
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent syntheses of 5- and 6-membered aromatic heterocycles using group 4–8 transition metal catalysts
Daniel N.
Huh
 a,
Yukun
Cheng
a,
Connor W.
Frye
a,
Dominic T.
Egger
b and
Ian A.
Tonks
*a
a,
Yukun
Cheng
a,
Connor W.
Frye
a,
Dominic T.
Egger
b and
Ian A.
Tonks
*a
aUniversity of Minnesota Twin Cities, USA. E-mail: itonks@umn.edu
bETH Zurich, D-CHAB, Switzerland
First published on 29th June 2021
Abstract
In this Perspective, we discuss recent syntheses of 5- and 6-membered aromatic heterocycles via multicomponent reactions (MCRs) that are catalyzed by group 4–8 transition metals. These MCRs can be categorized based on the substrate components used to generate the cyclized product, as well as on common mechanistic features between the catalyst systems. These particular groupings are intended to highlight mechanistic and strategic similarities between otherwise disparate transition metals and to encourage future work exploring related systems with otherwise-overlooked elements. Importantly, in many cases these early- to mid-transition metal catalysts have been shown to be as effective for heterocycle syntheses as the later (and more commonly implemented) group 9–11 metals.
1. Introduction
Atom- and step-economy are increasingly important measures in the development of new synthetic processes.1 One strategy that can rapidly increase synthetic efficiency is the use of multicomponent reactions (MCRs). These reactions incorporate three or more separate or tethered functional groups to form multiple bonds in a single chemical process to generate a new product.2 MCRs can dramatically increase chemical complexity in a single step and often benefit from readily accessible, simple, and modular starting materials. Importantly, key classes of heterocycles (in particular 5- and 6-membered monocyclic aromatic heterocycles) can be synthesized by MCRs. The imperative to develop efficient routes to 5- and 6-membered heterocycles results from their ubiquitous application in pharmaceuticals,3–14 electronic materials,15–18 polymer science,5,13,19 pesticides,14,20 and dyes.5,13,14,21In this Perspective, we will discuss recent (10–15 years) group 4–8 catalyzed MCRs of 5- and 6-membered aromatic heterocycles. Many such catalytic MCRs have been demonstrated by group 9–11 metals.13,22–25 In recent years, the earlier group 4–8 metals have increasingly emerged as useful catalysts for 5- and 6-membered heterocycle syntheses, and often have advantages with respect to cost and earth abundance.26,27 Although groups 4–8 are often considered quite disparate in their properties, there is strikingly significant overlap in multicomponent synthetic strategies using these elements, in particular in the synthesis of pyrroles and pyridines (Fig. 1). While Ru catalysts have perhaps been most heavily investigated (owing to historically significant early reactions), earth-abundant26,27 Fe and Ti are also well-represented.
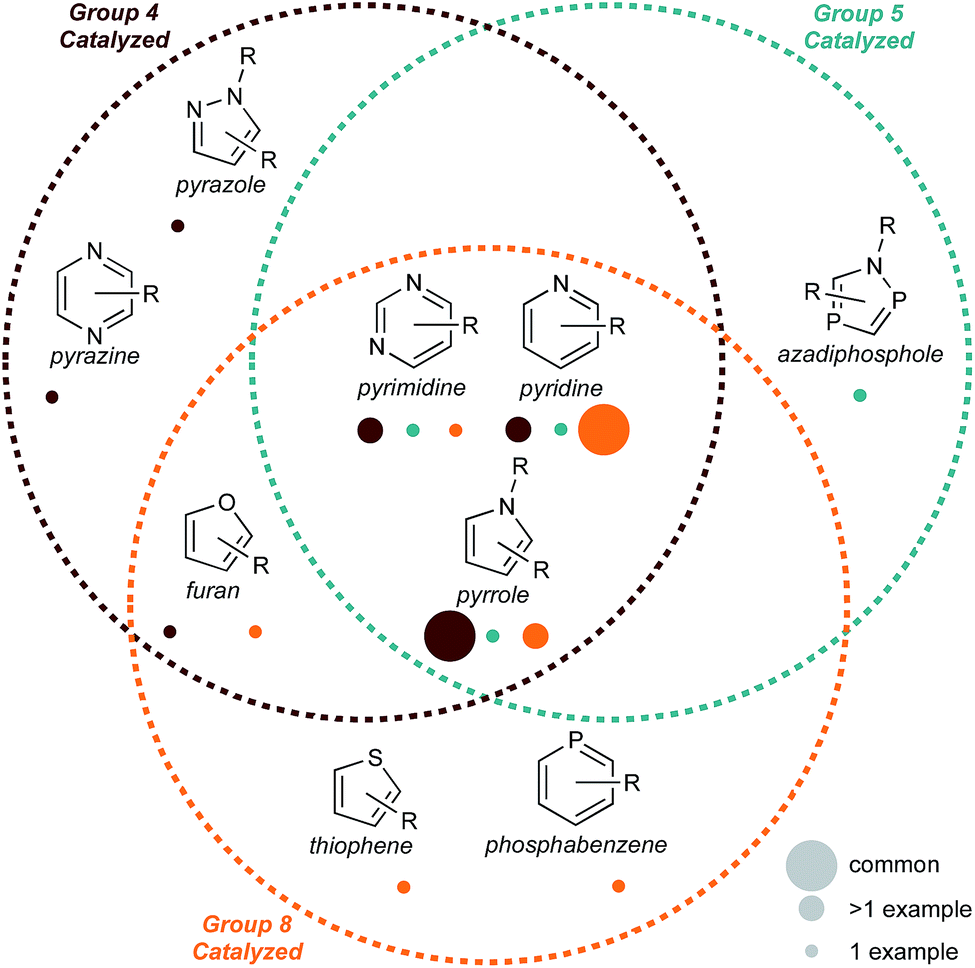 | ||
| Fig. 1 Summary of recent group 4–8 catalyzed multicomponent reactions for aromatic heterocycle synthesis discussed in this Perspective. Circle color is correlated to methods of synthesis from a particular catalyst group (4, 5, or 8), while size is correlated to the prevalence of methods (common, multiple examples, or 1 example). There are no examples of group 6 catalysts and only 1 example of group 7. | ||
This Perspective is organized by retrosynthetic strategies for each 5- and 6-membered heterocycle. There is a preponderance of examples of pyrrole and pyridine syntheses, which is likely a function of the importance of these heterocycles in bioactive molecules.20,28 Importantly, although pyridine synthesis is dominated by group 8 and pyrrole synthesis by group 4, there are similar mechanistic or retrosynthetic demonstrations with the other elements. These early hints into new and complementary reactivity demonstrate that there remains a substantial opportunity to explore the catalytic syntheses of other 5- and 6-membered heterocycles using group 4–8 transition metals. We hope that in highlighting these examples, common mechanistic threads will ultimately lead to new methods development across the early- and mid-transition metal series.
2. 5-Membered rings
2.1 Pyrroles, furans, and thiophenes
Monocyclic aromatic heterocycles such as pyrroles, furans, and thiophenes are typically prepared through condensation reactions like the Paal–Knorr cyclization of a 1,4-diketone with the appropriate heteroatomic nucleophile. However, the scope and efficacy of these condensation reactions are often limited by the need for specific substitution patterns or complex carbon skeleton preconstruction. As a result, there has been considerable effort in developing multicomponent approaches for the convenient construction of these heterocycles, some of which have been summarized previously.29,30 Because of their similar structures, approaches to the synthesis of the monocyclic aromatic heterocycles are often similar. In fact, several of the MCR strategies catalyzed by groups 4–8 can be applied across all three heterocycles, although the majority of demonstrated examples have been aimed toward the synthesis of pyrroles. Most methods involve combinations of on-metal cycloadditive or insertion reactions, combined with final heterocycle formation through electrocyclization (either on-metal or off-metal).When amines are used as the nitrogen source, pyrrole formation can be achieved from a cascading sequence of alkyne hydroamination followed by electrocyclization. For example, Ti-catalyzed hydroamination of 1,4-diynes (1) or 1,5-diynes (4)31via azatitanacyclobutene intermediates32,33 results initially in the formation of monoiminated 4-iminoalkyne (2) and 5-iminoalkyne (5) products, respectively (Fig. 2). These iminoalkyne intermediates can then undergo 5-endo-dig (4-iminoalkyne) or 5-exo-dig (5-iminoalkyne) cyclization to ultimately form 2,5-disubstituted pyrrole products. Either Ti(NMe2)2(dpma) (CAT1) or Ti(NMe2)2(dmpm) (dpma = di(pyrrolyl-α-methyl)methylamine; dmpm = 5,5-dimethyldipyrrolylmethane) (CAT2) can be used as the hydroamination catalyst to form the 4- or 5-iminoalkyne. In both reactions, the Ti catalyst is only responsible for the formation of the iminoalkyne product that subsequently cyclizes to form the pyrrole product. These reactions are compatible with both terminal and internal diynes featuring phenyl and alkyl (Me, nBu) groups. CAT1 was used exclusively for terminal diynes, while CAT2 was used exclusively for internal diynes. Unsubstituted 1,4-pentadiyne resulted in a double hydroamination product rather than pyrrole. However, substituted 1,4-diynes slowed the rate of the second hydroamination, allowing for pyrrole formation. Terminal substituted diynes (R1 = H) resulted in formation of the Markovnikov amination product in all cases. Similarly, high regioselectivity is obtained in reactions with unsymmetrical 1,4-diynes with aryl and alkyl substituents, due to selective amination β to the phenyl moiety. With some substrates such as 1-phenyl-1,4-pentadiyne, oligomerization of the diyne fragment prevented productive catalysis.
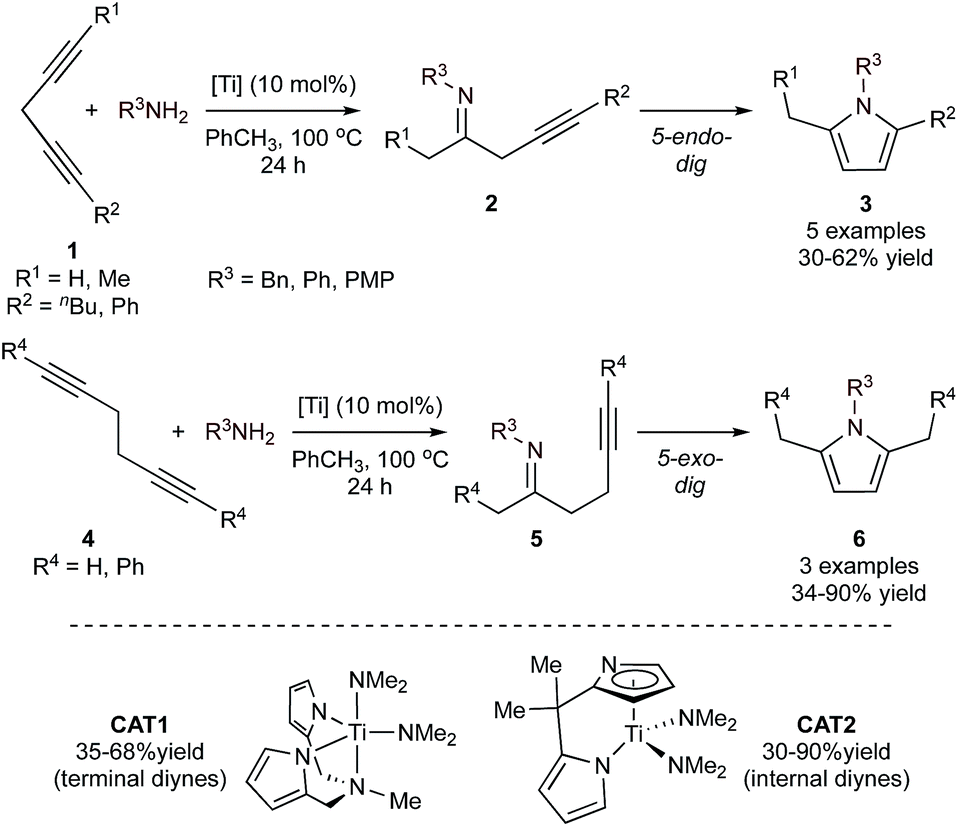 | ||
| Fig. 2 Ti-catalyzed [2 + 2 + 1] synthesis of pyrroles from diynes and amines through monohydroamination and subsequent 5-endo-dig or 5-exo-dig cyclization. | ||
Nitrene group transfer reactions catalyzed by Ti, V, and Ru have also been broadly used in the [2 + 2 + 1] synthesis of pyrroles. The first example of a nitrene group transfer strategy was reported in 2016,34 in the [2 + 2 + 1] multicomponent cycloaddition of alkynes and aryl diazenes catalyzed by simple Ti imido halides (Fig. 3), such as CAT3. The proposed catalytic cycle for this process, determined through combined experimental and computational studies,35–38 is highlighted in Fig. 3. First, a Ti![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NR imido (IM1) undergoes [2 + 2] cycloaddition with an alkyne to form azatitanacyclobutene IM2, identical to the first step of Ti-catalyzed hydroamination.32 Next, a second alkyne equivalent undergoes alkyne insertion into the reactive Ti–C bond of IM2 to form an azatitanacyclohexadiene (IM3),39 which then undergoes C–N reductive elimination to afford pyrrole and TiII. The Ti imido is then regenerated through disproportionation of the diazene oxidant. This reaction can be further extended to the synthesis of N-benzyl or 3° alkyl pyrroles using azides as the nitrene oxidant.37
NR imido (IM1) undergoes [2 + 2] cycloaddition with an alkyne to form azatitanacyclobutene IM2, identical to the first step of Ti-catalyzed hydroamination.32 Next, a second alkyne equivalent undergoes alkyne insertion into the reactive Ti–C bond of IM2 to form an azatitanacyclohexadiene (IM3),39 which then undergoes C–N reductive elimination to afford pyrrole and TiII. The Ti imido is then regenerated through disproportionation of the diazene oxidant. This reaction can be further extended to the synthesis of N-benzyl or 3° alkyl pyrroles using azides as the nitrene oxidant.37
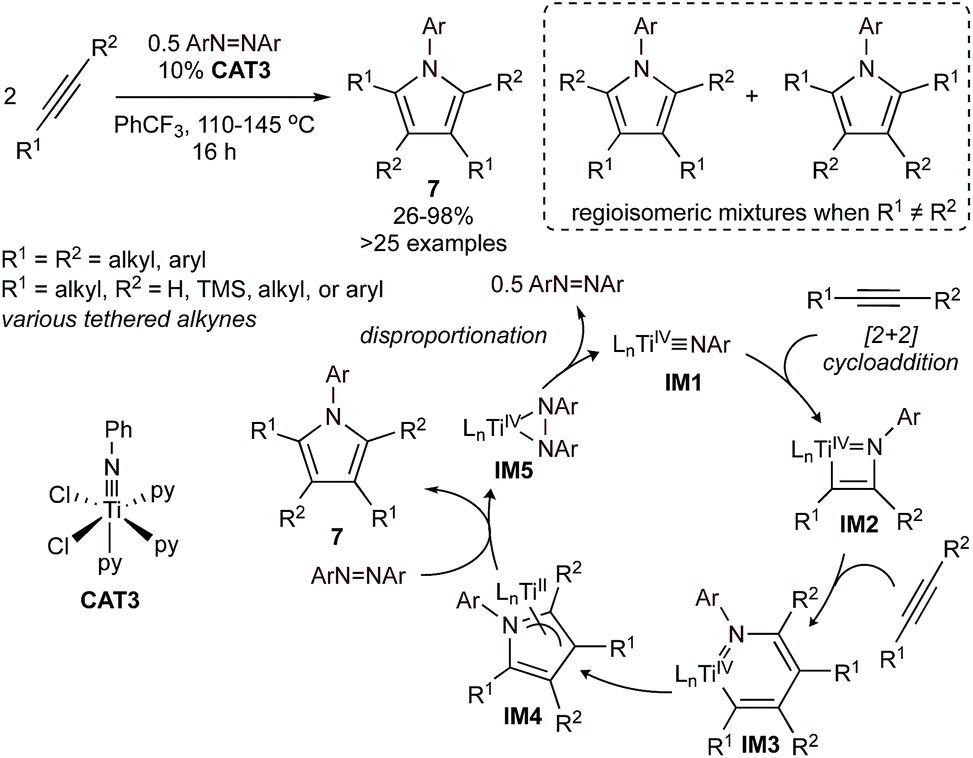 | ||
| Fig. 3 Top: Ti-catalyzed formal [2 + 2 + 1] synthesis of highly substituted N-aryl pyrroles from alkynes and diazenes. Bottom: simplified mechanism of Ti-catalyzed [2 + 2 + 1]. | ||
The initial substrate scope of this reaction included both terminal and internal alkynes, yielding access to otherwise synthetically inaccessible highly substituted electron-rich pyrroles. However, unsymmetrical alkynes typically yielded statistical distributions of potential regioisomers. Follow-up studies found that heteroatom-substituted alkynes 8 (ECCR, where E = SiR3, SnR3, 9-BBN) are suitable partners for highly chemo- and regioselective heterocoupling (Fig. 4) either with38 or without40,41 chelating directing groups. The resulting heteroatom-substituted pyrroles can be further functionalized, allowing for the synthesis of a wide variety of multi-substituted pyrroles, including natural products such as Lamellarin R. Catalyst control of chemo- and regioselectivity has proven more challenging, although a recent report used statistical analysis (ISPCA)42 to identify pyridine-substituted catalysts that could selectively homocouple phenylpropyne to 2,5-Me2-1,3,4-Ph3-1H-pyrrole.
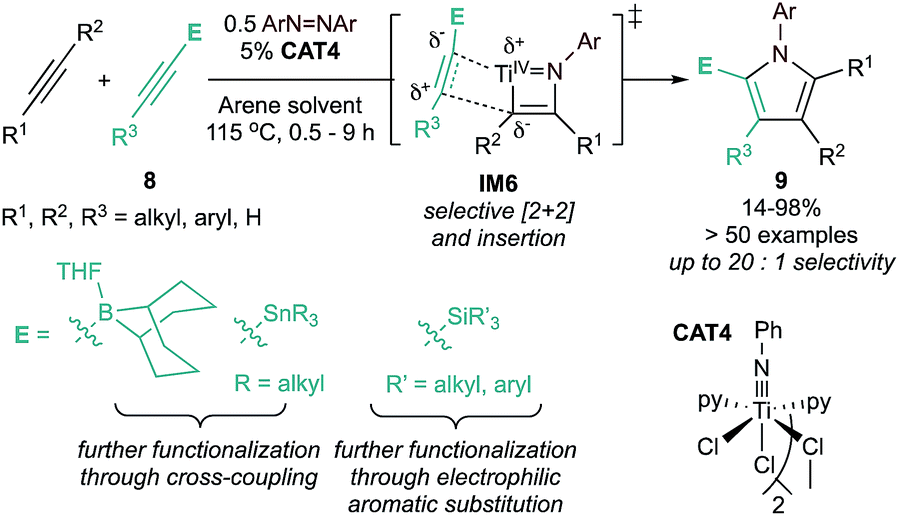 | ||
| Fig. 4 Strategies for regioselective pyrrole synthesis through alkyne heterocoupling. | ||
VIII/VV catalysis can also be used to form pyrroles via multicomponent group transfer of azobenzene with alkynes (Fig. 5).43 Here, VCl3(THF)3 and N,N-bis(trimethylsilyl)aniline are used to form a mono(imido)vanadium(III) species in situ that can be oxidized by azobenzene to a reactive bis(imido)vanadium(V) species (IM7). The key difference between the Ti and V systems is the need to π-overload44 V to engender reactivity of the otherwise less-reactive V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR bond toward alkyne [2 + 2] cycloaddition. While the overall scope of the V-catalyzed method is similar to Ti, the V-catalyzed methods appear to be slightly more regioselective and sensitive to steric effects; likely owing to the smaller metal center coupled with steric pressure from the 2nd imido ligand.
NR bond toward alkyne [2 + 2] cycloaddition. While the overall scope of the V-catalyzed method is similar to Ti, the V-catalyzed methods appear to be slightly more regioselective and sensitive to steric effects; likely owing to the smaller metal center coupled with steric pressure from the 2nd imido ligand.
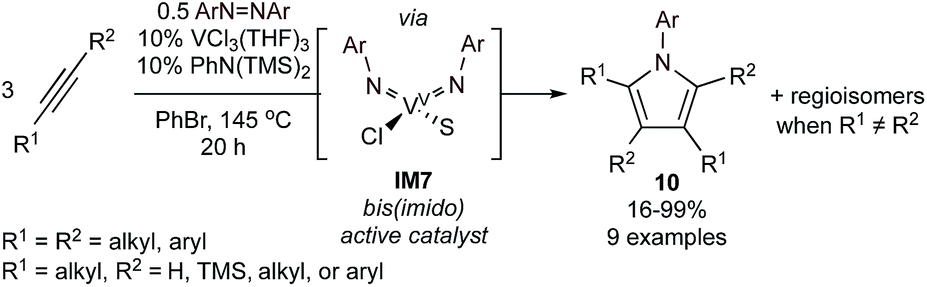 | ||
| Fig. 5 V-catalyzed [2 + 2 + 1] synthesis of highly substituted N-aryl pyrroles from alkynes and azobenzene. S = substrate or solvent. | ||
Sulfoximines (12) can also be used as nitrene sources in combination with α,ω-diynes (11) for [2 + 2 + 1] synthesis of pyrroles (Fig. 6).45 This reaction proceeds by forming a key biscarbenoid ruthenacycle (IM8) by initial oxidative cyclization of the α,ω-diyne (this intermediate will be discussed in more detail in the context of pyridine synthesis, vide infra). Subsequent nitrogen transfer from a sulfoximine occurs to produce an azaruthenacyclohexatriene complex (IM9), which then undergoes cycloisomerization to a η5-pyrrole complex (IM10). Release of the pyrrole and oxidative cyclization of another diyne substrate regenerates IM8. In contrast to the Ti- and V-catalyzed [2 + 2 + 1] nitrene transfer reactions, in this Ru example the diyne reacts first, followed by nitrogen transfer. Because of the challenging formation of IM8, the substrate scope of this reaction is limited to tethered diynes. However, in many ways, the Ru catalyzed substrate scope is complementary to that of Ti and V: tethered heteroatoms are tolerated here but not with Ti, and Ru provides access to N-alkyl (vs. N-aryl) pyrroles. A preliminary screen of S=NMe sulfoximines showed that symmetric diphenyl or dialkyl sulfoximines were inefficient, resulting in low yields (2–25%). However, through optimization it was found that using a p-fluorophenyl substituent (Fig. 6, bottom left) produced drastically increased yields (67%). Varying the N-substituent of the sulfoximine (e.g. n-decyl) to more sterically demanding groups required larger sulfoximine loadings (2 equiv.).
 | ||
| Fig. 6 Top: Ru-catalyzed [2 + 2 + 1] synthesis of bicyclic N-alkyl pyrroles from diynes and sulfoximines. Bottom: mechanism for the Ru-catalyzed [2 + 2 + 1] synthesis of pyrroles from diynes and sulfoximines. | ||
Similar Ru-catalyzed MCRs have also been reported for the synthesis of furans and thiophenes. For example, reaction of tethered diynes (11) and 5 equiv. of dimethylsulfoxide (DMSO) (14) or 1 equiv. of a nitrone (15) in the presence of a cyclopentadienyl–Ru catalyst yields bicyclic furans (17) (Fig. 7),46–48 while reaction with thiocarbonyls (16) yields bicyclic thiophenes (18).49 The mechanisms of these reactions closely mirror the similar Ru-catalyzed pyrrole reactions, wherein IM8 undergoes reaction with the “E1” source prior to cyclization.
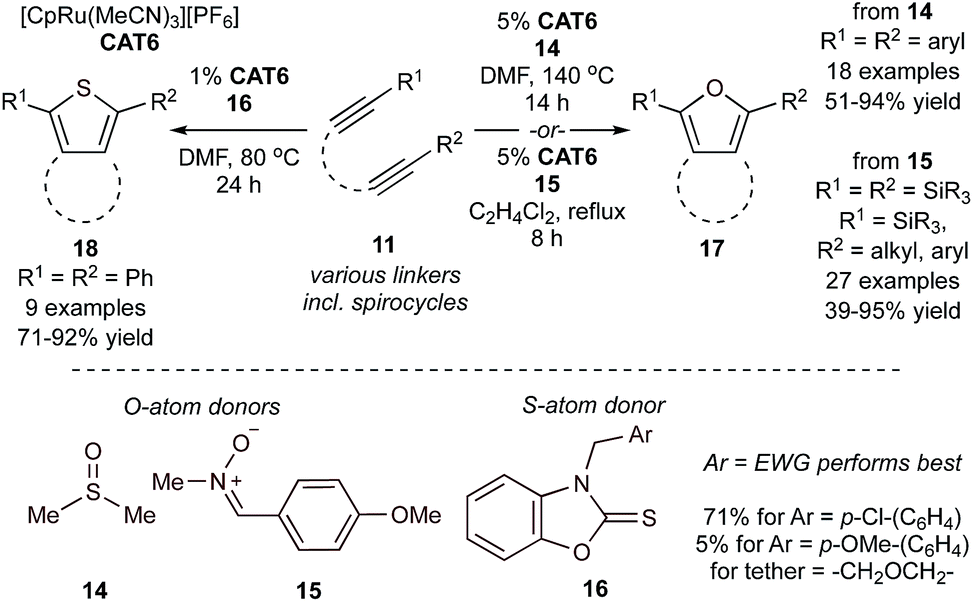 | ||
| Fig. 7 Ru-catalyzed [2 + 2 + 1] synthesis of furans and thiophenes from O- and S-atom donors. | ||
Catalytic syntheses of furans (17) using DMSO (14) were possible using both electron-donating and -deficient aryl diynes, providing >90% conversion. Notably, the electron-deficient aryl diynes underwent shorter reaction times. These reactions were also found to tolerate ester, ketone, nitrile, and sulfone functional groups. Reactions using the nitrone group transfer reagent 15 expanded functional group tolerance on the diyne fragment to include bulky silyl groups. This increased tolerance was attributed to the milder reaction conditions (refluxing 1,2-Cl2C2H4, approx. 84 °C).
In the catalytic syntheses of thiophenes (18) using thiocarbonyl group transfer reagents (16), it was found that electron-deficient thiocarbonyls (Ar = p-Cl-C6H4, 71%) generated thiophenes in higher yields than electron-rich thiocarbonyls (Ar = p-OMe-C6H4, 5%). Like the furan [2 + 2 + 1] reactions, both electron-rich and -deficient aryl diynes generated thiophenes in excellent yields ranging from 71–92%. Alkyl diynes also generated thiophenes, however at more moderate yields (50–62%). These reactions demonstrated similar functional group tolerance to the furan and pyrrole reactions, although TMS-substituted (TMS = trimethylsilyl) diynes were desilylated upon thiophene formation. Diynes containing the bulkier TBS (TBS = tert-butyldimethylsilyl) group generated a mixture of desilylated and silylated thiophene in 36% and 49% yield, respectively.
 | ||
| Fig. 8 Top: Cp2TiCl2-catalyzed [2 + 2 + 1] synthesis of multi-substituted 1H-pyrroles from terminal alkynes and nitriles. Bottom: Cp2TiCl2-catalyzed [2 + 2 + 1] synthesis of multi-substituted furans from alkynes and carboxylic esters. | ||
This reaction can be further used for the synthesis of furans (22) from alkynes and esters (Fig. 8, bottom).52,53 Here, the metallacyclopropene (M = Ti, Zr) intermediate undergoes insertion with two equivalents of ester forming a dioxametallacycloheptene, which is then converted to 21 following transmetallation with EtAlCl2. The proposed mechanism of cyclization upon transmetallation to Al/aqueous workup is different than that of the pyrrole reaction: here, Al is directly involved in elimination of the alkoxide substituents from 21 prior to cyclization. Catalysis with Cp2ZrCl2 was also successful, but compared to smaller Cp2TiCl2 (CAT7) the use of the larger Zr resulted in formation of side products from the dimerization or trimerization of the alkyne. Unlike in the synthesis of 1H-pyrroles, symmetrical internal alkynes with various alkyl substituents were shown to be applicable for furan synthesis. A number of esters were examined and it was found that conjugated substituents will not react under the examined conditions, but the reaction does proceed with alkyl, aryl, and heteroaryl rings that are separated from the carbonyl by a methylene unit (not conjugated).
1,2,3,5-Tetrasubstituted pyrroles (25) can be synthesized through FeCl3 (Lewis acid)-catalyzed domino Michael addition/cyclization of amines, dialkyl acetylenedicarboxylates (23), and phenacyl bromides (24) (Fig. 9, top).54 This reaction is proposed to occur through initial hydroamination of the alkyne (Michael addition) to an enamine (IM13), followed by FeCl3-promoted nucleophilic addition to phenacyl bromide to form an enaminone (IM14). Spontaneous cyclization then produces the pyrrole product. This protocol was later extended to include β-nitrostyrenes (26) in place of phenacyl bromide (Fig. 9, bottom) through a similar mechanism.55 These protocols provide complementary regioselectivity, with phenacyl bromides yielding 1,2,3,5-substituted pyrroles (25) and β-nitrostyrene derivatives yielding 1,2,3,4-substituted pyrroles (27).
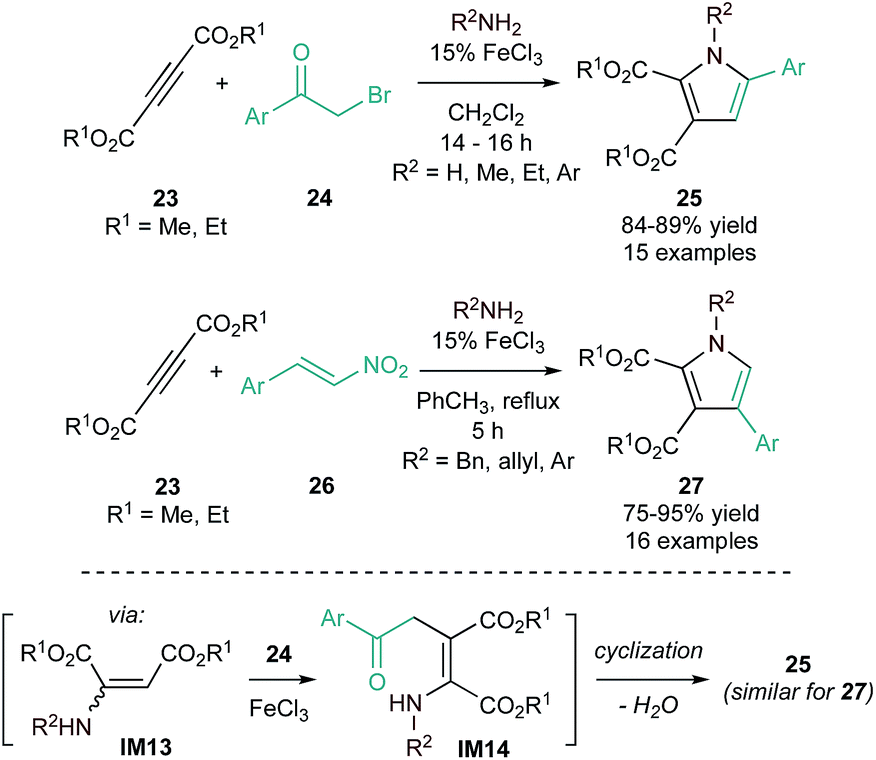 | ||
| Fig. 9 [2 + 2 + 1] synthesis of tetra-substituted pyrroles catalyzed by FeCl3 using highly activated alkynes and amines with either phenacyl bromides (top) or β-nitrostyrenes (middle), along with a proposed mechanism (bottom). | ||
NR imidos and alkynes can also be intercepted in other ways to generate pyrroles via “interrupted hydroamination” reactions. For example, reaction of alkynes, isonitriles, and arylamines catalyzed by a Ti indolyl catalyst (CAT8) yields 2,3-diaminopyrroles (28) via a [2 + 1 + 1 + 1] iminoamination reaction (Fig. 10).56 Here, the key reaction step is sequential 1,1-insertions of an isonitrile into the azatitanacyclobutene intermediate (IM16) to form first IM17 then IM18. Subsequent protolytic cleavage by an amine releases an enamine formimine (IM19) that undergoes cyclization and tautomerization to the final product. This method is suitable for both terminal and internal alkynes (symmetrical and unsymmetrical), however, internal alkynes require harsher conditions. A number of aniline derivates were successfully implemented in this reaction, but ortho-substituted anilines resulted in the formation of formamidine or iminoamination products from competing side reactions.
 | ||
| Fig. 10 Top: Ti-catalyzed [2 + 1 + 1 + 1] synthesis of N-aryl 2,3-diaminopyrroles from isonitriles, arylamines, and unactivated alkynes. Bottom: mechanism for Ti-catalyzed synthesis of 2,3-diaminopyrroles via an iminoamination reaction that occurs through “interrupted alkyne hydroamination”. | ||
Although it only involves two components (and is thus not formally an MCR), an interesting example of Ru-catalyzed synthesis of multi-substituted 1H-pyrroles (31) is the formal [3 + 2]-cycloaddition between 2H-azirines (30) and activated alkynes (29) (Fig. 11).57 This reaction was proposed to occur through the initial ring-opening of 2H-azirines by a Ru catalyst (CAT9) to form an azaruthenacyclobutene intermediate (IM21). This intermediate (and its subsequent reactivity) is similar to the intermediates of TiNR and alkyne [2 + 2] cycloaddition (IM2, Fig. 3). A follow-up computational study58 revealed that IM21 has significant nitrene character and undergoes RuN + alkyne [2 + 2] cycloaddition to IM22 rather than Ru–C alkyne insertion (Ti–C insertion occurs in IM2 to IM3). Subsequent C–N reductive elimination and isomerization affords the pyrrole product. This method is limited to activated alkynes (29) with electron-drawing substituents such as esters and ketones. The use of activated alkynes ensures insertion is selective, resulting in a single regioisomer. The reaction is sensitive to the sterics of both the carbonyl substituents and alkyne substituent when R1 = aryl, where ortho substitution was observed to greatly hinder reactivity. In contrast, a wide range of 2H-azirenes was found to be compatible, with minimal impact from electronics or sterics of the substituents.
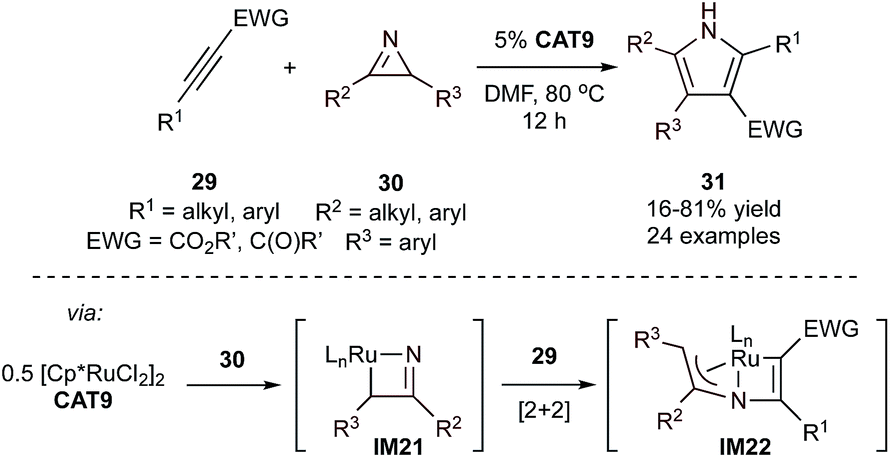 | ||
| Fig. 11 Top: Ru-catalyzed [3 + 2] synthesis of multi-substituted 1H-pyrroles from activated alkynes and 2H-azirines. Bottom: DFT-proposed mechanism proceeds through C–N oxidative addition followed by Ru–N + alkyne [2 + 2] cycloaddition. | ||
2.2 Pyrazoles
Knorr condensation of hydrazines with 1,3-diketones or 1,3-dipolar cycloaddition of hydrazones with alkynes are the most commonly used synthetic routes to multi-substituted pyrazoles.7 Multicomponent strategies toward pyrazoles have mirrored these methods, and include several strategies for generating C3 frameworks in situ that can undergo ring closure via hydrazine condensation. Unlike pyrroles, there are only a few examples of MCRs with group 4–8 catalysts.An early example of a MCR for pyrazoles synthesis includes a 3-component reaction using alkynes that proceeds through a 1-pot, 2-step Ti-catalyzed iminoamination/hydrazine condensation sequence (Fig. 12, top).59 In the first step, alkynes undergo iminoamination to 1,3-diimine tautomers (IM23) via an interrupted alkyne hydroamination sequence (vide supra), where the [2 + 2] cycloaddition intermediate is intercepted by 1 equiv. of isonitrile prior to protonolysis. IM23 can then undergo Knorr-like cyclization through condensation with hydrazine in a second step. Both terminal and internal alkynes are tolerated, however, the method is complicated by the potential for mixtures of regioisomers to be formed during hydrazine condensation when using terminal or unsymmetrical internal alkynes. Typically, the reactions were generally found to favor 1,4-disubstituted pyrazoles with terminal alkynes (28, where R1 = alkyl or aryl, R2 = H).60 Similarly, for internal alkynes such as 1-phenylpropyne, there is preferential formation of the regioisomer with the aryl group in the 4-position (28, R2 = aryl). The formation of 1,3-disubstituted pyrazoles is not generally favored due to preferential attack onto the aldimine carbon by the hydrazine NH2 during the condensation step. However, a 3-substituted pyrazole was favored (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) over the 4-substituted product with CAT1 if H2NNH2 and 1-hexyne are used.
:1) over the 4-substituted product with CAT1 if H2NNH2 and 1-hexyne are used.
 | ||
| Fig. 12 Formal [2 + 2 + 1] syntheses of pyrazoles from alkynes, isonitriles, and hydrazines. Top: 2-step Ti-catalyzed iminoamination/condensation sequence. Bottom: improved 1-step Ti-catalyzed iminohydrazination/cyclization strategy. | ||
This 2-step sequence was later improved to a multicomponent synthesis of pyrazoles starting from terminal alkynes, isonitriles, and hydrazines (Fig. 12, bottom).61 The key advance of this reaction was using hydrazine (rather than cyclohexylamine) to perform an interrupted alkyne hydrohydrazination, wherein the resultant 1,3 imine-hydrazone (IM24) can undergo direct cyclization with loss of H2NR to form the pyrazole product. Unlike the 2-step process, this single-step reaction is limited to terminal alkynes featuring small alkyl and aryl groups. A single 1,3-disubstituted regioisomer (29) was able to be obtained in all cases due to the selectivity of the [2 + 2]-cycloaddition in which the substituent of the terminal alkyne is always opposite to the metal center.
2.3 Imidazoles
Simple imidazoles can be synthesized by condensation of glyoxal, formaldehyde, and ammonia. Syntheses of more complex imidazoles from MCRs vary widely,62 although there are no formal MCRs catalyzed by group 4–8 metals. However, catalytic [3 + 2] annulations with Ti provide insights into potential strategies that could be extended to MCRs. For example, highly regioselective formation of multisubstituted imidazoles (31) from propargylamines (30) and nitriles can be accomplished using a titanacarborane amide catalyst (CAT11) (Fig. 13).63 This reaction is proposed to proceed through an initial amine-exchange reaction between the amide catalyst and the propargylamine substrate to afford a titanacarborane amide (IM25). Coordination of a nitrile (IM26) and subsequent migratory insertion (IM27) is followed by intramolecular insertion of the alkyne into the Ti–N bond to form IM28. This then undergoes protonolysis to regenerate the initial Ti-amido species and liberate the imidazole product upon isomerization of IM29. This reaction is notable in that it does not proceed through a TiNR imido, unlike most of the other Ti-catalyzed N-heterocycle syntheses herein. An extensive scope revealed that alkyl-, aryl- and heteroaryl-nitriles are all tolerated. Arylnitriles were observed to be more reactive than alkylnitriles, especially when featuring donor ortho-substituents. Terminal and internal propargylamines, as well as N-functionalized propargylamines, are also tolerated. This reaction was further extended for the synthesis of 2-aminoimidazoles by replacing the nitrile fragment with carbodiimines.64
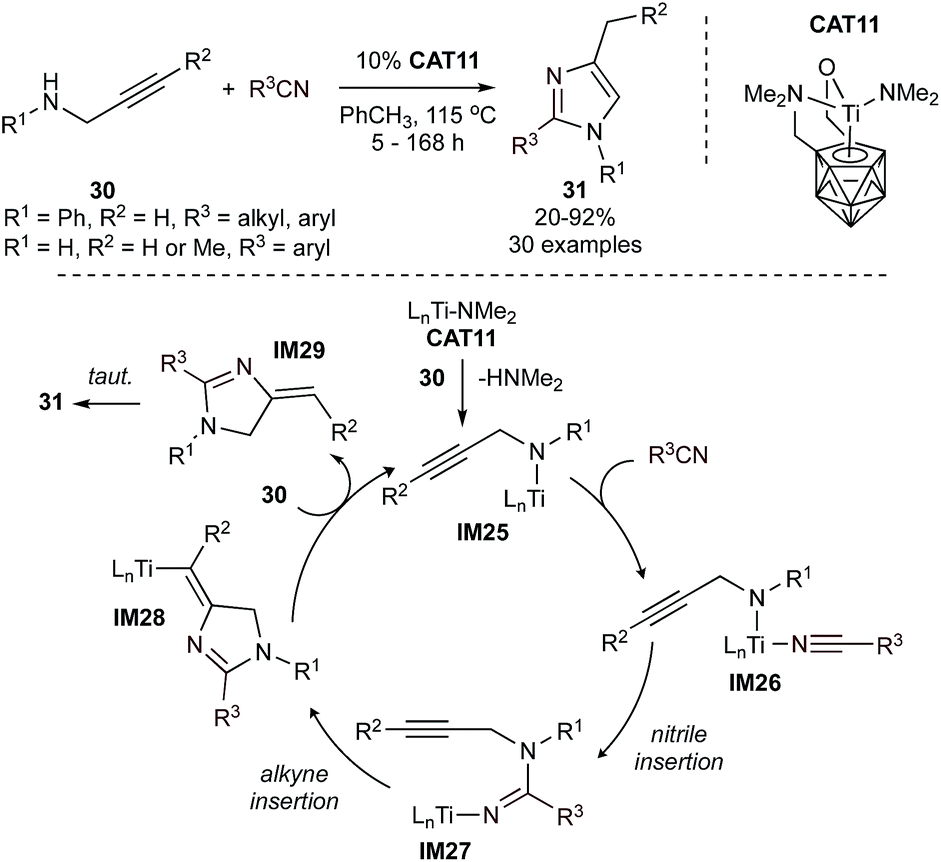 | ||
| Fig. 13 Ti-catalyzed [3 + 2] annulation of propargylamines with nitriles yields imidazoles. | ||
2.4 Azadiphospholes
Azadiphospholes are 5-membered aromatic heterocycles containing 2 P atoms and 1 N atom. The general metal-mediated synthesis of these heterocycles was, until recently, limited to only a handful of stoichiometric couplings of phosphaalkynes with simple V and Ti imido complexes,65,66 yielding 1,2,4-azadiphosphole.67 Building off of this stoichiometric example, a V-catalyzed [2 + 2 + 1] synthesis of multisubstituted azadiphospholes (33) from phosphaalkynes (32) and azobenzene was recently reported (Fig. 14).68 Similarly to Ti- and V-catalyzed [2 + 2 + 1] pyrrole syntheses (Fig. 3 and 5), the mechanism of this reaction is proposed to go through initial [2 + 2] cycloaddition of the phosphaalkyne with a VNR imido, followed by further phosphaalkyne insertion before reductive elimination. Phosphaalkynes are highly reactive functional groups, and as such, the scope was limited to bulky tertiary phosphaalkynes such as adamantyl or dimethylbenzyl phosphaalkyne. A variety of substituted azobenzenes were examined with adamantyl phosphaalkyne giving yields of 47–66%.
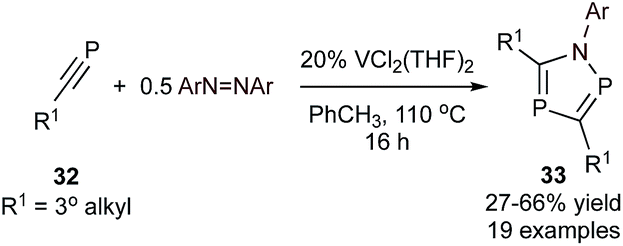 | ||
| Fig. 14 [2 + 2 + 1] V-catalyzed synthesis of azadiphospholes from phosphaalkyne and diazene substrates. | ||
3. 6-Membered heterocycles
3.1 Pyridines
The multicomponent synthesis of pyridines has been extensively studied, in particular strategies involving the formal [2 + 2 + 2] cycloaddition of alkynes and nitriles.11,13,22,69–74 This topic has been the subject of numerous recent reviews, so this perspective will focus primarily on comparing and contrasting mechanistic and scope aspects of the group 4–8 catalysts. In the context of group 4–8 metal-catalyzed [2 + 2 + 2] syntheses, methods are primarily dominated by Ru and Fe, although there are several emergent examples with group 4 and 5 that suggest they may have unrealized potential for pyridine synthesis as well. These formal [2 + 2 + 2] reactions can be categorized into 2 classes: (1) partially intramolecular tethered substrates such as diynes or alkynylnitriles that avoid inherent problems of chemo- and regioselectivity; and (2) fully intermolecular 3-component reactions. The reactions typically occur through either a metallacyclopentadiene-like intermediate (IM30, alkyne + alkyne first) or an azametallacyclopentadiene-like intermediate (IM31, alkyne + nitrile first) (Fig. 15). Depending on the metal, IM30 can be considered a true metallacyclopentadiene (wherein the metal has undergone 2-electron oxidative coupling—typically invoked for groups 4 or 5) or a biscarbenoid metallacycle (typically invoked for Ru). These pathways are also believed to be operative in the analogous [2 + 2 + 2] reactions catalyzed by late transition metals such as Co,75 Ir76 (alkyne + alkyne) and Ni77,78 (alkyne + nitrile) respectively.12,79 | ||
| Fig. 15 Comparison of potential first steps in pyridine synthesis via [2 + 2 + 2] cycloaddition of alkynes and nitriles. | ||
Ru-catalyzed [2 + 2 + 2] diyne + nitrile methods are typified by the Cp*RuCl(COD) (CAT12)-catalyzed reaction shown in Fig. 16. In initial work from 2001,80 unsymmetric 1,6-diynes were reacted with several nitriles to yield pyridine products, 35 and 35′. The regiochemistry can be influenced by reacting 34 with electron-deficient nitriles to favor 35. Alternatively, 35′ can be isolated if both the diyne and nitrile contain an electron-withdrawing group (EWG). Compared to other transition metals, Ru-based catalysts provide complementary access to pyridines with electron-deficient nitriles,80–82 which are often challenging substrates for later transition metals like Co.83 In contrast, cycloaddition using simple nitriles such as MeCN and PhCN is challenging with Ru due to often endothermic nitrile insertion,81 although examples of partially or fully intermolecular [2 + 2 + 2] cycloaddition with nitriles containing coordinating groups have been reported.81,84,85
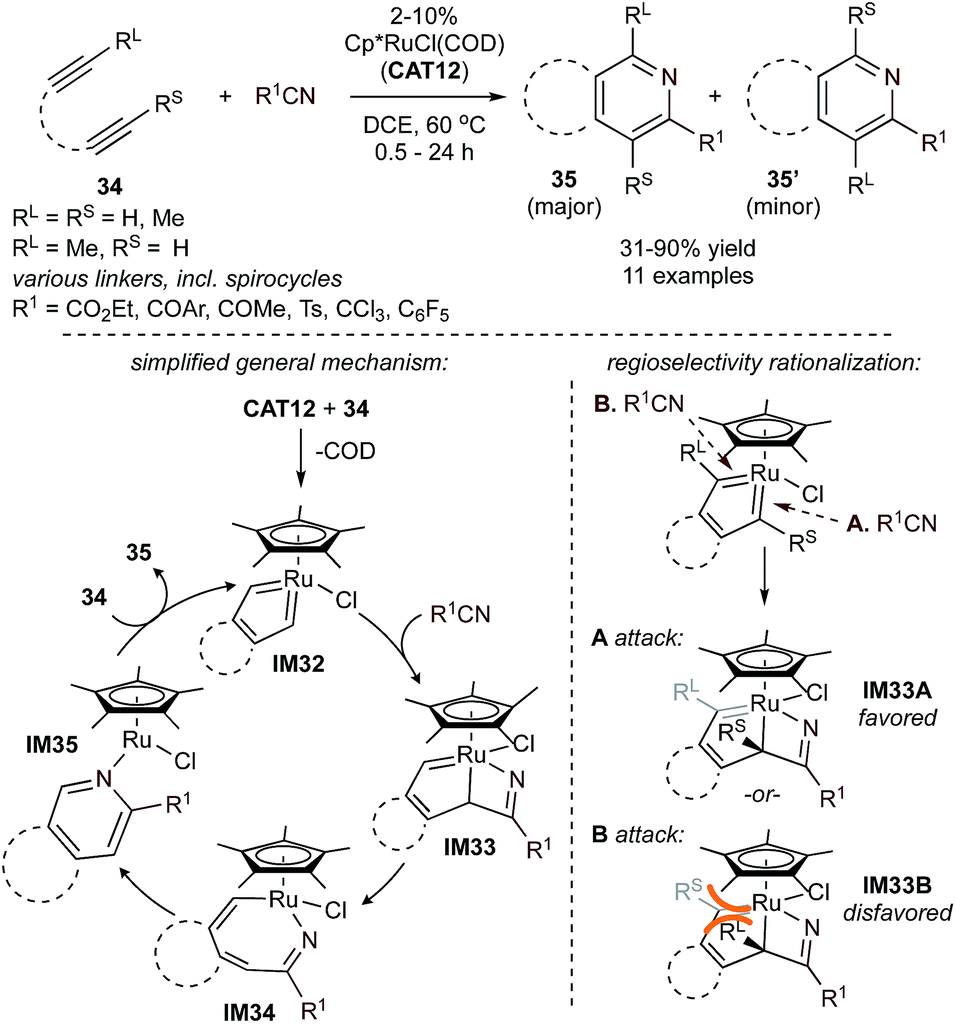 | ||
| Fig. 16 CAT12-catalyzed [2 + 2 + 2] pyridine synthesis from tethered diynes and nitriles. Bottom: initial mechanistic proposal (left, RL and RS omitted for clarity) and regioselectivity rationalization (right).80 | ||
Detailed experimental and computational studies (both of [2 + 2 + 2] alkyne cyclotrimerization and pyridine formation)22,86 have revealed that Cp*RuCl(COD)-catalyzed reactions of α,ω-diynes likely proceed first through initial oxidative cyclization of the diyne to form ruthenacyclic biscarbenoid IM32. This intermediate, which related complexes have been structurally characterized via X-ray crystallography and NMR in several instances,87 is shared between [2 + 2 + 2] pyridine formation, alkyne cyclotrimerization, and [2 + 2 + 1] pyrrole/furan/thiophene synthesis (vide supra). Nitrile then coordinates to the ruthenacycle to form a η1-or η2-nitrile complex (see Fig. 17, IM36B′ or IM36B), with the η2-nitrile being the productive intermediate despite being less thermodynamically stable. Insertion of nitrile leads to the Ru-bridged intermediate azaruthenabicyclo[3.2.0]heptatriene IM33, followed by Ru–C bond cleavage to afford an azaruthenacycloheptatriene IM34. Finally, reductive elimination yields pyridine and regenerates the Ru catalyst. Calculations on the coupling of acetylene with trifluoroacetonitrile by CpRuCl(COD) (CAT13) agree with this general mechanistic picture (Fig. 17). In this case, the biscarbenoid ruthenacycle route (path A) is both thermodynamically and kinetically favorable with ΔΔG‡ = −5.5 kcal mol−1 between TS1A and TS1B and ΔΔG = −17.1 kcal mol−1 between IM37A and IM37B. Later computational analysis further corroborates the predominance of the “biscarbenoid ruthenacycle” path A over path B.22,86
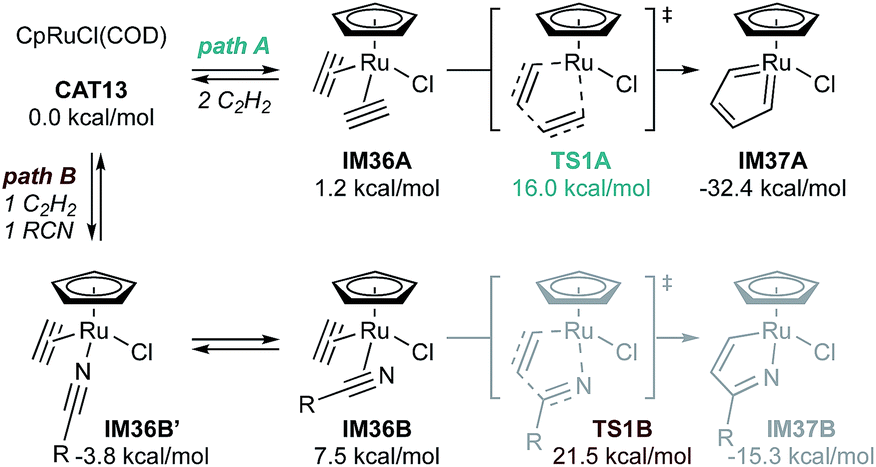 | ||
| Fig. 17 Computed free energies of potential alkyne + alkyne coupling (path A) and alkyne + nitrile coupling (path B) in Ru-catalyzed [2 + 2 + 2] pyridine synthesis, demonstrating kinetically and thermodynamically favored formation of IM36A. B3LYP; single point free energies calculated with SDD (Ru) and 6-311++G(d,p) for all other elements. R = CF3. | ||
Although mechanistic investigations point primarily to the “biscarbenoid ruthenacycle” mechanism for these Ru-catalyzed partially intramolecular couplings, the regiochemical outcome of the intermolecular [2 + 2 + 2] of ethyl cyanoformate with 2 equiv. of ethyl propiolate indicates that the “azametallacyclopentadiene” route may still play a role (Fig. 18). In this instance, only 2 of the 4 possible regioisomers of 36 are produced, which are hypothesized to arise solely from IM38. Here, selective formation of IM38 is proposed to arise from the polarization of the propiolate. Indeed, given the relatively small ΔΔG‡ = 5.5 kcal mol−1 calculated for acetylene (Fig. 17), it is reasonable to expect that either pathway may play a role depending on stereoelectronic modifications to the substrates or the catalyst structure.
 | ||
| Fig. 18 Regiochemical outcome of CAT12-catalyzed [2 + 2 + 2] cycloaddition of ethyl propiolate supports an “alkyne + nitrile” mechanism through IM38.81 | ||
Further studies have revealed subtle catalyst structure effects on reaction selectivity and mechanism. In a demonstration of switchable selectivity, it was found that CAT12-catalyzed [2 + 2 + 2] reactions of tethered diynes (37) with conjugated alkynylnitriles (38) could yield either cyano-substituted benzenes (39) or alkynylpyridines (40), in either the absence or presence of AgOTf, respectively (Fig. 19).88 In this example, DFT calculations indicated that the more electron-rich neutral Cp*Ru(metallacycle) (IM39) preferred coordination of the π-acid alkyne of the alkynylnitrile, leading to formation of cyanobenzene, while the electron-deficient cationic Cp*Ru(metallacycle) (IM40), generated by halide abstraction by Ag+, preferred coordination of the nitrile, leading to pyridine formation.
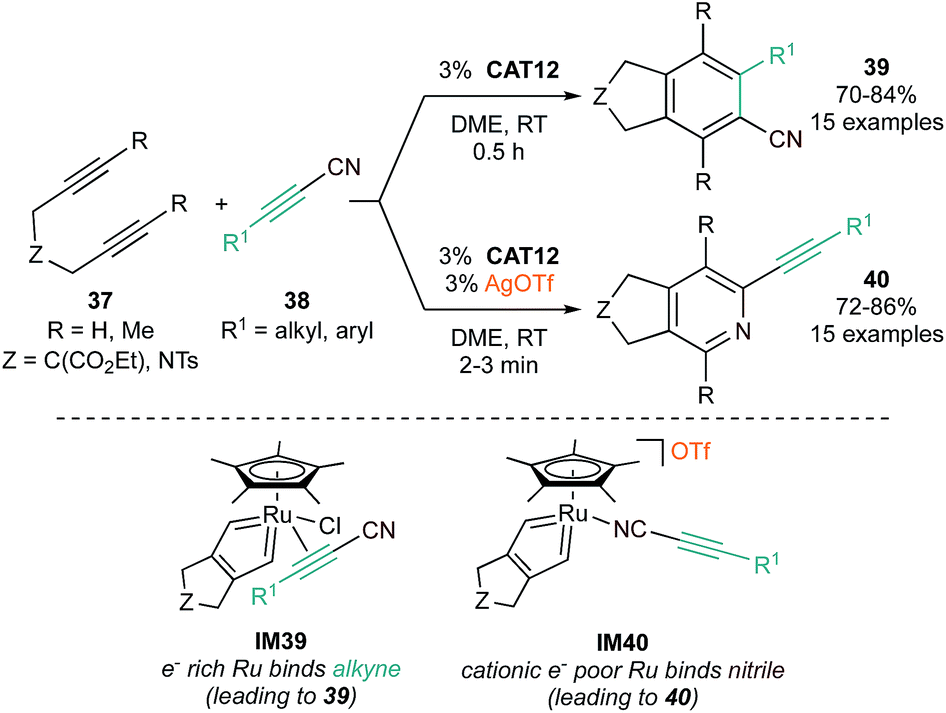 | ||
| Fig. 19 Top: example of AgOTf-induced switchable chemoselectivity in Ru-catalyzed [2 + 2 + 2] cycloaddition of diynes and alkynylnitriles. Bottom: potential mechanistic rationale for selectivity change with AgOTf. | ||
In recent years, there has been an increased interest in generating 2-heteroatom-substituted pyridines using heteroatom-substituted nitriles. For example, [Cp*Ru(MeCN)3]PF6 (CAT14) is an efficient catalyst in the synthesis of 2-aminopyridines (42-NR2) by [2 + 2 + 2] cycloaddition of tethered diynes and cyanamides (Fig. 20).89,90 Heterocumulene-like cyanamides are highly reactive partners in [2 + 2 + 2] cycloaddition reactions. The system could be further modified to use simple solvent-free conditions by using a RuCl3·nH2O catalyst.91 Applications were sought in the synthesis of related fluorescent azafluorenones with promising results.92 Further studies have extended 2-heteroatom-substituted pyridine synthesis across a diverse array of heteroatoms, leading to efficient syntheses of 1-(2-pyridyl)indoles (42-Ind),93 2-aryloxy pyridines (42-OR),94 including 3-(2-thiopyridyl)indoles (42-SR),95 and 2-selenopyridines (42-SeR),96,97 variously demonstrated with either CAT12 or CAT14 (Fig. 20). Mono- and dihalodiynes can also be transformed to halopyridines via reaction with nitriles, providing a platform for further functionalization through cross-coupling reactions.98
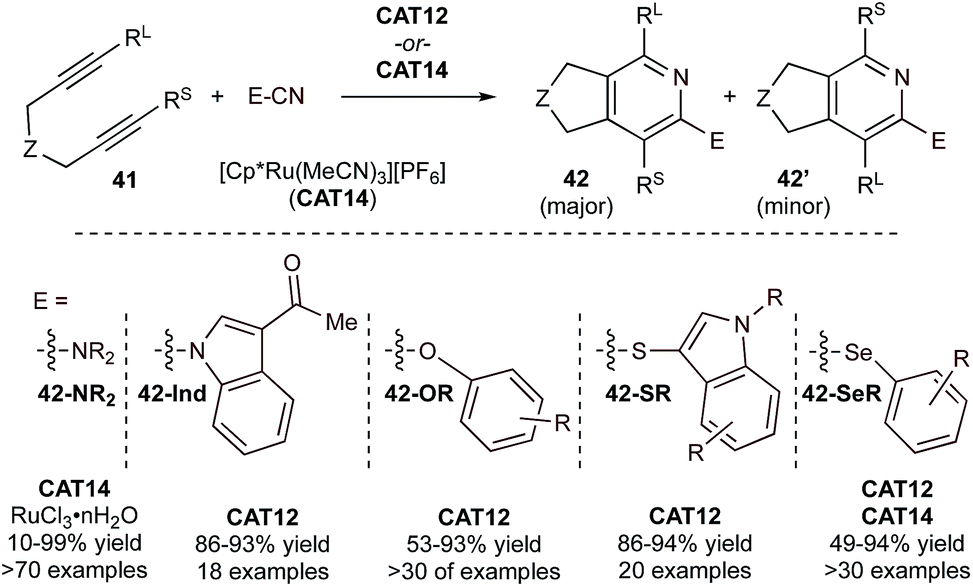 | ||
| Fig. 20 Examples of Ru-catalyzed [2 + 2 + 2] cycloaddition of tethered diynes with various heteroatom-substituted nitriles. | ||
Cationic CAT14 was also found to be a selective catalyst for the coupling of alkynyl triazenes (44) and alkynylnitriles (43) (Fig. 21, top)99 yielding 5,6-tethered 3-triazenylpyridines (45) with excellent regioselectivity. Conveniently, synthesis of the other isomer, 5,6-tethered 3-triazenylpyridines (47) could be achieved via reaction of triazenyldiyne 46 with nitriles catalyzed by CAT15 (Fig. 21, bottom), albeit with slightly lower regioselectivities resulting from misinsertion of the nitrile. Here, the synthetic versatility of the triazene group could be put to use, resulting in facile subsequent access to a variety of functionalized pyridines.
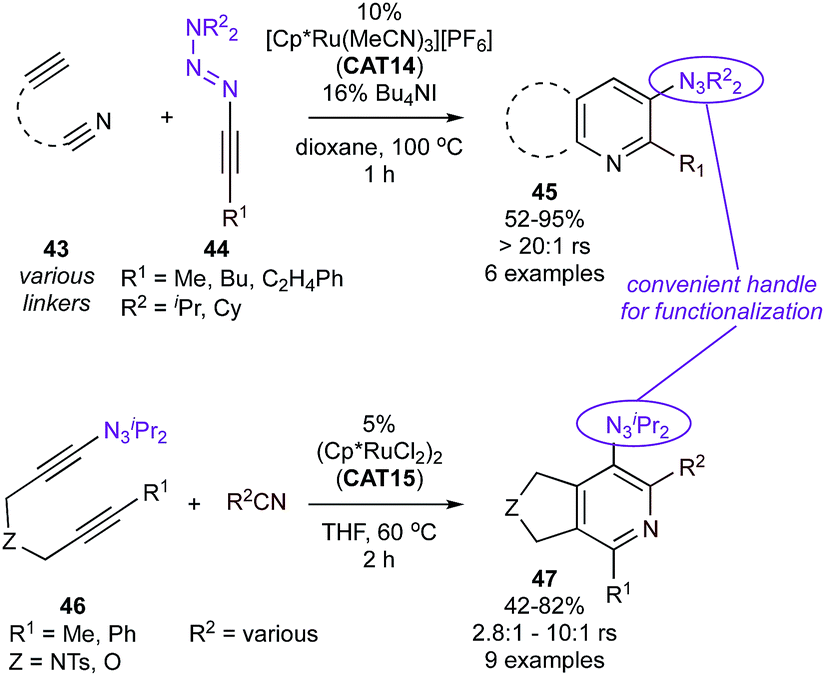 | ||
| Fig. 21 Ru-catalyzed [2 + 2 + 2] cycloaddition reactions with alkynyl triazene components. rs = regioselectivity over other regioisomers. | ||
Given the success found with Ru, there has been increased attention on Fe-catalyzed [2 + 2 + 2] pyridine synthesis as well. Fe-catalyzed [2 + 2 + 2] methods go back decades, including a notable report on the [2 + 2 + 2] synthesis of 1,2,4-triazenes from adiponitriles with nitriles.100 Following up on early [2 + 2 + 2] pyridine syntheses from the 1990s,101,102 several advanced Fe-based systems with good chemoselectivity have been developed in the past decade. For example, reduced bis(phosphine)Fe complexes can be made in situ through reduction of Fe halide salts by Zn. These complexes are competent for the formal [2 + 2 + 2] coupling of internal tethered diynes (48) with a variety of nitriles with yields ranging from 43–98% (Fig. 22).103 Regioselective cycloadditions of unsymmetrical alkynes, in combination with competition experiments of phenylacetylene with nitrile, suggest that azaferracyclopentadienes (IM41), rather than the two-alkyne coupled ferracyclopentadiene, are likely the key intermediate prior to the formation of pyridine. Both bis(diphenylphosphino)ethane (dppe) and 1,3-bis(diphenylphosphino)propane (dppp) ligands yielded pyridine formation, however, reactions with dppp could be conducted at lower catalyst loadings (5% FeI2, 10% dppp) without erosion of the yield.
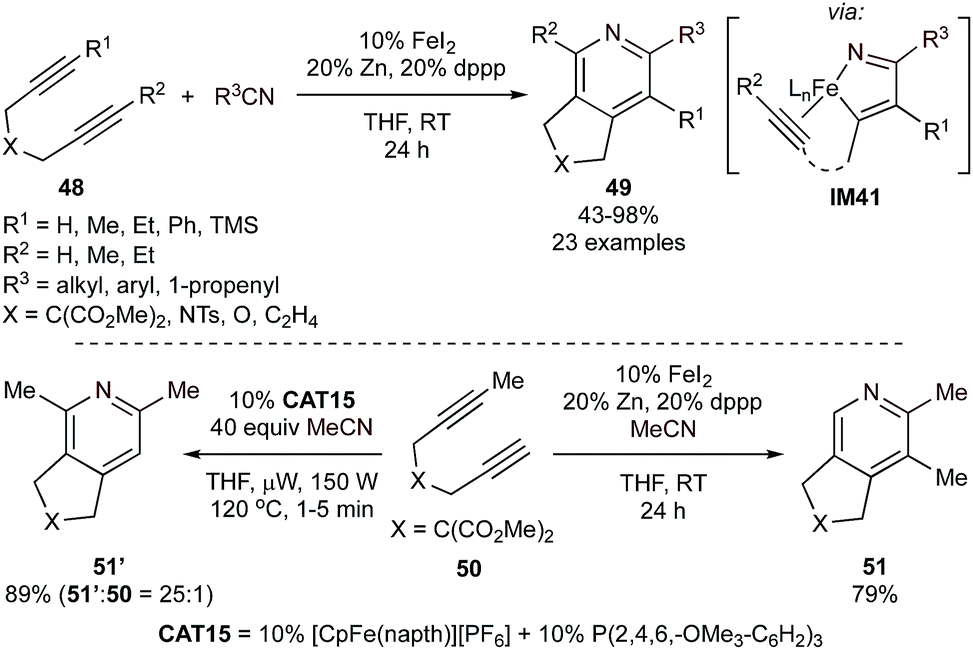 | ||
| Fig. 22 Top: Fe-catalyzed [2 + 2 + 2] synthesis of pyridine using diynes and nitriles in the presence of Zn and 1,3-bis(diphenylphosphino)propane (dppp). Bottom: comparison of the regioselectivity between CAT15 microwave pyridine synthesis (left) and reduced Fe/dppp-catalyzed pyridine synthesis (right). | ||
Interestingly, the regiochemistry of the [2 + 2 + 2] reaction could be inverted by employing a system catalyzed by CpFe(napth)PF6/P(2,4,6-OMe3C6H2)3 (CAT15) under microwave irradiation (Fig. 22, bottom left).104 Although a specific rationale for the inversion of the regioselectivity was not provided, it was noted that the yields of reactions catalyzed by the Cp-derived system are very sensitive to sterics, which may also play a role in the alkyne chemoselectivity of the initial alkyne/nitrile oxidative coupling to form IM41.
Louie and co-workers also reported a Fe-catalyzed synthesis of pyridine using sterically encumbered pyridyl bis(aldimine) (PDAI) ligands in the presence of tethered alkynylnitriles and both symmetric or unsymmetric alkynes (Fig. 23).105 Several pyridyl bis(ketimine) and bis(aldimine) ligands were screened, and it was found that pyridyl bis(aldimine) ligands gave higher conversion than pyridyl bis(ketimine) ligands, with PDAI-1 giving the best yield.
 | ||
| Fig. 23 (PDAI-1)-Fe catalyzed [2 + 2 + 2] coupling of alkynylnitriles with alkynes. | ||
This PDAI-based ligand system was then extended to the coupling of diynes (54) with highly reactive cyanamides using modified PDAI-2, leading to the regioselective synthesis of 2-aminopyridines (55) (Fig. 24).106,107 In this instance, the mechanism of the reaction was proposed to proceed through initial diyne coupling (IM42) on the basis of the regiochemistry, which mirrors that of related Co-catalyzed pyridine syntheses.108,109 Interestingly, reactions of diynes with cyanamides catalyzed by an in situ-generated (dppp)Fe catalyst resulted in the opposite regioisomer 55′.110 In these cases, the catalyst-controlled regioselectivity is proposed to be a function of a change in mechanism (IM42 for PDAI-2, IM43 for (dppp)Fe), although detailed comparative mechanistic studies are not available. Nonetheless, the development of initial examples of catalyst-controlled selectivity is an important step toward broader method development.
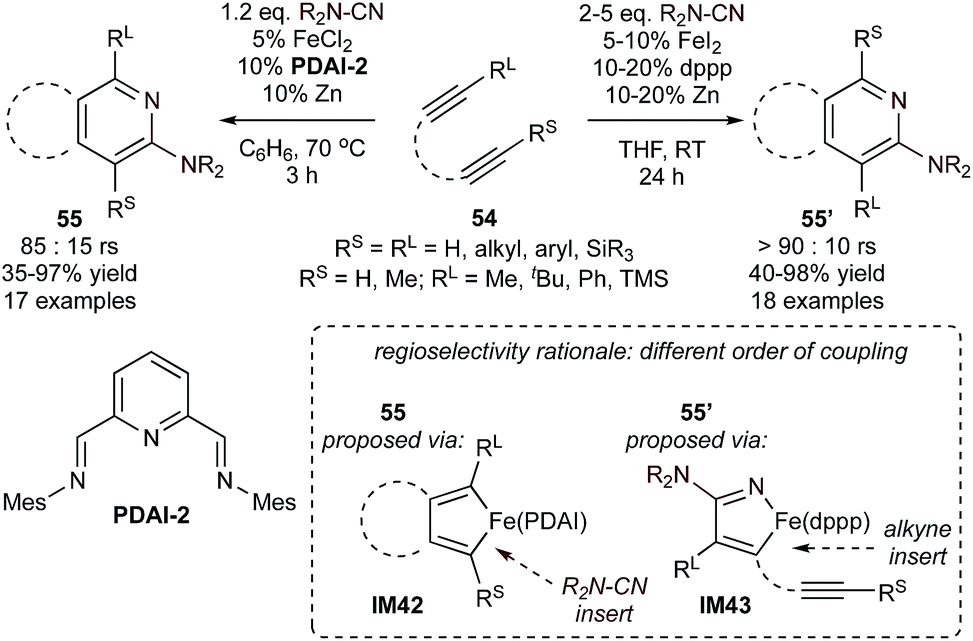 | ||
| Fig. 24 Comparison of regiochemical outcomes of cyanamide and diyne [2 + 2 + 2] cycloaddition catalyzed by pyridyl bis(aldimine) or bis(phosphine)-based Fe catalysts. rs = regioselectivity for the major product (55 or 55′) relative to the minor product (55′ or 55). | ||
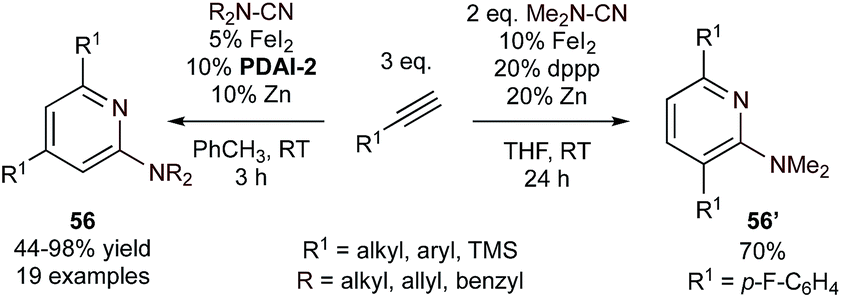 | ||
| Fig. 25 Intermolecular Fe-catalyzed [2 + 2 + 2] cyclization of cyanamides and alkynes. | ||
Additionally, in situ formed low-valent Nb complexes catalyze the [2 + 2 + 2] cycloaddition of tert-butylacetylene with arylnitriles, yielding 2,3,6-trisubstituted pyridines (57) (Fig. 26).111,112 This example is an extension of earlier work where low valent Nb complexes (made in situ from reduction with LiAlH4, Zn, or other reductants) were used to catalyze alkyne cyclotrimerization.112,113 In this case, it was found that alkoxysilane additives such as Ph2Si(OMe)2 can dramatically improve the chemoselectivity against benzene formation. Mechanistic studies suggest that niobacyclopentadiene IM44, formed from oxidative coupling of alkynes on NbIII, is a key intermediate. IM44 can then undergo nitrile insertion followed by reductive elimination. The intermediacy of IM44 was identified via reaction of tBuCCH and benzonitrile with the in situ reduced Nb complex under ambient conditions, which led solely to the formation of diene 58 rather than aminated (59) or oxygenated (60) products that would result from azaniobacyclopentadiene intermediate IM45. Interestingly, the regiochemistry of these Nb-catalyzed [2 + 2 + 2] reactions is the same as in the (dppp)Fe-catalyzed intermolecular example (56’, Fig. 25) although via a different proposed intermediate.
 | ||
| Fig. 26 Nb-catalyzed [2 + 2 + 2] synthesis of tri-substituted pyridines with a minor formation of cyclotrimerization product. | ||
Although there are no catalytic reports of [2 + 2 + 2] cycloaddition, group 4 metals can also produce pyridines via coupling of metallacyclopentadienes with nitriles,114–116 somewhat analogous to the mechanisms seen with Ru, Fe, and Nb (Fig. 27). For example, there are several well-established protocols117,118 for the stepwise addition of alkynes to “TiII” synthons, resulting in selective formation of titanacyclopentadienes (IM46). Treatment of in situ generated IM46 with TolSO2CN results in IM48, either from IM47 or IM47′. Elimination of –SO2Tol forms titanated pyridine IM49, which can then be trapped with several electrophiles, resulting in tetrasubstituted pyridines (61) in good yield.119 An analogous zirconacyclopentadiene intermediate was also recently isolated and shown to yield pyridines upon reaction with nitrile (vide infra).120
 | ||
| Fig. 27 Stoichiometric stepwise coupling of alkynes with “TiII” and subsequent cascading reactivity with nitriles. | ||
 | ||
| Fig. 28 Formal [3 + 2 + 1] syntheses of pyridines via multistep, one-pot hydroamination/functionalization sequences. | ||
3.2 Pyrimidines
Pyrimidines are core structures in a wide variety of pharmaceuticals and materials.14,16,17,28 Much like pyridines, formal [2 + 2 + 2] cycloaddition is a common strategy to form pyrimidines, in this case from the head-to-tail coupling of two nitriles with an alkyne. The strategies for [2 + 2 + 2] pyrimidine synthesis mainly focus on Lewis acid-promoted cyclization and low valent metal-catalyzed cycloaddition, catalyzed primarily by the harder early transition metals and first row metals like Fe. Beyond these strategies, condensation strategies featuring formal [3 + 2 + 1] coupling schemes with amidines as the 3-atom component have been variously reported with alcohols, alkynes, and isonitriles. | ||
| Fig. 29 Fe-assisted, Nb (Lewis acid)-catalyzed [2 + 2 + 2] pyrimidine synthesis. rs = regioselectivity of 64 relative to 64′. | ||
Masked low valent early transition metals were also found to be efficient catalysts for cycloadditive pyrimidine synthesis. The cumulene-masked “ZrII” zirconocene species CAT17 can serve as the catalyst for the synthesis of tetrasubstituted 4-silylethynyl-3-silylpyrimidine 66via the coupling of diyne 65 with benzonitrile (Fig. 30).126 Under elevated temperature the precatalyst can be activated to IM54 by releasing one equivalent of diyne. The resulting zirconacyclopentene can undergo twofold nitrile insertion to IM56, followed by reductive elimination of 66 and regeneration of the low-valent zirconocene. Excellent chemoselectivity of up to 50:1 against the formation of pyridine has been observed in this reaction. Stoichiometric studies revealed that the formation of azazirconacyclopentadiene from the cycloaddition of an alkyne and a nitrile was the key intermediate to the selectivity, while the other possible intermediate, zirconacyclopentadiene from two alkynes, was minor.
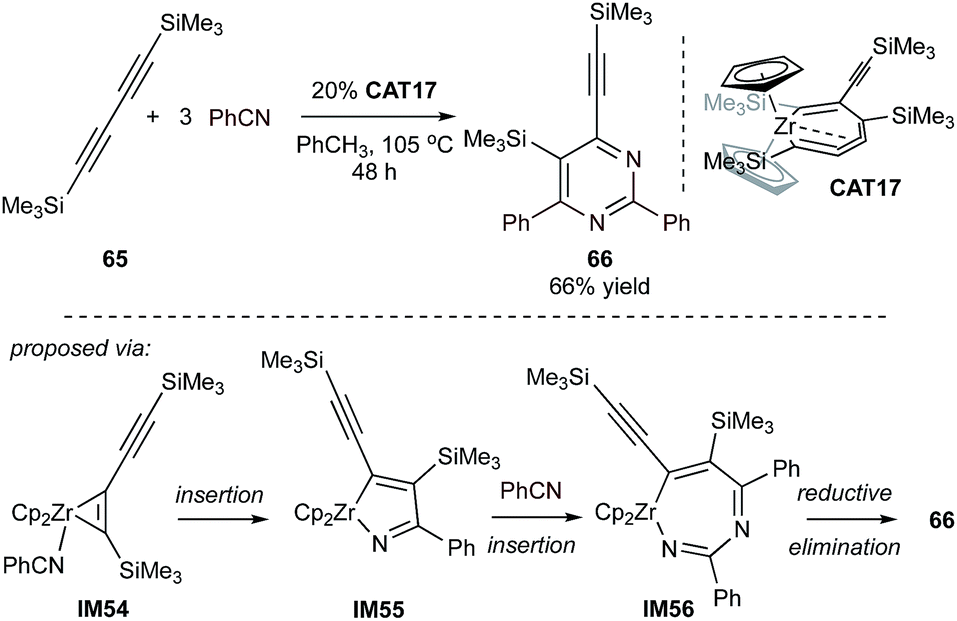 | ||
| Fig. 30 [2 + 2 + 2] pyrimidine synthesis from 1,4-bis(trimethylsilyl)-1,3-diyne and nitrile catalyzed by zirconocene-based complex. | ||
Similarly, the anthracene-masked zirconium(II) complex CAT18 was found to be an efficient catalyst for the synthesis of tri- and tetrasubstituted pyrimidines (67) from terminal or internal alkynes with benzonitrile derivatives (Fig. 31).120 When terminal alkynes were used as substrates, the major product was the 2,4,6-trisubstituted pyrimidine, similar to the selectivity observed in NbCl5-catalyzed reactions (Fig. 29).124,125IM58 could be as isolated as the major product by reacting 2 equiv. of TolCN and 1 equiv. of PhCCH with the 2-electron reduced IM57. The pyrimidine product 67 could then be isolated from reaction of IM58 with an equivalent of PhCN. Alternatively, when IM57 is treated with only 2 equivalents of PhCCH, the zirconacyclopentadiene IM59 forms. Treatment of IM59 with PhCN results in the formation of pyridine 68. Based on this reactivity, facile precoordination of nitrile to Zr appears to be key for selective pyrimidine (67) formation during catalysis. Interestingly, neither IM58 nor IM59 react with an equivalent of PhCCH to generate trisubstituted pyridine (68) or benzene (69), respectively.
 | ||
| Fig. 31 [2 + 2 + 2] pyrimidine synthesis from alkynes and nitriles catalyzed by anthracene-masked Zr(II) complex. | ||
In addition to the early transition metal-catalyzed reactions above, low-valent (PDAI-3)Fe complexes are also competent for the synthesis of bicyclic 2-aminopyrimidines (71) through formal [2 + 2 + 2] cycloaddition from tethered alkynylnitriles (70) and cyanamides (Fig. 32).127 The system is a modification from previous work on Fe-catalyzed 2-aminopyridine synthesis from tethered diynes and cyanamides (Fig. 24).106 Interestingly, catalysis with Fe(OAc)2 was unproductive compared to FeI2, leading to speculation on the potential role of ZnI2 as a Lewis acid in catalysis.
 | ||
| Fig. 32 Fe-catalyzed [2 + 2 + 2] synthesis of bicyclic 2-aminopyrimidines from tethered alkynylnitriles and cyanamides. | ||
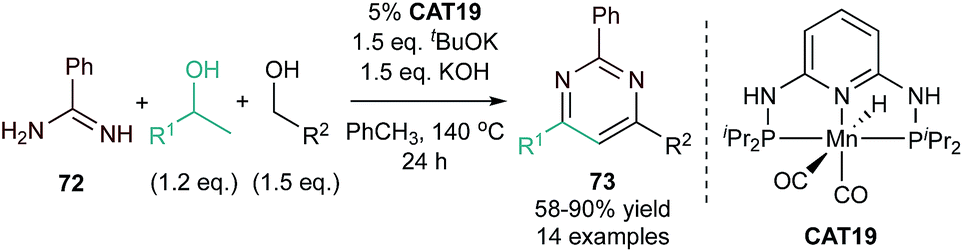 | ||
| Fig. 33 Mn-catalyzed [3 + 2 + 1] synthesis of pyrimidine from alcohols and benzamidine. | ||
Additionally, interrupted alkyne iminoamination (cf.Fig. 12) can be used to generate 1,3-diimines (IM23) that can then similarly undergo condensation with amidines to generate pyrimidines (74 ) (Fig. 34).131 In these examples, two different Ti-catalysts (CAT1 (ref. 132) or CAT2 (ref. 133)) were found to be most effective for the 2-step iminoamination/condensation sequence.
 | ||
| Fig. 34 Ti-catalyzed [3 + 2 + 1] synthesis of pyrimidine from alkynes, tert-butylisonitrile, amidines, and amines. | ||
3.3 Pyrazine
Despite the wide utility of pyrazines,134,135 there are few examples of catalytic MCRs for their synthesis. Hypothetically, pyrazine could be divided into one alkyne and two nitriles in [2 + 2 + 2] cycloaddition just like pyrimidines. However, because of the polarized nature of the nitrile, “head-to-tail” coupling (resulting in pyrimidines) is more common than “tail-to-tail” coupling (resulting in pyrazines).However, a recent report has capitalized on the tail-to-tail coupling of nitriles on “Cp2TiII”,136,137 resulting in a formal [2 + 2 + 1 + 1] coupling of nitriles to tetrasubstituted pyrazines (Fig. 35) via Ti-to-Al transmetallation (cf.Fig. 8).138 In this case, a single nitrile serves as both the 2-atom component and the 1-atom component. This reaction is proposed to occur first through oxidative cyclization of 2 nitriles to yield a 2,5-diazatitanacyclopentadiene (IM60). Two subsequent nitrile insertions lead to the formation of a ring-expanded 1,3,6,8-tetraazatitanacyclononatetraene (IM61), which then undergoes transmetallation with EtAlCl2 to form IM62 (or linear oligomers thereof). Hydrolysis likely results in subsequent cyclization to 75. This interesting 4-component cyclization results in good yields of the resulting pyrazine, although it is currently limited to aryl and benzyl nitriles and symmetric products.
 | ||
| Fig. 35 Ti-catalyzed [2 + 2 + 1 + 1] synthesis of pyrazine. | ||
3.4 Phosphabenzene
Extending [2 + 2 + 2] heterocycle synthesis to the heavier congeners, the first catalytic cycloadditive [2 + 2 + 2] syntheses of phosphabenzenes were recently reported using FeI2 as a catalyst (Fig. 36).139 Tri- and tetrasubstituted phosphabenzenes 77 can be synthesized from tethered diynes 76 and phosphaalkynes with either tertiary alkyl or bulky siloxide (TBDPS) substituents—the latter of which could be deprotected to 2-phosphaphenols.140 Notably, group 9 metal catalysts also exhibited catalytic activity, however in much lower yields and with poorer chemoselectivity against trimerization of alkyne. The intermediacy of the two alkyne-coupled intermediate IM63 was proposed, although alkyne/phosphaalkyne coupled intermediates analogous to IM43 (Fig. 24) could not be ruled out. Interestingly, computational analysis (B3LYP/6-311G**) of the Gibbs free energy change for the [2 + 2 + 2] cycloaddition processes were similar for benzenes (−119.4 kcal mol−1), pyridines (−83.6 kcal mol−1), and phosphabenzenes (−101.2 kcal mol−1), despite the fact that phosphaalkynes are significantly less thermodynamically stable than alkynes and nitriles.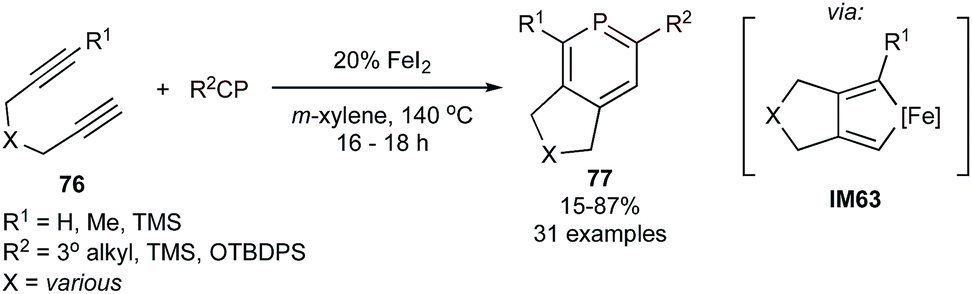 | ||
| Fig. 36 Fe-catalyzed [2 + 2 + 2] synthesis of phosphabenzenes from diynes and phosphaalkynes or siloxyphosphaethynes. | ||
4. Conclusions and future outlook
The above examples illustrate the limited instances of group 4–8 metal catalyzed syntheses of 5- and 6-membered aromatic heterocycles. Of the examples summarized in this Perspective, catalysis is dominated by group 4 (5-membered rings) and group 8 (6-membered rings), and can be broadly classified as “on-metal” cycloadditive processes (e.g. formal [2 + 2 + 2] cycloaddition) or “off-metal” cycloadditive processes that take place after catalysis (e.g. interrupted hydroamination/cyclization). Although partially intramolecular tethered syntheses have now been demonstrated with exquisite selectivity, there remains a significant challenge in controlling the selectivity of fully intermolecular reactions for nearly all heterocycle classes.Within the 5-membered ring MCRs, it is interesting to note overlapping [2 + 2 + 1] strategies between Ti/V and Ru: each metal can couple alkynes and nitrene equivalents to pyrrole, albeit through a different order of addition. The extension of Ru catalysis into [2 + 2 + 1] thiophene and furan syntheses raises the question of potential compatibility of the earlier (and more oxophilic) transition metals for similar reactions, which are ubiquitous in pyrrole synthesis.
For [2 + 2 + 2] cycloaddition reactions, early examples have been dominated by Ru, and method development with Ru catalysts remains an active area of research. However, significant advances with Fe have brought potentially earth-abundant alternatives to the fore, along with the potential for different mechanisms (order of component addition, for example) and subsequent potential for control of regio- and chemoselectivity in intermolecular examples. Nascent similar transformations have now also been reported for the early transition metals as well, where the harder, more electropositive metals appear well-poised for incorporating additional electronegative moieties (e.g. 2 nitriles in pyrimidine synthesis) into MCRs.
Notably absent in recent examples are catalytic variants with groups 6 and 7. However, stoichiometric syntheses with both Mo141 and W exist,142,143 and given the complementary reactivity between groups 4/5 and group 8, there is compelling evidence to explore these elements for catalysis, as well.
Author contributions
C. W. F. contributed to Section 2 and Y. C. and D. T. E. contributed to Section 3. D. N. H. and I. A. T. conceived and contributed to all portions of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Institutes of Health (R35GM119457, F32GM137547 to D. N. H.). Y. C. is supported by a University of Minnesota Wayland E. Noland Fellowship. D. T. E. kindly acknowledges financial support from the ETH Zürich Foundation (D. T. E. is a 2019 ESOP scholar).Notes and references
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- J. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC.
- M. Balasubramanian and J. G. Keay, Compr. Heterocycl. Chem. II, 1996, 245–300 CAS.
- L. Zhang, X.-M. Peng, G. L. V. Damu, R.-X. Geng and C.-H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1883, 16, 2597–2599 CrossRef.
- V. L. M. Silva, A. M. S. Silva, D. C. G. A. Pinto, J. A. S. Cavaleiro and J. Elguero, Eur. J. Org. Chem., 2004, 4348–4356 CrossRef CAS.
- S. T. Heller and S. R. Natarajan, Org. Lett., 2006, 8, 2675–2678 CrossRef CAS PubMed.
- H. Kwiecień and A. Wodnicka, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, 2020, vol. 31, pp. 281–323 Search PubMed.
- P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS.
- N. E. Schore, Chem. Rev., 1988, 88, 1081–1119 CrossRef CAS.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- T. P. Selvam, C. R. James, P. V. Dniandev and S. K. Valzita, Res. Pharm., 2012, 2, 1–9 Search PubMed.
- T. V. Vernitskaya and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443–457 CrossRef.
- S. Achelle and N. Plé, Curr. Org. Chem., 2012, 9, 163–187 CAS.
- R. Komatsu, H. Sasabe and J. Kido, J. Photonics Energy, 2018, 8, 032108 Search PubMed.
- W. Sun, Y. Shen, Y. Guo, Y. Chen and Q. Wang, Appl. Surf. Sci., 2021, 554, 149499 CrossRef CAS.
- T. Kato and J. M. Fréchet, Macromol. Symp., 1995, 98, 311–326 CrossRef CAS.
- P. Jeschke, Bioact. Heterocycl. Compd. Cl., 2012, 209–223 CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- A. Roglans, A. Pla-Quintana and M. Solà, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed.
- D. M. D'Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC.
- T. Nagata and Y. Obora, Asian J. Org. Chem., 2020, 9, 1532–1547 CrossRef CAS.
- G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2310 CrossRef CAS PubMed.
- A. J. Hunt, T. J. Farmer and J. H. Clark, in Element Recovery and Sustainability, The Royal Society of Chemistry, 2013, pp. 1–28 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed.
- I. M. Lagoja, Chem. Biodivers., 2005, 2, 1–50 CrossRef CAS PubMed.
- V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC.
- V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- B. Ramanathan, A. J. Keith, D. Armstrong and A. L. Odom, Org. Lett., 2004, 6, 2957–2960 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- A. L. Odom, Dalton Trans., 2005, 225–233 RSC.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2015, 8, 63 CrossRef PubMed.
- Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267–7281 CrossRef CAS PubMed.
- Z. W. Davis-Gilbert, K. Kawakita, D. R. Blechschmidt, H. Tsurugi, K. Mashima and I. A. Tonks, Organometallics, 2018, 37, 4439–4445 CrossRef CAS PubMed.
- A. J. Pearce, X. Y. See and I. A. Tonks, Chem. Commun., 2018, 54, 6891–6894 RSC.
- H.-C. Chiu, X. Y. See and I. A. Tonks, ACS Catal., 2019, 9, 216–223 CrossRef CAS PubMed.
- N. Vujkovic, B. D. Ward, A. Maisse-François, H. Wadepohl, P. Mountford and L. H. Gade, Organometallics, 2007, 26, 5522–5534 CrossRef CAS.
- H.-C. Chiu and I. A. Tonks, Angew. Chem., Int. Ed., 2018, 57, 6090–6094 CrossRef CAS PubMed.
- Y. Cheng, C. K. Klein and I. A. Tonks, Chem. Sci., 2020, 11, 10236–10242 RSC.
- X. Y. See, X. Wen, T. A. Wheeler, C. K. Klein, J. D. Goodpaster, B. R. Reiner and I. A. Tonks, ACS Catal., 2020, 10, 13504–13517 CrossRef CAS PubMed.
- K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194–4198 CrossRef CAS PubMed.
- J. De With, A. D. Horton and A. G. Orpen, Organometallics, 1993, 12, 1493–1496 CrossRef CAS.
- K. Matsui, M. Shibuya and Y. Yamamoto, Commun. Chem., 2018, 1, 21 CrossRef.
- K. Yamashita, Y. Yamamoto and H. Nishiyama, J. Am. Chem. Soc., 2012, 134, 7660–7663 CrossRef CAS PubMed.
- K. Matsui, M. Shibuya and Y. Yamamoto, ACS Catal., 2015, 5, 6468–6472 CrossRef CAS.
- Y. Yamamoto, Synlett, 2017, 28, 1250–1257 CrossRef CAS.
- K. Matsui, M. Shibuya and Y. Yamamoto, Angew. Chem., Int. Ed., 2016, 55, 15397–15400 CrossRef CAS PubMed.
- X. Yan and C. Xi, Acc. Chem. Res., 2015, 48, 935–946 CrossRef CAS PubMed.
- L. O. Khafizova, M. G. Shaibakova, N. A. Rikhter, T. V. Tyumkina and U. M. Dzhemilev, Tetrahedron, 2019, 75, 906–911 CrossRef CAS.
- M. G. Shaibakova, L. O. Khafizova, N. M. Chobanov, R. R. Gubaidullin, N. y. R. Popod’ko and U. M. Dzhemilev, Tetrahedron Lett., 2014, 55, 1326–1328 CrossRef CAS.
- L. O. Khafizova, M. G. Shaibakova, N. M. Chobanov, R. R. Gubaidullin, T. V. Tyumkina and U. M. Dzhemilev, Russ. J. Org. Chem., 2015, 51, 1277–1281 CrossRef CAS.
- B. Das, G. C. Reddy, P. Balasubramanyam and B. Veeranjaneyulu, Synthesis, 2010, 2010, 1625–1628 CrossRef.
- E. Ghabraie, S. Balalaie, M. Bararjanian, H. R. Bijanzadeh and F. Rominger, Tetrahedron, 2011, 67, 5415–5420 CrossRef CAS.
- E. Barnea, S. Majumder, R. J. Staples and A. L. Odom, Organometallics, 2009, 28, 3876–3881 CrossRef CAS.
- T. Li, H. Yan, X. Li, C. Wang and B. Wan, J. Org. Chem., 2016, 81, 12031–12037 CrossRef CAS PubMed.
- Y. Yamamoto, Bull. Chem. Soc. Jpn., 2019, 92, 619–628 CrossRef CAS.
- S. Majumder, K. R. Gipson, R. J. Staples and A. L. Odom, Adv. Synth. Catal., 2009, 351, 2013–2023 CrossRef CAS.
- A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS PubMed.
- A. A. Dissanayake and A. L. Odom, Chem. Commun., 2012, 48, 440–442 RSC.
- D. A. Shabalin and J. E. Camp, Org. Biomolec. Chem., 2020, 18, 3950–3964 RSC.
- H. Shen and Z. Xie, J. Am. Chem. Soc., 2010, 132, 11473–11480 CrossRef CAS PubMed.
- Y. Wang, H. Shen and Z. Xie, Synlett, 2011, 969–973 Search PubMed.
- F. Tabellion, C. Peters, U. Fischbeck, M. Regitz and F. Preuss, Chem. -Eur. J., 2000, 6, 4558–4566 CrossRef CAS PubMed.
- F. G. N. Cloke, J. C. Green, N. Hazari, P. B. Hitchcock, P. Mountford, J. F. Nixon and D. J. Wilson, Organometallics, 2006, 25, 3688–3700 CrossRef CAS.
- F. G. N. Cloke, P. B. Hitchcock, J. F. Nixon, D. James Wilson, F. Tabellion, U. Fischbeck, F. Preuss and M. Regitz, Chem. Commun., 1999, 2363–2364 RSC.
- W. Liang, K. Nakajima and Y. Nishibayashi, RSC Adv., 2020, 10, 12730–12733 RSC.
- J. A. Varela and C. Saá, Synlett, 2008, 2571–2578 CAS.
- B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC.
- D. L. Broere and E. Ruijter, Synthesis, 2012, 44, 2639–2672 CrossRef CAS.
- G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC.
- J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787–3802 CrossRef CAS PubMed.
- J. S. S. Neto and G. Zeni, Tetrahedron, 2020, 76, 130876 CrossRef CAS.
- R. S. Senaiar, D. D. Young and A. Deiters, Chem. Commun., 2006, 1313–1315 RSC.
- G. Onodera, Y. Shimizu, J.-n. Kimura, J. Kobayashi, Y. Ebihara, K. Kondo, K. Sakata and R. Takeuchi, J. Am. Chem. Soc., 2012, 134, 10515–10531 CrossRef CAS PubMed.
- R. M. Stolley, H. A. Duong, D. R. Thomas and J. Louie, J. Am. Chem. Soc., 2012, 134, 15154–15162 CrossRef CAS PubMed.
- R. M. Stolley, H. A. Duong and J. Louie, Organometallics, 2013, 32, 4952–4960 CrossRef CAS PubMed.
- N. D. Staudaher, R. M. Stolley and J. Louie, in Organic Reactions, 2019, vol. 97, pp. 1–202 Search PubMed.
- Y. Yamamoto, S. Okuda and K. Itoh, Chem. Commun., 2001, 1102–1103 RSC.
- Y. Yamamoto, K. Kinpara, T. Saigoku, H. Takagishi, S. Okuda, H. Nishiyama and K. Itoh, J. Am. Chem. Soc., 2005, 127, 605–613 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, R. Ogawa, H. Nishiyama and K. Itoh, Chem. -Eur. J., 2006, 12, 5618–5631 CrossRef CAS PubMed.
- A. Naiman and K. P. C. Vollhardt, Angew. Chem., Int. Ed., 1977, 16, 708–709 CrossRef.
- Y. Yamamoto, R. Ogawa and K. Itoh, J. Am. Chem. Soc., 2001, 123, 6189–6190 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, H. Nishiyama and K. Itoh, Adv. Synth. Catal., 2005, 347, 1913–1916 CrossRef CAS.
- G. Dazinger, M. Torres-Rodrigues, K. Kirchner, M. J. Calhorda and P. J. Costa, J. Organomet. Chem., 2006, 691, 4434–4445 CrossRef CAS.
- R. Schmid and K. Kirchner, Eur. J. Inorg. Chem., 2004, 2609–2626 CrossRef CAS.
- D. Bhatt, N. Patel, H. Chowdhury, P. V. Bharatam and A. Goswami, Adv. Synth. Catal., 2018, 360, 1876–1882 CrossRef CAS.
- F. Ye, M. Haddad, V. Ratovelomanana-Vidal and V. Michelet, Org. Lett., 2017, 19, 1104–1107 CrossRef CAS PubMed.
- F. Ye, F. Boukattaya, M. Haddad, V. Ratovelomanana-Vidal and V. Michelet, New J. Chem., 2018, 42, 3222–3235 RSC.
- F. Ye, M. Haddad, V. Michelet and V. Ratovelomanana-Vidal, Org. Chem. Front., 2017, 4, 1063–1068 RSC.
- F. Ye, C. Tran, L. Jullien, T. Le Saux, M. Haddad, V. Michelet and V. Ratovelomanana-Vidal, Org. Lett., 2018, 20, 4950–4953 CrossRef CAS PubMed.
- H. Chowdhury and A. Goswami, Adv. Synth. Catal., 2017, 359, 314–322 CrossRef CAS.
- P. Kalaramna, D. Bhatt, H. Sharma and A. Goswami, Adv. Synth. Catal., 2019, 361, 4379–4385 CrossRef CAS.
- H. Chowdhury and A. Goswami, Org. Biomolec. Chem., 2017, 15, 5824–5830 RSC.
- P. Kalaramna, D. Bhatt, H. Sharma and A. Goswami, Eur. J. Org. Chem., 2019, 4694–4700 CrossRef CAS.
- C. Tran, M. Haddad and V. Ratovelomanana-Vidal, Synthesis, 2019, 51, 2532–2541 CrossRef CAS.
- E. Bednářová, E. Colacino, F. Lamaty and M. Kotora, Adv. Synth. Catal., 2016, 358, 1916–1923 CrossRef.
- J.-F. Tan, C. T. Bormann, F. G. Perrin, F. M. Chadwick, K. Severin and N. Cramer, J. Am. Chem. Soc., 2019, 141, 10372–10383 CrossRef CAS PubMed.
- E. R. F. Gesing, U. Groth and K. P. C. Vollhardt, Synthesis, 1984, 351–353 CrossRef CAS.
- U. Schmidt and U. Zenneck, J. Organomet. Chem., 1992, 440, 187–190 CrossRef CAS.
- F. Knoch, F. Kremer, U. Schmidt, U. Zenneck, P. Le Floch and F. Mathey, Organometallics, 1996, 15, 2713–2719 CrossRef CAS.
- C. Wang, X. Li, F. Wu and B. Wan, Angew. Chem., Int. Ed., 2011, 50, 7162–7166 CrossRef CAS PubMed.
- V. Richard, M. Ipouck, D. S. Mérel, S. Gaillard, R. J. Whitby, B. Witulski and J.-L. Renaud, Chem. Commun., 2014, 50, 593–595 RSC.
- B. R. D'Souza, T. K. Lane and J. Louie, Org. Lett., 2011, 13, 2936–2939 CrossRef PubMed.
- T. K. Lane, M. H. Nguyen, B. R. D'Souza, N. A. Spahn and J. Louie, Chem. Commun., 2013, 49, 7735–7737 RSC.
- N. A. Spahn, M. H. Nguyen, J. Renner, T. K. Lane and J. Louie, J. Org. Chem., 2017, 82, 234–242 CrossRef CAS PubMed.
- R. Diercks, B. E. Eaton, S. Gürtzgen, S. Jalisatgi, A. J. Matzger, R. H. Radde and K. P. C. Vollhardt, J. Am. Chem. Soc., 1998, 120, 8247–8248 CrossRef CAS.
- H. Bönnemann, Angew. Chem., Int. Ed., 1985, 24, 248–262 CrossRef.
- C. Wang, D. Wang, F. Xu, B. Pan and B. Wan, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed.
- Y. Satoh and Y. Obora, J. Org. Chem., 2013, 78, 7771–7776 CrossRef CAS PubMed.
- Y. Satoh and Y. Obora, Eur. J. Org. Chem., 2015, 5041–5054 CrossRef CAS.
- K. Yamamoto, H. Nagae, H. Tsurugi and K. Mashima, Dalton Trans., 2016, 45, 17072–17081 RSC.
- T. Takahashi, F.-Y. Tsai and M. Kotora, J. Am. Chem. Soc., 2000, 122, 4994–4995 CrossRef CAS.
- T. Takahashi, F.-Y. Tsai, Y. Li, H. Wang, Y. Kondo, M. Yamanaka, K. Nakajima and M. Kotora, J. Am. Chem. Soc., 2002, 124, 5059–5067 CrossRef CAS PubMed.
- Z. Xi, K. Sato, Y. Gao, J. Lu and T. Takahashi, J. Am. Chem. Soc., 2003, 125, 9568–9569 CrossRef CAS PubMed.
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed.
- N. F. O'Rourke, M. J. Kier and G. C. Micalizio, Tetrahedron, 2016, 72, 7093–7123 CrossRef PubMed.
- R. Tanaka, A. Yuza, Y. Watai, D. Suzuki, Y. Takayama, F. Sato and H. Urabe, J. Am. Chem. Soc., 2005, 127, 7774–7780 CrossRef CAS PubMed.
- C. H. Low, J. N. Rosenberg, M. A. Lopez and T. Agapie, J. Am. Chem. Soc., 2018, 140, 11906–11910 CrossRef CAS PubMed.
- Y. Shi, C. Hall, J. T. Ciszewski, C. Cao and A. L. Odom, Chem. Commun., 2003, 586–587 RSC.
- A. A. Dissanayake, R. J. Staples and A. L. Odom, Adv. Synth. Catal., 2014, 356, 1811–1822 CrossRef CAS.
- E. K. J. Lui, D. Hergesell and L. L. Schafer, Org. Lett., 2018, 20, 6663–6667 CrossRef CAS PubMed.
- M. Fuji and Y. Obora, Org. Lett., 2017, 19, 5569–5572 CrossRef CAS PubMed.
- Y. Satoh, K. Yasuda and Y. Obora, Organometallics, 2012, 31, 5235–5238 CrossRef CAS.
- V. V. Burlakov, L. Becker, V. S. Bogdanov, M. V. Andreev, P. Arndt, A. Spannenberg, W. Baumann and U. Rosenthal, Eur. J. Inorg. Chem., 2014, 5304–5310 CrossRef CAS.
- T. K. Lane, B. R. D'Souza and J. Louie, J. Org. Chem., 2012, 77, 7555–7563 CrossRef CAS PubMed.
- M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543–15546 CrossRef CAS PubMed.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734–7741 CrossRef CAS PubMed.
- N. Deibl and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 1663–1666 CrossRef CAS PubMed.
- S. Majumder and A. L. Odom, Tetrahedron, 2010, 66, 3152–3158 CrossRef CAS.
- S. A. Harris, J. T. Ciszewski and A. L. Odom, Inorg. Chem., 2001, 40, 1987–1988 CrossRef CAS PubMed.
- A. Novak, A. J. Blake, C. Wilson and J. B. Love, Chem. Commun., 2002, 2796–2797 RSC.
- I. J. Krems and P. E. Spoerri, Chem. Rev., 1947, 40, 279–358 CrossRef CAS PubMed.
- F. B. Mortzfeld, C. Hashem, K. Vranková, M. Winkler and F. Rudroff, Biotechnol. J., 2020, 15, 2000064 CrossRef CAS.
- L. Becker, P. Arndt, H. Jiao, A. Spannenberg and U. Rosenthal, Angew. Chem., Int. Ed., 2013, 52, 11396–11400 CrossRef CAS PubMed.
- M. Reiß, F. Reiß, A. Spannenberg, P. Arndt and T. Beweries, Organometallics, 2018, 37, 4415–4423 CrossRef.
- L. O. Khafizova, M. G. Shaibakova and U. M. Dzhemilev, ChemistrySelect, 2018, 3, 11451–11453 CrossRef CAS.
- K. Nakajima, S. Takata, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2015, 54, 7597–7601 CrossRef CAS PubMed.
- K. Nakajima, W. Liang and Y. Nishibayashi, Org. Lett., 2016, 18, 5006–5009 CrossRef CAS PubMed.
- K. S. Lokare, J. T. Ciszewski and A. L. Odom, Organometallics, 2004, 23, 5386–5388 CrossRef CAS.
- N. Iwasawa, K. Maeyama and M. Saitou, J. Am. Chem. Soc., 1997, 119, 1486–1487 CrossRef CAS.
- N. Iwasawa, T. Ochiai and K. Maeyama, J. Org. Chem., 1998, 63, 3164–3165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |