cells.29
1.4. ADC linker technology
As demonstrated in the structures of FDA-approved ADCs in the above section, the linkers connecting ‘A’ and ‘D’ can consist of different groups. (1) chemical conjugation moiety that binds to the antibody, (2) protease- or acid-cleavable group with self-elimination spacer, and (3) hydrophilic poly(ethylene glycol). The approved ADCs selectively have different combinations of each. Depending on the existence of protease- or acid-cleavable group with self-elimination spacer, the linker is classified as either cleavable or non-cleavable. 2 out of 11 FDA-approved ADCs, T-DM1 (Fig. 4) and Belamaf (Fig. 7) use non-cleavable linkers, as they only have chemical conjugation moiety. In T-DM1, trastuzumab (T) and DM1 are linked via a succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), a heterobifunctional linker with NHS ester group and a maleimide reactive group at each end of a cyclohexyl spacer. In Belamaf, MMAF is attached to the antibody via maleimide with a hexyl spacer. It is believed that the drugs DM1 and MMAF are released with an amino acid appendage from the antibody in tumour cells via the catabolism of antibody itself.28In contrast, the other 9 FDA-approved ADCs utilise cleavable linkers that contain peptide- or acid-cleavable groups to release drugs in the lysosomes by exploiting the tumour-associated intracellular conditions. GO and InO (Fig. 2) have a hydrazone moiety that is stable at neutral pH in blood, but becomes more susceptible to hydrolysis inside tumour cells.30 The disulfide adjacent to the hydrazone undergoes thiol–disulfide exchange with abundant intracellular glutathiones (GSH) to release the payloads.31 SG (Fig. 6) also contains pH-sensitive benzyl carbonate bond.32 Alongside, it has a lysine unit, which undergoes cleavage by cysteine protease cathepsin B,33,34 which is overexpressed in invasive and metastatic cancers.35
In cases of SG and LT, polyethylene glycol (PEG) groups were inserted to increase solubility and reduce aggregation of hydrophobic cytotoxic drugs in blood plasma.36 BV, EV and PV (Fig. 3) and LT (Fig. 8) have a dipeptide valine–citrulline and valine–alanine linked with p-aminobenzyl oxycarbonyl (PABC) electronic cascade spacer. Upon the cleavage by cathepsin B, the PABC undergoes 1,6-elimination to release the free drug with the expulsion of carbon dioxide.37 T-Dxd, on the other hand, has a tetrapeptide Gly–Gly–Phe–Gly as a lysozyme-cleavable group linked with self-immolative amino methylene (AM) spacer.38
While a variety of protease cleavable groups and self-elimination spacer are employed in the FDA-approved ADCs, the chemical conjugation strategies are limited to only two kinds; N-hydroxysuccinimidyl ester and maleimide.
2. Bioconjugation methods for ADC linker
In this review, recent advancements in native amino acid conjugation methods will be analysed in light of the applicability as ADC linker. With aforementioned aspects of ADC and general rules of bioconjugation taken into consideration, criteria for ADC linker molecules can be summarized as follows: (1) they must bind to proteins in a highly site-selective manner, so that the number of payloads conjugated to antibody is well-controlled with no side products (2) they must efficiently bind to antibodies under biocompatible conditions (aqueous medium, pH neutral, no transition-metal catalyst, room temperature), without perturbing the ternary structure of proteins (3) the chemistry of resulting protein–linker conjugate should be highly stable in blood plasma, with no retro-reaction (4) it should be synthetically easy to attach functional payloads on the linker.Site-selectivity comes in two categories: chemoselectivity and regioselectivity. Chemoselectivity can be achieved by exploiting the intrinsic reactivity of a certain functional group in mAbs. Of 20 kinds of canonical amino acids constructing a mAb, the cysteine thiol (–SH) group and the lysine amino (ε-NH2) are the most common targets as conjugation sites, due to their nucleophilicities and availabilities in solvent-accessible regions.30 Three of FDA-approved ADCs link cytotoxic drugs to lysine residues, and the rest, including the most recently approved ADCs, use cysteine modifications. While being widely used in these clinical ADCs, the issue of regioselectivity (i.e. conjugating to a specific cysteine or lysine residue among many) remains unsolved and research efforts to address and resolve it will be analysed in this review. Along with this, recent developments and selectivity aspects of bioconjugation to other amino acids including tyrosine (Tyr), tryptophan (Trp), and methionine (Met) will be explored. Last but not least, the conjugation should ideally avoid generating a new stereocentre. The stereochemical heterogeneity of the conjugation linker complicates the product analysis.
Linker molecules must be reactive enough to bind to mAb rapidly under biocompatible conditions, because mAbs are highly sensitive to reaction conditions. Transition-metal ions are reported to perturb a strictly defined 3-dimensional structure of mAb by cleaving the hinge region (shown in Fig. 1), forming highly stable pluridentate with nucleophilic amino acids, or by catalysing redox reactions of amino acid side chains.39,40 Hence, bioconjugation methods involving transition-metal catalysts will not be reviewed. Moreover, the mechanical stress caused by stirring is reported to lead to the aggregation of mAbs.41 For one example, the percentage of aggregation of anticancer mAb cetuximab after 24 h stirring-period was about 25%.42 Therefore, the reaction duration of each bioconjugation method will be addressed in details. As summarised in Scheme 1, ADC linker molecules should bind to mAb in pH neutral aqueous system at room temperature with the minimal mechanical stress. The detailed effects of organic solvent, pH variance, and light (ultraviolet or fluorescence) on monoclonal antibodies are discussed in the related reviews.43,44
 | ||
| Scheme 1 Requirements for bioconjugation strategies for ADC chemical conjugation technology. | ||
Recently, many methods to achieve site-selective linker conjugation for homogenous ADC production were reported. One is an enzymatic conjugation strategy, such as transglutaminase-driven glutamine modification.45 Another approach involves engineered carbohydrates or engineered amino acids to provide an orthogonal handle for conjugation. One of the most advanced approaches is the THIOMAB® technology,46 which involves mutating amino acid residues at particular sites in the constant domains of the antibody to Cys residues that can then be selectively conjugated via their thiol groups. An excellent summary on such methods are recently published.47
Although these newly developed chemical conjugation technologies have shown clear advantages in DAR control, an important consideration is that these strategies introduce mutations to antibodies and these can be potentially problematic in immunocompetent patients.48 Furthermore, a full replacement or introduction of an unnatural amino acid into proteins is not straightforward, where a new tRNA, new tRNA-amino acids transferase, and genetic codon modifications are needed to cheat the ribosome complexes.49 The mutation site can have drastic effects on the stability of resulting antibodies.50 In spite of the fact that recent advances have addressed stability issues, early-stage clinical ADCs rely on mutations or inefficient conjugation chemistries often requiring extensive engineering or multiple steps.
This review will focus on how establishing new organic synthesis principles can solve the existing challenge of ADC chemical conjugation technology. Protein engineering methods or enzymatic ligation methods will not be discussed. Rather, this review will highlight novel and relatively obscure conjugation chemistries which use native amino acids. In addition, the review will cover in-depth the problems of traditional methods. A wider choice of suitable conjugation linkers with different synthetic scopes will provide an orthogonal and complementary approach to preparing the desired protein conjugates.
2.1. Lysine functionalisation
Lysine modification is used to produce three of eleven FDA-approved ADCs, GO,2,3 T-DM1,51 and InO.52 A typical immunogloblin G1 (IgG1) antibody has roughly 90 lysine residues, meaning that non-selective conjugation strategies have the potential to give up to 106 distinct species statistically possible when targeting the DAR value of 2 to 4.53 In industry, the process condition is optimized that the conjugation occurs at 8–10 kinetically preferred lysine sites out of 90.5450% of anti-CD33 mAbs are reported to be unconjugated in GO, with the remainder being a mixture ranging from 4 to 6 drugs per mAb.2 Such heterogenicity was further studied by its developer with size-exclusion chromatography – the species with higher average drug loading were found to be responsible for forming aggregates.55 Physical instability such as aggregation is a stability limiting parameter for ADC; the product was voluntarily withdrawn from the US market in 2010 due to safety concerns.56 InO has an average DAR of 6, and the drug load distribution varies from 0 to 9.54
T-DM1 has an average 3.5 DM-1 molecules conjugated to every trastuzumab molecule, and at least 70 lysine sites are partially conjugated with the drug.57 The statistical study demonstrated that the drug load distribution varies from 0 to 8 and fits to a Poisson distribution.57 For a DAR of 3.48 this leads to a relatively large variance (λ) of about 3.28 in the Poisson model, demonstrating a drug load with high heterogeneity.
Another noteworthy aspect of lysine modification strategy is a potential conjugation of an electrophilic linker to the N-termini of mAb. In other words, lysine conjugation strategy does not display good levels of chemoselectivity. The pKa of the N-terminus α-amino group of the protein is lower (pKa ∼ 8) than the pKa of ε-amino group of the lysine side chain (pKa ∼ 10–11). At the conjugation pH 7.5–8, the α-amino groups of the N-termini of the mAb molecules are more likely to be deprotonated compared to the ε-amino groups of the lysine side chains. The higher reactivity of the N-terminal amines at the conjugation pH along with other factors such as better accessibility to the electrophilic linker leads to some conjugation at the N-termini of the mAb molecules. While it is unclear whether the conjugation of the drug to the N-termini of the mAb directly leads to the formation of aggregates, the structural characterization studies have shown that the level of conjugation to the N-termini of the mAb was higher in aggregates than in the monomeric ADC.55
However, it is debatable whether they directly lead to inferior therapeutic profiles. It is worth introducing a preclinical study on cervical cancer, where a lysine conjugated ADC afforded a greater efficacy on a molar payload basis than a site-specific engineered cysteine conjugated ADC.58 Establishing a general rule about ADC chemical conjugation technology is not as simple, due to the complexity of cellular mechanisms of cancer and the mode of action of ADC. Hence, a more meticulous study is required to further investigate the causal relationship between the linker conjugation selectivity and therapeutic efficacy.
Moreover, there have been recent developments in a regioselective approach for lysine conjugation, suggesting novel synthetic methodologies can overcome the existing drawbacks of lysine conjugation. Scheme 2 shows an overall summary of reported lysine conjugation methods. While not covered explicitly in the following section, the research work by Kelly and co-workers is also worth noting, as they have demonstrated that a stilbene derivative molecule can selectively label 1 of the 8 lysine ε-amino groups displayed on the surface of the plasma protein transthyretin.59
 | ||
| Scheme 2 Lysine conjugation methods. | ||
In the production of GO and InO, succinimidyl-activated calicheamicin derivative (linker-payload) is synthesized prior to conjugation to mAb (Scheme 3).
 | ||
| Scheme 3 Synthesis of NHS linker-Calicheamicin derivative payload and conjugation to antibody. | ||
The NHS ester moiety in activated calicheamicin derivative is susceptible to hydrolysis under aqueous conditions, so the pH of the aqueous medium is well-controlled in the purification step.54 The conjugation reaction reaches completion within 2 min, at 10 mg mL−1 mAb concentration, demonstrating a high reactivity of the NHS ester towards lysine.
The resulting 4-(4-acetylphenoxy)butanoic acid (AcBut) linker is stable in physiologic buffers (pH 7.4) and the acid-labile hydrazone moiety releases calicheamicin in an acidic pH environment such as found in lysosomes and endosomes (pH 4–6).64,65 However, it is reported that even in human plasma, the hydrazone hydrolyses at a rate of 1.5–2% per day.66 The premature release of ozogamicin resulting from the instability of hydrazone67,68 is hypothesized to be a primary reason for the poor efficacy of Mylotarg, which was withdrawn from the market in 2010.
Whilst also employing an acid-sensitive N-acyl hydrazone linkage, the linker contains a reducible disulfide that is stable at physiologic pH but susceptible to nucleophilic attack from thiols. This disulfide linkage has been stabilized by two methyl groups to prevent premature release of calicheamicin.28 It breaks down selectively in tumour cells via a thiol–disulfide exchange reaction with thiol-containing glutathiones (GSH), which are elevated in cancer tissues (up to 1000-fold higher) due to stress conditions such as hypoxia.31 With these chemically labile groups, the AcBut linker is classified as cleavable.
In the production of T-DM1, the NHS-based linker is conjugated to the antibody first, and the DM1 covalently attaches to maleimide on the other side of the linker via a thioether bond formation (Scheme 4).11 The resulting conjugate exists in diastereomers, as two stereochemical configurations are possible with the C–S bond formed.
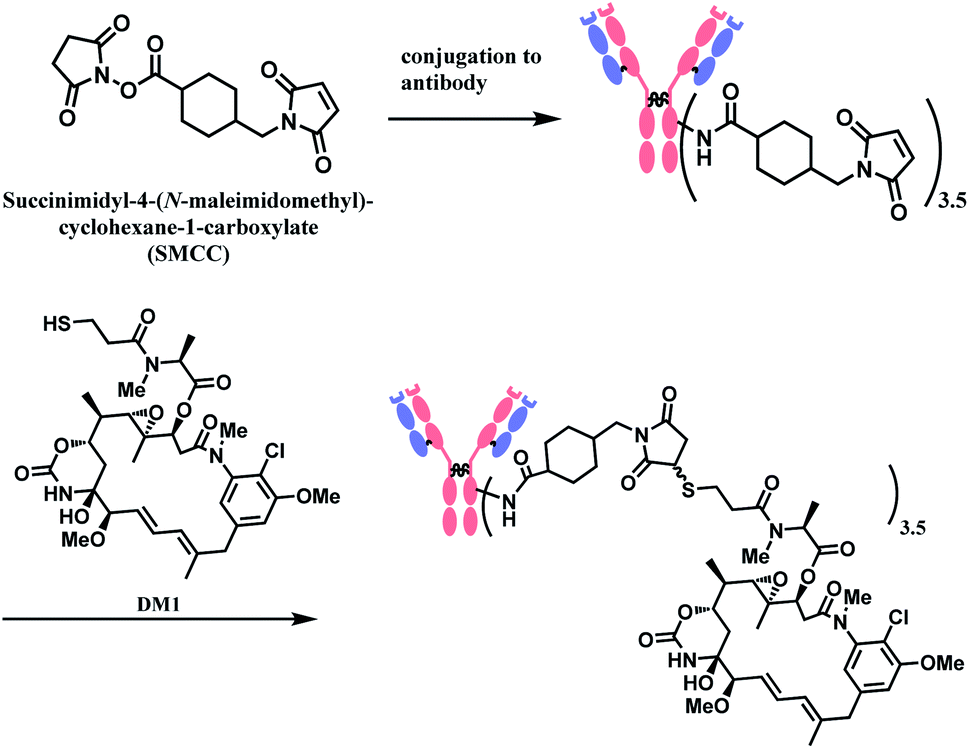 | ||
| Scheme 4 Conjugation of SMCC linker to antibody followed by DM1 attachment. | ||
NHS esters are observed to react with cysteines, serines, tyrosines, and threonines, not just lysines.69 The mass spectra analysis showed that they also carry out methionine oxidation, and deamidation at asparagine and glutamine residues.69 Not just the lack of chemoselectivity, their inherent issue is low regioselectivity. Therefore, research attempts on enhancing its regioselectivity are worth introducing. Weil and co-workers have achieved a more selective lysine by using a substoichiometric amount of NHS. With 0.5 equiv. of NHS relative to RNase A, only a mono-adduct was yielded, as only the most reactive and accessible residue reacted. However, such approach decreases the conjugation yield, and an optimal equiv. with each mAb should be first found out to efficiently use such approach.70
In 2019, Yamada and co-workers have developed a platform termed ‘AJICAP’ to achieve the regioselective modification of antibody even with NHS ester, through the use of Fc affinity peptide.71,72 A trastuzumab–DM1 conjugate was generated with an average DAR of 1.9.
Isothiocyanate is reported to be more stable than isocyanate in many literatures,78,79 and hence was used in constructing an antibody–radionuclide conjugate of 211At and CA12.10C12, an IgG1 isotype mAb.80 The conjugation was carried out in HEPES buffer (100 mM with 150 mM NaCl, pH 8.6) and required overnight stirring (Scheme 5). It has provided high labelling yields ranging from 72 to 92%. The isothiocyanato-conjugate was reported to have a lower renal toxicity as compared to its maleimido-conjugate analogue.
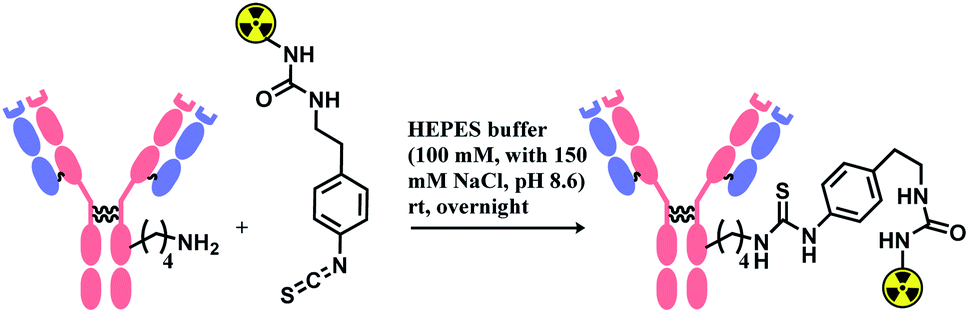 | ||
| Scheme 5 Construction of antibody–radionuclide conjugate using isothiocyanate linker. | ||
However, it should be noted that benzyl isothiocyanate has been recently reported as a cysteine-selective conjugation molecule.81 Keserü and co-workers have exploited the effect of reaction pH on the protonation states of amino acid residues; at pH 7.4–9.1, amino groups of lysine residues are protonated (–NH2 → –NH3+), and thus, thiol reactivity is higher.
 | ||
| Scheme 6 ‘Plug’ and ‘Play’ strategy for antibody conjugation with 4-azidobenzoyl fluoroide linker. | ||
AFB-antibody conjugation studies were performed on Trastuzumab monoclonal antibody with varying equivalence of ABF in PBS 1× M buffer (pH 7.3) at 25 °C for 18 h to conclude a 72% efficacy of this technique. This method displays superior efficiency to an NHS ester of 4-azidobenzoic acid (ABNHS) of 49% efficacy. This could be attributed to the better reactivity of ABF with the amine groups of lysine residue. Evidence of their relative kinetics suggest that the bioconjugation with benzoyl fluoride is faster than the electrophile hydrolysis, as shown by the second-order rate constant for benzylamine acylation with ABF and ABNHS to be k2 = 87.9 M−1 s−1 (t1/2 = 1.9 min) and k2 = 2.72 M−1 s−1 (t1/2 = 62 min) respectively. The efficiency of the conjugation with trastuzumab was further tested starting with the experiment of trastuzumab (1 mg mL−1) with ABF (4 equiv.) in PBS buffer 1× (pH 7.3). The trastuzumab–ABF (T–N3) conjugate then undergoes SPAAC conjugation with TAMRA-BCN (1.5 equiv. for every azide group). At a room temperature of 25 °C, it was demonstrated to have a rapid and near complete conversion of ABF. The completion of reaction was reported to be after 15 min with a degree of conjugation (DoC) of 2.9 (72% efficacy).
Later in the study, this conjugation strategy was performed on the drug monomethyl auristatin BCN derivative (MMAE-BCN) linked with a valine–citrulline linker. This SPAAC modification was done with T–N3 conjugate (DoC = 2.16) starting with 3 equiv. of ABF, which produced the following trastuzumab–MMAE conjugates with a maintained DoC of 2.10.
Besides RGD, the research group has attached zanamivir (Relenza™), a neuraminidase inhibitor, to the β-lactam moiety.84 They successfully formed a 38C2–zanamivir conjugate (DAR = 2) by incubating 4 equiv. of the functionalised linker with the mAb for 2 h in PBS buffer at room temperature (Scheme 7). The conjugate showed inhibitory activity (IC50 = 26.6 nM and 38.9 nM depending on x group) similar to zanamivir alone (IC50 = 16.6 nM). It was deemed that the mAb conjugation improved the binding with neuraminidase, as each 38C2 was loaded with two molecules of zanamivir, and the target enzyme is a tetramer. In the mice study, the 38C2-zanamivir conjugate had a significantly longer half-life of 72 h as compared to the zanamivir alone (10 min).
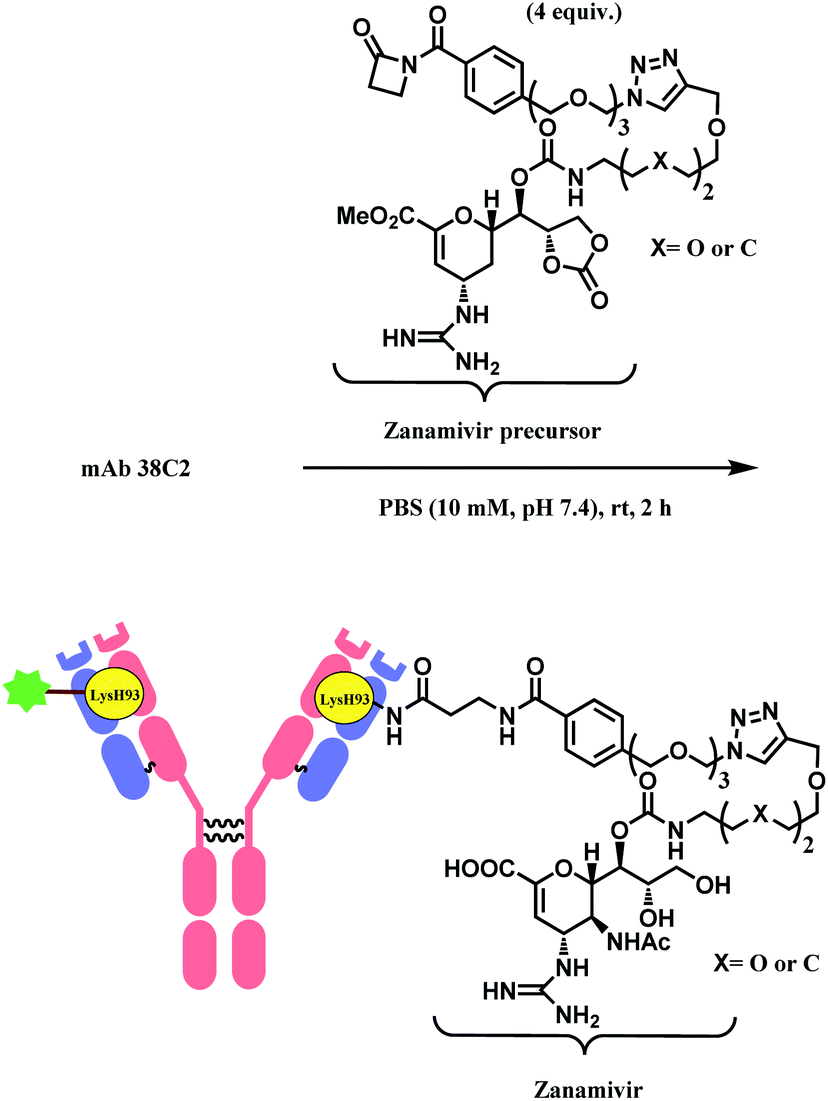 | ||
| Scheme 7 Formation of 38C2–zanamivir conjugate, which targets influenza neuraminidase. | ||
Rader and co-workers have used the β-lactam linker to conjugate 2 MMAF moieties to a lysine residue in a dual variable domain (DVD) format.85 The DVDs were produced by combining the variable domains of h38C2, which is an anti-hapten mAb, and trastuzumab. Just like the LysH93 on mAb 38C2, the DVD format has two catalytic lysine residues that are located at the bottom of 11 Å deep hydrophobic pocket, that are more nucleophilic (pKa ∼6). These two lysine sites were labelled with 4 equiv. of β-lactam-MMAF (two equivalents per Lys residue) in PBS buffer within 4 h (Scheme 8). No conjugation on lysines on light chain or conjugates with higher DAR were observed.
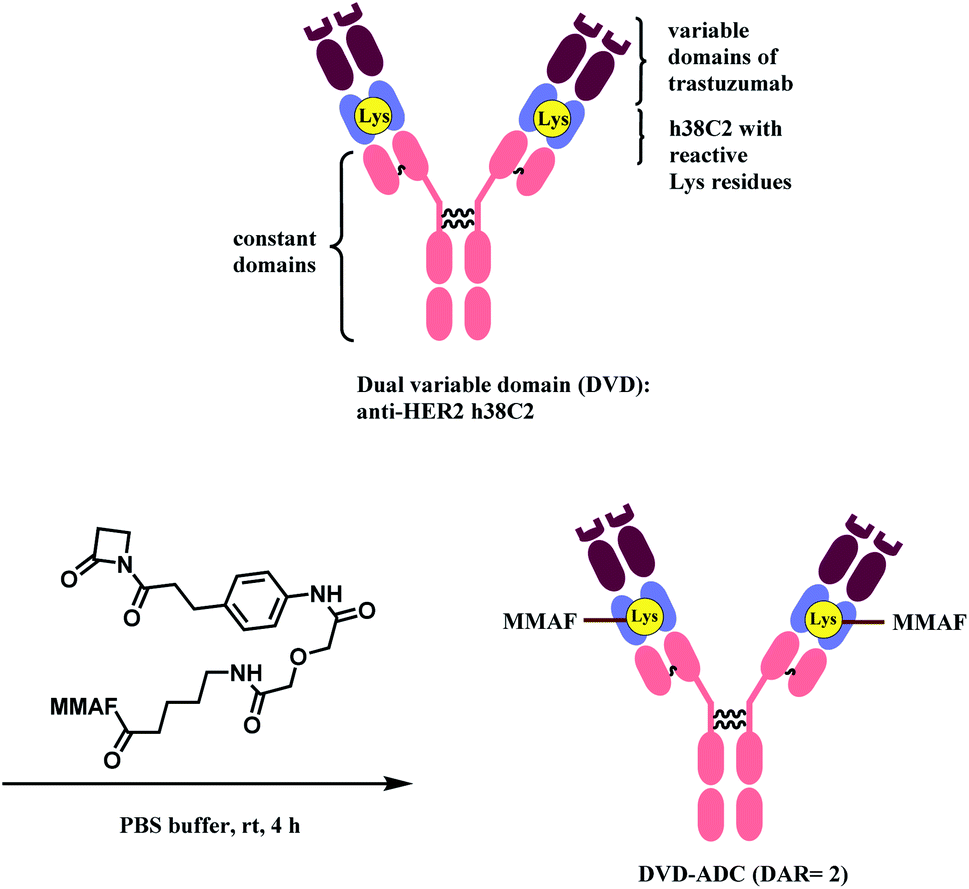 | ||
| Scheme 8 Conjugation of anti-HER2 DVD-ADC with MMAF via the β-lactam linker. | ||
The resulting DVD-ADC was evaluated against HER2+ (SK-BR-3) and HER2− (MDA-MB-468) breast cancer cells. It showed high potency against HER2+ cells (IC50 = 68 pM), with no reactivity towards HER2− cells. In addition, the cytotoxicity did not degrade even after incubation in human plasma at 37 °C for 3 days, demonstrating the high linker stability of the conjugate.
Followed by this work, Rader and co-workers expanded the use of β-lactam linker to various applications.86–88 In one report, instead of the DVD-IgG1 format (h38C2), they have used triple variable domain Fab (TVD-Fab) format that uses a tandem inner Fv domain based on h38C2 and generated a conjugate with DAR of 2.86 The TVD-Fab antibody carrier-MMAF conjugate had a circulatory half-life of 13.3 ± 2.5 h and deeper tumour tissue distribution as compared to the DVD-IgG1–MMAF conjugate. In another instance, with Alnylam Pharmaceuticals, they have generated a h38C2–siRNA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) conjugate via the β-lactam-based linker.89 The conjugate successfully targeted human β-catenin (CTNNB1) mRNA in multiple myeloma cells.
:2) conjugate via the β-lactam-based linker.89 The conjugate successfully targeted human β-catenin (CTNNB1) mRNA in multiple myeloma cells.
:1). They confirmed that the enzymatic activity of proteins was not affected by lysine labelling through the phospha–Mannich reaction.
The phospha–Mannich reaction was applied on several proteins containing multiple lysine residues, including the Fab fragment of trastuzumab, and consistently yielded single lysine-conjugated adducts with the conversion ratio 20–42%. It is noteworthy that carbon-based nucleophiles showed poor site-selectivity, in contrast to triethylphosphite. The resulting mono-adducts could be further functionalized via the oxime formation of the aromatic substituent of benzaldehyde. This strategy was used to produce an ADC of trastuzumab and doxorubicin with the DAR of 0.92 (Scheme 9). However, the exact site of conjugation in the trastuzumab was not definitively given. In addition, unlike the Fab fragment conjugation, an extended reaction time of 8 h was required to modify the lysine residues of the full antibody. An additional 14 h was further needed for the post-functionalization.
 | ||
| Scheme 9 Synthesis of trastuzumab–doxorubicin conjugate using Phospha–Mannich reaction. | ||
 | ||
| Fig. 9 Structure of TAK-242. | ||
TAK-242 contains a cyclohexene group where aza-Michael addition reaction occurs, and it features a sulfonamide moiety, which is found to be an important trigger for conjugation. Among several analogues, 2,4-difluorophenyl sulfonamide had the highest reactivity (Scheme 10). In human plasma, 2 equiv. of TAK-242 derivative (50 μM) was reacted with a recombinant HSA at 37 °C within 2 h.
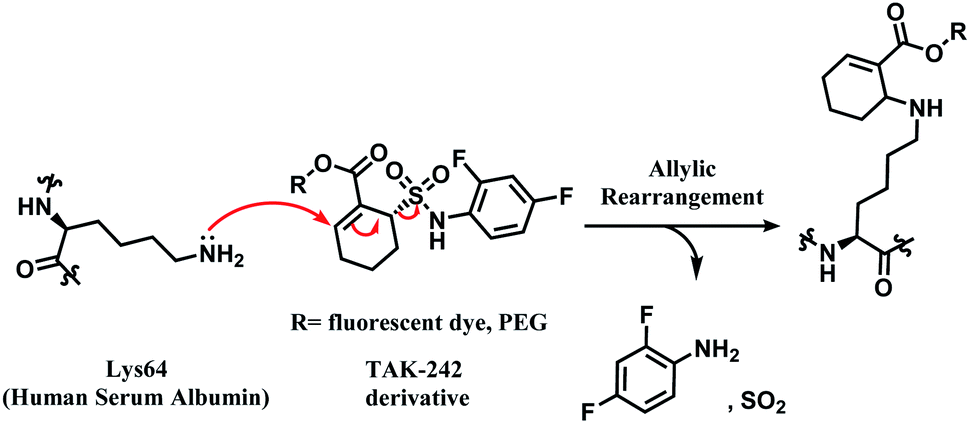 | ||
| Scheme 10 Proposed mechanism of the conjugation reaction between HSA Lys64 and TAK-242 derivative. | ||
TAK-242 derivative was incubated with lysine-blocked HSA, and showed no reaction with cysteines, further establishing a high chemoselectivity towards lysine. Barbas and co-workers later used this conjugation strategy to establish a synthetic method for homogenous antibody conjugate production.92 A fusion protein of trastuzumab and HSAdIC34S, the domain I of HSA, was generated using a (GlySer)3 spacer. Then, a TAK-242 derivative (R = rhodamine dye) was reacted with the Lys64 of the HSA. No reaction between TAK-242 derivative and lysine or N-termini on trastuzumab was observed, but a longer reaction time was required than previously reported for HSA alone.91
Although this strategy was only limited to HSA, not antibodies, this work is highly noteworthy since it is the first reported regioselective and chemoselective lysine labelling, to our knowledge. A linker molecule that is only reactive towards a certain lysine residue among many opens a new approach in selectivity control for the ADC chemical conjugation technology.
This conjugation reaction occurs rapidly within 1–2 h and, with only 1 equiv. of the designed sulfonyl acrylate reagent at near physiological conditions of 37 °C and pH 8.0. At this pH, the proportion of neutral, nucleophilic lysines was minimal. This was done without having to alter the proteins genetically preceding lysine conjugation or targeting specific residues located in a hydrophobic pocket. Post-functionalisation to the resulting methyl acrylate moiety on the conjugate was easily achieved via aza-Michael reactions, while preserving their original secondary structures and functionalities.
Lysine conjugation studies were performed on rHSA, C2Am, Lysozyme, Annexin-V, and Trastuzumab. Remarkably, all the proteins were singly modified on the lysine with the lower pKa. Further modification of the conjugated trastuzumab–acrylate conjugate (10 μM) with crizotinib anticancer drug in 1000 equivalence (10 mM) to completely generate the respective antibody–drug conjugate (Scheme 11). Cell-line studies with the trastuzumab–crizotinib conjugate on both SKBR3 cells and HEK293 cells indicated a conserved functionality of its antigen binding properties. This is as observed of its specificity for SKBR3 cells with high Her2 antigen expression, and unlike HEK293 cells with low expression of HER2 antigen.
 | ||
| Scheme 11 Preparation of trastuzumab–crizotinib conjugate using sulfonyl acrylate linker. | ||
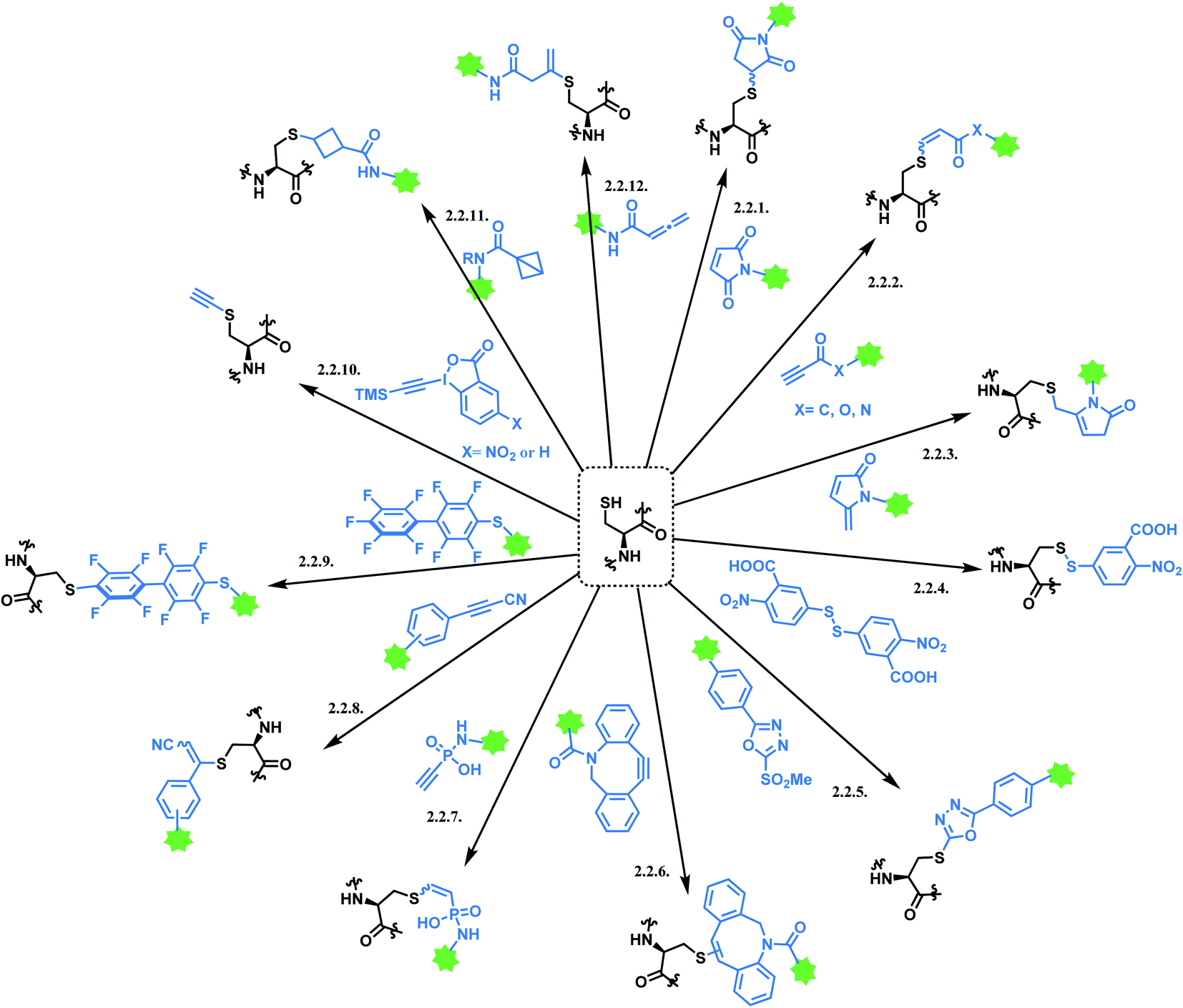
2.2. Cysteine: single thiol functionalisation
Cysteine is the most commonly targeted amino acid in current ADC clinical developments. 7 out of 11 FDA-approved ADCs exploit a cysteine conjugation strategy. The following aspects make cysteine an attractive target. Firstly, its nucleophilicity is distinguished from other amino acids, which facilitates a chemoselective conjugation. Hard and Soft, Acids and Bases (HSAB) principles by Pearson classify sulfur-based nucleophiles as soft, relative to the nitrogen-based nucleophiles in ε-amine of lysine or N-terminus with higher charge density.94 The theory suggests that soft nucleophiles thermodynamically prefer to associate with soft electrophiles, and so are hard nucleophiles and hard electrophiles. Indeed, a range of Michael acceptors (soft electrophiles) have shown to selectively conjugate to cysteine residues of protein. Furthermore, under neutral conditions, the thiol group of cysteine (pKa = 8) is more acidic than other common nucleophiles in proteins, such as the ε-amine of lysine (pKa = 10). Secondly, its relative rarity compared to lysine allows a better control of DAR in the ADC production. In practice, interchain disulfide bonds of antibodies are reduced by agents such as TCEP or DTT.95 Both IgG1 and IgG4 mAbs have four interchain disulfide bonds in total, so reducing them generates 8 thiol moieties in each mAb.96However, the issue of regioselectivity (selectively targeting one –SH among 8 thiols) still remains, and there seems to be mixed conclusions around the effect of disulfide reduction on the ADC stability.97 An overall summary of the reported cysteine bioconjugation methods is shown in Scheme 12.
 | ||
| Scheme 12 Cysteine conjugation methods. | ||
 | ||
| Fig. 10 Maleimide-based linkers in FDA-approved ADCs; (A) MC linker used in Belamaf; (B) MC-VC-PABC linker used in BV, EV, and PV; (C) MC-GGFG-AM linker used in T-Dxd; (D) CL2A linker in SG; and (E) maleimide-VA-PABC linker in LT. | ||
In a large-scale preparation of anti-Notch 3 ADC and BV (Adcetris™),98,99 6 equiv. of maleimide linker-payload is reacted with mAb (anti-Notch3 mAb or brentuximab) within 1 h, in DMSO (11% v/v) and pH 7.0 buffer to afford a yield of 85–88% with an average DAR of 3.6. Maleimide is able to react with a cysteine thiol in mAb in a neutral aqueous solution rapidly with a good yield, in large-scale production.
However, the maleimide–thiol reaction generates two diastereomers, not a single product (Fig. 11). The resulting diastereomers make the profile of their chromatograms complicated,100 and readily undergo isomerisation to each other.101
 | ||
| Fig. 11 Two diastereomers of the thiosuccinimide adduct. | ||
In addition, the resulting thiosuccinimide adduct is reported to be prone to deconjugation via a retro-Michael reaction,102 or to undergo ring-opening by hydrolysis.103 The retro-Michael reaction leads to premature drug release in blood plasma. Regenerated maleimide linker-payload then can rapidly react with other thiol groups in blood, such as human serum albumin (HSA) or glutathione (Scheme 13).50 The characterization study conducted by Pfizer further supports this in vitro study; trastuzumab–mc-vc-PABC-drug was incubated with human plasma for 144 h, and almost 100% of the DAR loss happened.104 Intact protein mass analysis showed that at the 144 h time point, the mass of albumin in human plasma had an additional 1347 Da over the native albumin extracted from human plasma, exactly matching the mass of the maleimide linker-payload. Such deconjugation and exchange reaction results in off-target toxicity, and reduced therapeutic efficacy, as less than desired amounts of cytotoxins get delivered to tumour cells.
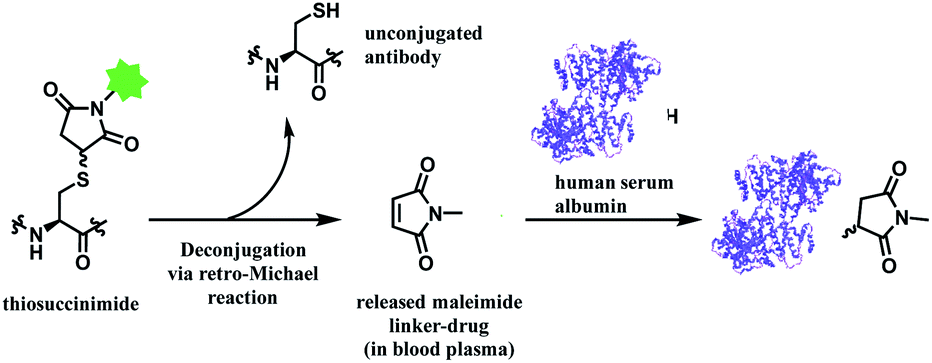 | ||
| Scheme 13 Deconjugation of thiosuccinimide via retro-Michael process and subsequent attachment of maleimide on human serum albumin (pdb: 1AO6). | ||
Although the hydrolysis of maleimides was shown to improve conjugate stability and generates ADCs with increased in vivo potency, the occurrence of incomplete hydrolysis of many maleimides may limit this approach.
Deconjugation can be attenuated by hydrolysis of the thiosuccinimide ring to the succinamic acid derivatives. Hydrolysis leads to heterogenous mixtures of two succinamic acids, each existing in diastereomers (Scheme 14). Once the ring-opening by hydrolysis occurs, exchange reactions with HSA or glutathione no longer takes place,105 stabilising the conjugate and thus avoiding drug loss. Companies such as Roche attempted to promote hydrolysis by using engineered antibodies with unnatural conjugation site50 or by incorporation of an amine group on the N-alkyl spacer.106
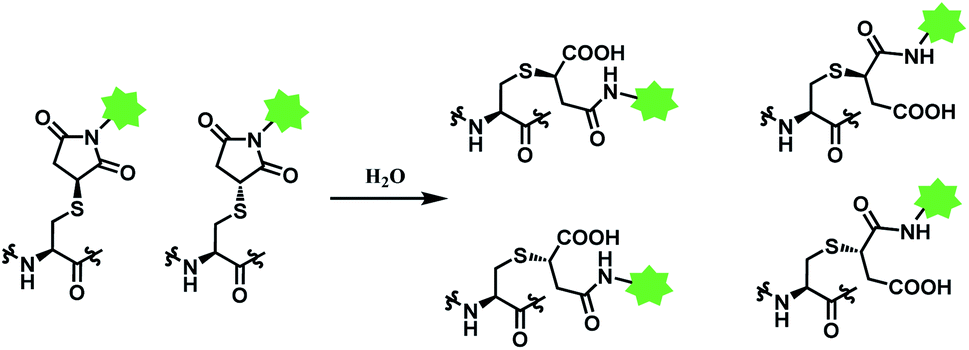 | ||
| Scheme 14 Hydrolysis of thiosuccinimide conjugate. | ||
Promoting hydrolysis, however, does not solve the fundamental issue of having heterogenous mixtures in vivo. Maleimide-based ADCs administered to patients therefore consist of both less potent low drug-loaded species and high drug-loaded species with hydrolysed linkers, resulting in suboptimal therapeutic indexes.
A side reactivity of maleimide with the imidazole group of histidine is observed.107 In addition, maleimide undergoes an irreversible reaction with the reducing agent TCEP to form an ylene adduct.108
:1). While alkynones have the highest reactivity, they have the least stereoselectivity; the Z/E ratios of alkynoic amides, alkynoic esters, and alkynones are 22:1, 8:1, and 5:1 respectively. The electron-withdrawing ability of the activating group leads to the isomerisation of the carbon–carbon double bond of the vinyl sulfide via intermediate such as allenol and ketene hemiacetal. The resulting vinyl sulfide conjugate is susceptible to the nucleophilic attack of other thiol groups (Scheme 15B).
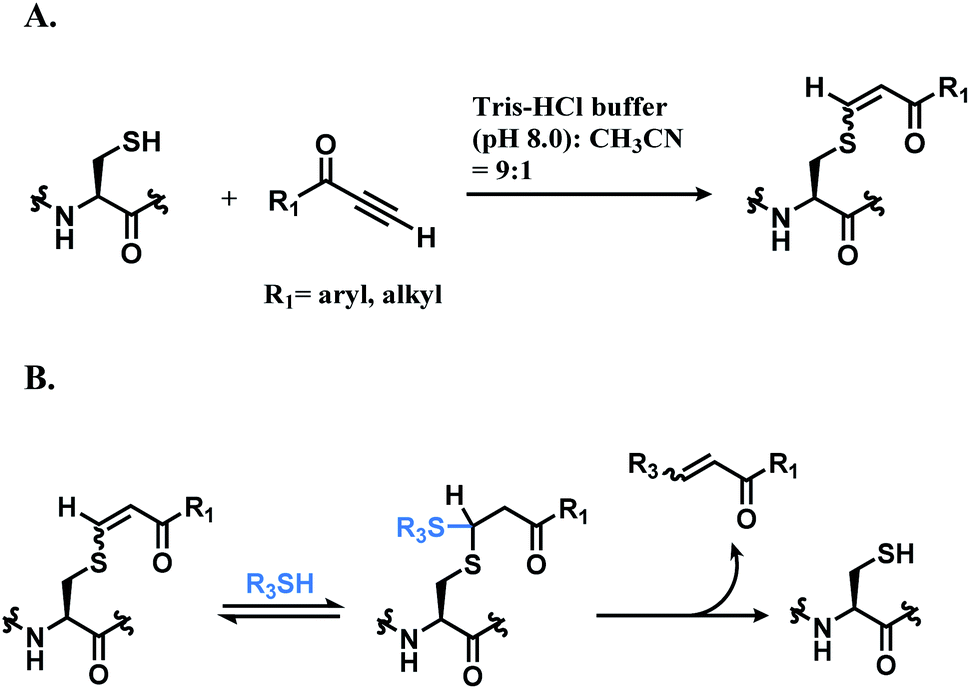 | ||
| Scheme 15 Conjugation of alkynone linker with cysteine; (A) formation of regioisomeric mixtures of vinyl sulfide; (B) secondary addition of another thiol group followed by deconjugation. | ||
In 2019, Vilarrasa and co-workers have reported N-substituted propynamide as a stereoselective, cysteine-selective linker (Scheme 16).110 With the co-solvent system of H2O and tBuOH (1:1 v/v), the resulting vinyl sulfide was mostly Z-isomer (Z/E = >99:1). The conjugate was more resistant towards hydrolysis or secondary thiol addition, than the counterpart formed with alkynoic ester linker.
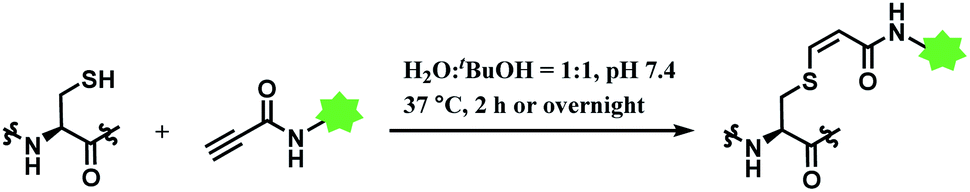 | ||
| Scheme 16 Labelling cysteine residue with (Z)-oxopropene-1,3-diyl linker. | ||
The alkynoic amide linker selectively labelled cysteines on reduced oxytocin and glutathione, at concentrations of the reactants of ≤0.1 M. The linker was inert towards amino groups, even at pH 10.0 or pH 12.0. With glutathione, around an equimolar amount of the propynamide linker afforded the adduct with the yield >95% within 2 h, at pH 7.4 and 37 °C. With oxytocin, a nonapeptide with two cysteine residues, an overnight stirring at 37 °C was required for labelling.
The thiol–5MP conjugate does not undergo ring-opening hydrolysis, in contrast to the thiol–maleimide conjugate (shown in Scheme 17). However, the thiol–5MP conjugate underwent retro-Michael reaction at pH 9.5 or thiol-exchange at pH 7.5, just like the maleimide-modified conjugate.
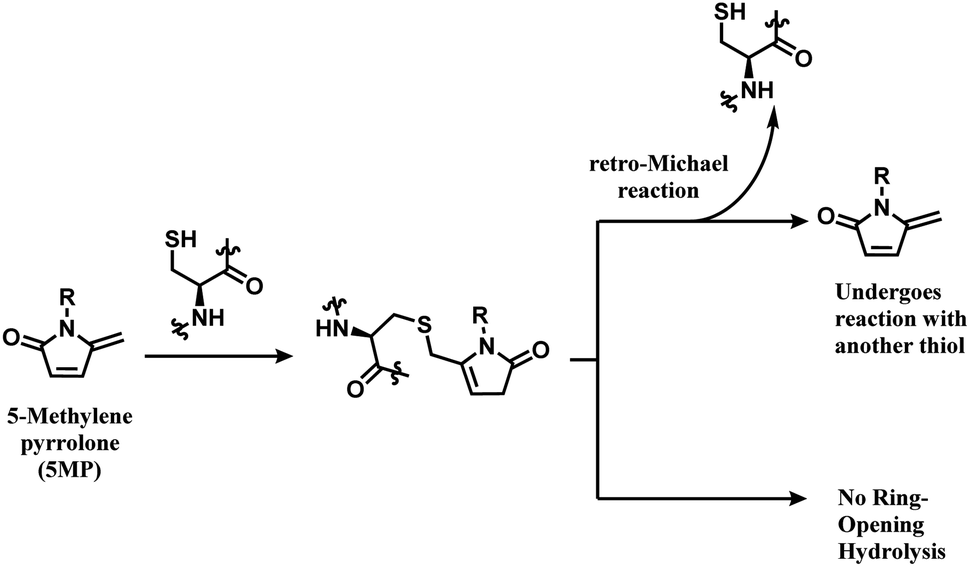 | ||
| Scheme 17 5MP conjugation to cysteine and the resulting adduct's stability. | ||
The research group later found that having a bromine atom at 3-position of the 5MP molecule could also serve as a thiol modification agent.112 The efficiency and specificity of the 3-bromo-5-methylene pyrrolone (3Br-5MP) in a single thiol modification, relative to maleimide was tested with a peptide with only one cysteine. It showed a comparable reaction efficiency to maleimide, where 2 equiv. of 3Br-5MP gave cysteine-modified products in more than 96% yield within 5 min in pH 7.5 buffer at both 37 °C and 4 °C. The 3Br-5MP showed a higher cysteine-specificity compared to maleimide. It did not show any reactivity towards N-terminal amino acids, when maleimide did. The 3Br-5MP also serves a disulfide rebridging conjugation molecule and such reactivity is explored in Section 2.3.3.
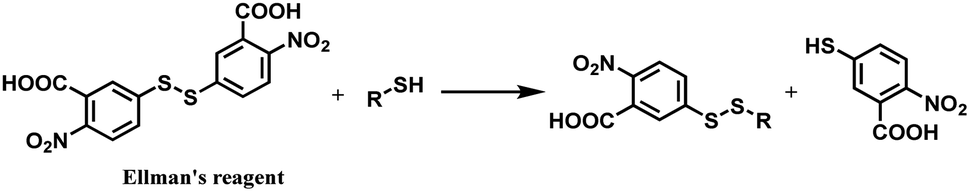 | ||
| Scheme 18 Thiol–disulfide exchange of DTNB. | ||
In 2012, Neri and co-workers have used the DTNB reagent as a linker to construct an ADC.114 This ADC was designed to release the cytotoxic drug, a thiol-containing cemadotin analogue (CemCH2-SH), into the extracellular region. Human antibody F8 was selected to target tumour cells and was used in a small immune protein (SIP) format. The disulfide bond between the C-terminal cysteine residues of the SIP(F8) antibodies was first reduced using TCEP before the thiol groups were activated by Ellman's reagent. After CemCH2-SH was synthesized, SIP(F8)-Ellman's antibodies were incubated with CemCH2-SH (10 equiv.) in PBS (pH 7.4) at room temperature for 10 min to yield the desired ADC, SIP(F8)-SS-CH2Cem, with 50% yield. In addition, the ADC was stable when it was stored at −80 °C and 4 °C and was still present in mouse plasma after incubation for 72 h at 37 °C (Scheme 19).
 | ||
| Scheme 19 Generation of ADC, SIP(F8)-SS-CH2Cem. | ||
In 2014, the research group used the Ellman's reagent to design a new immunocytokine drug conjugate (IDC) known as F8-IL2-SS-DM1, by linking a F8-IL2 fusion protein to DM1 maytansinoid.115 After this new IDC was administered to the mice that were bearing subcutaneously grafted F9 and C1498 tumours, F8-IL2-SS-DM1 was observed to selectively accumulate in the tumours and also showed stronger tumour growth inhibition than the single-payload immunocytokine F8-IL2.
:1) with good yields of 78–99%. It is noteworthy that the reaction conversion was lower when pure water was used instead of the buffer. The resulting thioether conjugate was shown to have higher stability in varying pH levels as compared to a maleimide–cysteine conjugate. It did not have a thiol-exchange with glutathione.
A functional payload could be loaded on the linker as shown in the Scheme 20, and 10 equiv. of the azide-functionalised phenyloxadiazole sulfone specifically labelled Cys34 of human serum albumin (HSA), and a single cysteine on maltose-binding protein (MBP-C-HA), in pure PBS, within 2 h. A conjugate of fluorescein and MBP-C-HA linked via PODS had a superior stability in human plasma (t1/2 = 117 h) to the maleimide-linked conjugate (t1/2 = 59.9 h).
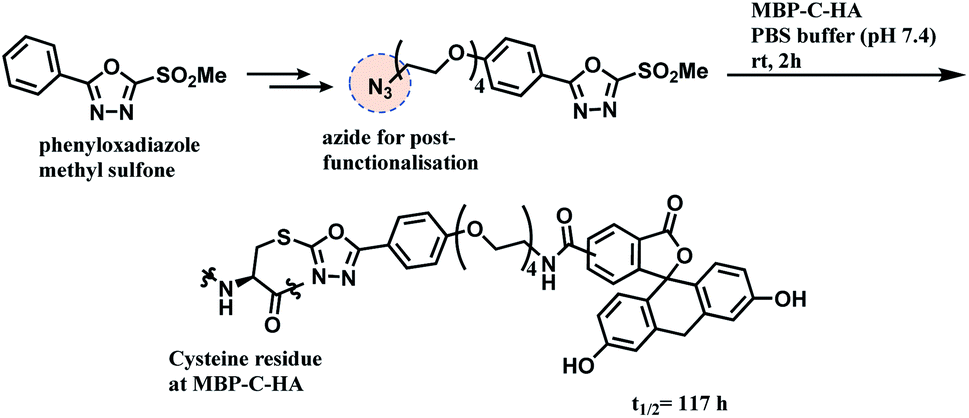 | ||
| Scheme 20 Conjugation of MBP-C-HA with phenyloxadiazole sulfone (PODS) linker. | ||
In 2019, Zeglis and co-workers adopted this linker to generate a conjugate of trastuzumab and a radionuclide chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA).118 The antibody was incubated with 10 equiv. of PODS–DOTA for 2 h at room temperature and yielded the conjugate with 80% yield. The average degree of labelling was found to be ∼1.8 DOTA/mAb (Scheme 21). The resulting conjugate exhibited immunoreactivities similar or better relative to the counterpart conjugate prepared by using maleimide linker.
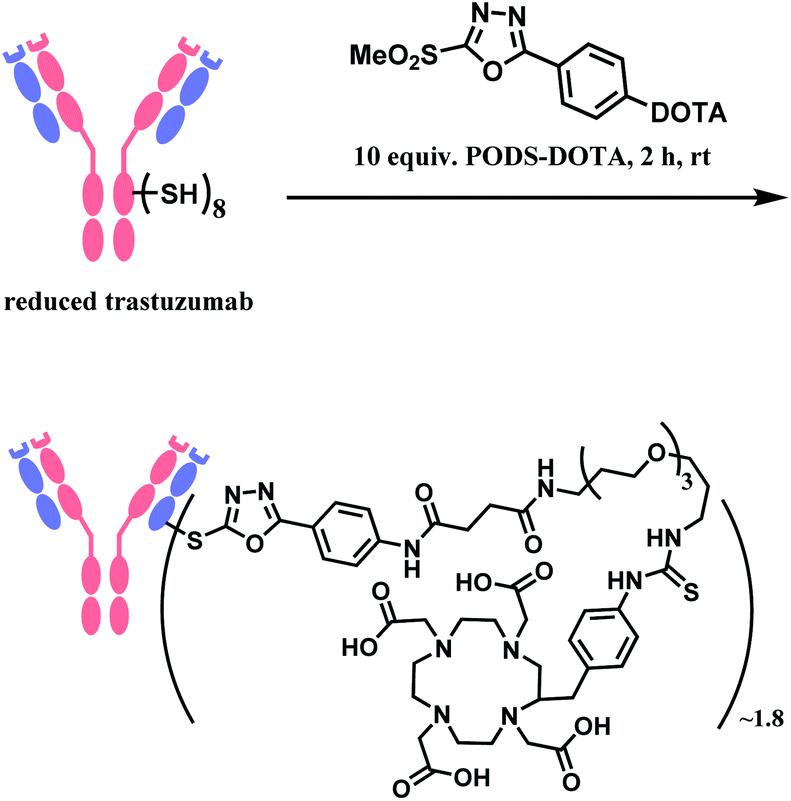 | ||
| Scheme 21 Conjugation of trastuzumab with PODS–DOTA. | ||
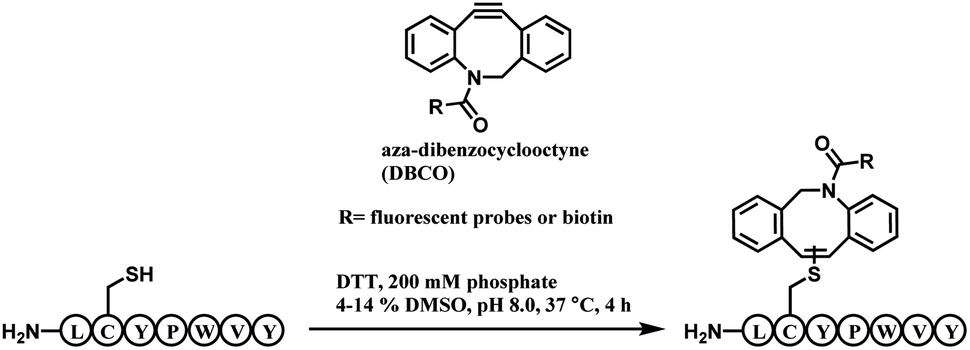 | ||
| Scheme 22 DBCO-tag mediated thiol–yne reaction. | ||
A unique heptapeptide sequence Leu–Cys–Tyr–Pro–Trp–Val–Tyr (LCYPWVY) conjugated with cyclooctyne rapidly with a rate constant of 0.81 ± 0.02 M−1 s−1 in pH 8.0 phosphate buffer at 37 °C. Other peptide sequences do not react as rapidly or are inert to DBCO (ex. Phe–Cys–Pro–Phe), allowing a selectivity control. The researchers produced a trastuzumab–biotin conjugate by incorporating the peptide tag onto the C terminus of the heavy chains of trastuzumab and reacting it with DBCO-(PEG)4-biotin. The reaction only generated 90% heavy-chain mono-adduct and no reaction between liberated thiols and cyclooctyne was observed. The resulting conjugate exists in isomers generated from addition of the thiol to either one of the two alkyne carbons.
 | ||
| Scheme 23 Phosphonamidate electrophile for cysteine bioconjugation; (A) Staudinger-phosphonite reaction (SPhR) to generate electron-poor ethynylphosphonamidate linker; (B) generation of ADC using ethynylphosphonamidate linker via thiol addition. | ||
The linkage stability of the biotin-modified antibody was compared to the maleimide adduct in a head-to-head fashion in bovine serum albumin (BSA) solution under physiological conditions. A significant transfer of the biotin to BSA was observed for the maleimide linkage whereas the phosphonamidite linkage was stable under the tested conditions. The P–N bond hydrolysis only happened under the extreme condition of 24 h incubation at pH 0.
In 2019, the research group expanded the scope by producing a structural analogue of brentuximab vedotin (BV, Adcetris™) by substituting the maleimide linker with the phosphonamidate linker (Scheme 23B).122 Its in vivo activity was tested in a Karpas 299 derived tumour xenograft model in immunodeficient female CB17-SCID mice. A superior stability of the phosphonamidate linker allowed the lower dosing of ADC to be still active, with a prolonged drug delivery in circulation. The BV analogue with the phosphonamidate linker had a median survival of 48 days, whereas the FDA-approved BV had 21 days.122
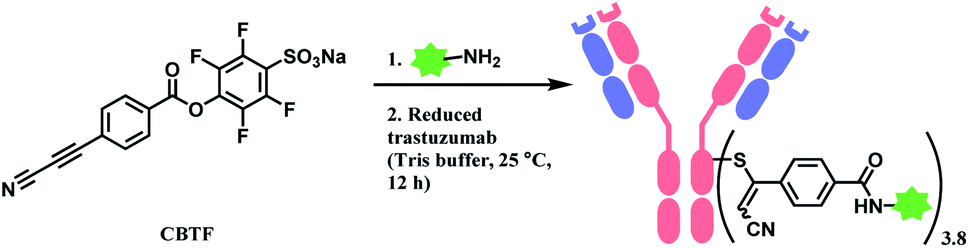 | ||
| Scheme 24 Labelling cysteine residues on trastuzumab with CBTF bifunctional linker containing APN moiety. | ||
In terms of the stability of the APN-cysteine adduct, no degradation was observed in aqueous medium of pH 0–14, in the presence of excess nucleophiles such as thiophenol, cysteine, thioethanol and glutathione (100–1000 equiv., 10–100 mM), as well as reducing agents that include TCEP or DTT (0.1–1 M) at pH 7.4. However, the FRET-based APN conjugate was observed to be less stable when it was incubated at 37 °C in human plasma and when it was incubated for different times in living cells. Nonetheless, the FRET-based APN conjugate displayed a higher stability compared to the FRET-based maleimide–thiol conjugate. Notably, the addition of a cysteine derivative to APN produces a stereoisomeric product mixture that contains mostly Z-isomers.
In the chemoselectivity assessment of APN, APN–TMPP was attached to tris(2,4,6-trimethoxymethyl)-phosphonium (TMPP), a highly responsive MS tag. The conjugation of the APN-TMPP probe to a peptide increases retention time in LC-MS. Experiments to prove the chemoselectivity of APN for cysteine were carried out by reacting APN-TMPP with tryptic digest at a 200:1 molar ratio of APN-TMPP to protein at 25 °C for 1 h. The peptide mixtures before and after tagging were analysed with LC-MS. It was found that all peptide mixtures with cysteine-containing peptides reacted with APN-TMPP probes since these mixtures had a higher retention time after tagging. On the other hand, mixtures that contained cysteine-free peptides did not react with APN-TMPP and thus did not exhibit any change to their retention time.
Followed by this initial discovery, Wagner and co-workers have developed a heterobifunctional linker, sodium 4-((4-(cyanoethynyl)benzoyl)oxy)-2,3,5,6-tetrafluoro benzene sulfonate (CBTF), where one end is cysteine-selective APN and the other end is amino-reactive activated ester (Scheme 24).124 The research group reacted CBTF with TAMRA-NH2 and later conjugated the resultant product to reduced trastuzumab. The CBTF conjugated antibodies yielded DAR values of 1.9 and 3.8 when the antibody was reduced with 1.1 and 2.2 equiv. of TCEP. For comparison, SMCC was used in place of CBTF as the bifunctional linker, the SMCC conjugates provided DAR values of 2.3 and 3.9 respectively. In addition, CBTF conjugates were observed to be more stable in human plasma compared to SMCC conjugates. The toxicity test of a CBTF model linker on HeLa and HuH-7 cell lines revealed that the linker did not show any toxicity up to 100 μM.
Even when a lower equivalence of cysteine derivatives was used, disubstitution was observed, specifically at 1,4 positions. It is proposed that the stabilisation of the substituted intermediate by sulfur, favours a second thiol substitution in the para-position. A similar reactivity was observed with decafluorobiophenyl species. Chemoselectivity of the perfluoroaromatic linker was assessed by incubating it with model peptide systems containing various nucleophilic amino acid residues such as histidine and lysine, and was shown that no reactivity was observed between amino acids other than cysteine.
However, the reactivity of perfluoroarene is significantly lower in water, limiting its application to bioconjugation. In order to adapt the method to aqueous media, the research group has used a promiscuous enzyme, glutathione S-transferase (GST) along with the perfluoroaromatic reagents.126 GST is a cytosolic enzyme that catalyses conjugation reactions between the Cys residue of glutathione (GSH, γ-Glu–Cys–Gly) and various electrophiles. With the presence of 5–10 mol% of GST, the quantitative labelling of cysteine in GSH with the perfluorobiphenyl moiety was achieved within 30 min in pH 8.0 phosphate buffer at 37 °C. No reactivity was observed in the absence of GST. Due to the nature of enzyme (GST)-substruate (GSH) interaction, this conjugation method is GSH-exclusive, where other Cys residues in different peptides or the thiol in 100-excess of 4-mercaptophenylacetate were not conjugated. Applying this strategy for the construction of ADC would therefore require an incorporation of GSH motif into antibody.
In 2016, the research group developed a specific four-amino-acid sequence (Phe–Cys–Pro–Phe), dubbed as ‘π-clamp’, that promotes the SNAr reaction in water, without the aid of GST.127 The tetrapeptide sequence was inserted into the C termini of the heavy chains of trastuzumab, C225 antibody, and the N-termini of Sortase A, and payload-perfluoroaryl compounds facily formed adducts. The MMAF loading on trastuzumab was carried out in 0.2 M phosphate with 5% DMSO, 20 mM TCEP for 16 h at 37 °C (Scheme 25). Proteins without the π-clamp showed no reactivity towards perfluoroaryl-payload, highlighting a high site-selectivity of this conjugation strategy. A conjugate of trastuzumab and MMAF connected via π-clamp and decafluorobiphenyl linker selectively killed BT474 cells (HER2 positive) but showed no cytotoxicity towards CHO cells (HER2 negative), demonstrating that the π-clamp conjugation does not affect the binding ability of trastuzumab.
 | ||
| Scheme 25 Construction of ADC of trastuzumab and MMAF via π-clamp and decafluorobiphenyl linker. | ||
Among all the TMS–EBX reagents, the one with an electron-withdrawing NO2 substituent showed the highest reactivity towards thiols. Many different tetrapeptide systems containing nucleophilic residues were reacted with 2 equiv. of nitro-substituted TMS–EBX in 0.2 M pH 8.2 PBS buffer at 37 °C for 15 min, in order to assess the chemoselectivity of this method (Scheme 26). Steric hindrance by branched hydrocarbon chains of isoleucine or valine did not slow down the reaction, and the TMS–EBX showed selective reactivity towards cysteine in the presence of serine, lysine, tryptophan or histidine, producing the ethynylated cysteine with >90% yields. However, undesired side-reactivity was observed with arginine and tyrosine.
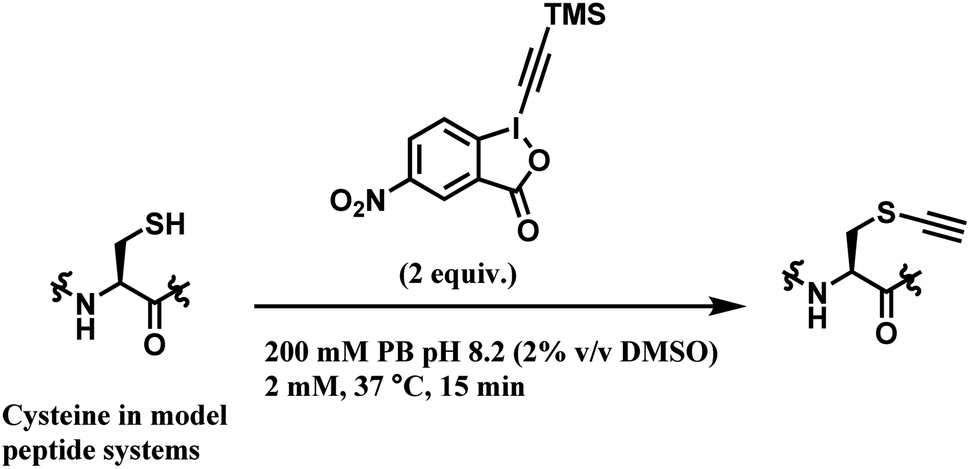 | ||
| Scheme 26 Cysteine conjugation with TMS–EBX. | ||
When applied to disulfide-reduced trastuzumab, a complex mixture was formed after the CuAAC step, suggesting a partial degradation of the antibody or side-reactions of ethynylation or cycloaddition. When a non-reduced trastuzumab was reacted with nitro-substituted TMS–EBX, the antibody degraded even in the absence of liberated thiols and the formation of a terminal alkyne was not observed. Without the nitro-substituent, side-reactivity was decreased. 2 equiv. of TMS–EBX gave the average degree of conjugation (DoC) of 0.8, and with 16 equiv., the average DoC of 4.4 was achieved. The conversion rate and the average DoC was dependent on the pH, where pH < 7.5 led to suboptimal DoC and yield. The antibody subsequently is reacted with an azide-functionalised TAMRA probe via CuAAC (Scheme 27).
 | ||
| Scheme 27 Labelling trastuzumab with TAMRA with TMS–EBX reagent. | ||
The thiol group undergoes strain-driven nucleophilic addition with BCB, yielding diastereomeric mixtures of 1,3-disubstituted cyclobutane (cis:trans ratio of ∼7:3). Its reactivity towards cysteine was only studied with glutathiones (GSHs), in pH 7.2 0.1 M potassium phosphate buffer at 37 °C. The reaction rates were significantly slower than those of acrylamides, where first-order reaction kinetic analysis showed that the half-time (t1/2) of phenyl BCB carboxyl amide was 32.7 h, whereas that of crylamide analogue (N-phenylacrylamide) was 1.2 h.
The chemoselectivity of BCB carboxylic amide towards cysteine was confirmed by reacting it with other nucleophilic amino acids (Lys, Ser, His, Tyr, and Trp) in 0.1 M K2CO3 DMF/H2O. No reaction was observed even under such basic conditions.
The reactivity of cysteine thiols in proteins with allenamide and the stability of the formed conjugates under peptic digestion conditions were also tested using bovine serum albumin (BSA) proteins by incubating the protein with benzyl allenamide at 37 °C for 30 min. The cysteine residues were successfully conjugated by the allenamide. The research group also showed that labelling probes fluorescein isothiocyanate (FITC), Cy3 and Cy5 dyes that were modified with allenamide handles were capable of labelling BSA.
The allenamide–thiol click reaction was adopted by many research groups as a robust, cysteine-targeting conjugation linker.133–135 In 2016, Roush and co-workers have prepared a conjugate of selenocysteine-incorporated mAb (SELENOMAB) and fluorescein with allenamide and phenyloxadiazole sulfone (Section 2.2.5) linkers.136 The reduced SELENOMAB reacted with fluorescein-allenamide within 2 h at room temperature. In human plasma at 37 °C, and SELENOMAB–allenamide–fluoroscein conjugate had the half-life of over 28 days whereas SELENOMAB–PODS–fluoroscein conjugate had a half-day of 12 h. These results demonstrate allenamide linker's high resistance against hydrolysis, thiol–disulfide exchange or deconjugation. In the in vitro test with SK-BR-3 breast cancer cells, it was shown that the fluoroscein conjugation with the allenamide linker did not degrade the binding ability of the antibody towards HER2 receptors.
2.3. Cysteine: disulfide functionalisation
A bis-reactive linker can undergo reactions with both thiol residues, which are produced by reducing a disulfide bond in antibody. Such method has a competitive advantage in controlling DAR near to 4 when modifying IgG1 and IgG4 antibodies, as they have four reducible interchain disulfides. Moreover, such bioconjugation strategy reanneals the cleaved disulfide bridges between antibody chains via covalent bonds. OBI-999, a clinical ADC that is currently being studied in phase I/II for advanced solid tumours, uses such disulfide rebridging linker.137 The linker in OBI-999 is bissulfone moiety-based, and is explored in Section 2.3.1. The DAR of OBI-999 was well-controlled to be 4 and showed good pharmacokinetic profiles.138However, one drawback of disulfide rebridging linkers is the formation of ‘half-antibody’. It is formed by intrachain bridging of the hinge region in heavy chain cysteines (Scheme 28).
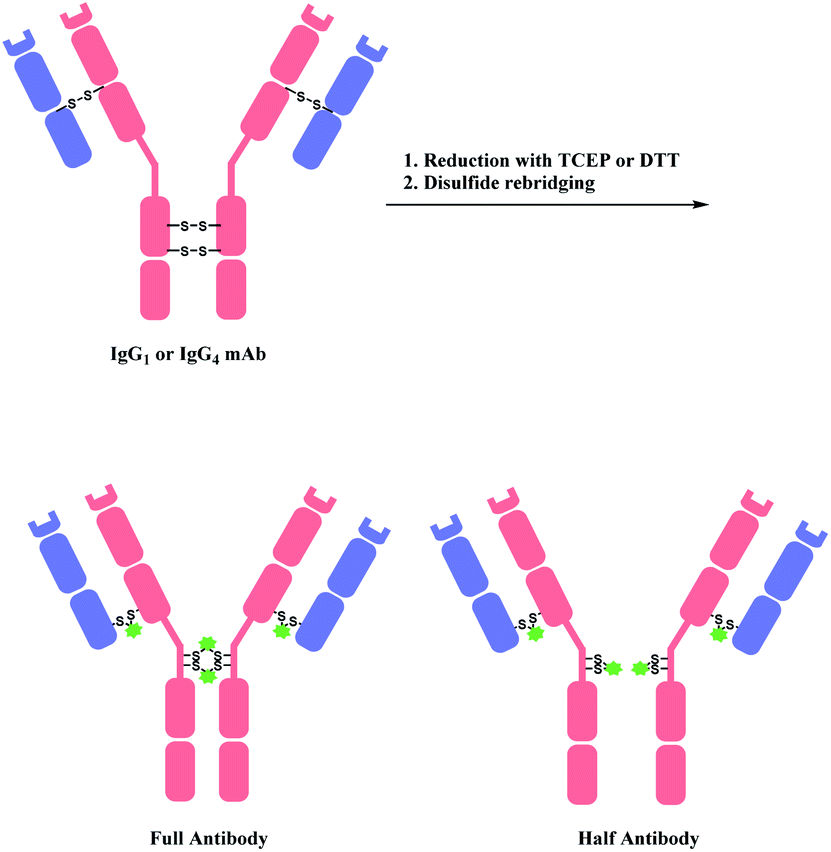 | ||
| Scheme 28 Disulfide reduction followed by rebridging with linker, and the formation of undesired half-antibody. | ||
An overview of reported disulfide rebridging linker molecules is shown in Scheme 29.
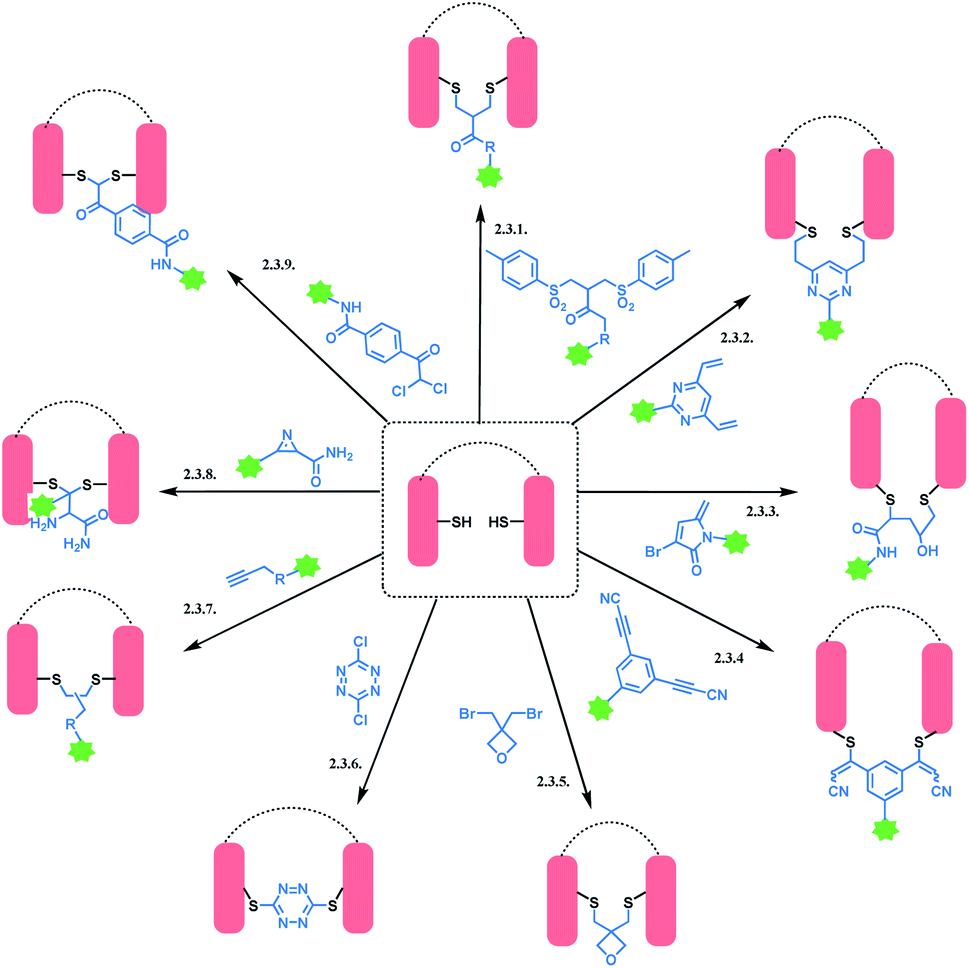 | ||
| Scheme 29 Disulfide rebridging methods. | ||
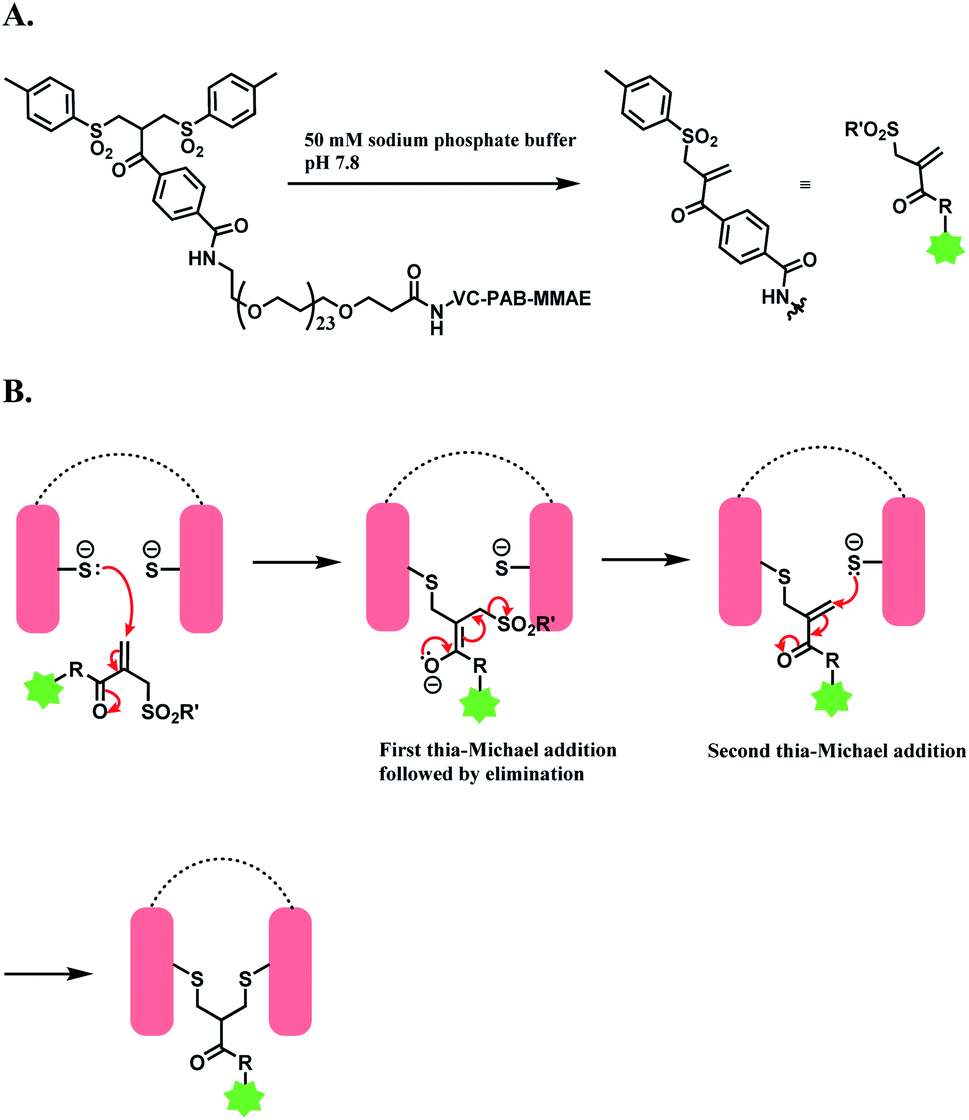 | ||
| Scheme 30 Bis-sulfone reagent as a disulfide rebridging linker; (A) Formation of mono-sulfone Michael acceptor; (B) disulfide rebridging via a sequence of Michael addition and elimination reactions. | ||
The trastuzumab–MMAE conjugate with this sulfone linker was stable in human serum containing albumin, where no DAR loss was observed after being incubated for 120 h. The ADC of trastuzumab-derived Fab and MMAE linked via the sulfone linker was found to have potent antiproliferative activity in both SK-BR-3 and BT-474 cell lines, which are HER2-positive.
10 equiv. of alkyne-functionalised DVP per disulfide was reacted with a reduced trastuzumab for 2 h at 37 °C in pH 8 borate buffered saline (BBS), and yielded >90% re-bridging of the Fab regions. Analysis by LC-MS showed that 41% was half-antibody, and 49% was correctly bridged full-antibody. The DVP linker did not react with an unreduced trastuzumab Fab, even after 2 h at 37 °C, demonstrating an exclusive reactivity towards free thiols.
The DVP linker could either be conjugated to an antibody as a synthetic handle for post-conjugation functionalization (Scheme 31A), or form a linker-payload moiety prior to antibody conjugation (Scheme 31B).
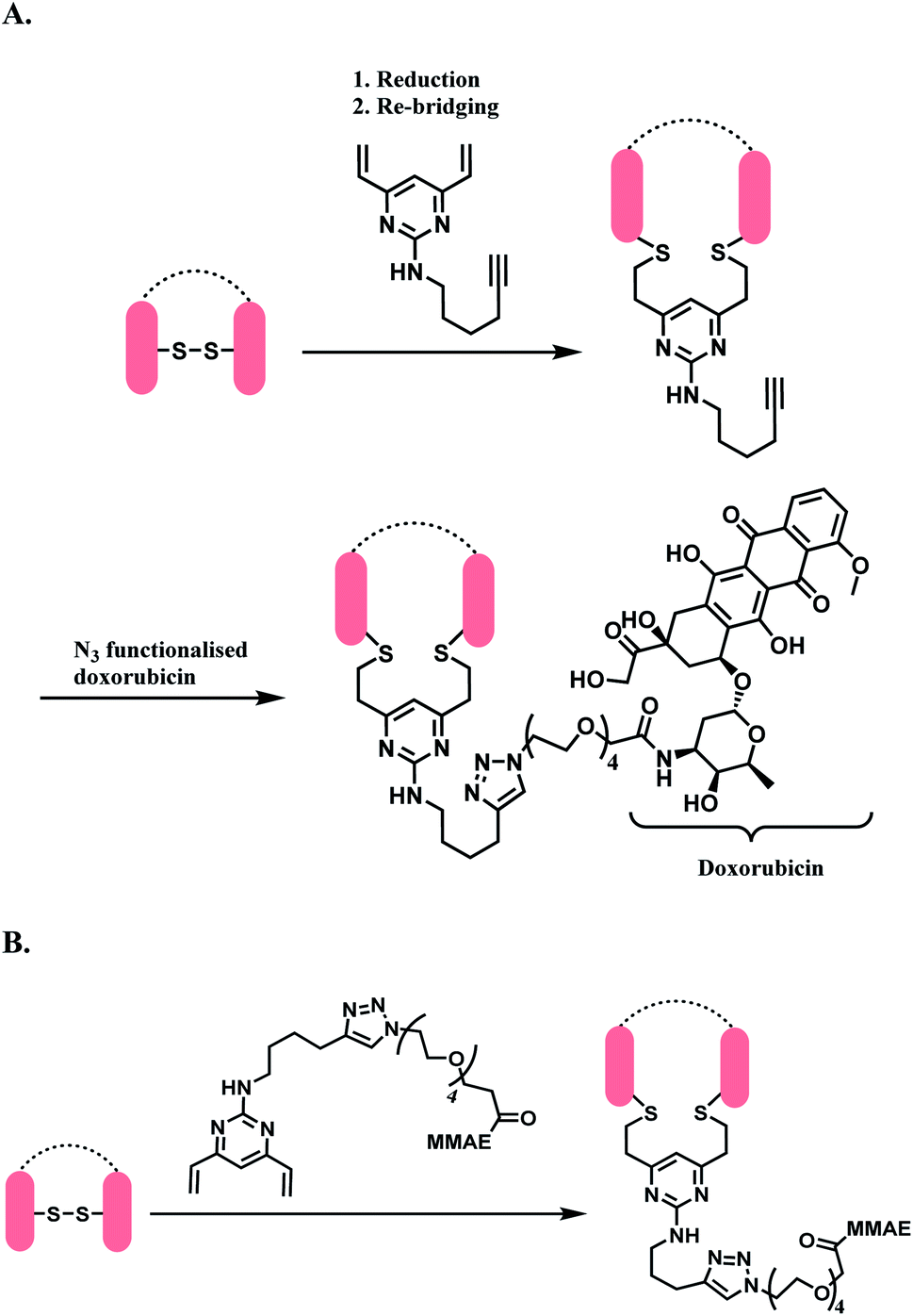 | ||
| Scheme 31 DVP as a disulfide rebridging linker; (A) conjugation of linker to antibody, followed by the attachment of drug payload; (B) conjugation of DVP linker-payload moiety to antibody. | ||
In Scheme 31A, the trastuzumab–DVP linker conjugate was reacted with an azide-functionalized doxorubicin in the presence of CuSO4·5H2O, (3-hydroxypropyltriazolylmethyl) amine (THPTA) and sodium ascorbate, at 37 °C for 2 h. LC-MS and UV-vis analysis revealed a high conversion to an ADC with an average DAR of 4.0. In Scheme 31B, a reduced trastuzumab was reacted with the linker-payload moiety, DVP–PEG4–MMAE (10 equiv. per disulfide). The reaction was carried out in 0.25 M Tris buffer, at 37 °C for 3 h. LC-MS analysis demonstrated >95% conversion to a half–antibody conjugate.
The correctly bridged DVP-MMAE ADC was tested against both HER2-positive and HER2-negative breast cancer cell lines and it was shown that it only shows cytotoxicity in HER2-positive cell lines, and that the DVP linker did not affect the cell-killing ability of MMAE.
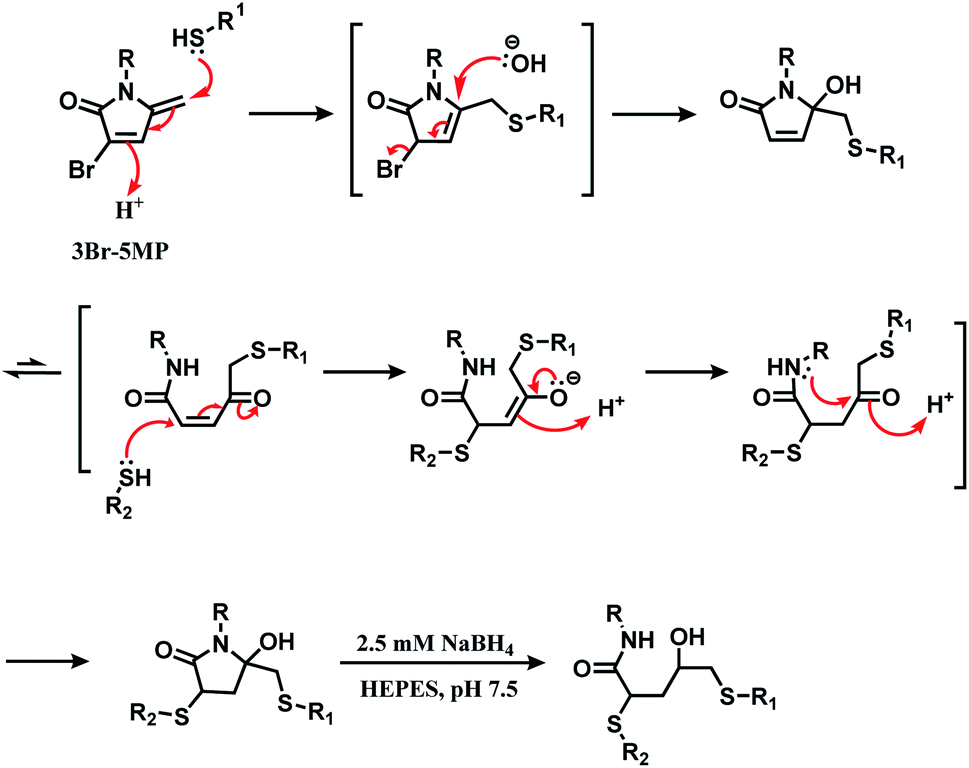 | ||
| Scheme 32 Mechanism of the reaction of 3Br-5MP with two thiol groups. | ||
Its reactivity as a disulfide rebridging agent was investigated on the endogenous hormone somatostatin (SST) and Fab fragment of goat anti-human antibody IgG. With SST, 1.3 equiv. of 3Br-5MP per reduced disulfide led to the rebridged product in a quantitative yield in HEPES buffer (pH 7.5) at 37 °C within 1 h. The rebridged product was a mixture of regioisomers, depending on which cysteine reacted first with 3Br-5MP.
The Fab fragment has one interchain disulfide bond, and 1.4 equiv. of 3Br-5MP-functionalised fluoroscein modified the reduced Fab fragment within 1 h at 37 °C. The degree of labelling determined by UV-Vis spectrum showed was 1.1 approximately. The modified fragment and the intact Fab fragment migrated to the same place on SDS-PAGE gel analysis.
Two ADCs were prepared in this study-both consist of trastuzumab (T), ADPN, a galactoside (Gal) and monomethyl auristatin E (MMAE), with one ADC containing an additional short PEG linker (PEG4). For trastuzumab, its interchain disulfide bonds were reduced after incubation with 5 equiv. of TCEP in PBS at 37 °C. After ADPN–Gal–MMAE and ADPN–PEG4–Gal–MMAE fragments were synthesized, the resultant products were incubated with reduced trastuzumab for 12 h at 25 °C separately. The average DAR values obtained for T–ADPN–Gal–MMAE and T–ADPN–PEG4–Gal–MMAE were 4.5 and 4.0 respectively. An in vitro cytotoxicity assessment of the new ADCs on SK-BR-3 HER2-positive and MDA-MB-231 HER2-negative cancer cell-lines showed that both ADCs strong activity on HER2-positive cancer cells with IC50 values that are comparable to FDA-approved T-DM1. Both T–ADPN–Gal–MMAE and T–ADPN–PEG4–Gal–MMAE were more potent towards MDA–MB–231 HER2-negative cancer cells than T-DM1.
The disulfide rebridging with 3,3-bis(bromomethyl)oxetane was applied on somatostatin, octreotide, Fab arms of trastuzumab, DesA-Aß, and CRM197. Somatostatin is a homing peptide used in Lutathera™, a peptide–drug conjugate (PDC) for the treatment of gastroenteropancreatic neuroendocrine tumours (GEP-NETs),146 and octreotide (Sandostatin™) is used to treat acromegaly.147 Both polypeptides contain one disulfide bond. After the reduction with TCEP, reacting the peptides with 1.2 equiv. of 3,3-bis(bromomethyl)oxetane in a 1:9 mixture of DMF/H2O at 25 °C yielded the grafted cyclic peptides (yield > 70%) after 12 h. Both resulting peptides showed a similar affinity against this receptor somatostatin receptor 2 (SSTR2) to the unconjugated peptides, and the oxetane-grafted somatostatin had an improved binding property with a 4-fold increment in KD value.
The oxetane-stapled Fab–trastuzumab was stable under reducing conditions and in human plasma, unlike the disulfide native antibody. In addition, bio-layer interferometry (BLI) experiments showed that the oxetane-stapled Fab fragment had a higher binding affinity to the HER2 receptor than the native antibody. DesAb-Aß3–9-1, which targets the region 3–9 of human Amyloid-β (Aβ42) peptide, has a hindered intra-domain disulfide bond. By reacting the reduced antibody with 20 equiv. of 3,3-bis(bromomethyl)oxetane at 37 °C for 3 days, a complete conversion to the disulfide-rebridged antibody was achieved. The oxetane grafted antibody had a slightly increased flexibility.
CRM197 is used as a carrier protein for vaccines, for epitope presentation.148 The in vivo study showed that the antibodies generated by the oxetane-stapled CRM197 had a 3-fold higher avidity (avidity index = 0.8:0.4 M) for the protein antigen compared to the intact CRM 197 (avidity index = 0.3:0.1 M).
While this work did not involve post-functionalisation or synthesis of a linker construct incorporating functionalised molecules (e.g. cytotoxins), the oxetane grafting was applied on a diverse range of therapeutic proteins, and the biological activity of the resulting protein–oxetane conjugates were analysed in vivo. Such research results provide valuable insights into the effect of covalent re-bridging of disulfide bonds on therapeutic biomolecules.
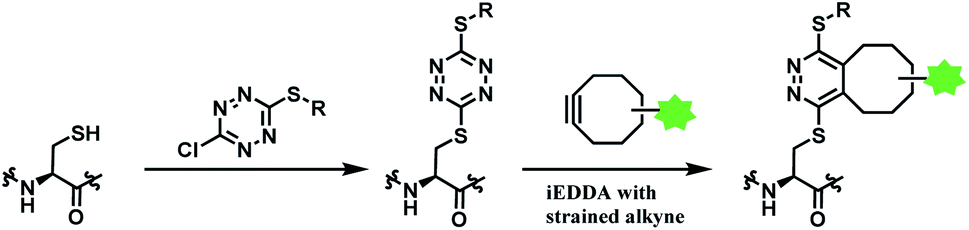 | ||
| Scheme 33 Using dichlorotetrazine as a disulfide conjugation reagent, with post-functionalisation via iEDDA. | ||
Monosubstituted chlorotetrazine, (R)-NODAGA–S-tetrazine–Cl, was reacted with two model peptides, Ac-CKYWGRGDS-NH2, and Ac-MKYWGRGDS-NH2 to investigate the chlorotetrazine linker's cysteine selectivity (Scheme 34). 3 equiv. of (R)-NODAGA–S-tetrazine–Cl linker conjugated to the cysteine residue of Ac-CKYWGRGDS-NH2 (20 μM) within 1 min at room temperature in pH 7.13 phosphate buffer, leading to full conversion. The S-tetrazine–Cl linker did not react at all with the other peptide with no cysteine, demonstrating its high selectivity towards cysteine. (R)-NODAGA–NH-Tz–Cl did not lead to conjugation with both peptides at biocompatible temperature. The necessity to have a sulfur atom para to chloro-highlights the dichlorotetrazine's potential as a double thiol conjugation linker.
 | ||
| Scheme 34 Reaction between (R)-NODAGA–X–Cl (X = S or NH) and peptide. | ||
The method was only applied on bovin serum albumin (BSA), not monoclonal antibodies. Disulfide reduction was not carried out, and in a slightly acidic buffer (pH 5.15), 10 equiv. of (R)-NODAGA–S-tetrazine–Cl was used to target a free thiol of Cys34. A mono-adduct was yielded after 1 h at room temperature. Post-functionalisation attached a fluorophore Sulfo-Cyanine5 (sulfoCy5) to the adduct, via iEDDA with a sufloCy5-bicyclo[6.1.0]nonyne (BCN). The iEDDA with BCN took 12 h at 37 °C and the protein recovery yield was 68%. The resulting BSA–sulfoCy5 conjugate was stable in human plasma at 37 °C for 48 h.
In 2015, Bräse and co-workers have reported a direct insertion of an alkyne into a disulfide bond. After reduction of the disulfide bond with TCEP, thiol–yne reaction in a photochemical batch reactor, yielded the dithioether with 49% isolated yield as a mixture of diastereomers in about a 1:1 ratio. The solvent system in the reactor was water/methanol (v/v = 1:1).152
This method was applied on terlipressin and pressinoic acid, polypeptides with a single disulfide bond. After the reduction of the disulfide bond, the substrates were treated with 6-heptynoic acid, and the resulting mixture was then irradiated with UV-light (365 nm) of an LED UV-pen, in the presence of lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) as radical initiator. Both peptides yielded a mixture of two regioisomers, and each regioisomer existed in two diastereomers. LC/MS analyses showed that the thiol–yne reaction proceeds via a monoaddition of a thiyl radical on the alkyne in the first 1 h, and a subsequent intramolecular thiol–ene coupling between the alkene of the monoadduct and the second cysteine thiol group (Scheme 35). Sufficient reaction time of 3 h was needed to completely yield the desired bis-addition products.
 | ||
| Scheme 35 Sequential steps of thiol–yne coupling to rebridge reduced disulfide. | ||
Using this thiol–yne coupling, a disulfide bond in Fab fragment M14-G07 was successfully bridged with 6-heptynoic acid as the alkyne (1 equiv.) and LAP. The reaction was carried out over 4 h, and 20 mol% of LAP was added each hour of UV irradiation (4 × 20 mol%). However, the method was not applied on a full antibody. The concentration of the protein solution was shown to be critical, where a concentration lower than 46.1 mg mL−1 did not lead to the desired conjugation.
The optimal conditions for the diaddtion of thiophenol to 2H-azirine Ph(CNCH)CONH2, forming the thiocetal products selectively over monothiol adducts, was determined to be performed with an 8.0:1.0 equivalence of PhSH and 2H-azirine respectively, in a cosolvent system of PBS and EtOH (v/v = 1:1) at pH 7.4 under nitrogen and 37 °C. Utilizing these conditions, various 2-substituted 2H-azirines, such as carboxylic acid, esters and amides showed efficient synthesis of the dithiol product. Further studies on the reactivity of 2-amide-2H-azirines concluded that aryls that have electron donating substituents, possesses a quaternary carbon, are heterocyclic-, napthalenyl-, or cyclohexyl-substituted yielded the desired product, except for the aryls with electron withdrawing substituents which remain unreacted. On the other hand, coupling reactions with alkyl dithiols, napthalenyl- and heterocyclic substituted thiols proceeded smoothly to yield the desired thioacetals. Similarly, thiolphenols that possess electron rich groups gave a higher yield of thiolacetals than electron deficient groups, whereas those with active hydrophilic amino and hydroxyl groups achieved complete chemoselectivity without N- or O- adducts detected.
Lastly, the reactivity of cysteine derivatives with multiple 2H-azirines containing MeO, Cl and CN functional groups in solely aqueous PBS solvent at room temperature was investigated, and the most reactive methoxy 2H-azirine was subsequently used to couple with different peptides with free thiol moieties. Single cysteine residues generate dimerized thioacetals as the linkers function in a linchpin mode, while peptides having 2 cysteine residues will result in cyclic thioacetal adduct bridges. In both situations, it is selective to only to amino acids residues of thiol groups.
In general, the thioacetalization reaction pathway for 2H-azirine was proposed to undergo via an aziridine intermediate, specific to thiol functional groups.
The strategy was applied onto 2 different peptides, lanreotide and N-Ac-oxytocin. Disulfide reduction was first performed with tris[2-carboxyethyl]phosphine (TCEP, 1.1 equiv.) for 30 min to release two free thiol groups derived from cysteine residues. They are then subjected to the dichloroacetophenone substrate for 2 h at 15 °C, in NH4HCO3 (0.1 M) buffer solution and MeCN (10%, v/v). Both the peptides were able to successfully produce the respective disulfide bridged conjugates.
2.4. Tyrosine functionalisation
Tyrosine residues are attractive conjugation targets, as the reactivity of the phenol substituent is mostly orthogonal to that of cysteine or lysine. In addition, tyrosine residues are often partially buried in the surface of the proteins, because of the amphiphilic nature of the phenolic group155 with the hydroxyl group being solvent exposed. Appearing relatively scarce on protein surfaces, one of several tyrosine residues could be targeted selectively through a proper linker molecule design. While this provides an attractive alternative to lysine or cysteine-targeting strategies with a higher homogeneity of resulting conjugates, it can be translated as lack of conjugation sites. This can deter the oncological application of ADCs, as the drug loading less than 4 was observed to give less potent antitumor effects.156 However, tyrosine conjugated ADCs could be applied in non-cancer indication, such as human immunodeficiency virus (HIV) infection. This shows the wide applicability of tyrosine conjugation methods in ADC, beyond oncological drugs.Despite its potential, only a relatively small number of biocompatible transformations have been developed, as shown in Scheme 36. Aside from the reactions covered in this section, palladium-catalysed O-alkylation of tyrosine by Trost allylation,157 or metalloenzyme-catalysed tyrosine conjugations158–160 are reported. However, they involve toxic transition-metal ions and therefore, will not be reviewed.
 | ||
| Scheme 36 Tyrosine conjugation methods. | ||
 | ||
| Scheme 37 Reaction between tyrosine and in situ generated imine. | ||
The method selectively conjugated a fluorescent rhodamine dye with various proteins, including chymotrypsinogen A, lysosome and RNase A, within 18 h in pH 6.5 0.1 M phosphate buffer at room temperature. Chymotrypsinogen has four tyrosines in the primary sequence,162 and it has been shown that 34% of chymotrypsinogen A molecules were unconjugated. Of the other 66%, only one tyrosine residue was conjugated to the dye. They further expanded the scope to the conjugate of chymotrypsinogen A and short peptide sequences with useful properties, such as aniline-terminated Myc, FLAG epitopes, an HIV-Tat derived peptide and sequences that bind terbium(III) ions or nucleate gallium arsenide nanocrystals.163 The protein labelling with these peptides was carried out at physiological temperature 37 °C for 20–24 h.
The chemoselectivity of this three-component coupling was explored with isotopic labelling and nuclear magnetic resonance (NMR)-based studies.162 It was revealed that side reactions between reactive formaldehydes and the nucleophilic indole ring of tryptophan residues occurred. Aside from this selectivity aspect, it should also be noted that no reactivity has been observed for horse heart myoglobin, a protein that has relatively low number of surface-accessible tyrosine residues. Furthermore, a relatively long reaction time (20–24 h) and the requirement of a large excess of coupling reagents make this method less attractive.
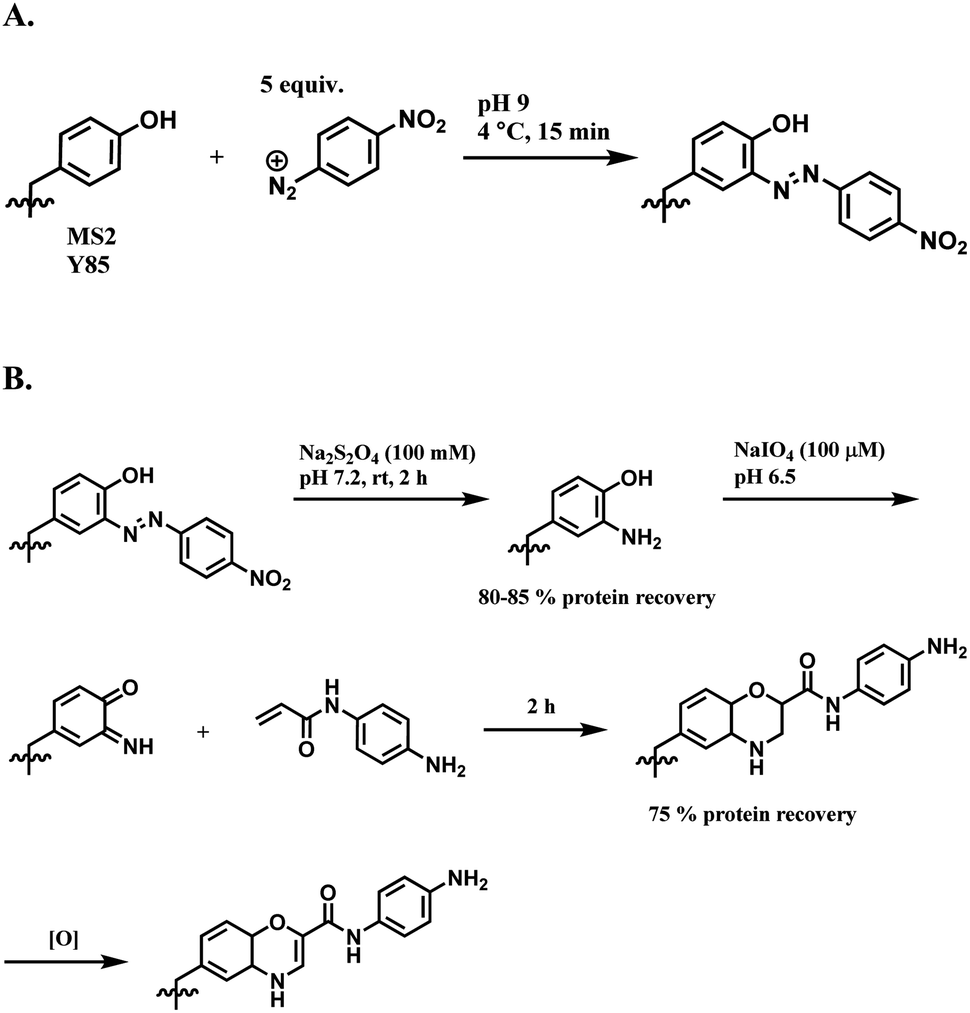 | ||
| Scheme 38 (A) Reaction of 4-nitrobenzenediazonium salt with tyrosine linings of MS2 bacteriophage; (B) post-functionalisation of the resulting o-imino-quinone conjugate. | ||
The nitro substituent in the diazonium salt reagent was irreplaceable in the reaction, thus limiting the substrate scope. Therefore, the reactive functional group o-imino-quinone was introduced by redox reactions (Scheme 38B) in order to react with another exogenous moiety, acrylamide, through a novel hetero-Diels–Alder reaction. After the reaction with acrylamide, 75% of the MS2 was recovered.
In later work, Francis and co-workers used 35 equiv. of diazonium salts to selectively label tyrosine residues in the exterior protein shell of the tobacco mosaic virus (TMV).165 In this work, they reported a decrease in the efficiency of diazonium coupling to TMV, from >90 to ∼30%, by replacing the para-substituent of the diazonium salt from NO2 group to a less electron withdrawing C(O)NH2 (Scheme 39).
 | ||
| Scheme 39 Varying yields of diazonium salt labelling of tyrosine based on the para-substituent of the diazonium salt. | ||
This strategy to use diazonium salts was accommodated by many other research groups and brought significant developments in tyrosine bioconjugation. In 2012, Haddleton and co-workers selectively conjugated monomethoxy poly(ethylene glycol)s (mPEGs) to tyrosine residues of [D-ala2] leucine enkephalin (enkeph) and salmon calcitonin (sCT), by using diazonium derivatives as coupling agents (Scheme 40).166 The pH was adjusted to 4 for this PEGylation reaction, not 9. This was to prevent diazonium derivatives from reacting with other nucleophilic amino acids. Tryptophan has been reported to react efficiently with diazonium salts such as 3-diazonium-1,2,4,-triazole (3-DT) at pH lower than 3. A general trend for the N-nucleophiles (Lys, Arg, and N-terminal amine) is that with lower pH, higher proportions of the nucleophilic nitrogen centres will be in their protonated form, which is non-reactive toward electrophilic reagents such as diazonium salt derivatives. Therefore, the pH of the reaction medium was adjusted to 4, where tyrosine residues could react to diazonium salts selectively.
Instead of using diazonium salts directly, aniline-terminated polymers were generated and converted to the required diazonium derivatives in situ, by trifluoroacetic acid and NaNO2 at 0 °C, during the peptide conjugation. The pH was then adjusted to 4.5 with 0.5 M acetate buffer, and the reaction was carried out for 24 h. With the 10 equiv. of diazonium derivatives, enkeph and sCT, both containing a single tyrosine residue, yielded mono-adducts. At pH 7, cross-reactivity with His, N-terminal amine, and Lys in sCT was observed. When the number of PEG groups in the diazonium derivative linker molecule was increased from 6 to 45, a longer reaction time of 56 h, and a higher molar equivalence (20 equiv.) were required to yield 78% conversion ratio with sCT.
 | ||
| Scheme 40 Synthetic strategy to label tyrosine residue with in situ generated diazonium salt. | ||
In the same year, Barbas and co-workers reported that 4-formylbenzene diazonium hexafluorophosphate (FBDP) selectively reacts with tyrosine residues of trastuzumab antibody in 0.1 M phosphate buffer pH 8, within 30 minutes at room temperature (Scheme 41).167 An excess amount of FBDP (10 equiv.) was required. With the aldehyde functionality instead of nitro group at 4-position, the introduction of payloads was easier. The aldehyde tag was then modified with 20 equiv. of biotin–oxyamine (pH 6.0, 4 °C). Each trastuzumab molecule was found to have an average of 1.8 biotin tags.
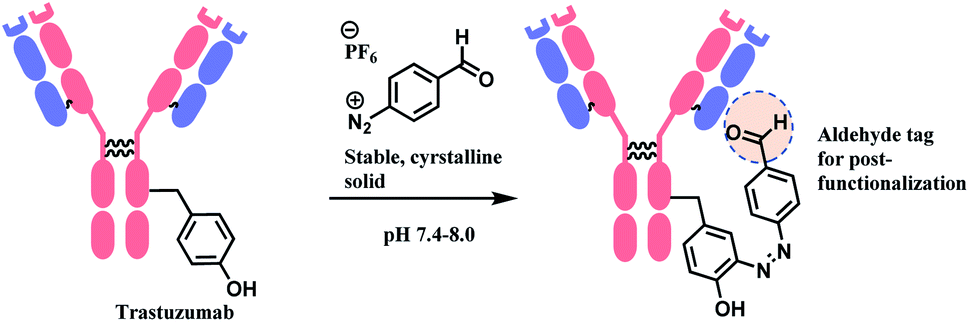 | ||
| Scheme 41 Labelling tyrosine residues of trastuzumab with 4-formylbenzene diazonium hexafluorophosphate (FBDP). | ||
In 2013, the research group used the same method to produce trastuzumab–aplaviroc conjugates, as well as conjugates with broadly neutralizing antibodies (Scheme 42), and tested them in HIV cell line.168 Aplaviroc is CCR5 antagonist developed for the treatment of HIV infection.169 It was found that 1 aplaviroc moiety was loaded per trastuzumab molecule on average. With broadly neutralising monoclonal antibodies (BNmAbs b12, 2G12, PG9, PG16) and CD4-IgG, a relatively homogenous DAR value of 0.5 to 2 was observed. The PG9–aplaviroc conjugate was tested against a panel of 117 HIV-1 strains and was found to neutralise 100% of the viruses.
 | ||
| Scheme 42 Formation of mAb–aplaviroc conjugate. | ||
:1), 2.5 equivalents of PTAD was reacted with a tripeptide containing Tyr within 30 min at room temperature to provide the conjugate in 85% yield. It was shown that aqueous conditions played a critical role, and that the reaction could not proceed in pure organic solvent. In the chemoselectivity study, PTAD was shown to react with trytophan and lysine as well. However, unlike tyrosine, tryptophan reacted with PTAD equally sluggish in organic solvent or in mixed aqueous media.
PTAD labelling was further studied with chymotrypsinogen A, bovine serum albumin (BSA) and horse heart myoglobin, in pH 7.4 phosphate buffer with a minimal amount (1–5%) of DMF. The conjugation efficiency (yield) was dependent on the protein type, and the electronic properties of the PTAD reagent. With an electron withdrawing group adjacent to the phenyl ring in PTAD (Fig. 12A), 60% of chymotrypsinogen A, 68% of BSA, and 16% of myoglobin were labelled. With the –OR group substituted PTAD (Fig. 12B), 81% of chymotrypsinogen A, 96% of BSA, 6–8% of myoglobin were labelled with the payload. In the substrate scope expansion done by the same research group suggested that an electron donating substitution on the phenyl ring in PTAD provides stability in water without compromising electrophilicity towards the phenol group of tyrosine.171 In the further study, the research group selectively PEGylated tyrosine residues in chymotrypsinogen with 10 equiv. of PTAD-PEG within 30 min at room temperature, in pH 7.4 buffer. There are 4 tyrosine residues present in chymotrypsinogen, and mono-PEG adducts were produced predominantly.
 | ||
| Fig. 12 PTAD reagents substituted with (a) electronically poor moieties; (b) electronically rich moieties. | ||
Tyrosine click reaction with PTAD was further expanded to the synthesis of ADC of trastuzumab and aplaviroc (Scheme 43). An excess of phenyl urazole (Ph-Ur)-aplaviroc was oxidised by 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione (DBDMH) to produce the PTAD moiety, then immediately reacted with tyrosine residues of trastuzumab in 0.1 M phosphate buffer (pH 7) at room temperature, for less than 5 min. A trastuzumab mAb has 62 tyrosine residues in solvent-accessible regions, and the average DAR of the resulting trastuzumab–aplaviroc conjugate was 1.3. The neutralization activity and IC50 value of this conjugate were very similar to those of aplaviroc alone.
 | ||
| Scheme 43 Generation of trastuzumab–aplaviroc conjugate with the DAR of 1.3. | ||
There are limitations of this conjugation method. Firstly, PTAD was shown to be stable in acetonitrile but to easily decompose in water to form isocyanate, an active electrophile (Scheme 44).172 The resulting isocyanate undergoes undesired side reactions with lysine residues and the N-terminus. Berti and co-workers at Novartis used the PTAD reagent to selectively label tyrosine residues on CRM197, a protein carrier in vaccines, and observed that 38% of CRM197 was unmodified, 33% was a phenyl isocyanate–lysine conjugate, and only a trace amount was a desired, PTAD-tyrosine conjugated product.173
 | ||
| Scheme 44 Side reaction with amine groups via isocyanate generation. | ||
Barbas and co-workers attempted to resolve this issue by using tris-(hydroxymethyl) aminomethane (Tris) buffer (0.1 M). The primary amine of the Tris buffer scavenges the isocyanate. However, using even high concentrations of the Tris buffer was not always fully effective and amine groups modifications were still observed.158 It should be noted that the decomposition itself lowers the reaction's atom economy, even if the undesired isocyanate can be removed. Using acetonitrile as a co-solvent to stabilize PTAD limits the expansion of this reaction to protein conjugations; adding acetonitrile to an aqueous solvent is reported to enhance the peptide–peptide hydrogen bonds, leading to the denaturation of proteins.174,175
In order to overcome these limitations, Gouin and co-workers devised a strategy to electrochemically activate Ph-Ur species in the presence of proteins (Scheme 45), rather than oxidising them by DBDMH.176
 | ||
| Scheme 45 e-Y-Click reaction. | ||
In this ‘e-Y-Click’ reaction, Ph-Ur undergoes anodic oxidation at 0.36 V (versus saturated calomel electrode as the reference electrode) in pure phosphate buffer without the isocyanate scavenger, and PTAD is gradually produced in the reaction system. Because the PTAD generated by anode oxidation reacts with tyrosine instantaneously, side reactions with nucleophilic residues and N-terminus via isocyanate formation can be suppressed. A relatively low potential 0.36 V allowed peptides and proteins to be labelled without severe oxidative damage. Using glucose oxidase enzyme (GOx) as the substrate, they confirmed that the enzymatic activity of GOx was not affected by tyrosine labelling through the e-Y-Click reaction. This method was also applied in the antibody conjugation; 11.2 equiv. of N3-Ph-Ur was reacted with epratuzumab (LymphoCide™), a humanized anti-CD22 mAb for the treatment of inflammatory autoimmune disease,177 giving the conversion yield 95% within 2 h. The average Ph-Ur molecule loading per mAb was 2.3 (Scheme 46).
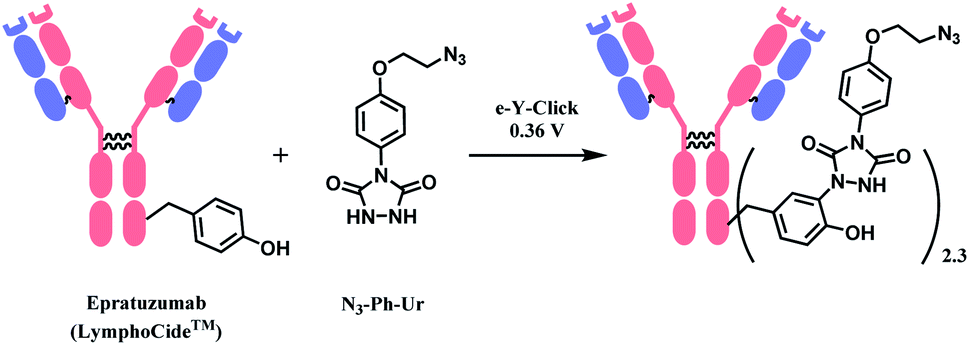 | ||
| Scheme 46 Tyrosine conjugation of epratuzumab with N3-Ph-Ur. | ||
The mechanism was proposed to proceed with an initial single-electron oxidation (SET) of PTZ, generating a nitrogen radical, and a subsequent site-selective addition of this radical to the ortho-position of the phenol ring on tyrosine residues (Scheme 47). As the ortho-positions are more electron rich as compared to the remaining para- and meta-positions, this favours the radical addition onto the ortho-position with excellent selectivity. With the optimized reaction conditions utilizing a graphite anode rod and a nickel cathode plate at a constant current of 10 mA for 75 min in Na2SO4 electrolyte and CH3CN/H2O (6.0:4.0 v/v ratio) at room temperature, protected tyrosine (1 equiv.) and an unsubstituted phenothiazine (1 equiv.) gave the product in an 85% yield. The solvent system with >50% CH3CN was necessary.
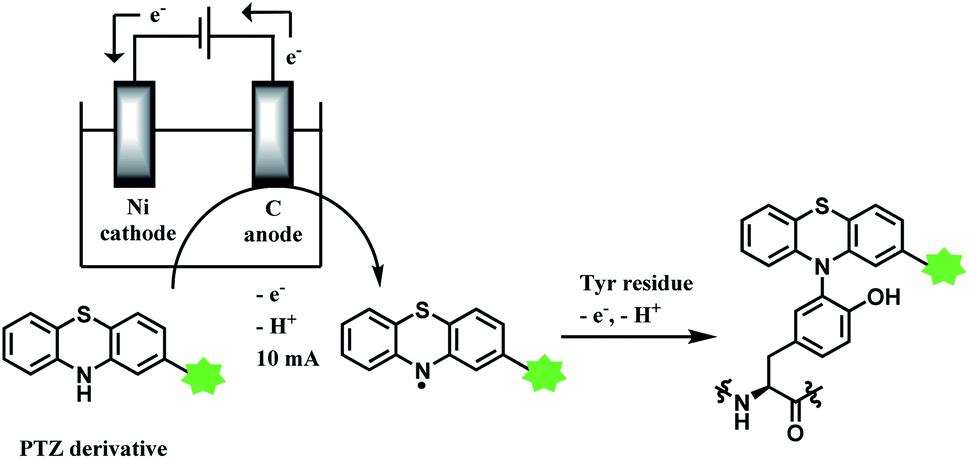 | ||
| Scheme 47 Tyrosine-selective conjugation by PTZ derivatives. | ||
In a further study, the group investigated the effects of various electron-withdrawing and electron-donating substituted phenothiazine and it was concluded that they were all decent substrates but relatively lower yields of 33–48% were obtained. Although the PTZ reagent is unreactive towards other aromatic amino acid residues, namely Trp, His and Phe, it is still able to react with cysteine residues probably due to it being able to be easily oxidized.
The study also included the synthesis of PTZ-modified unprotected polypeptides from 5-mers to 29-mers Tyr-containing polypeptides, such as RGD peptide, acyclic pentapeptide and other endogenous peptides with biological activity. With the above optimized reaction conditions but in CH3CN/PBS (1:1 v/v ratio), it was observed that phenothiazine chemoselectively conjugated to Tyr in 30 min at room temperature, even in the presence of other amino acid residues. Moreover, the research was extended to include insulin and myoglobin proteins and the results indicate a successful tagging of PTZ onto the Tyr residues.
2.5. Tryptophan functionalisation
There are only two reported methods which achieve tryptophan functionalisation without metal catalysts, in aqueous medium. They are summarised in Scheme 48.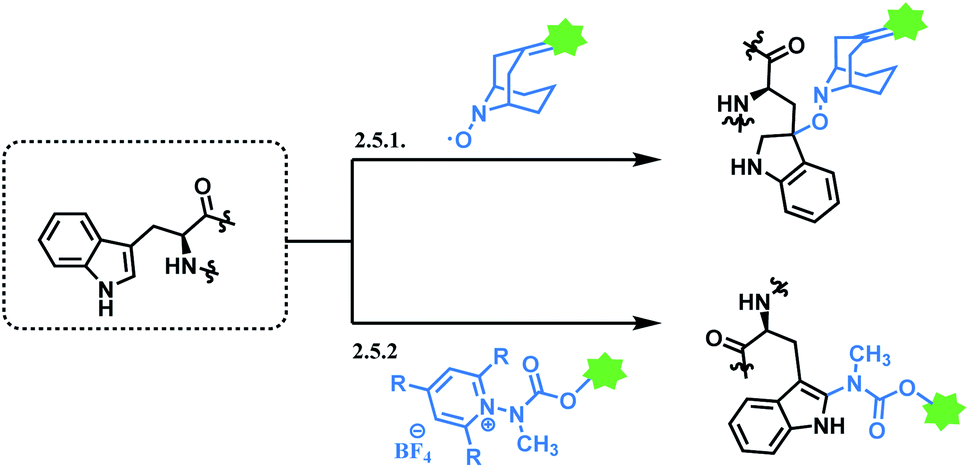 | ||
| Scheme 48 Tryptophan conjugation methods. | ||
Initially, keto-ABNO is oxidised to oxoammonium cation via disproportionation and oxidation by NOx, which is generated from NaNO2 and acetic acid (Scheme 49A). The enamine nucleophile in the indole ring of a Trp residue then attacks the oxoammonium, followed by the attack of H2O or an adjacent nitrogen in the amide bond towards the resulting imine. This generates heterogenous products as shown in Scheme 49B.
 | ||
| Scheme 49 (A) Oxidation of keto-ABNO by NOx to generate oxoammonium electrophile; (B) proposed mechanism tryptophan conjugation with keto-ABNO. | ||
The conjugation strategy was also applied to produce a conjugate of anti-Aβ1–16 antibody (6E10) and fluorescein. Dot-blot assay showed that the binding ability of the antibody towards Aβ was retained after ABNO labelling, demonstrating the method's high applicability in the ADC construction.
The research group expanded the scope of this tryptophan modification strategy by building an ofatumumab-folic acid conjugate with the keto-ABNO linker.179 Folic acid is a ligand for cancer cells and ofatumumab is a CD20-targeting mAb. The resulting conjugate showed an antibody-dependent cellular cytotoxicity (ADCC) and induced an enhanced cytotoxic activity of natural killer (NK) cells against folic receptor-α positive cancer cells. 10 equiv. of ABNO-PEG-folic acid reagent in the presence of NaNO2 and acetic acid yielded the degree of labelling of folic acid of 1–2 (Scheme 50).
 | ||
| Scheme 50 Formation of ofatumumab-folic acid conjugate. | ||
Simultaneous PET coupling and Trp labelling can be achieved with the utility of a Katritsky-type N-substituted pyridinium salt that possesses a carbamate label. The pyridinium salt having a positively charged π-electron system enables reduction via single electron transfer, while varying the substituents on the pyridinium ring and the substituted N atom facilitates homolytic cleavage of the reactive N–N bond. As such, the aromaticity of the pyridinium ring will be restored, and the corresponding radical species produced are reactive for a subsequent formation of bond. N-substituted pyridinium salt triggered by the single electron reductant Trp forms an N-centred and a positively charged Trp radicals, which then both recombine to give a selectively labelled Trp.
This selective labelling process is kinetically feasible and at the same time, prevents photodegradation especially when done in the optimised conditions with NH4OAc buffer, UV-B light and in 1 mM of glutathione (GSH) to yield a complete conversion >95% (mono/di label ratio > 20:1). GSH functions as a reactive oxygen species (ROS) scavenger to promote the reaction restrictedly on the Trp residue for modification, where both the labels are found on Trp. On the other hand, the 2,4,6-tri-methylated pyridinium ring helps to prevent nucleophilic addition and thus, more stable and less prone to decomposition. Furthermore, with the carbamate functional group present on the pyridinium salt, it allows the salt to be derivatisable with other useful functional groups and subsequently, easily transferred onto Trp. These factors contribute to the high conversion of selective Trp labelling in moderately photolabile proteins, the exclusive reactive site, even in the presence of other amino acids of contrasting topologies and redox/photoionization potentials.
However, in a more photolabile protein (14.3 kDa) with many redox active residues, the undesired cys-glutathionylated products are observed as a result of thiyl radical exchange or TCEP reduction of disulfides in close vicinity to [Trp]*. Nonetheless, intraprotein PET degradation can be avoided by ensuring that the donation of a radical from Trp to an excited pyridinium salt is the primary route, which is achieved with a phenyl-substituted pyridinium salt, to produce a >90% conversion of 6:1 mono/di labelled conjugation, and a higher GSH concentration for irradiation not surpassing 320 nm.
Overall, PET is the key mechanism propelling the reaction of Trp labelling forward based on observations and results. Complete deuteration experiments of Trp at C4-position indicate that initiation of the reaction is due to the photoexcitation of Trp and pyridinium salt, while the quantum yield indicates that the mechanism consists of [Trp]* as the electron source for transfer. Additionally, the NMR data proves that labelling does not involve the methyl group of the indole, but at the C2-position via a formation of C–N bond. Precomplexation and amination mechanisms for Trp selectivity were ruled out as such behaviours are also displayed in phenylalanine and tyrosine.
2.6. Methionine functionalisation
Protein conjugation methods traditionally rely on the nucleophilicity of amino acid residues. Thus, methionine has not been a popular target for conjugation, as the thioether (-S-CH3) side chain has a relatively weak nucleophilicity.181 Moreover, with its hydrophobic side chain, methionine is mostly found buried in proteins, which makes it difficult to modify with labelling reagents.182However, being an important cellular antioxidant, methionine is readily oxidised, rendering it susceptible to a modification via redox reactivity.183 It should be noted, however, that the oxidation of methionine to methionine sulfoxide (MetO) is a major oxidative degradation pathway of IgG1 monoclonal antibodies.184 When oxidised to MetO, the apolar chain transforms into a highly polar one, causing a disruptive change in the ternary structure of protein (Scheme 51). Thus, an important criterion for a methionine labelling agent is to be able to conjugate functional molecules to a methionine residue without forming MetO, while exploiting the redox reactivity.
 | ||
| Scheme 51 Oxidation of methionine residue. | ||
An overall summary of reported methionine conjugation methods is shown in Scheme 52.
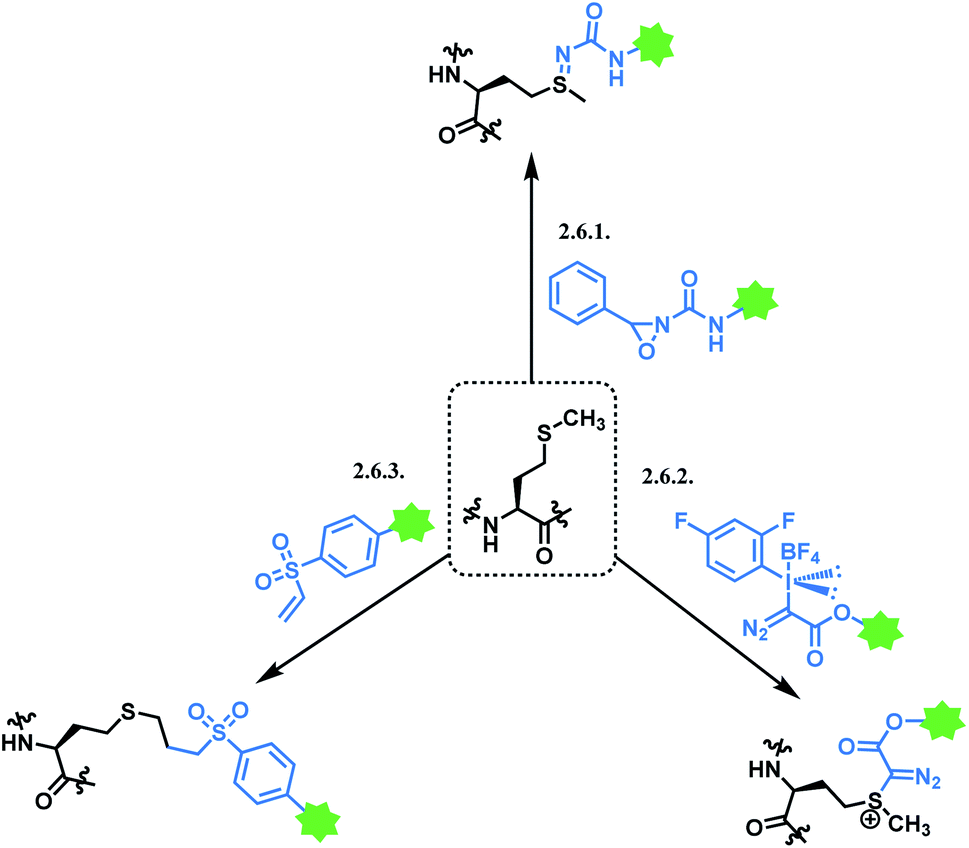 | ||
| Scheme 52 Methionine conjugation methods. | ||
:1) involving a strained oxaridine molecule (Ox1) managed to provide a 95% conversion of the desired sulfimide product (NTP) and the undesired sulfoxide compound (OTP) in a 5:1 ratio, without the presence of any catalyst in a short time frame of 2.5 min, observed with 1H NMR. The selectivity of the reaction can, however, be improved by replacing the carbamate moiety in Ox1 to a weaker electron withdrawing urea moiety in Ox2 yielded the products NTP:OTP in a 12:1 ratio with a similar conversion rate of 93%. Increasing the water content in the cosolvent system CD3OD/D2O to 1:19, the NTP:OTP selectivity improved significantly to 18:1. This could be accounted for by the better stabilization of the transition state of the NTP alkoxy anion intermediate due to better hydrogen bonding and thus, solvation.
This method of methionine functionalization with the ReACT reagent displayed exceptional selectivity over its sulfur analogues. Even though cysteine and selenocysteine were observed to be oxidised, no NTP product was formed. However, in a further study of oxaziridine probe functionalization with Ox4 and a whole proteome, HeLa cell lysate, it was verified with LC-MS/MS that not only 235 methionine residues were labelled, but an additional lysine residue was also conjugated. Similarly, the methionine bioconjugation strategy was shown to be successful with bovine serum albumin (BSA) protein. It was done with a two-step approach utilizing BSA (15 μM) was conjugated through ReACT with the Ox4 oxaziridine probe (100 μM) consisting of a bioorthogonal alkyne group, which can then undergo a second conjugation with Cy3-azide via a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. In-gel fluorescence imaging revealed a >95% yield and that the ReACT modification underwent completion in 1–2 min. In a separate ReACT modification of calmodulin (CaM, 100 μM) with various alkyne- and azide-containing oxaziridine probe molecules (1 mM), it demonstrated selective conjugation on the entire 9 methionine residues present on the CaM protein with good selectivity of NTP:OTP (25:1). Experiments on a variety of biomolecules such as biotin and polyethylene glycol (PEG) were also successful.
Moving on to the synthesis of ADCs, ReACT modification was performed on an antibody fragment to a green fluorescent protein (GFP-Fab). However, as the existing endogenuous methionine residues are inaccessible from the surface, they were unable to be conjugated unless additional methionine sites are engineered on the surface. Supporting evidence from experiments which involve the methionine-substituted antibody fragment at surface-accessible positions of heavy chain (HC)-A114 or light chain (LC)-V205 residues reported effective oxaziridine labelling with ReACT. Furthermore, in the attempt of methoinine conjugation through ReACT of an engineered Herceptin–Fab (Her–Fab) conjugate, enabled to possess one or two methionine residues on its light chain at the C terminus, the respective methionine labelled products generated exhibited an excellent DAR ratio with good purity. Notably, the ADC which was then produced from a subsequent conjugation reaction of the Her–Fab, installed with an azide or alkyne handle from methionine conjugation, and MMAE displayed five times more efficient toxicity to Her2-positive BT474 breast cancer cells, in contrast to the individual wild-type Her–Fab and free MMAE or when mixed.
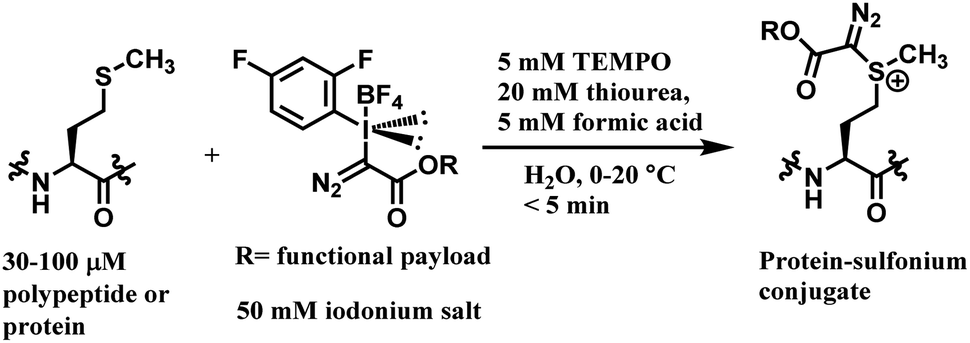 | ||
| Scheme 53 Labelling methionine residue with hypervalent iodine reagent. | ||
The conjugation strategy was applied on exenatide (Byetta™), aviptadil, and tetracosactide, the therapeutic polypeptides with a single methionine residue. The addition of a low concentration of thiourea (20 mM) improved reaction kinetics, but it was accompanied by unselective modifications and oxidative by-products. 10 mM of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) and aqueous formic acid solution (5 mM, approximately pH 3) were required to minimise such side-reactions. The labelling happened almost instantaneously in water, with 500 equiv. to >1600 equiv. of the hypervalent iodine, at 0 to 20 °C.
Methionine residues on more complex protein systems such as teriparatide, α-lactalbulmin, aprotonin, and ubiquitin were also successfully labelled with the iodonium salt, with the conversion yields ranging from 84% to >95%. Teriparatide, containing two methionines, yielded a bis–sulfonium conjugate (di:mono-labelling ratio 12:1).
The resulting protein–sulfonium conjugate can undergo post-functionalisation via a photoredox radical cross-coupling with 2000 equiv. of o-tolyl Hantzsch ester. C-benzylation product is obtained with a good yield (Scheme 54). This radical–radical cross-coupling is orthogonal to native amino acids, and allows two distinct functionalities to be introduced sequentially at methionine residue. However, it must be noted that a transition metal catalyst (Ru(II)) was used for this secondary modification, which may deter the direct application in therapeutic protein modifications.
 | ||
| Scheme 54 Post-functionalisation via photoredox radical cross-coupling between the diazo sulfonium–protein conjugate and the C-4 substituted Hantzsch ester. | ||
:1) at a micromolar concentration of proteins, and gives the conjugates with good conversion yields within 30 min. The 10 equiv. of lumiflavin and 200 equiv. of Michael acceptors relative to a protein system were required.
This method's selectivity towards methionine was tested with a wide range of peptides that do not contain methionine residues. The peptide substrates were recovered (>90%) with no conjugate product. However, thiol radicals generated from liberated cysteines readily underwent LF-catalysed Michael reactions.
It should be noted that iridium- and ruthenium-based complexes, the traditional metal-based photocatalysts, gave an undesired methionine sulfoxide (MetO), whereas lumiflavin did not, likely owing to its ability to act as a base in its radical anion form (LF−). Followed by a single electron oxidation, the LF− rapidly deprotonates the methyl group in sulfur radical cation to generate α-thio radical, preventing the formation of MetO (Scheme 55).
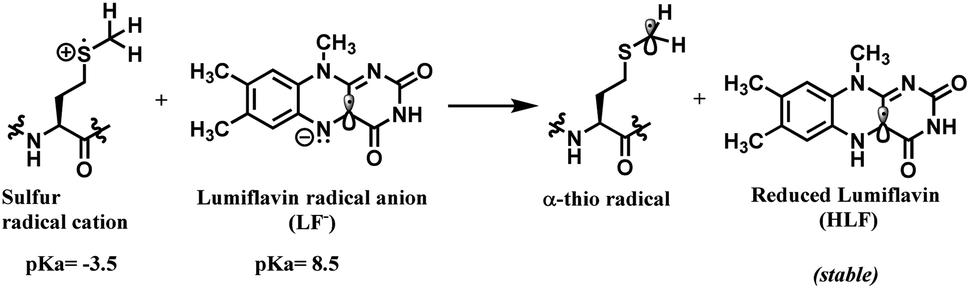 | ||
| Scheme 55 Formation of α-thio radical from methionine radical cation via hydrogen abstraction by lumiflavin radical anion. | ||
The mechanism of lumiflavin-catalysed methionine conjugation is shown in Scheme 56. Lumiflavin, photoexcited to its triplet state (Ered1/2 [3LF/LF−] = 1.5 V vs. SCE)188 by blue-light, achieves a single-electron oxidation of the thioether. The α-thio radical then undergoes radical addition with the electrophilic alkene which has a high affinity towards SOMO, to generate α-sulfone radical. This radical intermediate is then reduced by HLF to give the methionine conjugate product, regenerating the ground state lumiflavin.
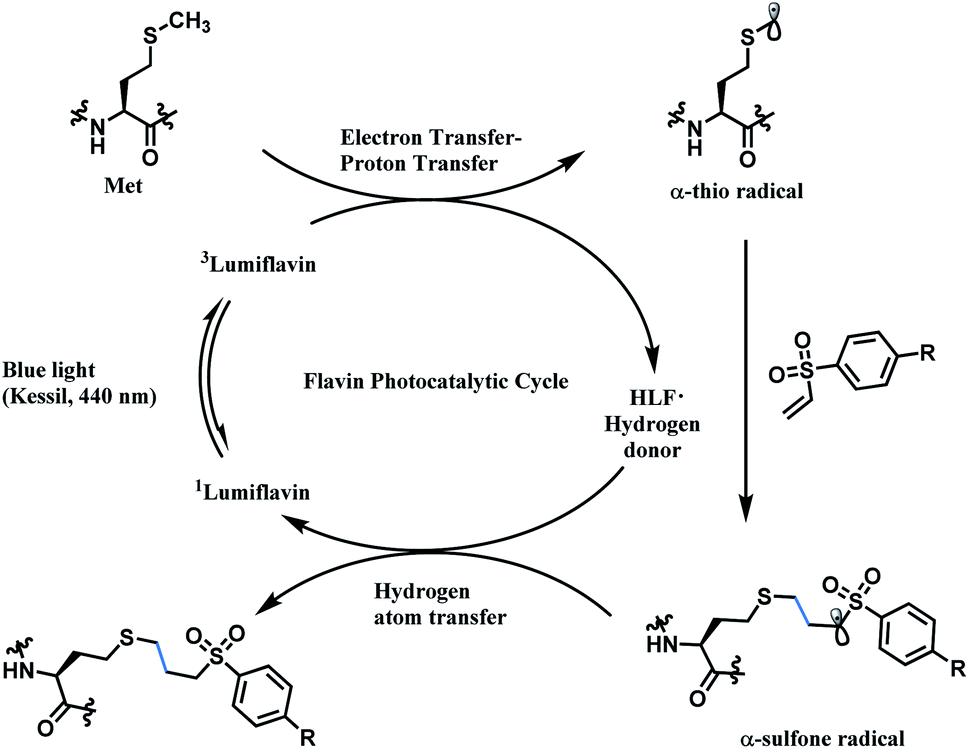 | ||
| Scheme 56 Proposed mechanism of lumiflavin-catalysed methionine conjugation. | ||
The conjugation strategy was applied on various functional proteins with different sizes, including aprotinin (6.5 kDa), α-lactalbumin (14.2 kDa), myloglobin (16.7 kDa) and enhanced green fluorescent protein. The fluorescence activity of enhanced green fluorescent protein (EGFP) was almost preserved (>95%) even after the methionine labelling, demonstrating this method's high applicability in biomolecule conjugation. No work on IgG monoclonal antibodies (∼150 kDa)189 was carried out.
Among many Michael acceptors, phenyl vinyl sulfones served particularly useful for post-functionalisation. A phenyl vinyl sulfone with an alkyne moiety in the R group was conjugated to EGFP with 45% conversion, and the conjugate subsequently undewent copper-catalysed alkyne–azide cycloaddition (CuAAC) with an azide-functionalised biotin or nonaarginine.
3. Summary and future outlook
In the era of immunotherapy, antibody–drug conjugate (ADC) is rising as a promising therapeutic modality. One attractive aspect of this modality is that a new linker leads to the registration of a new biologics entity in terms of intellectual property rights,190 allowing a huge advantage in patent space and novelty.Nanobodies,191 non-Ig-Based protein scaffolds such as DARPins,192 and peptide aptamers193 are rising as alternatives to monoclonal antibodies with their competitive advantages (higher affinity binding, lower cost, and lower immunogenicity relative to mAbs). A wider toolbox of native amino acid conjugation methods which are orthogonal thus complementary to existing ones' synthetic scopes will open wider avenues to prepare biomolecule conjugates.
Orthodox bioconjugation methods are rooted in the nucleophilicity of substituents on amino acids and therefore, the functionalisation of non-nucleophilic amino acids, such as valine, leucine, or phenylalanine, remains a challenge. An extension of metal-free, rapid and aqueous synthetic methodologies in C–H activation will provide a wider pool of potential designs for antibody conjugation linkers.
While many reactions are reported for native amino acid functionalisation to produce antibody or protein–payload conjugate, only few has succeeded to do it in nearly 100% aqueous medium, within a short enough duration, without any potentially denaturing agents. Last but not least, atom-economy is highly important for the antibody conjugation efficiency. While disulfide rebridging linker is emerging as a promising approach to control Drug-to-Antibody Ratio (DAR), a recent preclinical study suggests that a strict control of DAR may not be the most critical factor in the therapeutic efficacy.58 It seems that more thorough and detailed studies on different combinations of antibody, drug, linker's chemistry and conjugation site are required in order to better understand ADC's therapeutic profile. We envisage that antibody–payload conjugates will expand their indications beyond cancer, with the development of good linkers.
Author contributions
All of the authors participated in this review work. T. P. L. and M. S. K. proposed and designed the review. M. S. K., T. K., and J. K. prepared the draft. T. P. L. finally revised the review.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully acknowledge the financial support from Distinguished University Professor grant, Nanyang Technological University, Singapore, and AcRF Tier 1 grants from the Ministry of Education of Singapore (RG11/20 and RT14/20). We thank Tianhu Li for his valuable insights and advice.References
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- P. Bross, J. Beitz, G. Chen, X. Chen, E. Duffy, L. Keiffer, S. Roy, R. Sridhara, A. Rahman, G. Williams and R. Pazdur, Clin. Cancer Res., 2001, 7, 1490–1496 CAS.
- P. Hamann, L. Hinman, I. Hollander, C. Beyer, D. Lindh, R. Holcomb, W. Hallent, H. Tsou, J. Upeslacis, D. Shorchat, A. Mountain, D. Flowers and I. Bernstein, Bioconjug. Chem., 2002, 13, 47–58 CrossRef CAS PubMed.
- Y. N. Lamb, Drugs, 2017, 77, 1603–1610 CrossRef CAS PubMed.
- R. A. de Claro, K. McGinn, V. Kwitkowski, J. Bullock, A. Khandelwal, B. Habtemariam, Y. Ouyang, H. Saber, K. Lee, K. Koti, M. Rothmann, M. Shapiro, F. Borrego, K. Clouse, X. H. Chen, J. Brown, L. Akinsanya, R. Kane, E. Kaminskas, A. Farrell and R. Pazdur, Clin. Cancer Res., 2012, 18, 5845–5849 CrossRef CAS PubMed.
- C. Deng, B. Pan and O. A. O'Connor, Clin. Cancer Res., 2013, 19, 22–27 CrossRef CAS PubMed.
- P. M. Challita-Eid, D. Satpayev, P. Yang, Z. An, K. Morrison, Y. Shostak, A. Raitano, R. Nadell, W. Liu, D. R. Lortie, L. Capo, A. Verlinsky, M. Leavitt, F. Malik, H. Aviña, C. I. Guevara, N. Dinh, S. Karki, B. S. Anand, D. S. Pereira, I. B. J. Joseph, F. Doñate, K. Morrison and D. R. Stover, Cancer Res., 2016, 76, 3003–3013 CrossRef CAS PubMed.
- D. P. Petrylak, A. V. Balar, P. H. O'Donnell, B. A. McGregor, E. I. Heath, E. Y. Yu, M. D. Galsky, N. M. Hahn, E. M. Gartner, J. Pinelli, A. Melhem-Bertrandt and J. E. Rosenberg, J. Clin. Oncol., 2019, 37, 4505 Search PubMed.
- E. D. Deeks, Drugs, 2019, 79, 1467–1475 CrossRef PubMed.
- L. Amiri-Kordestani, G. M. Blumenthal, Q. C. Xu, L. Zhang, S. W. Tang, L. Ha, W. C. Weinberg, B. Chi, R. Candau-Chacon, P. Hughes, A. M. Russell, S. P. Miksinski, X. H. Chen, W. D. McGuinn, T. Palmby, S. J. Schrieber, Q. Liu, J. Wang, P. Song, N. Mehrotra, L. Skarupa, K. Clouse, A. Al-Hakim, R. Sridhara, A. Ibrahim, R. Justice, R. Pazdur and P. Cortazar, Clin. Cancer Res., 2014, 20, 4436–4441 CrossRef CAS PubMed.
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- S. Modi, C. Saura, T. Yamashita, Y. H. Park, S. B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S. A. Im, K. S. Lee, S. A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver and I. Krop, N. Engl. J. Med., 2020, 382, 610–621 CrossRef CAS PubMed.
- B. L. Staker, K. Hjerrild, M. D. Feese, C. A. Behnke, A. B. J. Burgin and L. Stewart, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15387–15392 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2020, 80, 1019–1025 CrossRef CAS PubMed.
- S. Lonial, H. C. Lee, A. Badros, S. Trudel, A. K. Nooka, A. Chari, A. O. Abdallah, N. Callander, N. Lendvai, D. Sborov, A. Suvannasankha, K. Weisel, L. Karlin, E. Libby, B. Arnulf, T. Facon, C. Hulin, K. M. Kortüm, P. Rodríguez-Otero, S. Z. Usmani, P. Hari, R. Baz, H. Quach, P. Moreau, P. M. Voorhees, I. Gupta, A. Hoos, E. Zhi, J. Baron, T. Piontek, E. Lewis, R. C. Jewell, E. J. Dettman, R. Popat, S. D. Esposti, J. Opalinska, P. Richardson and A. D. Cohen, Lancet Oncol, 2020, 21, 207–221 CrossRef CAS PubMed.
- A. Lee, Drugs, 2021, 81, 1229–1233 CrossRef CAS PubMed.
- T. Linke, M. T. Aspelund, C. Thompson, G. Xi, A. Fulton, M. Wendeler, T. M. Pabst, X. Wang, W. K. Wang, K. Ram and A. K. Hunter, Biotechnol. Prog., 2014, 30, 1380–1389 CrossRef CAS PubMed.
- R. J. Kreitman, Hematology Am. Soc. Hematol. Educ. Program, 2012, 2012, 660–666 CrossRef PubMed.
- S. Dhillon, Drugs, 2018, 78, 1763–1767 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- R. M. Lu, Y. C. Hwang, I. J. Liu, C. C. Lee, H. Z. Tsai, H. J. Li and H. C. Wu, J. Biomed. Sci., 2020, 27, 1–30 CrossRef CAS PubMed.
- G. J. Weiner, Immunol. Res., 2007, 39, 271–278 CrossRef CAS PubMed.
- J. Christiansen and A. K. Rajasekaran, Mol. Cancer Ther., 2004, 3, 1493–1501 CAS.
- J. M. Reichert, C. J. Rosensweig, L. B. Faden and M. C. Dewitz, Nat. Biotechnol., 2005, 23, 1073–1078 CrossRef CAS PubMed.
- H. M. Abdolahi, A. S. Asiabar, S. Azami-Aghdash, F. Pournaghi-Azar and A. Rezapour, Asia-Pac. J. Oncol. Nurs., 2018, 5, 57–67 Search PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- G. Von Minckwitz, C. S. Huang, M. S. Mano, S. Loibl, E. P. Mamounas, M. Untch, N. Wolmark, P. Rastogi, A. Schneeweiss, A. Redondo, H. H. Fischer, W. Jacot, A. K. Conlin, C. Arce-Salinas, I. L. Wapnir, C. Jackisch, M. P. DiGiovanna, P. A. Fasching, J. P. Crown, P. Wülfing, Z. Shao, E. R. Caremoli, H. Wu, L. H. Lam, D. Tesarowski, M. Smitt, H. Douthwaite, S. M. Singel and C. E. Geyer, N. Engl. J. Med., 2019, 380, 617–628 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- Y. V. Kovtun, C. A. Audette, Y. Ye, H. Xie, M. F. Ruberti, S. J. Phinney, B. A. Leece, T. Chittenden, W. A. Blättler and V. S. Goldmacher, Cancer Res., 2006, 66, 3214–3221 CrossRef CAS PubMed.
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS PubMed.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- S.-J. Moon, S. V Govindan, T. M. Cardillo, C. A. D'Souza, H. J. Hansen and D. M. Goldenberg, J. Med. Chem., 2008, 51, 6916–6926 CrossRef CAS PubMed.
- D. M. Goldenberg, T. M. Cardillo, S. V Govindan, E. A. Rossi and R. M. Sharkey, Oncotarget, 2015, 6, 22496–22512 CrossRef PubMed.
- T. M. Cardillo, S. V. Govindan, R. M. Sharkey, P. Trisal and D. M. Goldenberg, Clin. Cancer Res., 2011, 17, 3157–3169 CrossRef CAS PubMed.
- H. Ruan, S. Hao, P. Young and H. Zhang, Horizons cancer Res, 2015, 56, 23–40 CAS.
- T. Tedeschini, B. Campara, A. Grigoletto, M. Bellini, M. Salvalaio, Y. Matsuno, A. Suzuki, H. Yoshioka and G. Pasut, J. Control. Release, 2021, 337, 431–447 CrossRef CAS PubMed.
- F. M. H. De Groot, W. J. Loos, R. Koekkoek, L. W. A. Van Berkom, G. F. Busscher, A. E. Seelen, C. Albrecht, P. De Bruijn and H. W. Scheeren, J. Org. Chem., 2001, 66, 8815–8830 CrossRef CAS PubMed.
- Y. Ogitani, T. Aida, K. Hagihara, J. Yamaguchi, C. Ishii, N. Harada, M. Soma, H. Okamoto, M. Oitate, S. Arakawa, T. Hirai, R. Atsumi, T. Nakada, I. Hayakawa, Y. Abe and T. Agatsuma, Clin. Cancer Res., 2016, 22, 5097–5108 CrossRef CAS PubMed.
- Z. K. Glover, L. Basa, B. Moore, J. S. Laurence and A. Sreedhara, MAbs, 2015, 7, 901–911 CrossRef CAS PubMed.
- M. J. Tamás, S. K. Sharma, S. Ibstedt, T. Jacobson and P. Christen, Biomolecules, 2014, 4, 252–267 CrossRef PubMed.
- S. Kiese, A. Papppenberger, W. Friess and H.-C. Mahler, J. Pharm. Sci., 2008, 97, 4347–4366 CrossRef CAS PubMed.
- A. Lahlou, B. Blanchet, M. Carvalho, M. Paul and A. Astier, Ann. Pharm. Françaises, 2009, 67, 340–352 CrossRef CAS PubMed.
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS PubMed.
- Y. Le Basle, P. Chennell, N. Tokhadze, A. Astier and V. Sautou, J. Pharm. Sci., 2020, 109, 169–190 CrossRef CAS PubMed.
- J. L. Spidel, B. Vaessen, E. F. Albone, X. Cheng, A. Verdi and J. B. Kline, Bioconjug. Chem., 2017, 28, 2471–2484 CrossRef CAS PubMed.
- J. P. M. Nunes, V. Vassileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- Q. Zhou, Biomedicines, 2017, 5, 64 CrossRef PubMed.
- S. Smolskaya and Y. A. Andreev, Biomolecules, 2019, 9, 255 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- P. M. LoRusso, D. Weiss, E. Guardino, S. Girish and M. X. Sliwkowski, Clin. Cancer Res., 2011, 17, 6437–6447 CrossRef CAS PubMed.
- H. Kantarjian, E. Vandendries and A. Advani, N. Engl. J. Med., 2016, 375, 2100–2101 CrossRef PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- X. Hu, E. Bortell, F. W. Kotch, A. Xu, B. Arve and S. Freese, Org. Process Res. Dev., 2017, 21, 601–610 CrossRef CAS.
- Q. Jiang, B. Patel, X. Jin, D. Di Grandi, E. Bortell, B. Czapkowski, T. F. Lerch, D. Meyer, S. Patel, J. Pegg, A. Arbuckle, J. Lagliva, V. Sriskanda, L. Letendre, H. Li, E. Thomas and D. Nadkarni, ACS Omega, 2019, 4, 6468–6475 CrossRef CAS.
- C. Selby, L. R. Yacko and A. E. Glode, J. Adv. Pract. Oncol., 2019, 10, 68–82 Search PubMed.
- M. T. Kim, Y. Chen, J. Marhoul and F. Jacobson, Bioconjug. Chem., 2014, 25, 1223–1232 CrossRef CAS PubMed.
- N. C. Yoder, C. Bai, D. Tavares, W. C. Widdison, K. R. Whiteman, A. Wilhelm, S. D. Wilhelm, M. A. McShea, E. K. Maloney, O. Ab, L. Wang, S. Jin, H. K. Erickson, T. A. Keating and J. M. Lambert, Mol. Pharm., 2019, 16, 3926–3937 CrossRef CAS PubMed.
- S. Choi, S. Connelly, N. Reixach, I. A. Wilson and J. W. Kelly, Nat. Chem. Biol., 2010, 6, 133–139 CrossRef CAS PubMed.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, J. Am. Chem. Soc., 1964, 86, 1839–1842 CrossRef CAS.
- D. E. Ames and T. F. Grey, J. Chem. Soc., 1955, 631–636 RSC.
- M. Mentinova and S. A. McLuckey, J. Am. Chem. Soc., 2010, 132, 18248–18257 CrossRef CAS PubMed.
- S. Dou, J. Virostko, D. L. Greiner, A. C. Powers and G. Liu, Nucleosides, Nucleotides Nucleic Acids, 2015, 34, 69–78 CrossRef CAS PubMed.
- A. D. Ricart, Clin. Cancer Res., 2011, 17, 6417–6427 CrossRef CAS PubMed.
- K. Ulbrich and V. Subr, Adv. Drug Deliv. Rev., 2004, 56, 1023–1050 CrossRef CAS PubMed.
- J. F. DiJoseph, K. Khandke, M. M. Dougher, D. Y. Evans, D. C. Armellino, P. R. Hamann and N. K. Damle, Haematol. Meet. Reports, 2008, 2, 74–77 Search PubMed.
- V. H. J. van der Velden, J. G. te Marvelde, P. G. Hoogeveen, I. D. Bernstein, A. B. Houtsmuller, M. S. Berger and J. J. M. van Dongen, Blood, 2001, 97, 3197–3204 CrossRef CAS PubMed.
- E. R. Boghaert, K. M. Khandke, L. Sridharan, M. Dougher, J. F. Dijoseph, A. Kunz, P. R. Hamann, J. Moran, I. Chaudhary and N. K. Damle, Cancer Chemother. Pharmacol., 2008, 61, 1027–1035 CrossRef CAS PubMed.
- S. Kalkhof and A. Sinz, Anal. Bioanal. Chem., 2008, 392, 305–312 CrossRef CAS PubMed.
- X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjug. Chem., 2012, 23, 500–508 CrossRef CAS PubMed.
- K. Yamada, N. Shikida, K. Shimbo, Y. Ito, Z. Khedri, Y. Matsuda and B. A. Mendelsohn, Angew. Chem., Int. Ed., 2019, 58, 5592–5597 CrossRef CAS PubMed.
- Y. Matsuda, K. Yamada, T. Okuzumi and B. A. Mendelsohn, Org. Process Res. Dev., 2019, 23, 2647–2654 CrossRef CAS.
- R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47–76 CrossRef CAS.
- A. V Wisnewski, R. Srivastava, C. Herick, L. Xu, R. Lemus, H. Cain, N. M. Magoski, M. H. Karol, K. Bottomly and C. A. Redlich, Am. J. Respir. Crit. Care Med., 2000, 162, 2330–2336 CrossRef PubMed.
- G. R. Stark, Biochemistry, 1965, 4, 1030–1036 CrossRef CAS PubMed.
- J. M. Hettick, T. B. Ruwona and P. D. Siegel, J. Am. Soc. Mass Spectrom., 2009, 20, 1567–1575 CrossRef CAS PubMed.
- M. S. Rolph, A. L. J. Markowska, C. N. Warriner and R. K. O'Reilly, Polym. Chem., 2016, 7, 7351–7364 RSC.
- T.-T. Liu and T.-S. Yang, J. Food Sci., 2010, 75, C445–C451 CrossRef CAS PubMed.
- T. Nakamura, Y. Kawai, N. Kitamoto, T. Osawa and Y. Kato, Chem. Res. Toxicol., 2009, 22, 536–542 Search PubMed.
- D. S. Wilbur, M. K. Chyan, H. Nakamae, Y. Chen, D. K. Hamlin, E. B. Santos, B. T. Kornblit and B. M. Sandmaier, Bioconjug. Chem., 2012, 23, 409–420 CrossRef CAS PubMed.
- L. Petri, P. A. Szijj, Á. Kelemen, T. Imre, Á. Gömöry, M. T. W. Lee, K. Hegedus, P. Ábrányi-Balogh, V. Chudasama and G. M. Keseru, RSC Adv., 2020, 10, 14928–14936 RSC.
- I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjug. Chem., 2017, 28, 1452–1457 CrossRef CAS PubMed.
- J. I. Gavrilyuk, U. Wuellner and C. F. Barbas, Bioorganic Med. Chem. Lett., 2009, 19, 1421–1424 CrossRef CAS PubMed.
- M. Hayakawa, N. Toda, N. Carrillo, N. J. Thornburg, J. E. Crowe Jr. and C. F. Barbas III, ChemBioChem, 2012, 13, 2191–2195 CrossRef CAS PubMed.
- A. R. Nanna, X. Li, E. Walseng, L. Pedzisa, R. S. Goydel, D. Hymel, T. R. Burke Jr., W. R. Roush and C. Rader, Nat. Commun., 2017, 8, 1112 CrossRef PubMed.
- D. Hwang and C. Rader, Biomolecules, 2020, 10, 764 CrossRef CAS PubMed.
- D. Hwang, N. Nilchan, A. R. Nanna, X. Li, M. D. Cameron, W. R. Roush, H. J. Park and C. Rader, Cell Chem. Biol., 2019, 26, 1229–1239.e9 CrossRef CAS PubMed.
- J. Qi, K. Tsuji, D. Hymel, T. R. Burke, M. Hudecek, C. Rader and H. Peng, Angew. Chem., Int. Ed., 2020, 59, 12178–12185 CrossRef CAS PubMed.
- A. R. Nanna, A. V. Kel’in, C. Theile, J. M. Pierson, Z. X. Voo, A. Garg, J. K. Nair, M. A. Maier, K. Fitzgerald and C. Rader, Nucleic Acids Res., 2020, 48, 5281–5293 CrossRef CAS PubMed.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- S. Asano, J. T. Patterson, T. Gaj and C. F. Barbas, Angew. Chem., Int. Ed., 2014, 53, 11783–11786 CrossRef CAS PubMed.
- J. T. Patterson, H. D. Wilson, S. Asano, N. Nilchan, R. P. Fuller, W. R. Roush, C. Rader and C. F. Barbas, Bioconjug. Chem., 2016, 27, 2271–2275 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- M. M. C. Sun, K. S. Beam, C. G. Cerveny, K. J. Hamblett, R. S. Blackmore, M. Y. Torgov, F. G. M. Handley, N. C. Ihle, P. D. Senter and S. C. Alley, Bioconjug. Chem., 2005, 16, 1282–1290 CrossRef CAS PubMed.
- H. Liu and K. May, MAbs, 2012, 4, 17–23 CrossRef PubMed.
- T. Wang, Y. D. Liu, B. Cai, G. Huang and G. C. Flynn, J. Pharm. Biomed. Anal., 2015, 102, 519–528 CrossRef CAS PubMed.
- D. V. Nadkarni, Q. Jiang, O. Friese, N. Bazhina, H. Meng, J. Guo, R. Kutlik and J. Borgmeyer, Org. Process Res. Dev., 2018, 22, 286–295 CrossRef CAS.
- R. P. Lyon, D. L. Meyer, J. R. Setter and P. D. Senter, Methods Enzymol., 2012, 502, 123–138 CAS.
- D. G. Smyth, O. O. Blumenfeld and W. Konigsberg, Biochem. J., 1964, 91, 589–595 CrossRef CAS PubMed.
- T. Kuninori and J. Nishiyama, Agric. Biol. Chem., 1985, 49, 2453–2454 CAS.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjug. Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- M. R. Lewis and J. E. Shively, Bioconjug. Chem., 1998, 9, 72–86 CrossRef CAS PubMed.
- C. Wei, G. Zhang, T. Clark, F. Barletta, L. N. Tumey, B. Rago, S. Hansel and X. Han, Anal. Chem., 2016, 88, 4979–4986 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjug. Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- D. G. Smyth, A. Nagamatsu and J. S. Fruton, J. Am. Chem. Soc., 1960, 82, 4600–4604 CrossRef CAS.
- T. Kantner and A. G. Watts, Bioconjug. Chem., 2016, 27, 2400–2406 CrossRef CAS PubMed.
- H. Y. Shiu, T. C. Chan, C. M. Ho, Y. Liu, M. K. Wong and C. M. Che, Chem. - Eur. J., 2009, 15, 3839–3850 CrossRef CAS PubMed.
- E. Petit, L. Bosch, A. M. Costa and J. Vilarrasa, J. Org. Chem., 2019, 84, 11170–11176 CrossRef CAS PubMed.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef CAS PubMed.
- Y. Zhang, C. Zang, G. An, M. Shang, Z. Cui, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- G. J. L. Bernardes, G. Casi, S. Trüssel, I. Hartmann, K. Schwager, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2012, 51, 941–944 CrossRef CAS PubMed.
- T. List, G. Casi and D. Neri, Mol. Cancer Ther., 2014, 13, 2641–2652 CrossRef CAS PubMed.
- D. Zhang, N. O. Devarie-Baez, Q. Li, J. R. Lancaster and M. Xian, Org. Lett., 2012, 14, 3396–3399 CrossRef CAS PubMed.
- N. Toda, S. Asano and C. F. Barbas, Angew. Chem., Int. Ed., 2013, 52, 12592–12596 CrossRef CAS PubMed.
- M. Davydova, G. D. Le Roi, P. Adumeau and B. M. Zeglis, J. Vis. Exp., 2019, e59063 CAS.
- R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjug. Chem., 2012, 23, 392–398 CrossRef CAS PubMed.
- C. Zhang, P. Dai, A. A. Vinogradov, Z. P. Gates and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 6459–6463 CrossRef CAS PubMed.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS PubMed.
- M. A. Kasper, A. Stengl, P. Ochtrop, M. Gerlach, T. Stoschek, D. Schumacher, J. Helma, M. Penkert, E. Krause, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11631–11636 CrossRef CAS PubMed.
- O. Koniev, G. Leriche, M. Nothisen, J. S. Remy, J. M. Strub, C. Schaeffer-Reiss, A. Van Dorsselaer, R. Baati and A. Wagner, Bioconjug. Chem., 2014, 25, 202–206 CrossRef CAS PubMed.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J. Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjug. Chem., 2015, 26, 197–200 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 14001–14005 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- N. Shindo, A. Ojida, K. Tokunaga, M. Sato, K. Kuwata, C. Miura, H. Fuchida, N. Matsunaga, S. Koyanagi and S. Ohdo, J. Am. Chem. Soc., 2020, 142, 18522–18531 CrossRef PubMed.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103–8112 CrossRef CAS.
- C. Schneider, K. Niisuke, W. E. Boeglin, M. Voehler, D. F. Stec, N. A. Porter and A. R. Brash, Proc. Natl. Acad. Sci., 2007, 104, 18941–18945 CrossRef CAS PubMed.
- A. Abbas, B. Xing and T. P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- T. N. Tiambeng, D. S. Roberts, K. A. Brown, Y. Zhu, S. D. Mitchell, T. M. Guardado-alvarez, S. Jin and Y. Ge, Nat. Commun., 2020, 11, 3903 CrossRef CAS PubMed.
- A. J. Cameron, P. W. R. Harris and M. A. Brimble, Angew. Chem., Int. Ed., 2020, 59, 18054–18061 CrossRef CAS PubMed.
- M. R. Jafari, J. Lakusta, R. J. Lundgren and R. Derda, Bioconjug. Chem., 2016, 27, 509–514 CrossRef CAS PubMed.
- L. Pedzisa, X. Li, C. Rader and W. R. Roush, Org. Biomol. Chem., 2016, 14, 5141–5147 RSC.
- A. M. Tsimberidou, J. A. Ajani, P. Hsu, I.-J. Chen and T. E. Pearce, J. Clin. Oncol., 2020, 38, TPS3657 Search PubMed.
- M.-C. Yang, Y.-J. Chen, C.-S. Shia, H.-W. Chang, W.-F. Li, C.-D. T. Yu and I.-J. Chen, Cancer Res., 2019, 79, 4815 Search PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjug. Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- S. Balan, J. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjug. Chem., 2007, 18, 61–76 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chemie Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Medchemcomm, 2018, 9, 827–830 RSC.
- N. Martínez-Sáez, S. Sun, D. Oldrini, P. Sormanni, O. Boutureira, F. Carboni, I. Compañón, M. J. Deery, M. Vendruscolo, F. Corzana, R. Adamo and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 14963–14967 CrossRef PubMed.
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS PubMed.
- U. Hennrich and K. Kopka, Pharmaceuticals, 2019, 12, 114 CrossRef CAS PubMed.
- P. Grass, P. Marbach, C. Bruns and I. Lancranjan, Metabolism, 1996, 45, 27–30 CrossRef CAS.
- V. Vingtdeux, H. Zhao, P. Chandakkar, C. M. Acker, P. Davies and P. Marambaud, Mol. Med., 2016, 22, 841–849 CAS.
- R. Guermazi, L. Royer, L. Galmiche, G. Clavier, P. Audebert and A. Hedhli, J. Fluoresc., 2016, 26, 1349–1356 CrossRef CAS PubMed.
- C. Canovas, M. Moreau, C. Bernhard, A. Oudot, M. Guillemin, F. Denat and V. Goncalves, Angew. Chem., Int. Ed., 2018, 57, 10646–10650 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chemie Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- N. Griebenow, S. Bräse and A. M. Dilmac, RSC Adv., 2015, 5, 54301–54303 RSC.
- Y. Chen, W. Yang, J. Wu, W. Sun, T. P. Loh and Y. Jiang, Org. Lett., 2020, 22, 2038–2043 CrossRef CAS PubMed.
- L. H. Wu, S. Zhou, Q. F. Luo, J. S. Tian and T. P. Loh, Org. Lett., 2020, 22, 8193–8197 CrossRef CAS PubMed.
- A. Hung, M. Mager, M. Hembury, F. Stellacci, M. M. Stevens and I. Yarovsky, Chem. Sci., 2013, 4, 928–937 RSC.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- S. Sato, K. Nakamura and H. Nakamura, ACS Chem. Biol., 2015, 10, 2633–2640 CrossRef CAS PubMed.
- S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475–478 CrossRef CAS PubMed.
- S. Sato, K. Nakane and H. Nakamura, Org. Biomol. Chem., 2020, 18, 3664–3668 RSC.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- J. M. McFarland, N. S. Joshi and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 7639–7644 CrossRef CAS PubMed.
- D. W. Romanini and M. B. Francis, Bioconjug. Chem., 2008, 19, 153–157 CrossRef CAS PubMed.
- J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 Search PubMed.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134(17), 7406–7413 CrossRef CAS PubMed.
- J. Gavrilyuk, H. Ban, M. Nagano, W. Hakamata and C. F. Barbas, Bioconjug. Chem., 2012, 23, 2321–2328 CrossRef CAS PubMed.
- J. Gavrilyuk, H. Ban, H. Uehara, S. J. Sirk, K. Saye-Francisco, A. Cuevas, E. Zablowsky, A. Oza, M. S. Seaman, D. R. Burton and C. F. Barbas, J. Virol., 2013, 87, 4985–4993 CrossRef CAS PubMed.
- H. Nakata, K. Maeda, T. Miyakawa, S. Shibayama, M. Matsuo, Y. Takaoka, M. Ito, Y. Koyanagi and H. Mitsuya, J. Virol., 2005, 79, 2087–2096 CrossRef CAS PubMed.
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, Bioconjug. Chem., 2013, 24, 520–532 CrossRef CAS PubMed.
- H. Wamhoff and K. Wald, Chem. Ber., 1977, 110, 1699–1715 CrossRef CAS.
- Q. Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827–3832 RSC.
- E. L. Kovrigin and S. A. Potekhin, Biophys. Chem., 2000, 83, 45–59 CrossRef CAS PubMed.
- K. Gekko, E. Ohmae, K. Kameyama and T. Takagi, Biochim. Biophys. Acta, 1998, 1387, 195–205 CrossRef CAS.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- T. Dörner and D. M. Goldenberg, Ther. Clin. Risk Manag., 2007, 3, 953–959 Search PubMed.
- C. Song, K. Liu, Z. Wang, B. Ding, S. Wang, Y. Weng, C. W. Chiang and A. Lei, Chem. Sci., 2019, 10, 7982–7987 RSC.
- H. Tagawa, K. Maruyama, K. Sasaki, N. Konoue, A. Kishimura, M. Kanai, T. Mori, K. Oisaki and Y. Katayama, RSC Adv., 2020, 10, 16727–16731 RSC.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC.
- C. Zhu, Y. Gao, H. Li, S. Meng, L. Li, J. S. Francisco and X. C. Zeng, Proc. Natl. Acad. Sci., 2016, 113, 12946–12951 CrossRef CAS PubMed.
- J. C. Aledo, Protein Sci., 2019, 28, 1785–1796 CrossRef CAS PubMed.
- E. Folzer, K. Diepold, K. Bomans, C. Finkler, R. Schmidt, P. Bulau, J. Huwyler, H.-C. Mahler and A. Koulov, J. Pharm. Sci., 2015, 104, 2824–2831 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- J. Kim, B. X. Li, R. Y. C. Huang, J. X. Qiao, W. R. Ewing and D. W. C. Macmillan, J. Am. Chem. Soc., 2020, 142, 21260–21266 CrossRef CAS PubMed.
- C. Lu, W. Lin, W. Wang, Z. Han, S. Yao and N. Lin, Phys. Chem. Chem. Phys., 2000, 2, 329–334 RSC.
- J. T. Ryman and B. Meibohm, CPT pharmacometrics Syst. Pharmacol., 2017, 6, 576–588 CrossRef CAS PubMed.
- U. Storz, MAbs, 2015, 7, 989–1009 CrossRef CAS PubMed.
- E. Y. Yang and K. Shah, Front. Oncol., 2020, 10, 1182 CrossRef PubMed.
- O. N. Shilova and S. M. Deyev, Acta Naturae, 2019, 11, 42–53 CrossRef CAS PubMed.
- R. Seigneuric, J. Gobbo, P. Colas and C. Garrido, Oncotarget, 2011, 2, 557–561 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |