
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning product selectivity in CO2 hydrogenation over metal-based catalysts
Ling-Xiang
Wang
a,
Liang
Wang
*b and
Feng-Shou
Xiao
 *b
*b
aDepartment of Chemistry, Zhejiang University, Hangzhou 310028, China
bKey Lab of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: fsxiao@zju.edu.cn; liangwang@zju.edu.cn
First published on 7th September 2021
Abstract
Conversion of CO2 into chemicals is a promising strategy for CO2 utilization, but its intricate transformation pathways and insufficient product selectivity still pose challenges. Exploiting new catalysts for tuning product selectivity in CO2 hydrogenation is important to improve the viability of this technology, where reverse water-gas shift (RWGS) and methanation as competitive reactions play key roles in controlling product selectivity in CO2 hydrogenation. So far, a series of metal-based catalysts with adjustable strong metal–support interactions, metal surface structure, and local environment of active sites have been developed, significantly tuning the product selectivity in CO2 hydrogenation. Herein, we describe the recent advances in the fundamental understanding of the two reactions in CO2 hydrogenation, in terms of emerging new catalysts which regulate the catalytic structure and switch reaction pathways, where the strong metal–support interactions, metal surface structure, and local environment of the active sites are particularly discussed. They are expected to enable efficient catalyst design for minimizing the deep hydrogenation and controlling the reaction towards the RWGS reaction. Finally, the potential utilization of these strategies for improving the performance of industrial catalysts is examined.
 Ling-Xiang Wang | Ling-Xiang Wang received his B.S. degree (2016) from Wuhan University, and his PhD degree (2021) in Chemistry from Zhejiang University, China. His research is mainly focused on metal/oxide catalysts toward CO2 conversion. |
 Liang Wang | Liang Wang received his B.S. degree (2008) in chemistry from Jilin University, China. He obtained his PhD degree from the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University in 2013. He then joined the Institute of Catalysis in Zhejiang University for postdoctoral work. Currently, he is a research professor in the College of Chemical and Biological Engineering in Zhejiang University. His research is focused on nanoporous catalysts and the conversion of low-carbon molecules. |
 Feng-Shou Xiao | Prof. Feng-Shou Xiao received his B.S. and M.S. degrees in chemistry from Jilin University, China. From there he moved to the Catalysis Research Center, Hokkaido University, Japan, where he was involved in collaborative research between China and Japan. He was awarded his PhD degree at Jilin University in 1990. After postdoctoral work at the University of California at Davis, USA, he joined Jilin University in 1994, where he is a distinguished professor of chemistry. He moved to Zhejiang University in 2009, and is now a distinguished professor at the College of Chemical and Biological Engineering. His research is mainly focused on zeolites, porous materials, and catalysis. |
1. Introduction
Carbon dioxide (CO2), a major greenhouse gas, has been paid much attention recently due to the consumption of massive amounts of fossil fuel and increase of atmospheric CO2 level, and a solution for this challenge is to suppress CO2 emission.1 To this end, transformation of CO2 into chemicals is extremely promising, which not only benefits the CO2 elimination but also provides carbon resources for industrial processes.2–6 In these transformations, CO2 hydrogenation over metal-based catalysts is a critical route, but the intricate transformation network and multiple active sites strongly influence the product selectivity.7–11 In recent years, various chemicals have been achieved via CO2 hydrogenation, including CO,12–22 methane,23–27 methanol,28–37 olefins,38–41 gasolines,42–46 aromatics,47–51 and alcohols.52–58Among these products, CO formed by reverse water-gas shift (RWGS) and CH4 formed by CO2 methanation are the most fundamental products, which are usually chosen as model products for investigations due to their strong competitiveness. From the viewpoint of chemical transformation, CO is preferred because of its potential for further applications, such as Fischer–Tropsch synthesis for hydrocarbons and oxygenates which possess higher economic value.59–62 In contrast, methane is relatively undesirable because of the limited applications and transformation routes. On the other hand, CO is a primary product and/or intermediate, which could be further transformed into other products. In contrast, methane is the completely hydrogenated product, which is basically stable in CO2 hydrogenation. Therefore, studying the selectivity control between CO and methane could provide deep understanding of reaction mechanisms of CO2 hydrogenation, which should be helpful for designing highly efficient catalysts. This understanding even helps to improve the catalysis in methanol and C2+ product synthesis from CO2 hydrogenation.17
As shown in Fig. 1, the RWGS is thermodynamically favorable at high temperature because of its endothermic nature, while CO2 methanation is thermodynamically favorable at relatively low temperature. However, the eight-electron transfer process of CO2 to CH4 is hindered by the high kinetic barrier. To overcome the kinetic limitation, a large number of catalysts have been employed for efficient CO2 hydrogenation.7,9,23,24
| CO2 + H2 = CO + H2O, ΔH298 K = 42.1 kJ mol−1 | (1) |
| CO2 + 4H2 = CH4 + 2H2O, ΔH298 K = −165.0 kJ mol−1 | (2) |
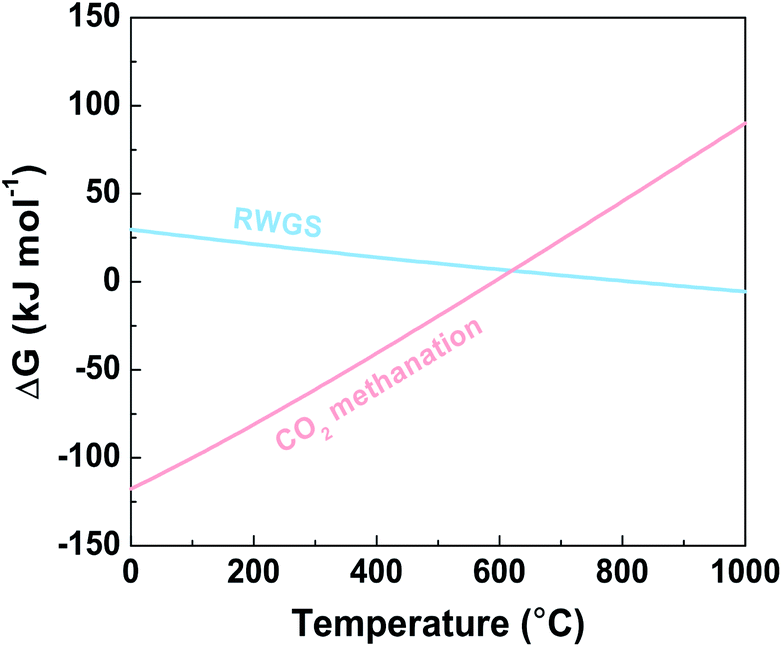 | ||
| Fig. 1 Gibbs free energy of the RWGS reaction and CO2 methanation. | ||
The CO2-to-CO/CH4 transformation is very complex. In most cases, *CO is an inevasible intermediate during CO2 hydrogenation.9,32 The CO2 hydrogenation proceeds via primary hydrogenation to *CO, and deep hydrogenation of *CO to methane. Based on this knowledge, the adsorption strength of *CO on the catalyst surface is regarded as a crucial factor (Fig. 2a). For example, Cu catalysts prefer to catalyze the RWGS reaction,34 while Co and Ni catalysts are favorable for CO2 methanation.24,63 These results are attributed to the fact that Co and Ni exhibit stronger adsorption for the *CO intermediate than Cu, thus leading to efficient C–O bond cleavage to form methane.9,16 The surface electronic states of the supported metal nanoparticles could optimize the *CO adsorption, which could be significantly controlled by strong metal–support interactions (SMSI) on reducible oxide supports. With the discovery of SMSI on non-oxides, the strategy of *CO-adsorption control for tuning product selectivity in CO2 hydrogenation is expanded to phosphates. Based on the transformation routes of *CO to CH4, as far as inhibiting C–O dissociation and deep hydrogenation of *CO species is concerned, new catalysts including bimetallic alloys and carbides are exploited (Fig. 2b). In addition, during SMSI construction, O vacancies easily form on the reducible oxide supports and play a crucial role in a series of charge transfer processes. The O vacancies could result in positively charged metal nanoparticles, which reduces the back-donation of d-electrons to the 2π antibonding orbital of CO, and the interaction between metal and *CO species is weakened.64 Based on this understanding, the advantages of alloy and carbide catalysts are maximized, because alloy catalysts have adjustable electronic structures for optimizing reaction intermediate adsorption, and carbide catalysts can provide a functional catalytic surface for new reaction routes (Fig. 2c). These unique structures and surface properties show more opportunities for selective CO2 hydrogenation. In addition to *CO-adsorption, *H spillover on the catalytic surface is equally remarkable.9,10,28 Under the precondition of moderate H2 dissociation, inhibiting *H spillover efficiently avoids deep hydrogenation of *CO, which might provide new insights for selectivity control in CO2 hydrogenation.
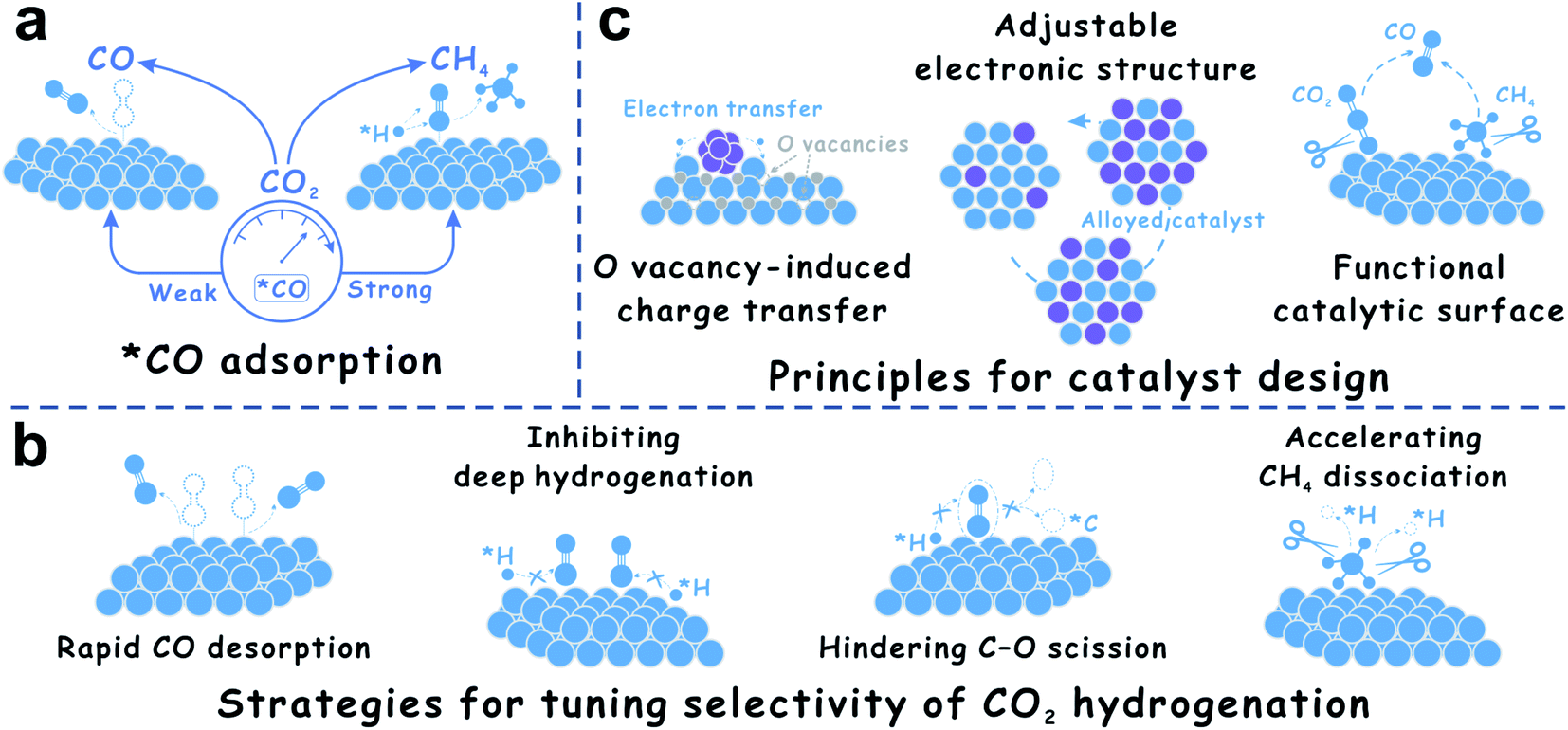 | ||
| Fig. 2 (a) Importance of *CO adsorption in CO2 hydrogenation. (b) Strategies for tuning the selectivity of CO2 hydrogenation. (c) Principles for catalyst design for CO2 hydrogenation. | ||
With regard to the rapid growth of investigations in selective CO2 hydrogenation, and the requirements for in-depth understanding of reaction mechanisms, we believe that it is time to summarize recent achievements in tuning product selectivity in CO2 hydrogenation. Previous reviews have focused on applications of the catalysts and the reaction mechanisms from CO2 to specific products,7–10,65,66 but strategies for selectivity control and principles for catalyst design are rarely discussed. In this perspective, the structural features of oxide, phosphate, metal alloy, and carbide-based catalysts are briefly summarized. Furthermore, the principles for controlling the product selectivity are proposed.
2. Oxide-supported metal nanoparticle catalysts
2.1 Crystal phase of oxides
A typical phenomenon is observed on titania-supported cobalt catalysts,67–69 where Co/r-TiO2 (rutile) selectively catalyzes CO2 methanation, but CO is predominant on the Co/a-TiO2 (anatase) catalyst.69 Calcination at 800 °C results in a partial transition from anatase to rutile, enhancing the adsorption of the *CO intermediate that leads to deep hydrogenation to CH4.16,34,70,71 Similarly, CO selectivity in In2O3 catalyzed CO2 hydrogenation can be improved by crystal phase transition from hexagonal In2O3 (h-In2O3) to cubic In2O3 (c-In2O3).72 The h-In2O3 is reduced by H2 and oxidized by CO2 to form c-In2O3. The rearrangement of surface O species makes it more active for H2 dissociation to form O vacancies. CO2 adsorbs on the O vacancies and heals the vacancies by desorbing CO, resulting in higher RWGS activity.73–76 Yang et al.17 reported the transformation from Co3O4 rhombic dodecahedra (denoted as Co3O4-0 h) with the (111) plane to Co3O4 nanorods (Co3O4-2 h) with the (110) plane by prolonging hydrothermal aging during synthesis, leading to different catalytic performances. For example, CO selectivity of Co3O4-2 h exceeds 90%, while the catalyst without aging (Co3O4-0 h) gives a CH4 selectivity of 85% in CO2 hydrogenation. Density functional theory (DFT) calculations reveal that the formation of O vacancies on Co3O4(111) (0.96 eV) is much easier than that on the Co3O4(110) surface (2.20–2.79 eV). The O vacancies lead to low-coordinated Co atoms, followed by the formation of a metallic Co cluster, which is highly active for CO2 methanation (Table 1).| Catalyst | H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratio :CO2 ratio |
Temperature (°C) | Pressure (MPa) | CO2 conversion (%) | Selectivity (%) | Rate (mmol gcat−1 h−1) | ||
|---|---|---|---|---|---|---|---|---|
| CO | CH4 | CO | CH4 | |||||
| Rh/TiO2 (ref. 13) | 1:1 |
200 | 0.1 | 0.4 | 92.3 | 7.7 | 0.48 | 0.04 |
| Ir/CeO2 (ref. 14) | 4:1 |
300 | 1.0 | 6.8 | 100 | 0 | 6.9 | 0 |
| Ru/CeO2 (ref. 15) | 4:1 |
240 | 0.1 | <5 | 92.1 | 7.9 | 21.0 | 1.8 |
| PtCo/TiO2 (ref. 16) | 2:1 |
300 | 0.1 | 8.2 | 98.8 | 1.2 | 43.4 | 0.5 |
| PtCo/CeO2 (ref. 16) | 2:1 |
300 | 0.1 | 9.1 | 92.3 | 7.7 | 40.5 | 3.4 |
| PtCo/ZrO2 (ref. 16) | 2:1 |
300 | 0.1 | 7.8 | 89.5 | 10.5 | 39.3 | 4.6 |
| Co3O4 (ref. 17) | 3:1 |
350 | 0.1 | 10.0 | 95.0 | 5.0 | 38.1 | 2.0 |
| Mo2C18 | 2:1 |
300 | 0.1 | 8.7 | 93.5 | 6.5 | 43.6 | 3.0 |
| Co/Mo2C18 | 2:1 |
300 | 0.1 | 9.5 | 98.1 | 1.9 | 49.9 | 1.0 |
| Rh@S-1 (ref. 19) | 3:1 |
500 | 1.0 | 51.6 | 79.8 | 20.2 | 13.2 | 3.4 |
| Ni-in-Cu20 | 3:1 |
550 | 0.1 | 50.7 | 100 | 0 | 181.1 | 0 |
| Ni–Au21 | 3:1 |
600 | 0.1 | 18.0 | 95.0 | 5.0 | 109.9 | 5.8 |
| Rh/NbOPO4 (ref. 22) | 3:1 |
500 | 2.0 | 39.9 | 98.9 | 1.1 | 58.1 | 0.6 |
| Co/r-TiO2 (ref. 69) | 4:1 |
400 | 3.0 | 85.0 | 1.0 | 99.0 | 0.5 | 54.1 |
| Co/a-TiO2 (ref. 69) | 4:1 |
400 | 3.0 | 15.0 | 90.0 | 10.0 | 8.7 | 1.0 |
| Ru/r-TiO2 (ref. 85) | 4:1 |
400 | 0.1 | 57.0 | 3.0 | 97.0 | 0.05 | 1.6 |
| Ru/a-TiO2 (ref. 85) | 4:1 |
400 | 0.1 | 23.0 | 100 | 0 | 0.66 | 0 |
| Ni3Fe9/ZrO2 (ref. 101) | 2:1 |
400 | 0.1 | 18.6 | 95.8 | 3.7 | 22.1 | 0.9 |
| Cu/β-Mo2C109 | 2:1 |
600 | 0.1 | 40.0 | 99.2 | 0.8 | 1771.4 | 14.3 |
| InNi3C0.5 (ref. 111) | 3:1 |
500 | 0.1 | 53.0 | 97.0 | 3.0 | 117.6 | 3.6 |
| Ni/SiO2 (ref. 112) | 4:1 |
750 | 0.1 | 58.0 | 100 | 0 | 2071.4 | 0 |
The distinct selectivity for CO2 hydrogenation of oxides with different phases can be explained by the arrangement of O atoms in the lattice, and the activation of CO2 and H2 is affected simultaneously. Rearrangement of O atoms benefits H2 dissociation to form O vacancies, which accelerate CO2 adsorption and transformation. However, the unstable oxide surface can form excessive O vacancies, which might lead to low-coordinated metal atoms for CO2 methanation.
2.2 SMSI on oxides
SMSI was firstly reported by Tauster and Fung77,78 in the 1970s to study the suppressed CO and H2 adsorption on the supported metals.79–84 In these cases, the geometric and electronic modulation of the metal nanoparticles by the oxides plays an important role in optimizing the CO2 hydrogenation.12–15,85–88TiO2- and CeO2-supported Rh, Ru and Ir catalysts with high loadings can selectively catalyze CO2 methanation. With lower metal loadings to reduce the nanoparticle size, these catalysts yield CO as the predominant product. These results are reasonably attributed to the chemical features of the small nanoparticles. Li et al.14 reported the SMSI on an Ir/CeO2 catalyst, where the partially oxidized Ir nanoparticles exhibit relatively weak CO adsorption, resulting in rapid CO desorption rather than hydrogenation to CH4 (Fig. 3a–d).89 Similarly, the atomically dispersed RuOx species, which might be generated during the oxidative treatment, could maintain the oxidized state even under the reaction conditions with a reductive atmosphere, because of the strong bonding with the CeO2 support.15
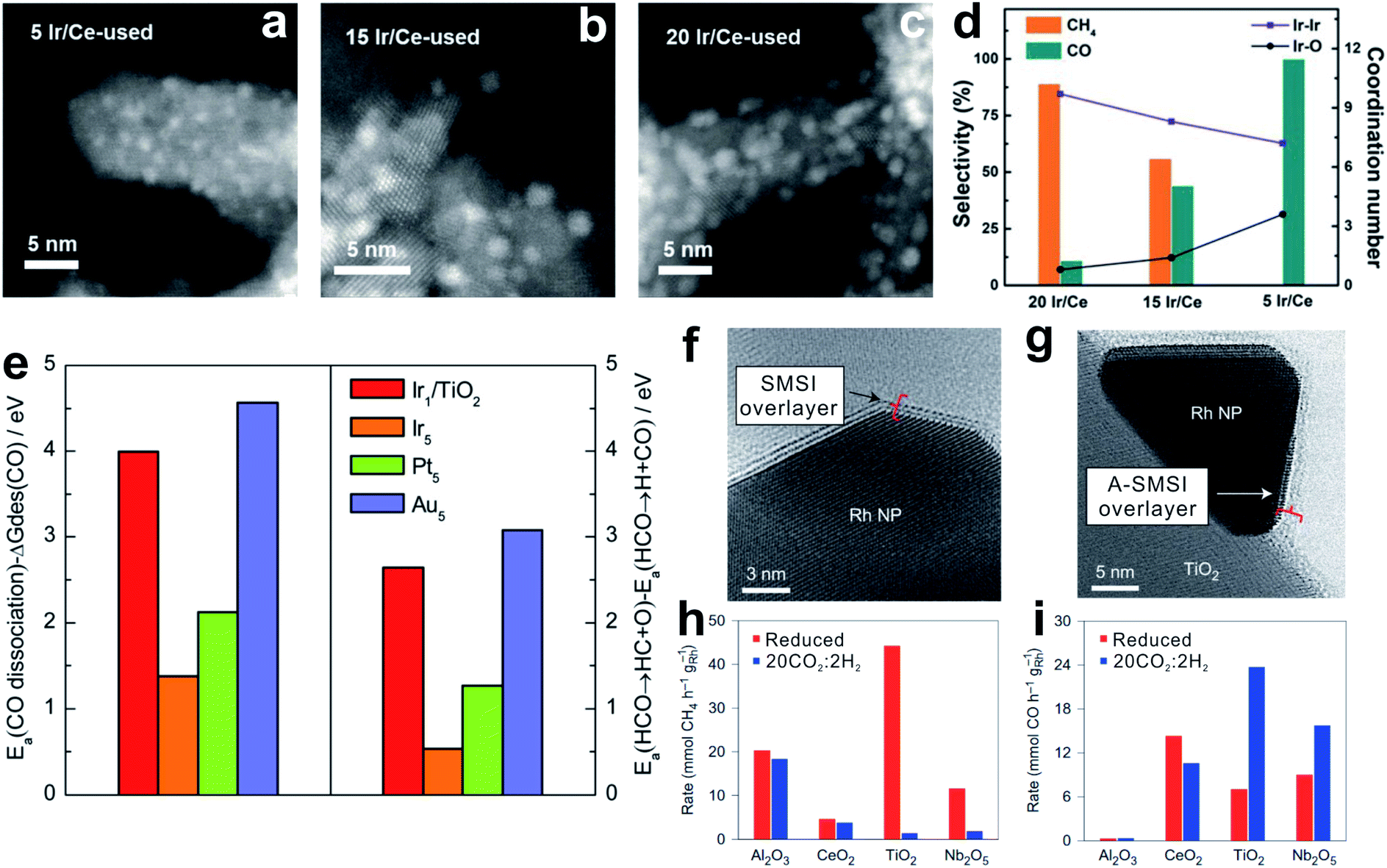 | ||
| Fig. 3 (a–c) HAADF-STEM images of catalysts: (a) 5 Ir/Ce used, (b) 15 Ir/Ce used, and (c) 20 Ir/Ce used. (d) The coordination number of Ir–Ir and Ir–O shells (data, right axis) relative to catalytic selectivity (bars, left axis) of Ir/Ce catalysts with different Ir loadings. Reproduced with permission from ref. 14. Copyright (2017) Wiley-VCH. (e) Left: difference between activation energies for CO dissociation and desorption free energies of CO. Right: difference in activation energies between HCO → HC + O and HCO → H + CO on Ir1/TiO2, and stepped Ir (Ir5), Pt (Pt5), and Au (Au5) surfaces. Reproduced with permission from ref. 88. Copyright (2017) American Chemical Society. (f and g) In situ STEM images of (f) an SMSI overlayer, a TiOx crystalline bilayer containing exclusively Ti3+, and (g) an A-SMSI overlayer, an amorphous TiOx overlayer containing a mixture of Ti3+ and Ti4+, on the surface of Rh nanoparticles. (h) CH4 and (i) CO generation rates on 2% Rh with various supports after reduction or 20CO2:2H2 treatment. Reproduced with permission from ref. 13. Copyright (2017) Springer Nature. | ||
DFT calculations provide mechanistic understanding of the SMSI-controlled product selectivity. Fig. 3e shows the difference between CO dissociation barriers and CO desorption free energies of single-atom Ir (Ir1) and stepped Ir (Ir5).88 The stepped Ir shows a much lower value than that of the single atom Ir, suggesting preferentially occurring CO desorption on the single-atom Ir, which could explain the highly selective RWGS reaction. In addition, the difference between C–O dissociation to *CH and dehydrogenation of *CHO to *CO on the single-atom Ir is greatly increased compared to that of the stepped Ir. C–O bond cleavage of the main intermediates (*HCOO, *COOH, and M–CO) strongly determines the CO2 hydrogenation selectivity.
In addition, *H spillover also plays a significant role in this reaction.85 For example, Ru/a-TiO2 and Ru/r-TiO2 could selectively catalyze RWGS and methanation, respectively. In addition to the influence of the crystalline phase on *CO adsorption in the aforementioned discussion, it is found that the hydrogen spillover is important for the reaction. The H atoms from H2 dissociation at metallic sites could spill to the TiO2 surface and form Ti–O(H)–Ti species, leading to electron donation into shallow trap states in the band gap of TiO2.90,91 Identified by the band at 1740 cm−1 in diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), *H spillover is more likely to occur on the Ru/a-TiO2 compared with Ru/r-TiO2. That leads to charge transfer from Ru to a-TiO2, reducing the π back-donation from Ru to adsorbed *CO, which results in quick desorption of *CO and hinders deep hydrogenation to methane.
Besides the classical SMSI, a new type of SMSI was developed by Matsubu et al.,13 which is realized by pretreating the Rh/TiO2 catalyst in mixed gases of CO2 and H2 with a ratio of 10:1 to form carbonate-containing species in the overlayer (Fig. 3f and g). It is denoted as adsorbate-induced SMSI (A-SMSI). The amorphous overlayer on the Rh nanoparticles contains a mixture of Ti species (Ti4+/Ti3+ at 7/3), different from the classical SMSI overlayer on the TiO2 support by H2 treatment, where the Ti species are dominantly in the Ti3+ state.92,93 It is proposed that the adsorbed HCOx species might coordinate with TiOx in the overlayer, and change the surface properties of the Rh nanoparticles. The redshift and decreased intensity of the linear CO in DRIFTS indicate the weakened CO adsorption, because of the physically blocked Rh nanoparticles and the polarization of CO bonds induced by charge transfer to Rh. In the CO2 hydrogenation, the Rh/TiO2 catalyst with A-SMSI shows 90% selectivity for CO formation, which is different from the general Rh catalysts with dominant CO2 methanation (Fig. 3h and i).
As observed in these examples, the adjustments of crystal phases of oxides and construction of SMSI on supported catalysts are efficient routes for hindering the CO2 deep hydrogenation. The features of SMSI, including weakening *CO adsorption, inhibiting C–O dissociation and deep hydrogenation, and optimizing *H spillover, are emphasized.
These examples for SMSI show great success on the selectivity control in CO2 hydrogenation over oxide-supported catalysts. However, the formation of the SMSI still relies on the reducible oxides. In addition, high CO selectivity is always obtained at low conversion of CO2 (<5%). Also, the classical SMSI could be destroyed by re-oxidation from water or oxygen at high temperature.12–15 To overcome this limitation, exploiting new supports or catalysts for this reaction is always important.
3. Phosphate-based catalysts
SMSI has been reported on phosphates such as hydroxyapatite (HAP) and LaPO4 for CO oxidation.94–96 It also plays a role in tuning the selectivity of CO2 hydrogenation. Wang et al.22 reported a Rh/NbOPO4 catalyst with phosphate-based SMSI for highly active and selective RWGS reaction, which is quite different from the general Rh catalyst in CO2 methanation. The Rh nanoparticles with small size at 1.1 nm are uniformly dispersed on the NbOPO4 support, exhibiting CO as a predominant product in CO2 hydrogenation in a wide temperature range of 200–500 °C (Fig. 4a). The catalyst with Rh loading at 0.7% gives CO2 conversion of 39.9% with CO selectivity of 98.9%. Such performance remarkably outperforms the Rh nanoparticles on CeO2, TiO2, and Nb2O5 supports with SMSI.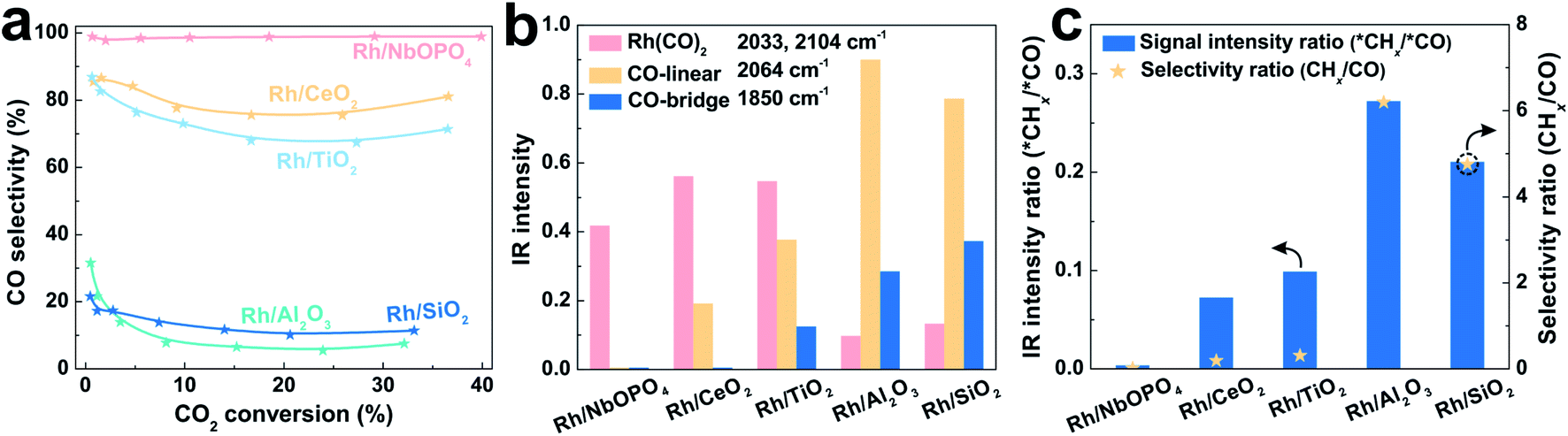 | ||
| Fig. 4 (a) Dependence of CO selectivity on CO2 conversion over various catalysts at 200–500 °C. (b) DRIFTS peak intensities characterizing the different CO species adsorbed on various catalysts. (c) Correlation of DRIFTS peak intensity and selectivity in CO2 hydrogenation over various catalysts. Reproduced with permission from ref. 22. Copyright (2020) Wiley-VCH. | ||
The SMSI between Rh and a NbOPO4 support is explored by H2-TPR and CO-TPD tests, where the reduction temperatures of Rh+ → Rh0 are increased and the desorption temperatures of CO are decreased, compared with the generally supported Rh catalysts. In addition, the Rh/NbOPO4 sample gives the lowest Rh0/Rh3+ ratio among these catalysts identified by XPS, due to the electronic interactions on the Rh–NbOPO4 interface to stabilize the positively charged Rh.82 The Rh/NbOPO4 catalyst also exhibits decreased CO adsorption, where only the weak stretches of gem-dicarbonyl Rh(CO)2 species12,13,97,98 are observed in CO-adsorption DRIFTS (Fig. 4b). In the CO2 hydrogenation (Fig. 4c), in situ DRIFTS shows that the *CHx species, a crucial intermediate for CH4 formation, can be observed on the other catalysts, but is undetectable on the Rh/NbOPO4 catalyst, in agreement with the highly selective RWGS.
Therefore, the high CO selectivity for the Rh/NbOPO4 catalyst in CO2 hydrogenation could be explained by the decreased *CO adsorption, resulting from the SMSI between Rh and NbOPO4, which is similar to the phenomena on the general oxide-supported catalysts with SMSI. Although it is observed that phosphate-based SMSI tunes the Rh catalyst from CO2 methanation to RWGS, the influence on CO2 hydrogenation still needs further operando characterization and theoretical investigations. This observation still provides a new type of catalyst for CO2 hydrogenation with optimized selectivity.
4. Alloy-based catalysts
Inspired by the catalysts with SMSI for weak adsorption of the *CO intermediate to selectively obtain the CO product in CO2 hydrogenation, it is reasonable that construction of alloyed metal catalysts with variable adsorption of the *CO intermediate could be adjusted by the alloyed compositions and supports.99,100 Recently, alloys of PtCo, NiFe, CuNi and NiAu16,20,21,101,102 have been reported for tuning product selectivity in CO2 hydrogenation.Kattel et al.16 reported CO2 hydrogenation over PtCo bimetallic catalysts supported on oxides of CeO2, ZrO2, and TiO2, which all give RWGS as a dominant reaction. Particularly, PtCo/TiO2 gives a much higher CO/CH4 ratio than those of PtCo/CeO2 and PtCo/ZrO2. On the PtCo/TiO2 catalyst, the energy barrier of *CO desorption is much lower than that for hydrogenation to *CHO, leading to the generation of gas phase CO (Fig. 5a and b). In contrast, energy for *CO hydrogenation is comparable with that of *CO desorption on the PtCo/ZrO2 catalyst, resulting in the formation of CH4 or CH3OH as competitive reactions, evidenced by DRIFTS and ambient-pressure X-ray photoelectron spectroscopy (AP-XPS).
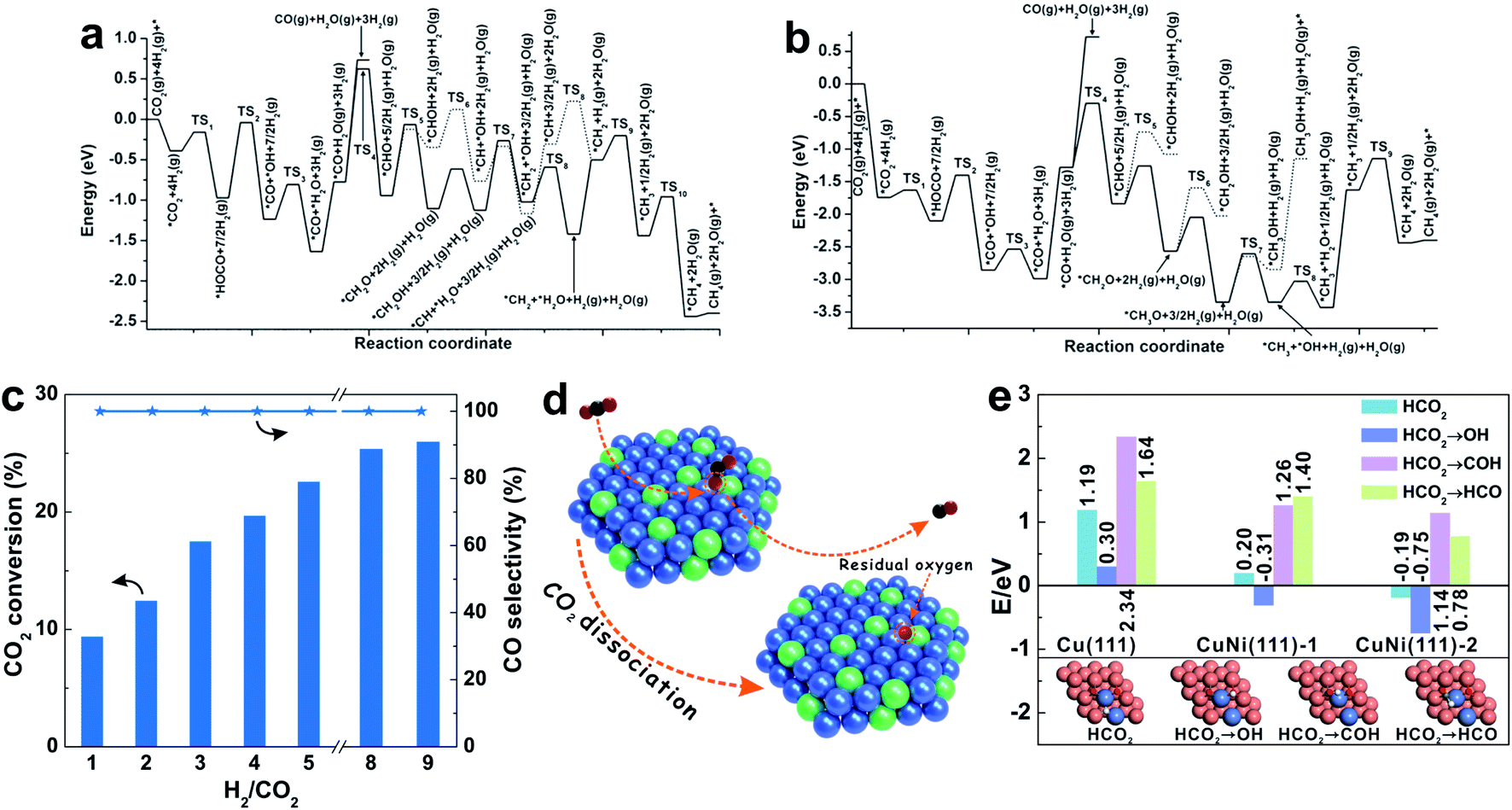 | ||
| Fig. 5 (a and b) Potential energy diagrams for the synthesis of CO and CH4 through the RWGS and CO hydrogenation pathway, on the hydroxylated (a) Ti3O6/PtCo(111) and (b) Zr3O6/PtCo(111) surfaces. Reproduced with permission from ref. 16. Copyright (2016) Wiley-VCH. (c) Dependence of CO2 conversion and CO selectivity on the feed H2/CO2 ratio with the Ni-in-Cu catalyst at 673 K. Various H2/CO2 ratios balanced with 10% Ar in the feed gas. (d) Schematic illustration of the CO2 dissociation on the Ni-in-Cu catalyst. (e) Enthalpies of the reaction of H migration on various surfaces: H adsorbed on catalyst surfaces (HCO2), H adsorbed on oxygen atoms (HCO2 → OH), H adsorbed on the oxygen of CO (HCO2 → COH), and H adsorbed on the carbon of CO (HCO2 → HCO). Reproduced with permission from ref. 20. Copyright (2020) American Chemical Society. | ||
These PtCo catalysts provide an example for tuning the product selectivity in CO2 hydrogenation, which combines the advantages of alloys and oxide supports. Investigations on ZrO2-supported NiFe catalysts reveal the structure–performance relationship of catalyst interfaces.101 Ni3/ZrO2 is highly active for CO2 methanation (CO2 conversion of 34.2% and CH4 selectivity of 84.7%), while Fe3/ZrO2 shows low activity, but is highly selective for CO production (CO2 conversion of 3.1% and CO selectivity of 100%). Interestingly, upon introducing Ni to an Fe-based catalyst, the activity is markedly increased, and the CO selectivity can be tuned from 11.5% to 91.8% by adjusting the Ni/Fe ratios. Generally, the Ni–ZrO2 interface is regarded as the active site for CO2 methanation. However, when a large amount of Fe species is introduced, the dispersed Fe species would cover the Ni particles to form Ni–FeOx interfaces, which changes the product selectivity in the CO2 hydrogenation. Therefore, CO2 methanation occurs on the Ni/ZrO2 catalyst via the RWGS + CO hydrogenation pathway, but fails on the NiFe/ZrO2 catalyst which gives CO as the predominant product.
Because of the structural nonuniformity of the catalysts, it is still difficult to investigate the relationship between the catalyst structure and catalytic performance. Moreover, addition of promoters/additives might result in the formation of new active sites and interfaces for CO2 adsorption and transformation. Wang et al.20 reported a CuNi alloy-based catalyst (Ni-in-Cu), showing highly dispersed Ni incorporated into the Cu lattice, which combines the advantages of high activity of Ni and high selectivity of Cu. The Ni-in-Cu catalyst gives a CO2 conversion of 1.1–50.7%, and the CO selectivity always remains at 100%. The superior CO selectivity is obtained even under conditions with H2/CO2 ratios in a wide range of 1–9 (Fig. 5c). In comparison, the general Cu (ref-Cu) and Ni (ref-Ni) catalysts show much lower CO2 conversion and CO selectivity than those of the Ni-in-Cu catalyst. In these cases, the atomic dispersion of Ni in the Cu lattice is crucial for such performance, and the ref-CuNi catalyst with the same composition but partially separated Cu and Ni phases yields the methane product under equivalent reaction conditions.
More importantly, the simple and uniform structure of the Ni-in-Cu catalyst provides a model for mechanistic investigation to identify the reaction routes and active sites. By in situ DRIFTS and XPS studies, the CO32−, CO2δ− and HCO3− species are observed on the catalyst surface, giving decreased signals during the CO2 hydrogenation, which indicate their important roles as the intermediates for CO formation. In contrast, *HCOO is formed and remains unchanged during this process, which is attributed to the fact that this species is stable and difficult to be hydrogenated.33 Combining various characterization techniques, it has been found that the CO2 molecules simultaneously interact with Cu and Ni sites on the surface of the CuNi alloy. When CO is formed from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O cleavage of CO2, it rapidly desorbs from the catalyst surface to avoid deep hydrogenation (Fig. 5d). DFT calculations reveal the easy CO cleavage of CO2 to form CO on the CuNi(111) surface. Because CO can easily desorb from the catalytic surface, it is difficult to form *HCO by hydrogenation (Fig. 5e), which is due to the fact that H atoms preferentially remove the isolated O atoms on the CuNi(111) surface via an exothermic process with energy barriers of −0.31/−0.75 eV. In contrast, *CO hydrogenation to *HCO is an endothermic step with energy barriers of 1.40/0.78 eV. These results demonstrate the multifunctionality of CuNi alloy sites on the Ni-in-Cu catalyst, which embodies the efficient CO2 activation and H2 dissociation, and accelerates the CO desorption, benefiting the RWGS reaction but switching off the methanation.
O cleavage of CO2, it rapidly desorbs from the catalyst surface to avoid deep hydrogenation (Fig. 5d). DFT calculations reveal the easy CO cleavage of CO2 to form CO on the CuNi(111) surface. Because CO can easily desorb from the catalytic surface, it is difficult to form *HCO by hydrogenation (Fig. 5e), which is due to the fact that H atoms preferentially remove the isolated O atoms on the CuNi(111) surface via an exothermic process with energy barriers of −0.31/−0.75 eV. In contrast, *CO hydrogenation to *HCO is an endothermic step with energy barriers of 1.40/0.78 eV. These results demonstrate the multifunctionality of CuNi alloy sites on the Ni-in-Cu catalyst, which embodies the efficient CO2 activation and H2 dissociation, and accelerates the CO desorption, benefiting the RWGS reaction but switching off the methanation.
Zhang et al.21reported a Ni–Au bimetallic catalyst with a core–shell structure, where the Au shell is always in contact with the Ni core. The core–shell structure kinetically transforms to a NiAu alloy during the CO2 hydrogenation, and reverses after the reaction. In this process, CO is a dominant product with selectivity higher than 95%. In the environmental transmission electron microscopy (ETEM) characterization (Fig. 6a) at near-ambient pressure (9 ± 0.1 mbar, 25% CO2/75% H2), the ultrathin Au shell is observed around the Ni@Au nanoparticles at 400–500 °C, and disappears to form a NiAu alloy at 600 °C. The segregation energy (Eseg) of the Ni atom from the bulk to the surface Au layer was calculated. The Eseg can be reduced by the adsorbed species, such as H2, *H, *OH and *CO, on the Au surface (Fig. 6b). Particularly, the minimized Eseg is obtained under CO adsorption, which helps Ni transfer to the Au layer to form a NiAu alloy. These results are also in agreement with the fact that the NiAu alloy is detected during the CO2 hydrogenation, but disappears after the reaction. Moreover, the CO2 hydrogenation on the NiAu alloy undergoes a two-step pathway. In the first step, CO2 hydrogenation to CO occurs on Ni sites with an energy barrier of 0.89 eV. In the second step, CO prefers to diffuse from Ni to Au sites and desorbs, with energy barriers of 1.23 and 0.45 eV, respectively. In contrast, both dissociation and deep hydrogenation of CO need to overcome higher energy barriers. Therefore, it is a virtuous circle that the NiAu alloy is a selective catalytic surface for the RWGS reaction. Notably, CO could benefit the formation of the NiAu alloy, which is evidenced by NiAu alloy formation after quenching in CO rather than H2 or N2 (Fig. 6c).
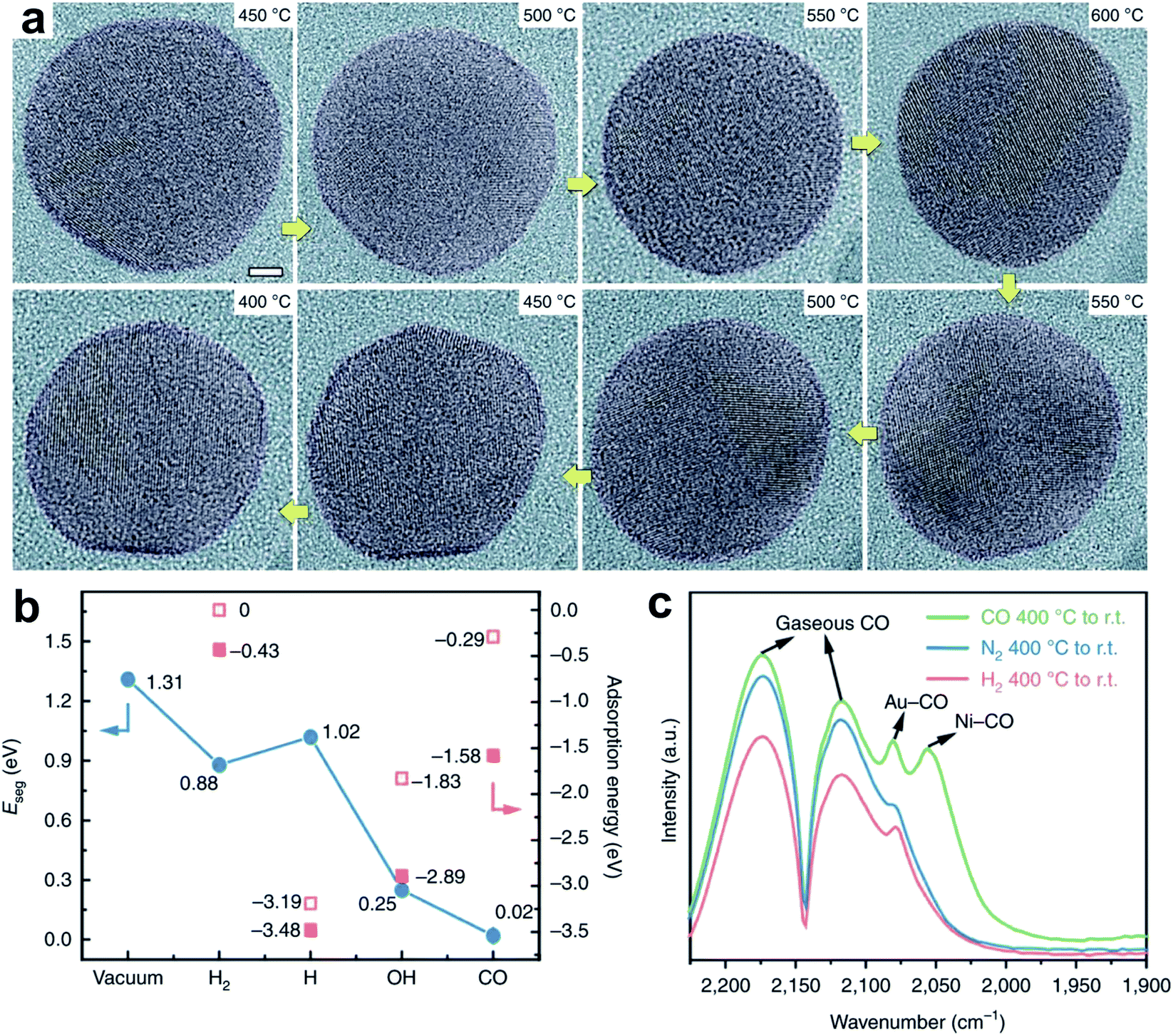 | ||
| Fig. 6 (a) In situ TEM images of the alloying and dealloying evolution of an individual NiAu particle during the CO2 hydrogenation reaction. Scale bar, 2 nm. (b) Segregation energy (Eseg) of the adsorption of H2, H, OH and CO under vacuum (blue dots), and the adsorption energies of different adsorbates when all the Ni atoms are located in the bulk (hollow pink squares) and when a single Ni atom is moved to the Au surface (solid pink squares). (c) FTIR spectra using CO as the probing molecule after fast quenching in H2, N2 and CO. Reproduced with permission from ref. 21. Copyright (2020) Springer Nature. | ||
The metal alloy based-catalysts play a role in decreasing the energy barriers of the *CO desorption, to a level below *CO dissociation or deep hydrogenation. Compared to the SMSI that also has similar functions in weakening CO adsorption, the alloyed interfaces exhibit more adjustable properties, because of their precisely controllable metal compositions and electronic structures. These observations on the alloy based-catalysts convincingly demonstrate that an appropriate binding strength of intermediates, throughout the CO2 hydrogenation, is a key to controlling product selectivity.
5. Carbide-based catalysts
Transition metal carbides (TMCs) are well known to have excellent catalytic properties, which are similar to those of noble metal catalysts. The high activity of carbides originates from the carbon, and results in modulating the electronic properties, and tuning the binding energies of reaction intermediates.103,104 Metal carbides have been extensively used in reforming105,106 and WGS107,108 reactions. Also, they are promising for CO2 hydrogenation because of the dual functions of H2 dissociation and CO bond scission.109–112
Porosoff et al.18 reported CO2 hydrogenation on defined Mo2C surfaces, which are highly active and selective for CO production. The Mo2C catalyst shows a CO2 conversion of 8.7% and CO/CH4 ratio of 14.5 for CO2 hydrogenation at 300 °C, outperforming noble metal bimetallic catalysts. The catalytic performance can be further improved by modification with Co, a well-known catalyst for methanation or Fischer–Tropsch synthesis to produce alkanes. CO2 conversion and CO selectivity of 9.5% and 51.3% (CO/CH4 product ratio of 51.3) were obtained on the Co–Mo2C catalyst. By employing temperature-programmed surface reaction (TPSR), the Mo2C surface is proved to be the active phase. In AP-XPS experiments (Fig. 7), when CO2 gas is introduced into the Mo2C catalyst, a signal assigned to O–Mo–C at 283.6 eV appears,113 rather than CO32−, CO2δ− and *HCOO species, suggesting a different pathway for CO2 activation on the Mo2C. It is proposed that CO2 directly reacts with Mo2C through the lone-pair electrons on the O atom to produce CO and an oxycarbide surface (Mo2C–O), which is subsequently reduced by H2 to regain the Mo2C surface. Notably, the unreduced MoOx species always exists in the Mo2C catalyst, with a ratio of 16.8% identified by in situ X-ray absorption near edge spectroscopy (XANES). Introducing Co into the Mo2C catalyst leads to the formation of a new CoMoCyOz phase during the reduction process. The CoMoCyOz phase is highly active for CH4 dissociation,114 which further increases the CO selectivity, in agreement with the much higher CO/CH4 ratio obtained on the Co–Mo2C catalyst.
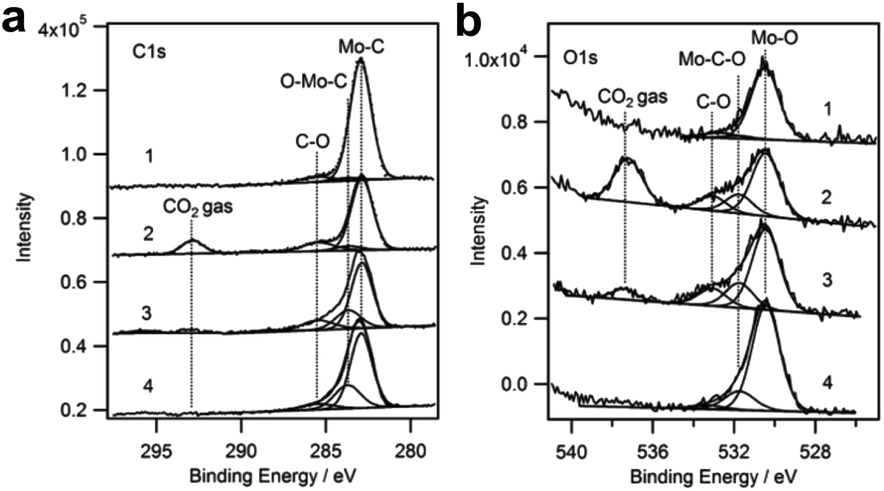 | ||
| Fig. 7 AP-XPS of (a) C 1s and (b) O 1s of Mo2C under various treatment conditions for CO2 activation. (1) Clean Mo2C; (2) 150 mTorr CO2 at room temperature; (3) 150 mTorr CO2 + 550 mTorr H2 with annealing to 523 K, followed by cooling to room temperature; (4) 150 mTorr CO2 + 550 mTorr H2 at 523 K. Reproduced with permission from ref. 18. Copyright (2014) Wiley-VCH. | ||
Because of the excellent properties, Mo2C was further coupled with other metals, such as Cu, a classical catalyst for CO2 hydrogenation. Zhang et al.109 reported a Cu/β-Mo2C catalyst, which shows extraordinary RWGS activity, selectivity, and stability. The Cu/β-Mo2C exhibits acceptable deactivation after six-cycle start-up–cool-down tests, and maintains 85% of its initial activity after 40 h reaction at a high reaction temperature of 600 °C. Cu+ species are detected on the Cu/β-Mo2C catalyst, suggesting a strong interaction between Cu and Mo2C, resulting in electron transfer from Cu to Mo2C. Such interaction helps in Cu nanoparticle stabilization, modulates the electronic structure for efficient CO2 activation and hinders Cu sintering. In the CO2 dissociation experiments without H2, the Cu/β-Mo2C catalyst exhibits much higher CO production than that of β-Mo2C and Cu/ZnO/Al2O3. These results support the mechanism of the RWGS reaction on Mo2C catalysts involving two steps, CO2 dissociation on the catalytic surface and H2 reduction of the residual O species.18,115
Moreover, Zhang et al.110 coupled the high activity of Mo2C and the non-thermal plasma (NTP) technique to produce CO. The TOF activity of β-Mo2C nanorods in NTP-catalysis (applying NTP and the catalyst, without heating) is two orders of magnitude higher than that obtained under catalysis-only conditions (applying the catalyst and heating) (Fig. 8a and b), for example, 26.0 s−1 and 0.55 s−1 for NTP catalysis and thermal catalysis-only conditions, respectively. In the designed reaction between CO2 and the catalyst surface, CO was detected immediately upon introducing a CO2/Ar flow. It is suggested that the CO originates from direct CO2 dissociation, which facilitates the high CO selectivity, in agreement with reports on carbide-based catalysts. In the NTP-catalysis, CO2 and H2 can be vibrationally excited and dissociated by plasma. In the first-step CO2 dissociation test (Fig. 8c–e), β-Mo2C nanorods under NTP-only conditions (applying NTP, without catalyst and heating) show a 20 times higher CO signal than that under catalysis-only conditions, indicating that NTP can promote CO2 dissociation. In the meanwhile, an abundant O2 signal is detected, which originates from the three-body (M) recombination of dissociative O atoms from split CO2. NTP-catalysis also exhibits a stronger CO signal, in agreement with the high activity and weaker O2 signal due to the O affinity of carbides,18 evidenced by abundant H2O generated in the second-step H2 treatment. In addition, NTP can help the decomposition of *HCOO adsorbed on the catalyst, facilitating the CO production and regaining the catalytic surface.116 Overall, the NTP-catalysis exhibits a synergetic enhancement for the RWGS reaction. The NTP induces vibration, excitation and dissociation of reactants, which subsequently interact with β-Mo2C. In this process, β-Mo2C exists as a platform for various intermediates to accelerate the reaction. The highly porous structure of β-Mo2C nanorods provides a large accessible surface, modifies the electron energy distribution, and expands the discharge region, which not only promote the formation of charge-induced intermediates, but also change the adsorption and desorption.117 The molecule–surface interactions on the β-Mo2C lead to not only superior CO selectivity but also high productivity.
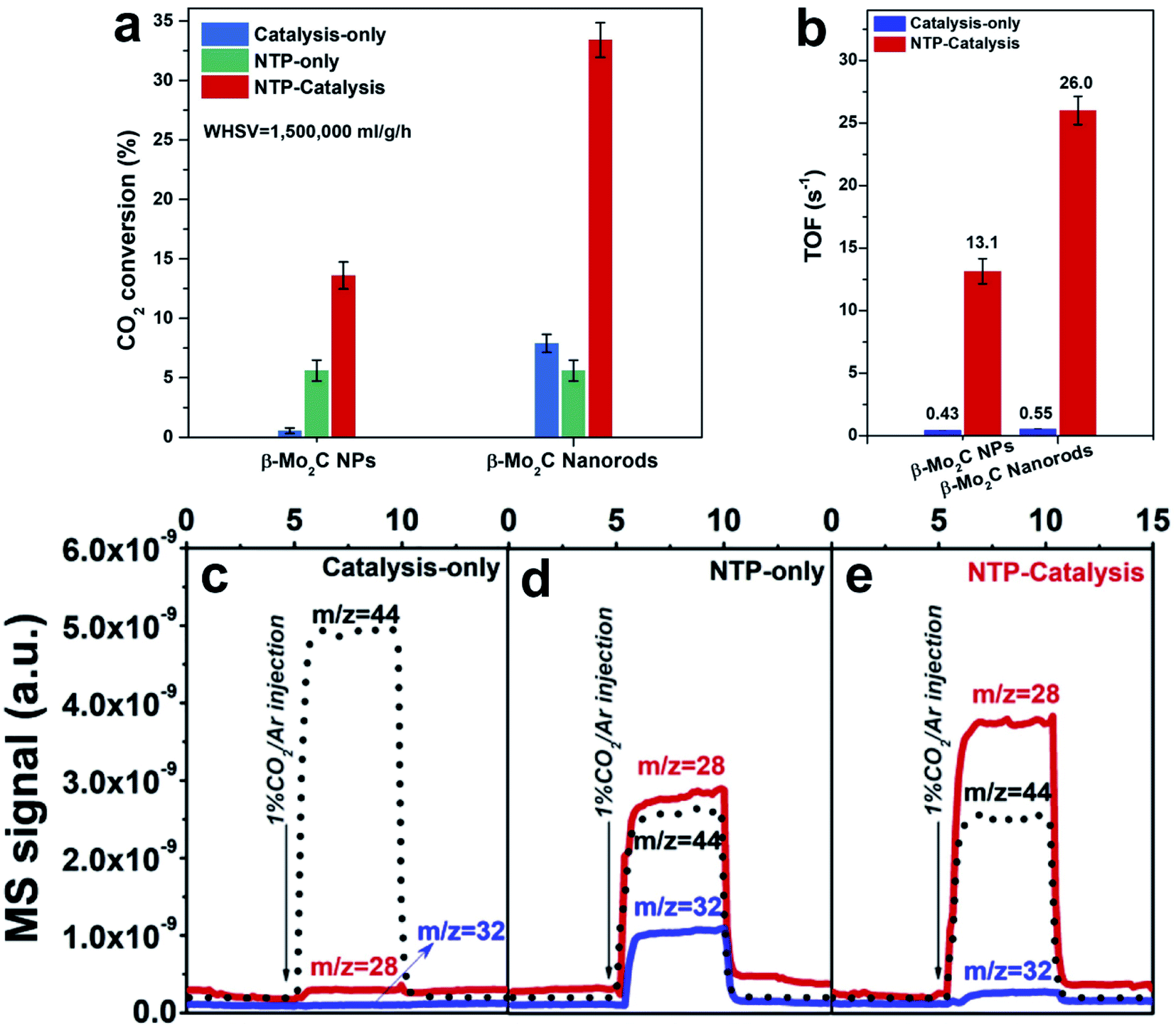 | ||
| Fig. 8 (a) CO2 conversion under catalysis-only (at 350 °C), NTP-only, and NTP catalysis conditions (input power of ca. 36 W) over β-Mo2C NP and β-Mo2C nanorod catalysts (AP, CO2:H2 = 1:2, WHSV = 1500000 mL g−1 h−1). (b) TOF comparison over β-Mo2C NP and β-Mo2C nanorod catalysts under catalysis-only (at 350 °C) and NTP-catalysis conditions (input power of ca. 36 W). (c–e) Surface reaction experiment with injection of 1% CO2/Ar under (c) catalysis-only, (d) NTP-only and (e) NTP-catalysis conditions. Reproduced with permission from ref. 110. Copyright (2020) Elsevier. | ||
In addition to the Mo2C, other carbides such as Ni3C and InNi3C0.5 were explored for CO2 hydrogenation.111,112 Although Ni is highly selective for methanation, both Ni3C and InNi3C0.5 with carbide structures exhibit superior RWGS features. Chen et al.111 reported that InNi3C0.5 supported on Al2O3/Al-fibers shows over 97% CO selectivity in CO2 hydrogenation under wide reaction conditions. For example, the CO2 conversion is 53% at 540 °C, which is close to the equilibrium value of 54%. The InNi3C0.5 has an anti-perovskite-type structure containing a stable (111) surface with a hexagonal shape. DFT calculations reveal the dual active sites of 3Ni–In (h1) and 3Ni–C (h2), which give a richer electron density distribution, facilitating activated *H formation and CO2 dissociation to CO via a redox mechanism. CO2 prefers to adsorb on the h1 site, and the dissociated *CO and *O are adsorbed on h2 and h1 sites, respectively. The dissociated *H species are adsorbed on both h1 and h2 sites. The *O on the h1 site could react with H* to form *OH, and two *OH easily convert to H2O. The dual sites always provide lower energy barriers than those of the sole h1 site, demonstrating the advantages of the dual sites on InNi3C0.5.
Carbide phases, such as Ni3C, easily form in Ni catalysts at high reaction temperature, because carbon is highly miscible on the Ni surface.118 Galhardo et al.112 reported that the Ni3C phase, which forms in CO2 hydrogenation, can switch the selectivity from CH4 to CO. The fresh Ni/SiO2 catalysts show suppressed methanation activity in CO2 hydrogenation with CO as a dominant product at a wide temperature range (100–800 °C, Fig. 9a). Catalysts with different Ni loadings, particle sizes, or supports show similar catalytic features of methanation in Run 1, and suppressed selectivity of CH4 in Run 2. Under operando conditions, energy-dispersive X-ray absorption spectroscopy (ED-XAS) and EXAFS (Fig. 9b and c) reveal that the Ni–C scattering belongs to the Ni3C structure, which contributes to the selectivity changes. The Ni/SiO2 catalyst exhibits much lower CO-adsorption intensity in DRIFTS after Run 1, suggesting the weak CO-binding ability of the formed Ni3C surface, which benefits CO desorption. It is further evidenced by DFT calculations that various CO-adsorption modes give higher adsorption energies on the Ni3C(001) surface, compared to those on the Ni(111) surface (Fig. 9d and e). These results help to explain the suppressed methanation: because CO2 adsorption always occurs on the oxide supports, and the activated *H species can spill to reduce CO2, the CO2-to-CO process is not affected by the C atoms covered on Ni. However, the subsequent CO-to-CH4 process is suppressed, due to the weakened CO adsorption on the Ni3C phase.
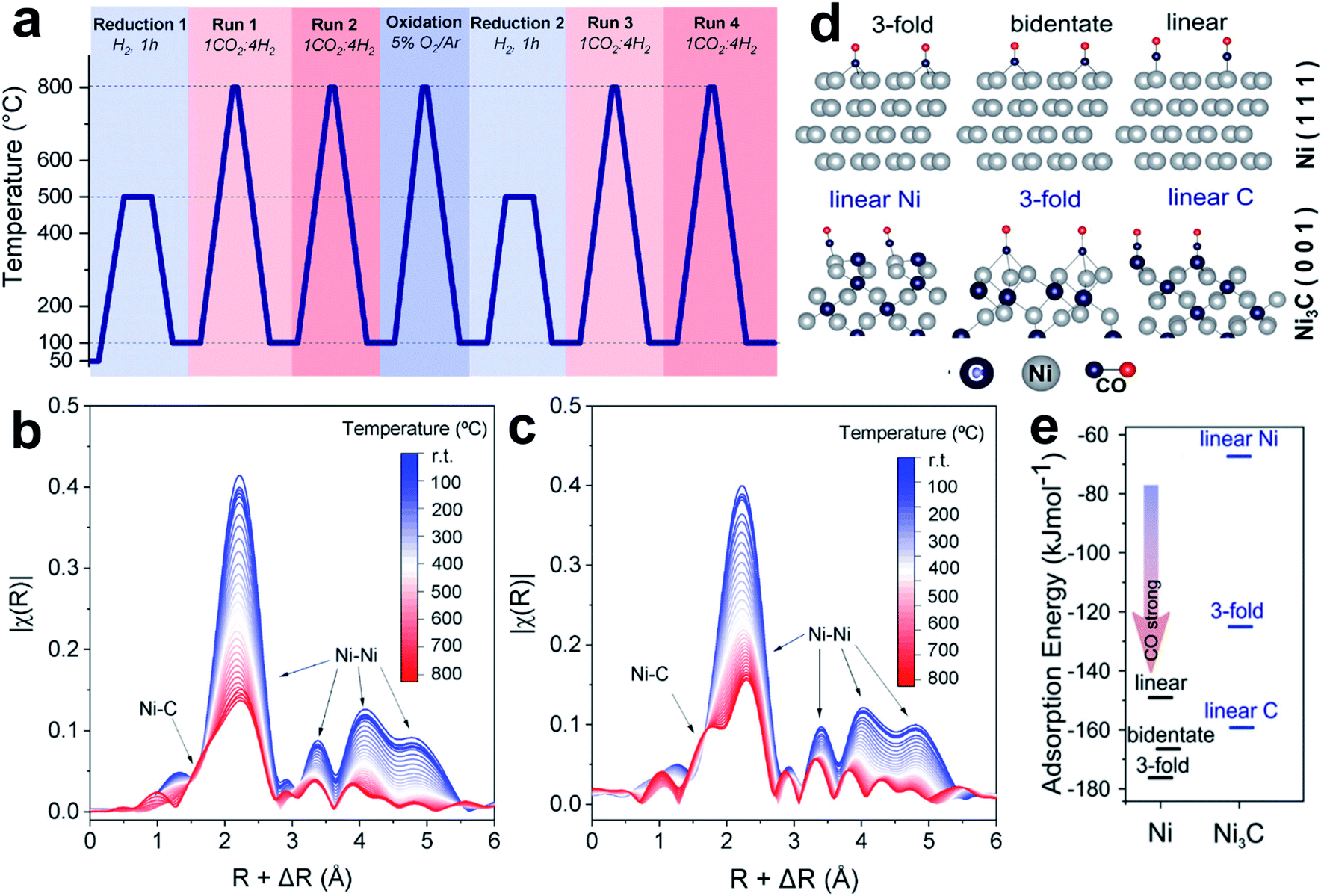 | ||
| Fig. 9 (a) Experimental setup used to investigate the activity–structure relationships. (b and c) Temperature-resolved Ni K-edge ED-XAS spectra for Ni/SiO2 under CO2 hydrogenation conditions during (b) Run 1 and (c) Run 2. (d) CO adsorption structure models at Ni(111) and C-terminated Ni3C(001). (e) CO adsorption energy at Ni(111) (dark gray lines) and C-terminated Ni3C(001) (blue lines). Reproduced with permission from ref. 112. Copyright (2021) American Chemical Society. | ||
Carbide-based catalysts effectively control the transformation pathways of *CO species. When the *CO adsorption is weakened, the C–O bond cleavage and *CO deep hydrogenation are hindered. The CO formation on the carbides always follows the pathway of direct dissociation of CO2 to CO via a redox mechanism, and because of the strong interaction between the carbide surface and oxygen, an oxycarbide surface could form and subsequently be reduced by H2. Sometimes, the unique active phases of carbides even show new functions of catalyzing CH4 dissociation, further benefiting the selective formation of CO.
6. Conclusions and perspectives
In conclusion, the developments for metal-based catalysts to tune product selectivity in CO2 hydrogenation are briefly summarized. Oxide-supported metal catalysts with classical SMSI show an effective strategy for weakening *CO and H2 adsorption to hinder deep hydrogenation. Phosphate-supported metal catalysts with similar phenomena to classical SMSI show even more excellent catalytic performances. Alloy- and carbide-based catalysts exhibit multifunctionality, contributing to reducing the CO desorption energy barrier to a level lower than CO dissociation or deep hydrogenation. Alloy-based catalysts also exhibit satisfactory controllability of the structure–performance relationship by easily adjusting the metal compositions. Carbide-based catalysts can strongly bond with the O atom of CO2, facilitating direct CO2 dissociation. Sometimes, the unique CH4 dissociation ability further inhibits CH4 formation and improves the CO selectivity.Based on this knowledge, active sites for tuning the CO adsorption and transformation are rationally designed. However, the local environments of the active sites are sometimes overlooked. The activation and diffusion of H2, which determine the hydrogenation of the carbon-containing intermediates, could be controlled to optimize the reaction. Wang et al.19 showed a representative example of tuning the selectivity of CO2 hydrogenation via controlling H spillover around the metal nanoparticles. The Rh nanoparticles fixed within siliceous zeolite (Rh@S-1) enable high CO selectivity in CO2 hydrogenation, which is beyond the general expectation of Rh-catalyzed CO2 methanation (Fig. 10a–d). The referenced catalyst of Rh@HZSM-5 prefers to produce CH4, while both Rh@KZSM-5 (introducing K+ by ion exchange) and Rh@S-1–OH (introducing silanol groups to the zeolite micropores) catalysts mainly give CH4 at high CO2 conversion. Because of the same content and size of Rh nanoparticles, these different catalytic performances are attributed to the nanoporous environment of zeolite sheaths. Experiments of a WO3-probe, H–D exchange and D2O treatment demonstrate that the stronger H spillover in the zeolite micropores with protons or silanols (Rh@HZSM-5 and Rh@S-1–OH) could provide active *H species for deep hydrogenation, but studies on Rh@S-1 catalysts with weak hydrogen spillover ability are scarce (Fig. 10e–g).119–121 In addition, the weakened CO adsorption on the S-1 zeolite fixed Rh nanoparticles also contributes to the hindered methanation in CO2 hydrogenation.
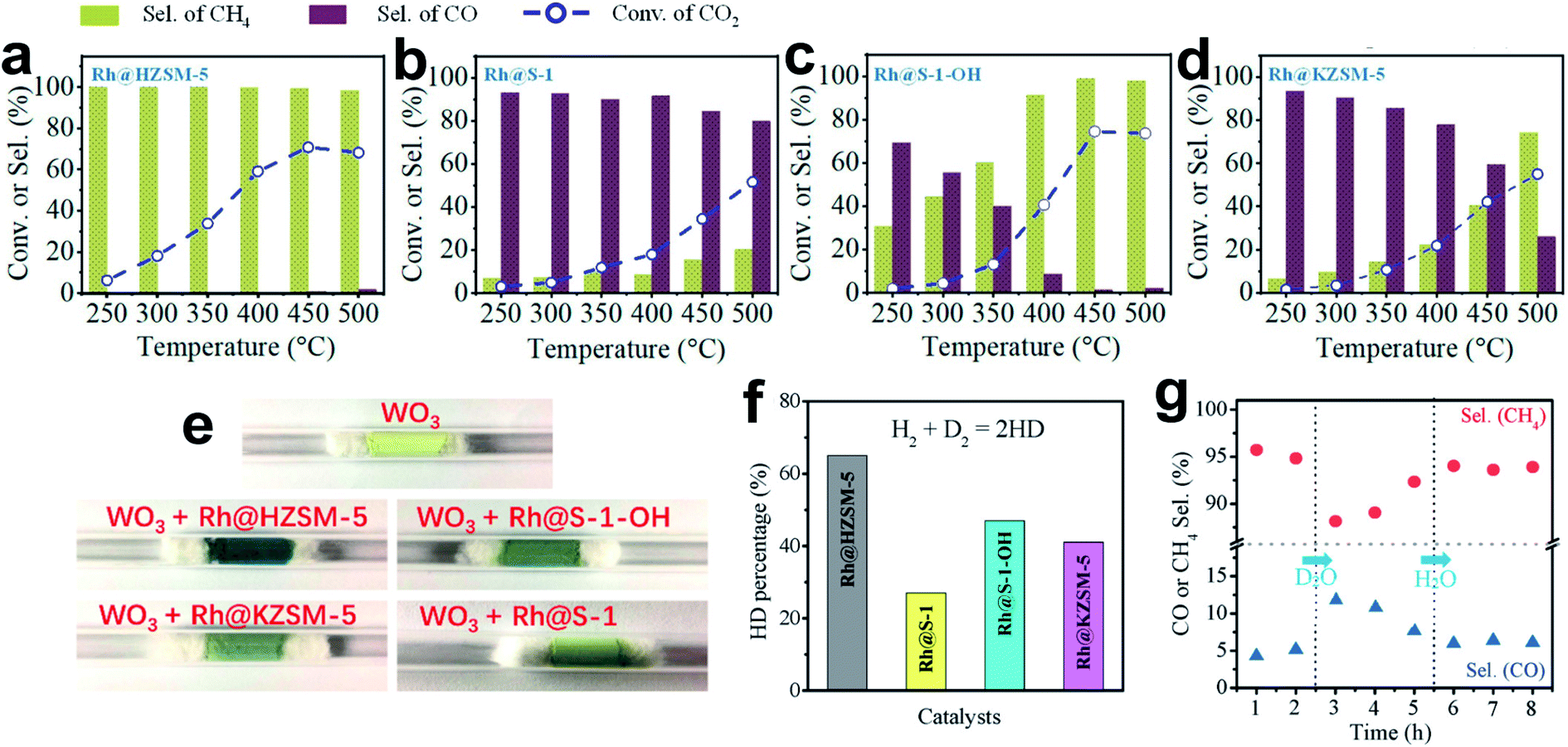 | ||
| Fig. 10 (a–d) (a) Catalytic performance of (a) Rh@HZSM-5, (b) Rh@S-1, (c) Rh@S-1–OH and (d) Rh@KZSM-5 in CO2 hydrogenation. Reaction conditions: 0.5 g of catalyst; 1 MPa feed gas pressure, CO2/H2/Ar = 1/3/1, molar; feed flow rate 30 mL min−1. (e) Photographs of samples made with 1 g of WO3 mixed with 0.02 g of various catalysts after treatment with H2 at 30 °C for 10 min. (f) Results of H–D exchange experiments with various catalysts. (g) CH4 and CO selectivity in CO2 hydrogenation catalyzed by Rh@HZSM-5 treated with D2O and H2O. Reproduced with permission from ref. 19. Copyright (2019) American Chemical Society. | ||
It is also expected that investigations on the completive processes of the RWGS reaction and CO2 methanation will help to elucidate the reaction mechanism of CO2 hydrogenation, and guide the preparation and optimization of industrial catalysts. Compared with the simple products of CO and CH4, the synthesis of methanol and even C2+ compounds with higher economic value is more desired. However, in practice, the inevitable CO or CH4 formation in CO2 hydrogenation will not only consumes hydrogen feed, but will also lead to insufficient yield of target products. As a successful example, Yang et al.17 reported ethanol synthesis from CO2 hydrogenation over a Cu/Co3O4 catalyst at high pressure (1–30 bar). These achievements expand the applications of the model reactions (RWGS and CO2 methanation) to CO2-to-valuable chemical processes, offering good opportunities for industrial applications in the future, particularly in carbon neutralization for global environmental protection.
Author contributions
L. W. and F.-S. X. conceived the topic and structure of the article. All authors reviewed and contributed to this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21932006 and 21822203).References
- Global Greenhouse Gas Reference Network, Trends in Atmospheric Carbon Dioxide, National Oceanic and Atmospheric Administration, Earth System Research Laboratory, Global Monitoring Division, U. S. Department of Commerce, Global, March 5, 2020 Search PubMed.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- X. Wang, H. Shi and J. Szanyi, Nat. Commun., 2017, 8, 513 CrossRef PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS PubMed.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS PubMed.
- C. Yang, S. Liu, Y. Wang, J. Song, G. Wang, S. Wang, Z. J. Zhao, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 11242–11247 CrossRef CAS PubMed.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- L. X. Wang, E. Guan, Z. Wang, L. Wang, Z. Gong, Y. Cui, Z. Yang, C. Wang, J. Zhang, X. Meng, P. Hu, X. Q. Gong, B. C. Gates and F.-S. Xiao, ACS Catal., 2020, 10, 9261–9270 CrossRef CAS.
- X. Zhang, S. Han, B. Zhu, G. Zhang, X. Li, Y. Gao, Z. Wu, B. Yang, Y. Liu, W. Baaziz, O. Ersen, M. Gu, J. T. Miller and W. Liu, Nat. Catal., 2020, 3, 411–417 CrossRef CAS.
- L. Wang, W. Fang, L. Wang and F.-S. Xiao, ChemSusChem, 2020, 13, 6300–6306 CAS.
- J. Cored, A. García-Ortiz, S. Ibora, M. J. Climent, L. Liu, C. H. Chuang, T. S. Chan, C. Escudero, P. Concepción and A. Corma, J. Am. Chem. Soc., 2019, 141, 19304–19311 CrossRef CAS PubMed.
- A. Parastaev, V. Muravev, E. H. Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal., B, 2016, 196, 108–116 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D. J. Wang, H. C. Liu, C. H. Yan and Y. W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- F. Sha, Z. Han, S. Tang, J. Wang and C. Li, ChemSusChem, 2020, 13, 6160–6181 CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- S. Kattle, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef PubMed.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F.-S. Xiao, Nat. Commun., 2020, 11, 1033 CrossRef CAS PubMed.
- D. Wang, Z. Xie, M. D. Porosoff and J. G. Chen, Chem, 2021, 7, 2277–2311 CAS.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, ACS Catal., 2016, 6, 913–927 CrossRef CAS.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- P. Gao, L. Zhang, S. Li, Z. Zhou and Y. Sun, ACS Cent. Sci., 2020, 6, 1657–1670 CrossRef CAS PubMed.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- J. Wei, R. Yao, Q. Ge, Z. Wen, X. Ji, C. Fang, J. Zhang, H. Xu and J. Sun, ACS Catal., 2018, 8, 9958–9967 CrossRef CAS.
- Z. He, M. Cui, Q. Qian, J. Zhang, H. Liu and B. Han, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12654–12659 CrossRef CAS PubMed.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- Y. Wang, L. Tan, M. Tan, P. Zhang, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, ACS Catal., 2019, 9, 895–901 CrossRef CAS.
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2020, 10, 302–310 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2020, 7, 849–881 Search PubMed.
- Z. He, Q. Qian, J. Ma, Q. Meng, H. Zhou, J. Song, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS PubMed.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- L. Wang, L. Wang, J. Zhang, X. Liu, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J. G. Kim, X. Meng and F.-S. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- L. Wang, S. He, L. Wang, Y. Lei, X. Meng and F.-S. Xiao, ACS Catal., 2019, 9, 11335–11340 CrossRef CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CrossRef CAS.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov and J. Liu, Chem. Soc. Rev., 2021, 50, 2337–2366 RSC.
- S. Kasipandi and J. W. Bae, Adv. Mater., 2019, 31, 1803390 CrossRef PubMed.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- M. Xu, S. He, H. Chen, G. Cui, L. Zheng, B. Wang and M. Wei, ACS Catal., 2017, 7, 7600–7609 CrossRef CAS.
- J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2019, 9, 3026–3053 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- Y. Lin, T. Zhu, X. Pan and X. Bao, Catal. Sci. Technol., 2017, 7, 2813–2818 RSC.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef CAS.
- W. Li, G. Zhang, X. Jiang, Yi. Liu, J. Zhu, F. Ding, Z. Liu, X. Guo and C. Song, ACS Catal., 2019, 9, 2739–2751 CrossRef CAS.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, J. Catal., 2016, 343, 115–126 CrossRef CAS.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CrossRef CAS.
- J. Wang, C. Y. Liu, T. P. Senftle, J. Zhu, G. Zhang, X. Guo and C. Song, ACS Catal., 2020, 10, 3264–3273 CrossRef CAS.
- A. Posada-Borbón and H. Grönbeck, Phys. Chem. Chem. Phys., 2019, 21, 21698–21708 RSC.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- Y. Zhou, C. Jin, Y. Li and W. Shen, Nano Today, 2018, 20, 101–120 CrossRef CAS.
- L. Wang, L. Wang, X. Meng and F.-S. Xiao, Adv. Mater., 2019, 31, 1901905 CrossRef CAS PubMed.
- J. Zhang, H. Wang, L. Wang, S. Ali, C. Wang, L. Wang, X. Meng, B. Li, D. S. Su and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 2975–2983 CrossRef CAS PubMed.
- M. E. Strayer, J. M. Binz, M. Tanase, S. M. K. Shahri, R. Sharma, R. M. Rioux and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 5687–5696 CrossRef CAS PubMed.
- M. Xu, S. Yao, D. Rao, Y. Niu, N. Liu, M. Peng, P. Zhai, Y. Man, L. Zheng, B. Wang, B. Zhang, D. Ma and M. Wei, J. Am. Chem. Soc., 2018, 140, 11241–11251 CrossRef CAS PubMed.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang, X. Liu, Y. Zhou, H. Wang, A. Wang, B. Qiao, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Green Chem., 2020, 22, 6855–6861 RSC.
- Y. Guo, Z. Liu, F. Zhang, D. Wang, K. Yuan, L. Huang, H. C. Liu, S. D. Senanayake, J. A. Rodriguez, C. H. Yan and Y. W. Zhang, ChemCatChem, 2021, 13, 874–881 CrossRef CAS.
- X. Chen, X. Su, H. Y. Su, X. Liu, S. Miao, Y. Zhao, K. Sun, Y. Huang and T. Zhang, ACS Catal., 2017, 7, 4613–4620 CrossRef CAS.
- F. J. C. M. Toolenaar, A. G. T. M. Bastein and V. Ponec, J. Catal., 1983, 82, 35–44 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, J. Phys. Chem. C, 2007, 111, 2959–2964 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, Chem. Phys. Lett., 2007, 436, 204–208 CrossRef CAS.
- M. Bowker, P. Stone, P. Morrall, P. Smith, R. Smith, R. Bennett, N. Perkins, R. Kvon, C. Pang, E. Fourre and M. Hall, J. Catal., 2005, 234, 172–181 CrossRef CAS.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16, 4528–4534 CrossRef CAS PubMed.
- H. Tang, F. Liu, J. Wei, B. Qiao, K. Zhao, Y. Su, C. Jin, L. Li, J. Liu, J. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10606–10611 CrossRef CAS PubMed.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS PubMed.
- H. Tang, Y. Su, Y. Guo, L. Zhang, T. Li, K. Zang, F. Liu, L. Li, J. Luo, B. Qiao and J. Wang, Chem. Sci., 2018, 9, 6679–6684 RSC.
- C. Yang and C. W. Garl, J. Phys. Chem., 1957, 61, 1504–1512 CrossRef.
- J. T. Yates, T. M. Duncan, S. D. Worley and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1219 CrossRef CAS.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- B. Yan, B. Zhao, S. Kattel, Q. Wu, S. Yao, D. Su and J. G. Chen, J. Catal., 2019, 374, 60–71 CrossRef CAS.
- N. Nityashree, C. A. H. Price, L. Pastor-Perez, G. V. Manohara, S. Garcia, M. M. Maroto-Valer and T. R. Reina, Appl. Catal., B, 2020, 261, 118241 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Chem. Phys., 2004, 120, 5414–5423 CrossRef CAS PubMed.
- J. G. Chen, Chem. Rev., 1996, 96, 1477–1498 CrossRef CAS PubMed.
- A. Zhang, A. Zhu, B. Chen, S. Zhang, C. Au and C. Shi, Catal. Commun., 2011, 12, 803–807 CrossRef CAS.
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu, B. Chen, E. Crumlin, J. Guo, Z. Zuo, W. Li, J. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389–393 CrossRef CAS PubMed.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Nat. Catal., 2018, 1, 349–355 CrossRef CAS.
- J. Dong, Q. Fu, Z. Jiang, B. Mei and X. Bao, J. Am. Chem. Soc., 2018, 140, 13808–13816 CrossRef CAS PubMed.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y. W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
- P. Chen, G. Zhao, X. R. Shi, J. Zhu, J. Ding and Y. Lu, iScience, 2019, 17, 315–324 CrossRef CAS PubMed.
- T. S. Galhardo, A. H. Braga, B. H. Arpini, J. Szanyi, R. V. Gonçalves, B. F. Zornio, C. R. Miranda and L. M. Rossi, J. Am. Chem. Soc., 2021, 143, 4268–4280 CrossRef CAS PubMed.
- X. Deng, A. Verdaguer, T. Herranz, C. Weis, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474–9478 CrossRef CAS PubMed.
- S. Izhar, H. Kanesugi, H. Tominaga and M. Nagai, Appl. Catal., A, 2007, 317, 82–90 CrossRef CAS.
- L. Dietz, S. Piccinin and M. Maestri, J. Phys. Chem. C, 2015, 119, 4959–4966 CrossRef CAS.
- L. Wang, Y. Yi, H. Guo and X. Tu, ACS Catal., 2018, 8, 90–100 CrossRef CAS.
- L. Liu, Z. Zhang, S. Das and S. Kawi, Appl. Catal., B, 2019, 250, 250–272 CrossRef CAS.
- C. Mirodatos, J. A. Dalmon and G. A. Martin, Stud. Surf. Sci. Catal., 1984, 19, 505–512 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Nat. Commun., 2014, 5, 3370 CrossRef PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |